

Abstract
The newest generation of drug delivery systems (DDSs) exploits ligands to mediate specific targeting of cells and/or tissues. However, studies investigating the link between ligand density and nanoparticle (NP) uptake are limited to a small number of ligand-receptor systems. C-type lectin-like molecule-1 (CLL1) is uniquely expressed on myeloid cells, which enables the development of receptors specifically targeting treat various diseases. This study aims to investigate how NPs with different CLL1 targeting peptide density impact cellular uptake. To this end, poly(styrene-alt-maleic anhydride)-b-poly(styrene) NPs are functionalized with cyclized CLL1 binding peptides (cCBP) ranging from 240 ± 12 to 31 000 ± 940 peptides per NP. Unexpectedly, the percentage of cells with internalized NPs is decreased for all cCBP-NP designs regardless of ligand density compared to unmodified NPs. Internalization through CLL1 receptor-mediated processes is further investigated without confounding the effects of NP size and surface charge. Interestingly, high density cCBP-NPs (>7000 cCBP per NP) uptake is dominated by CLL1 receptor-mediated processes while low density cCBP-NPs (≈200 cCBP per NP) and untargeted NP occurred through non-specific clathrin and caveolin-mediated endocytosis. Altogether, these studies show that ligand density and uptake mechanism should be carefully investigated for specific ligand-receptor systems for the design of targeted DDSs to achieve effective drug delivery.
Keywords: cell uptake, C-type lectin-like molecule-1 binding peptides, ligand density
1. Introduction
Selective targeting of drugs to cells and tissues remains a critical challenge in the drug delivery field.[1,2] Recent studies have focused on targeting receptors over-expressed by cells or proteins uniquely or abundantly expressed within tissues.[3] To this end, targeting ligands have been introduced to nanoparticles (NPs) to facilitate cell-specific drug delivery via affinity-based binding and uptake.[3] The density of cell-targeting ligands has been varied to improve binding affinity based on the premise that multivalent ligand-receptor interactions increase binding affinity and uptake through receptor-dependent processes.[4] Interestingly, though, there are few studies that investigate the effect of ligand density on uptake mechanism of NPs.[5] NPs can be internalized via clathrin-dependent and independent endocytosis, caveolin-dependent and independent endocytosis, and macropinocytosis.[6,7] The most common uptake processes observed for NPs are clathrin and caveolin-dependent endocytosis, which result in NP trafficking via endo-lysosomal routes.[7] However, various receptors (i.e., T cell receptor, major histocompatibility complex I, etc.) are taken up through clathrin and caveolin-independent mechanisms, resulting in unique subcellular trafficking patterns.[8–10] Therefore, the aim of this work was to vary receptor targeting ligand density on NPs to improve uptake efficiency and direct NP uptake toward receptor mediated uptake mechanisms rather than non-specific uptake processes. To do so, NPs were functionalized with increasing densities of a peptide that selectively binds to the c-type lectin-like molecule-1 (CLL1) receptor.
CLL1 is a type II glycoprotein transmembrane receptor preferentially expressed on myeloid cells and acute myeloid leukemia (AML) leukemic stem cells (LSCs).[11–15] As LSCs evade traditional chemotherapies, exploiting CLL1 overexpression and internalization may improve drug delivery strategies in the development of AML treatments. Unlike other LSC surface proteins such as CD33, the CLL1 receptor protein is not expressed on hematopoietic stem cells,[16] which makes it an attractive target for selective delivery to myeloid cells in the bone marrow generally, beyond treating AML. The intracellular domain of the alpha (α) and gamma (γ) isoforms of CLL1 contains an immunotyrosine-based inhibition motif, which is responsible for internalization via clathrin-independent processes, and a YXXM motif, which is responsible for lysosomal trafficking.[17–20] To exploit the CLL1 receptor for drug delivery, many studies have designed antibody-drug conjugates, bispe-cific antibodies, and immunotherapies toward CLL1 expressing AML, LSCs, and dendritic cells.[16,21–27]
Complementary to these efforts, Zhang et al. identified a cyclized CLL1 binding peptide (cCBP), which increased uptake of a dendrimer-based drug delivery system in epithelial cells transfected with the CLL1 receptor and in patient AML samples.[23,24] To further investigate the cCBP for targeting, multivalent peptide functionalization of polymeric NPs was explored, with the hypothesis that increasing ligand densities would enhance cellular uptake by CLL1 expressing cells due to specific ligand-receptor engagement for selective uptake. Herein, self-assembled NPs composed of poly(styrene-alt-maleic anhydride)-b-poly(styrene) (PSMA-b-PS) diblock copolymers were used due to their ease of peptide functionalization and myriad advantages for downstream drug delivery purposes.[28–30] By incorporating different densities of cCBPs, control over uptake efficiency and CLL1 receptor mediated internalization versus non-specific uptake of NPs was explored.
2. Results
2.1. CLL1 Binding Peptides Uptake is Dominated by Energy-Dependent Processes
To test uptake of cCBP-functionalized NPs, the human AML cell line, MV411, was used. Confocal microscopy was used to demonstrate that CLL1 is expressed on the surface of MV411 cells (Figure 1A), which corroborated previous studies.[15,16,31–33] Quantification of CLL1 expression via flow cytometry revealed that 99.7 ± 0.03% of MV411 cells expressed CLL1 (Figure 1B).[34] In a variety of control non-leukemic cells, CLL1 expression was absent, thereby supporting previous reports that show that CLL1 protein is restricted to the myeloid lineage (Figure S1, Supporting Information).[11,35] Based on these findings, CBP uptake was examined to establish ligand-receptor interactions.
Figure 1.
