

SUMMARY
The bacterium, Francisella tularensis (Ft), is one of the most infectious agents known. Ft virulence is controlled by a unique combination of transcription regulators: the MglA-SspA heterodimer, PigR, and the stress signal, ppGpp. MglA-SspA assembles with the σ70-associated RNAP holoenzyme (RNAPσ70), forming a virulence-specialized polymerase. These factors activate Francisella pathogenicity island (FPI) gene expression, which is required for virulence, but the mechanism is unknown. Here we report FtRNAPσ70-promoter-DNA, FtRNAPσ70-(MglA-SspA)-promoter-DNA and FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-promoter-DNA cryo-EM structures. Structural and genetic analyses show MglA-SspA facilitates σ70 binding to DNA to regulate virulence and virulence-enhancing genes. Our Escherichia coli RNAPσ70-homodimeric EcSspA structure suggests this is a general SspA-transcription regulation mechanism. Strikingly, our FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-DNA structure reveals ppGpp binding to MglA-SspA tethers PigR to promoters. PigR in turn recruits FtRNAP αCTDs to DNA UP elements. Thus, these studies unveil a unique mechanism for Ft pathogenesis involving a virulence-specialized RNAP that employs two (MglA-SspA)-based strategies to activate virulence genes.
INTRODUCTION
Francisella tularensis (Ft), the causative agent of tularemia, is a Gram-negative γ-proteobacterium that is one of the most infectious pathogens known, with as few as 10 organisms constituting an infectious dose (Dennis et al., 2001; Maurin, 2015). Due to its high infectivity and ease of aerosolization, several countries have developed the bacterium as a bioweapon and the CDC has listed it as a category A bioterrorism agent (Dennis et al., 2001; Maurin, 2015). Multidrug-resistant forms of Ft can be readily generated in the laboratory, further heightening concern of this pathogen (Gestin et al., 2010; Loveless et al., 2010; Sutera et al., 2013). The factors and molecular mechanisms that control Ft pathogenicity are not fully understood. However, a cluster of genes present on the Francisella pathogenicity island (FPI) has been shown to be necessary for phagosomal escape by this bacterium into the cytosol of infected macrophages, a key step in Ft replication and pathogenesis (Lauriano et al., 2004; Nano et al., 2004; Larsson et al., 2005; Weiss et al., 2007; Barker et al., 2009; Bröms et al., 2010; Eshraghi et al., 2016). The FPI cluster, which encodes a type VI secretion system (Nano et al., 2004; Barker et al., 2009; Bröms et al., 2010; Russell et al., 2014), is present in two chromosomal copies in the most virulent Ft subspecies F. tularensis tularensis and F. tularensis holarctica.
Recent studies have revealed the Ft transcription regulatory system that mediates FPI activation. This system is composed of the stringent starvation protein A (SspA), the macrophage growth locus protein A (MglA) and the pathogenicity island gene regulator (PigR) (Lauriano et al., 2004; Charity et al., 2007; Brotcke and Monack, 2008; Charity et al., 2009; Rohlfing and Dove, 2014; Cuthbert et al., 2015; Cuthbert et al., 2017). Insight into how these proteins sense infection to activate the FPI was revealed by studies showing that the stress alarmone, ppGpp, produced upon infection binds directly to the MglA-SspA complex (Cuthbert et al., 2017). The only component of the Ft virulence circuitry with homologs in other Gram-negative bacteria is SspA, which is homodimeric in these bacteria and is involved in stress and virulence responses (Hansen et al., 2005). Interestingly, our recent data showed that MglA is an SspA-like protein that heterodimerizes with the FtSspA (Cuthbert et al., 2015; Cuthbert et al., 2017) and studies revealed that MglA and SspA, which are expressed during Ft infection, are constitutively associated with the FtRNA Polymerase (RNAP) (Ramsey et al., 2015). Bacterial RNAP core enzymes consist of large β and β′ subunits that combine to form the active site. Their assembly is facilitated by the homodimeric N-terminal domains of the α subunit (αNTDs) (Murakami and Darst, 2003; Feklistov et al., 2014; Murakami, 2015). Bacterial σ factors are dissociable RNAP subunits that dictate DNA promoter specificity (Feklistov et al., 2014).
Unlike most bacteria, Ft encodes only two σ factors, the general “housekeeping” σ70 and σ32. Notably, Ft utilizes the σ70 containing holoenzyme (FtRNAPσ70) for activation of its virulence genes. Intriguingly, Ft also encodes two distinct α RNAP subunits, which heterodimerize (Charity et al., 2007; Mukhamedyarov et al., 2011). Thus, Ft utilizes a regulatory circuitry to activate pathogenesis that includes a unique RNAP and unique combination of regulatory factors. How MglA-SspA interacts with the FtRNAPσ70 is unknown. Moreover, while other SspA-like proteins are well-known activators of virulence genes, their mechanisms have remained enigmatic. The molecular basis by which PigR, FtRNAPσ70-(MglA-SspA) and the stress signal, ppGpp collaborate to activate transcription of FPI genes is also not known. Here we address these key issues by employing cryo-electron microscopy (cryo-EM), X-ray crystallography and cellular analyses. Our studies unveil specific molecular mechanisms for activation of Ft virulence genes while also revealing a conserved interaction mechanism of SspA proteins for RNAPσ70 holoenzymes.
RESULTS
Overall FtRNAPσ70-(MglA-SspA)-DNA structure
To elucidate the FtRNAPσ70 structure and determine how it interacts with MglA-SspA, we used single-particle cryo-EM. FtRNAPσ70-(MglA-SspA) complexes, consisting of (ββ′α1α2ω)σ70-(MglA-SspA), were isolated from cells of the live vaccine strain (LVS) (Fortier et al., 1991) an attenuated form of Ft subspecies holarctica, which contained an RNAP β′ subunit with an engineered TAP-tag (Figure S1A). Previous studies showed the resultant RNAP is fully active and that MglA and SspA are isolated in stoichiometric amounts with the cellular FtRNAPσ70 (Figure S1A) (Charity et al., 2009). Notably, the FtRNAPσ70-(MglA-SspA) complex can also be pulled down if only MglA is tagged (Figure S1A). Using the FtRNAPσ70-(MglA-SspA) complex purified with the β′-TAP tag, we determined the 3.46 Å resolution cryo-EM structure of FtRNAPσ70-(MglA-SspA) bound to a 53-mer DNA site that encompassed base pairs (bps) −51 to 1 bp beyond the transcription start site (+1) of the FPI-housed iglA promoter (Figure 1A) (Ramsey et al., 2015). The structure revealed unambiguous density for most of the protein subunits. The promoter DNA, from bps −35 to +1, was also visible while the DNA upstream of the −35 element was disordered (Table S1; Figure 1A). The MglA-SspA heterodimer is well resolved in the structure and makes extensive interactions with the FtRNAPσ70 holoenzyme.
Figure 1. Structures of FtRNAPσ70-(MglA-SspA)-DNA and FtRNAPσ70-DNA complexes.
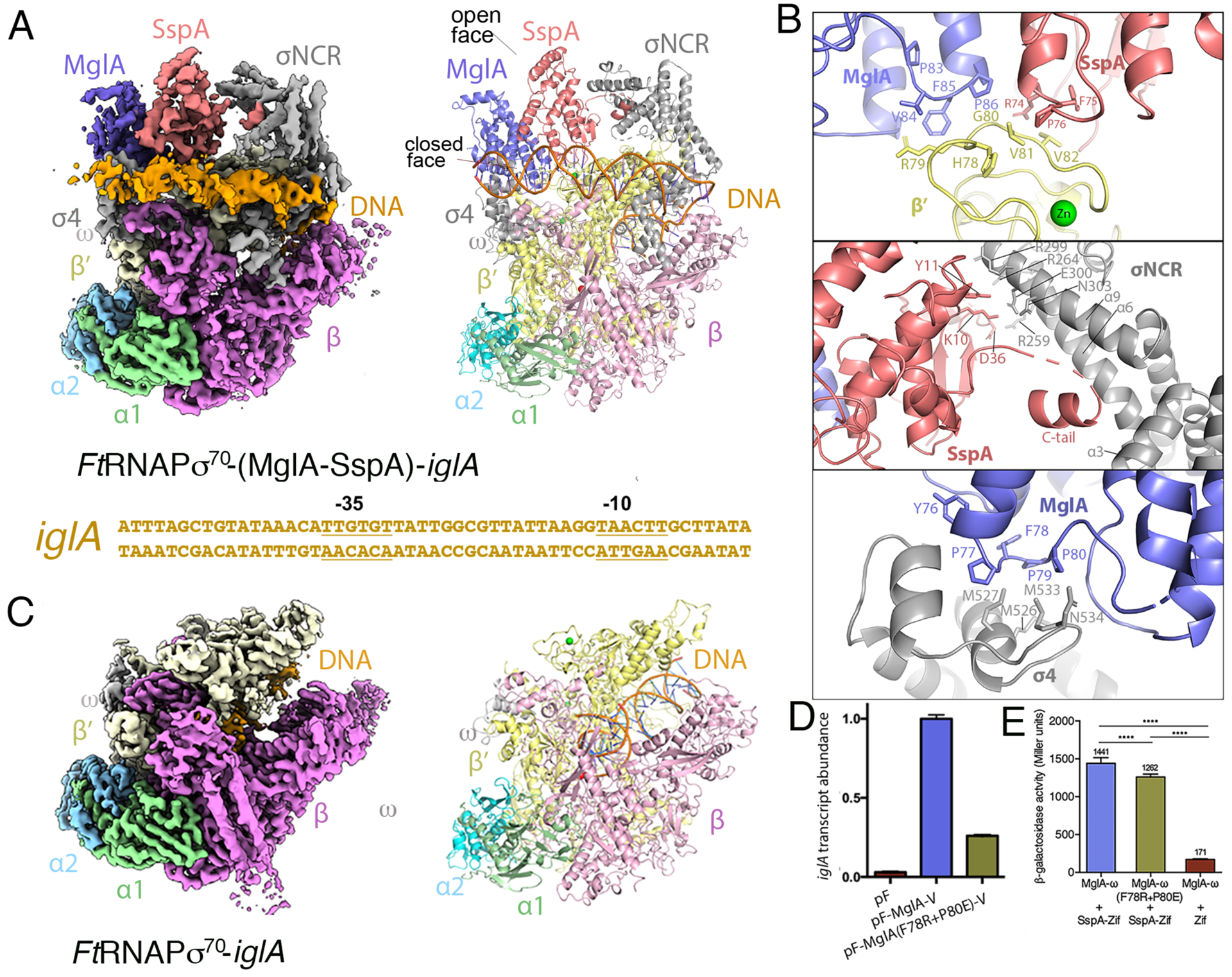
A FtRNAPσ70-(MglA-SspA)-iglA promoter DNA cryo-EM structure. Left, density map with protein subunits and DNA labeled. Right, ribbon diagram of the complex (obtained in the absence of ppGpp). Below the structure is the iglA promoter sequence used in the structure with −35 and −10 elements labeled. B Close up views of the three main MglA-SspA and FtRNAPσ70 contact points. C FtRNAPσ70-iglA promoter DNA complex. Left, density map and right is the corresponding ribbon diagram; density is absent for promoter DNA and σ70 in this structure. D qRT-PCR data showing the relative abilities of VSV-G tagged WT and mutant MglA, supplied by vectors pF-MglA-V and pF-MglA(F78R-P80E)-V respectively, to promote iglA expression in ΔmglA mutant cells. Transcripts were normalized to tul4, whose expression is independent of MglA. pF is an empty vector control. Error bars represent standard deviations of the mean. Statistical significance was assessed in Prism using a two-tailed t-test assuming equal variance; ***P<0.001, ****P<0.0001. E Bacterial two-hybrid assay of the ability of MglA(F78R-P80E)-ω to interact with the SspA-Zif fusion protein. Assays were performed with cells of the E. coli reporter strain KDZif1ΔZ containing compatible plasmids directing the IPTG-controlled synthesis of the indicated proteins. Cells were grown in LB supplemented with IPTG (50 μM) and then assayed for β-galactosidase activity. Statistical significance was assessed in Prism using one-way ANOVA with Tukey’s multiple comparisons; **** P<0.0001.
FtRNAP contains a distinct α heterodimer
The FtRNAP core enzyme harbors a similar overall organization as observed in E. coli and other structurally characterized bacterial RNAPs (Murakami and Darst, 2003; Murakami, 2015). The Cα atoms of the individual Ft β, β′ and ω subunits in the structure can be superimposed onto those of the E. coli RNAP with root mean squared deviations (rmds) of 1.4 Å, 1.3 Å, and 1.4 Å, respectively. But while the Ft α subunits also superimpose well with the E. coli α subunit (rmsd = 1.4 Å), instead of an α homodimer, the FtRNAP core contains an a heterodimer that is composed of two subunits, which share only 32% sequence identity. The presence of two Ftα subunits suggested the possibility that four FtRNAP core enzymes might form, (α1)2ββ′, (α2)2ββ′, (α1α2)ββ′, and (α2α1)ββ′, with the latter enzyme species differing in which α contacts β and which α contacts β′ (Charity et al., 2007; Mukhamedyarov et al., 2011). RNAP α subunits consist of N-terminal and C-terminal domains connected by a long linker. Density is not visible for the linkers or the α C-terminal domains (αCTDs) in the FtRNAPσ70-(MglA-SspA)-DNA structure, but the α1 and α2 N-terminal domains (αNTDs) are well resolved and the data clearly show only one form of the FtRNAP core, which contains a specific α1-α2 heterodimer with α1 contacting β and α2 interfacing with β′ (Figure S1B–C).
The FtαNTDs from each α subunit make distinct contacts important for formation of the RNAP core, burying 2670 Å2 of protein surface from solvent. The α1 subunit uses residues that are not conserved in α2 to form a network of selective hydrogen bonds with the β subunit (Figure S1B–C). Specific contacts to β are provided from α1 residues Asn137, Lys76, and Asp174 to β residues Glu729, Val773 and Arg823 (Figure S1C). Residues in α2, Arg183, Thr188, Phe174 and Tyr47 which are not found in α1, contact β′ residues Glu411, Glu441, the Cβ of Glu529 and the carbonyl atom of Arg533, respectively (Figure S1B). Interestingly, α2 also contributes a few contacts to the β subunit; residues Arg31, Tyr35, Phe39 and Asn43 interact with β residues Glu822, Arg1228, Thr1229, and Lys1231, respectively (Figure S1B). Thus, the two α subunits in Ft are not interchangeable and form a selective heterodimer that makes specific interactions with β and β′, critical for correct Ft RNAP core assembly.
MglA-SspA contacts with FtRNAPσ70
The mechanism by which MglA-SspA interacts with FtRNAPσ70 has been unknown. One hypothesis has been that because MglA-SspA is a heterodimer, it would interact with the distinct subunits of the FtRNAP α heterodimer (Rohlfing and Dove, 2014; Cuthbert et al., 2017). However, in the FtRNAPσ70-(MglA-SspA)-DNA structure the MglA-SspA heterodimer docks opposite the α heterodimer and near the promoter DNA binding region (Figure 1A–B; Figure S2A–G). The interactions between MglA-SspA and RNAP, which buries ~2200 Å2 of protein surface from solvent, are consistent with it existing as a constitutive subunit of the polymerase (Ramsey et al., 2015). MglA-SspA interacts with the RNAP β′ subunit, but primarily interfaces with σ70 (which dissociates during transcription elongation), consistent with the heterodimer specifically forming a complex with the FtRNAPσ70 holoenzyme. Indeed, the structure shows that the function of the MglA-SspA complex as a transcription activator does not involve DNA binding. Rather, the heterodimer stabilizes σ70 contacts with the RNAP core and promoter DNA, permitting the σ2 and σ4 domains, which are conserved with the corresponding E. coli σ70 domains, to bind the −10 and −35 promoter elements, respectively. The iglA promoter bound in this complex adopts an open conformation as ascertained by the presence of a transcription bubble (Figure 1A).
The FtRNAPσ70-(MglA-SspA)-DNA structure reveals three contact points between MglA-SspA and the FtRNAPσ70 holoenzyme. The interface involving the β′ subunit includes interactions from both MglA and SspA. In this interface, the so-designated “closed face” of the MglA-SspA heterodimer, which includes regions 83-PVFP-86 and 74-RFP-76 of MglA and SspA, respectively, interacts with β′ residues 77-KHRGVV-82 (Figure 1B, top panel; Movie S1). The remaining contact surfaces are between MglA-SspA and σ70. Each subunit of the MglA-SspA heterodimer interacts with a different region of σ70, thereby stabilizing the extended form of σ70 on the DNA. SspA interacts with the σ70 nonconserved region (σNCR) and MglA contacts the σ4 domain (Figure 1A). The σNCR, which is located between σ70 regions 1 and 2, is found in larger, type 1 σ proteins such as σ70 (Feklistov et al., 2014). SspA contacts two regions of the σNCR. One interaction interface includes SspA residues Lys10 and Tyr11 contacting σNCR residues Glu300, Asn303 and Arg299 and SspA residue Asp36 making electrostatic interactions with σNCR residue Arg259 (Figure 1B, middle panel). The second, more extensive contact region involves the SspA C-terminal tail (C-tail), residues 202–209 (Movie S1). These SspA residues, which are disordered in MglA-SspA crystal structures (Cuthbert et al., 2017), adopt a helical structure in the RNAP complex that docks into a cavity between σNCR helices α3, α6, and α9 (Figure 1B, middle panel). With the exception of the SspA C-tail, there are no large structural changes in MglA-SspA upon FtRNAPσ70 binding.
