

Abstract
Background
Beckwith-Wiedemann Syndrome (BWS) is characterised by overgrowth and tumour predisposition. While multiple epigenetic and genetic mechanisms cause BWS, the majority are caused by methylation defects in imprinting control regions on chromosome 11p15.5. Disease-causing methylation defects are often mosaic within affected individuals. Phenotypic variability among individuals with chromosome 11p15.5 defects and tissue mosaicism led to the definition of the Beckwith-Wiedemann Spectrum (BWSp). Molecular diagnosis of BWSp requires use of multiple sensitive diagnostic techniques to reliably detect low-level aberrations.
Methods
Multimodal BWS diagnostic testing was performed on samples from 1057 individuals. Testing included use of a sensitive qRT-PCR-based quantitation method enabling identification of low-level mosaic disease, identification of CNVs within 11p15.5 via array comparative genomic hybridisation or qRT-PCR, and Sanger sequencing of CDKN1C.
Results
A molecular diagnosis was confirmed for 27.4% of individuals tested, of whom 43.4% had mosaic disease. The presence of a single cardinal feature was associated with a molecular diagnosis of BWSp in 20% of cases. Additionally, significant differences in the prevalence of mosaic disease among BWS molecular subtypes were identified. Finally, the diagnostic yield obtained by testing solid tissue samples from individuals with negative blood testing results shows improved molecular diagnosis.
Conclusion
This study highlights the prevalence of mosaic disease among individuals with BWSp and the increases in diagnostic yield obtained via testing both blood and solid tissue samples from affected individuals. Additionally, the results establish the presence of a molecular diagnosis in individuals with very subtle features of BWSp.
INTRODUCTION
Beckwith-Wiedemann Syndrome (BWS (MIM: 130650)) is an overgrowth disorder characterised by a range of clinical phenotypes, including lateralised overgrowth, macroglossia, omphalocele, hyperinsulinism and cancer predisposition.1 Multiple epigenetic and genetic defects can give rise to BWS, and each defect results in altered expression and/or function of one or more genes within the 11p15.5 imprinted gene cluster.2 Clinical manifestations of the disease include wide variation and constitute the Beckwith-Wiedemann Spectrum (BWSp), and the phenotypic presentation of affected individuals ranges from ‘classical BWS’ (eg, omphalocele and macroglossia) to isolated lateralised overgrowth.1
There are two differentially methylated imprinting control regions, H19/IGF2:IG DMR (hereafter termed IC1) and KCNQ1OT1:TSS DMR (hereafter termed IC2), within 11p15.5 that regulate parent-of-origin-specific expression of IGF2 and KCNQ1OT1 from the paternally inherited allele and CDKN1C and H19 from the maternally inherited allele, respectively (figure 1).3 Epigenetic and genetic changes resulting in increased expression of IGF2 and/or loss of CDKN1C expression are thought to cause the majority of BWS phenotypes.4 Molecular defects are identified in approximately 80% of individuals with BWS and epigenetic defects represent the most commonly identified cause of disease.2 Multiple distinct epigenetic defects are known to cause BWS; loss of methylation on the maternally inherited allele of IC2 (IC2 LOM) is found in ~50% of affected individuals, gain of methylation on the paternally inherited allele (IC1 GOM) in ~10% of affected individuals and chromosome 11 paternal uniparental disomy (upd(11)pat) is detected in ~20% of affected individuals.5 Epigenetic defects are often present at mosaic levels within affected individuals, and the proportion of defect-containing cells within the same individual can differ across sites.6–8 Additionally, multiple types of genetic defects also cause BWS. Sequence changes resulting in CDKN1C loss-of-function are identified in approximately 5% of sporadic and ~40% of familial cases.9 Deletions, duplications and chromosomal rearrangements involving either or both IC1 and IC2 are identified in ~5% of cases.10
Figure 1.
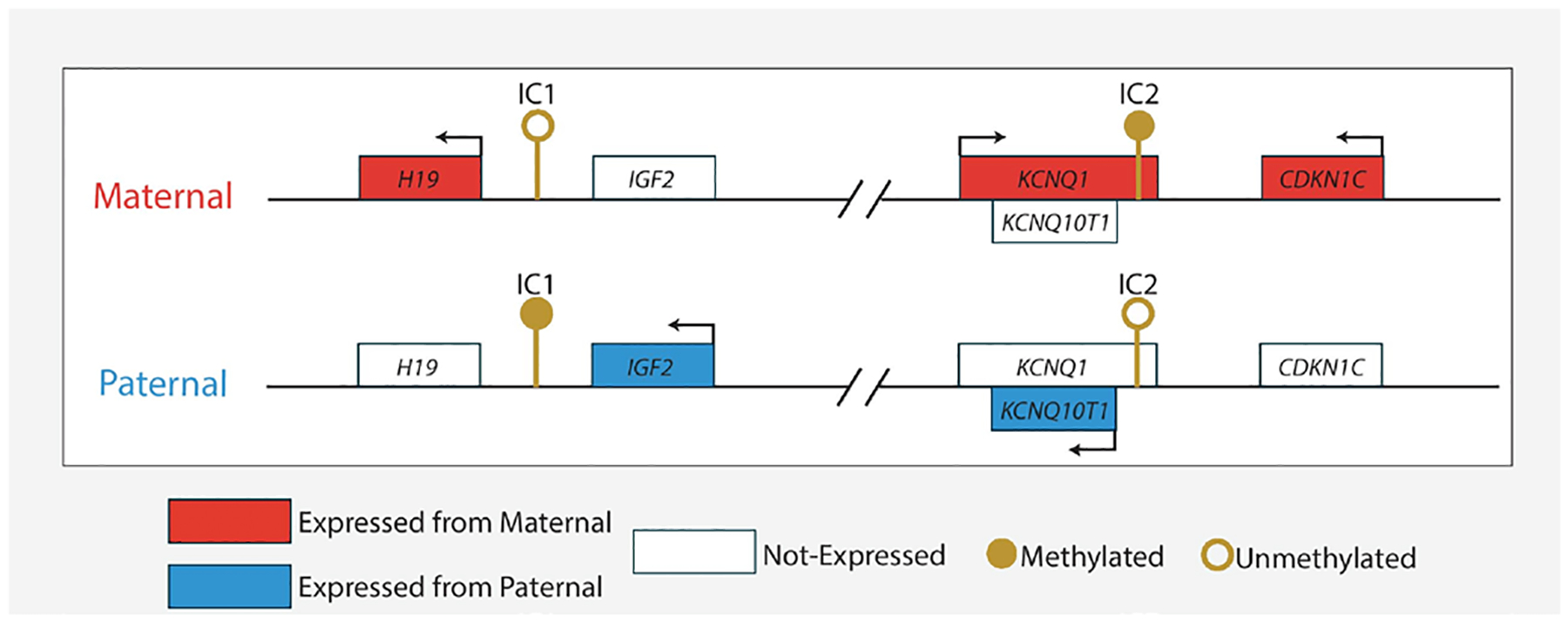
Chromosome 11p15.5 Beckwith-Wiedemann Syndrome locus.This locus is divided into two distinct regions, termed IC1 and IC2, each of which contains multiple imprinted genes. Filled/unfilled boxes represent genes and the direction of transcription is indicated by arrows above gene boxes. The direction of antisense transcription is represented by arrows below gene boxes. Note, this locus is not drawn to scale.
