

SUMMARY
Low-grade mitochondrial stress can promote health and longevity, a phenomenon termed mitohormesis. Here, we demonstrate the opposing metabolic effects of low-level and high-level mitochondrial ribosomal (mitoribosomal) stress in hypothalamic proopiomelanocortin (POMC) neurons. POMC neuron-specific severe mitoribosomal stress due to Crif1 homodeficiency causes obesity in mice. By contrast, mild mitoribosomal stress caused by Crif1 heterodeficiency in POMC neurons leads to high-turnover metabolism and resistance to obesity. These metabolic benefits are mediated by enhanced thermogenesis and mitochondrial unfolded protein responses (UPRmt) in distal adipose tissues. In POMC neurons, partial Crif1 deficiency increases the expression of β-endorphin (β-END) and mitochondrial DNA-encoded peptide MOTS-c. Central administration of MOTS-c or β-END recapitulates the adipose phenotype of Crif1 heterodeficient mice, suggesting these factors as potential mediators. Consistently, regular running exercise at moderate intensity stimulates hypothalamic MOTS-c/β-END expression and induces adipose tissue UPRmt and thermogenesis. Our findings indicate that POMC neuronal mitohormesis may underlie exercise-induced high-turnover metabolism.
Keywords: mitochondria, stress, ribosome, hypothalamus, metabolism, thermogenesis, exercise, proopiomelanocortin, adipose, obesity, Crif1, MOTS-c, β-endorphin, mitohormesis
Graphical Abstract
eTOC blurb
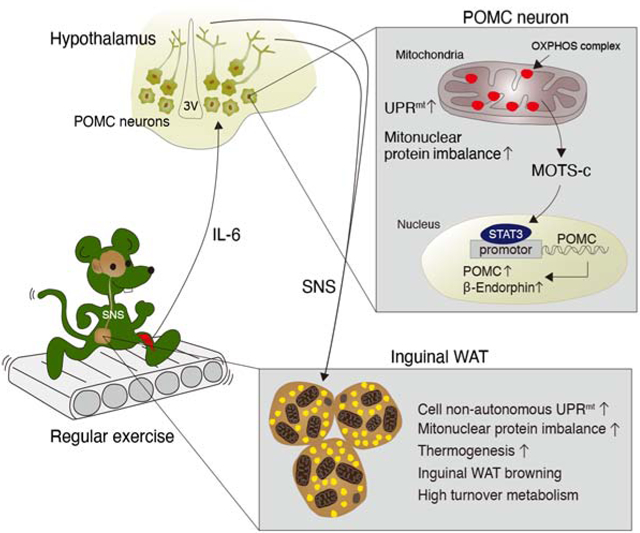
Kang et al. demonstrate that high-level mitochondrial stress in POMC-producing neurons causes severe obesity. In contrast, low-level mitochondrial stress in the same neurons enhances thermogenesis in the adipose tissue and protects against obesity via interorgan mitochondrial stress responses between the brain and adipose tissue.
INTRUDUCTION
Mitochondria are multifunctional organelles that serve as a major energy production site and communication hub for adaptive responses. Mitochondrial dysfunction can disrupt insulin signaling, in part, through the generation of reactive oxygen species (ROS) and accumulation of toxic lipids, which potentially predispose one to type 2 diabetes mellitus. However, recent evidence indicates that low-levels of mitochondrial stress may be cytoprotective rather than harmful (Ristow and Zarse, 2010; Yun and Finkel, 2014). Repeated exposure to low-levels of mitochondrial stress induces various cytosolic and nuclear responses that build resilience against higher levels of stress. This adaptive response, largely known as mitohormesis, has been shown to extend healthspan and/or lifespan in several model organisms (Yun and Finkel, 2014).
The mitochondrial unfolded protein response (UPRmt) is an adaptive stress response that promotes survival and recovery from various mitochondrial insults (Mottis et al., 2019; Shpilka and Haynes, 2018). Unfolded or misfolded proteins that accumulate inside mitochondria are degraded by mitochondrial proteases and exported to the cytosol (Haynes et al., 2010; Yun and Finkel, 2014). Further, mitochondrial-derived signals communicate mitochondrial proteotoxic stress to the nucleus to induce an adaptive response to resolve the insult. Although UPRmt has been most well studied in C. elegans, its mechanisms are increasingly being identified in vertebrates, especially in the context of the integrated stress response (ISR) that regulates an adaptive translational program (Anderson and Haynes, 2020; Costa-Mattioli and Walter, 2020; Mottis et al., 2019).
Mitochondria have their own ribosomes (mitoribosomes), which are required to synthesize the 13 mitochondrial DNA (mtDNA)-encoded electron transport chain (ETC) proteins (Sharma et al., 2003). Mitochondrial ETC complexes, except for complex-II, are composed of proteins encoded by nuclear DNA (nDNA) and mtDNA. Hence, appropriate stoichiometric ratios between nDNA- and mtDNA-encoded ETC proteins are critical for mitochondrial respiration (Houtkooper et al., 2013). Deficiencies in nuclear-encoded mitoribosomal proteins causes mitonuclear protein imbalance, which in turn activates the UPRmt in C. elegans (Houtkooper et al., 2013) and yeast (Suhm et al., 2018). Notably, in addition to acting cell-autonomously, UPRmt can signal cell-non-autonomously to neighboring or distal tissues (Durieux et al., 2011). For example, in C. elegans, knocking down ETC components in the nervous system can trigger UPRmt responses in the intestine and extend life span (Durieux et al., 2011).
Mitochondrial communication is key to adaptive stress response following mitochondrial perturbation. Recently, small functional peptides that are encoded within the mtDNA, known as mitochondrial-derived peptides (MDPs), have been identified (Reynolds et al., 2020). MDPs represent a unique class of mitochondrial-encoded signaling factors that respond to mitochondrial stress and promote health (Galluzzi et al., 2018; Mottis et al., 2019; Quiros et al., 2016; Tan and Finkel, 2020). Notably, MOTS-c (mitochondrial open-reading-frame of the 12S rRNA-c) directly mediates mitonuclear communication by translocating to the nucleus upon metabolic stress and regulating adaptive nuclear gene expression to promote cellular homeostasis (Kim et al., 2018). In mice, MOTS-c improves insulin resistance and glucose intolerance induced by consumption of a high fat diet (HFD), aging, and ovariectomy (Lee et al., 2015; Lu et al., 2019b) and restores physical capacity in aged mice (Reynolds et al., 2019)
The hypothalamus is a pivotal governing center for various metabolic processes in the body (Roh et al., 2016; Waterson and Horvath, 2015). Proopiomelanocortin (POMC)-producing neurons in the hypothalamic arcuate nucleus (ARH) have key roles in the regulation of energy balance. POMC neuronal activity is strongly tied to mitochondria. In mice, POMC neuronal activity can be upregulated by mitochondrial-derived ROS (Diano et al., 2011). Moreover, mitochondrial dynamics (i.e. fusion and fission) in POMC neurons are essential for maintaining whole-body energy and glucose homeostasis under altered metabolic conditions (Ramirez et al., 2017; Santoro et al., 2017). Despite the evident importance of mitochondria-originated signals in POMC neurons (Mishra et al., 2014), the details of the underlying mechanisms remain largely unknown.
In our current study, we investigated the metabolic impact of mitoribosomal stress in hypothalamic POMC neurons by depleting mitoribosomal protein CR6-interacting factor 1 (Crif1). Our study shows that POMC neurons exhibit a dimorphic response to mitoribosomal stress in a dose-dependent manner; homozygous deletion of Crif1 was detrimental (i.e. severe stress), whereas heterozygous disruption was beneficial (i.e. mild stress). We found that low-levels of mitoribosomal stress in POMC neurons induced high metabolic turnover and resistance to obesity through cell-non-autonomous mitochondrial stress signaling between the hypothalamus and adipose tissues.
RESULTS
Severe mitoribosomal stress in POMC neurons causes maturity-onset obesity
CRIF1 is a mitoribosomal protein that coordinates the insertion of mitochondrial-encoded proteins into the ETC complexes (Kim et al., 2012). To study mitoribosomal stress in POMC neurons, we generated mice lacking Crif1 in POMC neurons using the Cre-loxP system. FACS sorting of POMC-eGFP cells and real time PCR analysis confirmed successful Crif1 depletion in the hypothalamic POMC neurons, but not in non-POMC cells (Figures S1A and S1B). We compared the metabolic phenotype of Pomc-Cre; Crif1f/f mice to the Crif1f/f littermates (i.e. control group) as Crif1f/f and Pomc-Cre mice were metabolically indistinguishable (Figure S1C). Pomc-Cre; Crif1f/f mice were born and developed normally until around 10 weeks of age when they started to show accelerated weight gain under chow diet (CD)-fed conditions (Figure 1A). By 25 weeks, all Pomc-Cre; Crif1f/f mice gained considerably more weight, but the effect was more pronounced in females compared to males (Figure 1A). Body composition analysis revealed that both male and female Pomc-Cre; Crif1f/f mice had normal lean mass at 8 and 14 weeks of age, but increased fat mass at 14 weeks, but not 8 weeks, of age (Figures 1B and 1C). These findings suggest that Crif1 homodeficiency in POMC neurons leads to maturity-onset obesity in both males and females.
Figure 1. Severe mitoribosomal stress in POMC neurons causes maturity-onset obesity and glucose intolerance.
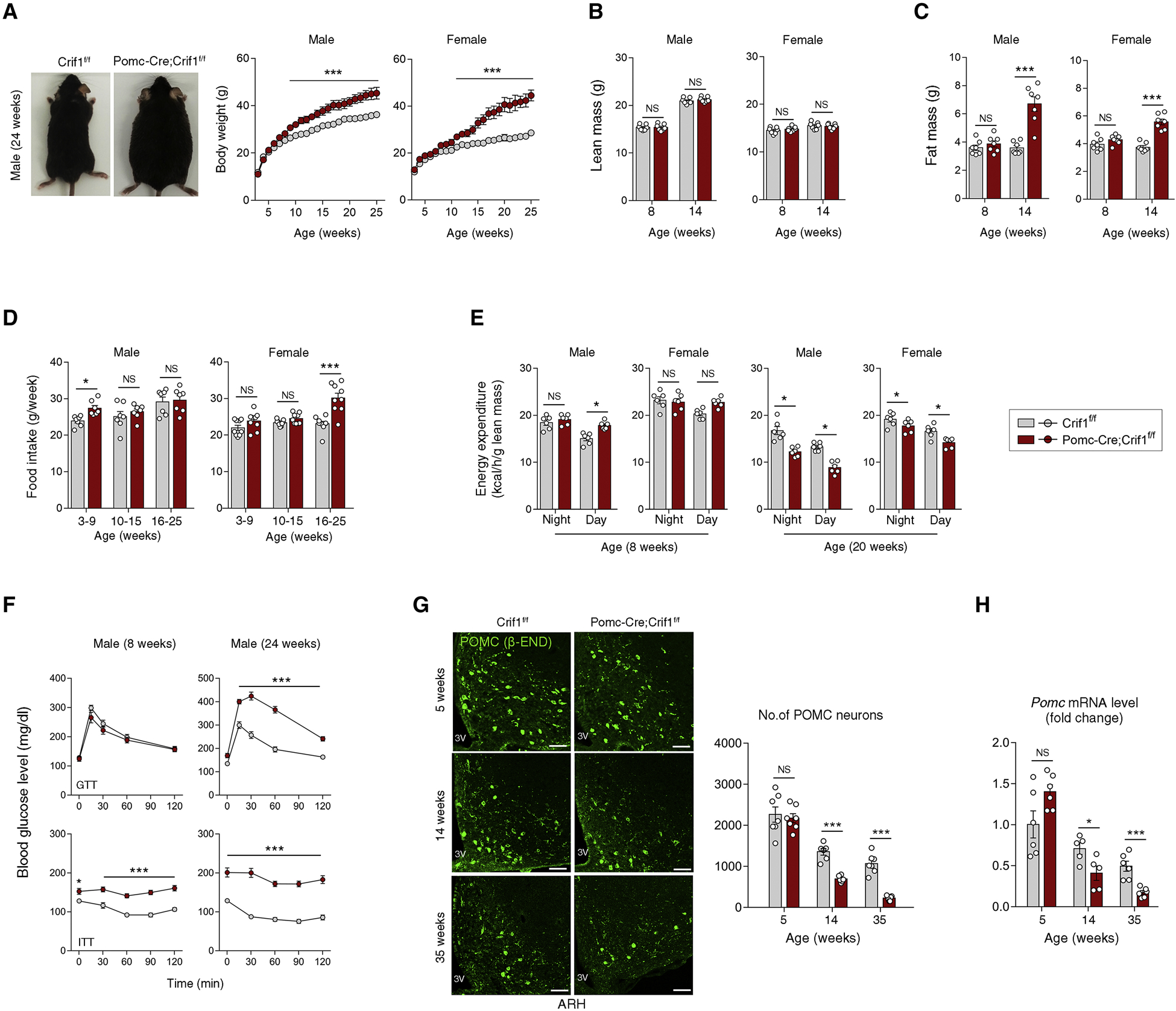
(A–C) Representative body images, body weights, lean mass, and fat mass in Crif1f/f and Pomc-Cre; Crif1f/f mice at the indicated ages (n = 7–8).
(D, E) Food intakes and energy expenditure measured at the indicated ages (n = 6–9).
(F) Results of glucose tolerance tests (GTT) and insulin tolerance tests (ITT) (n = 5–8).
(G, H) Immunohistochemistry of POMC neurons using β-endorphin (β-END) antibody and hypothalamic Pomc mRNA expression measurement at 5, 14 and 35 weeks of age (n = 6–7). 3V, third ventricle; ARH, hypothalamic arcuate nucleus. Scale bars, 50 μm.
Results are presented as a mean ± SEM. *p < 0.05, and ***p < 0.001 vs. Crif1f/f littermates. NS, not significant. See also Figure S1
Monitoring of food intake revealed that Pomc-Cre; Crif1f/f males consumed more food before 10 weeks of age but showed normal food intake thereafter (Figure 1D), whereas females showed delayed-onset hyperphagia starting after 15 weeks of age. Energy expenditure (EE) was significantly higher in 8-week old male, but not female, Pomc-Cre; Crif1f/f mice (Figure 1E). However, at 20 weeks of age, both sexes exhibited significantly reduced EE (Figure 1E).
Glucose and insulin tolerance tests showed that male Pomc-Cre; Crif1f/f mice were insulin resistant by 8 weeks, but also developed hyperglycemia by 24 weeks of age (Figures 1F and S1D). Similarly, female Pomc-Cre; Crif1f/f mice were also insulin resistant and hyperglycemic at 15 weeks of age (Figure S1E). Hence, hypothalamic POMC neuron-specific Crif1 homodeficiency leads to glucose intolerance resembling features of human type 2 diabetes.