The third contact surface between MglA-SspA and FtRNAPσ70 involves interactions between MglA and σ70. In this interface, MglA makes multiple contacts to the σ4 region, which recognizes the −35 element of the promoter via a helix-turn-helix (HTH) motif. MglA residues 76-YPFPP-80 form a network of contacts with σ4 helices α2 and α3 and the loop connecting the two helices. In particular, MglA residue Phe78 binds near a shallow hydrophobic cavity within this long loop composed of Met526, Met527 and Met533. MglA residue Pro80 also inserts into this hydrophobic cleft and interacts with residues Met533, Thr535 and the Cβ of Asn534 (Figure 1B, bottom panel; Movie S1). The structure explains why MglA-SspA might not associate with the FtRNAPσ32 holoenzyme, as the smaller Ft σ32 protein does not contain an NCR and the σ4 loop region contacted by MglA differs in sequence and length between the two Ft σ factors.
PigR-independent virulence role for MglA-SspA
Our FtRNAPσ70-(MglA-SspA)-promoter DNA structure indicates that MglA-SspA functions to facilitate the interaction of σ70 with both the core RNAP and promoter DNA, leading to open complex formation. Notably, the promoter DNA used to obtain the structure did not contain a pre-formed bubble (from non-complementary DNA) (Figure 1A) and thus promoter opening was mediated de novo by the FtRNAPσ70-(MglA-SspA) complex. This finding suggests the possibility that MglA-SspA might activate Ft transcription in a PigR-independent manner. To investigate this possibility further we next obtained a 3.0 Å cryo-EM structure of FtRNAPσ70 with iglA promoter DNA in the absence of MglA-SspA (Table S1; Figure S3A–G). Strikingly, in this structure, density was absent for both σ70 and the promoter DNA, consistent with data showing FPI transcription is dependent on MglA-SspA (Charity et al., 2007; Rohlfing and Dove, 2014) (Figure 1C). Interestingly, DNA and some RNA density were present in the active site channel of the RNAP, presumably carried over from the two step purification of the holoenzyme from Francisella cells. Hence, the FtRNAPσ70 purified in this manner captures the RNAP in complex with non-specific DNA and the addition of iglA promoter DNA is insufficient to allow promoter binding or formation of a stable open promoter complex by the FtRNAPσ70 without MglA-SspA.
Examination of the iglA promoter shows that while it harbors a strong −10 element, required for binding the σ2 domain, the −35 element is not optimal, which suggests a role for the MglA-σ4 interaction in the formation of a stable transcription initiation complex. To test further the importance of MglA contacts to σ4 in transcription, we constructed an MglA mutant with substitutions F78R and P80E, as the structure shows these substitutions should impede MglA-SspA binding to Ft σ4. qRT-PCR analyses revealed that the VSV-G tagged MglA(F78R-P80E) mutant in LVS ΔmglA mutant cells significantly impaired transcription activation of the iglA gene compared to wild type (WT) VSV-G tagged MglA (Figure 1D). Notably, the MglA(F78R-P80E) mutant interacts with SspA as well as the WT MglA protein in a bacterial two-hybrid assay indicating that the impact on expression was not due to impaired MglA-SspA heterodimer formation (Figure 1E). MglA-SspA binding to FtRNAPσ70 does not induce structural changes in polymerase subunits nor does the heterodimer directly interact with σ2 to influence melting of the −10 element. Therefore, the mechanism by which MglA-SspA favors open promoter complex appears similar to that of the Thermus thermophilus TAP activator, which promotes the generation of the TtRNAP open complex by forming stabilizing adhesive protein-protein interactions with the polymerase (Feng et al., 2016). Unlike TtTAP, MglA-SspA appears to act as a σ70 activator by making direct contacts to σ70 and the RNAP β′ subunit as well as facilitating the interaction of σ70 with promoter DNA.
The structural data showing that the presence of MglA-SspA at the iglA promoter is required to drive open complex formation suggested that the heterodimer alone might impact Ft transcription of virulence genes even in the absence of PigR (Ramsey et al., 2015). To test this hypothesis and obtain information on genes that may be regulated by MglA-SspA but not PigR, we performed RNA-Seq studies (STAR Methods). In these experiments, the transcriptomes of LVS WT cells, LVS ΔmglA mutant cells and LVS ΔpigR mutant cells were analyzed using biological triplicate samples. These studies revealed that the MglA-SspA regulon (as defined by genes whose expression changes by a factor of 2 or more in ΔmglA mutant cells compared to WT with an adjusted p-value <0.05) is larger than previously described (Figure 2A–B; Table S2). They also revealed that although most of the PigR-regulated genes (those whose expression changes by a factor of 2 or more in ΔpigR mutant cells compared to WT with an adjusted p-value <0.05) are also controlled by MglA-SspA, many MglA-SspA regulated genes are not regulated by PigR (Figure 2A–B).
Figure 2. MglA-SspA mediated-PigR independent transcription and general binding mode of SspA proteins for RNAPσ70.
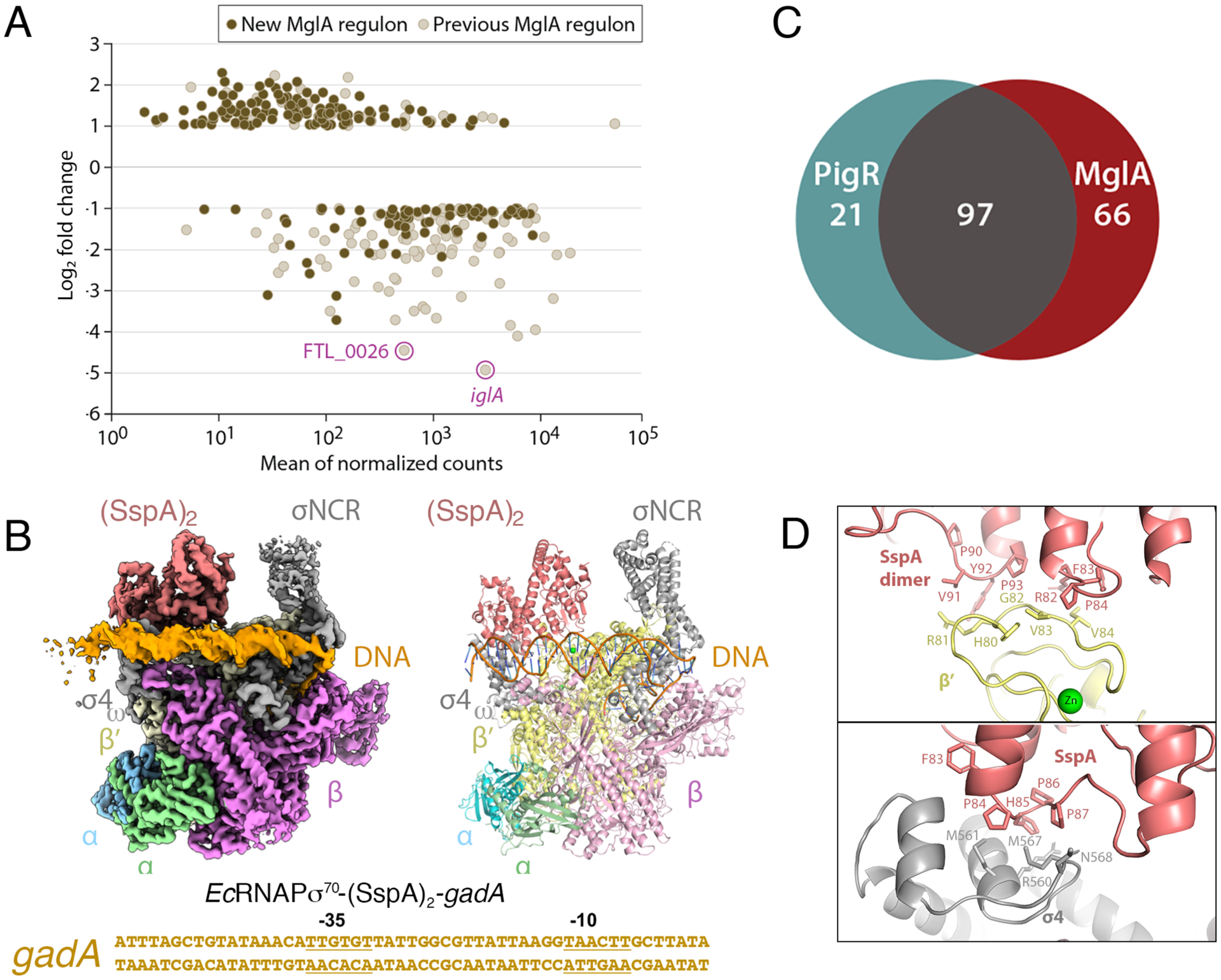
A DESeq2 was used to conduct differential gene expression analysis between WT and ΔmglA mutant RNA-Seq libraries. The graph shows log2 fold change in transcript abundance in ΔmglA compared to WT cells. Previously described MglA-controlled transcripts (such as iglA and FTL_0026) are in beige (with the iglA and FTL_0026 transcripts highlighted). Newly identified MglA-transcripts are in brown. B Venn diagram indicating MglA-regulated genes that are controlled by PigR and those that are not. Most genes regulated by PigR are also regulated by MglA-SspA but MglA-SspA regulates multiple genes independently of PigR. C Cryo-EM EcRNAPσ70-(SspA)2-gadA DNA structure. Left is cryo-EM map, right is ribbon diagram and below is the sequence of the gadA promoter used in the structure, with −35 and −10 elements labeled. The complex is shown in the same orientation as Figure 1A. D Close up views of contact points between EcSspA and EcRNAPσ70.
Conserved SspA-binding mode with RNAPσ70
The finding that MglA-SspA alone controls transcription of multiple genes led us to question whether homodimeric SspA proteins might utilize a similar mechanism. SspA proteins play key roles in starvation responses and virulence in multiple bacteria, including E. coli, Yersinia enterocolitica and Neisseria gonorrhoreae (Ishihama and Saitoh, 1979; Williams et al., 1994; De Reuse and Taha, 1997; Badger and Miller, 1998; Hansen et al., 2003; Hansen et al., 2005). Interestingly, like MglA-SspA, the transcription regulation functions of EcSspA transcription are mediated through the RNAPσ70 holoenzyme (Hansen et al., 2003). In addition, the region encompassing EcSspA residues 83-FPHPP-87, which has been shown to be key for its transcription function (Hansen et al., 2005), corresponds to MglA residues 76-YPFPP-80 and FtSspA residues 74-RFPAP-78, which our cryo-EM structure revealed contact σ4 and β′ (Figure 1A). As the E. coli protein is the best studied of the SspA homologs, we turned to cryo-EM studies to examine the interaction of the EcSspA homodimer with EcRNAPσ70.
EcSspA was shown to be important in E. coli acid stress resistance during stationary phase; in a microarray study using an SspA mutant, acid-resistance glutamate-dependent (gad) genes were downregulated, with the greatest effect observed for gadA (Hansen et al., 2005). Thus, we obtained a 3.2 Å cryo-EM structure of the E. coli RNAPσ70(SspA) complex bound to the gadA promoter. The structure shows that the EcSspA homodimer forms essentially the same complex with EcRNAPσ70 as MglA-SspA does with FtRNAPσ70. Specifically, EcSspA contacts the σ4 domain of σ70 and the RNAP β′ subunit using corresponding surfaces and the structure adopts an open promoter complex as in the FtRNAPσ70-(MglA-SspA)-DNA structure (Figure 2C; Figure S4A–G; Table S1). The SspA dimer is also juxtaposed near the σNCR but unlike FtMglA-SspA does not contact the σNCR in this complex. Similar to the MglA-SspA complex with FtRNAPσ70, residues 74-PHP-76 of one SspA subunit contacts the β′ subunit, while these residues in the other subunit of the EcSspA homodimer interact with the open loop in σ4 (Figure 2D). The σ4 loop of the Ecσ70 is different from other Ecσ factors, explaining the preference of SspA proteins for the RNAPσ70 holoenzyme. Residues 90-PVYP-93 of one SspA subunit also interact with Ecβ′, analogous to the interaction between MglA residues 83-PVFP-86 and Ftβ′. The N-terminal helix of Ecβ′ contributes additional contacts to EcSspA as does the C-terminal helix of ω, which is disordered in most RNAP structures (Figure S5). Similar to the Ft iglA promoter, Ec gadA lacks an optimal −35 element and EcSspA appears to stabilize the RNAP σ4 domain onto promoter DNA. Notably, the phage P1 Ps late promoter, which requires EcSspA for transcription activation, lacks a recognizable RNAPσ70 −35 element (Hansen et al., 2003).
Ft(MglA-SspA)-ppGpp-PigR crystal structure
A pivotal step in Ft pathogenicity is escape of Francisella cells from the phagosome into the cytosol. This step depends on transcription of FPI encoded genes, the full activation of which requires RNAPσ70-(MglA-SspA), the Francisella-specific protein, PigR and the stress alarmone, ppGpp (Lauriano et al., 2004; Brotcke et al., 2006; Charity et al., 2007; Brotcke and Monack, 2008; Charity et al., 2009; Rohlfing and Dove, 2014; Cuthbert et al., 2017). MglA-SspA binds ppGpp in the absence of PigR, but ppGpp is required for high affinity binding of the C-terminal 22 residues of PigR to MglA-SspA (Cuthbert et al., 2017). Although unknown, the structure of the 111 residue PigR protein is predicted to contain a MerR-like winged HTH (wHTH) (Heldwein and Brennan, 2001; Brown et al., 2003) followed by the C-terminal tail, composed of residues 90–111. To delineate the details of the PigR interaction with (MglA-SspA)-ppGpp we solved the (MglA-SspA)-ppGpp-PigR(90–111) crystal structure to 2.95 Å resolution (Table S3; Figure 3A; STAR Methods). In the structure, the vast majority of contacts to the ppGpp are from the Ft-specific MglA protein, which shares only ~25 sequence identity with SspA homodimeric proteins. Consistent with this, previous studies showed that MglA-SspA binds ppGpp with a Kd of 12 μM while the EcSspA homodimer showed very weak binding to ppGpp (Cuthbert et al, 2017). The (MglA-SspA)-ppGpp-PigR structure reveals that the PigR C-tail binds within the ppGpp binding pocket or “open face” (Figure 1A). The N-terminal region of the PigR fragment forms a strand that extends into the (MglA-SspA)-ppGpp binding pocket and the remaining PigR residues fold into an amphipathic helix (Figure 3A–B).
Figure 3. X-ray structure of (MglA-SspA)-ppGpp-PigR peptide complex.
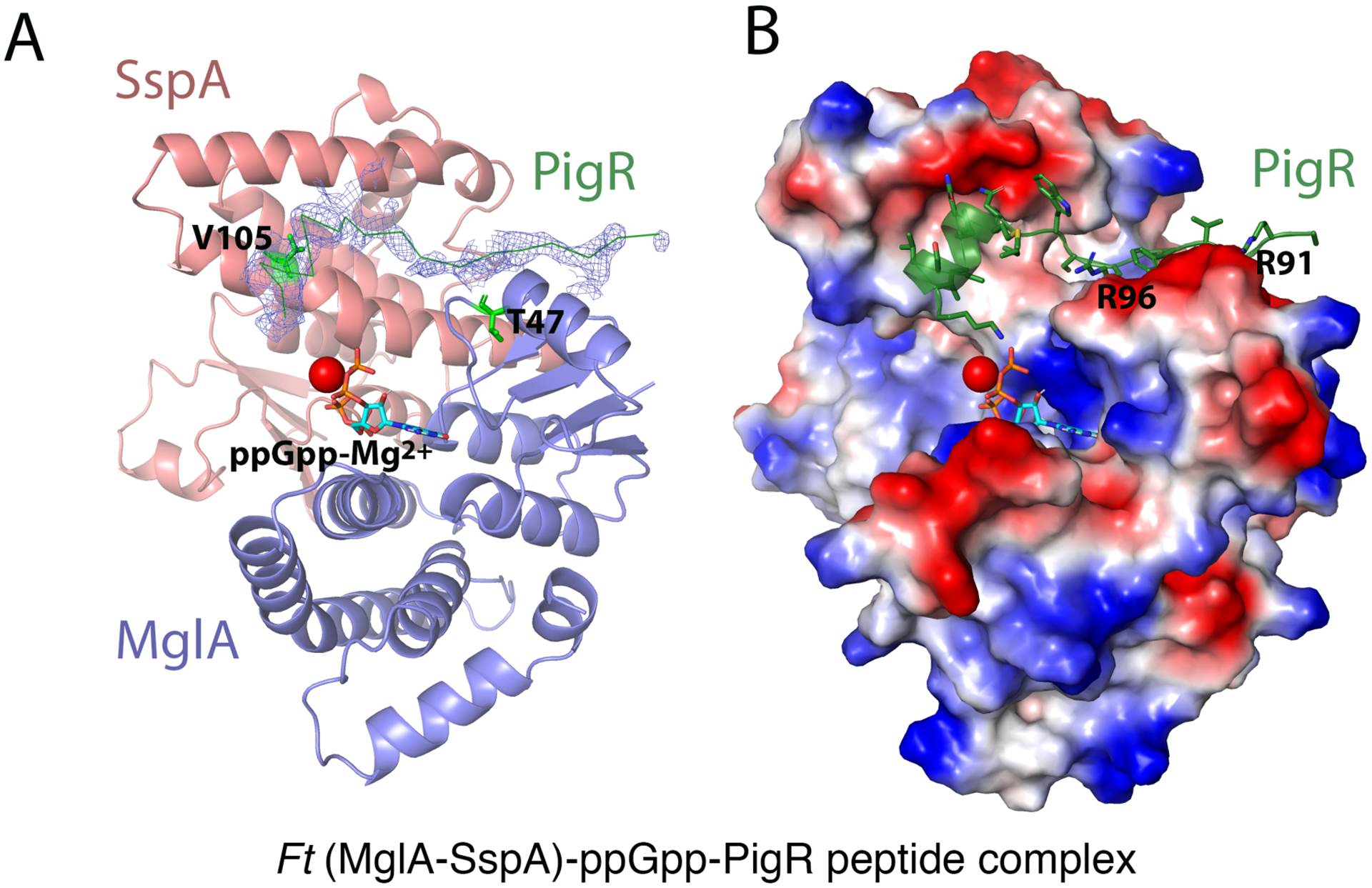
A Ribbon diagram of MglA-SspA with 2Fo-Fc electron density (0.65 σ) shown before the PigR peptide was added. Shown also are the locations of SspA V105 and MglA T47 (green sticks). The PigR peptide is shown as a green ribbon, ppGpp as sticks and Mg2+ as a red sphere. B Electrostatic surface of MglA-SspA with the PigR peptide depicted as a green ribbon. Positive and negative regions are blue and red, respectively.
