

Abstract
One of the consistent pathologies associated with both clinical and experimental traumatic brain injury is axonal injury, especially following mild traumatic brain injury (or concussive injury). Several lines of experimental evidence have demonstrated a role for NAD+ metabolism in axonal degeneration. One of the enzymes that metabolizes NAD+ in axons is Sarm1 (Sterile Alpha and TIR Motif Containing 1), and its activity is thought to play a key role in axonal degeneration. Using a Sarm1 knock-out mouse, we examined if loss of Sarm1 offers axonal injury protection and improves cognitive outcome after repeated mild closed head injury (rmCHI). Our results indicate that rmCHI caused white matter damage that can be observed in the corpus callosum, cingulum bundle, alveus of the hippocampus, and fimbria of the fornix of wild-type mice. These pathological changes were markedly reduced in injured Sarm1−/− mice. Interestingly, the activation of astrocytes and microglia was also attenuated in the areas with white matter damage, suggesting reduced inflammation. Associated with these improved pathological outcomes, injured Sarm1−/− mice performed significantly better in both motor and cognitive tasks. Taken together, our results suggest that strategies aimed at inhibiting Sarm1 and/or restoring NAD+ levels in injured axons may have therapeutic utility.
INTRODUCTION.
The length of axons in a human nervous system can range from a millimeter to as long as a meter. Because of their length, axons are particularly vulnerable to mechanical injuries such as traumatic brain injury (TBI). Both clinical and experimental studies have repeatedly reported that mild, moderate, and severe forms of TBI can all cause axonal injury (Bramlett and Dietrich, 2007; Sharp and Ham, 2011; Smith et al., 2003). The transmission of traumatic force from the skull into the brain results in axonal stretching, which in turn initiates neurochemical changes within the axons that can alter axonal transport and lead to axonal severance. Transported proteins such as amyloid precursor protein (APP) can accumulate at focal sites where transport is impaired after injury, allowing axonal damage to be assessed by immunostaining for these proteins (Kelley et al., 2006; Stone et al., 2004, 2000). Removal of axonal debris in the mammalian CNS can last from days-to-weeks (Bignami and Ralston, 1969; Miklossy et al., 1991; Miklossy and Van der Loos, 1991, 1987; Perry et al., 1987). This is thought to be due to a relatively slower activated microglia clearance of axonal fragments than occurs in the peripheral CNS (Bignami and Ralston, 1969; George and Griffin, 1994; Perry et al., 1987). Contrary to the notion that axonal degeneration is passive, studies of a naturally occurring mutation in the C57BL6 strain of mice referred to as slow Wallerian degeneration (WldS) have revealed that axonal degeneration is an active process that involves nicotinamide adenine dinucleotide (NAD) metabolism (Lunn et al., 1989; Raff et al., 2002; Tsao et al., 1999). The mutation underlying WldS generates a chimeric protein (fusion of the N-terminus of UBE4B and the C-terminus of the nuclear localized NAD synthesizing enzyme Nmnat1) (Conforti et al., 2000) that is translocated to the axon where it supports local NAD synthesis and flux (Wang et al., 2015). These and other studies have indicated that NAD+ metabolism may dictate axon survival versus degeneration following an injury (Mack et al., 2001; Sasaki et al., 2016).
Experiments carried out in both Drosophila and mice have shown that loss of sterile alpha and TIR motif-containing protein 1 (Sarm1) can protect axons against degeneration (Gerdts et al., 2015; Gilley et al., 2017, 2015; Osterloh et al., 2012). Sarm1 was found to be a NAD+ degrading enzyme that cleaves NAD+ to generate ADP-ribose, cyclic ADP ribose, and nicotinamide, further supporting a role for NAD+ in axonal preservation (Gerdts et al., 2015; Horsefield et al., 2019). The localized depletion of NAD+ in injured axons by Sarm1 is thought to be an essential component of axonal degeneration, suggesting that Sarm1 may play a role in TBI-triggered axonal injury. Consistent with this, a study by Henninger et al., has reported that Sarm1−/− mice have reduced axonal injury and acute preservation of neurological function after controlled cortical impact (CCI) injury as compared to C57BL6 injured mice (Henninger et al., 2016). Additionally, Ziogas and Koliatsos used CLARITY microscopy to visualize individual axons in Thy1-eYFP-Sarm1−/− mice and found a reduction of injured axons in Sarm1−/− mice as compared to wild-type mice after CCI (Ziogas and Koliatsos, 2018). While these studies suggest a role for Sarm1 in mediating axonal injury after TBI, it has not been examined if the observed axonal protection is associated with improved cognitive function in brain injured Sarm1−/− mice.
In this study, we used a mouse model of repeated mild closed head injury (rmCHI) to examine if loss of Sarm1 lessens cognitive dysfunction. Previous studies have shown that rmCHI causes white matter damage in the absence of overt neuronal loss (Hylin et al., 2013; Ojo et al., 2016). Similar to what was observed following CCI (Henninger et al., 2016), we found evidence of decreased axonal injury in Sarm1−/− mice subjected to rmCHI. Injured Sarm1−/− mice also exhibited reduced markers of inflammation as compared to injured wild-type mice. Associated with these changes, Sarm1−/− injured mice had significantly improved learning and memory, suggesting that strategies to inhibit Sarm1 to preserve axonal NAD+ levels may be beneficial in the treatment of TBI.
Materials and Methods
Materials.
Male Sarm1 knockout mice (B6.129X1-Sarm1tm1Aidi/J; strain 018069) were purchased from Jackson Laboratories (Bar Harbor, ME). The following antibodies were used in these studies: astrocytic marker GFAP (Bethyl, custom), microglia marker Iba1 (Synaptic Systems, cat# 234004), myelin marker MBP (Millipore, cat# MAB386), dendritic marker MAP2 (Millipore, cat# AB5622), neuronal marker NeuN (Millipore, cat# ABN91), and axonal injury marker amyloid precursor protein APP (Abcam, cat# ab32136). Species-specific secondary antibodies linked to AlexaFluor dyes (AlexaFluor-430, AlexaFluor-488, AlexaFluor-54, AlexaFluor-594, and AlexaFluor-647) were purchased from Thermo Fisher Scientific (Waltham, MA).
Repeated mild closed head injury (rmCHI).
The Sarm1−/− mice purchased from Jackson Labs have been backcrossed to C57BL/6J mice for at least 16 generations and maintained using a homozygous × homozygous breeding scheme. Therefore, wild-type C57BL/6J mice are recommended by Jackson Labs as the appropriate control strain. Twenty male Sarm1−/− mice and twenty C57BL/6J mice (aged 24 weeks) were single housed on a 12-hour light/dark cycle, with ad libitum access to food and water. Experiments were performed during the light cycle. All experimental procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and approved by the Institutional Animal Care and Use Committee (IACUC). Repeat mild closed head injury was delivered essentially as described previously (Hylin et al., 2013; Ojo et al., 2016). Mice were initially anesthetized with 5% isoflurane in a 1:1 O2/air mixture, mounted on a stereotaxic frame, then maintained with a 2.5% isoflurane and 1:1 O2/air mixture via a face mask. A midline incision was made and the soft tissue reflected to expose the skull. A stereotaxic frame was used to immobilize the animal during the surgical incision and ensure a smooth cut was made. The mouse was then transferred to a foam pad designed to support the head level with the body. At 40 sec after discontinuation of anesthesia (a time point at which mice typically regain their tail withdrawal reflex after anesthesia), a single impact was applied to the skull. A metal impactor tip (5 mm in diameter) was driven at a velocity of 5.0 m/sec to a depth of 1.0 mm. The center of the tip was located midway between lambda and bregma, over the sagittal suture. Immediately following the injury, the animals were monitored for apnea, and when normal breathing was observed, the scalp was closed using sterile surgical staples. Mice received one injury/day for 4 consecutive days. Minor skull fractures were observed in 15% of the injured mice independent of genotype. Mice with skull fractures, noted either during surgery or during euthanasia, were removed from the study. Sham mice received daily anesthesia, but were not injured.
Vestibulomotor and Motor Function.
The foot-fault, or grid walking task, and Rotarod tasks were used to assess motor function and coordination. Motor testing began 24 hr after the last injury, and was carried out for three consecutive days. For each day of testing, animals were brought to the testing room and allowed to acclimate for an hour. Following the acclimation period, animals were first tested in the foot fault task, followed by a 1–2 hour break. Animals were then tested on the Rotarod task. This testing order was chosen to avoid any fatigue or distress from the Rotarod task that could have altered performance in the foot fault task.
Foot-faults were evaluated by placing the animal on a wire grid (1×1 cm) and the number of foot misplacements out of a total of 50 steps were counted. A fault was defined as when a front paw missed and appeared below the plane of the wire grid. Right and left forepaws were scored independently on three trials/day which were averaged. Animals were trained on the Rotarod for the first two days after injury by placing the animal upon the stationary Rotarod for a period of 5 sec, after which a slow rotation (4 rpm) was initiated and then incrementally increased by 4 rpm every 10 sec until a maximum of 40 rpm was reached. On the third day after the last injury, locomotor function was tested by placing the animal upon the stationary Rotarod for a period of 5 sec, after which the rotation was initiated at a constant speed of 32 rpm. Rotarod testing was repeated three times and averaged. Animals were allowed 10 to 15 minutes between trials to ensure ample time for the animals to rest and recover.
Novel Objection Recognition (NOR).
Beginning on day 4 post-injury, animals were habituated to an empty training chamber (30 cm × 30 cm) for two 10-minute periods per day for two days. On the third day, training was carried out by placing two identical objects in the chamber and the animal allowed to freely explore them for 10 minutes. Twenty four hours later, the mouse was tested for its long-term memory by replacing one of the objects with a new object of a different shape and allowing it to again explore the objects for 10 minutes. Trials were video recorded and the time spent exploring each object was measured during both the familiarization period and memory test by an experimenter blind to the animals’ experimental group.
Context Fear Discrimination.
The context fear discrimination task assesses an animal’s ability to distinguish between two similar contexts (one “safe” and one “shock”), and has been shown to be dependent on hippocampal function (Frankland et al., 1998). On day 28 post-injury, mice were pre-exposed (without shock) to two contexts sharing certain features (background noise, animal handling to and from the room) while differing in others (different floor surface, scent, and wall colors). During training, animals were given two trials each day, one in each chamber (“safe” and “shock”). For the “shock” chamber, mice were placed in the chamber and allowed to freely explore their surroundings for 148 sec. A 2-sec, 0.70 mA shock was then given, followed by an additional 30 sec during which the animal remained in the chamber. In the “safe” chamber, animals were allowed to freely explore the chamber for the entire 3 min and no shock was given. Animals were exposed to the “shock” and “safe” chambers once a day for 4 days. Discrimination of the two contexts was assessed by comparing the time spent freezing in each chamber during training. Trials were recorded and freezing behavior analyzed using tracking software (Ethovision, Noldus Information Technology, Leesbury VA, USA).