For each individual with BWS, identification of their specific disease-causing aberration is critical, as this information informs risks for cancer predisposition in the same individual and risk of familial recurrence.1 However, for any given individual with BWS, finding such an aberration requires the use of multiple assays, each of which is designed to identify a subset of the epigenetic and genetic changes that cause BWS.8 Methylation-sensitive multiplex ligation-dependent probe amplification (MS-MLPA) is the most commonly used method for BWS epimutation molecular diagnosis and has been demonstrated to reliably detect methylation level changes of ≥20% relative to normal 50% values. However, MS-MLPA may have difficulty identifying methylation level changes of <20% relative to normal 50% values.1–3,8,11–17 Thus, the use of sensitive assays capable of ascertaining low-level mosaic disease may increase the diagnostic utility of BWS molecular testing.
The results of molecular testing of 1057 sequentially tested individuals with concern for BWSp are reported here. The use of a multimodal testing strategy allowed identification of disease-causing epigenetic and genetic defects, and the use of sensitive quantitative methods of methylation level measurement enabled enhanced molecular diagnosis of individuals with mosaic disease.
MATERIALS AND METHODS
Cohort source
Cohort members were referred to the University of Pennsylvania Genetic Diagnostic Laboratory (GDL) for molecular testing by an outside physician on the basis of suspicion for BWS. Results from testing performed between February 2012 and February 2018 were analysed retrospectively after communication of results. Note, the same kits and reagents were used to run all clinical assays performed during this period. Clinical and phenotype data for each tested individual was extracted from test requisition forms filled out by ordering physicians. Molecular data for all tested individuals was extracted from test result records. Study data were anonymised by removing all personal identifiers.
DNA isolation
Two independent DNA isolates were collected from each sample submitted for testing as previously described.18 A quality control check, consisting of an evaluation of polymorphic short tandem repeats to verify that both independent DNA isolates were collected from the same individual, was performed as described.19
Measurement of IC1 and IC2 methylation level values
Duplicate allele-specific methylated multiplex quantitative real-time PCR (ASMM)-qRT-PCR experiments were performed using DNA isolated from two separate blood collection tubes, separate aliquots from the same collection tube if only a single collection tube was provided, or bisected tissue samples. Bisulfite conversion of 200 ng of genomic DNA was performed using the EZ DNA Methylation Kit (Zymo, Irvine, California, USA). ASMM-qRT-PCR was performed as previously described.20 ASMM-qRT-PCR reactions were run using a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific, Waltham, Massachusetts, USA), and data were analysed using the cycle threshold (Ct) method.21 Data from one of four technical replicate wells was selected for exclusion from methylation level calculations. Wells selected for exclusion represent Ct value outliers, or, if no outlier is identified, the first replicate position within the ASMM-qRT-PCR plate. Three previously run samples with known normal methylation levels were run simultaneously and used as normal controls. Final reported methylation level values represent the combined results from two ASMM-qRT-PCR experiments compared relative to the normal controls.
Calculation of the fraction of cells with altered level of methylation
The per centage of BWS cells in an individual with upd(11)pat is the absolute value of the difference between the IC1 methylation per centage and 50% plus the absolute value of the difference between the IC2 methylation per centage and 50%. For patients with IC1 GOM or IC2 LOM, the per cent of BWS cells is the absolute value of the difference between the methylation per centage for IC1 or IC2, respectively and 50%, multiplied by 2. Methylation level values obtained via ASMM-qRT-PCR were validated using samples from patients with upd(11)pat with known levels of mosaicism ascertained via chromosomal SNP microarray testing, as previously described, data not shown.22
Array comparative genomic hybridisation
Array comparative genomic hybridisation (aCGH) was performed using a custom-designed Agilent Technologies (Santa Clara, California, USA) 180 k array that included increased probe density within chromosome 11p15.5. DNA digestion, labelling and hybridisation within an Agilent G2545a Hybridisation Oven were performed following manufacturer’s guidelines. Scanning was performed using an Agilent Technologies Surescan Scanner. Agilent Cytogenomics 4.0 was used for analysis. CNVs and copy neutral regions of homozygosity (cnROH) were classified following American College of Medical Genetics (ACMG) recommended guidelines.23
Maternal cell contamination
Maternal cell contamination in prenatal samples was evaluated as reported previously.19
CDKN1C variant characterisation and classification
For methylation negative cases, CDKN1C sequence was interrogated by Sanger sequencing. Variants were reported only if identified in both independent genomic DNA isolates collected from the same individual. Variants in CDKN1C were annotated using the NM_000076.2 transcript and classified following ACMG recommended guidelines.24 Variants classified as a variant of unknown significance, likely pathogenic, or pathogenic were included in the diagnostic report. Testing of maternal and paternal samples was performed, when available, to aid in classification of CDKN1C variants.
Statistical analysis
Data were analysed using R V.3.5.0. Summary statistics including frequencies and descriptives were performed on all variables. Two-tailed Fisher’s exact and χ2 tests were performed as indicated, to evaluate for significant differences between factors analysed. Test result p values <0.05 were considered to be statistically significant.
RESULTS
Cohort description
A total of 1057 individuals were referred to the GDL for BWS clinical molecular diagnosis. Of these, 53 were tested prenatally. Median age of the remaining 1004 individuals was 9 months (range 0–41 years), with 51.8% being female and 48.2% being male. Sex information was not available for individuals tested prenatally. A total of 1174 samples representing 21 tissue types were tested (online supplementary table 1). The most common tissue types submitted for testing included blood, skin and amniocytes.