Previous data showed that mutations in SspA and MglA residues that directly contact ppGpp impaired PigR binding, in line with data showing that ppGpp is required for high affinity PigR binding (Rohlfing and Dove, 2014; Cuthbert et al., 2017). However, SspA(V105E) and MglA(T47A) substitutions do not impact ppGpp binding but significantly impede the interaction of PigR with MglA-SspA (Rohlfing and Dove, 2014). Consistent with these data, our structure reveals that SspA residue Val105 and MglA residue Thr47 line the PigR binding pocket, with the hydrophobic face of the PigR C-terminal helix making extensive interactions with SspA residue Val105 (Figure 3A). Complementary electrostatic interactions also occur between PigR residues Arg91 and Arg96 and the negatively charged surface of the α2 helix of MglA (Figure 3B). The ppGpp binding pocket of MglA-SspA and the C-terminus of the PigR helix (sequence: KAKS) docked in this pocket are both highly positively charged; PigR helix residue Lys108 is positioned to contact ppGpp. Thus, ppGpp appears to facilitate PigR binding to MglA-SspA via direct contacts to ppGpp as well as by preventing unfavorable charge-charge clashes between PigR C-terminal residues and the basic ppGpp binding pocket in MglA-SspA.
FtRNAPσ70-(MglA-SspA)-ppGpp-PigR complex
Our (MglA-SspA)-ppGpp-PigR crystal structure revealed that PigR is anchored to the (MglA-SspA)-ppGpp complex via its C-terminal tail. To understand how PigR enhances FPI transcription in the context of the full FtRNAP complex, we obtained a 4.5 Å resolution cryo-EM structure of the FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-iglA promoter DNA complex (Figure 4A; Figure S6A–H). In this complex, MglA-SspA is bound in the identical location as in the FtRNAPσ70-(MglA-SspA)-DNA structure. Density for ppGpp in the MglA-SspA pocket was evident. Unlike the FtRNAPσ70-(MglA-SspA)-DNA complex, in this structure density is also present for the upstream DNA, including the DNA binding site for PigR, the PigR response element (PRE), and DNA upstream and downstream of the PRE (Figure 4A). Density for the PigR wHTH near the PRE site was too weak to be readily modelled. But there are ~10 residues between the MglA-SspA binding region of PigR and the wHTH-PRE binding site, which would allow the connection of the two PigR regions (Movie S2). These data indicate that PigR binds the (MglA-SspA)-ppGpp complex as a monomer, bringing its wHTH motif within proximity of the PRE.
Figure 4. Cryo-EM structure of the FtRNAPs70-(MglA-SspA)-ppGpp-PigR-DNA complex.
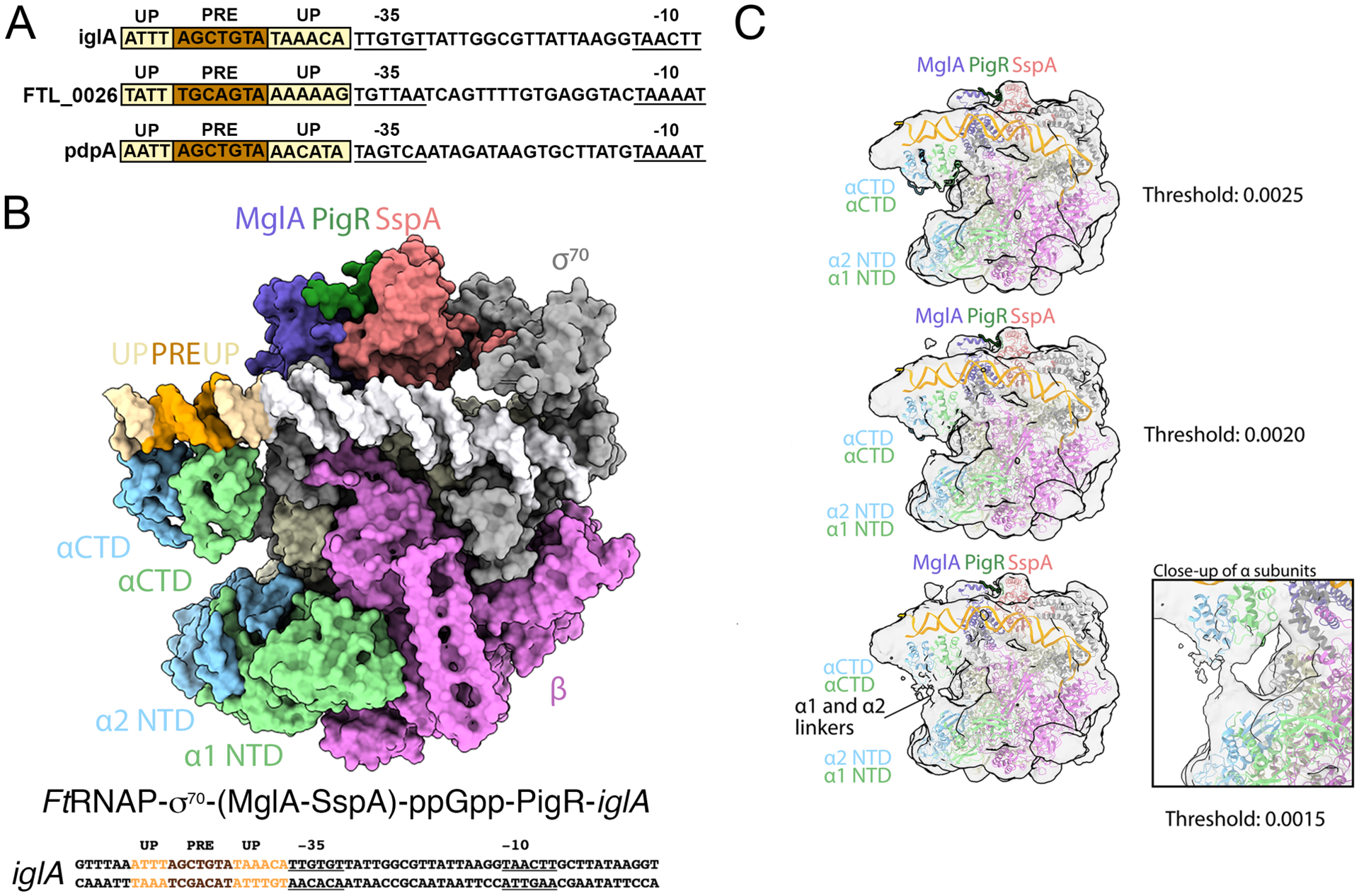
A Cryo-EM structure of the FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-DNA complex. The PigR C-tail is modeled from the crystal structure. Above the structure is the full promoter sequence used in the structure, with promoter elements labeled. B DNA sequences of PigR controlled genes showing just the promoter regions. −35 and −10 elements are indicated, PRE elements are colored brown and AT-rich UP elements are light yellow. C Images of the FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-DNA density map at various contour levels. Right shows a close up of the linker region, which leaves ambiguous which NTD is linked to which CTD.
Unexpectedly, in the FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-DNA structure we also observed two globular regions of density contacting the DNA upstream and downstream of the PRE. Both these PRE flanking DNA sites are very AT-rich (Figure 4A). Strikingly, examination of the DNA sequences of promoters known to be regulated by PigR, such as the iglA, pdpA and FTL_0026 promoters, revealed all contain AT-rich regions flanking their PRE sites (Figure 4B). Studies in model bacteria have revealed the importance of AT-rich regions located upstream of the −35 element (Newlands et al., 1991; Ross et al., 1993; Estrem et al., 1998; Ross and Gourse, 2009), termed Upstream (UP) elements, as contact sites for the C-terminal domains of the α subunits (αCTDs) of RNAP (Newlands et al., 1991; Ross et al., 1993; Estrem et al., 1998; Ross and Gourse, 2009). Consistent with the assignment of these densities in our structure as FtαCTDs, lower contoured density maps revealed a connection between the densities bound to the AT-rich DNA sites and the FtRNAP αNTDs and the EcαCTD structure could be readily docked into these densities (Figure 4C). At the current resolution, however, we were unable to assign which αCTD, α1 or α2, is bound to which UP element (upstream or downstream of the PRE). But both αCTDs are positioned on the DNA as in previously observed αCTD-UP structures with conserved DNA-binding residues proximal to the DNA (Benoff et al., 2002; Ross and Gourse, 2009). Notably, density was not visible for the αCTDs in our FtRNAPσ70-iglA, EcRNAPσ70-SspA-gadA or FtRNAPσ70-(MglA-SspA)-iglA structures indicating that PigR recruits the αCTDs to the DNA. Precisely how PigR facilitates UP element binding by the FtαCTDs is currently unclear due to the weak density of the PigR wHTH. However, the proximity of the PRE to the UP elements suggests that PigR may recruit the αCTDs through direct protein-protein contacts. The possibility that distinct αCTDs may bind separate surfaces of PigR and be directed to different UP elements would provide another rationale for the presence of two different Ft α subunits, in addition to the importance of the Ft αNTDs in RNAP core assembly. Alternatively PigR binding to the PRE may help order and stabilize the upstream DNA, damping its thermal motions, thereby favoring αCTD binding.
Our structural data suggest that in the presence of ppGpp, PigR binds to MglA-SspA and PRE DNA to enable the recruitment to and/or stabilization of the αCTDs on the PRE-adjacent UP elements, forming stable initiation complexes (Figure 4A; Movie S2). To test this hypothesis, we generated reporter constructs in which the iglA and FTL_0026 promoters were transcriptionally fused to lacZ on a chromosomal integration vector, thus allowing us to assess the effects of mutations within the proposed UP promoter elements in their native contexts (Ramsey et al., 2015). Using these constructs, we showed that substitutions of bps within the PRE as well as within the proposed UP elements downstream of the PRE in these promoters significantly impaired or abrogated PigR-dependent transcription (Figure 5A–D). Thus, these data support the structural model demonstrating the importance of UP elements in Ft virulence gene activation.
Figure 5. Test of importance of UP elements in activity of PigR-regulated promoters.
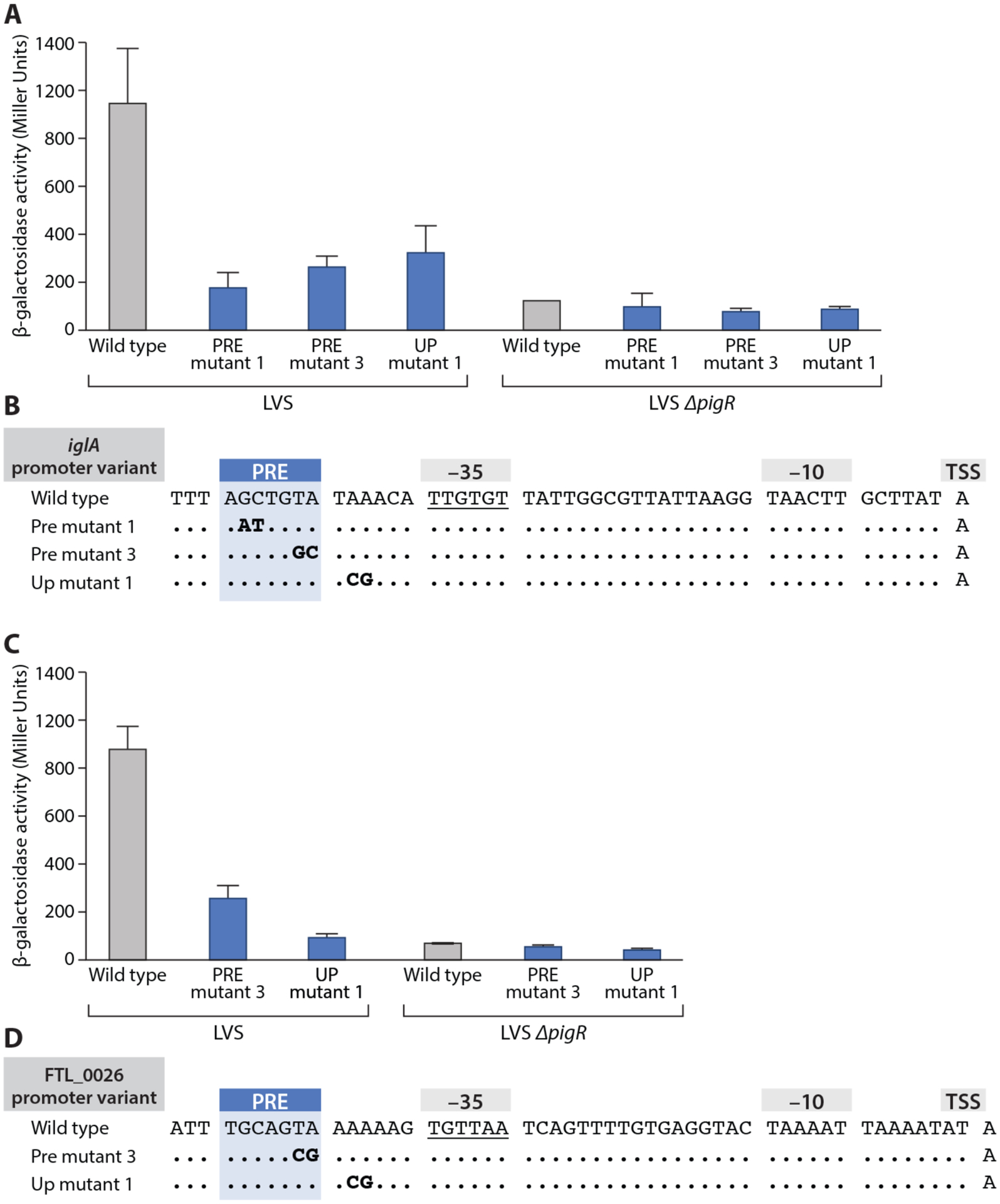
A Mutations were made in the PRE (PRE mutant 1 and PRE mutant 3 in bold) as well as in the UP element downstream of the PRE (UP mutant 1, in bold). Wild type is also labeled. In PRE mutant 3, the TA bps of the PRE at positions 6 and 7 are mutated to CG. In UP mutant 1, bps 2 and 3 in the UP element downstream of the PRE were changed from AA to GC. All mutations impaired PigR-dependent regulation. B Quantification of iglA-lacZ expression in LVS wild-type (LVS) and ΔpigR mutant (LVS ΔpigR) cells containing the indicated iglA promoter variants (X-axis) by β-galactosidase assay (Miller units). Promoter variants linked to a lacZ reporter gene were integrated into the FTL_0111 locus. Statistical significance was assessed in Prism using one-way ANOVA with Tukey’s multiple comparisons; **** P<0.0001. C Double mutations were made in the PRE (PRE mutant 3, as indicated in bold) and in the UP element downstream of the PRE (UP mutant 1, as indicated in bold). Wild type is labeled. In PRE mutant 3, the TA bps of the PRE at positions 6 and 7 are mutated to CG. In UP mutant 1, bps 2 and 3 in the FTL_0026 promoter downstream UP element were changed from AA to GC. All mutations impaired PigR-dependent regulation. D Quantification of FTL_0026-lacZ expression in LVS wild-type (LVS) and ΔpigR mutant (LVS ΔpigR) cells containing the indicated FTL_0026 promoter variants (X-axis) by β-galactosidase assay (Miller units). Promoter variants linked to a lacZ reporter gene were integrated into the FTL_0026 locus. Statistical significance was assessed in Prism using one-way ANOVA with Tukey’s multiple comparisons; **** P<0.0001.
DISCUSSION
Francisella tularensis is the causative agent of the zoonotic disease tularemia (Barker and Klose, 2007). Due to its low infectious dose, ability to be transmitted to humans via multiple routes and the potential to cause life-threatening infections, Ft has been developed as a bioterrorist agent. Indeed, the potential of Ft to be exploited deliberately to cause disease was recognized by the WHO as early as the 1970s (Dennis et al., 2001; Maurin, 2015). In addition, there is growing concern over the increase in reported outbreaks of tularemia in Europe over the past two decades (Eliasson and Bäck, 2007; Faber et al., 2018; Schroll et al., 2018). While Ft can infect more than 200 different organisms and multiple cell types, it primarily invades macrophages, where it is initially contained within a phagocytic vacuole. A necessary step in Ft virulence is escape from the phagosome into the cytosol, where the bacterium is able to rapidly divide and spread to surrounding tissues. What controls this step was unclear until a spontaneous Ft mutant with altered colony formation and inability to replicate in macrophages was identified. The mglA gene was subsequently identified as a mutated locus key for this step (Baron and Nano, 2002). Later studies revealed that, in addition to the MglA protein, the Ft SspA and PigR proteins and the stress molecule, ppGpp are essential for robust transcription of Francisella pathogenicity island (FPI) genes (Lauriano et al., 2004; Charity et al., 2007; Brotcke and Monack, 2008; Charity et al., 2009; Rohlfing and Dove, 2014; Cuthbert et al., 2017).