Immunohistochemistry.
After the completion of behavioral testing (six months after the last injury) mice were given an overdose of sodium pentobarbital (100 mg/kg), followed by exsanguination with ice-cold phosphate-buffered saline (PBS) followed by PBS containing 4% paraformaldehyde. Brains were carefully removed and post-fixed in PBS/4% paraformaldehyde overnight, and then cryoprotected in PBS with 30% sucrose. Brains were sectioned on a cryostat in the coronal plane at 20 μm through the rostral-caudal extent of the brain. Selected sections were rinsed in PBS and then permeabilized in PBS containing 0.25% Triton X-100 two times for 10 min each. After washing in PBS, sections were blocked in PBS containing 2.5% BSA and 2.5% normal goat serum for 1 h, then incubated overnight in blocking buffer supplemented with primary antibody (0.5–1.0 μg/ml). After extensive washing in PBS, sections were incubated for 1 h in PBS containing species-specific secondary antibodies linked to AlexaFluor dyes. After additional washing, sections were rinsed for 2 min in PBS containing 0.1 μg/ml Hoechst 33258 (Sigma Aldrich, St. Louis, MO). Sections were mounted onto glass Apex Superior Adhesive Slides (Leica, Wetzlar, Germany) and coverslipped with ProLong Gold (Life Technologies, Carlsbad, CA) to reduce fading.
Fluorescence Intensity Quantification.
Immunofluorescence imaging was performed using an Axio Scan.Z1 slide scanning microscope (Zeiss, Thornwood, NY) equipped with an X-Cite XYLIS LED light source (Waltham, MA) and custom filter sets for AlexaFluor-430, AlexaFluor-488, AlexaFluor-546, AlexaFluor-594, and AlexaFluor-647 (Semrock). Images were captured using a 20X/0.8 Plan-Apochromat objective (Zeiss) and ORCA-Flash4.0 sCMOS digital camera (Hamamatsu) at 325 nm/pixel resolution, and montaged using Zen Blue 2.3 software. Exposure and gain settings were adjusted using a section from a sham animal and then kept constant across all groups. Four sections from each animal, ~250 μm apart (Bregma −1.22 mm to −2.18 mm), were used to quantify fluorescence intensity signals for each of the markers in regions of interest (ROIs) in both hemispheres. For each marker, the ROI was traced in ImageJ. Myelin basic protein (MBP) immunoreactivity was used as a guide to accurately differentiate between the alveus and the overlying corpus callosum and external capsule. ROIs were identified using a mouse brain atlas and regions included the corpus callosum (750 μm traced at the midline between the cingulum bundle of either hemisphere), the entirety of the cingulum bundle, the alveus (traced from middle of the cingulum bundle to the lateral ventricle), and the entirety of the fimbria. The off-target fluorescence intensity (measurement of region without specific signal) for each fluorochrome was measured for each section, averaged and subtracted from the target signal for quantification. The corrected fluorescent intensity was then averaged across the four sections for each animal and data (mean ± standard error) is presented relative to wild-type sham (set to 100%). For presentation purposes, a magnified image from within the ROI is shown to illustrate staining within axon bundles (for APP) or in astrocytes (for GFAP) or microglia (for Iba1).
Statistical Analysis.
Data were analyzed using Sigma Plot 14.0 (Systat Software, San Jose, CA). Acute neurological measures (duration of apnea and suppression of pain reflexes, and latency to return of righting response) and the number of foot missteps in foot fault were analyzed using three-way repeated measures ANOVAs for Injury, Genotype, and Time. Latency to fall from the Rotarod, and fluorescence intensity were analyzed using two-way ANOVAs for Injury and Genotype. Percent time freezing in contextual fear discrimination task was analyzed within each group with two-way repeated measures ANOVAs for Context and Time. Percent time spent with the novel compared to familiar objects in the novel object recognition task was analyzed within groups using two-sample paired t-tests. All data was tested for normality using a Shapiro-Wilk test. Data not having a normal distribution was statistically compared using an appropriate nonparametric test. Data was not transformed to satisfy normality. Significant omnibus differences were further analyzed using Bonferroni corrected post-hoc analyses. All data are reported as means ± standard error of the mean.
Results
Acute neurological deficits after rmCHI.
After each injury (or exposure to anesthesia), acute neurological responses were recorded. rmCHI resulted in a significantly longer duration of apnea (injury group main effect F=135.536, p < 0.001). There was an interaction between the injury-induced apnea and genotype (F=5.054, p=0.027), with the apnea after the first and second injuries in the rmCHI Sarm1−/− animals being significantly longer than was recorded for the rmCHI wild-type animals (Fig. 1A). The duration of suppression of the pain reflex (tail-pinch) showed a significant injury-related difference (F=170.806, p < 0.001; Fig. 1B), though there was no significant difference related to genotype. Similarly, there was a significant injury-related difference in the latency to recover the righting response when compared to sham-operated controls (F=185.354, p < 0.001; Fig. 1C), but no genotype effect was observed. Across the repeated injuries, the latency to recover the righting response decreased, indicated by a significant main effect of time (F=6.628, p < 0.001), suggesting the possibility of pre-conditioning. Taken together, the acute neurological data suggested that the injury level was equivalent between the rmCHI wild-type and Sarm1−/− groups.
Figure 1. Acute neurological deficits after rmCHI.
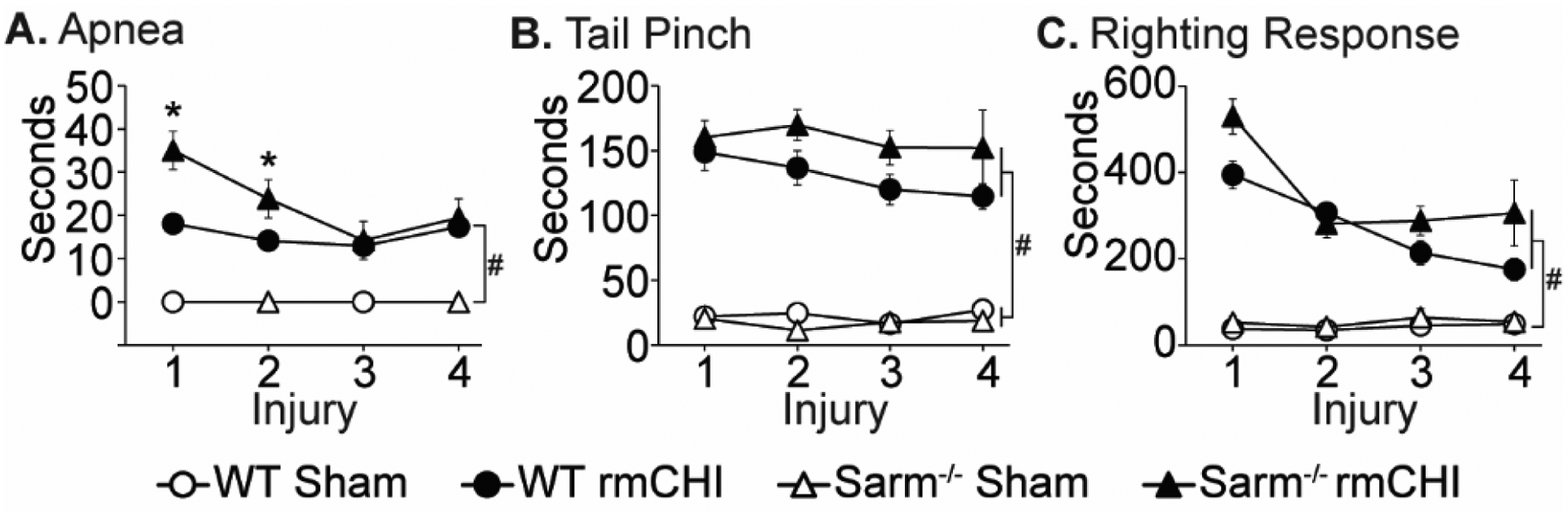
The durations of post-injury (A) apnea, (B) suppression of pain reflex (tail pinch), and (C) suppression of righting reflex were recorded immediately after each injury. Data are presented as the mean ± SEM. #significant difference between sham and injury group, p < 0.05; * significant difference between WT and knockout injury groups, p<0.05 post-hoc.
Axonal injury is attenuated in Sarm1−/− mice after rmCHI.
Figure 2 shows representative low magnification montaged images of coronal sections from sham and injured wild-type C57BL6J and Sarm1−/− mice immunostained with the neuronal marker NeuN (top panel; shown in green). No overt tissue damage or neuronal loss was apparent six-months after injury. Similarly, microtubule-associated protein 2 (MAP2) immunoreactivity showed no gross perturbation of dendrites in either sham or injured C57BL6J or Sarm1−/− mice (bottom panel; shown in red).
Figure 2. Repeated mild CHI does not result in gross tissue or cellular pathology.
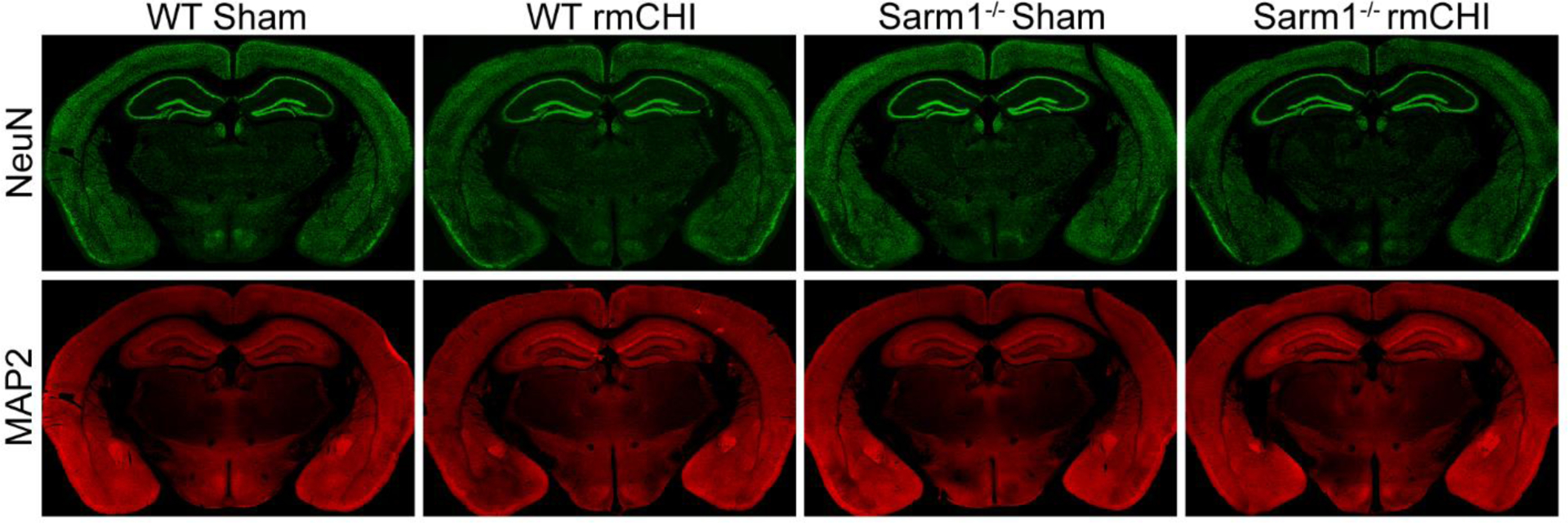
Representative montaged images of coronal brain sections taken six months after injury or sham surgery from a WT sham, WT rmCHI, Sarm1−/− sham, and Sarm1−/− rmCHI immunostained for the neuronal marker NeuN (top panel; green) and the dendritic marker Map2 (bottom panel; red).