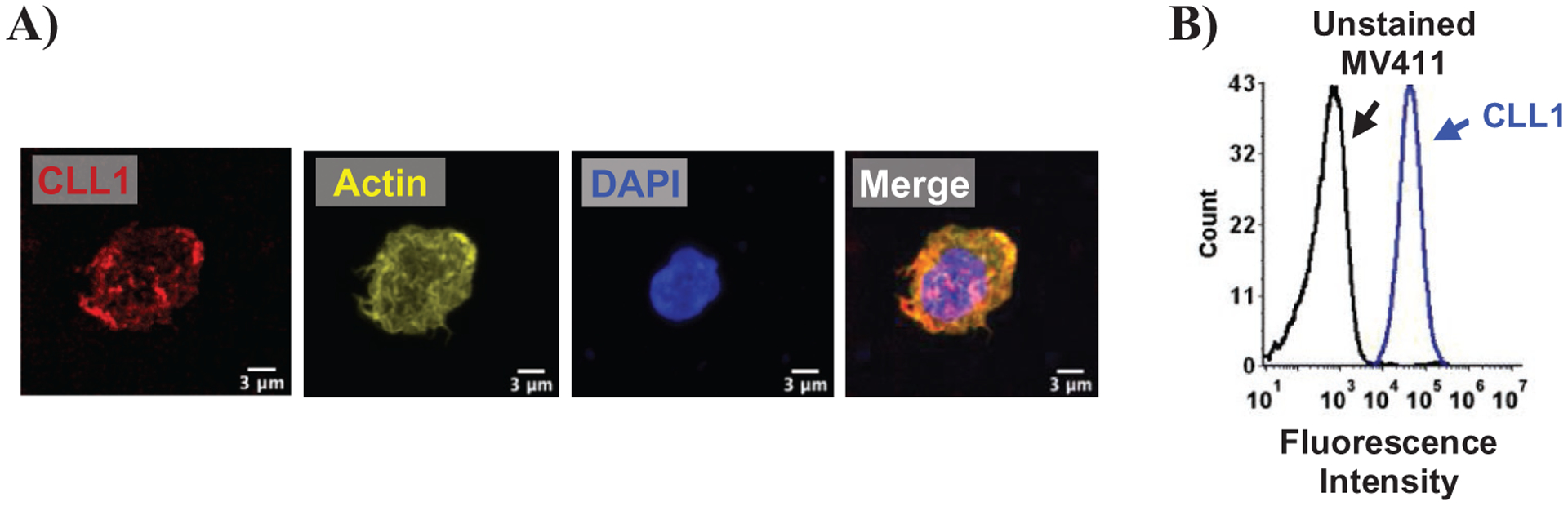
MV411 cells express C-type lectin-like molecule-1 (CLL1). A) Confocal microscopy of MV411 cells stained for DAPI, CLL1, and actin. Scale bar = 3 μm. B) Histogram showing expression of CLL1 receptor in MV411 cells as detected by flow cytometry. Black = unstained MV411 cells, blue = CLL1-antibody treated MV411 cells.
Cyclized CBPs (cCBPs) were formed via intramolecular binding between cysteine residues (Figure 2A).[23,24] MV411-cCBP binding was measured by flow cytometry and revealed minimal extracellular binding of cCBP at 4 °C (Figure S2, Supporting Information) and dose dependent internalization of cCBP with receptor saturation at 500 μg mL−1 cCBP (Figure S2, Supporting Information). To determine whether cCBP uptake was driven by endocytosis, an energy-dependent process, and cells were treated with cCBP at 37 °C. After 30 min, 40 ± 3% of cells were positive for cCBP with threefold less uptake observed at 4 °C (Figure 2B). A reduction in cCBP-positive cells at 4 °C suggested that cCBP internalizes via endocytosis. Further investigation focused on identifying specific uptake pathways of cCBP. cCBP internalization was unaffected in the presence of inhibitors for clathrin mediated endocytosis (chlorpromazine), caveolin mediated endocytosis (genistein), and macropinocytosis (rottlerin), suggesting that cCBP uptake occurred via clathrin independent processes, consistent with uptake mechanisms reported for the CLL1 receptor (Figure 2C).[15] These data were important for establishing ligand-receptor interactions of cCBP and the CLL1 receptor, and corroborating cCBP uptake in MV411 cells.
Figure 2.
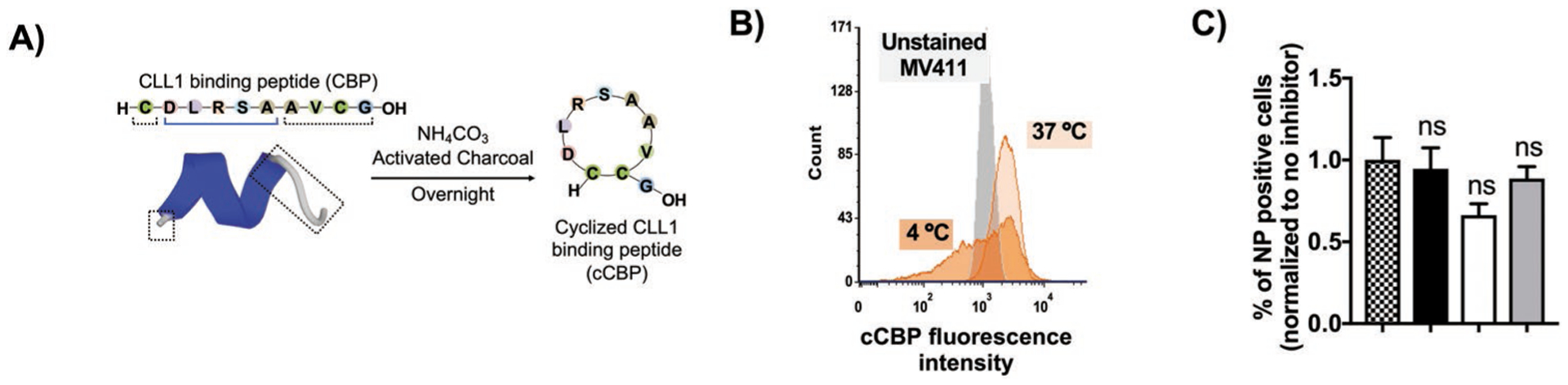
CLL1 binding peptide uptake is dominated by energy dependent processes. A) Schematic showing cyclized CLL1 binding peptides (cCBP) and 3D structure of CBP as determined via PEP-FOLD3.0. B) Histogram showing 50 μg mL−1 cCBP uptake after 30 min in 4 and 37 °C. MFI = median fluorescence intensity, NT = no treatment. C) cCBP uptake in the presence of endocytosis inhibitors. Legend: pattern = no inhibitor, black = chlorpromazine (clathrin-endocytosis), white = genistein (caveolin-endocytosis), and grey = rottlerin (macropinocytosis). Data represents mean ± standard error mean (SEM) (n = 6).
2.2. Poly(styrene-alt-maleic anhydride)-b-poly(styrene) Nanoparticles as a Platform to Study cCBP-Mediated Targeting of MV411 Cells
PSMA-b-PS-based NPs were utilized to investigate the role of cCBP multivalency on NP uptake. Prior to functionalizing with cCBP, PSMA-b-PS diblocks and NPs were characterized. PSMA-b-PS diblock copolymers synthesized via reversible addition-fragmentation chain transfer (RAFT) polymerization were monodisperse (polydispersity index of < 1.01) (Scheme 1). Self-assembled PSMA-b-PS NPs had hydrodynamic diameters of 86 ± 2 nm and overall negative surface charges of −36 ± 1 mV. MV411 cells were treated with a range of NP concentrations to assess cytocompatibility. After incubating MV411 cells for 2 h with 6.25–200 μg mL−1 PSMA-b-PS NPs, cell viability was largely unaffected compared to untreated controls. Though treatments at the highest concentration 200 μg mL−1 showed statistically significant decreases in viability to 82 ± 3%, this level is still above the 70% threshold required by cytocompatibility standards (Figure 3A).[36]
Scheme 1.