In our previous study, Crif1 deficiency in CaMKII-expressing neurons caused neuronal death in the hippocampus, leading to locomotor disability and early death prior to 24 weeks of age (Kim et al., 2012). Here, we show that, by 14 weeks of age, Crif1 deficiency in POMC neurons (Pomc-Cre; Crif1f/f male mice) significantly reduced (i) the number of POMC neurons in the hypothalamic ARH as determined by β-endorphin (β-END) staining (Figure 1G), (ii) the level of Pomc transcripts (Figure 1H), and (iii) POMC neuronal activity as determined by c-Fos expression (Figure S1F). These findings suggest that Crif1 homodeficiency in POMC neurons reduces Pomc expression and neuronal activity after ~10 weeks of age and accelerates the loss of POMC neurons, resulting in severe obesity.
Mild mitoribosomal stress in POMC neurons promotes a high-turnover metabolism and resistance to diet-induced obesity
We next assessed the metabolic phenotype of mice with POMC-specific Crif1 heterodeficiency. In the CD-fed condition, Pomc-Cre; Crif1f/+ males and females showed no alteration in body weight, lean mass, nor fat mass (Figures 2A–2C). Yet, young male Pomc-Cre; Crif1f/+ mice exhibited higher food intake, night-time EE, and overall locomotor activity compared to Crif1f/f littermates (Figures 2D–2F) while maintaining normal body weight/composition (Figures 2A–2C), glucose tolerance, and insulin sensitivity (Figure S2A). Because POMC neurons control adipose tissue thermogenesis (Dodd et al., 2015), we assessed the thermogenic capacity of Pomc-Cre; Crif1f/+ mice and found that they held rectal temperature much better during a 3-hour cold (4°C) challenge (Figure 2G). Infrared thermal imaging also displayed higher skin temperature in these mice (Figure 2H).
Figure 2. Mild mitoribosomal stress in POMC neurons promotes a high-turnover metabolism and resistance to diet-induced obesity.
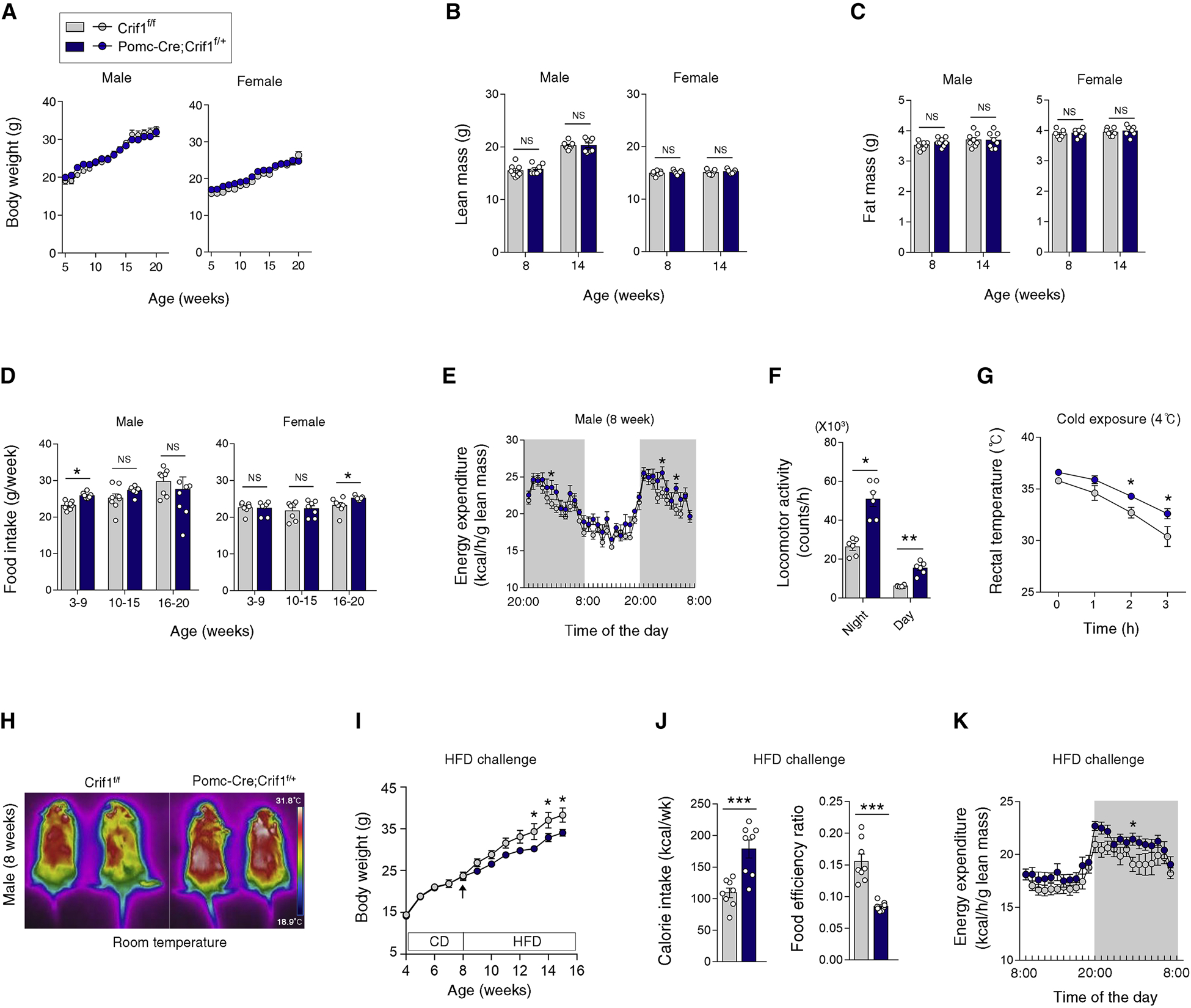
(A–C) Body weights, lean mass and fat mass of Crif1f/f and Pomc-Cre; Crif1f/+ mice, determined at the indicated ages (n = 6–8).
(D) Food intakes in the mice at the indicated ages (n = 7–8).
(E, F) Energy expenditure and locomotor activity in Crif1f/f and Pomc-Cre; Crif1f/+ males at 8 weeks of age (n = 6).
(G, H) Rectal temperatures during 4°C cold exposure and skin temperature in young Crif1f/f and Pomc-Cre; Crif1f/+ males (n = 5–8).
(I–K) Comparison of body weights, food intake, and energy expenditure during a high fat diet (HFD, 60% fat) challenge between Pomc-Cre; Crif1f/+ males and Crif1f/f controls (n = 7–8).
Results are presented as a mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 vs. Crif1f/f controls. NS, not significant. See also Figure S2
We metabolically challenged Pomc-Cre; Crif1f/+ males by feeding them a 60% high fat diet (HFD) starting at 8 weeks of age. Pomc-Cre; Crif1f/+ mice on a HFD gained significantly less weight compared to Crif1f/f controls despite consuming more calories, indicating a reduction in food efficiency (Figures 2I and 2J). Pomc-Cre; Crif1f/+ mice had higher EE during the night (Figure 2K), which may have contributed to the resistance to diet-induced obesity.
Gene deletion using Pomc-Cre transgenic mice starts during the mid-embryonic period (Padilla et al., 2010). Moreover, during embryogenesis, POMC is transiently expressed in immature hypothalamic neurons, some of which adopt a non-POMC fate later in life (Padilla et al., 2010). To avoid Crif1 deletion in non-POMC neurons and the developmental defects caused by embryonic gene deletion, we induced Crif1 depletion specifically in POMC neurons during young adulthood by injecting tamoxifen in 7 week-old Pomc-ERT2-Cre; Crif1f/+ mice. Notably, Pomc-ERT2-Cre; Crif1f/+ mice recapitulated the metabolic phenotype of the Pomc-Cre; Crif1f/+ mice (Figures S2B–S2F). Thus, the induction of POMC-specific partial Crif1 deficiency in young adult mice also caused high turnover metabolism with enhanced thermogenesis.
Mild mitoribosomal stress in POMC neurons activates a thermogenic program and UPRmt in adipose tissues
To further decipher the mechanisms underlying increased EE and thermogenesis in the Pomc-Cre; Crif1f/+ mice, we first histologically examined brown adipose tissue (BAT), the primary site of non-shivering thermogenesis in rodents. Hematoxylin and eosin staining revealed increased eosin staining and smaller fat droplets in the Pomc-Cre; Crif1f/+ mice (Figure 3A), indicating increased mitochondrial mass and enhanced lipolysis and/or lipid oxidation. Indeed, electron micrographs (EM) of Pomc-Cre; Crif1f/+ BAT showed increased mitochondrial density (number) and coverage (Figure 3B), suggesting increased mitochondrial biogenesis. Consistently, mtDNA/nDNA ratio was increased in BAT, reflecting increased mitochondrial mass (Figure S3A). The aspect ratio (AR), an index of mitochondrial elongation, was also increased (Figure 3B), consistent with increases in mitochondrial fusion and OXPHOS (Mishra et al., 2014). A histological examination of inguinal white adipose tissue (iWAT) revealed BAT-like multilocular fat droplets in Pomc-Cre; Crif1f/+ mice, reflecting inducible thermogenesis through the “browning/beigeing” phenotype (Ikeda et al., 2018) (Figure 3A). Mitochondrial biogenesis determined by mtDNA/nDNA ratio was also increased in iWAT (Figure S3A). In line with these findings, the mRNA expression levels of thermogenesis-related genes such as Cidea1, Ppargc1a, and Ucp1 were markedly elevated in iWAT of Pomc-Cre; Crif1f/+ mice and in BAT to a lesser degree (Figure 3C). Epididymal WAT (eWAT) showed a significant reduction in adipocyte size without browning (Figure 3A). Taken together, our results indicate that POMC neuron-Crif1 heterodeficiency induces remarkable changes in iWAT mitochondrial context, lipid utilization, and thermogenesis.
Figure 3. Mild mitoribosomal stress in POMC neurons activates a thermogenic program and UPRmt in adipose tissues.
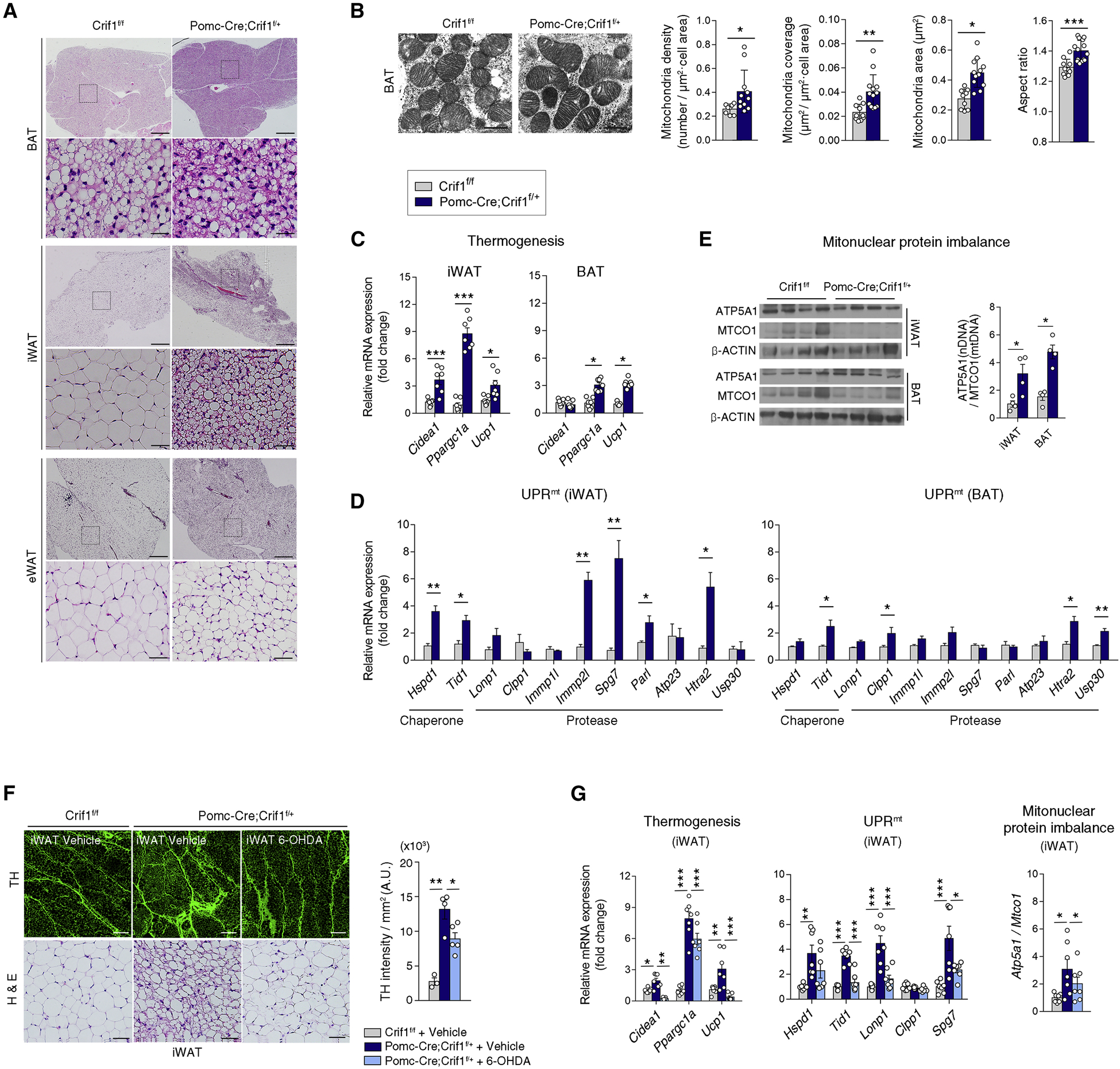
(A) Histology of brown adipose tissue (BAT), inguinal white adipose tissue (iWAT) and epididymal white adipose tissue (eWAT) in Crif1f/f and Pomc-Cre; Crif1f/+ males. Scale bars, 500 μm (upper) and 50 μm (lower).
(B) Electron microscopic examination of the BAT from 8 week-old Crif1f/f and Pomc-Cre; Crif1f/+ males (n = 8–13 cells). Scale bars, 0.5 μm.
(C) Thermogenesis-related gene expression in the iWAT and BAT at 8 weeks (n = 7).
(D) Increased mitochondrial chaperone and protease expression in the iWAT and BAT of Pomc-Cre; Crif1f/+ males, indicating a mitochondrial unfolded protein response (UPRmt) (n = 4).
(E) Mitonuclear protein imbalance between nuclear DNA (nDNA)-encoded ETC protein (ATP5A1) versus mitochondrial DNA (mtDNA)-encoded protein (MTCO1) in the iWAT and BAT (n = 4).