Unlike many bacteria, which employ specialized s factors for activation of virulence genes, Ft utilizes the FtRNAPσ70 holoenzyme, which contains the housekeeping σ70 protein, to transcribe FPI genes (Kazmierczak et al., 2005; Ramsey et al., 2015). As noted, MglA was established early on as a central activator of Ft virulence and our studies have revealed that MglA harbors an SspA-like fold and specifically heterodimerizes with FtSspA. MglA-SspA interacts with FtRNAPσ70 to form a virulence-specialized RNAP holoenzyme. Here we undertook cryo-EM, X-ray crystallography and genetic studies to elucidate the molecular underpinnings of this Ft-specialized virulence activating transcription machinery. Our FtRNAPσ70-(MglA-SspA)-DNA cryo-EM structure revealed that the MglA-SspA heterodimer does not interact with DNA, as do typical transcription activators, but instead makes extensive contacts with the RNAPσ70 holoenzyme that fastens the enzyme to the promoter (Lee et al., 2012; Browning and Busby, 2016). Specifically, the MglA-SspA heterodimer primarily interacts with σ70 and makes additional contacts to the core RNAP β′ subunit. Through contacts to the σNCR, located at the σ70 N-terminal region of the σ70 protein, and the σ4 domain, which is located at the C-terminus of σ70, MglA-SspA properly orients the malleable σ70 to key DNA promoter elements, i.e. the −10 and −35 elements, as well as affixes it to the RNAP core, leading to the formation of an open promoter complex (Bae et al., 2015; Murakami, 2015).
Interestingly, our structures showed that the MglA-SspA heterodimer is similar in its overall oligomeric organization and subunit structure to SspA homodimers (Hansen et al., 2005; Cuthbert et al., 2017). Our cryo-EM analyses of an E. coli RNAPσ70-(SspA)2-promoter DNA structure revealed the same overall organization of SspA with the EcRNAP. Moreover, both FtRNAPσ70-(MglA-SspA)-DNA and EcRNAPσ70-(SspA)2-DNA structures form open promoter complexes (Figure 6). Promoters of the genes activated by MglA-SspA in Ft and by the SspA homodimer in E.coli are not ideal. Indeed, while they contain near optimal −10 motifs (both contain key bps A-11 and T-7 found in the T-12A-11T-10A-9A-8T-7 consensus motif), their −35 motifs poorly match the ideal −35 consensus element. In particular, the Ps promoter activated by EcSspA lacks any recognizable −35 element. Previous structural studies revealed that key base contacts to the −35 element are made by residues Gln414, Glu410 and Arg409 from the Taq σA (Campbell et al., 2002). These residues, which are conserved in the Ec and Ft σ70 proteins, interact with bases within the T-35T-34G-33 and the C-31 bps. The −35 element of the iglA promoter, T-35T-34G-33T-32G-31T-30, departs from the optimal −35 motif (T-35T-34G-33A-32C-31A-30), and notably lacks the guanine of the C-31-G31 bp, which is specifically read by Arg409 (Campbell et al., 2002). While phosphate contacts to the −35 motif are made by residues in the σ4 domain in the FtRNAPσ70-(MglA-SspA)-DNA complex, base specific contacts are missing. Further, the iglA lacks an extended −10 motif (T-17G-16T-15G-14), which data have shown can substitute for weak −35 elements (Kumar et al., 1993; Barne et al., 1997). Analyses of promoters of genes shown to be activated by MglA-SspA in a PigR-independent manner by RNA-Seq revealed that all contained poor −35 elements with a few (FTL_1012, FTL_1511 and FTL_0361) possibly also harboring poor matches to the −10 element; we note the −35 and −10 elements of these genes are predictions based on our annotated transcription start site (Figure S7A). Thus, our structural data as well as genetic studies indicate MglA-SspA functions as an activator of weak promoters. This was also supported by our reporter assays, which demonstrated that the interactions between MglA-SspA and σ4 that enable the latter domain to contact the −35 element were necessary for activation of the iglA promoter (Figure 1D). Future transcription studies on homodimeric SspA proteins will be needed to assess their roles in transcription at specific promoters.
Figure 6. Open-promoter complexes are formed in Ft(MglA-SspA)- and Ec(SspA)2-containing transcription complexes.
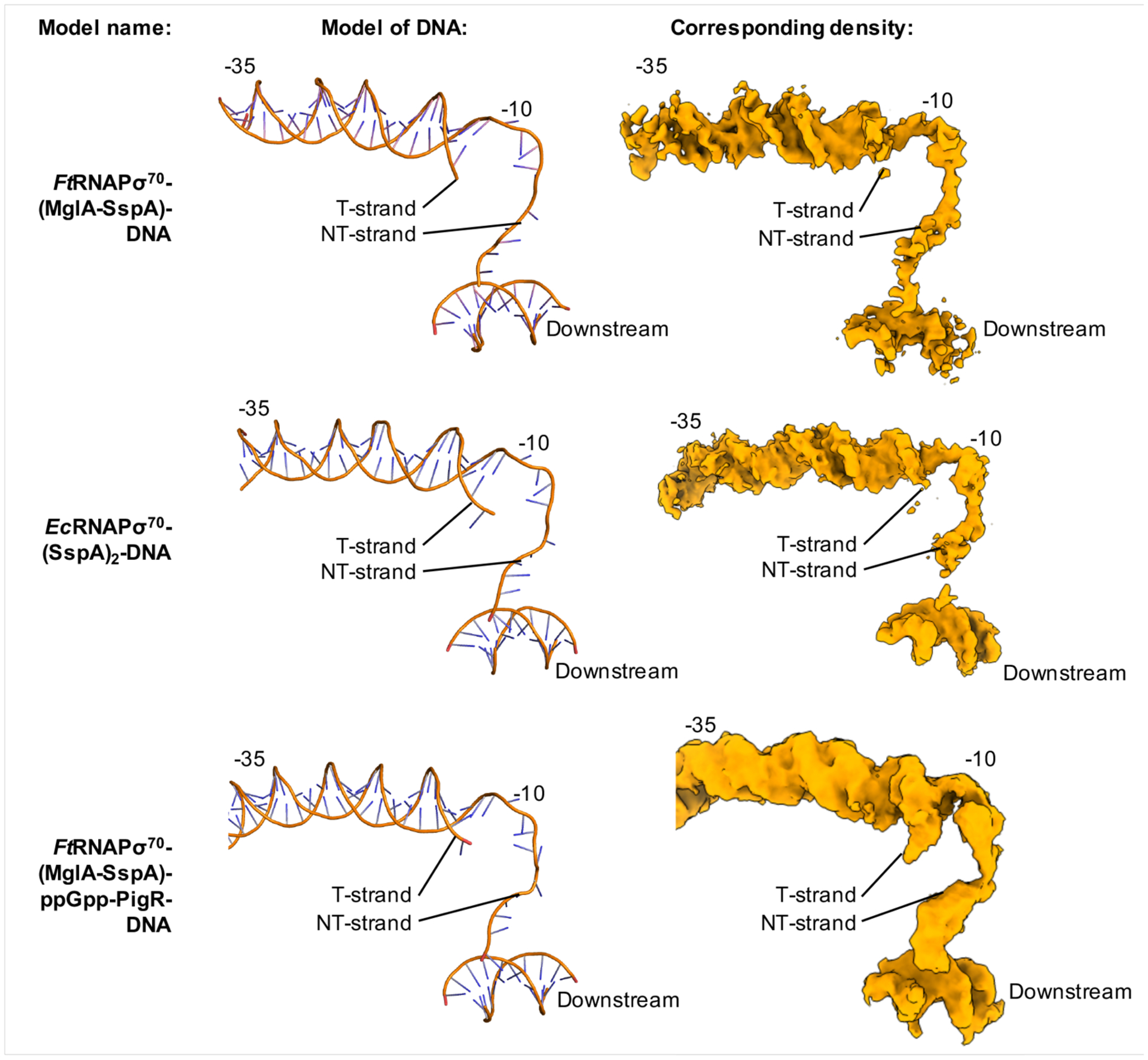
Protein subunits were removed from each model for clarity. Left shows ribbon diagrams of the DNA and right shows the corresponding DNA density.
Thus, based on our findings the MglA-SspA heterodimer appears to function as a σ activator, a term recently coined by Cartagena et al. to describe the Salmonella enterica Crl protein (Cartagena et al., 2019). The authors proposed that Crl, which is a small arc-shaped monomeric protein composed of β strands, acts as a σS activator through its interactions with σS region 2 and the RNAP β′ subunit, which enable σS-RNAP complex formation (Cartagena et al., 2019). MglA-SspA stabilizes primarily the σ4 region of σ70 along with β′, enabling σ70 binding to both the FtRNAP core and promoter DNA. These data indicate that σ activators may comprise multiple structural families and employ distinct interactions to aid in the assembly or stabilization of σ subunits with RNAP core enzymes and promoter DNA. Given that the FtRNAPσ70-(MglA-SspA)-DNA structure assumed an open promoter complex, we hypothesized that this complex alone might be capable of transcription activation in a manner independent of PigR. We thus performed RNA-seq analyses to delineate and compare the MglA-SspA and PigR regulons. Although these studies indicate that MglA-SspA and PigR activate many of the same genes, they also revealed that MglA-SspA activates the expression of a number of genes independently of PigR, the roles of which in virulence remain to be elucidated.
Collectively, our structural and genetic analyses indicate that the MglA-SspA heterodimer contributes to Ft virulence in a PigR-independent manner. Regardless, optimal activation of the FPI requires both the Ft-specific PigR protein and ppGpp. Unlike MglA-SspA, PigR has a conventional DNA-binding activator domain, a central winged HTH motif. While the wHTH of PigR places it in the MerR family of proteins, which are known to act as dimers (Heldwein and Brennan, 2001; Brown et al., 2003), our studies indicate that PigR functions as a monomer. Indeed, our (MglA-SspA)-ppGpp-PigR peptide crystal structure demonstrated that only one C-terminal tail of PigR inserts into the ppGpp binding pocket of MglA-SspA. Binding of ppGpp to MglA-SspA facilitates its interaction with the C-tail of PigR, thus revealing a second function of MglA-SspA; recruitment of an additional transcription activator. This interaction depends on the presence of ppGpp. ppGpp has emerged as a central second messenger in bacteria and shown to bind a variety of protein targets (Zhang et al., 2017; Wang et al., 2019) and crucially, to play an important role in reprogramming transcription (Kanjee et al. 2012; Kransy and Gourse, 2004; Liu et al., 2015). This latter process has been extensively studied in E. coli. In E. coli RNAP ppGpp binds to two sites: site 1, which is at the interface of the ω and β′ subunits, and site 2, which is a pocket formed by the interaction of β′ and the transcription factor DksA (Ross et al., 2013; Ross et al., 2016; Zuo et al., 2013). Binding of ppGpp to either RNAP site 1 or site 2 can inhibit transcription, but only binding of ppGpp to site 2 can stimulate transcription (Ross et al., 2013; Ross et al., 2016). Notably, the amino acid residues that form this EcRNAP pocket are not found in FtRNAP and DksA does not appear to be present in Ft. Thus, Ft employs ppGpp as a potentiator of virulence through its interaction with the MglA-SspA heterodimer. The ppGpp-binding pocket of MglA-SspA is also distinct from the ppGpp binding sites that have been characterized in multiple proteins from other bacteria (Kanjee et al., 2012).
To understand how PigR collaborates with FtRNAPσ70-(MglA-SspA) to drive FPI activation requires we solved the structure of the FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-iglA complex (Figure 4). Because PigR functions as a monomer its specific binding to the PRE, on its own, would likely contribute only weakly to the affinity of the RNAP complex for regulated promoters. However, the structure uncovered an unexpected role for PigR; the recruitment of the FtRNAP αCTDs to DNA sites that directly flank the PRE. Examination of these DNA sites revealed they are AT-rich. Such AT-rich sites upstream of the −35 element have been termed upstream (UP) elements and were discovered by Ross and Gourse as key contributors to transcription activation in E. coli through their interactions with the αCTDs of RNAP (Ross et al., 1993; Ross et al., 2003). Ft is unusual in encoding two distinct α RNAP subunit genes. The only other bacterium with two α subunits that has been characterized experimentally is Streptomyces granaticolor, where the presence of two α subunits with molecular weights of 40 kDa and 43 kDa was reported (Najmanová et al., 2000). However, it was revealed that the smaller α subunit is produced from proteolytic cleavage that occurred during sporulation and was not encoded by a separate gene (Najmanová et al., 2000). Our structures have revealed that a distinct Ft αNTD heterodimer is critical for proper assembly of β and β′ into a functional RNAP core (Figure S1B). The presence of two αCTDs in Ft also presents the opportunity for their different surfaces to be utilized in interactions with, for example distinct regions of PigR, or potentially other transcriptional activators, thus endowing additional versatility in Ft αCTD function.
Early studies suggested that αCTD interactions with UP elements function to not only enhance binding of RNAP to promoters but also to stimulate isomerization from the closed to the open complex (Rao et al., 1994; Saecker et al., 2011). However, recent single molecule experiments from the Gelles lab examining E. coli RNAP transcription from the rrnB P1 promoter revealed that αCTD-UP interactions stimulated RNAP binding, with no effect on open promoter formation (Mumm et al., 2020). Our FtRNAPσ70-(MglA-SspA)-DNA complex revealed that MglA-SspA alone facilitated open promoter complex formation when in complex with FtRNAPσ70 and thus is consistent with the Gelles study in that the αCTD-UP element interaction does not appear to be required for open promoter complex formation. The addition of the FtαCTD-UP contacts, which occurs only in the presence of PigR, thus likely functions to enhance RNAP binding. Notably, these are the first data revealing the utilization of UP elements in Ft transcription. Underscoring the importance of this interaction, the known PigR regulated promoters all contain AT-rich sites flanking their PRE sites (Figure 4B). Comparison of our RNA-Seq data of additional genes regulated in a PigR-dependent manner showed that, again, all have relatively weak predicted −35 elements (Figure S7B). However, the most strikingly findings from MEME analyses of these PigR regulated promoters was that not only is the PRE conserved, but also that the AT-rich motifs that directly flank the PRE are remarkably conserved (Figure S7C). These data indicate that αCTD recruitment to the Ft UP elements is a key and previously unrecognized mechanism for PigR-mediated activation of virulence genes.
In conclusion, our studies have elucidated the molecular basis for virulence activation of the dangerous pathogen Ft. Specifically, these collective studies have unveiled MglA-SspA as the linchpin in Ft virulence, whereby this heterodimer employs two roles in the activation of virulence genes; one involving a σ70 activation function and a second mechanism whereby binding of a small molecule alarmone, ppGpp, produced during infection, to the MglA-SspA heterodimer anchors a unique transcription activator, PigR, to virulence promoters, which in turn allows the recruitment and stable binding of the two Ft αCTDs to previously uncharacterized UP elements (Figure 7). These data thus provide the foundation for the future development of specific therapeutics that directly target virulence activation in Francisella tularensis.
Figure 7. Two roles of MglA-SspA in Ft virulence activation.
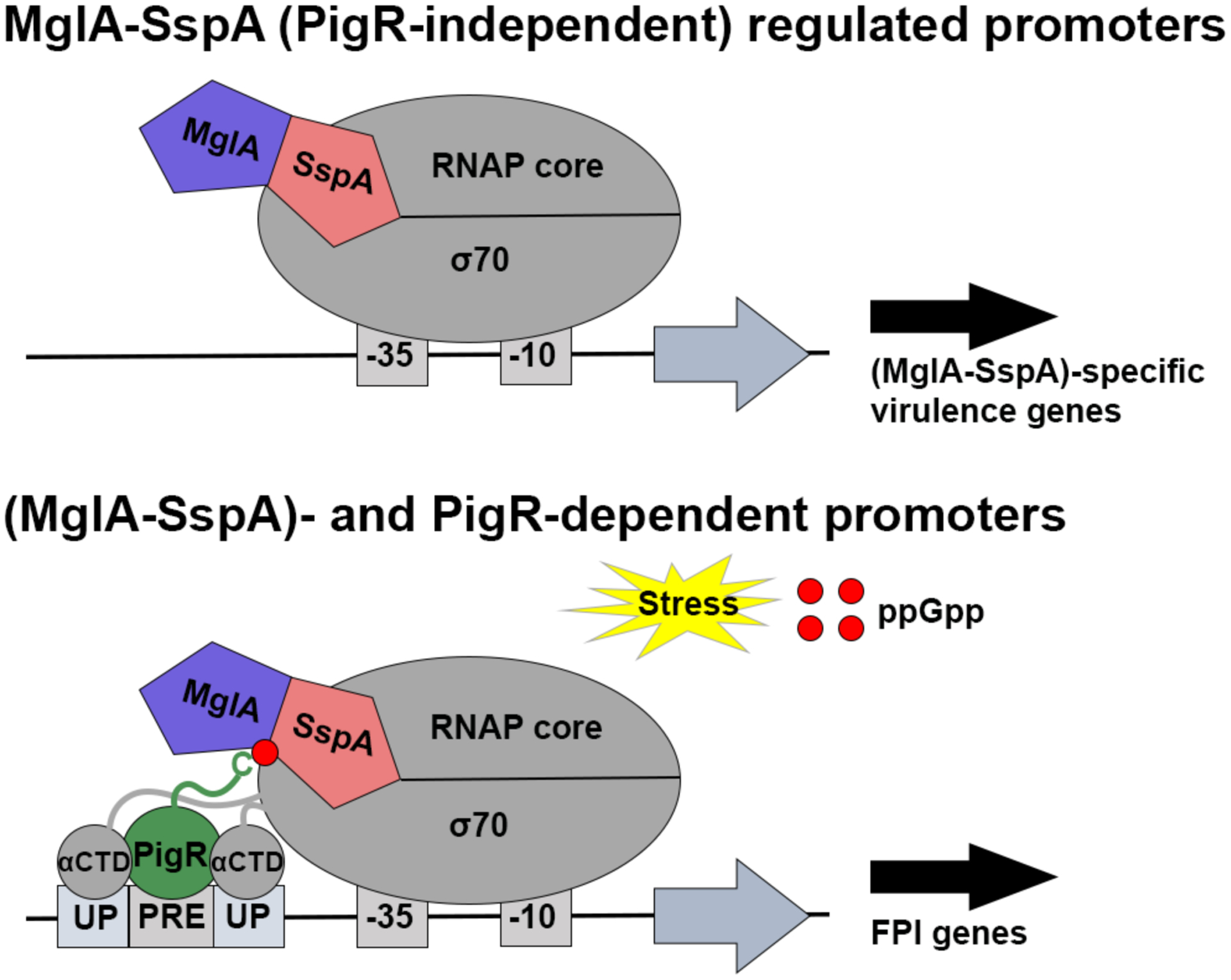
Schematic model for Ft virulence gene activation. Upper, PigR-independent genes are activated by MglA-SspA stabilizing σ70 and DNA interactions with FtRNAP. Lower, PigR-dependent genes involve PigR binding to (MglA-SspA)-ppGpp to recruit αCTDs to UP elements.