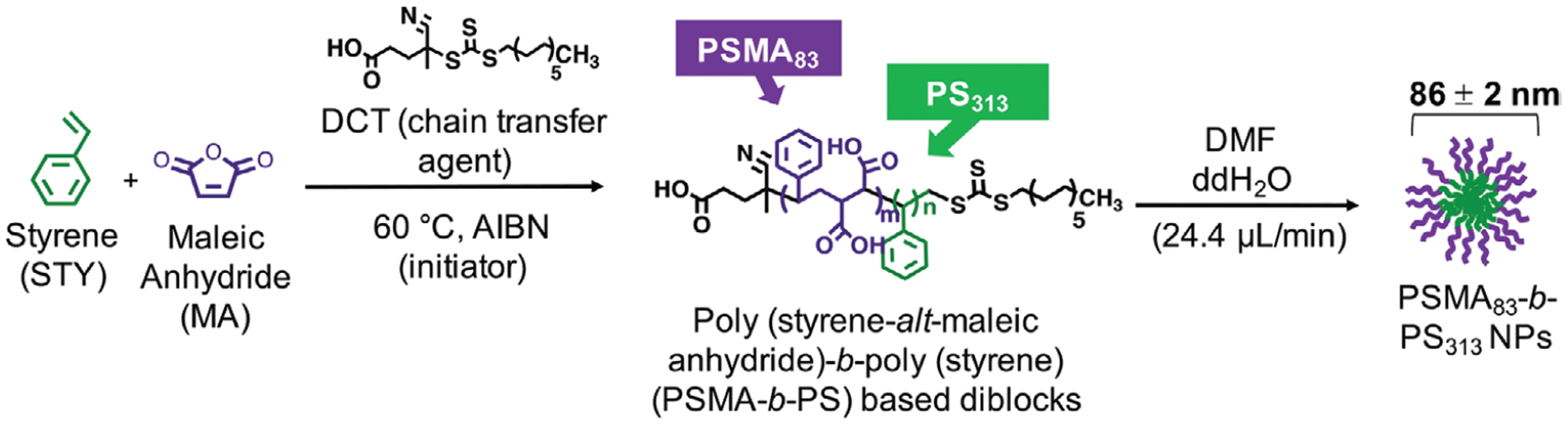
Synthesis of poly(styrene-alt-maleic anhydride)-b-poly(styrene) (PSMA-b-PS) based diblock copolymers via one-step RAFT polymerization.
Figure 3.
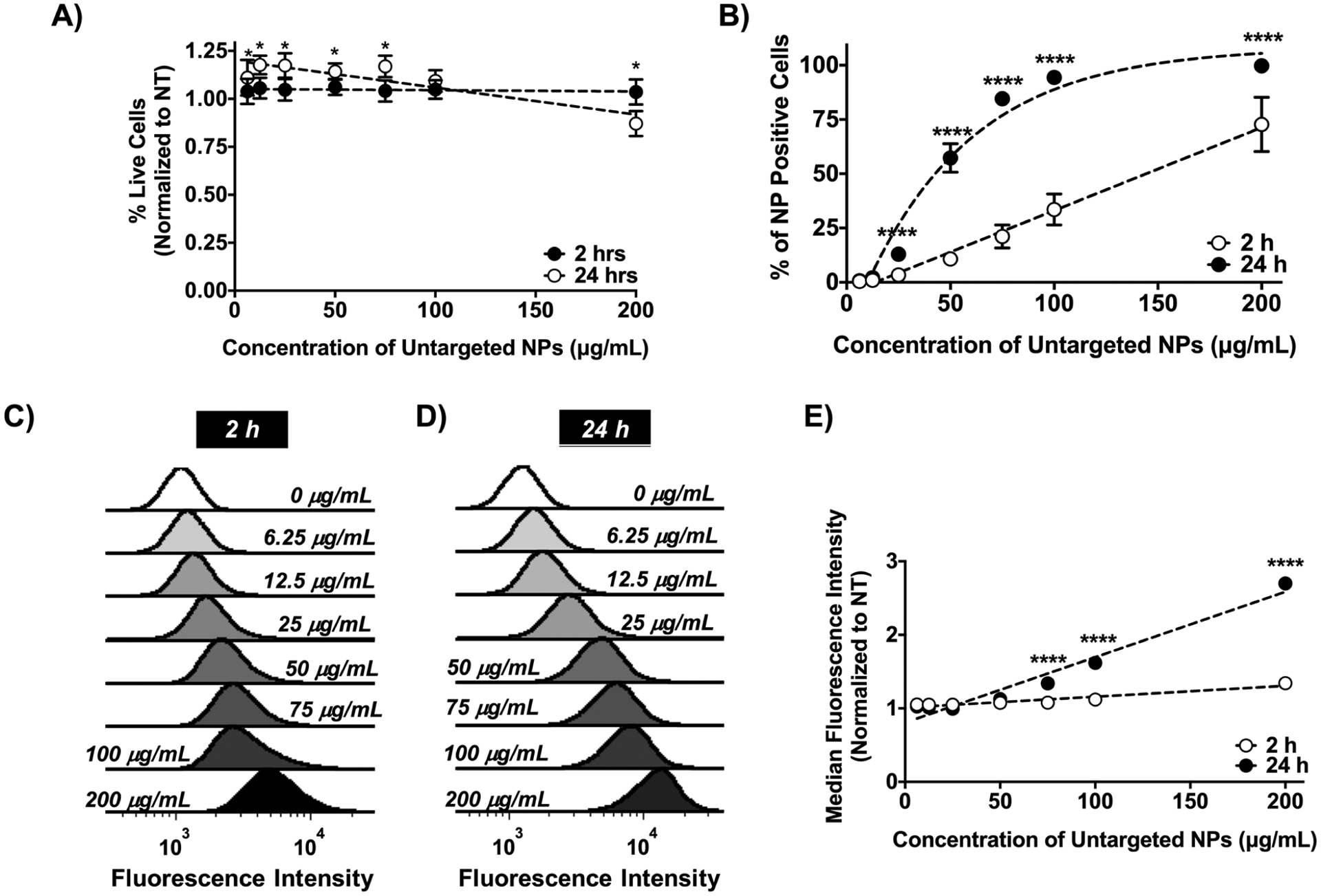
Unmodified PSMA-b-PS NP MV411 are cytocompatible and are taken up in a concentration dependent manner. A) Treatment of MV411 cells with untargeted PSMA-b-PS NPs for 2 and 24 h normalized to the no treatment (NT) group. Two-way ANOVA followed by Dunnett’s multiple comparisons was used to compare NT group with each concentration at all-time points (*p < 0.05). B) Percent of NP positive MV411 cells after treating with increasing concentrations of untargeted PSMA-b-PS NPs. Data represents mean ± standard error mean (N = 3, n = 3). Two-way ANOVA followed by Sidak’s multiple comparisons was used to compare 2 and 24 h time points (****p < 0.0001). C,D) Representative histogram of uptake at 2 and 24 h. E) Median fluorescence intensity (MFI) normalized to no treatment. Data represents mean ± standard (n = 9). Two-way ANOVA followed by Sidak’s multiple comparisons was used to compare 2 and 24 h time points (****p < 0.0001).