Establishment and reporting of diagnostic methylation level values
In order to define the lower limit of detection for methylation level differences measured at IC1 and IC2, methylation levels in 100 individuals with normal methylation at IC1 and IC2 were examined. Mean methylation values at IC1 and IC2 were between 47% and 53% (figure 2A). Using these results, criteria for all IC1 and IC2 diagnostic methylation level findings were defined as follows: (1) to be designated as positive for a methylation defect, methylation values must differ from normal 50% values by ±>3% and (2) the 95% CIs, defined as the mean±2 times the SD of the mean, for both patient and control methylation values must not overlap. The two horizontal dotted lines in figure 2A indicate the boundaries of mosaic results. Thus, methylation values that differed from normal by >±3% but ≤±20% were reported as mosaic gain of methylation or mosaic loss of methylation, respectively. Such mosaic findings indicate the presence of postzygotic changes in a fraction of cells within a tissue and that such cells may not be uniformly distributed in all tissues as demonstrated later. The upper and lower thresholds between which methylation level changes were reported as mosaic represent the limit of detection of the ASMM-qRT-PCR assay used by our laboratory and reported lower limit of detection of MS-MLPA, the most common method used in the molecular diagnosis of BWS epimutations.1–3,8 Methylation values that differed from normal by ±>20% were reported as gain of methylation or loss of methylation, respectively. Methylation values were reported as mosaic upd(11)pat if IC1 methylation values differed from normal by >+3% but ≤+20% and IC2 methylation values differed from normal by >−3% but ≤−20%. Methylation values were reported as upd(11)pat if either the IC1 or IC2 measurements were above the mosaic range.
Figure 2.
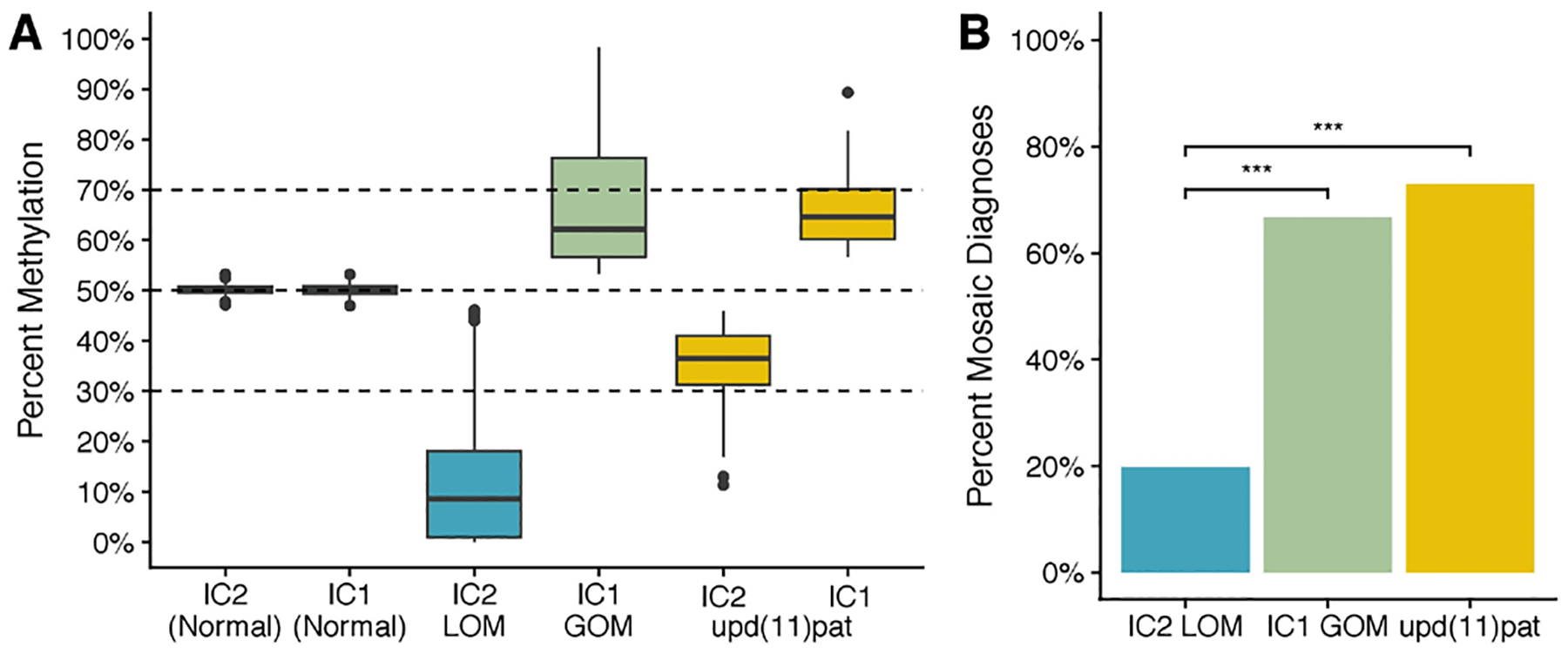
Mosaic methylation findings. (A) Distribution of measured mosaic methylation defects among BWS molecular subtypes. Normal n=100. IC2 LOM n=126, IC2 GOM n=45, upd(11)pat n=48. (B) Prevalence of mosaic methylation defects among BWS molecular subtypes. ***P<0.0001. BWS, Beckwith-Wiedemann Syndrome.
Molecular testing results
BWS molecular testing performed consisted of up to three assays: (1) measurement of methylation levels at IC1 and IC2, (2) assessment of CNVs and cnROH at chromosome 11p15.5 via either aCGH or qRT-PCR-based methods and (3) Sanger sequencing of the CDKN1C gene. While each assay was individually orderable, the majority of individuals received chromosome 11p15.5 methylation and qRT-PCR copy number assays; that, if negative, reflexed to Sanger sequencing of CDKN1C (see online supplementary table 2 for details).
Molecular testing of all 1174 samples resulted in confirmation of clinical suspicion and a molecular diagnosis for 290/1057 (27.4%) individuals (figure 3A). Testing of methylation levels at IC1 and IC2 on chromosome 11p15.5 detected disease-causing methylation defects in 282/290 (97.2%) individuals (figure 3B). Overall, IC2 LOM was identified in 141/290 (48.6%) individuals, IC1 GOM identified in 57/290 (19.7%) individuals and upd(11)pat identified in 73/290 (25.2%) individuals (figure 3B). In total, 271/282 (96.1%) individuals with a methylation defect received a molecular diagnosis of BWS. Significant differences in the number of individuals diagnosed with IC2 LOM, IC1 GOM and upd(11)pat were noted (p<0.001, χ=20.6).
Figure 3.