Limitations of study
Our work provides structures of key Francisella tularensis virulence transcription complexes. The resolution of the FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-DNA structure limits the details that can be discerned from this complex and the location of PigR in this structure is not clearly resolved. An additional limitation of the FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-DNA structure is the inability to assign which αCTD is bound to which UP element.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information for resources and requests should be directed to and will be fulfilled by the Lead Contact, Maria A. Schumacher (maria.schumacher@duke.edu).
Materials Availability
All reagents generated in this study are available upon request from the Lead Contact without restriction.
Data and Code Availability
The (MglA-SspA)-ppGpp-PigR peptide crystal structure coordinates and structure factors have been deposited in the Protein Data Bank under the accession number, 6WEG. Cryo-EM density maps have been deposited in the Protein Data Bank with the accession numbers 6WMR, 6WMP, 6WMU and 6WMT and EMD with accession codes EMD-21851, EMD-21850, EMD-21853 and EMD-21852. RNA-Seq data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) under accession number GSE150932.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Growth conditions
F. tularensis subps. holarctica LVS and its derivatives were grown aerobically at 37°C in either Mueller Hinton broth (BD Diagnostic Systems), supplemented with 0.1% glucose, 0.025% ferric pyrophosphate, and 2% Isovitalex (BD Diagnostic Systems) or on cysteine heart agar (BD Diagnostic Systems) supplemented with 1% hemoglobin solution (VWR). When appropriate, kanamycin (Research Products International) was used for selection at either 5 μg/mL or 10 μg/mL. Escherichia coli strains XL1 Blue and DH5αF′IQ were used for routine cloning. E. coli strains were grown aerobically at 37°C in LB medium.
METHODS DETAILS
Purification of FtRNAPσ70 complexes and PigR
FtRNAPσ70 and FtRNAPσ70-(MglA-SspA): FtRNAPσ70-(MglA-SspA) used for cryo-EM studies was purified directly from cells of the Francisella tularensis live vaccine strain (LVS) that synthesized the β′-subunit of RNAP with a tandem affinity purification (TAP)-tag at its C-terminus (LVS β′-TAP) (Charity et al., 2007). FtRNAPσ70-(MglA-SspA) was also purified from cells of LVS that synthesized MglA with a TAP-tag at its C-terminus (LVS MglA-TAP) (Charity et al., 2007). For Cryo-EM studies FtRNAP containing σ70 but lacking the MglA-SspA complex (FtRNAPσ70) was purified from LVS ΔmglA mutant cells that synthesize β′-TAP (LVS ΔmglA β′-TAP) (Rohlfing and Dove, 2014). In all cases, cells were grown with aeration at 37°C in 400 mL supplemented Mueller-Hinton Broth to mid-exponential phase (OD600 ~0.35) and harvested by centrifugation at 4°C. Cell pellets were resuspended in 5 ml lysis buffer [20 mM K-HEPES, pH 7.9, 50 mM KCl, 0.5 mM dithiothreitol (DTT), 1x BugBuster (Millipore Sigma), 10% glycerol, with one protease inhibitor tablet (Millipore Sigma) per 10 mL] and 10 U DNase I (Lucigen) was added to the cell lysis buffer. Cells were lysed by incubation at 37°C for 30 min and insoluble material was removed by centrifugation. Lysates were passed through a 0.22 micron filter and the buffer concentration was adjusted to 10 mM Tris-HCl pH 8, 150 mM NaCl, 0.1% NP-40 (Millipore Sigma). The lysates were incubated with rocking for 2 hr at 4°C in chromatography columns (Bio-Rad) with 200 μL IgG-Sepharose 6 Fast Flow beads (Millipore Sigma) that had been washed twice and resuspended in IPP150 buffer (10 mM Tris-HCl pH 8, 150 mM NaCl, 0.1% NP-40). Lysates were allowed to flow through the column and beads were washed three times with 10 mL IPP150 buffer and once with 10 mL Tobacco Etch Virus (TEV) cleavage buffer (10 mM Tris-HCl pH8, 150 mM NaCl, 0.1% NP-40, 0.5 mM EDTA, 1 mM DTT). Beads were incubated with rocking overnight at 4°C with 1 mL TEV cleavage buffer and 100 units AcTEV Protease (Life Technologies). Samples were eluted from the beads and combined with the eulate from a wash with 200 μL TEV cleavage buffer and the buffer concentration was adjusted to include 3 mM CaCl2. Samples were incubated with rocking for one hr at 4°C in clean chromatography columns with 200 μL calmodulin binding beads (Fisher Scientific) that had been washed twice and resuspended in calmodulin binding buffer (10 mM K-HEPES pH 7.9, 150 mM NaCl, 10 mM ß-mercaptoethanol, 1 mM Mg-acetate, 1 mM imidazole, 2 mM CaCl2). Unbound samples were allowed to flow through and beads were washed three times with 10 mL calmodulin binding buffer and once with 200 μL calmodulin elution buffer (10 mM KHEPES pH 7.9, 150 mM NaCl, 10 mM ß-mercaptoethanol, 1 mM Mg-acetate, 1 mM imidazole, 2 mM EGTA). Samples were eluted from beads using 1 mL calmodulin elution buffer. Ftσ70 copurified with the core enzyme in stoichiometric amounts in both cases as did MglA-SspA in the non ΔmglA cells.
PigR: An artificial gene, codon optimized for E. coli expression, encoding F. tularensis PigR was purchased from Genscript. The gene was cloned into the pET15b plasmid at the BamHI and NdeI restriction sites, providing a hexahistidine tag at the N-terminus of the expressed protein. E. coli C41(DE3) cells were transformed with the plasmid for protein expression. Cells were induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) when they reached an OD600 of ~0.6. After induction at 37°C for 3 h, cells were pelleted and resuspended in Buffer A [20 mM Tris-HCl pH 7.5, 300 mM NaCl, 5% glycerol, 7.5% sodium lauroyl sarcosinate (sarkosyl), 5 mM β-mercaptoethanol (BME)]. Cells were lysed by sonication and the soluble fraction was subjected to affinity chromatography using nickel-nitrilotriacetic acid (Ni-NTA) resin. Wash and elution steps were performed with Buffer B (20 mM Tris-HCl pH 7.5, 300 mM NaCl, 5% glycerol, 0.05% sarkosyl, 5 BME) supplemented with increasing concentrations of imidazole. Several milligrams of pure (>95%) PigR per liter of bacterial culture were obtained using this method.
Purification of EcRNAPσ70 and EcSspA
EcRNAPσ70 holoenzyme: The EcRNAP core enzyme (α dimer, β, β′, ω) and σ70 expression strains were a gift from Drs. Wilma Ross and Richard Gourse (UW-Madison). The core enzyme was expressed from a pIA900 vector in BL21(DE3) cells and purified by polyethylenimine (PEI) precipitation, ammonium sulfate precipitation, Ni-NTA affinity chromatography, and heparin-sepharose chromatography (Ross et al., 2003). For EcRNAP core expression, the cells were grown at 37°C until the OD600 reached 0.6 and were then induced with 1 mM IPTG for 3 hr at 30°C. Cells were pelleted and resuspended in Buffer C (50 mM Tris-HCl pH 8.0, 5% glycerol, 2 mM EDTA, 233 mM NaCl, 1 mM β-mercaptoethanol, 0.1 mM DTT, 0.26 mM PMSF). Lysozyme and deoxycholate were added to the mixture to a final concentration of 0.25 mg/mL and 0.07% respectively. Next, the cells were lysed by sonication and diluted with an equal volume of 0.2 mM NaCl + TGED (10 mM Tris-HCl pH 8.0, 5% glycerol, 0.1 mM EDTA, 0.1 mM DTT). After centrifugation to remove cell debris, the supernatant was cooled to 4°C. While stirring, PEI was added dropwise to a final concentration 0.6%. The resulting white precipitate was collected by centrifugation at 7500 rpm at 4°C and the supernatant was discarded. The pellet was extracted twice with 0.5 M NaCl + TGED by dissolving the pellet and repeating the centrifugation step. RNAP was eluted from the pellet with 1.0 M NaCl + TGED and the centrifugation step was repeated again. Next, 0.35 mg of ammonium sulfate was added per mL of supernatant and the mixture was stirred on ice for 30 min. The precipate was collected by centrifugation and the pellet was stored in 0.2 M NaCl + TGED overnight at 4°C. The next day, the pellet was resuspended in Buffer D (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 5% glycerol, 0.1% Tween, 10 mM imidazole). The resuspension was loaded onto a Ni-NTA column and washed with an equal volume of Buffer D. EcRNAP core was eluted from the column with Buffer F (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 5% glycerol) supplemented with increasing concentrations of imidazole. To remove nucleic acids that may have copurified with the EcRNAP core, the EcRNAP-containing elutions were pooled and subjected to heparin sepharose chromatography. This was performed by bringing the EcRNAP solution to 0.2 mM NaCl and 2 mM DTT, loading onto the heparin sepharose column pre-equilibrated with 0.2 M NaCl + TGED, washing with two column volumes of 0.2 M NaCl + TGED, and eluting with 15 mL 0.6 mM NaCl + TGED. The purified EcRNAP-containing fractions were dialyzed against 0.1 mM NaCl + TGED, flash frozen, and stored at −80°C until use. The σ70 subunit was expressed without an affinity tag from the LA4 expression vector in BL21 Gold (DE3)pLysS cells and purified from inclusion bodies. Cells expressing σ70 were grown at 37°C until reaching an OD600 of ~0.6. The cells were induced with 0.5 mM IPTG for 3 h at 37°C. The names and components of the buffers used for the following purification steps are the same as in Borukhov and Goldfarb (Borukhov and Goldfarb, 1993). σ70 containing cell pellets were suspended in 100 mL of Lysis Buffer (40 mM Tris pH 7.9, 0.3 M KCl, 10 mM EDTA, 0.1 mM PMSF), 0.2 mg/mL lysozyme, and 0.2% sodium deoxycholate. The cells were then lysed by sonication and centrifuged to pellet the insoluble inclusion bodies. The pellet was resuspended in 50 mL Lysis Buffer with 0.5% Triton X-100, sonicated, and pelleted two additional times. The inclusion bodies were solubilized by dissolving the pellet in 20 mL Denaturing Buffer (50 mM Tris pH 7.9, 6 M guanidine HCl, 10% glycerol, 10 mM MgCl2, 10 μM ZnCl2, 1 mM EDTA, 10 mM DTT) and centrifuged to remove insoluble particles. The supernatant was dialyzed against 2 L of Reconstitution Buffer (50 mM Tris pH 7.9, 20% glycerol, 10 mM MgCl2, 10 μM ZnCl2, 1 mM EDTA, 1 mM DTT) overnight. The next day, precipitate was removed by centrifugation and the supernatant was diluted 5-fold with Buffer E (40 mM Tris pH 8.3, 5% glycerol, 1 mM EDTA, 0.1 mM DTT) (Ross et al., 2003). Lastly, anion-exchange chromatography was performed using DE53 diethylaminoethyl cellulose (Whatman) pre-equilibrated in Buffer E. The protein was loaded onto the column, which was then washed with 15 mL Buffer E supplemented with 50 mM NaCl. The protein was eluted with 15 mL of Buffer E supplemented with 300 mM NaCl. Pure σ70 was obtained from this step. To form the EcRNAPσ70 holoenzyme, EcRNAP was mixed 1:3 with σ70 and incubated at room temperature (rt) in Buffer G (20 mM Tris-HCl pH 8.0, 5 mM MgCl2, 5% glycerol, 100 mM NaCl, 10 mM BME) for one hr followed by incubation at 4°C overnight. The holoenzyme was further purified by size exclusion chromatography using a HiLoad 26/600 Superdex 200 pg column (GE Healthcare) with Buffer G as the elution buffer. Fractions corresponding to purified holoenzyme were combined, flash frozen in liquid N2, and stored at −80°C until use.
EcSspA: An artificial gene encoding full-length E. coli SspA inserted into the pET15b plasmid at the BamHI and NdeI restriction sites was purchased from Genscript. C41(DE3) cells were transformed with the construct. For protein expression, the cells were grown and induced with 0.5 mM IPTG for 3 h at 37°C. For purification, the cells were pelleted and resuspended in 100 mL of Buffer H (20 mM Tris-HCl pH 7.5, 300 mM NaCl, 5% glycerol, 1 mM BME). Cells were lysed by sonication and the cleared lysate was loaded onto a Ni-NTA column pre-equilibrated with Buffer H. SspA was washed and eluted with Buffer H supplemented with increasing concentrations of imidazole, yielding purified SspA. SspA was buffer exchanged with Buffer H to remove imidazole and was flash frozen and stored at −80°C until use.
Cryo-EM specimen preparation
FtRNAPσ70-(MglA-SspA)-DNA and FtRNAPσ70-DNA: 0.4 mg/mL FtRNAPσ70±(MglA-SspA) was mixed 1:2 with iglA promoter DNA and incubated for one hr at 22°C. The iglA promoter DNA site (top strand: ATTTAGCTGTATAAACATTGTGTTATTGGCGTTATTAAGGTAACTTGCTTATA. Bottom strand: TATAAGCAAGTTACCTTAATAACGCCAATAACACAATGTTTATACAGCTAAAT) (IDT) were annealed by heating at 95°C for two min followed by slowly cooling to rt. For grid preparation, 3 μL of sample was applied to an UltrAufoil R1.2/1.3 Au 300 (Quantifoil) holey gold grid at 95% humidity and 22°C. The grid had been glow discharged for 30 s prior to sample application. After a 15.0 s incubation period, the grid was blotted for 2.0 s and plunge frozen into liquid ethane using a Leica EM GP2 (Leica Microsystems). All grids were stored in liquid nitrogen until data acquisition.
FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-DNA: To prepare this complex, 0.4 mg/mL FtRNAPσ70-(MglA-SspA) was mixed 1:2 with iglA DNA containing 6 additional upstream nucleotides and 4 additional downstream nucleotides (iglA top strand: GTTTAAATTTAGCTGTATAAACATTGTGTTATTGGCGTTATTAAGGTAACTTGCTTAT AAGGT. Bottom strand: ACCTTATAAGCAAGTTACCTTAATAACGCCAATAACACAATGTTTATACAGCTAAAT TTAAAC). Then, 1 mM ppGpp and 10 μM PigR were added and the mixture was incubated at 22°C for 1 h. For grid preparation, 3 μL of complex was applied to a glow discharged Quantifoil R1.2/1.3 Cu 300 holey carbon grid, incubated at 95% humidity and 22°C for 15.0 s, blotted for 2.5 s, and plunge frozen into liquid ethane using a Leica EM GP2.
EcRNAPσ70-(SspA)-DNA and EcRNAPσ70-DNA: Purified EcRNAPσ70 holoenzyme was thawed, concentrated to 6 mg/mL, and mixed 1:3 with SspA and 1:2 with gadA promoter DNA. The gadA promoter DNA site (top strand: CTGTAATGCCTTGCTTCCATTGCGGATAAATCCTACTTTTTTATTGCCTTC. Bottom strand: GAAGGCAATAAAAAAGTAGGATTTATCCGCAATGGAAGCAAGGCATTACAG) was annealed by heating at 95°C for two min followed by slowly cooling to rt. The complex was incubated at rt for 1 h. CHAPSO was added immediately prior to freezing at a concentration of 8 mM to alleviate orientation bias. Grids were prepared on Quantifoil R1.2/1.3 Cu 300 holey carbon grids as described for FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-DNA.