MV411 uptake of untargeted PSMA-b-PS NP exhibited a dose-dependent increase (slope = 0.4, R2 = 0.94) 2 h after treatment, with a maximum 73 ± 13% of MV411 cells positive for NP uptake at the highest NP dose investigated (200 μg mL−1) (Figure 3B,C). While the number of MV411 cells positive for NPs increased with dose, the median fluorescence intensity (MFI), indicative of the quantity of internalized NPs, was not significantly different for any NP dose tested. MFI did exhibit a dose-dependent increase at 2 h (slope = 0.001, R2 =0.66) (Figure 3E). By 24 h, 100% of MV411 cells were positive for NP at the highest dose (200 μg mL−1) with a linear correlation between MFI and dose (slope = 0.01, R2 = 0.95) (Figure 3B,D). This study confirmed the cytocompatibility of NPs, identified the relationship between NP dose and uptake, and rationalized cytocompatible NP doses to enable investigation of the effect of cCBP multivalency on uptake.
2.3. cCBP Multivalency does not Impact MV411 Uptake of NP
To test the relationship between ligand density and MV411 uptake, NPs were functionalized with various molar ratios of cCBP to NP diblock copolymer (Figure 4A).[28] Functionalization ranged from 240 ± 12 to 31 000 ± 940 cCBPs per NP with conjugation efficiencies of 91% with a correlation between predicted and actual peptide incorporation of 99% (Figure 4B). Note that subscripts are used to denote CBPs per NP, as quantified using detection of fluorescently labeled peptides after NP conjugation and purification. Hydrodynamic diameters of peptide-conjugated NPs ranged from 70 ± 1 to 102 ± 2 nm (Table 1). Additionally, the surface charge of cCBP-functionalized PSMA-b-PS NPs were largely unchanged (≈−30 mV) with exception to cCBP15 000-NPs (−25 ± 2 mV) (Table 1). NPs retained consistent morphology across untargeted, low (cCBP200-NPs), and high (cCBP30 000-NP) cCBP functionalization (Figure 4C). Altogether, these data suggest minimal physiochemical changes of NPs despite dramatic differences in cCBP densities.
Figure 4.
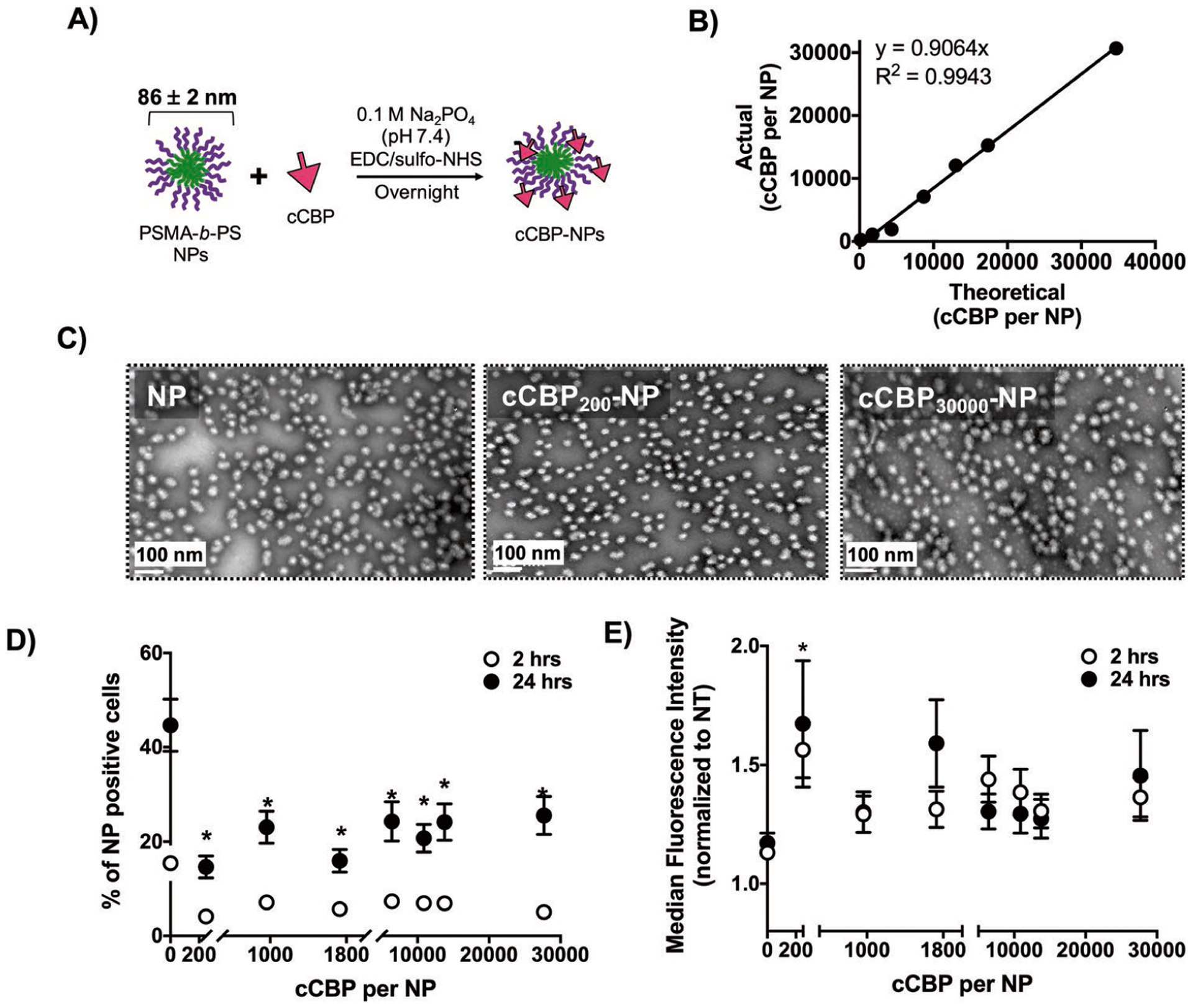
Multivalent presentation of cCBP on PSMA-b-PS NPs does not increase NP uptake in MV411 cells. A) Schematic showing cCBP incorporation on NPs. B) Functionalization efficiency of cCBP-NPs. C) Representative transmission electron microscopy (TEM) images of untargeted NP, cCBP200-NP, and CBP30000-NPs. Scale bar = 100 nm. D) Percent of cells positive for cCBP-NPs after incubation for 2 and 24 h in 37 °C. E) Median fluorescent intensity (MFI) normalized to untreated cells of cCBP-NPs after incubation for 2 and 24 h in 37°C. Data represents mean ± standard error mean (n = 9). Two-way ANOVA and Dunnett’s multiple comparisons indicate *p < 0.05 versus untargeted NP.