(F, G) Reversal of iWAT browning, increased sympathetic innervation and thermogenic gene expression, the UPRmt and the mitonuclear protein imbalance in Pomc-Cre; Crif1f/+ males following an intra-iWAT injection of neurotoxin 6-hydroxydopamine (OHDA). Tyrosine hydroxylase (TH) staining of sympathetic innervation in the iWAT is shown. Scale bars, 200 μm (upper) and 50 μm (lower) (n = 6–7).
Results are presented as a mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 between indicated groups. See also Figure S3.
In C. elegans, UPRmt can occur in a cell-non-autonomous fashion (Durieux et al., 2011; Lan et al., 2019; Taylor et al., 2014; Zhang et al., 2018). We therefore examined whether the Crif1 partial deletion-induced mitochondrial stress in POMC neurons can induce UPRmt in peripheral adipose tissues. The expression levels of mitochondrial proteases (Immp2l, Spg7, Parl and Htra2) and chaperones (Hspd1 and Tid1) were significantly elevated in iWAT and BAT of Pomc-Cre; Crif1f/+ mice (Figure 3D). Notably, UPRmt was more pronounced in iWAT compared to BAT. In contrast, UPRmt in eWAT was less distinct (Figure S3B). An imbalance in the stoichiometric ratio between nDNA- and mtDNA-encoded ETC proteins (i.e. mitonuclear protein imbalance) extends the lifespan in worms through UPRmt induction (Houtkooper et al., 2013). Therefore, we tested whether mitonuclear protein imbalance in iWAT and BAT occurred in our mouse model. The ratio between ATP5A1 (ATP synthase subunit α, nDNA-encoded ETC component) and MTCO1 (cytochrome c oxidase subunit 1, mtDNA-encoded ETC protein) expression was significantly higher in both fat depots of the Pomc-Cre; Crif1f/+ mice (Figure 3E). Consistently, Crif1 partial deficiency in POMC neurons induced during young adulthood caused iWAT browning, thermogenesis, and UPRmt (Figures S3C and S3D). Together, these data demonstrate that mild mitoribosomal stress in hypothalamic POMC neurons enhances thermogenesis and elicits mitochondrial stress responses in distal adipose tissues.
POMC neurons regulate adipose tissue thermogenesis through the sympathetic nervous system (SNS) (Yang and Ruan, 2015). We thus postulated that the SNS may mediate cell-non-autonomous mitochondrial stress responses in adipose tissues. Tyrosine hydroxylase (TH) staining, which labels sympathetic nerve terminals, revealed a dramatic increase in sympathetic fiber density in iWAT of Pomc-Cre; Crif1f/+ mice compared to the Crif1f/f littermates (Figure 3F). This data suggested that POMC-specific Crif1 partial deficiency may stimulate thermogenesis via increasing sympathetic innervation in iWAT. Consistently, multiple microinjections of neurotoxin 6-hydroxydopamine (OHDA) in one side of iWAT of Pomc-Cre; Crif1f/+ mice significantly reversed browning, thermogenic gene expression, UPRmt, and mitonuclear protein imbalance (Figures 3F and 3G). These findings demonstrate that the SNS mediates the communication of mitochondrial stress responses between the central nervous system and peripheral organs.
Mitochondrial changes and the UPRmt in POMC neurons with a Crif1 deficiency
To investigate the effects of Crif1 deficiency on mitochondria in hypothalamic POMC neurons, we conducted immuno-EM analysis on 11 week-old male Crif1f/f mice, Pomc-Cre; Crif1f/+ mice and Pomc-Cre; Crif1f/f mice. Crif1 heterodeficient POMC neurons showed no changes in mitochondrial mass and morphology except for a reduced mitochondrial coverage in the cytosol (Figure S3E). In contrast, Crif1 homodeficiency induced evident mitochondrial changes (Figure S3E). Mitochondrial density (number) was reduced without changes in mitochondrial coverage, indicating mitochondrial enlargement. Consistently, mitochondrial area and perimeter were proportionally increased without altering the aspect ratio. Notably, the fraction of mitochondria with morphological disruption of the inner membrane significantly increased and the mitochondria-endoplasmic reticulum contacts were reduced. Hence, POMC neurons that are Crif1 homodeficient already undergo mitochondrial dysfunction in 11 week-old mice.
We also evaluated the UPRmt and mitonuclear protein imbalance in the ARH POMC neurons. For this analysis, we generated RiboTag mice in which a hemagglutinin-tagged ribosomal subunit (Rpl22HA) was specifically expressed in POMC neurons to measure mRNA translation (Sanz et al., 2009). Crif1 homodeficient POMC neurons had apparent UPRmt activation and mitonuclear protein imbalance (Figure S3F). By contrast, Crif1 heterodeficiency caused a mild cell-autonomous mitochondrial stress response in POMC neurons (Figure S3F).
Mild mitoribosomal stress increases β-END expression in POMC neurons
We further investigated how POMC neurons promoted adipose tissue changes in Pomc-Cre; Crif1f/+ mice. Pomc in situ hybridization revealed no alteration in the total number of POMC neurons whereas the percentage of c-Fos+ activated POMC neurons were markedly increased in the ARH of Pomc-Cre; Crif1f/+ mice (Figure 4A). We next immunostained ARH POMC neurons with antibodies against β-END or α-melanocyte stimulating hormone (α-MSH), which are cleaved peptide products of POMC. The number of β-END+ neurons was increased but that of α-MSH+ neurons was unaltered in the Pomc-Cre; Crif1f/+ mice (Figure 4B). Moreover, Pomc-Cre; Crif1f/+ mice showed increased β-END+ axonal fiber intensity and unaltered α-MSH+ fiber intensity in the hypothalamic paraventricular nucleus (PVH) and dorsomedial nucleus (DMH) (Figures 4C and S4A). We also measured hypothalamic β-END and α-MSH contents using enzyme-linked immunosorbent assay (ELISA) and a radioimmunoassay (RIA), respectively, and confirmed selective elevation in β-END expression (Figure 4D). Similar changes in hypothalamic β-END and α-MSH expression were observed in Pomc-ERT2-Cre; Crif1f/+ mice (Figures S4B and S4C). Together, these data indicate that Crif1 heterodeficiency specifically increases β-END levels in the ARH POMC neurons.
Figure 4. Mild mitoribosomal stress increases β-END expression in POMC neurons.
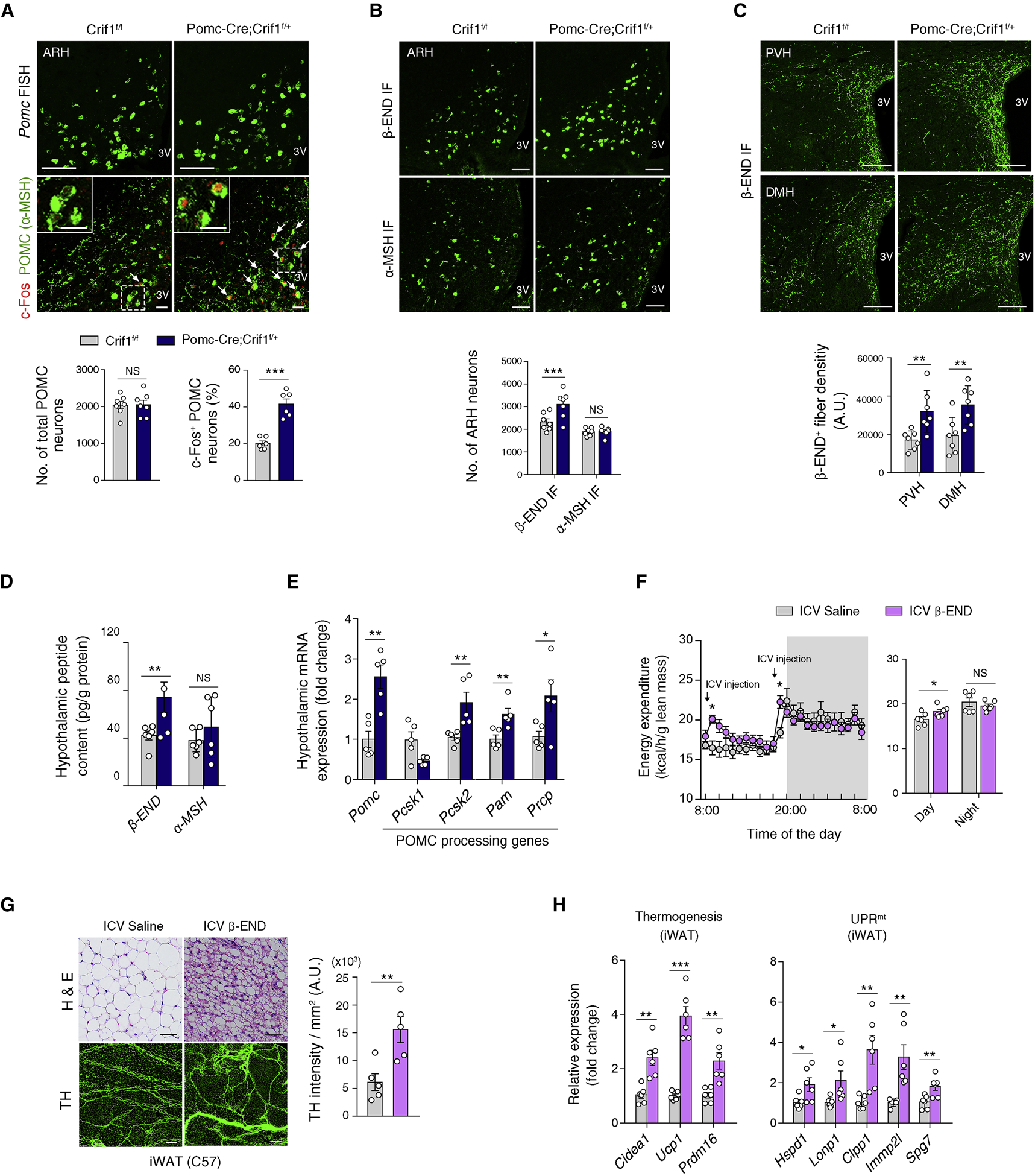
(A) Pomc fluorescence in situ hybridization (FISH) showing no alteration in ARH POMC neuron numbers (upper panel) and an increase in the percentage of c-Fos+ POMC neurons (lower panel) in 8 week-old Pomc-Cre; Crif1f/+ males compared to Crif1f/f controls (n = 6–7). Scale bars, 25 μm. Arrowheads indicate c-Fos+ POMC neurons.
(B) Increased numbers of β-END+ neurons, but no change in the α-MSH+ neuron population, in the hypothalamic ARH of Pomc-Cre; Crif1f/+ male mice at 8 weeks (n = 6–7). Scale bars, 50 μm. IF, immunofluorescence staining.
(C) Increase in β-END+ axonal projections in the hypothalamic paraventricular (PVH) and dorsomedial nucleus (DMH) of Pomc-Cre; Crif1f/+ males (n = 7). Scale bars, 100 μm.
(D) Quantitation of the hypothalamic β-END and α-MSH protein contents at 8 weeks of age (n =4–6).
(E) Changes in the hypothalamic mRNA expression levels of Pomc and of enzymes involved in the production and degradation of POMC-derived peptides in Pomc-Cre; Crif1f/+ mice (n = 5).
(F–H) Repeated ICV injection of β-END (0.1 μg twice a day for 4 days) increases EE and induces browning, enhanced sympathetic innervation, thermogenic gene expression, and the UPRmt in the iWAT of C57 mice (n = 5–6). Scale bars, 200 μm (G, upper) and 50 μm (G, lower).
Results are presented as a mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 between indicated groups. NS, not significant. See also Figure S4.
In addition, we found that Pomc mRNA expression was markedly elevated specifically in the hypothalamus of Pomc-Cre; Crif1f/+ mice (Figures 4E), but not in the brainstem and pituitary gland (Figure S4D), indicating region-specific differential Pomc expression. We next asked how α-MSH expression was unaltered despite higher Pomc mRNA expression. We assessed the alterations in the enzymes involved in α-MSH production and degradation. Biologically active α-MSH (1–13) is produced from precursor polypeptide POMC via multiple enzymatic steps catalyzed by prohormone convertase (PC)-1, PC-2 (encoded by Pcsk1 and Pcsk2), carboxypeptidase E (CPE, encoded by Cpe1), and peptidyl α-amidating monooxygenase (PAM). α-MSH (1–13) is degraded by prolyl carboxypeptidase (PRCP, encoded by Prcp) to the inactive α-MSH (1–12) form (Wallingford et al., 2009). We detected a significant elevation in the hypothalamic expression of Pcsk1, Pcsk2, Pam, and Prcp in Pomc-Cre; Crif1f/+ mice compared to the Crif1f/f controls (Figure 4E). Hence, α-MSH production and degradation may have both increased, providing a steady-state level of hypothalamic α-MSH.
We further tested the possibility that increased hypothalamic β-END expression may induce the WAT changes observed in the Pomc-Cre; Crif1f/+ mice. For this analysis, we administered β-END (1–13) (0.1 μg twice a day) intracerebroventricularly (ICV) into C57BL/6J (C57) mice for 4 days. Chronic β-END treatment did increase EE especially during daytime (Figures 4F). Moreover, it markedly increased sympathetic innervation, thermogenic gene expression, and UPRmt in iWAT and induced its browning (Figures 4G and 4H). Chronic β-END treatment did not significantly alter body weight nor food intake during the treatment period (Figure S4E). As the orexigenic effect of β-END are short-lasting (Dutia et al., 2012, Figure S4F), twice-a-day-injection may not induce a continuous hyperphagic behavior over the course of the chronic treatment period as determined by cumulative food intake. These data support the potential role of β-END in mediating the cell-non-autonomous mitochondrial stress response and inducible thermogenesis in the iWAT of Pomc-Cre; Crif1f/+ mice.
The mitochondrial-derived peptide MOTS-c coordinates mitoribosomal stress response in POMC neurons
We explored the downstream mediator of mitoribosomal stress in POMC neurons. We found that N1 hypothalamic neuronal cells significantly increased the expression of a mtDNA-encoded peptide, MOTS-c, upon Crif1 knockdown using small interfering RNA (siRNA) (Figure S5A). Moreover, MOTS-c and α-MSH double immunohistochemistry revealed that Pomc-Cre; Crif1f/+ mice showed increased MOTS-c expression in POMC neurons in ARH (Figure 5A). Pomc and MOTS-c double staining showed unaltered MOTS-c expression in the solitary tract nucleus POMC neurons of Pomc-Cre; Crif1f/+ mice whilst MOTS-c expression was negligible in mouse pituitary POMC cells (Figure S5B). The specificity of the MOTS-c antibody we used had been validated in our previous study (Kim et al., 2018) and also in hypothalamic tissues using a peptide competition antibody test (Figure S5C). MOTS-c ELISA also confirmed the elevation of hypothalamic MOTS-c content in both Pomc-Cre; Crif1f/+ mice and Pomc-ERT2-Cre; Crif1f/+ mice (Figure 5B). These findings suggested that mitoribosomal stress stimulates MOTS-c expression in hypothalamic POMC neurons.