Axonal injury was examined using amyloid precursor protein (APP) immunoreactivity, which has been previously shown to accumulate in injured axons (McGinn and Povlishock, 2016; Singleton et al., 2002; Stone et al., 2004; Weber et al., 2019). Representative high magnification images showing APP immunoreactivity within the white matter tracts of sham and injured wild-type and Sarm1−/− mice six months after injury or sham surgery are shown in Figure 3. Regions of interest were carefully outlined as described in the Materials and Methods section and shown in Supplemental Figure 1, and the fluorescence intensity measured. Data was compared using a two-way ANOVA. Summary data (presented as percent WT sham) of APP immunoreactivity within the corpus callosum shows that rmCHI (independent of genotype) caused a significant increase in APP immunoreactivity compared to uninjured controls (F=12.215, p=0.010). Although APP immunoreactivity in the corpus callosum of injured Sarm1−/− mice was reduced compared to injured WT mice, this reduction did not reach statistical significance (interaction of injury × genotype: F=3.163, p=0.119). Quantification of APP immunoreactivity in the cingulum revealed no significant effect of injury, nor genotype at the time point examined. However, APP quantification in the alveus revealed a significant interaction of injury and genotype, with injured Sarm1−/− mice having significantly reduced APP immunoreactivity compared to injured WT mice (interaction F=8.701; post hoc F=14.653, p = 0.006). Likewise, analysis of APP immunoreactivity in the fimbria of the fornix also revealed a significant interaction of injury and genotype (F=18.580, p=0.004), with the increase in APP immunoreactivity observed in WT mice not detected in injured Sarm1−/− mice. No significant differences were observed between uninjured wild-type and uninjured Sarm1−/− mice in any of the examined regions. These results suggest that loss of Sarm1 may offer axonal protection after injury.
Figure 3. Reduced amyloid precursor protein (APP) accumulation in white matter tracts of Sarm1−/− mice after injury.
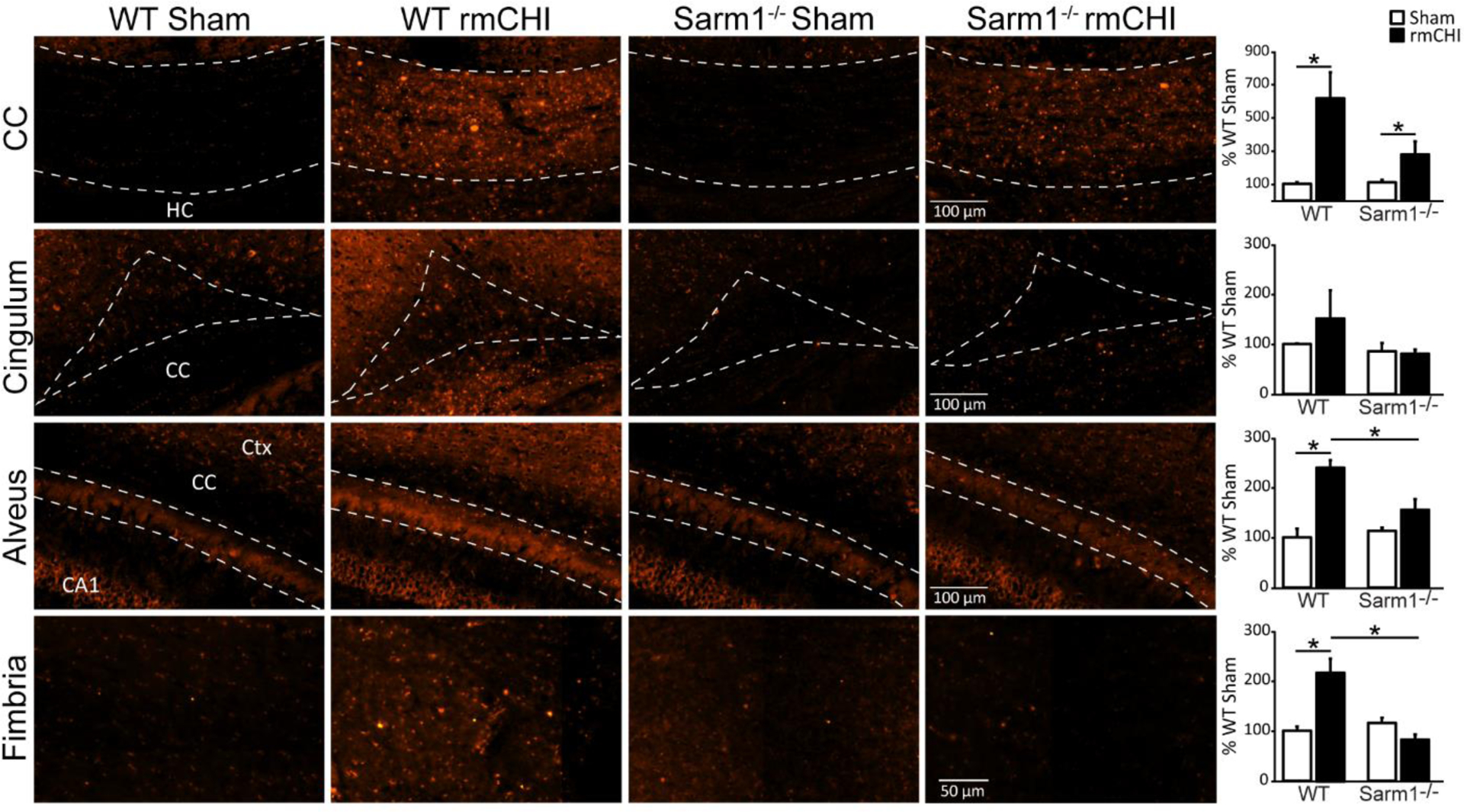
High magnification images of APP staining in white matter tracts (corpus callosum, cingulum, alveus, and fimbria of the fornix) from representative WT sham, WT rmCHI, Sarm1−/− sham, and Sarm1−/− rmCHI animals six months after injury or sham surgery. Summary data of the integrated fluorescent intensity of APP immunoreactivity in the white matter tracts are shown on the far right. Data (mean ± SEM) are shown as the percent change relative to the wild-type sham group. *p < 0.05 in two-way ANOVA with Bonferroni’s multiple comparisons. CA1: Cornu Ammonis 1; CC: corpus callosum; Ctx: cortex; HC: hippocampal commissure.
Loss of Sarm1 decreases white matter inflammation after rmCHI.
As glial populations have been implicated in clearing axonal debris after injury (Basiri and Doucette, 2010; Hosmane et al., 2012), we next examined the immunoreactivities of microglia (Iba1; Figure 4), and astrocyte (GFAP; Figure 5) markers in the damaged white matter tracts six months after injury or sham surgery. Regions of interest were carefully outlined as described in the Materials and Methods section and shown in Supplemental Figure 1, and the fluorescence intensity measured. Data was compared using a two-way ANOVA. The summary data is presented as percent WT sham.
Figure 4. Microglial activation after rmCHI is attenuated in Sarm1−/− animals.