Table 1.
Characterization of physicochemical properties from dynamic light scattering (DLS).
| Nanoparticle ID | Measured number of pep-tides per NP | Measured ligand density [per 100 nm2] | Hydrodynamic diameter [nm] | Polydispersity index [PDI] | Zeta potential [mV] |
|---|---|---|---|---|---|
| NP | 0 | N/A | 102 ± 2 | 0.03 ± 0.004 | ‒35 ± 2 |
| CCBP200-NP | 240 ± 12 | 0.9 ± 0.05 | 91 ± 6 | 0.15 ± 0.020 | ‒38 ± 2 |
| CCBP1000-NP | 1 100 ± 3 | 4 ± 0.01 | 99 ± 2 | 0.06 ± 0.009 | ‒34 ± 1 |
| CCBP2000-NP | 1 900 ± 60 | 6 ± 0.2 | 99 ± 4 | 0.01 ± 0.040 | ‒35 ± 2 |
| CCBP7000-NP | 7 100 ±170 | 38 ± 0.9 | 87 ± 12* | 0.04 ± 0.030 | ‒32 ± 1 |
| CCBP12000-NP | 12 000 ± 190 | 48 ± 0.7 | 90 ± 1 | 0.03 ± 0.020 | ‒32 ± 2 |
| CCBP15000-NP | 15 000 ± 320 | 66 ± 1.0 | 86 ± 4* | 0.10 ± 0.020 | ‒25 ± 2* |
| CCBP30000-NP | 31 000 ± 940 | 200 ± 6.0 | 72 ± 2* | 0.04 ± 0.020 | ‒31 ± 1 |
Data represents mean ± standard deviation. Statistical analysis was performed on hydrodynamic diameter and zeta potential results using a one-way ANOVA followed by Dunnett’s multiple comparison’s test between untargeted NP and cCBP-NP designs based on p-value of 0.001.
p < 0.001.
To examine whether cCBP multivalency enhances uptake, MV411 cells were treated with peptide-conjugated NPs at 50 ug mL−1 (Figure S3, Supporting Information). Discrimination of cells positive for intracellular versus the combination of intracellular and extracellularly-bound NPs was achieved using trypan blue exclusion.[37,38] After incubation at 2 or 24 h, cCBP was detrimental toward uptake of NPs on a population basis (Figure 4D) but at low doses, enhanced the overall amount of NP taken up on a per cell basis (Figure 4E). Specifically, after incubating MV411 cells with untargeted NPs for 2 h, 15 ± 13% of cells were positive for NPs compared to a range of 4 ± 3% to 7 ± 6% for various densities of cCBP (Figure 4D). Following 24 h of incubation, 45 ± 21% of cells were positive for untargeted NPs compared to 15 ± 9% to 26 ± 16% for cCBP-NPs (Figure 4D). MFI showed significant increases at 2 h only for cCBP-NP200 compared to untargeted NPs (Figure 4E). These data showed that cCBP functionalization reduced the overall number of NP-positive MV411 cells but increased the overall number of NPs taken up by MV411 at the lowest cCBP density tested. Altogether, cCBP functionalization of NPs resulted in consistent NP physicochemical properties and did not vary greatly in uptake as a function cCBP density. Therefore, these results were leveraged to investigate how cCBP-functionalization impacted NP uptake mechanism.
2.4. High Density cCBP Functionalization Shifts NP Uptake Mechanism Toward a Receptor-Mediated Uptake Mechanism
The uptake of cCBP-functionalized NPs was investigated using inhibitors of clathrin mediated endocytosis (chlorpromazine), caveolin mediated endocytosis (genistein), and macropinocytosis (rottlerin) (Figure S5, Supporting Information). The percent of cells positive for untargeted NPs was significantly reduced in the presence of chlorpromazine (3-fold) and switch twofold to 2-fold of untargeted NPs favors clathrin and caveolin-mediated endocytosis pathways. Of the cCBP-NPs tested, cCBP-NP200 did not differ from untargeted NP uptake behavior, whereby treatment with chlorpromazine and genistein resulted in significant 8-fold and 3-fold decreases in the percent of cells positive for cCBP-NP200, respectively (Figure 5). A more dramatic decrease of eight versus threefold when clathrin and caveolin mediated endocytosis were inhibited suggests cCBP-NP200 uptake occurred more efficiently through those processes than the untargeted NP design. With increasing densities of cCBP, the mode of uptake shifted to clathrin-independent endocytosis, as evidenced by no changes observed with clathrin inhibition and an increase in uptake when caveolin or macropinocytosis were inhibited. Despite clear shifts in uptake based on the percent of NP-positive cells, the intracellular fluorescence intensities of NP-positive cells were unchanged (Figure S6, Supporting Information). These findings indicate that increasing cCBP density on NPs triggered different modes of NP internalization, whereby low-density NPs were taken up similarly to unmodified NPs and high cCBP density uptake toward clathrin-independent mechanisms.
Figure 5.