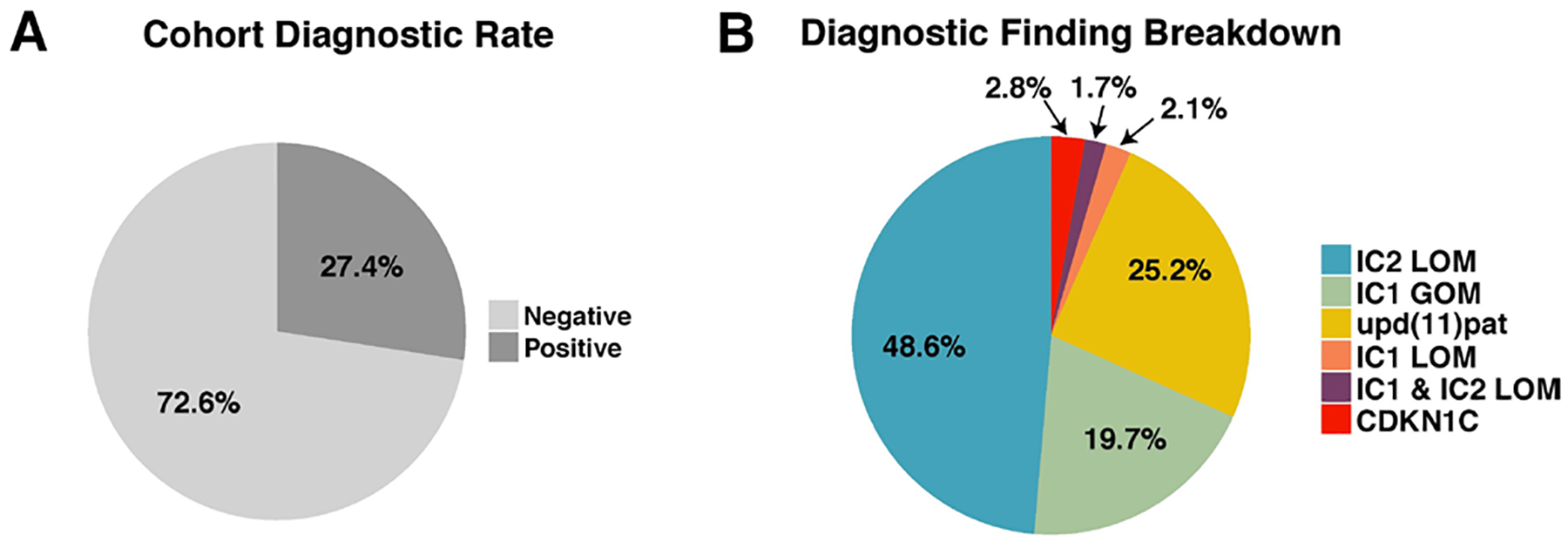
Summary of diagnostic findings. (A) Diagnostic yield of testing 1057 individuals, (B) breakdown of molecular diagnoses by defect molecular subtype.
Among the 271 individuals with BWS-causing methylation defects, concomitant pathogenic disease-associated findings were identified via aCGH in 2/141 (1.4%) individuals with IC2 LOM, 3/57 (5.3%) individuals with IC1 GOM and 2/73 (2.7%) individuals with upd(11)pat (online supplementary table 3). Genome-wide upd(11)pat was not identified in any individual within the cohort with methylation results positive for upd(11) pat for whom the aCGH assay was also performed (n=23). Additionally, gain of KCNQ1 copy number in 1/57 (1.8%) individuals with IC1 GOM and duplications on the paternally inherited allele inclusive of both IC1 and IC2 loci in 2/73 (2.7%) individuals were identified via a qRT-PCR-based copy number assay. Of note, both individuals had unbalanced translocations involving 11p15.5 via chromosome analysis (data not shown). A total of 11/282 (3.9%) individuals received an alternate molecular diagnosis of either Russell-Silver Syndrome (RSS (MIM: 180860)), caused by IC1 LOM (n=6), or a dual diagnosis of BWS/RSS subsequent to identification of IC1 LOM and IC2 LOM (n=5) (figure 3B). Similar to previously reported results, individuals who received BWS/RSS dual diagnoses had mixed BWS/RSS phenotypes and molecular testing of these individuals revealed similar levels of hypomethylation at IC1 and IC2 (data not shown).25
Sanger sequencing of CDKN1C identified pathogenic loss-of-function variants in 8/290 (2.8%) individuals (online supplementary table 4) (figure 3B).
Prevalence and magnitude of diagnostic 11p15.5 methylation level differences in BWS
Use of the sensitive ASMM-qRT-PCR method for quantification of methylation levels at IC1 and IC2 enabled detection of mosaic disease, defined by the GDL as methylation level differences of >±3% and <±20% relative to normal 50% values. Differences in the prevalence of mosaic disease among BWS molecular subtypes was assessed using data from 219 individuals who received a molecular diagnosis on the basis of peripheral blood testing. Mosaic disease was detected in 95/219 (43.4%) individuals and a significant association between the proportion of mosaic disease and BWS molecular subtype was noted (p<0.001, χ=67.15) (figure 2B). Accordingly, mosaic diagnoses were significantly more common among individuals diagnosed with either IC1 GOM versus IC2 LOM (p<0.001) and upd(11) pat versus IC2 LOM (p<0.001) (figure 2B).
Next, differences in the magnitude of diagnostic methylation level findings among BWS molecular subtypes were examined following the calculations described in the ‘Materials and methods‘ section. Individuals with IC2 LOM exhibited a median IC2 methylation level of 9%, corresponding to LOM in 82% of cells and 18% normal cells. A median methylation level of 62%, corresponding to GOM in 24% of cells and 76% normal cells, was observed among individuals with IC1 GOM. Within individuals with upd(11)pat, median methylation levels of 64% at IC1 and 36% at IC2 were observed, values corresponding to upd(11)pat in 28% of cells and 72% normal cells. Comparison of median methylation levels across BWS molecular subtypes indicated that the absolute value of median methylation level differences were significantly greater in individuals with IC2 LOM relative to individuals with either IC1 GOM (p<0.001) or upd(11)pat (p<0.001) (figure 2B).
Clinical features
The international BWS consensus group recently introduced a scoring system, based on the presence of both ‘cardinal’ and ‘suggestive’ features, to be used in the clinical diagnosis of BWSp.1 The GDL BWS test requisition form included 3/8 BWSp cardinal features (lateralised overgrowth, macroglossia and omphalocele), as well as Wilms tumour, a suggestive feature that was added to the requisition form subsequent to diagnosis of BWS in multiple individuals presenting with Wilms tumour.26,27 The BWS test requisition form can be found in online supplementary file 1.
Comparison of phenotype frequency among all individuals who received a molecular diagnosis revealed significant differences among individuals representing different BWS molecular subtypes (table 1). Clinical phenotypic data were not available for all cohort members, thus the following analyses include only individuals for whom such information was available. Lateralised overgrowth was significantly more common among those with either IC1 GOM or upd(11)pat relative to those with IC2 LOM (p<0.001 and p<0.001, respectively). Macroglossia was significantly more common among individuals with IC2 LOM relative to those with either IC1 GOM (p<0.001) or upd(11)pat (p<0.001). Omphalocele was significantly more common among individuals with IC2 LOM relative to those either IC1 GOM (p<0.001) or upd(11)pat (table 1, p<0.001). Wilms tumour was significantly more common in those with IC1 GOM relative to upd(11)pat (p<0.01) or IC2 LOM (p<0.001) and was also significantly more common in those with upd(11)pat relative to IC2 LOM (p<0.01). Phenotypic feature frequency information for individuals with pathogenic variants in CDKN1C can be found in online supplementary table 5.