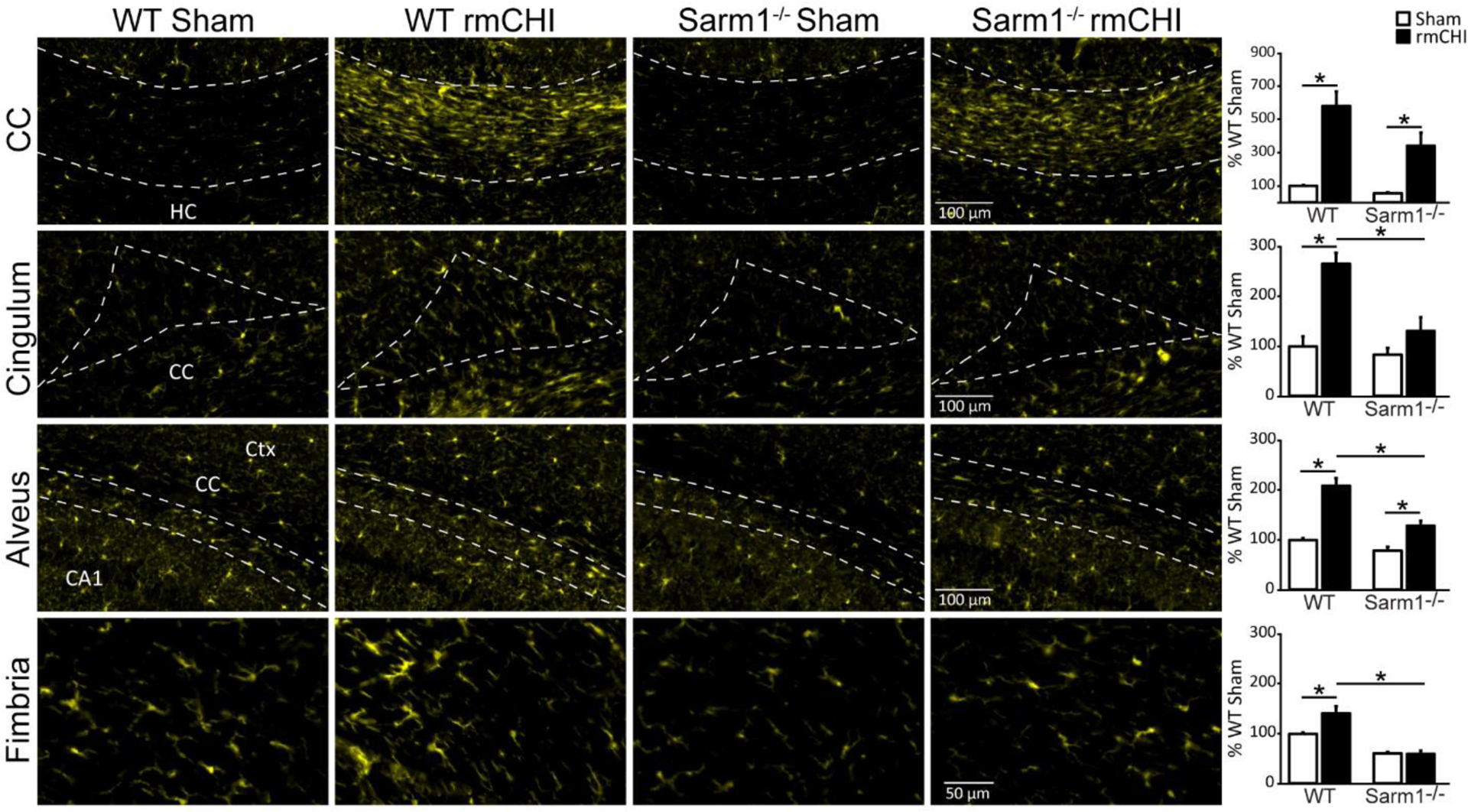
Representative high magnification images of Iba1 immunoreactivity from the white matter tracts (corpus callosum, cingulum, alveus, and fimbria of the fornix) of WT sham, WT rmCHI, Sarm1−/− sham, and Sarm1−/− rmCHI animals six months after injury or sham surgery. Summary data of the integrated fluorescent intensity of Iba1 immunoreactivity in the white matter tracts are shown on the far right. Data (mean ± SEM) are shown as the percent change relative to the wild-type sham group. *p < 0.05 in two-way ANOVA with Bonferroni’s multiple comparisons. CA1: Cornu Ammonis 1; CC: corpus callosum; Ctx: cortex; HC: hippocampal commissure.
Figure 5. Loss of Sarm1 reduces astrogliosis after rmCHI.
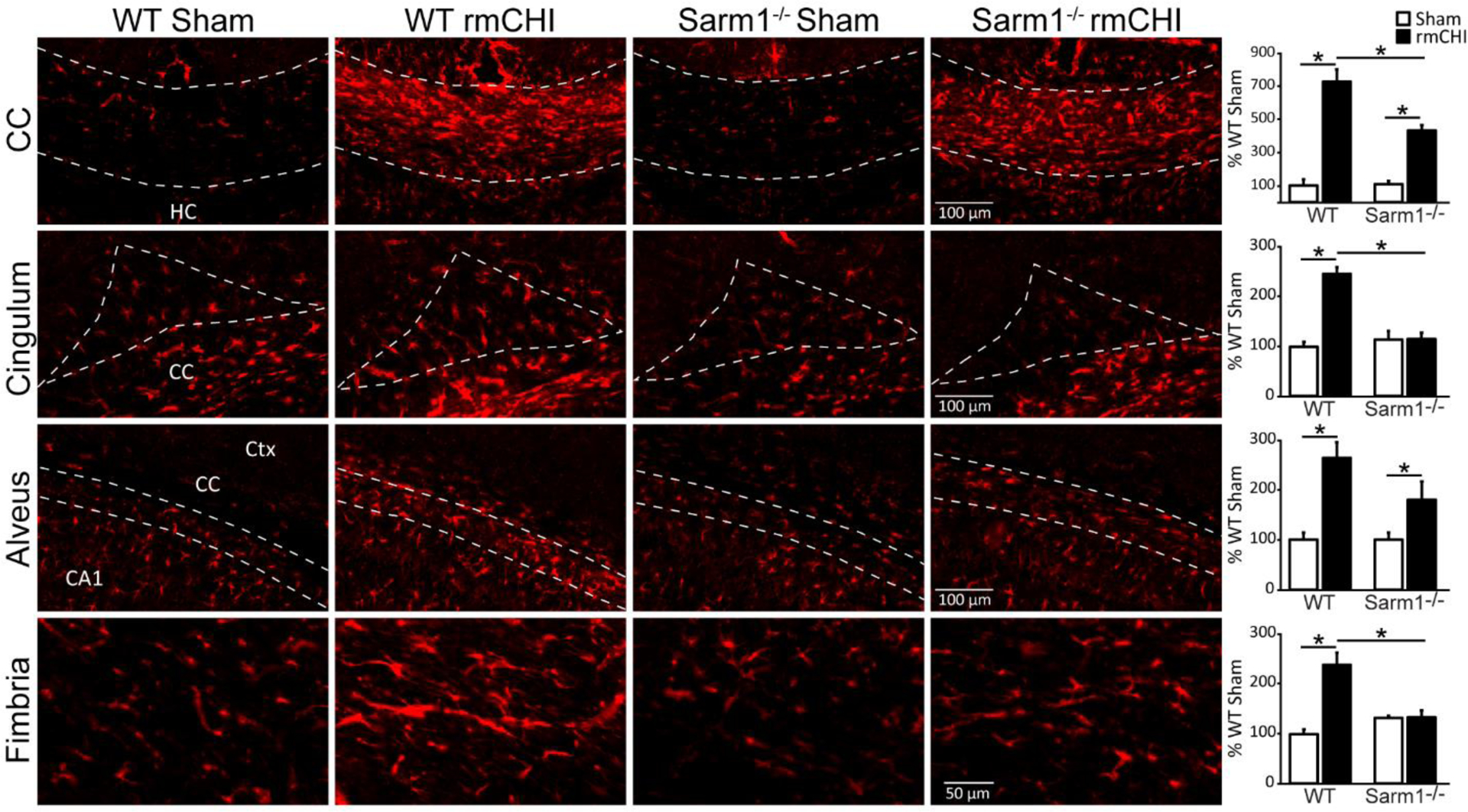
Representative high magnification images of GFAP immunoreactivity in the white matter tracts (corpus callosum, cingulum, alveus, and fimbria of the fornix) of WT sham, WT rmCHI, Sarm1−/− sham, and Sarm1−/− rmCHI animals six months after injury or sham surgery. Summary data of the integrated fluorescent intensity of GFAP immunoreactivity in the white matter tracts. Data (mean ± SEM) are shown as the percent change relative to the wild-type sham group. *p < 0.05 in two-way ANOVA with Bonferroni’s multiple comparisons. CA1: Cornu Ammonis 1; CC: corpus callosum; Ctx: cortex; HC: hippocampal commissure.
Similar to that seen for APP immunoreactivity, quantification of Iba1 in the corpus callosum revealed that rmCHI (independent of genotype) caused a significant increase in Iba1 immunoreactivity compared to uninjured controls (F=33.037, p = 0.001; Figure 4). However, no significant difference between injured Sarm1−/− mice and injured WT mice was detected. In contrast, a significant interaction of injury and genotype was detected in the cingulum (F=6.953, p = 0.034), the alveus (F=7.125, p = 0.032), and the fimbria of the fornix (F=5.680, p = 0.049) with post hoc analysis revealing that injured Sarm1−/− mice had significantly less Iba1 immunoreactivity than WT injured mice (Figure 4). In uninjured mice, no significant differences were detected between WT and Sarm1−/− mice.
In addition to microglial activation/proliferation, Figure 5 shows that rCHI causes a dramatic increase in GFAP immunoreactivity in all of the white matter tracts examined, suggesting astrocyte activation. Quantification of GFAP immunoreactivity revealed that, with the exception of the alveus, loss of Sarm1 resulted in a significant attenuation of injury-related GFAP immunoreactivity in the corpus callosum (F=9.793, p = 0.017), the cingulum (F=44.641, p = 0.0001), and the fimbria of the fornix (F=17.525, p = 0.004). This reduction was likely due to an attenuation of glial activation after injury, as no pre-existing differences were observed between uninjured WT and uninjured Sarm1−/− mice.
Loss of SARM1 improves post-injury motor function.
Our results showing that Sarm1−/− mice have reduced white matter damage after rmCHI are consistent with previous reports using moderate-severe models of TBI (Henninger et al., 2016; Marion et al., 2019; Ziogas and Koliatsos, 2018). However, whether loss of Sarm1 is associated with improved motor or cognitive outcome has not been examined. Motor function was assessed using the foot fault task beginning 24 hours after the last injury and continuing for an additional 2 days (Fig. 6A). Uninjured sham C57BL6J and Sarm1−/− animals, which had undergone anesthesia each day, showed an average of less than 1 misstep/foot (out of 50 steps; Fig. 6B). Wild-type C57BL6J mice that received rmCHI made significantly more left (F=315.2, p < 0.001; Fig. 6B) and right (F=425.2, p < 0.001; Fig. 6C) foot missteps than were recorded in wild-type sham controls. Interestingly, while there was an rmCHI-related increase in foot-faults in the Sarm1−/− mice compared to their genetic sham counterparts (left: F=8.549, p = 0.001; right: F=9.894, p = 0.001), a significant interaction of genotype and injury was detected indicating that they made significantly fewer left foot (F=6.709, p = 0.001; Fig. 6B) and right foot (F=7.603, p = 0.001; Fig.6C) missteps compared to the injured wild-type mice. Three days after the last mCHI was administered, animals were tested using the Rotarod task as described in the Methods section. Injured animals had a significantly shorter latency to fall when compared to sham controls regardless of genotype (F=9.316, p < 0.005; Fig. 6D). There was no significant difference between injured WT and injured Sarm1−/− mice in their ability to perform the Rotarod task.
Figure 6. Loss of Sarm1 improves motor performance after rmCHI.