Cryo-EM data acquisition and processing
FtRNAPσ70-(MglA-SspA)-DNA: A Titan Krios G3i (ThermoFisher Scientific) with Falcon 3EC direct electron detector at Duke University was used to collect 1,672 30-frame movies on grids of the FtRNAPσ70-(MglA-SspA)-DNA complex in counting mode. EPU software was used for automated data acquisition, the magnification was 75,000x to give a pixel size of 1.06 Å, and the dose rate and total exposure for each movie was 0.8 e−/pix/s and 42 e− Å−2, respectively. The defocus range was set from −1.0 to −2.25 μm. After manual curation of the raw data, 1,291 movies were imported into RELION-3.0 for processing (Zivanov et al., 2018). The wrapper to MotionCor2 was used for motion correction and dose weighting (Zeng et al., 2017). For frame alignment, 5 × 5 patches were used, and the applied B-factor was 150 Å2. GCTF was used for CTF estimation on unweighted summed images (Zhang, 2016). Three 2D templates generated from ~1,000 manually picked particles were used to autopick a total of 618,526 particles. The particles were extracted with a box size of 352 pixels and rescaled to 176 pixels (2.12 Å/pix). After sorting the particles by Z-score with the 2D templates as references, the 578,236 particles with the highest Z-scores were input to reference-free 2D classification. From this, 21 classes containing 478,764 particles showed clear structural features. These classes were combined and input to a 2nd round of reference-free 2D classification. Next, 29 classes containing 242,263 particles were used for 3D classification with an E. coli RNAP holoenzyme (PDB: 6CA0) (Narayanan et al., 2018) low-pass filtered to 60 Å as the initial model. One 3D class composed of 50,316 particles showed clear density that was unaccounted for after fitting an E. coli holoenzyme structure (Narayanan et al., 2018) that was well fit with the MglA-SspA heterodimer structure (PDB: 5U56) (Cuthbert et al., 2017). The particles in this class were then re-extracted without scaling and subjected to 3D refinement. After CTF refinement and Bayesian polishing, the global resolution improved to 3.46 Å. To improve the local resolution of MglA-SspA, a masked 3D classification was performed using a mask only around MglA-SspA. Two classes (21,457 particles) showing well-resolved MglA-SspA density were combined and refined. While the global resolution for the reconstruction was unchanged, the local resolution for MglA-SspA was improved. Lastly, this final polished particle set was exported to cisTEM (Grant et al., 2018) for a final round of 3D-refinement, which showed improved density for MglA-SspA. All local resolution calculations were performed in RELION-3.0 (Zivanov et al., 2018) and viewed in UCSF Chimera (Pettersen et al., 2004).
FtRNAPσ70-DNA: For this structure, data were collected at Duke University with a Titan Krios G3i operating at 300 keV and equipped with a K3 direct electron detector (Gatan, Inc.) and Latitude S automated data acquisition software. A total of 3,490 movies were collected in counting mode with a magnification of 22,500x to give a pixel size of 1.07 Å and a defocus range of −1.0 to −2.0 μm. The dose rate was 15 e−/pix/s and the total exposure for each movie was 60 e− Å−2. Manual curation resulted in the removal of 974 movies from the data. The remaining 2,516 movies were imported into RELION-3.0 for processing (Zivanov et al., 2018). MotionCor2 (Zheng et al., 2017) was used for motion correction and dose-weighting with a 1 × 1 patch and an applied B factor of 150 Å−2. CTF estimation was performed with GCTF (Zhang, 2016) on the unweighted summed images. Before autopicking, ~1000 particles were manually picked and subjected to reference-free 2D classification to generate templates. Two templates were used to autopick 1,503,586 particles from the summed images. The particles were extracted with a box size of 352 pixels and scaled to 88 pixels (4.28 Å/pix) for faster processing. Particles were sorted based on their similarity to the autopicking references and a set of the best 1,455,290 particles were input to reference-free 2D classification. From this, 30 classes with clear structural features were combined to form a particle set of 745,802 particles to use for 3D classification. The 3D classification was performed with a 60 Å map from the FtRNAPσ70-(MglA-SspA)-DNA data as the initial model. Class 7 was composed of 137,682 particles and showed the most well-resolved structural features. These particles were re-extracted without scaling (1.07 Å/pix) and input to 3D refinement, resulting in a 3.25 Å reconstruction. After two rounds of CTF refinement and Bayesian polishing, the global resolution of the refined map improved to 3.0 Å. Three-dimensional FSC calculations using 3DFSC showed that the best and worst directional resolutions are ~2.9 and ~3.3 Å (FSC 0.143), respectively, which is reflective of some particle orientation bias in the data.
FtRNAPσ70(MglA-SspA)-ppGpp-PigR-DNA: For this structure, 7,362 movies (60 frames per movie) were collected at Duke University with a Titan Krios G3i operating at 300 keV and equipped with a K3 direct electron detector. The data was acquired using Latitude S software. Movies were collected in counting mode at a magnification 22,500x with a pixel size of 1.07 Å. The defocus range was set to −1.25 to −2.75 μm and the dose rate for each movie was 15 e−/pix/s to give a total electron exposure of 60 e− Å−2. After manual curation, 7,081 movies were imported into RELION-3.0 for processing (Zivanov et al., 2018). MotionCor2 was used for beam-induced motion correction (Zhjeng et al., 2017) with dose-weighting for each dose-fractionated movie stack. The applied B-factor for frame alignment was 150 and 1 × 1 patches were used. The contrast transfer function was estimated for each unweighted summed imaged using a wrapper to GCTF (Zhang, 2016) in RELION-3.0 (Zivanov et al., 2018). ~1,000 particles were manually selected and classified using reference-free 2D classification. Subsequently, auto-picking was performed with 4 templates that each represented a different particle orientation to give a total particle set of 3,485,423 particles. Particles were extracted with a box size of 352 pixels, rescaled to 88 pixels (4.28 Å/pix), and were sorted by similarity to the 2D templates using the Particle Sort job. The 258,463 particles with the worst Z-scores (least similarity) were removed from the data after sorting. Next, the remaining particles were classified by reference-free 2D classification. After 2D classification, high-resolution classes representing 1,901,718 particles were combined and used for 3D classification. The initial model for 3D classification was created by generating a 60 Å density map from a EcRNAPσ70 holoenzyme structure (PDB: 6CA0) (Narayanan et al., 2018; Kang et al., 2017). Two holoenzyme classes were combined and re-extracted with a pixel size of 2.14 Å. Refinement of this particle set yielded a 5.0 Å reconstruction with extra density upstream of the −35 region of DNA. A focused classification of this upstream region revealed density for 2 RNAP α C-terminal domains. A focused classification of this upstream region revealed density for 2 RNAP α C-terminal domains and spurious density for PigR. To perform the focused classification, a spherical mask was created in RELION-3.0 to encompass the DNA upstream of −35. For a view of the mask, see the data processing flowchart (Figure S6C). The focused classification was performed with partial signal subtraction. One class composed of 22.9% of the particles revealed density for 2 αCTDs. After re-extraction, CTF refinement, and Bayesian polishing, a final map with a global resolution of 4.5 Å was obtained. Additional focused classifications were performed to further resolve the spurious PigR density but were unsuccessful likely due to heterogeneity.
EcRNAPσ70(SspA)2-DNA: Remote data collection was performed at the Pacific Northwest Cryo-EM Center (PNCC) using a Titan Krios G3i operating at 300 keV and equipped with a K3 direct electron detector. A total of 7,363 50-frame movies were collected in counting mode with a magnification of 81,000x, a pixel size of 0.53 Å, a dose per frame of 1 e− Å−2, and a defocus range of −1.0 to 3.0 μm. Following manual curation, 5,708 movies were imported to RELION-3.0 for processing. During beam-induced motion correction with MotionCor2, the movies were binned by a factor of 2. CTF estimation was performed using GCTF. For particle picking, ~1,000 particles were picked and exported to crYOLO (Wagner et al., 2019) to train a general model which was used to pick 672,043 particles from the full dataset. The particles were binned 4x and subjected to one round of reference-free 2D classification. Next, 40 2D classes comprising 393,711 particles were selected and input to 3D classification. All 4 classes represented holoenzymes with clear density for SspA. The class with the best occupancy for SspA contained 111,519 particles and was 3D auto-refined to 8.8 Å. Lastly, a focused 3D-classification of SspA with partial signal subtraction of the RNAP core and σ70 subunit was performed. Class 2 comprised 49,560 particles and had the best resolution for SspA. A final 3D refinement was performed on this class to give a map with a global resolution of 3.2 Å.
Model building and refinement
To build the FtRNAP core and σ70 subunits, a model of EcRNAPσ70 holoenzyme (PDB: 6CA0)4 was fit into the density map using Chimera Pettersen et al., 2004). The FtRNAP β, β′, ω, and σ70 subunits share 59%, 62%, 42% and 45% sequence identity with the corresponding subunits of EcRNAP and the two Ft α subunits share 39% and 41% sequence identity with the EcRNAP α subunit. After fitting, the RNAP core subunits and σ70 subunits were replaced by the Ft sequences and re-modelled to fit the map using Coot (Emsley et al., 2010). In the FtRNAPσ70-(MglA-SspA)-DNA structure, Chimera was also used to fit a model of MglA-SspA (PDB: 5U56) (Cuthbert et al., 2017) into the density map. The C-terminal helix of SspA, not visible in our previous crystal structure (Cuthbert et al., 2017) was built de novo to fit the electron density. Density was not present for DNA bps −52 to −36 and was not modeled. In FtRNAPσ70-DNA, nonspecifically bound DNA and RNA in the active site were fit into the map using Chimera using the corresponding DNA and RNA from PDB: 6ALH (Kang et al., 2017). Density was absent for the iglA promoter DNA in this structure. To build the EcRNAPσ70-(SspA)2-DNA structure, an EcRNAPσ70 holoenzyme (PDB: 6CA0) (Narayanan et al., 2018) was manually fit into the density map using Chimera. Next, a crystal structure of Y. pestis SspA (PDB: 1YY7) (Hansen et al., 2005) was fit into the density corresponding to SspA. Using Coot, the Y. pestis SspA sequence was replaced by the E. coli SspA sequence and all RNAP holoenzyme and SspA residues were inspected and remodeled as necessary. Some parts of the RNAP model, such as the N-terminus of β′, and the C-terminus of ω, were built de novo. Density was not present for nucleotides −45 to −38 in the upstream promoter DNA region and was not modeled. In the FtRNAPσ70(MglA-SspA)-ppGpp-PigR-DNA structure, density was evident for a ppGpp bound to MglA-SspA as previously observed (Cuthbert et al., 2017) and in our (MglA-SspA)-ppGpp-PigR crystal structure (below). Density was evident for most of the promoter in this structure except −58 to −53. In addition, density for a ppGpp molecule bound in the FtRNAP between the β′ and ω subunits, termed the site 1 ppGpp binding site in the E. coli RNAP, was observed. The role of this ppGpp binding site is unclear. Indeed, binding to site 2, which is between ppGpp, DksA and the RNAP channel in E. coli RNAP, results in the majority of ppGpp’s effects on transcription (Ross et al., 2013). There is no DksA in Ft and the residues involved in site 2 binding are also not present in FtRNAP. While the density for the FtαCTDs was evident in this structure, the side chains could not be fitted and hence the αCTDs were constructed as polyalanine models.
For all deposited cryo-EM coordinates, flexible regions and unstructured loops were removed from the model if the electron density was not well resolved. Real-space refinement was performed in Phenix (Afonine et al., 2012) using Ramachandran and secondary structure restraints. Refined coordinates were inspected and modified in Coot (Emsley et al., 2010), as needed.
Ft(MglA-SspA)-ppGpp-PigR determination
A his6-sspA-(his6-mbp-mglA) coexpression system was generated by cotransforming C41(DE3) cells with plasmids encoding F. tularensis sspA (cloned into the pMCSG21 vector using ligation-independent cloning) and the his6-mpb-mglA fusion (cloned into pET28A). C41(DE3) cells were transformed with both expression plasmids and cells grown to an OD600 of 0.5 at 37°C. The cells were then induced by addition of 0.5 mM IPTG overnight at 15°C. Cells were lysed in 20 mM Tris, pH 7.5, 200 mM NaCl, 10% glycerol, and 7.5 mM imidazole, 1 mM βME, 2 mg/L DNase I, and 1 mM PMSF with a microfluidizer. Cell debris was removed by centrifugation at 15,000 rpm and the resultant supernatant was loaded onto a Ni-NTA column and washed with increasing concentrations of imidazole in a buffer consisting of 20 mM Tris, pH 7.5, 200 mM NaCl, 10% glycerol, and 1 mM βME. The protein was eluted using the buffer containing 0.25 to 2.0 M imidazole. The MBP-tag was removed overnight at rt using his-tagged TEV protease. The treated protein sample was next applied to a Ni-NTA column, which removed the His6-TEV tag and any uncleaved fusion protein. The MglA-SspA was collected in the flow through and the protein further purified via size exclusion chromatography using a Superdex S75 column (GE Healthcare). The buffer used for size exclusion chromatography was 20 mM Tris, pH 7.5, 200 mM NaCl, 10% glycerol, and 1 mM DTT. Crystals were grown using the hanging drop vapor diffusion method by mixing the protein complex at 24 mg/mL with ppGpp (TriLink) and 22mer PigR C-terminal peptide (KRNVFSRCWINMNLYSVIKAKS) at final concentrations of 1 mM and 0.5 mM, respectively. This solution was mixed 1:1 with a crystallization reagent consisting of 0.1 M Tris-HCl pH 8.0 and 24% PEG 4000. Data collection revealed no peptide density, likely due to low peptide solubility. Thus, excess PigR peptide was added to the drops, which were then allowed to sit for 2 months prior to data collection. To collect data, the crystals were cryoprotected by dipping them for several seconds in a 1 μL drop containing the crystallization solution supplemented with 18% ethylene glycol. X-ray intensity data were collected at the Advanced Light Source (ALS) beamline 8.3.1. The data were processed and scaled using XDS. Phaser in CCP4 was used to solve the structure by molecular replacement using an MglA-SspA heterodimer from our previously solved structure (Cuthbert et al., 2017). The crystallographic ASU contained two MglA-SspA dimers. Each dimer has a bound ppGpp but packing only permits the interaction of the PigR peptide with one dimer. Density was found for all but the last three residues of the peptide in this dimer (though a low contour level of the map was required) and PigR peptide side chain density was also weak. After multiple rounds of rebuilding and refinement in Phenix (Emsley et al., 2010; Afonine et al., 2012), the model converged to Rwork/Rfree values of 21.8%/28.7% at 2.95 Å resolution.
Plasmids for qRT-PCR analyses
Plasmid pF-MglA-V directs the synthesis of LVS MglA with a vesicular stomatitis virus-glycoprotein (VSV-G) epitope-tag fused to its C-terminus (MglA-V) under the control of the groEL promoter and has been described previously (Rohlfing and Dove, 2014). Plasmid pF-MglA(F78R+P80E)-V directs the synthesis of mutant MglA-V containing amino acid substitutions F78R and P80E in MglA. This plasmid was made by introducing DNA specifying the corresponding mutations into pF-MglA-V. Plasmid pF was the empty vector control (Rohlfing and Dove, 2014).
qRT-PCR analyses
RNA was isolated from cells of the LVS ΔmglA mutant strain that contained plasmid pF, pF-MglA-V, or pF-MglA(F78R+P80E) that were grown with aeration at 37°C in supplemented Mueller-Hinton Broth to mid-log (OD600 = 0.35–0.4). Nucleic acids were purified using the Zymo Direct-zol RNA Miniprep Plus kit (Zymo Research) according to kit instructions and eluted in 80 μL RNase-free water. Purified nucleic acids were treated with 10 units of RQ1 DNase (Fisher Scientific) in 1x RQ1 DNase buffer at 37°C for one hr. RNA was purified from DNase-treated samples using the Zymo Direct-zol RNA Miniprep Plus kit according to kit instructions. cDNA was generated from RNA using Superscript III reverse transcriptase (Life Technologies). Relative transcript abundance for iglA was deteremined relative to the amount of tul4 trancript by qPCR using FastStart Essential DNA Green Master mix (Roche) with a Roche LightCycler 96 detection system and relative expression values were calculated by using the comparative threshold cycle (CT) method (2−ΔΔCT) (Livak and Schmittgen, 2001). The reported fold enrichment values are the means from three biological replicates, and error bars represent the standard deviation of the mean. The data shown are from one representative experiment.
Plasmids for Francisella reporter strains
Mutations in the iglA promoter were generated in plasmid pMO1, which harbors a transcriptional fusion between the iglA promoter and the lacZ gene (Ramsey et al., 2015). Plasmid pSP109 contains DNA specifying the iglA promoter containing the PRE 3 mutation and was used to generate strains LVS PiglA-M3-lacZ and LVS ΔpigR PiglA-M3-lacZ. Plasmid pSP111 contains DNA specifying the iglA promoter containing the UP 1 mutation and was used to generate strains LVS PiglA-UPM1-lacZ and LVS ΔpigR PiglA-UPM1-lacZ. Plasmid pSP113 harbors a transcriptional fusion between the wild-type FTL_0026 promoter and the lacZ gene and was made by replacing the iglA promoter region in plasmid pMO1 with 1100 bp of DNA upstream of the FTL_0026 gene by isothermal assembly. Plasmid pSP113 was used to generate strains LVS PFTL_0026-lacZ and LVS ΔpigR PFTL_0026-lacZ. Mutations in the FTL_0026 promoter in plasmid pSP113 were generated by site-directed mutagenesis. Plasmid pSP123 contains DNA specifying the FTL_0026 promoter containing the PRE 3 mutation and was used to generate strains LVS PFTL_0026-M3-lacZ and LVS ΔpigR PFTL_0026-M3-lacZ. Plasmid pSP125 contains DNA specifying the FTL_0026 promoter containing the UP 1 mutation and was used to generate strains LVS PFTL_0026-UPM1-lacZ and LVS ΔpigR PFTL_0026-UPM1-lacZ.
Francisella reporter strains
Reporter strains LVS PiglA-lacZ and LVS ΔpigR PiglA-lacZ contain lacZ integrated downstream of the wild-type iglA promoter at the FTL_0111 locus (Ramsey et al., 2015). Strains LVS PiglA-M1-lacZ and LVS ΔpigR PiglA-M1-lacZ contain lacZ integrated downstream of the iglA promoter containing the PRE 1 mutation at the FTL_0111 locus (Ramsey et al., 2015). Reporter strains described in this manuscript were created by electroporation of integration plasmids, described above, into LVS and selection of single homologous recombination events between the integration plasmid and the chromosome. Electroporations were performed using LVS cells washed three times with 10% sucrose and approximately 0.5–2.5 μg plasmid DNA in a 2 mm cuvette at 2.5 kV. Cells were allowed to recover, shaking aerobically at 37°C for 4–8 hr in supplemented Mueller-Hinton Broth and cells containing an integration event were selected on cysteine heart agar with 1% hemoglobin and 10 μg/mL kanamycin. Reporter strains containing the iglA-lacZ transcriptional fusion were integrated at the FTL_0111 iglA locus as determined by Southern blotting (Ramsey et al., 2015). Digested chromosomal DNA was transferred to a positively charged nylon membrane (Millipore Sigma). The blot was blocked and washed using the DIG Wash and Block buffer set (Millipore Sigma) following manufacturer’s instructions. The Southern blot probe was synthesized using the PCR DIG Probe Synthesis Kit (Millipore Sigma) and hybridized to the blot overnight in ULTRAhyb hybridization buffer (Life Technologies). The probe was detected after incubation with anti-digoxigenin-AP antibody (Millipore Sigma) and CDP-Star (Millipore Sigma). Reporter strains containing the FTL_0026 promoter-lacZ transcriptional fusion and its derivatives were integrated at the FTL_0026 locus as determined by PCR.