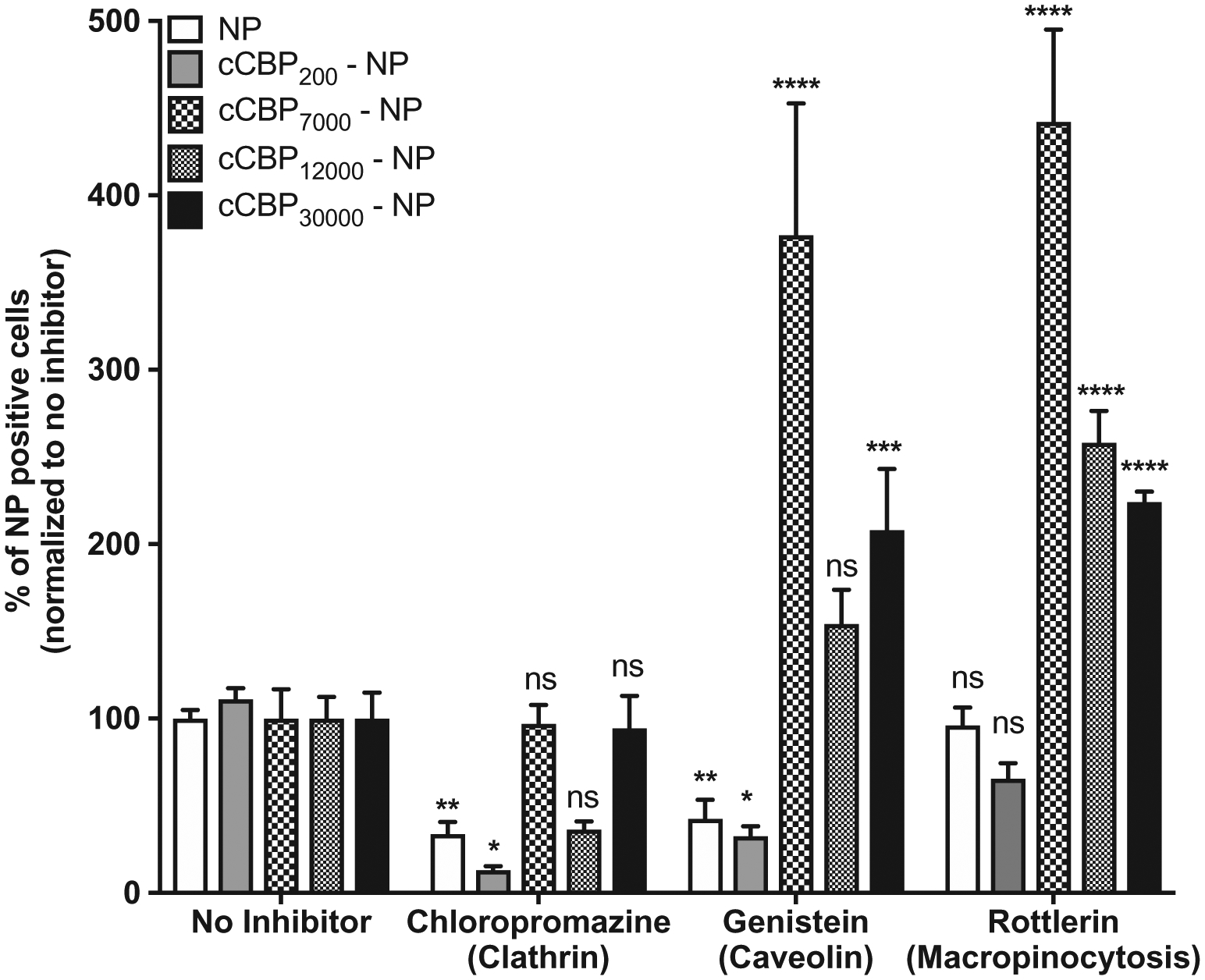
PSMA-b-PS NP uptake is altered with cCBP incorporation. Percent of NP positive cells after treating cells with Chropazamine (clathrin endocytosis), Genistein (caveolin endocytosis) and Rottlerin (macropinocytosis). Data represents mean ± standard error mean (SEM) (n = 6). One-way ANOVA followed by Dunnett’s multiple comparisons was used to compare treatments to the no inhibitor group (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant).
2.5. cCBP-NPs With Higher Ligand Density (30 000 cCBP per NP) are Taken Up Via the CLL1 Receptor
Blocking MV411 cells with CLL1 monoclonal antibody resulted in a significant fourfold decrease in cCBP30 000-NP positive cells but showed no impact on NP uptake at low cCBP densities (Figure 6A). Free cCBP was also introduced in a subset of experiments to competitively inhibit CLL1 receptor uptake. However, cCBP did not result in any significant differences in cell uptake, perhaps due to insufficient concentrations of free cCBP (Figure 6B). At the highest density of cCBP (cCBP30 000-NP), data trended to a 50% reduction in positive cells (Figure 6B), but this result was not statistically significant. Of note, the intracellular fluorescence intensities of cCBP-NPs were unchanged (Figure S7, Supporting Information). Overall, these data indicate that NP functionalization with low-density cCBP was internalized through non-specific processes while high-density cCBP facilitates uptake of NP by binding to the CLL1 receptor.
Figure 6.
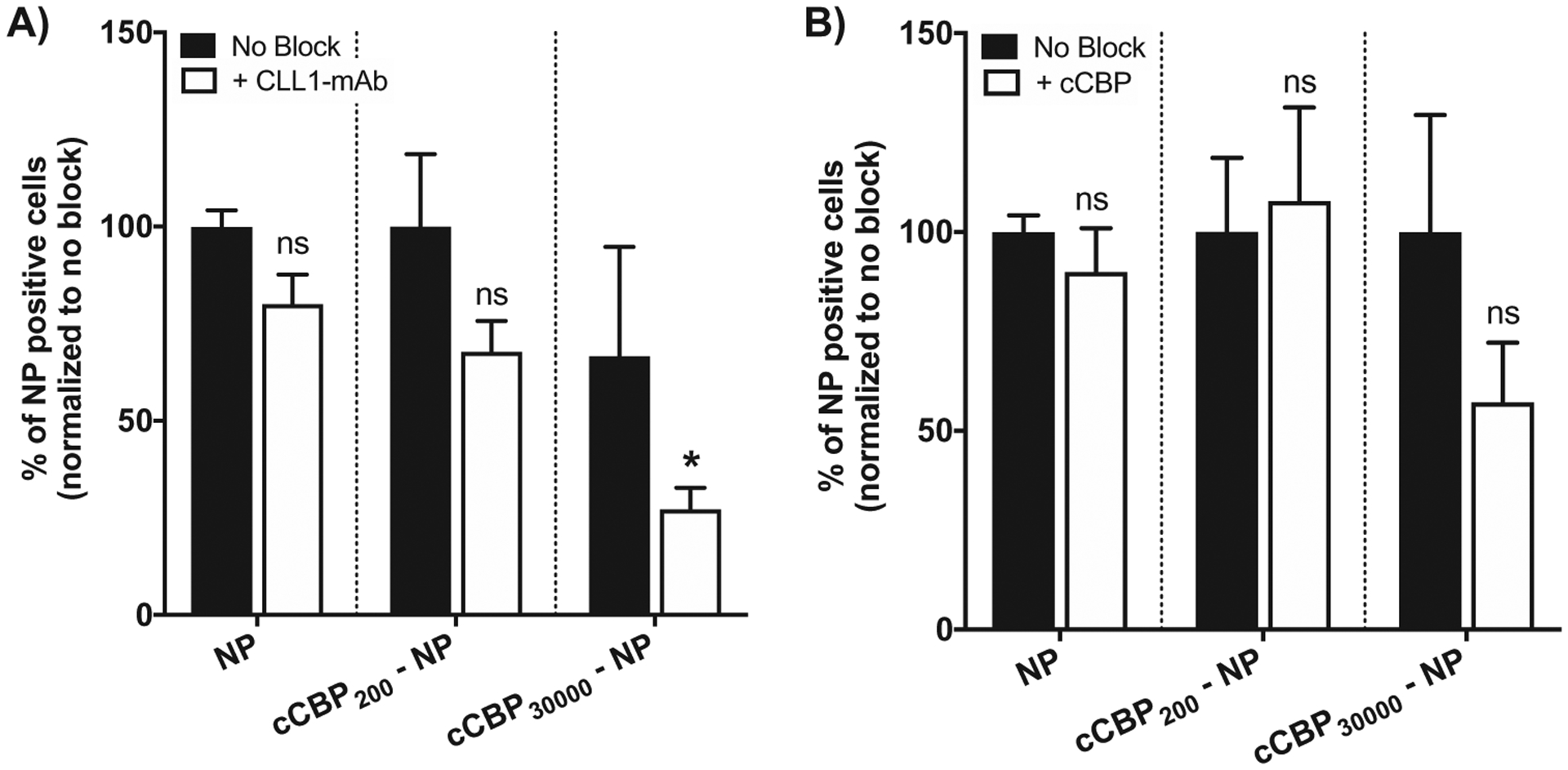
Percent of NP positive MV411 cells is reduced when monoclonal CLL1 antibody is used to block the receptor. A) Percent of NP positive cells after treatment with CLL1-mAb to block prior to NP incubation. B) Percent of NP positive cells after treatment with free cCBP to block prior to NP incubation. Data represents mean ± standard error mean (n = 6). Two-way ANOVA followed by Sidak’s multiple comparisons was used to compare treatment without blocking to CLL1-mab and cCBP groups, respectively (*p < 0.001, ns = not significant).