Table 1.
BWS phenotypic feature frequency
| Phenotypic feature | IC2 LOM n=124 | IC1 GOM n=53 | Upd(11)pat n=67 | Combined n=244 |
|---|---|---|---|---|
| Lateralised overgrowth | 29.0% (36) | 62.2% (33) | 65.7% (44) | 46.3% (113) |
| Macroglossia | 67.7% (84) | 32.1% (17) | 23.9% (16) | 52.0% (127) |
| Omphalocele | 37.1% (46) | 1.9% (1) | 10.4% (7) | 22.1% (54) |
| Wilms tumour | 0.0% (0) | 30.2% (16) | 9.0% (6) | 9.0% (22) |
BWS phenotypic feature frequency. The number of individuals across the IC2 LOM, IC1 GOM and upd(11)pat molecular subtypes with each phenotypic feature is shown. Combined represents the sum of all individuals in each of the three molecular subtype-specific columns. Note, only individuals for whom clinical information was available are included in table.
BWS, Beckwith-Wiedemann Syndrome.
Analysis of phenotype frequency among all individuals diagnosed with BWS based on methylation testing revealed significant differences between those with mosaic versus non-mosaic disease (table 2). Lateralised overgrowth was significantly more common among those with mosaic disease (p<0.001), whereas both macroglossia (p<0.001) and omphalocele (p<0.001) were significantly more common among those with non-mosaic disease. Within the IC2 LOM molecular subtype, lateralised overgrowth was significantly more common among those with mosaic disease (p<0.001), whereas macroglossia (p<0.01) and omphalocele (p<0.001) were more common among those with non-mosaic disease. Within the IC1 GOM molecular subtype, lateralised overgrowth was significantly more common among those with mosaic disease (p<0.001). No significant differences in phenotype frequency were observed among those with mosaic versus non-mosaic upd(11)pat. Overall, the frequencies of clinical phenotypes among individuals with non-mosaic disease are more similar to previously reported frequencies than are the phenotype frequencies observed among individuals with mosaic disease (table 2).1,27–29
Table 2.
BWS phenotypic feature frequency among individuals with mosaic vs non-mosaic disease
| IC2 LOM | IC1 GOM | Upd(11)pat | Combined | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phenotypic feature | Non-mosaic n=103 | Mosaic n=21 | Ibrahim et al n=321 | Maas et al | Non-mosaic n=12 | Mosaic n=41 | Ibrahim et al n=47 | Maas et al | Non-mosaic n=24 | Mosaic n=43 | Ibrahim et al n=135 | Maas et al | Non-mosaic n=139 | Mosaic n=105 | Total n=244 | Ibrahim et al n=507 | Maas et al |
| Lateralised overgrowth | 22.3% (23) | 61.9% (13) | 18.7% (60) | 33.0% (38/115) | 33.3% (4) | 70.7% (29) | 27.7% (13) | 57.9% (11/19) | 50.0% (12) | 74.4% (32) | 72.6% (98) | 85.7% (36/42) | 28.1% (39) | 70.5% (74) | 46.3% (113) | 33.7% (171) | 46.2% (103/223) |
| Macroglossia | 73.8% (76) | 38.1% (8) | 88.2% (283) | 86.2% (106/123) | 58.3% (7) | 24.4% (10) | 70.2% (33) | 85.0% (17/21) | 37.5% (9) | 39.5% (17) | 68.1% (92) | 79.1% (34/43) | 66.2% (92) | 33.3% (35) | 52.0% (127) | 80.5% (408) | 82.5% (198/240) |
| Omphalocele | 43.7% (45) | 4.8% (1) | 49.2% (158) | 32.0% (39/122) | 0.0% (0) | 2.4% (1) | 6.4% (3) | 0.0% (0/20) | 12.5% (3) | 9.3% (4) | 8.9% (12) | 12.8% (5/39) | 34.6% (48) | 5.7% (6) | 22.1% (54) | 34.1% (173) | 22.1% (52/235) |
| Wilms tumour | 0.0% (0) | 0.0% (0) | 1.8% 2/114 | 41.7% (5) | 26.8% (11) | 31.6% (6/19) | 20.8% (5) | 2.3% (1) | 6.8% (3/44) | 7.2% (10) | 11.4% (12) | 9.0% (22) | |||||
BWS phenotypic feature frequency among individuals with mosaic vs non-mosaic disease. The number of individuals with a molecular diagnosis representative of each of the IC2 LOM, IC1 GOM and upd(11)pat molecular subtypes indicated to have each phenotypic feature, broken down by identification of mosaic vs non-mosaic levels of disease is shown here. Also shown are phenotypic feature frequencies from two previously reported BWS cohorts.28,29 Combined represents the sum of all individuals represented in each of the three mosaic/non-mosaic molecular subtype-specific columns.
BWS, Beckwith-Wiedemann Syndrome.
Diagnostic yield
Overall, testing confirmed a molecular diagnosis for 27.4% (290/1,057) of individuals tested (figure 3A). In order to better understand the factors that contribute to a BWS diagnosis, differences in diagnostic yield among patients with varying numbers of cardinal phenotypic features indicated on test requisition forms were investigated. Additionally, the diagnostic utility of testing solid tissue samples in addition to blood was also evaluated.
One or more cardinal BWS phenotypic features was indicated for 82.0% (867/1057) of cohort members. Analysis of the diagnostic yield obtained from testing individuals with one, two or three cardinal features indicated a significant association between cardinal phenotypic feature number and diagnostic yield (p<0.001, X=13.71) (figure 4A). Accordingly, diagnostic yield among individuals with two or three cardinal phenotypic terms was significantly higher than that for individuals with one phenotypic term (figure 4A). A significant difference in diagnostic yield between individuals with two or three features was not noted. Of the 168 individuals with a single cardinal feature who received a molecular diagnosis, 59 (35.1%) had mosaic disease. Of the individuals with a single cardinal feature and mosaic disease, upd(11)pat and IC1 GOM were each identified in 24/59 (40.7%) individuals, with IC2 LOM was identified in 11/59 (18.6%) individuals. Additionally, the diagnostic yield of testing individuals with omphalocele was 36.2%, with a molecular diagnosis identified for 55/152 individuals.
Figure 4.