Plasmids for bacterial two-hybrid assays
Plasmids pBR-MglA-ω, pACTR-SspA-Zif, and pACTR-Ap-Zif were used for two-hybrid studies (Rohlfing and Dove, 2014). Plasmid pBR-MglA(F78R+P80E)-ω is identical to pBR-MglA-ω except that the MglA moiety of the MglA-ω fusion protein contains amino acid substitutions F78R and P80E. Plasmid pBR-MglA(F78R+P80E)-ω was made by introducing DNA specifying the F78R and P80E substitutions in MglA into plasmid pBR-MglA-ω.
β-Galactosidase assays
β-Galactosidase assays were performed with cells of the LVS and LVS ΔpigR reporter strains (Ramsey et al., 2015). Specifically, for β-galactosidase assays with LVS and LVS ΔpigR reporter strains, cells were grown with aeration at 37°C in supplemented Mueller-Hinton Broth to mid-log (OD600~0.3). Cells were lysed in Z-buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM βME) with SDS-chloroform and assayed for β-galactosidase using ONPG (O-nitrophenyl-β-D-galactoside) as substrate at 28°C. Bacterial two-hybrid assays were performed using E. coli reporter strain KDZif1ΔZ (Rohlfing and Dove, 2014). In detail, for bacterial two-hybrid assays, E. coli reporter strain KDZif1ΔZ was transformed with plasmid pBR-MglA-ω together with pACTR-SspA-Zif, plasmid pBR-MglA(F78R+P80E)-ω together with pACTR-SspA-Zif, and plasmid pBR-MglA-ω together with pACTR-Ap-Zif. Transformants were first grown overnight with aeration at 37°C in LB containing carbenicillin (100 μg/mL), tetracycline (10 μg/mL) and IPTG (50 μM). Cells were then back-diluted into LB containing carbenicillin (100 μg/mL), tetracycline (10 μg/mL) and IPTG (50 μM) and grown with aeration at 37°C to mid-log (OD600~0.3–0.5). Cells were lysed in Z-buffer with SDS-chloroform and assayed for β-galactosidase using ONPG as substrate at 28°C.
RNA-Seq
For RNA-Seq experiments, RNA was isolated from cells of strains LVS, LVS ΔmglA, and LVS ΔpigR (Charity et al., 2007; Charity et al., 2009). The cells were grown with aeration at 37°C in supplemented Mueller-Hinton Broth to mid-log (OD600 0.35–0.4) using the Direct-zol Miniprep kit (Zymo Research). RNA (5 μg) was treated with the Ribo-Zero Magnetic Kit (Bacteria) (Epicentre) and sequencing libraries were constructed using the KAPA stranded RNA-Seq library preparation kit (Kapa Biosystems) following the manufacturer’s instructions and sequenced on an HiSeq2500. Reads were trimmed with Trimmomatic (v 0.36) and aligned to the F. tularensis LVS genome (NC_007880) using bowtie2 (v 2.2.6) (Langmead and Salzberg, 2012). HTSeq (v 0.6.1) (Anders et al., 2014) was used to count reads that map to annotated genes and DESeq2 (v 1.14.1) (Love et al., 2014) was used for differential gene expression analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
qRT-PCR
In Figure 1D, experiments were performed with biological triplicate samples at least twice and data from a representative experiment are shown. Statistical significance was assessed in Prism using a two-tailed t-test assuming equal variance.
LVS reporter strain ß-galactosidease assays
In Figure 5, experiments were performed with biological triplicate samples at least twice and data from a representative experiment are shown. Statistical significance was assessed in Prism using one-way ANOVA with Tukey’s multiple comparisons.
Two-hybrid ß-galactosidease assays
In Figure 1E, experiments were performed with biological triplicate samples at least twice and data from a representative experiment are shown. Statistical significance was assessed in Prism using one-way ANOVA with Tukey’s multiple comparisons.
RNA-Seq
Transcriptomes were analzyed using biological triplicate samples and DESeq2 was used for differential gene expression analysis. Genes are considered differentially expressed if transcript abundance changed 2-fold or greater in mutant cells compared to WT with an adjusted p-value of less than 0.05.
Supplementary Material
Table S2: New MglA regulon revealed by RNA-Seq studies, related to Figure 2.
Supplemental Movie S1: Overview of the FtRNAPσ70-(MglA-SspA)-iglA complex, related to Figure 1.
Supplemental Movie S2: Overview of the FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-iglA complex, related to Figure 4.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Digoxigenin-AP, Fab fragments | Millipore Sigma | Cat# 11093274910 |
| Bacterial and Virus Strains | ||
| E. coli C41(DE3) | Lucigen | Cat# 60442–1 |
| E. coli BL21(DE3) | NEB | Cat# C25271 |
| E. coli XL1 Blue | Agilent | Cat# 200130 |
| E. coli DH5a F′ Iq | NEB | Cat# C2992I |
| E. coli BL21-Gold(DE3)pLysS | Agilent | Cat#230134 |
| E. coli KDZif1ΔZ | Vallet-Gely et al., 2005 | N/A |
| F. tularensis subsp. holarctica LVS β′-TAP | Charity et al., 2007 | N/A |
| F. tularensis subsp. holarctica LVS MglA-TAP | Charity et al., 2007 | N/A |
| F. tularensis subsp. holarctica LVS ΔmglA β′ -TAP | Rohlfing and Dove, 2014 | N/A |
| LVS | Fortier et al., 1991 | N/A |
| LVS ΔmglA | Charity et al., 2007 | N/A |
| LVS ΔpigR | Charity et al., 2009 | N/A |
| LVS PiglA-lacZ | Ramsey et al., 2015 | N/A |
| LVS ΔpigR PiglA-lacZ | Ramsey et al., 2015 | N/A |
| LVS PiglA-M1-lacZ | Ramsey et al., 2015 | N/A |
| LVS ΔpigR PiglA-M1-lacZ | Ramsey et al., 2015 | N/A |
| LVS PiglA-M3-lacZ | This paper | N/A |
| LVS ΔpigR PiglA-M3-lacZ | This paper | N/A |
| LVS PiglA-UPM1-lacZ | This paper | N/A |
| LVS ΔpigR PiglA-UPM1-lacZ | This paper | N/A |
| LVS PFTL_0026-lacZ | This paper | N/A |
| LVS ΔpigR PFTL_0026-lacZ | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| C-terminal PigR Peptide: KRNVFSRCWINMNLYSVIKAKS | Genscript | N/A |
| Guanosine-3’,5-bisdiphosphate (ppGpp) | TriLink Biotechnologies | Cat# N-6001 |
| Sarkosyl | Sigma | Cat# L5125–50G |
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma | Cat# 10837091001 |
| Lysozyme | Worthington Biochem | Cat# LS002933 |
| Sodium deoxycholate | Alfa Aesar | Cat# B20759 |
| EDTA | Sigma | Cat# EDS-100G |
| 1,4-Dithiothreitol (DTT) | Thermo Fisher | Cat# BP172–25 |
| Polyethyleneimine (PEI) | Spectrum | Cat# 46AT97 |
| Polysorbate 20 (Tween 20) | Thermo Fisher | Cat# BT337–500 |
| CHAPSO | Sigma | Cat# C3649–1G |
| Difco Mueller Hinton Broth | BD Diagnostic Systems | Cat# 275730 |
| IsoVitaleX Enrichment | BD Diagnostic Systems | Cat# 211876 |
| Cystine Heart Agar | BD Diagnostic Systems | Cat# 247100 |
| Bovine Hemoglobin | VWR | Cat# 90000–410 |
| BugBuster 10X Protein Extraction Reagent | Millipore Sigma | Cat# 70921 |
| AcTEV™ Protease | Life Technologies | Cat# 12575015 |
| Rnase-free DNase I | Lucigen Corporation | Cat# D9905K |
| cOmplete Mini EDTA-free Protease Inhibitor Cocktail Tablets | Millipore Sigma | Cat# 11836170001 |
| Millipore Millex Sterile Syringe Filters: PES Membrane | Millipore Sigma | Cat# SLGP033RS |
| NP-40 Alternative | Millipore Sigma | Cat# 492018 |
| FastStart Essential DNA Green Master | Roche | Cat# 06402712001 |
| Superscript III Reverse Transcriptase | Life Technologies | Cat# 18080093 |
| RQ1 DNase, Promega | Fisher Scientific | Cat# PR-M6101 |
| CDP-Star | Millipore Sigma | Cat# 11759051001 |
| ULTRAhyb Ultrasensitive Hybridization Buffer | Life Technologies | Cat# AM8670 |
| Kanamycin A | Research Products International | Cat# K22000 |
| Carbenicillin disodium salt | VWR | Cat# IC19509205 |
| Tetracycline hydrochloride | Fisher Scientific | Cat# BP912–100 |
| O-nitrophenyl-β-D-galactoside | Millipore Sigma | Cat# N1127 |
| Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Millipore Sigma | Cat# 420322 |
| Ferric pyrophosphate | Millipore Sigma | Cat# P6526 |
| Critical Commercial Assays | ||
| Direct-zol Miniprep kit | Zymo Research | Cat# R2070 |
| Ribo-Zero Magnetic Kit (Bacteria) | Illumina | Cat# MRZB12424 |
| KAPA Stranded RNA-Seq Kit | Kapa Biosystems | Cat# KK8400 |
| KAPA Single-Indexed Adapter Kit | Kapa Biosystems | Cat# KK8710 |
| PCR DIG Probe Synthesis Kit | Millipore Sigma | Cat# 11636090910 |
| DIG Wash and Block Buffer Set | Millipore Sigma | Cat# 11585762001 |
| Deposited Data | ||
| Coordinates/StructureFactors for the Ft(MglA-SspA)-ppGpp-PigR crystal structure | This paper | PDB: 6WEG |
| Cryo-EM map of FtRNAPσ70-(MglA-SspA)-iglA | This paper | EMD-21851 |
| Cryo-EM map of FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-iglA | This paper | EMD-21852 |
| Cryo-EM map of EcRNAPσ70-(SspA)2-gadR | This paper | EMD-21853 |
| Cryo-EM map of FtRNAPσ70-iglA | This paper | EMD-21850 |
| FtRNAPσ70-(MglA-SspA)-iglA coordinates | This paper | PDB: 6WMR |
| FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-iglA coordinates | This paper | PDB: 6WMT |
| EcRNAPσ70-(SspA)2-gadR coordinates | This paper | PDB: 6WMU |
| FtRNAPσ70-iglA coordinates | This paper | PDB: 6WMP |
| RNA sequencing | This paper | GEO: GSE150932 |
| Oligonucleotides | ||
| iglA-1 (top): ATTTAGCTGTATAAACATTGTGTTATTGGCGTTATTAAGGTAACTTGCTTATA | IDT | N/A |
| iglA-1 (bottom): TATAAGTATAAGCAAGTTACCTTAATAACGCCAATAACACAATGTTTATACAGCTAAAT | IDI | N/A |
| iglA-2 (top): GTTTAAATTTAGCTGTATAAACATTGTGTTATTGGCGTTATTAAGGTAACTTGCTTATAAGGT | IDI | N/A |
| iglA-2 (bottom): ACCTTATAAGCAAGTTACCTTAATAACGCCAATAACACAATGTTTATACAGCTAAATTTAAAC | IDI | N/A |
| gadA (top): CTGTAATGCCTTGCTTCCATTGCGGATAAATCCTACTTTTTTATTGCCTTC | IDI | N/A |
| gadA (bottom): GAAGGCAATAAAAAAGTAGGATTTATCCGCAATGGAAGCAAGGCATTACAG | IDI | N/A |
| Recombinant DNA | ||
| pET-15b vector | Novagen | Cat# 69661–3 |
| pIA900 vector | Svetlov & Artsimovitch, 2015 | Addgene plasmid #104401 |
| LA4 vector | Borukhov and Goldfarb, 1993 | N/A |
| pMCSG21 vector | DNASU | N/A |
| pET15-b-PigR | Genscript | This paper |
| pET28-a-His6MBP-MglA | Cuthbert et al., 2015 | N/A |
| pMCSG21-SspA | Cuthbert et al., 2015 | N/A |
| pF | Rohlfing and Dove, 2014 | N/A |
| pF-MglA-V | Rohlfing and Dove, 2014 | N/A |
| pF-MglA(F78R+P80E)-V | This paper | N/A |
| pSP109 | This paper | N/A |
| pSP111 | This paper | N/A |
| pSP113 | This paper | N/A |
| pSP125 | This paper | N/A |
| pBR-MglA-ω | Rohlfing and Dove, 2014 | N/A |
| pACTR-SspA-Zif | Rohlfing and Dove, 2014 | N/A |
| pACTR-Ap-Zif | Rohlfing and Dove, 2014 | N/A |
| pBR-MglA(F78R+P80E)- ω | This paper | N/A |
| Software and Algorithms | ||
| Coot v0.9 | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| MotionCor2 v1.2.2 | Zheng et al., 2017 | https://emcore.ucsf.edu/ucsf-software |
| Phenix v1.17 | Afonine et al., 2012 | https://www.phenix-online.org/download/ |
| RELION-3.0 | Zivanov et al., 2018 | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/MainPage |
| UCSF Chimera v1.13.1 | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera |
| UCSF ChimeraX v0.91 | Goddard et al., 2018 | https://www.rbvi.ucsf.edu/chimerax/ |
| HT-Seq v0.6.1 | Anders et al., 2014 | https://github.com/simon-anders/htseq |
| Bowtie2 v2.2.6 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| DESeq2 v1.14.1 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GCTF v1.06 | Zhang et al., 2016 | https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/#gctf |
| cisTEM v1.0.0 | Grant et al., 2018 | https://cistem.org/ |
| crYOLO v1.5.6 | Wagner et al., 2019 | https://sphire.mpg.de/wiki/doku.php?id=pipeline:window:cryolo |
| Trimmomatic v 0.36 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| XDS | Kabsch, 2010 | http://xds.mpimf-heidelberg.mpg.de |
| 3DFSC | Tan et al., 2017 | https://3dfsc.salk.edu/ |
| Other | ||
| Ni-NTA agarose | ThermoFisher | Cat# R90115 |
| HiLoad 26/600 Superdex 200 pg | GE Healthcare | Cat# GE28-9893-36 |
| HiLoad 26/600 Superdex 75 pg | GE Healthcare | Cat# GE28-9893-34 |
| DE53 diethylaminoethyl cellulose | Whatman | Cat# 4058200 |
| Heparin Sepharose 6 Fast Flow | GE Healthcare | Cat# GE17-0998-01 |
| Quantifoil R1.2/1.3 Cu 300 holey carbon grids | EM Sciences | Cat# Q350-CR1.3 |
| UltrAufoil R1.2/1.3 Au 300 holey gold grids | EM Sciences | Cat# Q350AR1.3A |
| Calmodulin Affinity Resin, Agilent Technologies | Fisher Scientific | Cat# 50-125-262 |
| IgG Sepharose® 6 Fast Flow | Millipore Sigma | Cat# GE17-0969-01 |
| KAPA Pure Beads | Kapa Biosystems | Cat# KK8000 |
| Poly-Prep Chromatography Columns | Bio-Rad | Cat# 7311550 |
| LightCycler 96 Instrument | Roche | Cat# 05815916001 |
| Nylon Membrane, positively charged | Millipore Sigma | Cat# 11209299001 |
ACKNOWLEDGEMENTS
This research was supported by National Institutes of Health grants (R35GM130290 to M.A.S., R21AI146641 to M.A.S. and R.G.B, AI081693 and AI145954 to S.L.D., F31AI150138 to B.A.T. and HD055148-08 to K.M.R.). We acknowledge beamline 8.3.1 for X-ray diffraction data collection; Beamline 8.3.1 at the Advanced Light Source (ALS) is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives grant MR-15-328599, the National Institutes of Health (R01GM124149 and P30GM124169), Plexxikin Inc. and the Integrated Diffraction Analysis Technologies program of the US Department of Energy Office of Biological and Environmental Research. The ALS (Berkeley, CA) is a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the US Department of Energy under contract number DE-AC02-05CH11231, Office of Basic Energy Sciences. We also acknowledge the Pacific Northwest Center for cryo-EM (PNCC). A portion of this research was supported by NIH grant U24GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research. Cryo-EM structural work was also performed at the Duke cryo-EM facility and the Harvard cryo-EM Center for Structural Biology at Harvard Medical School. The work at Duke was performed in part at the Duke University Shared Materials Instrumentation Facility (SMIF), a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), which is supported by the National Science Foundation (Grant ECCS-2025064) as part of the National Nanotechnology Coordinated Infrastructure (NNCI). We would like to thank Drs. Richard Gourse and Wilma Ross for the kind gifts of the EcRNAP and Ecσ70 expression constructs. We also thank Renate Hellmiss for artwork in Figure 2A–B and Figure 5 and Michael Gebhardt for helping with statistical analyses in Figure 1D.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Afonine PW, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2014). HTSeq--a Python framework to work with high throughput sequencing data. Bioinformatics 31,166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony LC, Foley KM, Thompson NE, and Burgess RR (2003). Expression, purification of, and monoclonal antibodies to sigma factors from Escherichia coli. Methods Enzymol. 370, 181–192. [DOI] [PubMed] [Google Scholar]
- Bae B, Feklistov A, Lass-Napiorkowska A, Landick R, and Darst SA (2015). Structure of a bacterial RNA polymerase open promoter complex. eLife 4, e08504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badger JL, and Miller VL (1998). Expression of invasion and motility are coordinately regulated in Yersnia enterocolitica. J. Bacteriol 180, 793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker JR, and Klose KE (2007). Molecular and genetic basis of pathogenesis in Ann. N.Y. Acad. Sci 1105, 138–159. [DOI] [PubMed] [Google Scholar]
- Barker JR, Chong A, Wehrly TD, Yu JJ, Rodriguez SA, Liu J, Celli J, Arulanandam BP, and Klose KE, (2009). The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol. Microbiol 74, 1459–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barne KA, Brown JA, Busby SJ, and Minchin SD (1997). Region 2.5 of the Escherichia coli RNAP polymerase sigma70 subunit is responsible for recognition of the ‘extended −10’ motif at promoters. EMBO J. 16, 4034–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron GS, and Nano FE (2002). MglA and MglB are required for intramacrophage growth of Francisella novicida. Microbiology 29, 247–259. [DOI] [PubMed] [Google Scholar]
- Benoff B, Yang H, Lawson CL, Parkinson G, Liu J, Blatter E, Berman HM, and Ebright RH (2002). Structural basis of transcription activation: the CAP-alpha CTD-DNA complex. Science 297, 1562–1566. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borukhov S, and Goldfarb A (1993). Recombinant Escherichia coli RNA polymerase: purification of individually overexpressed subunits and in vitro assembly. Protein Expr. Purif 4, 503–511. [DOI] [PubMed] [Google Scholar]