3. Discussion
Expression of the CLL1 receptor on myeloid cells, including AML cells and LSCs, has motivated the development of CLL1-targeted drug delivery systems (DDSs) and immunotherapies to reduce tumor burden.[15,17,22,26,27,35,39–41] The cCBP, provides an excellent opportunity to target drugs to CLL1 expressing cells. However, studies that investigate the relationship between cCBP density and receptor-mediated uptake have not been reported and are important in the development of CLL1-targeted drug delivery approaches.[23,24] Therefore, this work sought to identify how incorporating cCBP in a ligand density dependent manner impacted cellular uptake efficiency and receptor-mediated internalization processes. Notably, functionalization with cCBP maintained NP physiochemical properties, consistently reduced NP uptake irrespective of cCBP density, and did not grossly impact the extent of internalization of NPs on a per cell basis. Functionalization of NPs with high densities of peptide shifted internalization processes away from general clathrin and caveolin dependent mechanisms toward CLL1 receptor-mediated processes, which likely occurred via clathrin-independent pathways.[15] Altogether, these findings provide valuable insights for the design of targeted DDSs to ensure receptor mediated uptake to selectively target CLL1 receptor expressing cells.
The CLL1 receptor was chosen as the target for these studies due to multiple reports connecting its expression with LSCs.[11–14,42] We hypothesized that cCBP density would enhance NP uptake in CLL1 expressing cells based on improved receptor-target interactions. Ligand density is a design parameter that is commonly used to modulate uptake of targeted DDSs.[5] Ligand density typically results in a saturation[43] or inflection[44] of cellular uptake. While previous studies investigating cCBP did not interrogate how ligand density impacts uptake[23,24], dose-dependent increases in uptake of cCBP-targeted dendrimers were observed for epithelial cells transfected to express the CLL1 receptor and in primary AML cells.[23] In contrast, our studies show that the percent of cells positive for NPs was uniformly reduced for all targeted NP designs compared to untargeted controls. This unexpected result may be due to inefficient uptake of the high density cCBP designs,[20] heterogeneity in MV411 CLL1 receptor types, and/or different DDS physicochemical properties.
The CLL1 receptor is present in three isoforms (α, β, and γ) and additional splice variants that have not been fully characterized.[20,45] Detection of cellular CLL1 expression via antibodies, as used here, does not discriminate between isoforms. Therefore, while cells may express CLL1, the beta splice lacks a transmembrane region and has not been investigated for its ability to internalize.[20] In this study, the receptor isoform repertoires of MV411 cells were not determined. Therefore, it is possible that MV411 cells exhibit heterogeneity in expression of CLL1 receptor isoforms, thereby limiting uptake. cCBP-NPs that bind to non-internalizing isoforms of CLL1 would result in an overall reduction in NP-positive cells compared with unmodified NPs, which are taken up readily via non-specific mechanisms.
DDS physicochemical properties can impact ligand-receptor interactions and uptake efficiency.[46] In particular, carrier properties including size, morphology, and surface charge impact uptake efficiency.[47] The designs used in these studies were spherical 70–100 nm NPs, while the carrier used in previous studies was a 13.5 nm dendrimer.[23] The difference in DDS size and morphology may account for differences in ligand-dependent uptake. However, studies investigating the relationship of carrier size and/or morphology and uptake are often contradictory due to the complex and convoluted biological processes driving uptake.[46,48] In addition to size and morphological differences, PSMA-b-PS NPs are negatively charged. The dendrimers used previously to show cCBP ligand-dependent uptake bear a slight (+3.6 mV) positive charged and, therefore, have favorable electrostatic interactions with cell membranes, which may also lead to non-specific uptake.[49] Molecular dynamics modeling of positively charged NPs indicate that interactions with the cell membrane induce membrane wrapping and uptake while negatively charged NPs form fluid bilayers resulting in uptake through endocytosis.[50] Differences in simulated interactions between charged species and the cell membrane gives some rationale for observed divergence in uptake efficiencies observed herein.
Few studies have investigated how ligand density affects the uptake pathway utilized by NPs, although this information is vital for elucidating subcellular fates. For cCBP specifically, this is the first study to our knowledge that investigates uptake as a function of cCBP density. Uptake of NP is dependent on the carrier and the targeting ligand. For example, folic acid-functionalized quantum dots show that low density carriers (<20 ligands per NP) are preferentially taken up through caveolin dependent endocytosis while clathrin-mediated endocytosis dominates for high density carriers (>40 ligands per NP).[51] However, dramatic differences are observed with TAT peptide-targeted quantum dots whereby low (≈10 ligands per NP) and high (≈40 ligands per NP) ligand densities undergo lipid raft-mediated endocytosis but face different subcellular fates.[52] In another ligand-NP system, poly(styrene) (PS) NPs functionalized with low folic acid ligand densities resulted in clathrin-mediated processes and high densities (>5.5 folate/PS) were predominantly taken up via caveolin-mediated endocytosis.[43] While direct comparisons to previous studies are difficult due to different carriers and targeting ligands, our studies provide additional insight into ligand dependent changes in uptake processes. Our results suggest a shift away from clathrin- and caveolin-mediated uptake pathways when NPs are functionalized with high densities of cCBP (>7000 cCBP per NP) toward CLL1 receptor mediated uptake mechanisms. The lowest number of peptides tested in our system was cCBP200-NPs, which exhibited uptake similar to the untargeted NP. The shift from caveolin- and clathrin mediated uptake was observed at 7000 peptides per NP and greater, suggesting CLL1 receptor mediated clathrin independent uptake processes. The increase in uptake of >7000 cCBP per NP designs when caveolin and macropinocytosis pathways were blocked cannot be easily explained due to the complexities of uptake processes, however, it is a finding that should be further studied. Most importantly, the observed trends in uptake motivate the investigation of density dependent effects on uptake mechanisms of cCBP-NPs.