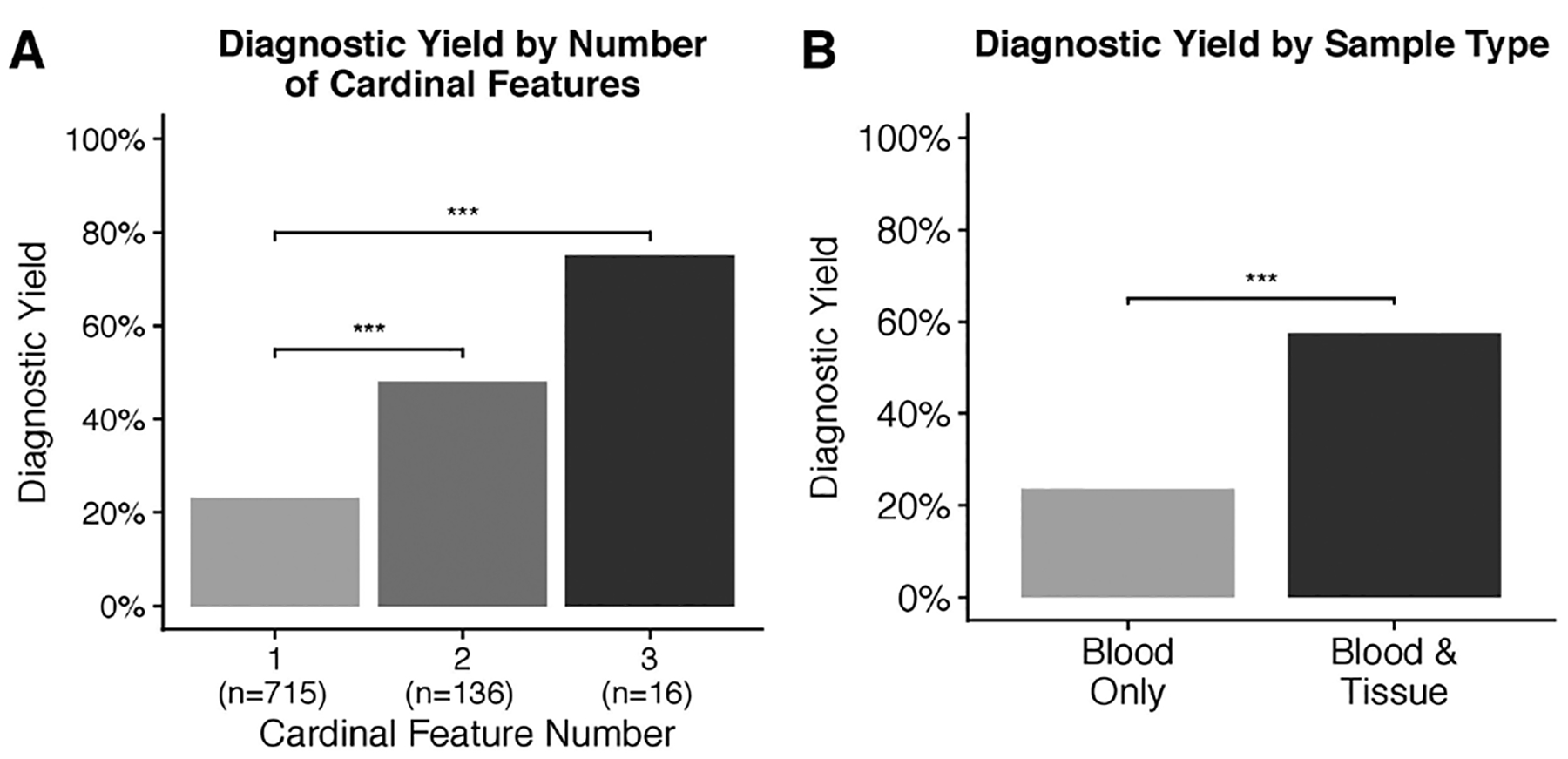
Factors influencing diagnostic yield. (A) Diagnostic yield by number of clinical features indicated on requisition form, (B) diagnostic yield by sample types tested. ***P<0.0001.
Dividing cohort members into two groups based on those who had only blood samples tested and those who had blood and tissue samples tested revealed significant intergroup differences (p<0.001, χ=32.32) (figure 4B). The diagnostic yield of testing blood samples was 23.6%, with a molecular diagnosis identified for 214/906 individuals. The diagnostic yield of testing blood and one or more solid tissue samples was significantly higher than that of testing blood samples alone, with a molecular diagnosis identified for 57.3% (35/61) individuals (p<0.001) (figure 4B). Of the 35 individuals diagnosed via blood and tissue testing, both blood and one or more tissue samples were positive in 21/35 individuals. Blood testing was negative and tissue testing was positive in 14/35 individuals; upd(11)pat was identified in pancreas samples from seven individuals presenting with hyperinsulinism; among seven individuals presenting with Wilms tumour, upd(11)pat was identified in six individuals and IC1 GOM identified in one individual. Additional clinical information for a subset of these patients and a discussion of progressive sample testing has been reported.7,26
DISCUSSION
Molecular diagnosis of BWS is crucial for proper clinical management and informs risk for tumour predisposition and familial recurrence. Here, the results of BWS molecular testing of 1057 individuals using a multimodal testing strategy that included use of sensitive quantitative methods for measurement of methylation levels at IC1 and IC2 were presented. Mosaic disease, defined by this laboratory as methylation level differences between >±3% and <±20% relative to normal 50% values, was a common finding within this cohort, with such findings identified in 95/219 (43.4%) of individuals tested. Mosaic disease was identified across the three most common BWS molecular subtypes, IC1 GOM, IC2 LOM and upd(11)pat, and notably, such mosaicism represented the majority of IC1 GOM and upd(11)pat molecular diagnoses.