Figure 5. The mitochondrial-derived peptide MOTS-c coordinates mitoribosomal stress response in POMC neurons.
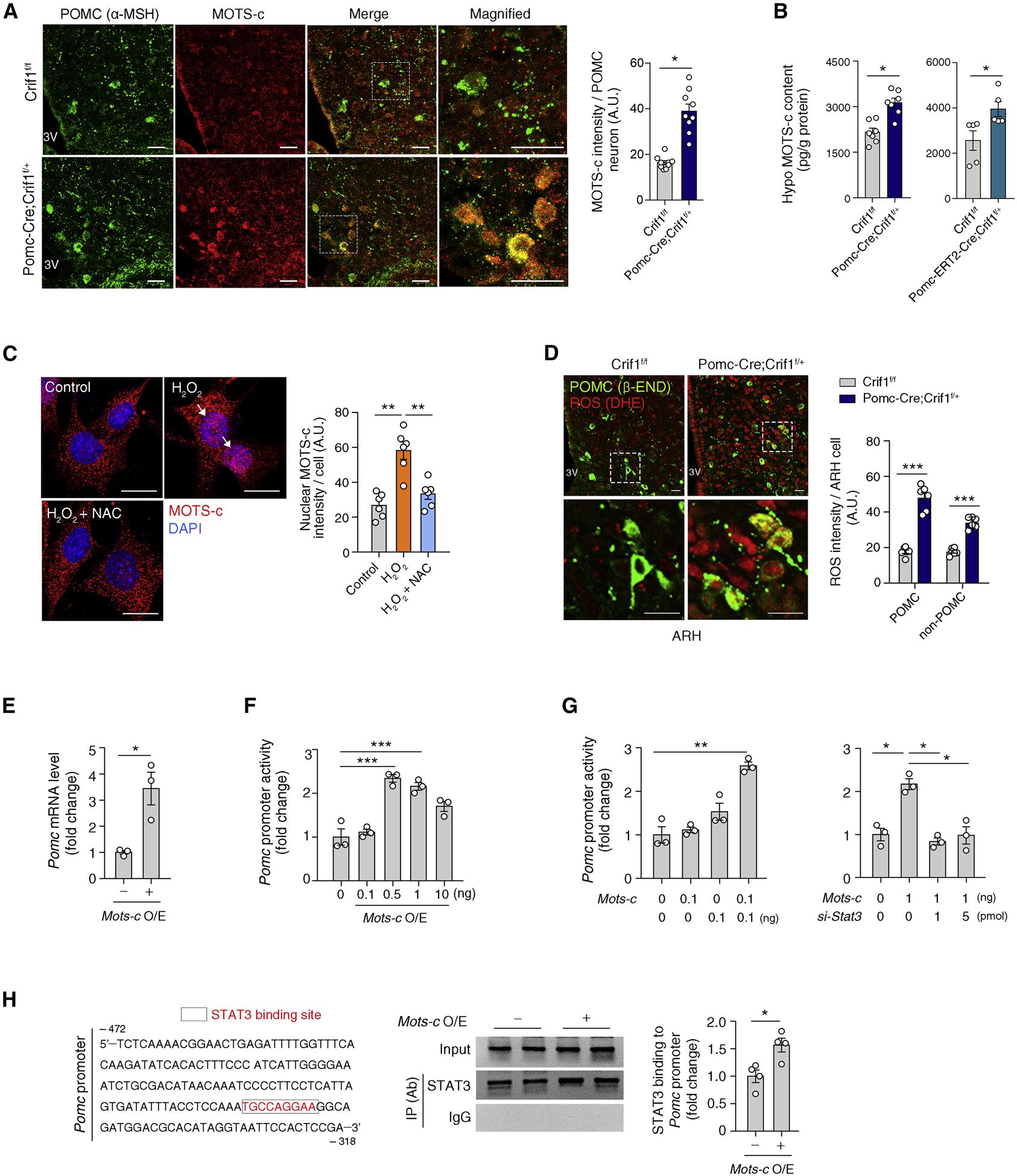
(A) αMSH and MOTS-c double immunohistochemistry showing increased MOTS-c expression in the POMC neurons of Pomc-Cre; Crif1f/+ mice compared to Crif1f/f controls (n = 9). Scale bars, 25 μm.
(B) Increased hypothalamic MOTS-c contents in Pomc-Cre; Crif1f/+ mice and Pomc-ERT2-Cre; Crif1f/+ mice compared to Crif1f/f mice (n = 5–7).
(C) Oxidative stress (H2O2 treatment) induces MOTS-c nuclear localization (white arrows) which is blocked by a cotreatment with antioxidant N-acetylcysteine (NAC) in N1 hypothalamic neurons (n = 6 wells). Scale bars, 25 μm.
(D) Dihydroethidium (DHE) and β-END double staining showing increased ROS production in the ARH of Pomc-Cre; Crif1f/+ mice (n = 6). Scale bars, 10 μm.
(E, F) Effect of Mots-c overexpression (O/E) on Pomc mRNA and Pomc transcriptional activity (n = 3).
(G) Effects of Stat3 coexpression and depletion via siRNA on the MOTS-c-mediated regulation of Pomc transcription (n = 3).
(H) Chromatic immunoprecipitation assay showing increased STAT3 binding to the Pomc promotor area under conditions of Mots-c overexpression (n = 4) IP, immunoprecipitation; Ab, antibody.
Results are presented as a mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 between indicated groups. See also Figure S5
Previously, we reported that MOTS-c translocates to the nucleus upon metabolic stress and regulates adaptive nuclear gene expression in an ROS-dependent manner (Kim et al., 2018). We confirmed that MOTS-c translocates to the nucleus in N1 hypothalamic neurons upon H2O2-induced oxidative stress, which was completely prevented by co-treatment with the antioxidant N-acetylcysteine (Figure 5C). Further, we assessed ROS levels in Crif1-depleted POMC neurons. For this, we intravenously injected dihydroethidium (DHE), a fluorescent probe used to detect tissue superoxide and hydrogen peroxide, into 14 week-old Crif1 heterodeficient mice and their age-matched controls 2 h prior to euthanasia. Indeed, the level of ROS in POMC neurons was significantly elevated in Crif1 heterodeficient mice (Figure 5D). Thus, mitoribosomal stress induced by Crif1 partial deficiency may increase ROS levels in POMC neurons. Interestingly, ROS production was also elevated in non-POMC cells in the ARH (Figure 5D). Thus, mitoribosomal stress in POMC neurons may cause oxidative stress in neighboring neurons and glia via currently unclear inter-cellular communication mechanisms.
Then, we tested whether MOTS-c regulates nuclear-encoded Pomc transcription. Indeed, Mots-c overexpression in POMC-producing AtT20 cells significantly increased Pomc mRNA expression and Pomc promoter activity (Figures 5E, 5F and S5D). Hence, elevated MOTS-c expression can stimulate Pomc transcription. We previously reported that, once in the nucleus, MOTS-c interacts with transcription factors (Kim et al., 2018). As STAT3 is a well-known transcriptional regulator of Pomc (Munzberg et al., 2003), we examined its involvement in MOTS-c-dependent Pomc transcription. Indeed, at sub-effective doses (0.1 ng each), Mots-c and Stat3 co-expression synergistically stimulated Pomc promotor activity whilst Stat3 depletion by siRNA treatment blocked MOTS-c-induced Pomc promotor activity (Figures 5G and S5E). Chromatin immunoprecipitation experiments revealed that STAT3 binding to the Pomc promotor was enhanced in cells overexpressing Mots-c (Figures 5H and S5F). These findings indicate that MOTS-c upregulates Pomc transcription in coordination with STAT3.
Central MOTS-c administration recapitulates the phenotypes of mice with mild mitoribosomal stress in POMC neurons
We tested whether hypothalamic MOTS-c treatment could recapitulate the metabolic and adipose phenotypes of the Pomc-Cre; Crif1f/+ mice. To this end, MOTS-c peptide (3 μg/day) was ICV-infused for 28 days using an osmotic minipump to young C57 mice. Chronic MOTS-c treatment increased nighttime EE and food intake without body weight changes (Figure 6A). In addition, ICV-infused MOTS-c caused significant iWAT browning, smaller BAT fat droplets, greater sympathetic innervation as well as increased thermogenic gene expression, UPRmt activation, and mitonuclear protein imbalance in the iWAT and BAT (Figures 6B–6D). These findings support the potential role of MOTS-c in mediating communication between the hypothalamus and adipose tissues as observed in Crif1 heterodeficient mice.
Figure 6. Central administration of MOTS-c recapitulates the phenotypes of mice with mild mitoribosomal stress in POMC neurons.
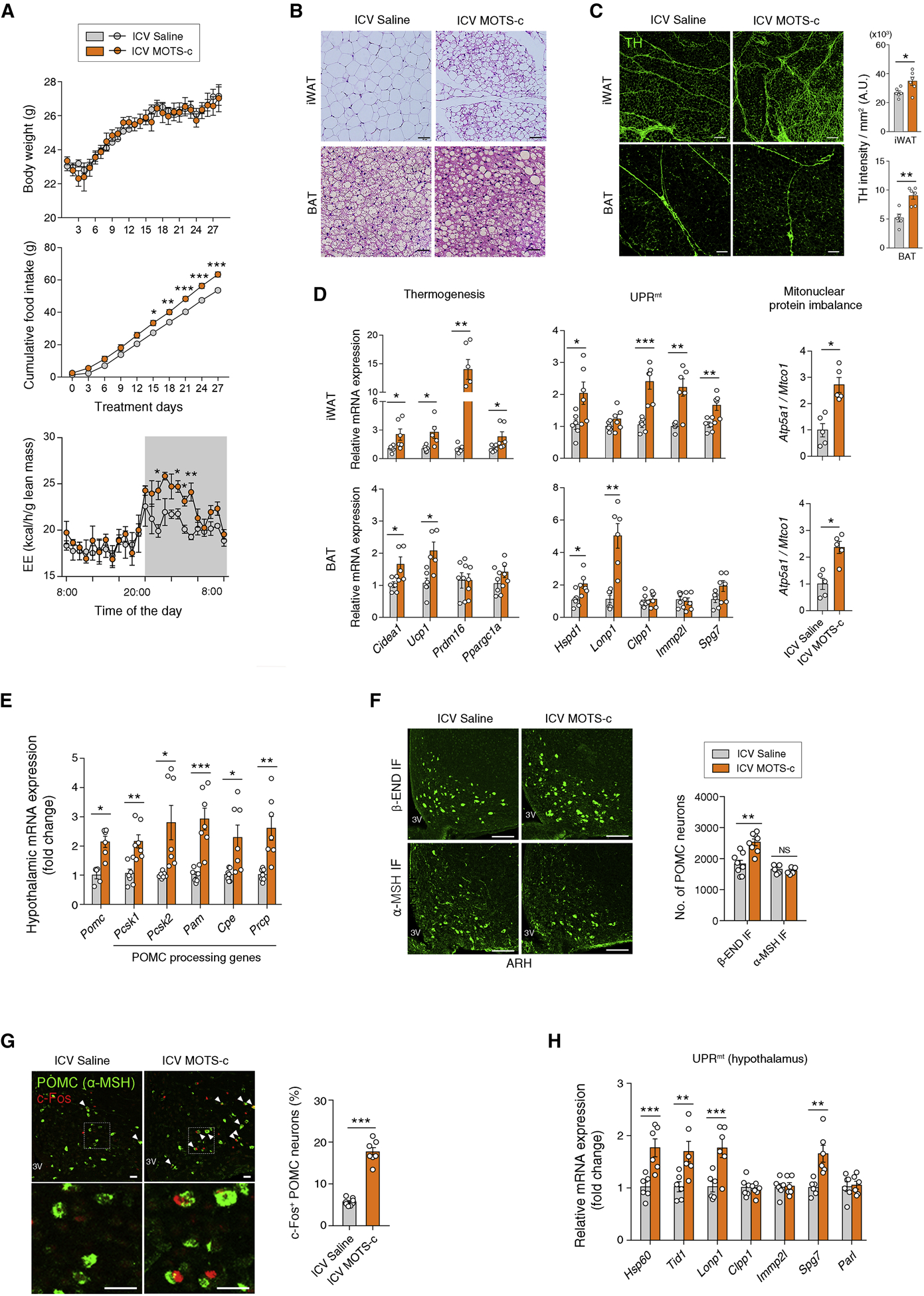
(A) Body weight, cumulative food intake, and energy expenditure during ICV infusion of MOTS-c in C57 mice (n = 6–9).
(B) ICV MOTS-c-treated mice show iWAT browning and smaller fat droplets in the BAT (n = 6). Scale bars, 50 μm.
(C, D) ICV MOTS-c-induces enhanced sympathetic innervation, thermogenesis, the UPRmt and a mitonuclear protein imbalance in the iWAT and BAT (n = 5–6). Scale bars, 200 μm (upper) and 50 μm (lower).
(E) Increased hypothalamic expression of Pomc and its processing enzymes in mice with ICV MOTS-c administration (n = 7).
(F) Increased numbers of the hypothalamic β-END+ neurons but no change in the α-MSH+ neuron numbers following ICV MOTS-c infusion (n = 5–8). Scale bars, 50 μm.
(G, H) Increase in the percentage of c-Fos+ POMC neurons and hypothalamic UPRmt gene expression in MOTS-c-treated mice (n = 6–8). Scale bars, 25 μm. Arrowheads indicate c-Fos+ POMC neurons.
Results are presented as a mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 between indicated groups. NS, not significant. See also Figure S6
We next examined whether the inhibition of hypothalamic MOTS-c expression or action could reverse the fat tissue changes in Pomc-Cre; Crif1f/+ mice. The specific inhibition of Mots-c transcript was technically challenging due to the short open reading frame within the mitochondrial 12S rRNA gene. As an alternative strategy, we infused MOTS-c neutralizing antibody into the bilateral ARH of Pomc-Cre; Crif1f/+ mice using an osmotic pump to block MOTS-c actions if it was secreted extracellularly from POMC neurons and acted in an autocrine and/or paracrine manner within the ARH. Intra-ARH infusion of MOTS-c antibody did not reverse iWAT browning in Pomc-Cre; Crif1f/+ mice (Figure S6A), supporting intracellular actions of hypothalamic MOTS-c.