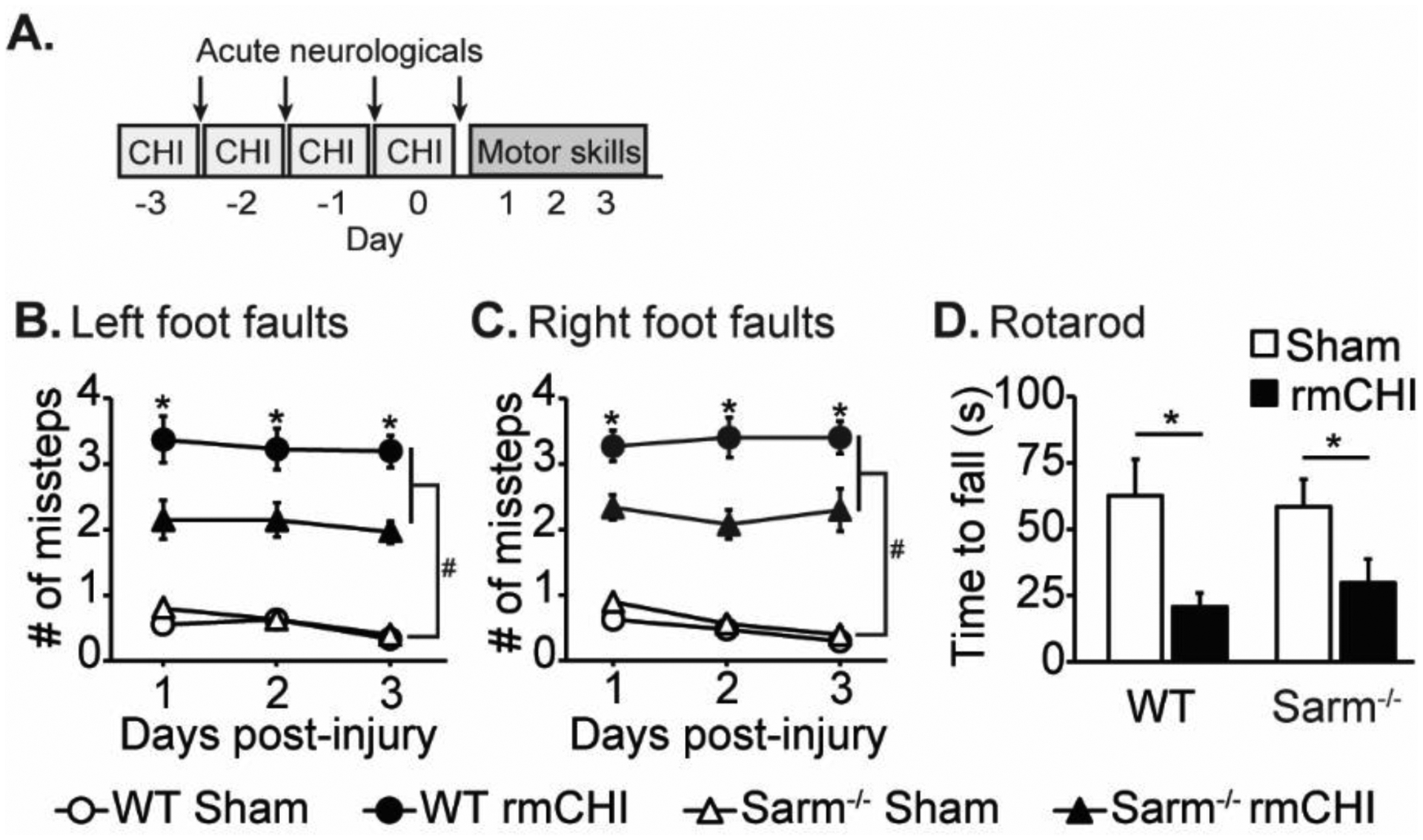
(A) Timeline for injury and motor assessments. Mice that received rmCHI made significantly more (B) left and (C) right foot fault errors (assessed using a 2 cm × 2 cm wire grid) than uninjured sham animals. Foot faults were significantly reduced in injured Sarm1−/− mice compared to injured WT mice. (D) rmCHI resulted in a significantly shorter latency to fall from the Rotarod compared to sham animals, regardless of genotype. Data are presented as mean ± SEM. #significant difference between sham and injury group, p < 0.05; *significant difference between WT and knockout injury groups, p<0.05 post-hoc.
rmCHI Sarm1−/− mice have improved recognition memory.
The integrity of the cingulum bundle, alveus, and the fimbria of the fornix are critical for communication between brain regions and have been implicated in learning and memory (Bubb et al., 2018; Harker and Whishaw, 2004; Neave et al., 1997; Warburton et al., 1998). We therefore tested the performance of sham and rmCHI-injured mice using the novel object recognition (NOR) task beginning 4 days after the last injury (Fig. 7A). The sequence of events for this task are depicted in Figure 7B. The exploratory times recorded for all 4 groups were equivalent (Fig. 7C) indicating there was no effect of genotype on exploratory behavior. Summary data (Fig. 7D) shows that during the testing phase of the NOR task, both WT sham (n=10; t=10.526, p < 0.0001) and Sarm1−/− sham (n=10; t=6.415, p < 0.0001) animals spent significantly more time exploring the novel object relative to the familiar object, indicating the gene knockout did not result in a baseline performance difference for recognition memory. Consistent with previous reports (Hylin et al., 2013), WT rmCHI mice (n=10) had a long-term recognition memory impairment and did not show a preference for the novel object in the testing phase (t=1.315, p = 0.207; Fig. 7D). In contrast, rmCHI Sarm1−/− mice spent significantly more time exploring the novel object as compared to the familiar one (t=3.145, p < 0.01; Fig. 7D), performing comparably to both uninjured Sarm1−/− and uninjured WT mice, suggesting that loss of Sarm1 was protective after injury.
Figure 7. Loss of Sarm1 improves object recognition memory after rmCHI.
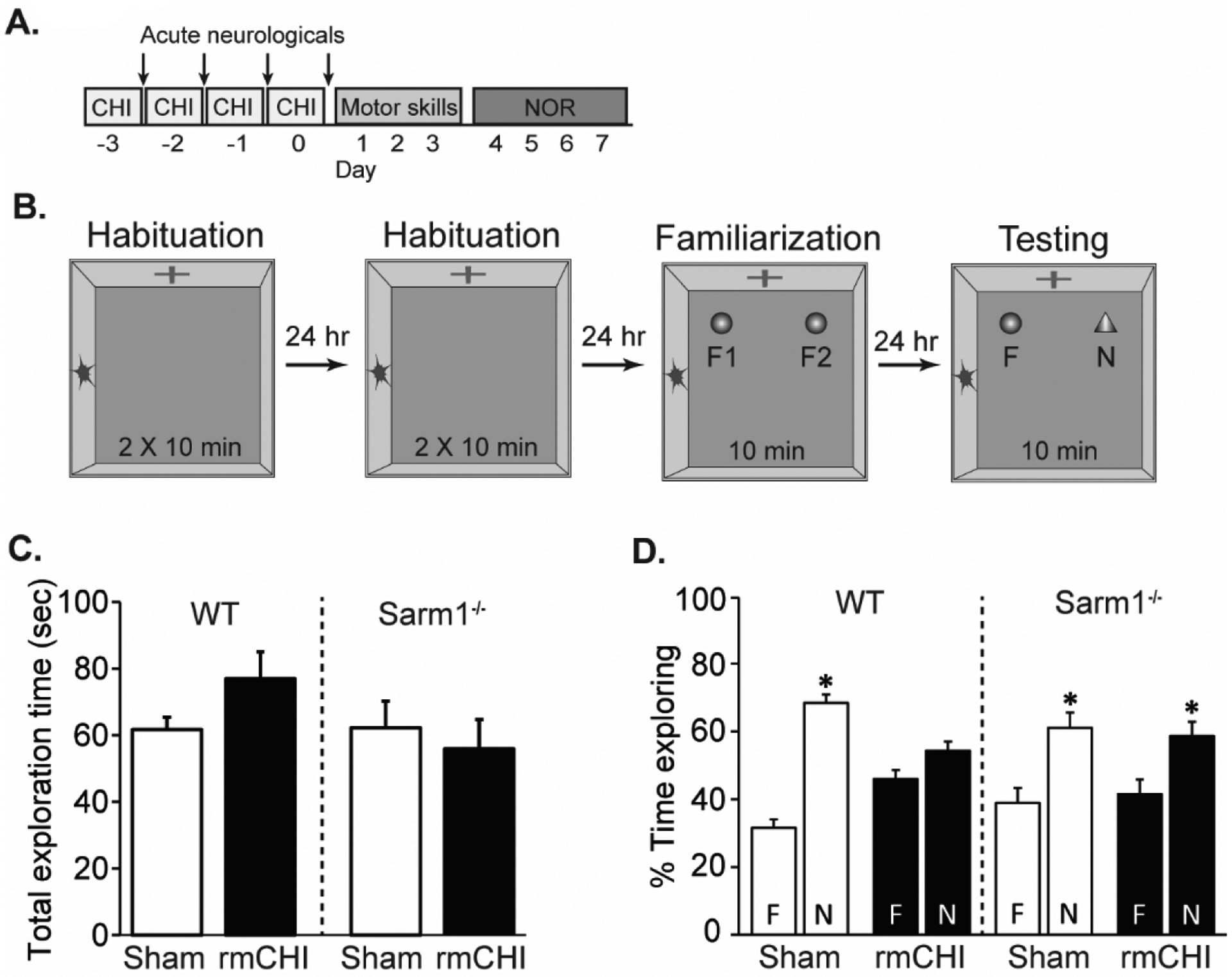
(A) Timeline of injury and cognitive assessments. Recognition memory was tested beginning four days after the last mCHI. (B) Drawing of the novel object recognition task. Animals were first exposed to the recording chamber with no objects. Two identical objects (F) were introduced to the chamber during the familiarization phase, and the animals allowed to freely explore for 10 min. Twenty-four hours later, one of the objects was exchanged for a novel (N) object and the animals again allowed to explore freely. (C) Total exploration time during the testing phase was equivalent across groups. (D) Relative exploration time for the familiar and novel objects in sham and injured wild-type and Sarm1−/− mice. In contrast to injured WT mice which showed no preference for the novel object, injured Sarm1−/− mice perform similar to both uninjured WT and SARM1−/− controls, indicating preservation of recognition memory. Data are presented as the mean ± SEM. *p<0.05 between novel and familiar object.
Contextual discrimination is improved in rmCHI Sarm1−/− mice.
The context discrimination task has been shown to require the function of the hippocampus (Frankland et al., 1998). Context fear discrimination was tested beginning 28 days after the last mCHI (Fig. 8A). In this task, animals learn to differentiate between two similar but distinct chambers (Fig. 8B). In one chamber, a mild foot shock is delivered to elicit a fear response, while no shock is administered in the other chamber. Figure 8C and 8E show that sham WT and Sarm1−/− mice both learn to differentiate between the “shock” and “safe” contexts as indicated by exhibiting significantly more freezing behavior in the “shock” chamber as compared to the “safe” chamber over the course of training (WT: F=17.350, p < 0.05; Sarm1−/−: F=12.337, p < 0.05). In contrast, injured WT animals display equivalent freezing behaviors in both the “safe” and “shock” contexts (F=0.938, p = 0.361), indicating impaired contextual discrimination (Fig. 8D). In contrast, injured Sarm1−/− mice froze significantly more in the “shock” context than the “safe” context (F=29.235, p < 0.005), indicating a preservation of their ability to discriminate between contexts (Fig. 8F).