Pathogenic variants in CDKN1C and pathogenic aCGH findings, including both pathogenic CNVs and pathogenic cnROH indicative of upd(11)pat, accounted for a small proportion of molecular diagnoses within the cohort, consistent with previously reported results.10 In some cases, ASMM-qRT-PCR detected low-level mosaicism for upd(11)pat when aCGH did not, highlighting both the increased sensitivity of ASMM-qRT-PCR and the inability of aCGH to reliably detect low-level mosaicism.22,30
These results support previously described epigenotype/phenotype correlations present among BWS molecular subtypes and extend these findings by describing significant differences in cardinal BWSp phenotype prevalence between individuals with mosaic and non-mosaic disease.7,31–34 Within this cohort, lateralised overgrowth was significantly more common among individuals with mosaic disease. Conversely, phenotypes associated with classical BWS, that is, macroglossia and omphalocele, were more common among individuals with non-mosaic disease. Recently published consensus guidelines recommended diagnostic testing for all individuals with one or more cardinal BWSp phenotypic features.1 A diagnostic yield of 23%, 48% and 75% was obtained from testing individuals with one, two and three cardinal features, respectively. These results both demonstrate a robust increase in diagnostic yield with increasing cardinal phenotype number and support the consensus statement diagnostic testing recommendations.1 Notably, diagnostic yield values reported here are greater than those reported for equivalent cardinal phenotype numbers from other cohorts and likely reflect the increased sensitivity of the ASMM-qRT-PCR method for methylation level measurement.20,28
The testing of blood and solid tissue samples from multiple individuals allowed the assessment of the diagnostic utility of testing solid tissues in addition to blood within this patient cohort. Overall, these results demonstrate testing blood and solid tissues offers a significantly increased diagnostic yield relative to the testing blood alone and support recent consensus recommendations advocating the testing of multiple tissues, when available.1
While the use of ASMM-qRT-PCR does allow for increased sensitivity in detecting mosaic disease-causing methylation changes, a molecular diagnosis was not identified for all patients. This work suggests that testing of solid tissue samples from individuals for whom blood testing was negative may result in identification of additional molecular diagnoses. Additionally, the use of techniques such as next-generation methylation sequencing may allow for both more sensitive methylation level measurement and identification of small sequence insertions and deletions within IC1 and IC2 that cannot readily be identified using either aCGH or chromosomal SNP microarray-based approaches.35
In summary, these findings indicate mosaic disease is a common cause of BWSp and help further define epigenotype/phenotype correlations present among individuals with mosaic disease. Additionally, our results support the recent consensus recommendations of testing of individuals across the BWSp as well as testing multiple sample types, when available. Finally, this work provides a strong rationale for the use of ASMM-qRT-PCR in routine BWS diagnostic testing.1
Supplementary Material
Acknowledgements
The authors would like to thank all patients and their families. The authors would like to thank referring institutions, particularly the Children’s Hospital of Philadelphia. The authors would also like to thank all members of the University of Pennsylvania Genetic Diagnostic Laboratory.
Funding This work was supported by the National Institute of Health K08 CA193915 (JMK), Alex’s Lemonade Stand Foundation (JMK) and St Baldrick’s Foundation (JMK).
Footnotes
Competing interests None declared.
Ethics approval This study was deemed exempt from IRB approval, as based on the Office of Human Research Protections Human Subject Decision Charts.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Study data are available on reasonable request.
REFERENCES
- 1.Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, Boonen SE, Cole T, Baker R, Bertoletti M, Cocchi G, Coze C, De Pellegrin M, Hussain K, Ibrahim A, Kilby MD, Krajewska-Walasek M, Kratz CP, Ladusans EJ, Lapunzina P, Le Bouc Y, Maas SM, Macdonald F, Õunap K, Peruzzi L, Rossignol S, Russo S, Shipster C, Skórka A, Tatton-Brown K, Tenorio J, Tortora C, Grønskov K, Netchine I, Hennekam RC, Prawitt D, Tümer Z, Eggermann T, Mackay DJG, Riccio A, Maher ER. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol 2018;14:229–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 2010;154C:343–54. [DOI] [PubMed] [Google Scholar]
- 3.Eggermann T, Perez de Nanclares G, Maher ER, Temple IK, Tümer Z, Monk D, Mackay DJG, Grønskov K, Riccio A, Linglart A, Netchine I. Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin Epigenetics 2015;7:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azzi S, Abi Habib W, Netchine I, Beckwith-Wiedemann NI. Beckwith-Wiedemann and Russell-Silver syndromes: from new molecular insights to the comprehension of imprinting regulation. Curr Opin Endocrinol Diabetes Obes 2014;21:30–8. [DOI] [PubMed] [Google Scholar]
- 5.Eggermann T, Algar E, Lapunzina P, Mackay D, Maher ER, Mannens M, Netchine I, Prawitt D, Riccio A, Temple IK, Weksberg R. Clinical utility gene card for: Beckwith-Wiedemann syndrome. Eur J Hum Genet 2014;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alders M, Maas SM, Kadouch DJM, van der Lip K, Bliek J, van der Horst CMAM, Mannens MMAM. Methylation analysis in tongue tissue of BWS patients identifies the (EPI)genetic cause in 3 patients with normal methylation levels in blood. Eur J Med Genet 2014;57:293–7. [DOI] [PubMed] [Google Scholar]
- 7.Kalish JM, Boodhansingh KE, Bhatti TR, Ganguly A, Conlin LK, Becker SA, Givler S, Mighion L, Palladino AA, Adzick NS, De León DD, Stanley CA, Deardorff MA. Congenital hyperinsulinism in children with paternal 11p uniparental isodisomy and Beckwith-Wiedemann syndrome. J Med Genet 2016;53:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russo S, Calzari L, Mussa A, Mainini E, Cassina M, Di Candia S, Clementi M, Guzzetti S, Tabano S, Miozzo M, Sirchia S, Finelli P, Prontera P, Maitz S, Sorge G, Calcagno A, Maghnie M, Divizia MT, Melis D, Manfredini E, Ferrero GB, Pecile V, Larizza L. A multi-method approach to the molecular diagnosis of overt and borderline 11p15.5 defects underlying Silver-Russell and Beckwith-Wiedemann syndromes. Clin Epigenetics 2016;8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatada I, Ohashi H, Fukushima Y, Kaneko Y, Inoue M, Komoto Y, Okada A, Ohishi S, Nabetani A, Morisaki H, Nakayama M, Niikawa N, Mukai T. An imprinted gene p57Kip2 is mutated in Beckwith-Wiedemann syndrome. Nat Genet 1996;14:171–3. [DOI] [PubMed] [Google Scholar]