Consistent with our in vitro findings (Figure 5), ICV MOTS-c infusion elevated hypothalamic Pomc mRNA and β-END expression in mice but α-MSH levels did not alter and were at a steady-state with increased expression of hypothalamic α-MSH–producing/degrading enzymes (Figures 6E and 6F). The percentage of c-Fos+ POMC neurons and the expression of ARH UPRmt genes were also upregulated by ICV MOTS-c treatment (Figures 6G and 6H). Therefore, ICV MOTS-c infusion recapitulated the phenotypes of POMC Crif1 heterodeficient mice. On the other hand, MOTS-c-treatment did not affect hypothalamic transcript levels of other neuronal markers (Agrp, Npy, Sf1), astrocyte marker (Gfap), microglia marker (Iba1) and leptin and insulin receptors (Lepr, Insr) (Figure S6B), supporting a specific regulatory role of MOTS-c on POMC neurons.
Regular exercise and the myokine interleukin-6 stimulate hypothalamic MOTS-c and β-END production
We next searched for a physiological regulator or condition related to POMC neuronal mitoribosomal stress. We first examined the involvement of leptin in this regard as it stimulates Pomc transcription via STAT3 and activates adipose tissue thermogenesis (Dodd et al., 2015). In contrast to our expectation, a single ICV leptin (1 μg) administration decreased MOTS-c expression in the hypothalamus of C57 mice as determined by immunohistochemistry (Figure S7A) and ELISA (Figure S7B), indicating that leptin may act as a negative regulator of hypothalamic MOTS-c.
We hypothesized that regular moderate-intensity exercise may induce high metabolic turnover and beneficial metabolic changes without marked weight changes via hypothalamic mitoribosomal stress responses. To test whether regular exercise increases hypothalamic MOTS-c expression, young C57 mice were subjected to moderate-intensity running (13 meters/min on a treadmill for 50 min/day) for 2 weeks. This regular exercise robustly increased hypothalamic MOTS-c expression (Figures 7A and 7B), whereas a single bout of high-intensity running until exhaustion did not cause this elevation (Figure S7C). These data highlighted the importance of exercise duration or intensity in inducing hypothalamic MOTS-c.
Figure 7. Regular exercise and myokine interleukin-6 stimulate hypothalamic MOTS-c and β-END production.
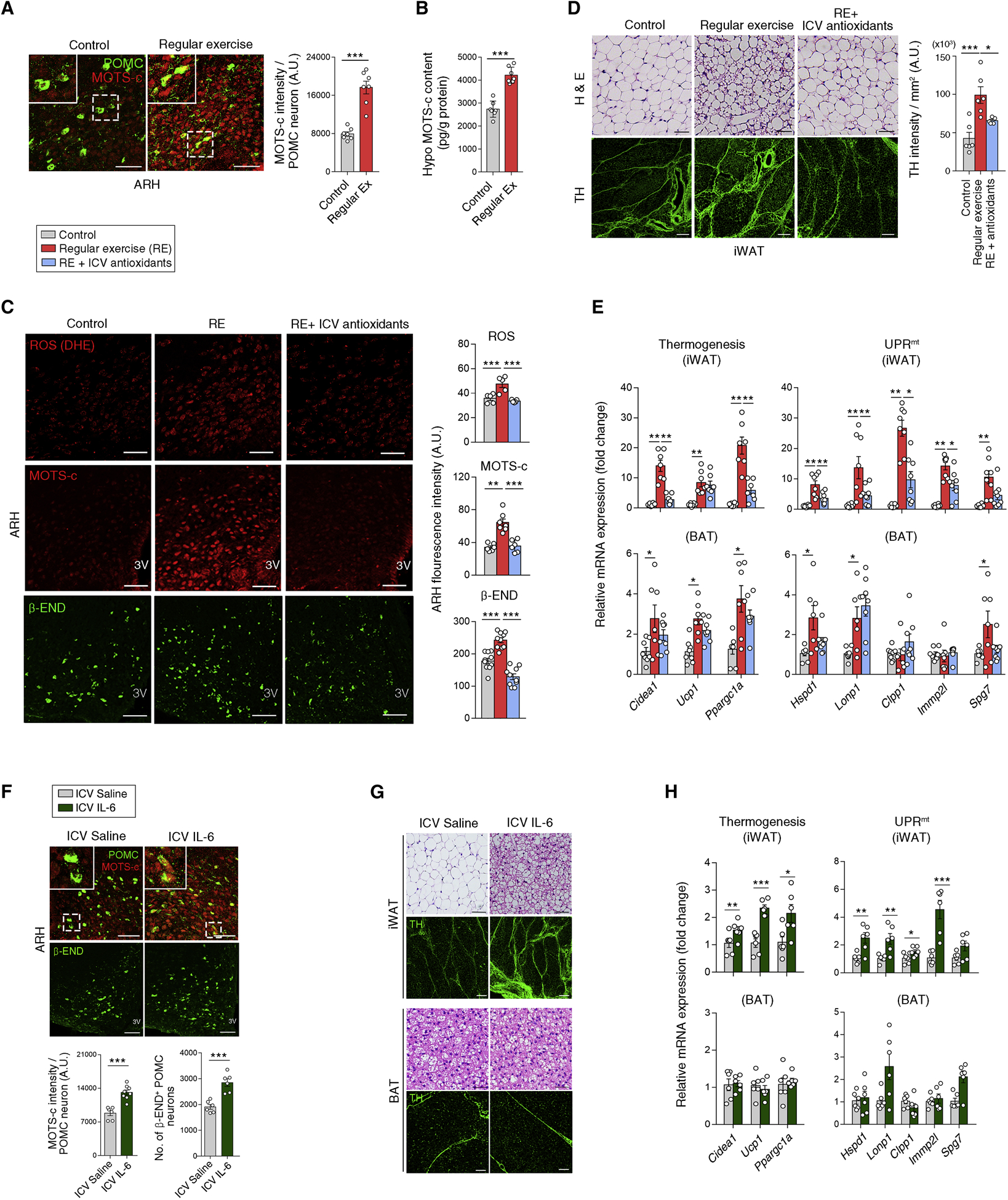
(A, B) Increased MOTS-c expression in the mouse hypothalamus, including POMC neurons following 2 weeks of regular treadmill exercise (n = 7). Scale bars, 50 μm.
(C) Regular running (RE)–induced increase in hypothalamic ROS production and MOTS-c/β-END expression, is reversed by an ICV injection of antioxidant cocktail prior to exercise (n = 5–10). Scale bars, 50 μm.
(D, E) Effects of RE with or without ICV antioxidant treatment on iWAT histology, sympathetic innervation, thermogenic gene expression, and UPRmt (n = 6–8). Scale bars, 50 μm (upper) and 200 μm (lower).
(F) Increased hypothalamic MOTS-c and β-END expression following an ICV IL-6 infusion (400 ng/day for 5 days) (n = 6–8). Scale bars, 50 μm.
(G, H) ICV IL-6-induced changes in the iWAT and BAT (n = 6). Scale bars, 50 μm (H & E and BAT TH image), and 200 μm (iWAT TH).
Results are presented as a mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 between indicated groups. See also Figure S7
Regular running also increased hypothalamic β-END expression and ROS production, particularly in POMC neurons (Figure 7C and S7D). It also induced histological changes, elevated sympathetic innervation, and increased thermogenic gene expression and UPRmt especially in iWAT of the mice (Figures 7D, 7E and S7E). Notably, a blockade of hypothalamic ROS production with a prior ICV injection of an antioxidant cocktail significantly inhibited regular exercise-induced hypothalamic MOTS-c/β-END expression as well as iWAT thermogenesis and UPRmt (Figures 7C–7E, and S7D), consistent with our prior results indicating coordination between ROS and MOTS-c (Figure 5C and 5D). Hence, hypothalamic ROS production may be critical for exercise-induced changes in the hypothalamic and adipose tissues.
We finally investigated bioactive molecules that mediate exercise-induced hypothalamic mitochondrial stress responses. Because interleukin-6 (IL-6) is an exercise-related myokine (Pedersen et al., 2001), we tested its ability to induce hypothalamic MOTS-c expression. For this purpose, we ICV infused IL-6 peptide (400 ng/day) in young C57 mice for 5 days and found that it significantly elevated hypothalamic MOTS-c and β-END expression (Figure 7F). Furthermore, ICV IL-6 treatment stimulated browning, sympathetic innervation, thermogenesis, and mitochondrial stress responses in iWAT (Figures 7G and 7H). Further, in N1 hypothalamic neurons, IL-6 (100 nM) treatment induced the nuclear translocation of MOTS-c in an ROS-dependent manner (Figure S7F). These findings collectively indicate that IL-6 promotes mitohormesis in hypothalamic neurons upon regular moderate-intensity exercise.
DISCUSSION
Our current study demonstrates the impact of mitoribosomal stress in hypothalamic POMC neurons on whole-body metabolism. Crif1 homodeficiency in POMC neurons leads to the development of adult-onset severe obesity and glucose dysregulation. As demonstrated in the hippocampal neurons of CamKII-Cre: Crif1f/f mice (Kim et al., 2012), Crif1 homodeficiency in POMC neurons may cause ETC dysfunction, defective ATP production, and eventually cell death. Supporting this possibility, EM examinations revealed disrupted inner mitochondrial membrane and reduced endoplasmic reticulum-mitochondria contacts in Crif1 homodeficient POMC neurons.
In contrast to the adverse outcomes of Crif1 homodeficiency, mice with Crif1 heterodeficiency exhibited higher metabolic turnover (i.e. a modest increase in energy intake and a higher EE) without changes in body weight. Interestingly, similar metabolic changes were observed in Crif1 homodeficient males before the development of obesity (prior to 10 weeks of age). During this period, POMC neurons with Crif1 homodeficiency may be under mitochondrial stress but not critically damaged. The high metabolic turnover induced by partial Crif1 deficiency may protect against diet-induced obesity. Indeed, Crif1 heterodeficient mice on a HFD, which consumed more food than the controls, were resistant to weight gain. These findings are in line with previous studies in lower organisms demonstrating that perturbations in mitochondrial ETC and ribosomes increase lifespan and delay ageing-related changes (Durieux et al., 2011; Houtkooper et al., 2013).
Interestingly, the induction of mitohormesis appears to be dependent on spatial and temporal contexts, whereby cell type and stage of life play critical roles. Neuronal, muscle, and intestinal cells have been shown to elicit mitohormesis (Berendzen et al., 2016; Durieux et al., 2011; Shao et al., 2016) and our findings provide evidence for POMC neurons. The timing of mitochondrial stress may also influence the induction of mitohormesis. In C. elegans, mitochondrial perturbation, introduced early in life during development and young adulthood, induced beneficial effects (Cox et al., 2018; Durieux et al., 2011; Schulz et al., 2007). Consistently, the embryonic and young adult-onset partial deletion of Crif1 in POMC neurons promoted similar metabolic phenotypes.
Mitochondrial stress during development increases resilience throughout life via a retrograde stress response from the mitochondria to the nucleus (Yun and Finkel, 2014). In C. elegans, the UPRmt constitutes an important part of mitochondrial stress responses as it helps in the recovery from mitochondrial perturbation by stimulating the production of nDNA-encoded mitochondrial chaperones and proteases. In C. elegans, the UPRmt can be induced cell-autonomously or cell-non-autonomously (Shao et al., 2016), mediated by secreted factors such as serotonin (neurotransmitter) and FLP-2 (neuropeptide) (Berendzen et al., 2016; Shao et al., 2016). We previously reported that in mammals, GDF15 acts as a secretory mediator of mitochondrial stress (Chung et al., 2017). Fibroblast growth factor 21, angiopoietin-like peptide 6, and adrenomedullin 2 are also potential candidate mediators of these processes (Kang et al., 2017; Kim et al., 2013; Lv et al., 2016).
In our current study, Crif1 partial deficiency in POMC neurons provoked remarkable cell-non-autonomous UPRmt in the iWAT and BAT, whereas it induced relatively mild cell-autonomous UPRmt. These data suggest that cell-non-autonomous UPRmt may have a more sensitive induction threshold than cell-autonomous UPRmt in hypothalamic POMC neurons. We also provide evidence of neural-mediated cell-non-autonomous UPRmt in the iWAT of mice, as shown by chemical sympathetic denervation, in response to mitoribosomal stress in hypothalamic POMC neurons. Although the detailed molecular mechanisms of SNS-mediated adipose tissue UPRmt remain elusive, a mitonuclear protein imbalance may be related to this process as it activates the UPRmt and extends the lifespan in worms (Houtkooper et al., 2013). Indeed, we found that POMC-specific Crif1 deficiency caused a mitonuclear protein imbalance in POMC neurons as well as in iWAT and BAT. Again, the cell-non-autonomous mitonuclear protein imbalance in iWAT of Crif1 heterodeficient mice was inhibited by disruption of sympathetic innervation. Hence, mitonuclear protein imbalance can be induced cell-non-autonomously through the autonomic nervous system.
Substantial evidence now indicates that MDPs serve as inherent mitochondrial signals that promote the cellular and organismal adaptation to mitochondria stress (Galluzzi et al., 2018; Kim et al., 2017; Lee et al., 2016; Mottis et al., 2019; Quiros et al., 2016; Tan and Finkel, 2020). In our current study, MOTS-c expression was significantly elevated in the POMC neurons with partial Crif1 deficiency. Furthermore, ICV MOTS-c infusion induced high turnover metabolism observed in the Pomc-Cre: Crif1f/+ mice. These data support a role for MOTS-c as a potential mitochondrial-encoded messenger that propagates mitochondrial stress from POMC neurons to adipose tissue to induce thermogenesis. MOTS-c is detected in circulation and its levels can decrease during aging, cold exposure and type 2 diabetes, but increase upon exercise (Lee et al., 2015; Lu et al., 2019a; Ramanjaneya et al., 2019; Reynolds et al., 2019), evidencing its role as a mitochondrial hormone (Zarse and Ristow, 2015). In addition, intracellular MOTS-c (endogenous and exogenous), directly relays mitochondrial stress conditions to the nucleus, where it coordinates with transcription factors (e.g. NRF2 and ATF1) to regulate adaptive nuclear gene expression (Kim et al., 2018; Reynolds et al., 2019). Consistently, in the hypothalamic neurons, MOTS-c entered the nucleus upon oxidative stress and stimulated Pomc transcription by enhancing STAT3 binding to the Pomc promotor.
POMC is cleaved by multiple enzymes to yield β-END and α-MSH. Intriguingly, we found that partial Crif1 deficiency and ICV MOTS-c infusion selectively increased hypothalamic β-END, but not α-MSH levels. This differential regulation of POMC processing by partial Crif1 deficiency or MOTS-c can be, in part, explained by a marked increase in the expression of PRCP, which degrades biologically active α-MSH (1–13). Interestingly, α-MSH and β-END, despite being derived from the same precursor POMC, have opposing effects on food intake; β-END has orexigenic effect although this effect is short-lasting and antagonizes the anorexigenic effect of α-MSH (Dutia et al., 2012). Indeed, POMC neurons mediate cannabinoid-induced hyperphagia by releasing β-END (Koch et al., 2015). Considering that ICV β-END administration induced iWAT browning, both β-END and α-MSH may have similar thermogenesis-promoting effects (Fan et al., 2005).