Figure 8. Performance in the context fear discrimination task is improved in rmCHI Sarm1−/− mice.
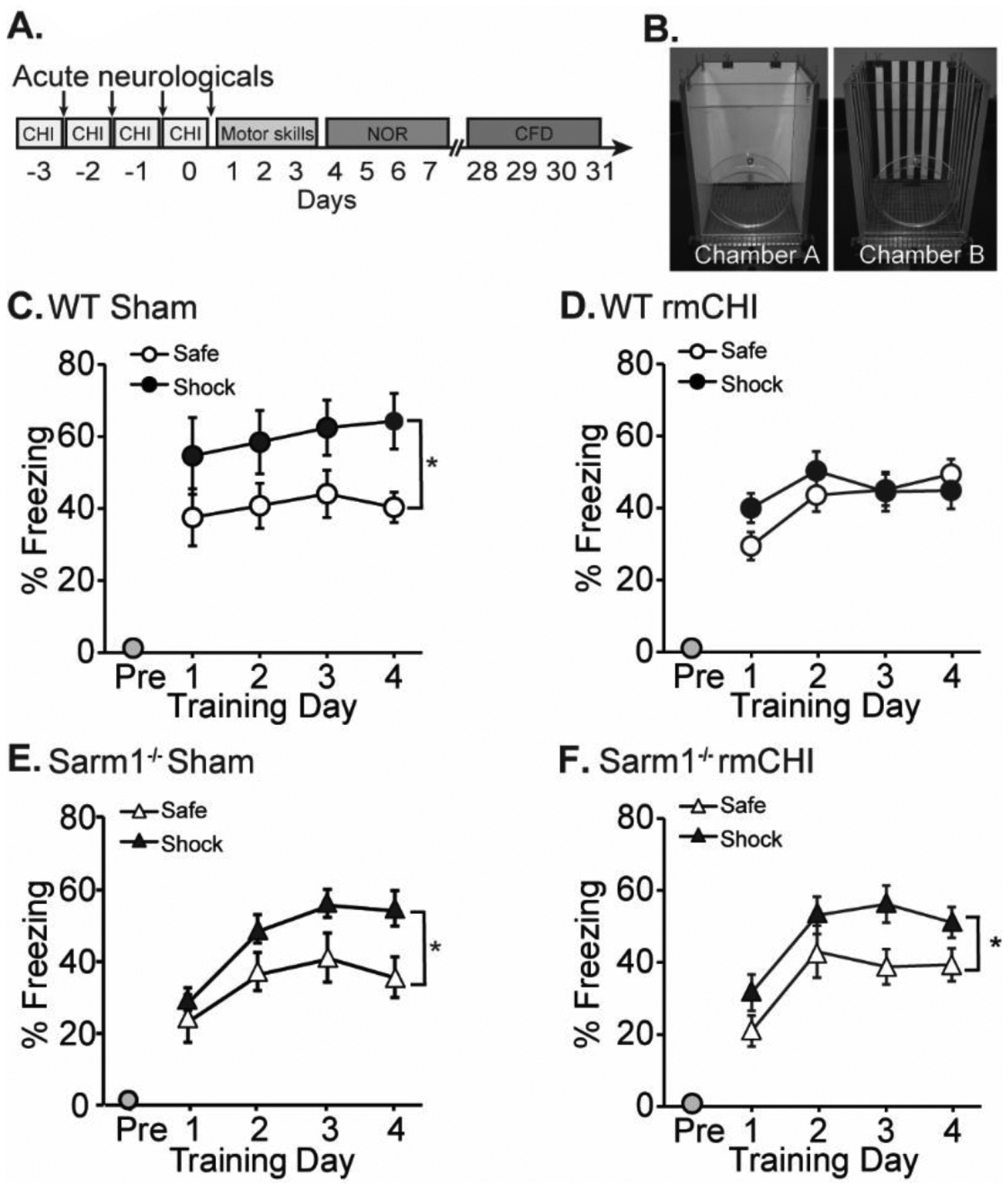
(A) Timeline of injury and cognitive assessments. Context fear discrimination was tested four weeks after the last injury. (B) Mice are trained to differentiate between “Shock” and “Safe” chambers that share some features, but differ in others. (C) Wild-type sham mice freeze more in the “shock” chamber versus the safe chamber. (D) Wild-type rmCHI mice are impaired in their ability to discriminate between the two contexts, as indicated by similar freezing behavior in both chambers. (E) Sham Sarm1−/− mice and (F) rmCHI Sarm1−/− mice are able to discriminate between the two contexts. *significant difference between freezing in “safe” and “shock” context. Data are presented as mean ± SEM. *p < 0.05.
DISCUSSION
Studies carried out in Drosophila and mutant mice have demonstrated that loss of Sarm1 attenuates axonal degeneration following axon severance (Gerdts et al., 2015; Gilley et al., 2017, 2015; Osterloh et al., 2012). Further, loss of Sarm1 has been reported to offer axonal protection following experimental TBI (Henninger et al., 2016; Marion et al., 2019; Ziogas and Koliatsos, 2018), although whether this protection is associated with improved cognitive function had yet to be examined. As axonal damage is a key pathology that has been observed in both experimental models of repeated mild TBI and in persons with concussion, we examined if loss of Sarm1 reduces white matter damage and improves cognitive outcome following rmCHI. Our results revealed four key findings: 1) rmCHI resulted in APP accumulation in white matter tracts including the corpus callosum, alveus of the hippocampus, and fimbria of the fornix; 2) Sarm1−/− mice exhibited significantly attenuated APP accumulation after injury, 3) brain inflammation resulting from repeated mild closed head injury was decreased in Sarm1−/− mice in the white matter tracts damaged by repeated injury, and 4) Sarm1−/− mice receiving four mild closed head injuries (once daily for 4 consecutive days) performed markedly better on motor and cognitive tests compared to wild-type injured mice. Taken together, these results suggest that the improved motor and cognitive outcome we observed in Sarm1−/− mice may have resulted from white matter preservation and/or reduced inflammation.
The first evidence that Wallerian degeneration can be delayed or attenuated was made by examining Wlds mice by Perry and colleagues (Perry et al., 1987). Sequence analysis of the Wlds gene found that the chimeric mutant gene encodes a fusion protein comprised of the N-terminal fragment of the ubiquitin conjugating factor 4b (Ube4b) fused to nicotinamide mononucleotide adenyl transferase 1 (Nmnat1) (Conforti et al., 2000). Nmnat1 is an enzyme that catalyzes the synthesis of nicotinamide adenine dinucleotide (NAD+) from nicotinamide mononucleotide (NAM) and ATP. While wild-type Nmnat1 is localized to the nucleus, the Wlds protein can also be found in axons where it synthesizes NAD+ and offers protection following axon transection (Conforti et al., 2014; Wang et al., 2015). Subsequent to this finding, Sarm1 was identified in a loss-of-function screen in Drosophila as a protein that suppresses Wallerian degeneration. Follow-up studies have revealed that Sarm1 is an enzyme that converts NAD+ to a number of metabolites, including NAM, ADP ribose, and cyclic ADP ribose (Essuman et al., 2017). These studies suggest that loss or inhibition of Sarm1 can protect axons after injury. Consistent with this, a study by Henninger et al., has reported that Sarm1 knockout mice have reduced APP accumulation in the corpus callosum and an acute preservation of neurological function assessed using the Neurological Severity Score (NSS) after cortical impact injury (Henninger et al., 2016). Recently, Ziogas and Koliatsos crossed Sarm1−/− mice with Thy1-eYFP-H mice (which express YFP sparsely in neurons) and used their offspring to visualize the effects of TBI on individual axons (Ziogas and Koliatsos, 2018). Using these mice, and similar mice obtained from crossing Sarm1−/− mice and B6.Cg-Tg(Thy1-YFP)16Jrs/J reporter mice, it has been observed that loss of Sarm1 is associated with reduced axonal injury and demyelination after TBI (Ziogas and Koliatsos, 2018)[31445042]. Our results that Sarm1−/− mice subjected to repeated mild closed head injury have less APP accumulation in multiple white matter tracts is consistent with these previous reports, and suggests that preservation of axonal NAD+ may be an effective strategy to reduce axonal injury following concussion.
NAD+ can be synthesized from its precursors nicotinamide, nicotinamide riboside, and nicotinic acid (Figure 9). Several studies have reported beneficial effects of nicotinamide administration following TBI in rodents. For example, a series of studies by Hoane and colleagues have reported that post-TBI administration of nicotinamide reduces contusion volume and improves learning and memory (Hoane et al., 2008, 2006b, 2006a). Furthermore, continuous infusion of nicotinamide for 7 days was found to reduce cortical contusion volume and attenuate sensory, motor and cognitive deficits after bilateral frontal controlled cortical impact (Anderson et al., 2011; Vonder Haar et al., 2011). However, a recent study by the Operation Brain Trauma Therapy consortium examined the therapeutic utility of low (50 mg/kg) and high (500 mg/kg) doses of nicotinamide following CCI, FPI, and a model of penetrating ballistic brain injury (PBBI), and found that treatment only exerted modest effects at high doses (Kochanek et al., 2016; Shear et al., 2016). Furthermore, a recent study has reported that post-injury administration of doses of nicotinamide ranging from 125 mg/kg up to 1000 mg/kg failed to reduce total cortical contusion volume or improve spatial memory in juvenile rats after CCI (Smith et al., 2019). The reason for these disparate results are unclear.
Figure 9. Simplified pathway for NAD metabolism.