- Bröms JE, Sjöstedt A, and Lavander M (2010). The role of the Francisella tularensis Pathogenicity Island in type VI secretion, intracellular survival, and modulation of host cell signaling. Front. Microbiol 1, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotcke A, Weiss DS, Kim CC, Chain P, Malfatti S, Garcia E, and Monack DM (2006). Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect. Immun 74, 6642–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotcke A, and Monack DM (2008). Identification of fevR, a novel regulator of virulence gene expression in Francisella novicida. Infect. Immun 76, 3473–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NL, Stoyanov JV, Kidd SP, and Hobman JL (2003). The MerR family of transcriptional regulators. FEMS Microbiol. Rev 27, 145–163. [DOI] [PubMed] [Google Scholar]
- Browning DF, and Busby SJW (2016). Local and global regulation of transcription initiation in bacteria. Nature Rev. Microbiol 14, 638–650. [DOI] [PubMed] [Google Scholar]
- Campbell EA, Muzzin O, Chlenov M, Sun JL, Olson CA, Weinman O, Trester-Zedlitz ML, and Darst SA (2002). Structure of the bacterial RNA polymerase promoter specific σ subunit. Mol. Cell 9, 537–539. [DOI] [PubMed] [Google Scholar]
- Cartagena AJ, Banta AB, Sathyan N, Ross W, Gourse RL, Campbell EA, and Darst SA (2019). Structural basis for transcription activation by Crl through tethering of σs. Proc. Natl. Acad. Sci. U.S.A 116, 18923–18927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charity JC, Blalock LT, Costante-Hamm MM, Kasper DL, and Dove SL (2009). Small molecule control of virulence gene expression in Francisella tularensis. PLoS Pathog. 5, e1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charity JC, Costance-Hamm MM, Balon EL, Boyd DH, Rubin EJ, and Dove SL (2007). Twin RNA polymerase-associated proteins control virulence gene expression in Francisella tularensis. PLoS Pathog. 3, e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert BJ, Brennan RG, and Schumacher MA (2015). Structural and biochemical characterization of the Francisella tularensis pathogenicity regulator, Macrophage Locus Protein A (MglA). PLoS One 10, e0128225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert BJ, Ross W, Rohlfing AE, Dove SL, Gourse RL, Brennan RG, and Schumacher MA (2017). Dissection of the molecular circuitry controlling virulence in Francisella tularensis. Genes Dev. 31,1549–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Freidlander AM, Hauer J, Layton M et al. (2001). Tularemia as a biological weapon: medical and public health management. JAMA 285, 2763–2773. [DOI] [PubMed] [Google Scholar]
- De Reuse H, and Taha MK (1997). RegF, an SspA homologue, regulates the expression of the Neisseria gonorrhoeae pilE gene. Res. Microbiol 148, 289–303. [DOI] [PubMed] [Google Scholar]
- Eliasson H, and Bäck E (2007). Tularemia in an emergent area in Sweden: an analysis of 234 cases in five years. Scand. J. Infect. Dis 39, 880–889. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshraghi A, Kim J, Walls AC, Ledvina HE, Miller CN, Ramsey KM, Whitney JC, Radey MC, Peterson SB, Ruhland BR, et al. (2016). Secreted effectors encoded within and outside of the Francisella pathogenicity island promote intramacrophage growth. Cell Host Microbe 20, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrem ST, Gaal T, Ross W, and Gourse RL (1998). Identification of an UP element consensus sequence for bacterial promoters. Proc. Natl. Acad. Sci. U.S.A 95, 9761–9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber M, Heuner K, Jacob D, and Grunow R (2018). Tularemia in Germany: a re-emerging zoonosis. Front. Cell Infect. Microbiol 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feklistov A, Sharon BD, Darst SA, and Gross CA (2014). Bacterial sigma factors: a historical, structural and genomic perspective. Annu. Rev. Microbiol 68, 357–376 [DOI] [PubMed] [Google Scholar]
- Feng Y, Zhang Y, and Ebright RH (2016). Structural basis of transcription activation. Science 252, 1330–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortier AH, Slayter MV, Ziemba R, Meltzer MS, and Nacy CA (1991). Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect. Immun 59, 2922–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestin B, Valade E, Thibault F, Schneider D, and Maurin M (2010). Phenotypic and genetic characterization of macrolide resistance in Francisella tularensis subsp. holarctica biovar I. J. Antimicrob. Chemother 67, 2359–2367. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Rohou A, and Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. Elife 7,10.7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AM, Gu Y, Li M, Andrykovitch M, Waugh DS, Jin DJ, and Ji X (2005). Structural basis for the function of stringent starvation proteins A as a transcription factor. J. Biol. Chem 280, 17380–1791. [DOI] [PubMed] [Google Scholar]
- Hansen AM, Qiu Y, Yeh N, Blattner FR, Durfee T, and Jin DJ (2005). SspA is required for acid resistance in stationary phase by downregulation of H-NS in Escherichia coli. Mol. Microbiol 56, 719–734. [DOI] [PubMed] [Google Scholar]
- Hansen AM, Lehnherr H Wang X, Mobely V, and Jin DJ (2003). Escherichia coli SspA is a transcription activator for bacteriophage P1 late genes. Mol. Microbiol 48, 1621–1631. [DOI] [PubMed] [Google Scholar]
- Heldwein EE, and Brennan RG (2001). Crystal structure of the transcription activator BmrR bound to DNA and a drug. Nature 409, 378–382. [DOI] [PubMed] [Google Scholar]
- Ishihama A, and Saitoh T (1979). Subunits of RNA polymerase in function and structure. IX. Regulation of RNA polymerase activity by stringent starvation protein (SSP). J. Mol. Biol 129, 517–530. [DOI] [PubMed] [Google Scholar]
- Kabsch W (2010). Integration, scaling, space-group assignment and post-refinement. Acta. Cryst. D 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JY, Olinares PDB, Chen J, Campbell EA, Mustaev A, Chait BT, Gottesman ME, and Darst SA (2017). Structural basis of transcription arrest by coliphage HK022 Nun in an Escherichia coli RNA polymerase elongation complex. Elife 6, 10.7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanjee U, Ogata K, and Houry WA (2012). Direct binding targets of the stringent response alarmone (p)ppGpp. Mol. Microbiol 85, 1029–1043. [DOI] [PubMed] [Google Scholar]
- Kazmierczak MJ, Wiedmann M, and Boor KJ (2005). Alternative sigma factors and their roles in bacterial virulence. Microbiol. Mol. Biol. Rev 69, 527–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasny L, and Gourse RL (2004). An alternative strategy for bacterial ribosome synthesis: Bacillus subtilis rRNA transcription regulation. EMBO J. 23, 4473–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Malloch RA, Fujita N, Smillie DA, Ishihama A, and Hayward RS (1993). The minus 35-recognition region of Escherichia coli sigma 70 is inessential for initation of transcription at “extended minus 10” promoter. J. Mol. Biol 232, 406–418. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson P, Oyston PC, Chain P, Chu MC, Duffield M, Fuxelius HH, Garcia E, Hälltorp G, Johansson D, Isherwood KE, et al. (2005). The complete genome sequence of Francisella tularensis, the causative agent of tularemia. Nat. Genet 37,153–159. [DOI] [PubMed] [Google Scholar]
- Lauriano CM, Barker JR, Yoon SS, Nano FE, Arulanandam BP, Hassett DJ, and Klose KE (2004). MglA regulates transcription of virulence factors necessary for Francisella tularensis intraamoebae and intramacrophage survival. Proc. Natl. Acad. Sci. U.S.A 101, 4246–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DJ, Minchin SD, and Busby SJW (2012). Activating transcription in bacteria. Annu. Rev. Microbiol 66, 125–152. [DOI] [PubMed] [Google Scholar]
- Liu K, Bittner AN, And Wang JD (2015). Diversity in (p)ppGpp metabolism and effectors. Curr. Opin. Microbiol 24, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real Time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveless BM, Yermakova A, Christensen DR, Kondig JP, Heine HS 3rd, Wasieloski LP, and Kulesh DA (2010). Identification of ciprofloxacin resistance by SimpleProbe, high resolution melt and pyrosequencing nucleic acid analysis in biothreat agents: Bacillus anthracis, Yersinia pestis and Francisella tularensis. Mol. Cell Probes 24, 154–160. [DOI] [PubMed] [Google Scholar]
- Maurin M (2015). Francisella tularensis as a potential agent of bioterrorism? Expert Rev. of Anti-infective Therapy 13,141–144. [DOI] [PubMed] [Google Scholar]
- McGuffie RA, Vallet-Gely T, and Dove S (2015). σ factor and anti-σ factor that control swarming and biofilm formation in Pseudomonas aeruginosa. J. Bacteriol 30, 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhamedyarov D, Makarova KS, Severinov K, and Kuznedelov K (2011). Francisella RNA polymerase contains a heterodimer of non-identical α subunits. BMC Mol. Biol 12, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumm JP, Friedman LJ, and Gelles J (2020). Mechanism of upstream promoter element stimulation of transcription at a ribosomal RNA promoter determined by single molecule imaging. BioRxiv. [Google Scholar]
- Murakami KS (2015). Structural biology of bacterial RNA polymerases. Biomolecules 5, 848–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami KS, and Darst SA (2003). Bacterial RNA polymerases: the wholo story. Curr. Opin. Struct. Biol 13,31–39. [DOI] [PubMed] [Google Scholar]
- Najmanová L, Janata J, Kalousek F, Novák P, Felsberg J, and Spížek J (2000). Two forms of DNA-dependent RNA polymerase a subunit in streptomyces. FEMS Microbiol. 187, 9–14. [DOI] [PubMed] [Google Scholar]
- Nano FE, Zhang N, Cowley SC, Klose KE, Cheung KK, Roberts MJ, Ludu JS, Letendre GW, Meierovics AI, Stephens G et al. (2004). A Francisella tularensis pathogenicity island required for intramacrophage growth. J. Bacteriol 186, 6430–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan A, Vago FS, Li K, Qayyum MZ, Yernool D, Jiang W, and Murakami KS (2018). Cryo-EM structure of Escherichia coli σ70 RNA polymerase promoter DNA complex revealed a role of the σ non-conserved region during the open complex formation. J. Biol. Chem 293, 7367–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlands JT, Ross W, Gosnick KK, and Gourse RL (1991). Factor-independent Activation of Escherichia coli rRNA Transcription II. Characterization of Complexes of rrnB P1 Promoters Containing or Lacking the Upstream Activator Region with Escherichia coli RNA polymerase. J. Mol. Biol 220, 569–583. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Ramsey KM, Osborne ML, Vvedenskaya IO, Su C, Nickels BE, and Dove SL (2015). Ubiquitous promoter-localization of essential virulence regulators in Francisella tularensis. PLoS Pathog. 11, e1004793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, and Dove SL (2016). A response regulator promotes Francisella tularensis intramacrophage growth by repressing an anti-virulence factor. Mol. Microbiol 101, 688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao L, Ross W, Appleman JA, Gaal T, Leirmo S, Schlax PJ, Record MT Jr., and Gourse RL (1994). Factor independent activation of rrnB P1. An “extended” promoter with an upstream element that dramatically increases promoter strength. J. Mol. Biol 235, 1421–1435. [DOI] [PubMed] [Google Scholar]
- Rohlfing AE, and Dove SL (2014). Coordinate control of virulence gene expression in Francisella tularensis involves direct interaction between key regulators. J. Bacteriol 196, 3516–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W, Gosnick KK, Salomon J, Igarashi K, Zou C, Ishihama A, Severinov K, and Gourse RL (1993). A third recognition element in bacterial promoters: DNA binding by the Alpha subunit of RNA polymerase. Science 262, 1407–1413. [DOI] [PubMed] [Google Scholar]
- Ross W, Schneider DA, Paul BJ, Mertens A, and Gourse RL (2003). An intersubunit contact stimulating transcription initiation by E. coli RNA polymerase: interaction of the alpha C-terminal domain and sigma region 4. Genes Dev. 17, 1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W, and Gourse RL (2009). Analysis of RNA polymerase-promoter complex formation. Methods 245,197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W, Vrentas CW, Sanchez-Vazquez P, Gaal T, and Gourse RL (2013). The magic spot: a ppGpp binding site responsible for regulation of transcription initiation. Mol. Cell 50, 420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W Sanchez-Vazquez P, Chen AY, Lee JH, Burgos HL, and Gourse RL (2016). ppGpp binding to a site at the RNAP-DksA interface accounts for its dramatic effects on transcription initiation during the stringent response. Mol. Cell 62, 811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AB, Wexler AG, Harding BN, Whitney JC, Bohn AJ, Goo YA, Tran BQ, Barry NA, Zheng H, Peterson SB, et al. (2014). A type VI secretion-related pathway in Bacteroidetes mediates interbacterial antagonism. Cell Host Microbe 16, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saecker RM, Record MT Jr., and Dehaseth PL (2011). Mechanism of bacterial transcription initiation: RNA polymerase-promoter binding, isomerization to initiation competent open complexes and initiation of RNA synthesis. J. Mol. Biol 412, 754–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroll AM, Theurl I, Gerogi E, Zange S, Rettenbacher T, Bellmann-Weiler R, and Weiss G (2018). Newly emerging ulceroglandular tularemia in Western Austria. Tick Borne Dis. 9, 1331–1333. [DOI] [PubMed] [Google Scholar]
- Sutera V Levert M, Burmeister WP, Schneider D, and Maurin M (2013). Evolution towards high-level fluoroquinoline resistance in Francisella tularensis. J. Antimicrob. Chemother 69,101–110. [DOI] [PubMed] [Google Scholar]
- Svetlov V, and Artsimovitch I (2015). Purification of bacterial RNA polymerase: tools and protocols. Methods Mol. Biol 1276, 13–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan YZ, Baldwin PR, Davis JH, Williamson JR, Potter CS, Carragher B, and Lyumkis D (2017). Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T, Merio F, Stabrin M, Moriya T, Antoni C, Apelbaum A, Hagel P, Sitsel O, Raisch T, Prumbaum D et al. (2019). SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol 2, 10.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Dai P, Ding D, Del Rosario A, Grant RA, Pentelute BL, and Laub MT (2019). Affinity-based capture and identification of protein effectors of the growth regulator, ppGpp. Nat. Chem. Biol 15, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss DS, Brotcke A, Henry T, Margolis JJ, Chan K, and Monack DM (2007). In vivo negative selection screen identifies genes required for Francisella virulence. Proc. Natl. Acad. Sci. U.S.A 104, 6037–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MD, Quyang TX, and Flickinger MC (1994). Glutathione S-transferase sspA Fusion binds to E. coli RNA polymerase and complements delta sspA mutation allowing phage P1 replication. Biochem. Biophys. Res. Commun 201, 123–127. [DOI] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zborníková E, Rejman D, and Gerdes K (2017). Novel (p)pp binding and metabolizing proteins of Escherichia coli. mBio 9, e02188–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJH, Lindahl E, and Scheres SHW (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, 10.7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Wang Y, and Steitz TA (2013). The structure of magic spot bound to RNA polymerase suggests how it is regulated by ppGpp. Mol. Cell 50, 430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2: New MglA regulon revealed by RNA-Seq studies, related to Figure 2.
Supplemental Movie S1: Overview of the FtRNAPσ70-(MglA-SspA)-iglA complex, related to Figure 1.
Supplemental Movie S2: Overview of the FtRNAPσ70-(MglA-SspA)-ppGpp-PigR-iglA complex, related to Figure 4.
Data Availability Statement
The (MglA-SspA)-ppGpp-PigR peptide crystal structure coordinates and structure factors have been deposited in the Protein Data Bank under the accession number, 6WEG. Cryo-EM density maps have been deposited in the Protein Data Bank with the accession numbers 6WMR, 6WMP, 6WMU and 6WMT and EMD with accession codes EMD-21851, EMD-21850, EMD-21853 and EMD-21852. RNA-Seq data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) under accession number GSE150932.