Moderate-intensity regular exercise increased hypothalamic MOTS-c expression through a ROS-dependent mechanism. Consistently, MOTS-c levels were increased in human skeletal muscle and plasma following exercise (Reynolds et al., 2019). Exercise-related myokine IL-6 elevated hypothalamic MOTS-c expression whereas leptin lowered it. Both leptin and IL-6 activate hypothalamic STAT3 signaling (Munzberg et al., 2003; Wrighting and Andrews, 2006). Thus, further studies are needed to elucidate the downstream signaling pathways leading to differential regulation of MOTS-c. It will also be worth examining whether ICV MOTS-c treatment regulates skeletal muscle metabolism and function as systemic MOTS-c treatment improved exercise performance and aging-related decline in physical capacity (Reynolds et al., 2019).
In conclusion, regular exercise-induced low-level mitoribosomal stress in POMC neurons causes high metabolic turnover via interorgan communication of mitochondrial stress. Our findings provide a novel neuroendocrine mechanism of exercise physiology that may provide new venues for future therapeutic development of exercise-mimetics.
Limitations of Study
While we provide strong evidence for the role of MOTS-c in communicating low-grade mitoribosomal stress in hypothalamic POMC neurons, the effect of Mots-c gene ablation remains unexplored. Targeted mtDNA editing tools that can precisely alter nucleotide sequences of interest are currently unavailable, but opportunities may emerge in the near future based on active ongoing research on mitochondrial genome editing technology (Bacman et al., 2013; Gammage et al., 2018; Mok et al., 2020). Further, whether mitoribosomal stress induces a differential metabolic effect in hypothalamic vs. hindbrain POMC neurons remains to be addressed.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Min-Seon Kim (mskim@amc.seoul.kr).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact. Crif1-floxed mice are available upon request under a material transfer agreement.
Data and Code Availability
This study did not generate datasets/code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
To generate POMC neuron-specific Crif1 deficient mice, Crif1 floxed mice (Kwon et al., 2008) were cross mated with Pomc-Cre mice (Jackson Laboratory) or Pomc-ERT2-Cre mice (MGI Mouse). Tamoxifen (75 mg/kg dissolved in DMSO) was then injected into the peritoneum of the Pomc-ERT2-Cre; Crif1f/+ mice at 7 weeks of age. To isolate POMC neurons or POMC neuron-specific transcripts, the mice were mated with Rosa26; eGFP-loxP mice or RiboTag (Rpl22HA) mice (both from Jackson Laboratory). C57BL/6J male mice at 7–8 weeks of age were purchased from Orient Bio. All mice had free access to a standard chow diet (Cargill Agri Purina) and water unless indicated. To induce diet-induced obesity, mice were fed a high-fat diet (60% fat, Research Diet). Animals were housed under controlled temperature conditions (22 ± 1°C) and subjected to a 12- h light-dark cycle, with light from 08:00 to 20:00 hours. All animal procedures were approved by the Institutional Animal Care and Use Committee of the Asan Institute for Life Sciences (Seoul, Korea).
Cell lines
N1 (Cedarlane) and AtT20 (Obtained from Dr. Byung Ju Lee, University of Ulsan) cells were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin.
METHOD DETAILS
FACS sorting of POMC cells
The medial basal hypothalamus of mice with eGFP-labeled POMC neurons were dissected, mechanically chopped into small pieces in lysis buffer (HBSS containing 0.002% Dnase 1) on ice using a scalpel. Chopped tissues were incubated in lysis buffer containing 0.01% trypsin in a 37 °C water bath for 30 min during which time the sample tubes were gently inverted every 5 min. After centrifugation (1000 rpm for 5 min), pellets were gently resuspended in 10% FBS-containing Neurobasal Media (Thermo Fisher Scientific) and dissociated into single cells by pipetting with 1 ml, 200 μl, and 10 μl tips (at least 10 times each). Pomc-eGFP+ cells and Pomc-eGFP– cells were gated and sorted by fluorescence-activated cell sorting (FACS) with the BD FACS Aria II Cell Sorter. Non-fluorescent hypothalamic cells were obtained from C57 mice to set the threshold of eGFP fluorescence. Successful FACS of eGFP cells and Crif1 depletion was confirmed by Pomc and Crif1 qRT-PCR analysis in eGFP+ cells and eGFP– cells (Figure S1A and S1B). For this, the hypothalami from two animals were pooled into one sample and total RNA was extracted using an RNeasy Plus mini kit (Qiagen).
Metabolic phenotyping
Food intake levels and body weights were monitored weekly after weaning until sacrifice. Body composition (lean mass and fat mass) was measured using dual x-ray absorptiometry (iNSiGHT VET DXA, OsteoSys) during the late light phase at the indicated ages. Energy expenditure (EE) and locomotor activity were determined using a comprehensive lab monitoring system (CLAMS) (Columbus Instruments). Mice were placed in the CLAMS chambers for 48 h to acclimatize to these conditions before measurement. During this period, day-night cycles were the same as those during the initial housing of the animals and food was provided as pellets on the floor of the CLAMS cages. For the glucose tolerance test, D-glucose (1 g/kg, Sigma) was orally administrated to overnight-fasted mice. For the insulin tolerance test, insulin (Humulin-R® 0.25 U/kg, Eli Lilly) were injected into the peritoneum following a 5 h-fasting. Blood samples were obtained from the tail vein for glucose measurement immediately before and at the indicated times after injections. Glucose levels were measured using a glucometer (ACCU-CHEK Aviva Plus System, Aviva).
Body temperature measurements
Skin temperature of mice was determined with thermal camera (FLIR T650SC, FLIR Systems). Rectal temperatures were measured with thermometers (BAT-12 Microprobe Thermometer, Physitemp Instruments) before and during 4°C cold exposure.
Immunostaining of the hypothalamus
Mice were anesthetized with an intraperitoneal injection of 40 mg/kg Zoletil® and 5 mg/kg Rompun®, and then perfused with 50 ml saline followed by 50 ml 1% paraformaldehyde (PFA) via the left ventricle of the heart. Whole brains were collected, fixed with 1% PFA for 2 h, and dehydrated in 30% sucrose solution until the tissues sank to the bottom of the container. Coronal brains including the hypothalamus were sectioned at a 30 μm thickness using a cryostat (Leica). One of every five slices (about 10 brain slices per animal) was collected. For β-END staining, hypothalamic slices were blocked with 3% bovine serum albumin (BSA) in 0.5% PBST and then incubated with anti-β-END antibody (1:1500, rabbit, Phoenix Pharmaceuticals) at 4°C for 48 h and then at room temperature (RT) for 1 h. For α-MSH staining, hypothalamic slices were pretreated with 1% NaOH/1% H2O2 for 20 min followed by 0.3% glycine for 10 min, blocked for 1 h with 5% normal donkey serum in 0.5% PBST and incubated with anti-α-MSH antibody (1:1000, sheep, MerckMillipore) in blocking solution for 48 h at 4°C. For MOTS-c staining, hypothalamic slices were blocked with 3% BSA in 0.1% PBST and subjected to treatment with anti-MOTS-c antibody (1:200, rabbit, produced by Dr. Changhan Lee) for 48 h at 4°C. For c-Fos staining, hypothalamic slices were blocked with 3% BSA in 0.5% PBST and incubated with anti-c-Fos antibody (1:1000, rabbit, Synaptic Systems) for 24 h at 4°C. After washing, slides were incubated with the appropriate Alexa-Flour 488- or 555-conjugated secondary antibodies (1:1000) at RT for 2 h. For nuclear staining, slides were treated with DAPI (1:10000) for 10 min before mounting. Immunofluorescence images were obtained using a confocal microscopy (Carl Zeiss 710, Germany). Quantification of the average fluorescence intensity of approximately 100 cells and the numbers of POMC neurons were analyzed on 5–7 hypothalamic slices or 3 brainstem and pituitary slices per mouse using ImageJ (NIH) or Photoshop version CS6 (Adobe Systems).
To determine β-END- and α-MSH-immunoreactive axonal projections, mice were cardiac-perfused with 4% PFA dissolved in borate buffer (38 g/L, pH 9.5) and post-fixed in 20% sucrose–4% PFA–borate solution at 4 °C for 5 h. Hypothalamic slices were blocked with 3% BSA in 0.5% PBST buffer for 1 h at RT, incubated with anti-β-END antibody (1:1000, rabbit, Phoenix Pharmaceuticals) or anti-α-MSH antibody (1:1000, sheep, MerckMillipore) in blocking solution at 4°C for 72 h and then with Alexa-Fluor-conjugated secondary antibodies (1:1000) at RT for 1 h. Quantitative analysis of the β-END+ or α-MSH+ axonal projection density was conducted in the entire area of PVH and DMH (3 brain sections per mouse) as previously described (Bouret et al., 2004) and the average values were presented. After adjustment of the brightness and contrast and elimination of background, we measured the density of immunoreactivity in these areas.
Pomc in situ hybridization
Mice were cardiac-perfused with 4% PFA dissolved in DEPC water. Whole brains were collected, post-fixed overnight, dehydrated in 30% sucrose solution for 48 h, and sectioned into 14 μm-thick slices. Fluorescence in situ hybridization (FISH) was performed using RNAscope® fluorescent multiplex reagents (Advanced Cell Diagnostics) according to the manufacturer’s instructions. RNA probes for mouse Pomc (Advanced Cell Diagnostics) was incubated with the brain slices and signal amplification was achieved using the multiplex reagents. For POMC and MOTS-c double staining, brain slices were subjected to Pomc in situ hybridization and then to MOTS-c immunostaining as described in the immunostaining section.
mRNA expression measurement
Total RNA was isolated from tissues or cells using Trizol reagent (Thermo Fisher), and cDNA was prepared from total RNA using M-MLV reverse transcriptase and oligo-dT primers (Invitrogen). The produced cDNA was then amplified on a 7500 Fast Real-Time PCR System (Applied Biosystems). Real-time PCR was performed in triplicate using SYBR Green premix (Enzynomics). All quantitative calculations were performed using the ΔΔCT method. All mouse primer sequences are listed in Table S1. To assay Crif1 and UPRmt gene expression specifically in POMC neurons, immunoprecipitation (IP) of POMC neuronal polyribosomes was conducted as described previously (Sanz et al., 2009). Hypothalamic samples from RiboTag mice were homogenized with a Dounce homogenizer on ice in 500 μl ice-cold homogenization buffer (50 mM Tris, pH 7.4, 100 mM KCl, 12 mM MgCl2, 1% NP-40, 1 mM dithiothreitol (DTT), 1:100 protease inhibitor, 200 units/ml Rnasin®, 0.1 mg/ml cycloheximide in 10% w/v Rnase-free DW). After centrifugation (4°C at 11.9 × g for 10 min), 50 μl of supernatant was immediately frozen and stored as an input sample. The remaining supernatant was split into 2 samples and subjected to IP using a hemagglutinin antibody (Covance) or mouse monoclonal IgG1 (Merck) and protein A/G magnetic beads (Promega) on a rotator at 4°C overnight. The following day, RiboTag-IP fractions were placed on a DynaMag-2 magnet (Life Technologies) on ice, and the bead pellet was washed with high salt buffer (HSB: 50 mM Tris, 300 mM KCl, 12 mM MgCl2, 1% NP-40, 1 mM DTT, and 100 μg/ml cyclohexamide). The beads were re-suspended in 400 μl of supplemented RLT buffer (10 μl beta-mercaptoethanol/10 ml of RLT Buffer) from the RNeasy plus micro kit (Qiagen) and vortexed vigorously. RLT buffer containing the immunoprecipitated RNA was removed from the magnetic beads prior to RNA purification using an RNeasy kit (Qiagen) in accordance with the manufacturer’s protocol. Likewise, 350 μl of RLT was added to the input fractions prior to RNA isolation. Both input and RiboTag-IP samples were eluted in 14 μl of water and subjected to real time PCR.
Immunoblotting
Fifty micrograms of tissue lysates were separated using 8% SDS-PAGE and transferred to PVDF membranes (GE Healthcare). After incubation in blocking buffer, the membranes were incubated overnight at 4 °C with antibodies against ATP5A1 (1:1000), MTCO1 (1:1000) and β-ACTIN (1:5000). Blots were developed using horseradish peroxidase (HRP)-linked secondary antibody and a chemiluminescent detection system (PerkinElmer). The band intensities of ATP5A1 and MTCO1 were normalized to that of β-ACTIN. The expression ratios of ATP5A1 versus MTCO1 were determined.
Adipose tissue histology and sympathetic innervation
The BAT, iWAT and gWAT were dissected, fixed in 4% PFA overnight and embedded in paraffin. Paraffin tissue sections were stained with hematoxylin and eosin (H&E) and observed by microscopy (BX53, Olympus). Sympathetic nerve terminals were stained as described previously (Vargovic et al., 2011). Paraffin sections of mouse adipose tissue were deparaffinized and rehydrated by washing twice for 10 min with xylene, followed by washes in 100%, 95% and 70% ethanol (2 min twice each). Sections were rinsed in distilled water, placed into a warmed citrate buffer (0.01 M, pH 6.0) for 5 min, followed by immersion in a hot citrate buffer (58 °C) and cooled at RT, washed with Tris-buffered saline (TBS) for 5 min three times, transferred into methanol solutions containing 0.5% H2O2 and washed again in TBS. Pre-blocking procedure with a 20 μg/ml solution of anti-rabbit IgG (Santa Cruz) for 1 h at RT was followed by blocking with 2% of normal horse serum for 1 h at RT. Slides were incubated with rabbit anti-tyrosine hydroxylase antibody (1:250, rabbit, MerckMillipore) overnight at 4 °C and then with Alexa-Flour 488-conjugated anti-rabbit secondary antibody (1:500) for 1 h at RT.
Chemical sympathetic denervation in the iWAT was performed as described previously (Harris, 2012). Briefly, under anesthesia, we exposed one side of the iWAT from a Pomc-Cre; Crif1f/+ male mouse (6–8 weeks of age) and microinjected 6-hydroxydopamine (6-OHDA) using a microsyringe [20 injections of 1 μl of 6-OHDA solution (9 mg dissolved in 1 ml of 0.15 M NaCl containing 1% (w/v) ascorbic acid)] as described previously (Chao et al., 2011). We microinjected the same volume of vehicle (20 injections) to the other side of the iWAT in the Pomc-Cre; Crif1f/+ mice or to both sides of iWAT of Crif1f/f controls. Two weeks after the 6-OHDA injections, both sides of the iWAT were collected, divided into two pieces, and subjected to histological and gene expression analysis.