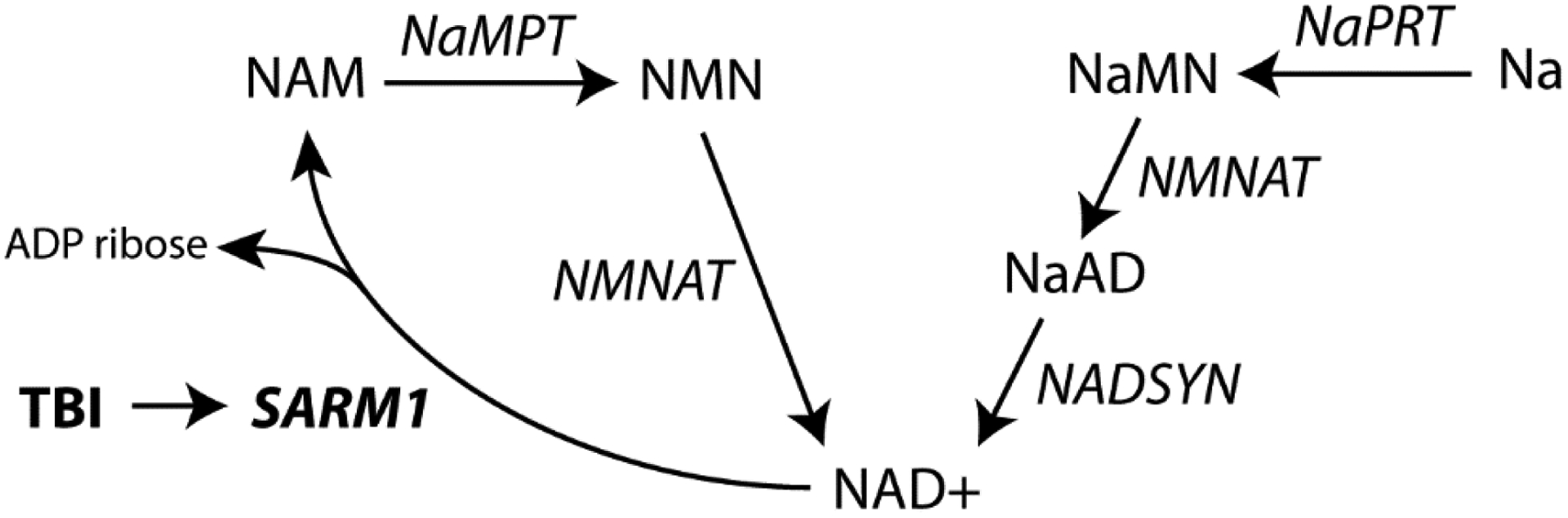
NAD+ is synthesized from two metabolic pathways: a de novo synthesis pathway from Na (and amino acids) or a recycling pathway. Sarm1 is a NAD+ consuming enzyme. Na: nicotinic acid; NaAD: nicotinic acid adenine dinucleotide; NAD+: nicotinamide adenine dinucleotide; NADSYN: Glutamine-dependent NAD(+) synthetase; NAM: nicotinamide; NaMN: nicotinic acid mononucleotide; NaMPT: Nicotinamide phosphoribosyltransferase; NaPRT: nicotinic acid phosphoribosyltransferase; NMN: nicotinamide mononucleotide; NMNAT: nicotinamide-nucleotide adenylyltransferase; Sarm1: Sterile Alpha and TIR Motif Containing 1; TBI: traumatic brain injury.
In addition to increasing axonal NAD+ levels as a means of axonal protection, recent studies have indicated that inhibition of Nampt can also reduce axonal damage. For example, Goodman et al., have demonstrated that administration of FK866 (an inhibitor of Nampt) protects dorsal root ganglion neuron axons against vincristine-induced degeneration (Liu et al., 2018). Nampt converts NAM to nicotinamide mononucleotide (NMN), which is subsequently used to generate NAD+. Therefore, inhibiting Nampt would be expected to result in an accumulation of NAM and depletion of NAD+ levels (Fig. 9) and exacerbate axonal damage. However, a recent study has reported that NMN can promote axon degeneration (Stefano et al., 2014). Thus, it is possible the protection seen following FK866 treatment may have resulted from a reduction in NMN levels, rather than an effect on axonal NAD+ levels. While these results have led to the hypothesis that increased in NMN, rather than depletion of NAD+, is critical for axonal degeneration, our results are consistent with previous findings that loss of NAD-consuming enzymes (i.e. Sarm1) is sufficient for reducing axonal damage, and that this reduction is associated with improved outcome after TBI. However, one limitation of this conclusion is that we did not directly measure NAD+ flux in individual white matter tracts after injury in wild-type and Sarm1−/− mice.
Our findings indicate that loss of Sarm1 may offer protection for some white matter tracts, but not others. For example, as shown in Figure 3, APP immunoreactivity was significantly decreased in the fimbria and alveus, but not the corpus callosum, of injured SARM1 KO mice compared to injured WT mice. Although the reason for this differential effect is unclear, this may be related to the degree of damage received, intrinsic vulnerability, and/or levels of Sarm1 expression in the various white matter tracts. For example, diffusion tensor imaging studies have shown that the genu of the corpus callosum is more often injured in concussed patients than the body of the corpus callosum [18272556]. Further, electrophysiological recordings of compound action potentials (CAP) evoked in the corpus callosum have suggested that the slow component of the CAP (due to unmyelinated axons) is persistently suppressed after TBI [16109409]. These findings, in conjunction with electron microscopic evaluation of white matter, suggest that unmyelinated axons may be preferentially vulnerable to TBI [16109409; 22318124; 17481596], and that the ratio of unmyelinated to myelinated axons within a white matter tract may contribute to overall susceptibility. It is interesting to speculate that this vulnerability may be related to decreased NAD+ flux in unmyelinated axons, possibly due to increased levels/action of Sarm1 in these axons. Evidence supporting this possibility has been previously observed in that Sarm1 deletion offered more protection on small unmyelinated fibers versus large myelinated fibers in response to paclitaxel-induced sensory neuropathy [28485482]. Unfortunately, our attempts to examine differential levels of NAD+ and Sarm1 using commercially available antibodies did not reveal any overt differences in myelinated versus unmyelinated axons, possibly due to the time point examined.
In addition to observing reduced axonal damage in injured Sarm1−/− mice, we also observed a concurrent decrease in astrocyte and microglia immunoreactivity in the white matter tracts damaged by rmCHI. Consistent with our findings, Sarm1 deletion has been reported to reduce the neuroinflammation caused by high fat diet (Pan and An, 2018). Although Sarm1 is expressed throughout the brain, it appears that this protein is not expressed by microglia, the primary immune cells found in the brain (Lin et al., 2014). Thus, it remains to be established how loss of Sarm1 in neurons reduces microglia activation following TBI. Associated with the reduction in white matter damage and inflammation, injured SARM1 mice performed significantly better in both motor (Rotarod and foot fault tasks) and cognitive (novel object recognition and context discrimination) tasks as compared to injured WT mice. As performance in these tasks requires communication both between neurons within a structure and across structures, preservation of axonal integrity may be an underlying cellular mechanism for the motor and cognitive improvements we observed. In addition, previous studies have shown that inflammation and white matter damage may be linked (Reeves et al., Exp Neurol. 2016 283:188–203). Thus, it is reasonable to conclude that the improvement in behavior we observed in injured SARM1 knockout mice may have arisen from a reduction in inflammation and subsequent white matter protection. However, at present, we cannot determine if the improved motor and cognitive outcome seen in injured knockout mice was due to decreased white matter damage, decreased inflammation, or a combination of both.
Supplementary Material
Acknowledgements.
The work described herein was supported by grants NS109118 and NS101686 from the National Institutes of Health.
References
- Anderson GD, Farin FM, Bammler TK, Beyer RP, Swan AA, Wilkerson H-W, Kantor ED, Hoane MR, 2011. The effect of progesterone dose on gene expression after traumatic brain injury. J. Neurotrauma 28, 1827–1843. 10.1089/neu.2011.1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basiri M, Doucette R, 2010. Sensorimotor cortex aspiration: a model for studying Wallerian degeneration-induced glial reactivity along the entire length of a single CNS axonal pathway. Brain Res. Bull 81, 43–52. 10.1016/j.brainresbull.2009.11.002 [DOI] [PubMed] [Google Scholar]
- Bignami A, Ralston HJ, 1969. The cellular reaction to Wallerian degeneration in the central nervous system of the cat. Brain Res. 13, 444–461. 10.1016/0006-8993(69)90259-5 [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD, 2007. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Prog. Brain Res 161, 125–141. 10.1016/S0079-6123(06)61009-1 [DOI] [PubMed] [Google Scholar]
- Bubb EJ, Metzler-Baddeley C, Aggleton JP, 2018. The cingulum bundle: Anatomy, function, and dysfunction. Neurosci. Biobehav. Rev 92, 104–127. 10.1016/j.neubiorev.2018.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Gilley J, Coleman MP, 2014. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci 15, 394–409. 10.1038/nrn3680 [DOI] [PubMed] [Google Scholar]
- Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP, 2000. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc. Natl. Acad. Sci. U. S. A 97, 11377–11382. 10.1073/pnas.97.21.11377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J, 2017. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 93, 1334–1343.e5. 10.1016/j.neuron.2017.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, Cestari V, Filipkowski RK, McDonald RJ, Silva AJ, 1998. The dorsal hippocampus is essential for context discrimination but not for contextual conditioning. Behav. Neurosci 112, 863–874. 10.1037//0735-7044.112.4.863 [DOI] [PubMed] [Google Scholar]
- George R, Griffin JW, 1994. Delayed macrophage responses and myelin clearance during Wallerian degeneration in the central nervous system: the dorsal radiculotomy model. Exp. Neurol 129, 225–236. 10.1006/exnr.1994.1164 [DOI] [PubMed] [Google Scholar]
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J, 2015. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 348, 453–457. 10.1126/science.1258366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Orsomando G, Nascimento-Ferreira I, Coleman MP, 2015. Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. 10, 1974–1981. 10.1016/j.celrep.2015.02.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Ribchester RR, Coleman MP, 2017. Sarm1 Deletion, but Not WldS, Confers Lifelong Rescue in a Mouse Model of Severe Axonopathy. Cell Rep. 21, 10–16. 10.1016/j.celrep.2017.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harker KT, Whishaw IQ, 2004. Impaired place navigation in place and matching-to-place swimming pool tasks follows both retrosplenial cortex lesions and cingulum bundle lesions in rats. Hippocampus 14, 224–231. 10.1002/hipo.10159 [DOI] [PubMed] [Google Scholar]
- Henninger N, Bouley J, Sikoglu EM, An J, Moore CM, King JA, Bowser R, Freeman MR, Brown RH, 2016. Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1. Brain J. Neurol 139, 1094–1105. 10.1093/brain/aww001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoane MR, Gilbert DR, Holland MA, Pierce JL, 2006a. Nicotinamide reduces acute cortical neuronal death and edema in the traumatically injured brain. Neurosci. Lett 408, 35–39. 10.1016/j.neulet.2006.07.011 [DOI] [PubMed] [Google Scholar]