- 10.Baskin B, Choufani S, Chen Y-A, Shuman C, Parkinson N, Lemyre E, Micheil Innes A, Stavropoulos DJ, Ray PN, Weksberg R. High frequency of copy number variations (CNVs) in the chromosome 11p15 region in patients with Beckwith-Wiedemann syndrome. Hum Genet 2014;133:321–30. [DOI] [PubMed] [Google Scholar]
- 11.Kalish JM, Conlin LK, Mostoufi-Moab S, Wilkens AB, Mulchandani S, Zelley K, Kowalski M, Bhatti TR, Russo P, Mattei P, Mackenzie WG, LiVolsi V, Nichols KE, Biegel JA, Spinner NB, Deardorff MA, Pheochromocytomas OB. Bilateral pheochromocytomas, hemihyperplasia, and subtle somatic mosaicism: the importance of detecting low-level uniparental disomy. Am J Med Genet A 2013;161A:993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Priolo M, Sparago A, Mammì C, Cerrato F, Laganà C, Riccio A. MS-MLPA is a specific and sensitive technique for detecting all chromosome 11p15.5 imprinting defects of BWS and SRS in a single-tube experiment. Eur J Hum Genet 2008;16:565–71. [DOI] [PubMed] [Google Scholar]
- 13.Scott RH, Douglas J, Baskcomb L, Nygren AO, Birch JM, Cole TR, Cormier-Daire V, Eastwood DM, Garcia-Minaur S, Lupunzina P, Tatton-Brown K, Bliek J, Maher ER, Rahman N. Methylation-Specific multiplex ligation-dependent probe amplification (MS-MLPA) robustly detects and distinguishes 11p15 abnormalities associated with overgrowth and growth retardation. J Med Genet 2008;45:106–13. [DOI] [PubMed] [Google Scholar]
- 14.Hömig-Hölzel C, Savola S. Multiplex ligation-dependent probe amplification (MLPA) in tumor diagnostics and prognostics. Diagn Mol Pathol 2012;21:189–206. [DOI] [PubMed] [Google Scholar]
- 15.van Veghel-Plandsoen MM, Wouters CH, Kromosoeto JNR, den Ridder-Klünnen MC, Halley DJJ, van den Ouweland AMW. Multiplex ligation-depending probe amplification is not suitable for detection of low-grade mosaicism. Eur J Hum Genet 2011;19:1009–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jennings LJ, Yu M, Fitzpatrick C, Smith FA. Validation of multiplex ligation-dependent probe amplification for confirmation of array comparative genomic hybridization. Diagn Mol Pathol 2011;20:166–74. [DOI] [PubMed] [Google Scholar]
- 17.Lee BH, Kim G-H, Oh TJ, Kim JH, Lee J-J, Choi SH, Lee JY, Kim J-M, Choi IH, Kim Y-M, Choi J-H, Yoo H-W. Quantitative analysis of methylation status at 11p15 and 7q21 for the genetic diagnosis of Beckwith-Wiedemann syndrome and Silver-Russell syndrome. J Hum Genet 2013;58:604–10. [DOI] [PubMed] [Google Scholar]
- 18.Lalonde E, Ebrahimzadeh J, Rafferty K, Richards-Yutz J, Grant R, Toorens E, Marie Rosado J, Schindewolf E, Ganguly T, Kalish JM, Deardorff MA, Ganguly A. Molecular diagnosis of somatic overgrowth conditions: a single-center experience. Mol Genet Genomic Med 2019;7:e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kessler L, Adams R, Mighion L, Walther S, Ganguly A. Prenatal diagnosis in haemophilia A: experience of the genetic diagnostic laboratory. Haemophilia 2014;20:e384–91. [DOI] [PubMed] [Google Scholar]
- 20.Azzi S, Steunou V, Rousseau A, Rossignol S, Thibaud N, Danton F, Le Jule M, Gicquel C, Le Bouc Y, Netchine I. Allele-Specific methylated multiplex real-time quantitative PCR (ASMM RTQ-PCR), a powerful method for diagnosing loss of imprinting of the 11p15 region in Russell silver and Beckwith Wiedemann syndromes. Hum Mutat 2011;32:249–58. [DOI] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 22.Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, Deardorff MA, Krantz ID, Hakonarson H, Spinner NB. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet 2010;19:1263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of medical genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 2011;13:680–5. [DOI] [PubMed] [Google Scholar]
- 24.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet Med 2015;17:405–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Azzi S, Rossignol S, Steunou V, Sas T, Thibaud N, Danton F, Le Jule M, Heinrichs C, Cabrol S, Gicquel C, Le Bouc Y, Netchine I. Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum Mol Genet 2009;18:4724–33. [DOI] [PubMed] [Google Scholar]
- 26.MacFarland SP, Duffy KA, Bhatti TR, Bagatell R, Balamuth NJ, Brodeur GM, Ganguly A, Mattei PA, Surrey LF, Balis FM, Kalish JM. Diagnosis of Beckwith-Wiedemann syndrome in children presenting with Wilms tumor. Pediatr Blood Cancer 2018;65:e27296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duffy KA, Cielo CM, Cohen JL, Gonzalez-Gandolfi CX, Griff JR, Hathaway ER, Kupa J, Taylor JA, Wang KH, Ganguly A, Deardorff MA, Kalish JM. Characterization of the Beckwith-Wiedemann spectrum: diagnosis and management. Am J Med Genet C Semin Med Genet 2019;181:693–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ibrahim A, Kirby G, Hardy C, Dias RP, Tee L, Lim D, Berg J, MacDonald F, Nightingale P, Maher ER. Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin Epigenetics 2014;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maas SM, Vansenne F, Kadouch DJM, Ibrahim A, Bliek J, Hopman S, Mannens MM, Merks JHM, Maher ER, Hennekam RC, Phenotype HRC. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A 2016;170:2248–60. [DOI] [PubMed] [Google Scholar]
- 30.Scott SA, Cohen N, Brandt T, Toruner G, Desnick RJ, Edelmann L. Detection of low-level mosaicism and placental mosaicism by oligonucleotide array comparative genomic hybridization. Genet Med 2010;12:85–92. [DOI] [PubMed] [Google Scholar]
- 31.Mussa A, Russo S, De Crescenzo A, Freschi A, Calzari L, Maitz S, Macchiaiolo M, Molinatto C, Baldassarre G, Mariani M, Tarani L, Bedeschi MF, Milani D, Melis D, Bartuli A, Cubellis MV, Selicorni A, Cirillo Silengo M, Larizza L, Riccio A, Ferrero GB. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome. Eur J Hum Genet 2016;24:183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalish JM, Biesecker LG, Brioude F, Deardorff MA, Di Cesare-Merlone A, Druley T, Ferrero GB, Lapunzina P, Larizza L, Maas S, Macchiaiolo M, Maher ER, Maitz S, Martinez-Agosto JA, Mussa A, Robinson P, Russo S, Selicorni A, Hennekam RC. Nomenclature and definition in asymmetric regional body overgrowth. Am J Med Genet A 2017;173:1735–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper WN, Luharia A, Evans GA, Raza H, Haire AC, Grundy R, Bowdin SC, Riccio A, Sebastio G, Bliek J, Schofield PN, Reik W, Macdonald F, Maher ER. Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur J Hum Genet 2005;13:1025–32. [DOI] [PubMed] [Google Scholar]
- 34.Prada CE, Zarate YA, Hopkin RJ. Genetic causes of macroglossia: diagnostic approach. Pediatrics 2012;129:e431–7. [DOI] [PubMed] [Google Scholar]
- 35.Barros-Silva D, Marques CJ, Henrique R, Jerónimo C. Profiling DNA methylation based on next-generation sequencing approaches: new insights and clinical applications. Genes 2018;9:429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.