Mitochondrial mass and morphology examinations
The mitochondrial morphology and network complexity in the POMC neurons were assessed by immunogold electron microscopy and analysis as described previously (Dietrich et al., 2013). Briefly, mice were transcardially perfused with 0.9% saline with heparin followed by fixative solution (4% PFA, 0.1% glutaraldehyde, 15% picric acid in 0.1 M phosphate buffer, pH 7.4). The brains were then removed and fixed overnight with the same fixative without glutaraldehyde. The brain were sliced at a 50 μm thickness using a vibrotome and subjected to POMC immunostaining using a primary antibody to β-END (1:1000, rabbit, Phoenix Pharmaceuticals, 48 h at 4 °C with gentle shaking) prior to the EM analysis. For mitochondria mass and morphology analysis in the BAT, mice were transcardially perfused as above. The BAT was cut into 1 mm3 fragments, washed in fresh 0.1 M phosphate buffer (pH 7.4) and fixed in the same buffer containing 2.5% glutaraldehyde at RT for 4 h. Tissues were then washed three times in fresh 0.1 M phosphate buffer (pH 7.4) for 10 min each. Subsequently, the tissues were fixed in 1% OsO4 for 1 h at RT and washed three times in 0.1 M phosphate buffer for 10 min each. Finally, the tissues were embedded in Epon using standard techniques following dehydration with ethyl alcohol and propylene oxide. Ultrathin sections (60 nm) were cut from the blocks using a Reichert–Jung Ultracut E ultramicrotome and a diamond knife. The sections were collected and stained with uranyl acetate, followed by lead citrate, and then observed using a JEM 1400 transmission electron microscope (JEOL Ltd.). To assess mitochondria biogenesis, DNA was extracted from the BAT and iWAT using DNA extraction kit (Qiagen) and the mtDNA content in the BAT and iWAT was quantitated using real time PCR analysis and normalized by the nDNA content as previously described (Weimer et al., 2014).
ELISA and radioimmunoassay
Mediobasal hypothalamic blocks were collected, weighed and quickly frozen in liquid nitrogen and stored at −80°C until use. Hypothalamic MOTS-c protein contents were measured using a MOTS-c commercial ELISA kit (MyBioSource) in accordance with the manufacturer’s protocol. Briefly, 50 μl of protein lysate from the mouse hypothalamus or N1 cells was added to the reaction and the absorbance of the sample was measured at 450 nm within 30 min. The hypothalamic α-MSH and β-END contents were measured using commercial RIA (Phoenix Pharmaceutical) and ELISA (MyBioSource) kits, respectively, according to the respective protocols provided by the manufacturer. Total protein amounts of samples were assayed using a BCA protein assay kit (Thermo Fisher) and hypothalamic peptide content was normalized by total protein content.
Intracerebroventricular cannulation and peptide administration
Stainless steel cannulae (26 gauge; Plastics One) were implanted into the third cerebral ventricle or intra-ARH of the mice as previously described (Kim et al., 2006). Peptides were dissolved in 0.9% saline solution just before use and were administered via the brain-implanted cannulae. To examine whether ICV β-END administration can recapitulate the metabolic phenotype of POMC Crif1 heterodeficient mice, β-END (0.1 μg, Sigma) was ICV-injected twice a day (in the early light phase and 1 h before light-off) in C57 mice for 4 days. Food intake and body weight were monitored just before injections and the EE was measured on treatment day 3. In β-END feeding study, ad libitum-fed C57 mice received a single ICV injection of either saline or β-END (0.03 μg/2 μl) during the early light phase and food intake was monitored for 24 h. For ICV MOTS-c continuous infusion, MOTS-c peptide (generated by Changhan Lee’s lab) was infused into HFD-fed mice via an ICV-implanted cannula using an Alzet osmotic minipump placed in the interscapular area (3 μg/2.6 μl per day for 28 days). Food intake and body weight were monitored daily whilst the EE were measured on treatment day 20. In an additional set of experiments, either anti-MOTS-c antibody (from Changhan Lee’ lab) or IgG (0.2 μg/μl, 1 μl injection each side) was injected daily into the bilateral ARH of 7–8 weeks-old Crif1f/f or Pomc-Cre; Crif1f/+ mice for 1 week. For ICV IL-6 treatment, IL-6 peptide (R&D Systems) was ICV-infused in C57 mice using an osmotic minipump (0.4 μg per day for 5 days). At the end of the treatments, the mice were sacrificed in the early light phase under freely-fed conditions. The hypothalamus, iWAT and BAT were collected for histological examination and for real time PCR analysis of thermogenic and UPRmt genes.
Exercise study
Mice belonging to the regular exercise group were subjected to treadmill running for 2 weeks between 1800 and 2000 hours. Each exercise section consisted of a 15 min warm-up (treadmill speed gradually increased from 5 to 18 meters/min over 15 min) followed by 50 min of running (18 meters/min, no incline) and a 5 min cooling-down period. The mice were euthanized by decapitation 24 h after last running training. In another group, the mice were acclimated on the treadmill as previously described (Reynolds et al., 2019) and four days later were subjected to high-intensity running till exhausted as follows: (Stage 1) 5 min run at 13 meters/min, (Stage 2) another 5 min run with increasing speed (1 meters/min), (Stage 3) 30 min run at a fixed speed of 18 meters/min, and (Stage 4) open running until exhaustion at 23 meters/min (no incline). Mice were sacrificed immediately after exercise.
Hypothalamic ROS measurement and blockade
Hypothalamic ROS production was assessed using dihydroethidium (DHE) following a previously described methodology (Andrews et al., 2008; Toda et al., 2016). Briefly, DHE (10 mg/kg, Invitrogen) was administered into the tail vein 2 h prior to sacrifice. To block hypothalamic ROS formation, we injected 2 μl of ROS-scavenging cocktail containing ascorbic acid (10 μM), N-acetyl-L-cysteine (NAC, 10 μM) and Tempo (1 μM) via a cerebroventricle-implanted cannulae 10 min before each running session in the second week of the regular exercise program.
MOTS-c nuclear translocation study
N1 cells were treated with H2O2 (100 nM) with or without NAC (1 mM) for 24 h before fixation. In separate experiments, the cells were treated with IL-6 (100 nM) with or without NAC (1 mM) for 24 h. Cells were fixed with 4% PFA for 10 min at RT, washed with PBS, permeabilized in 0.2% PBST at RT for 10 min. The samples were then blocked with 3% BSA in 0.1% PBST and incubated with anti-MOTS-c antibody (1:200) at 4°C overnight, followed by incubation with anti-rabbit secondary antibody for 1 h. Samples were then washed three times with 0.2% PBST and incubated with DAPI (1:10,000) for 10 min at RT to visualize the nuclei. After three washes in PBST, the coverslips were mounted in mounting medium. The cellular images were obtained using a Zeiss LSM700 confocal microscope system and software.
Gene overexpression and knockdown
Mots-c- or Stat3-expressing plasmids at indicated doses were transfected into 60~70% confluent cells using Lipofectamine. For small interfering RNA (siRNA)-mediated gene knockdown, cells were transfected using Lipofectamine with siRNA targeting murine Stat3 (Ambion) or Crif1 (targeting nucleotide positions +520 to +545: 5’-GGAGUGCUCGCUUCCAGGAACUAUU-3’). Controls were transfected with a non-targeting scrambled siRNA. Successful gene overexpression or knockdown was confirmed by real time PCR (for Crif1), immunoblotting (for STAT3) or ELISA (for MOTS-c) in cells collected at 48 h post-transfection.
Pomc promotor activity
AtT20 cells were cultured in 12-well plates and were transfected with plasmids expressing murine Pomc promoter-luciferase reporter (pPomc-luc; nucleotides −1040 to +60, 200 ng) or pCMV-β-gal (25 ng) with or without human MOTS-c-expressing plasmid (pMOTS-c, 0.1 to 10 ng) using Lipofectamine. To study the interaction between MOTS-c and STAT3 in regulating Pomc promotor activity, cells were transfected with pPomc-luc, pCMV-β-gal, pMOTS-c plus pStat3 or mock plasmid. In a separate experiment, cells were transfected with pMOTS-c and Stat3 siRNA or non-targeting siRNA. The total amount of DNA used for transfection in each condition was corrected by the addition of mock DNA. At 48 h after transfection, cells were harvested to measure luciferase activities, which were normalized to β-galactosidase activities. Transfections were performed in triplicate and the experiments were repeated at least three times.
Chromatin immunoprecipitation study
N1 cells were transfected with MOTS-c-expressing plasmid or mock plasmid. After 48 h of transfection, cells were fixed with formaldehyde, lysed, and sonicated. Soluble chromatin was immunoprecipitated with either 1 μg of anti-STAT3 antibody (Cell Signaling) or an equivalent amount of rabbit IgG. After de-crosslinking of the DNA, the samples were subjected to PCR to amplify the region containing a potential STAT3 binding site on the mouse Pomc promoter (nucleotide positions −679 to −688) (Figure 5) using the primers: 5’-TCTCAAAACGGAACTGAGAT-3’ and 5’-ATAGGTAATTCCACTCCGA-3’. The PCR band intensity of immunoprecipitated DNA sample was corrected by the band intensity of the input sample.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data values in this study are presented as the mean ± SEM. Statistical analysis was performed using SPSS-PC (version 22) or Graph Pad Prism software (version 7.0). Statistical significance between groups was tested using one-way, two-way or repeated measures analysis of variance (ANOVA) tests followed by a post-hoc LSD (least significant difference) test, or an unpaired Student’s t-test when appropriate. Significance was defined by a p value < 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-α-Melanocyte stimulating hormone, sheep polyclonal | MerckMillipore | Cat#: AB5087 RRID: AB_91683 |
| anti-Atp5a1, mouse monoclonal | Abcam | Cat#: ab14748 RRID: AB_301447 |
| Anti-β-Actin, mouse monoclonal | Santa Cruz | Cat#: sc-47778 RRID: AB_2714189 |
| anti-β-Endorphin, rabbit polyclonal | Phoenix Pharmaceuticals | Cat#: H-022–33 |
| anti-c-Fos, rabbit polyclonal | Synaptic Systems | Cat#: 226003 RRID: AB_2231974 |
| anti-Hemagglutinin, epitope-tag, mouse monoclonal | Bio Legend | Cat#: MMS-101R RRID: AB_10063630 |
| anti-Mots-c, rabbit polyclonal | Dr. Changhan Lee (Kim et al., 2018) | N/A |
| anti-Mtco1, mouse monoclonal | Abcam | Cat#: ab14705 RRID: AB_2084810 |
| anti-Stat3, rabbit monoclonal | Cell Signaling Technology | Cat#: 4904 RRID: AB_331269 |
| anti-Tyrosine hydroxylase, rabbit polyclonal | MerckMillipore | Cat#: AB152 RRID: AB_390204 |
| anti-Rabbit conjugated to HRP, Goat | Columbia Biosciences | Cat#: HRP004–18-005 |
| anti-Sheep IgG (H+L), Alexa Fluor 488, Donkey | Invitrogen | Cat#: A11015; RRID: AB_2534082 |
| anti-Rabbit IgG (H+L), Alexa Fluor 488, Donkey | Invitrogen | Cat#: A21206; RRID: AB_2535792 |
| anti-Rabbit IgG (H+L), Alexa Fluor 555, Donkey | Invitrogen | Cat#: A31572; RRID: AB_162543 |
| IgG, normal rabbit | Santa Cruz | Cat#: sc-2027 RRID: AB_737197 |
| IgG, goat | Sigma | Cat#: M8642 RRID: AB_260698 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ascorbic acid | Sigma | Cat#: 1043003 |
| β-Endorphin | Sigma | Cat#: E1142 |
| CellROX™ orange reagent | Thermo Fisher Scientific | Cat#: C10443 |
| Dihydroethidium | Thermo Fisher Scientific | Cat#: D1168 |
| Insulin (Humulin-R®) | Eli Lilly | Cat#: HI0210 |
| Interleukin-6 | R & D Systems | Cat#: 406-ML-005/CF |
| Leptin | R & D Systems | Cat#: 498-OB |
| MOTS-c peptide | Dr. Changhan Lee (Kim et al., 2018) | N/A |
| N-Acetyl cysteine | Sigma | Cat#: A7250 |
| Tamoxifen | Sigma | Cat#: T5648 |
| Tempo | Sigma | Cat#: 214000 |
| 6-Hydroxydopamine | Sigma | Cat#: H4381 |
| Critical Commercial Assays | ||
| α-MSH radioimmunoassay | Phoenix Pharmaceuticals | Cat#: RK-043–01 |
| β-Endorphin ELISA | MyBioSource | Cat#: MBS703919 |
| BCA protein assay kit | Thermo Fisher | Cat#: 23225 |
| DNA extraction kit | Qiagen | Cat#: 69504 |
| MOTS-c ELISA | MyBioSource | Cat#: MBS2090467 |
| RNeasy Plus mini kit | Qiagen | Cat#: 74136 |
| Diet | ||
| Standard chow diet | Cargill Agri Purina | Cat#: 38057 |
| High fat diet | Research Diets | Cat#: D12492 |
| Experimental Models: Cell Lines | ||
| Mouse: Hypothalamus cell line N1 (mHypoE-N1) | Cedarlane | Cat#: CLU101 |
| Mouse: AtT-20 cell line | Obtained from Dr. Byung Ju Lee (University of Ulsan, Korea) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Crif1-loxP | Dr. Minho Shong (Kwon et al., 2008) | N/A |
| Mouse: Pomc-Cre: B6.FVB-Tg (Pomc-Cre)1Lowl/J | Obtained from Joel K Elmquist (University of Texas Southwestern Medical Center, USA) | N/A |
| Mouse: Tg (Pomc-Cre/ERT2)#Jke | Obtained from Joel K Elmquist (University of Texas Southwestern Medical Center, USA) | N/A |
| Mouse: Rpl22tm1.1Psam: B6N.129-Rpl22<tm1.1Psam>/J | Jackson Laboratory | Cat#: JAX:029977 |
| Mouse: GFP-loxP: B6.129 (Cg)-Gt (ROSA) 26Sortm4 (ACTB-tdTomato,-EGFP)Luo | Jackson Laboratory | Cat#: JAX:007676 |
| Mouse: C57BL/6J | Orient Bio | N/A |
| Oligonucleotides | ||
| Primers for CHIP assay | In this paper | N/A |
| Mouse: Stat3 siRNA | Ambion | Cat#: AM16708 |
| Mouse: Crif1 siRNA | Thermo Fisher Scientific | Cat#: 5319236 |
| Non-targeting siRNA | Thermo Fisher Scientific | Cat#: AM4611 |
| RNA probe for mouse Pomc | RNAscope® Probe - form Advanced Cell Diagnostic | Cat#: 314081 |
| Primers for qRT-PCR, see Table S1 | N/A | |
| Recombinant DNA | ||
| Plasmid: Mots-c | Dr. Changhan Lee (Kim et al., 2018) | N/A |
| Plasmid: Stat3 | Dr. Minho Shong (Kwon et al., 2008) | N/A |
| Pomc promotor-luciferase construct | This paper | N/A |
| Software and Algorithms | ||
| BD FACS Aria II Cell Sorter | BD Biosciences | N/A |
| GraphPad Prism 7.0 | GraphPad Software | http://www.graphpad.com/scientific-software/prism/ |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| Photoshop version CS6 | Adobe | https://www.adobe.com/ |
| SPSS (version 22) | IBM | N/A |
| Other | ||
| Accu-Chek glucometer | Roche Diabetes Care | Cat#: GC04640323 |
| Alzet osmotic pump | Alzet | Cat#: 1004 |
| Comprehensive Lab Animal Monitoring System | Columbus Instruments | N/A |
| Dual x-ray absorptiometry (iNSiGHT VET DXA) | OsteoSys | N/A |
| Cryostat | Leica Biosystems | Product#: CM1850 |
| RNAscope® Fluorescent Multiplex Reagent Kit | Advanced Cell Diagnostics | Cat#: 320850 |
| Stainless steel dummy cannula | Plastics One | Cat#: C315DC |
| Stainless steel guide cannula | Plastics One | Cat#: C315G/Spc |
| Stainless steel internal cannula | Plastics One | Cat#: C315i/Spc |
| Thermal camera | FLIR | Product#: T650SC, |
| Thermometer | Physitemp Instruments | Product#: BAT-12 |
| Rompun® | Bayer | Cat#: 2138S |
| Zoletil® | Virbac | Cat#: 5G45 |
Highlights.