- Hoane MR, Pierce JL, Kaufman NA, Beare JE, 2008. Variation in chronic nicotinamide treatment after traumatic brain injury can alter components of functional recovery independent of histological damage. Oxid. Med. Cell. Longev 1, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoane MR, Tan AA, Pierce JL, Anderson GD, Smith DC, 2006b. Nicotinamide treatment reduces behavioral impairments and provides cortical protection after fluid percussion injury in the rat. J. Neurotrauma 23, 1535–1548. 10.1089/neu.2006.23.1535 [DOI] [PubMed] [Google Scholar]
- Horsefield S, Burdett H, Zhang X, Manik MK, Shi Y, Chen J, Qi T, Gilley J, Lai J-S, Rank MX, Casey LW, Gu W, Ericsson DJ, Foley G, Hughes RO, Bosanac T, von Itzstein M, Rathjen JP, Nanson JD, Boden M, Dry IB, Williams SJ, Staskawicz BJ, Coleman MP, Ve T, Dodds PN, Kobe B, 2019. NAD+ cleavage activity by animal and plant TIR domains in cell death pathways. Science 365, 793–799. 10.1126/science.aax1911 [DOI] [PubMed] [Google Scholar]
- Hosmane S, Tegenge MA, Rajbhandari L, Uapinyoying P, Ganesh Kumar N, Thakor N, Venkatesan A, 2012. Toll/interleukin-1 receptor domain-containing adapter inducing interferon-β mediates microglial phagocytosis of degenerating axons. J. Neurosci. Off. J. Soc. Neurosci 32, 7745–7757. 10.1523/JNEUROSCI.0203-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylin MJ, Orsi SA, Zhao J, Bockhorst K, Perez A, Moore AN, Dash PK, 2013. Behavioral and histopathological alterations resulting from mild fluid percussion injury. J. Neurotrauma 30, 702–715. 10.1089/neu.2012.2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BJ, Farkas O, Lifshitz J, Povlishock JT, 2006. Traumatic axonal injury in the perisomatic domain triggers ultrarapid secondary axotomy and Wallerian degeneration. Exp. Neurol 198, 350–360. 10.1016/j.expneurol.2005.12.017 [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Bramlett HM, Shear DA, Dixon CE, Mondello S, Dietrich WD, Hayes RL, Wang KKW, Poloyac SM, Empey PE, Povlishock JT, Mountney A, Browning M, Deng-Bryant Y, Yan HQ, Jackson TC, Catania M, Glushakova O, Richieri SP, Tortella FC, 2016. Synthesis of Findings, Current Investigations, and Future Directions: Operation Brain Trauma Therapy. J. Neurotrauma 33, 606–614. 10.1089/neu.2015.4133 [DOI] [PubMed] [Google Scholar]
- Lin C-W, Liu H-Y, Chen C-Y, Hsueh Y-P, 2014. Neuronally-expressed Sarm1 regulates expression of inflammatory and antiviral cytokines in brains. Innate Immun. 20, 161–172. 10.1177/1753425913485877 [DOI] [PubMed] [Google Scholar]
- Liu H, Smith CB, Schmidt MS, Cambronne XA, Cohen MS, Migaud ME, Brenner C, Goodman RH, 2018. Pharmacological bypass of NAD+ salvage pathway protects neurons from chemotherapy-induced degeneration. Proc. Natl. Acad. Sci 115, 10654–10659. 10.1073/pnas.1809392115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S, 1989. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur. J. Neurosci 1, 27–33. 10.1111/j.1460-9568.1989.tb00771.x [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP, 2001. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci 4, 1199–1206. 10.1038/nn770 [DOI] [PubMed] [Google Scholar]
- Marion CM, McDaniel DP, Armstrong RC, 2019. Sarm1 deletion reduces axon damage, demyelination, and white matter atrophy after experimental traumatic brain injury. Exp. Neurol 321, 113040. 10.1016/j.expneurol.2019.113040 [DOI] [PubMed] [Google Scholar]
- McGinn MJ, Povlishock JT, 2016. Pathophysiology of Traumatic Brain Injury. Neurosurg. Clin. N. Am 27, 397–407. 10.1016/j.nec.2016.06.002 [DOI] [PubMed] [Google Scholar]
- Miklossy J, Clarke S, Van der Loos H, 1991. The long distance effects of brain lesions: visualization of axonal pathways and their terminations in the human brain by the Nauta method. J. Neuropathol. Exp. Neurol 50, 595–614. [PubMed] [Google Scholar]
- Miklossy J, Van der Loos H, 1991. The long-distance effects of brain lesions: visualization of myelinated pathways in the human brain using polarizing and fluorescence microscopy. J. Neuropathol. Exp. Neurol 50, 1–15. 10.1097/00005072-199101000-00001 [DOI] [PubMed] [Google Scholar]
- Miklossy J, Van der Loos H, 1987. Cholesterol ester crystals in polarized light show pathways in the human brain. Brain Res. 426, 377–380. 10.1016/0006-8993(87)90892-4 [DOI] [PubMed] [Google Scholar]
- Neave N, Nagle S, Aggleton JP, 1997. Evidence for the involvement of the mammillary bodies and cingulum bundle in allocentric spatial processing by rats. Eur. J. Neurosci 9, 941–955. 10.1111/j.1460-9568.1997.tb01445.x [DOI] [PubMed] [Google Scholar]
- Ojo JO, Mouzon B, Algamal M, Leary P, Lynch C, Abdullah L, Evans J, Mullan M, Bachmeier C, Stewart W, Crawford F, 2016. Chronic Repetitive Mild Traumatic Brain Injury Results in Reduced Cerebral Blood Flow, Axonal Injury, Gliosis, and Increased T-Tau and Tau Oligomers. J. Neuropathol. Exp. Neurol 75, 636–655. 10.1093/jnen/nlw035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou Y-J, Nathan C, Ding A, Brown RH, Conforti L, Coleman M, Tessier-Lavigne M, Züchner S, Freeman MR, 2012. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. 10.1126/science.1223899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z-G, An X-S, 2018. SARM1 deletion restrains NAFLD induced by high fat diet (HFD) through reducing inflammation, oxidative stress and lipid accumulation. Biochem. Biophys. Res. Commun 498, 416–423. 10.1016/j.bbrc.2018.02.115 [DOI] [PubMed] [Google Scholar]
- Perry S, Cooper AM, Michels R, 1987. The psychodynamic formulation: its purpose, structure, and clinical application. Am. J. Psychiatry 144, 543–550. 10.1176/ajp.144.5.543 [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT, 2002. Axonal self-destruction and neurodegeneration. Science 296, 868–871. 10.1126/science.1068613 [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Nakagawa T, Mao X, DiAntonio A, Milbrandt J, 2016. NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD+ depletion. eLife 5. 10.7554/eLife.19749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp DJ, Ham TE, 2011. Investigating white matter injury after mild traumatic brain injury. Curr. Opin. Neurol 24, 558–563. 10.1097/WCO.0b013e32834cd523 [DOI] [PubMed] [Google Scholar]
- Shear DA, Dixon CE, Bramlett HM, Mondello S, Dietrich WD, Deng-Bryant Y, Schmid KE, Wang KKW, Hayes RL, Povlishock JT, Kochanek PM, Tortella FC, 2016. Nicotinamide Treatment in Traumatic Brain Injury: Operation Brain Trauma Therapy. J. Neurotrauma 33, 523–537. 10.1089/neu.2015.4115 [DOI] [PubMed] [Google Scholar]
- Singleton RH, Zhu J, Stone JR, Povlishock JT, 2002. Traumatically induced axotomy adjacent to the soma does not result in acute neuronal death. J. Neurosci. Off. J. Soc. Neurosci 22, 791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AC, Holden RC, Rasmussen SM, Hoane MR, Hylin MJ, 2019. Effects of nicotinamide on spatial memory and inflammation after juvenile traumatic brain injury. Behav. Brain Res 364, 123–132. 10.1016/j.bbr.2019.02.024 [DOI] [PubMed] [Google Scholar]
- Smith DH, Meaney DF, Shull WH, 2003. Diffuse axonal injury in head trauma. J. Head Trauma Rehabil 18, 307–316. [DOI] [PubMed] [Google Scholar]
- Stefano MD, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, Janeckova L, Vargas ME, Worrell LA, Loreto A, Tickle J, Patrick J, Webster JRM, Marangoni M, Carpi FM, Pucciarelli S, Rossi F, Meng W, Sagasti A, Ribchester RR, Magni G, Coleman MP, Conforti L, 2014. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 10.1038/cdd.2014.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JR, Okonkwo DO, Dialo AO, Rubin DG, Mutlu LK, Povlishock JT, Helm GA, 2004. Impaired axonal transport and altered axolemmal permeability occur in distinct populations of damaged axons following traumatic brain injury. Exp. Neurol 190, 59–69. 10.1016/j.expneurol.2004.05.022 [DOI] [PubMed] [Google Scholar]
- Stone JR, Singleton RH, Povlishock JT, 2000. Antibodies to the C-terminus of the beta-amyloid precursor protein (APP): a site specific marker for the detection of traumatic axonal injury. Brain Res. 871, 288–302. 10.1016/s0006-8993(00)02485-9 [DOI] [PubMed] [Google Scholar]
- Tsao JW, George EB, Griffin JW, 1999. Temperature modulation reveals three distinct stages of Wallerian degeneration. J. Neurosci. Off. J. Soc. Neurosci 19, 4718–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonder Haar C, Anderson GD, Hoane MR, 2011. Continuous nicotinamide administration improves behavioral recovery and reduces lesion size following bilateral frontal controlled cortical impact injury. Behav. Brain Res 224, 311–317. 10.1016/j.bbr.2011.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Vargas ME, Barres BA, 2015. Local axonal protection by WldS as revealed by conditional regulation of protein stability. Proc. Natl. Acad. Sci. U. S. A 112, 10093–10100. 10.1073/pnas.1508337112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton EC, Aggleton JP, Muir JL, 1998. Comparing the effects of selective cingulate cortex lesions and cingulum bundle lesions on water maze performance by rats. Eur. J. Neurosci 10, 622–634. 10.1046/j.1460-9568.1998.00074.x [DOI] [PubMed] [Google Scholar]
- Weber MT, Arena JD, Xiao R, Wolf JA, Johnson VE, 2019. CLARITY reveals a more protracted temporal course of axon swelling and disconnection than previously described following traumatic brain injury. Brain Pathol. Zurich Switz 29, 437–450. 10.1111/bpa.12677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziogas NK, Koliatsos VE, 2018. Primary Traumatic Axonopathy in Mice Subjected to Impact Acceleration: A Reappraisal of Pathology and Mechanisms with High-Resolution Anatomical Methods. J. Neurosci. Off. J. Soc. Neurosci 38, 4031–4047. 10.1523/JNEUROSCI.2343-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.