Mild mitoribosomal stress in POMC neurons leads to a high metabolic turnover.
POMC mitoribosomal stress activates thermogenesis and UPRmt in the adipose tissues.
mtDNA-derived MOTS-c coordinates mitoribosomal stress response in POMC neurons.
Regular exercise stimulates thermogenesis via hypothalamic neuronal mitohormesis.
ACKNOWLEDGEMENTS
This study was supported by grants from the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT of Korea (2017R1A2B3007123, 2017R1E1A1A01075126, 2020R1A2C3004843, 2020R1A4A3078962), the Asan Institute for Life Sciences (18-326, 2019-IP0855-1), and the National Institutes of Health (R01AG052558 and R01GM136837 to C.L. and R01DK123002 to Y.-B.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLALATION OF INTERESTS
C.L. is a consultant and shareholder of CohBar, Inc. All other authors declare no competing financial or other interests in relation to this study.
REFERENCES
- Anderson NS, and Haynes CM (2020). Folding the Mitochondrial UPR into the Integrated Stress Response. Trends in cell biology 30, 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschop MH, Shanabrough M, Cline G, Shulman GI, et al. (2008). UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 454, 846–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Pinto M, Peralta S, and Moraes CT (2013). Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nature medicine 19, 1111–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendzen KM, Durieux J, Shao LW, Tian Y, Kim HE, Wolff S, Liu Y, and Dillin A (2016). Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis. Cell 166, 1553–1563.e1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret SG, Draper SJ, and Simerly RB (2004). Trophic action of leptin on hypothalamic neurons that regulate feeding. Science (New York, N.Y.) 304, 108–110. [DOI] [PubMed] [Google Scholar]
- Chao PT, Yang L, Aja S, Moran TH, and Bi S (2011). Knockdown of NPY expression in the dorsomedial hypothalamus promotes development of brown adipocytes and prevents diet-induced obesity. Cell metabolism 13, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HK, Ryu D, Kim KS, Chang JY, Kim YK, Yi HS, Kang SG, Choi MJ, Lee SE, Jung SB, et al. (2017). Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. The Journal of cell biology 216, 149–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science (New York, N.Y.) 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CS, McKay SE, Holmbeck MA, Christian BE, Scortea AC, Tsay AJ, Newman LE, and Shadel GS (2018). Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling. Cell metabolism 28, 776–786.e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, Suyama S, Kelly K, Gyengesi E, Arbiser JL, et al. (2011). Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nature medicine 17, 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich MO, Liu ZW, and Horvath TL (2013). Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell 155, 188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd GT, Decherf S, Loh K, Simonds SE, Wiede F, Balland E, Merry TL, Munzberg H, Zhang ZY, Kahn BB, et al. (2015). Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 160, 88–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J, Wolff S, and Dillin A (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutia R, Meece K, Dighe S, Kim AJ, and Wardlaw SL (2012). beta-Endorphin antagonizes the effects of alpha-MSH on food intake and body weight. Endocrinology 153, 4246–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Voss-Andreae A, Cao WH, and Morrison SF (2005). Regulation of thermogenesis by the central melanocortin system. Peptides 26, 1800–1813. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Yamazaki T, and Kroemer G (2018). Linking cellular stress responses to systemic homeostasis. Nature reviews. Molecular cell biology 19, 731–745. [DOI] [PubMed] [Google Scholar]
- Gammage PA, Viscomi C, Simard ML, Costa ASH, Gaude E, Powell CA, Van Haute L, McCann BJ, Rebelo-Guiomar P, Cerutti R, et al. (2018). Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nature medicine 24, 1691–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RB (2012). Sympathetic denervation of one white fat depot changes norepinephrine content and turnover in intact white and brown fat depots. Obesity (Silver Spring, Md.) 20, 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Yang Y, Blais SP, Neubert TA, and Ron D (2010). The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Molecular cell 37, 529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, and Auwerx J (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Maretich P, and Kajimura S (2018). The Common and Distinct Features of Brown and Beige Adipocytes. Trends in endocrinology and metabolism: TEM 29, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SG, Yi HS, Choi MJ, Ryu MJ, Jung S, Chung HK, Chang JY, Kim YK, Lee SE, Kim HW, et al. (2017). ANGPTL6 expression is coupled with mitochondrial OXPHOS function to regulate adipose FGF21. The Journal of endocrinology 233, 105–118. [DOI] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK, et al. (2013). Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nature medicine 19, 83–92. [DOI] [PubMed] [Google Scholar]
- Kim KH, Son JM, Benayoun BA, and Lee C (2018). The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell metabolism 28, 516–524.e517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Pak YK, Jang PG, Namkoong C, Choi YS, Won JC, Kim KS, Kim SW, Kim HS, Park JY, et al. (2006). Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nature neuroscience 9, 901–906. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kwon MC, Ryu MJ, Chung HK, Tadi S, Kim YK, Kim JM, Lee SH, Park JH, Kweon GR, et al. (2012). CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell metabolism 16, 274–283. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Xiao J, Wan J, Cohen P, and Yen K (2017). Mitochondrially derived peptides as novel regulators of metabolism. The Journal of physiology 595, 6613–6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M, Varela L, Kim JG, Kim JD, Hernandez-Nuno F, Simonds SE, Castorena CM, Vianna CR, Elmquist JK, Morozov YM, et al. (2015). Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature 519, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon MC, Koo BK, Moon JS, Kim YY, Park KC, Kim NS, Kwon MY, Kong MP, Yoon KJ, Im SK, et al. (2008). Crif1 is a novel transcriptional coactivator of STAT3. The EMBO journal 27, 642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J, Rollins JA, Zang X, Wu D, Zou L, Wang Z, Ye C, Wu Z, Kapahi P, Rogers AN, et al. (2019). Translational Regulation of Non-autonomous Mitochondrial Stress Response Promotes Longevity. Cell reports 28, 1050–1062.e1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kim KH, and Cohen P (2016). MOTS-c: A novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free radical biology & medicine 100, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, Kim SJ, Mehta H, Hevener AL, de Cabo R, et al. (2015). The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell metabolism 21, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Tang S, Xue C, Liu Y, Wang J, Zhang W, Luo W, and Chen J (2019a). Mitochondrial-Derived Peptide MOTS-c Increases Adipose Thermogenic Activation to Promote Cold Adaptation. International journal of molecular sciences 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Wei M, Zhai Y, Li Q, Ye Z, Wang L, Luo W, Chen J, and Lu Z (2019b). MOTS-c peptide regulates adipose homeostasis to prevent ovariectomy-induced metabolic dysfunction. Journal of molecular medicine (Berlin, Germany) 97, 473–485. [DOI] [PubMed] [Google Scholar]
- Lv Y, Zhang SY, Liang X, Zhang H, Xu Z, Liu B, Xu MJ, Jiang C, Shang J, and Wang X (2016). Adrenomedullin 2 Enhances Beiging in White Adipose Tissue Directly in an Adipocyte-autonomous Manner and Indirectly through Activation of M2 Macrophages. The Journal of biological chemistry 291, 23390–23402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Carelli V, Manfredi G, and Chan DC (2014). Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell metabolism 19, 630–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, Hsu F, Radey MC, Peterson SB, Mootha VK, et al. (2020). A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottis A, Herzig S, and Auwerx J (2019). Mitocellular communication: Shaping health and disease. Science (New York, N.Y.) 366, 827–832. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Huo L, Nillni EA, Hollenberg AN, and Bjorbaek C (2003). Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology 144, 2121–2131. [DOI] [PubMed] [Google Scholar]
- Padilla SL, Carmody JS, and Zeltser LM (2010). Pomc-expressing progenitors give rise to antagonistic neuronal populations in hypothalamic feeding circuits. Nature medicine 16, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BK, Steensberg A, and Schjerling P (2001). Exercise and interleukin-6. Current opinion in hematology 8, 137–141. [DOI] [PubMed] [Google Scholar]
- Quiros PM, Mottis A, and Auwerx J (2016). Mitonuclear communication in homeostasis and stress. Nature reviews. Molecular cell biology 17, 213–226. [DOI] [PubMed] [Google Scholar]
- Ramanjaneya M, Bettahi I, Jerobin J, Chandra P, Abi Khalil C, Skarulis M, Atkin SL, and Abou-Samra AB (2019). Mitochondrial-Derived Peptides Are Down Regulated in Diabetes Subjects. Frontiers in endocrinology 10, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez S, Gomez-Valades AG, Schneeberger M, Varela L, Haddad-Tovolli R, Altirriba J, Noguera E, Drougard A, Flores-Martinez A, Imbernon M, et al. (2017). Mitochondrial Dynamics Mediated by Mitofusin 1 Is Required for POMC Neuron Glucose-Sensing and Insulin Release Control. Cell metabolism 25, 1390–1399.e1396. [DOI] [PubMed] [Google Scholar]
- Reynolds J, Lai RW, Woodhead JST, Joly JH, Mitchell CJ, Cameron-Smith D, Lu R, Cohen P, Graham NA, Benayoun BA, et al. (2019). MOTS-c is an Exercise-Induced Mitochondrial-Encoded Regulator of Age-Dependent Physical Decline and Muscle Homeostasis. bioRxiv, 2019.2012.2022.886432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JC, Bwiza CP, and Lee C (2020). Mitonuclear genomics and aging. Human genetics 139, 381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, and Zarse K (2010). How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Experimental gerontology 45, 410–418. [DOI] [PubMed] [Google Scholar]
- Roh E, Song DK, and Kim MS (2016). Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Experimental & molecular medicine 48, e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro A, Campolo M, Liu C, Sesaki H, Meli R, Liu ZW, Kim JD, and Diano S (2017). DRP1 Suppresses Leptin and Glucose Sensing of POMC Neurons. Cell metabolism 25, 647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, and Amieux PS (2009). Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proceedings of the National Academy of Sciences of the United States of America 106, 13939–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, and Ristow M (2007). Glucose Restriction Extends Caenorhabditis elegans Life Span by Inducing Mitochondrial Respiration and Increasing Oxidative Stress. Cell metabolism 6, 280–293. [DOI] [PubMed] [Google Scholar]
- Shao LW, Niu R, and Liu Y (2016). Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell research 26, 1182–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, and Agrawal RK (2003). Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell 115, 97–108. [DOI] [PubMed] [Google Scholar]
- Shpilka T, and Haynes CM (2018). The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nature reviews. Molecular cell biology 19, 109–120. [DOI] [PubMed] [Google Scholar]
- Suhm T, Kaimal JM, Dawitz H, Peselj C, Masser AE, Hanzén S, Ambrožič M, Smialowska A, Björck ML, Brzezinski P, et al. (2018). Mitochondrial Translation Efficiency Controls Cytoplasmic Protein Homeostasis. Cell metabolism 27, 1309–1322.e1306. [DOI] [PubMed] [Google Scholar]
- Tan JX, and Finkel T (2020). Mitochondria as intracellular signaling platforms in health and disease. The Journal of cell biology 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Berendzen KM, and Dillin A (2014). Systemic stress signalling: understanding the cell non-autonomous control of proteostasis. Nature reviews. Molecular cell biology 15, 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda C, Kim JD, Impellizzeri D, Cuzzocrea S, Liu ZW, and Diano S (2016). UCP2 Regulates Mitochondrial Fission and Ventromedial Nucleus Control of Glucose Responsiveness. Cell 164, 872–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargovic P, Ukropec J, Laukova M, Cleary S, Manz B, Pacak K, and Kvetnansky R (2011). Adipocytes as a new source of catecholamine production. FEBS letters 585, 2279–2284. [DOI] [PubMed] [Google Scholar]
- Wallingford N, Perroud B, Gao Q, Coppola A, Gyengesi E, Liu Z-W, Gao X-B, Diament A, Haus KA, Shariat-Madar Z, et al. (2009). Prolylcarboxypeptidase regulates food intake by inactivating alpha-MSH in rodents. The Journal of clinical investigation 119, 2291–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterson MJ, and Horvath TL (2015). Neuronal Regulation of Energy Homeostasis: Beyond the Hypothalamus and Feeding. Cell metabolism 22, 962–970. [DOI] [PubMed] [Google Scholar]
- Weimer S, Priebs J, Kuhlow D, Groth M, Priebe S, Mansfeld J, Merry TL, Dubuis S, Laube B, Pfeiffer AF, et al. (2014). D-Glucosamine supplementation extends life span of nematodes and of ageing mice. Nature Communications 5, 3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrighting DM, and Andrews NC (2006). Interleukin-6 induces hepcidin expression through STAT3. Blood 108, 3204–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, and Ruan HB (2015). Neuronal Control of Adaptive Thermogenesis. Frontiers in endocrinology 6, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, and Finkel T (2014). Mitohormesis. Cell metabolism 19, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarse K, and Ristow M (2015). A mitochondrially encoded hormone ameliorates obesity and insulin resistance. Cell metabolism 21, 355–356. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Wu X, Chen P, Liu L, Xin N, Tian Y, and Dillin A (2018). The Mitochondrial Unfolded Protein Response Is Mediated Cell-Non-autonomously by Retromer-Dependent Wnt Signaling. Cell 174, 870–883.e817. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets/code.