The findings herein may have broad applicability for the design of other targeted DDSs. The CLL1 receptor is also expressed on myeloid dendritic cells and may be useful for modulating immune responses and inducing tolerance in autoimmune disorders.[35,41] Previous studies show that CLL1 receptor mediated uptake results in efficient antigen cross-presentation to tumor reactive T cells, which may boost immunity in the context of cancer therapies.[15] Our findings directly contribute to these efforts as they show that modulating cCBP density is required for CLL1 receptor-specific uptake. For example, at the highest peptide density (30 000 cCBP per NP), the shift to CLL1 receptor mediated processes could result in early endosomal trafficking, followed by delivery to lysosomes, and finally, cross-presentation to immune cells in treated dendritic cells.[15] Alter-natively, for low density peptide NPs (200 cCBP per NP) and untargeted NPs, drug retention at lysosomes can be achieved through clathrin and caveolin-mediated uptake mechanisms.[53] The ability to modulate peptide ligand densities to bias uptake toward receptor mediated processes and downstream subcellular trafficking mechanisms provide an avenue to expand this work beyond traditional drug delivery, and toward development of effective vaccines and immunotherapies.
4. Conclusion
The study presented here explored how ligand density of cCBP on polymeric NPs modulates receptor-specific versus non-specific uptake. Unlike other studies investigating the link between ligand density and NP uptake, functionalization of polymeric NPs with cCBP reduced uptake is compared to untargeted NP controls, showcasing the complexities associated with NP mediated cell uptake. Notably, these studies found that modulating ligand density on NPs was necessary to shift uptake toward CLL1 receptor-mediated processes rather than non-specific uptake mechanisms. Altogether, this study stressed the importance of modulating ligand density on NPs to bias cell uptake toward receptor-mediated endocytosis processes rather than non-specific uptake mechanism. These findings are applicable to other targeting approaches and can be adapted to influence multivalent receptor interactions for other DDSs and immunotherapies for a wide variety of applications.
5. Experimental Section
Materials:
All chemicals were purchased from Sigma Aldrich and Alfa Aesar unless otherwise noted. Solvents used were spectroscopic grade. Distilled/deionized water (ddH2O) with a resistivity of 18 MΩ or greater was used for all studies. Styrene (STY) (99%, ACS grade) was purified by distillation, maleic anhydride (MA) was recrystallized from chloroform, and 2,2´-azo-bis(isobutylnitrile) (AIBN) was recrystallized from methanol prior to usage in polymerization reactions.
Preparation of Cyclized CLL-1 Binding Peptides:
Solid-phase peptide synthesis via a Liberty1 automated peptide synthesizer (CEM Corp.) was used to generate CBP (amino acid sequence: CDLRSAAVCG) on fluorenylmethyloxycarbonyl chloride (FMOC)-Gly-Wang resin (EMD Millipore). FMOC protected amino acids (AAPPTec and Peptides International) were dissolved in 0.2 M N-methyl pyrrolidone (NMP) and 5% piperazine/0.1 M hydroxybenzotriazole in dimethylformamide (DMF, Fisher Scientific) was used for deprotection of FMOC groups. Coupling was achieved using 0.5 M O-benzotriazole-N,N,N´,N´-tetramethyl-uronium-hexafluoro-phosphate (AnaSpec) in DMF as the activator, and 2 M diisopropylethylamine in NMP as the activator base. Peptides were cleaved from resin for 4 h in a cleavage cocktail composed of 90 vol% trifluoroacetic acid (TFA), 2.5 vol% triisopropylsilane, 2.5 vol% 3,6-dioxa-1,8-octanedithiol, 2.5 vol% ddH2O, and 2.5 vol% thioanisole. After cleavage, resin was removed by vacuum filtration and peptide solutions were precipitated by multiple washes in ice cold diethyl ether and dried in vacuum after precipitation. Peptide molecular weight was confirmed using a Bruker Daltonics Autoflex III Smart beam Matrix Assisted Laser Desorption Ionization-Time of Flight (MALDI-ToF) mass spectrometer. Peptides were dissolved in 50/50 ddH2O/acetonitrile with 0.1% TFA and combined with α-cyano-4-hydroxycinnamic acid (Tokyo Chemical Industry) as the matrix (1:1 [peptide]/[matrix]) and calibrations were performed using Care peptide standards (Bruker 206195). To predict the secondary structure of CBP, the sequence was entered into the PEP-FOLD3.0 module of Mobyle@RPBS.[54,55]
cCBP were produced by diluting 20 mg of peptide in 20 mL of 50 mM NH4HCO3 buffer (pH 8.0), adding 20 mg of activated charcoal and stirring at room temperature overnight.[23] Charcoal was separated via vacuum filtration, followed by syringe filtration using 0.2 μm cellulose acetate filters (VWR International, Cat #: 28145–477) and dialysis against ddH2O via (100–500 kDa MW cut-off dialysis tubing (Spectrum Laboratories)). Filtrate was lyophilized and elimination of thiols was verified using Ellman’s test and MALDI-ToF.
Carbodiimide chemistry was used to label cyclized peptides with Texas Red (TR) cadaverine (Thermo Fisher) prior to NP functionalization. Briefly, a mixture of peptide:1-ethyl-3-(3-dimethylamino) propyl carbodiimide (EDC, Thermo Fisher):TR (10:10:1 [peptide]/[EDC]/[TR]) was used for coupling with 5 mM hydroxysulfosuccinimide (sulfo-NHS, Thermo Fisher) in 0.1 M sodium phosphate buffer, pH 7.4. Fluorescent TR was then separated from peptides via dialysis (100–500 Da MW cut-off tubing (Spectrum laboratories)). Conjugation efficiency of labeled peptides was confirmed via fluorescence readings with TR excitation and emission of 583 and 603 nm, respectively accompanied with peptides concentrations determined via lyophilzation and the Fluoraldehyde o-Phthalaldehyde (OPA, Thermo Scientific) assay (Ex/Em = 360 nm/455 nm).
Synthesis and Characterization of PSMA-b-PS Nanoparticles:
One-step RAFT polymerization was used to synthesize PSMA-b-PS based amphiphilic diblock copolymers consisting of MA and STY monomers.[28,30,56] Excess STY was introduced into the polymerization to form block copolymers with alternating MA and STY in the first block (i.e., PSMA) and PS in the second block. STY was added in excess of MA (5:1 [STY]/[MA]) in the presence of the chain transfer agent (CTA) 4-Cyano-4-dodecylsulfanyltrithiocarbonyl sulfanyl pentanoic acid (84:1 [MA]/[CTA]), which was synthesized according to a previously published protocol.[57] AIBN was used as the radical initiator (2:1 [CTA]/[initiator]) in dioxane (50% w/w), and the reaction was purged with nitrogen for 45 min prior to being placed in a 60 °C oil bath for 72 h. Polymer solutions were exposed to air and dissolved in acetone prior to precipitation in petroleum ether. To confirm MA conversion, 100 μL of pre-polymerization and post-polymerization solutions (before precipitation) were dissolved in deuterated dimethyl sulfoxide and analyzed using 1H NMR (Bruker 300 MHz).
Molecular weights of polymerized diblock copolymers were determined using gel permeation chromatography on a Shimadzu system equipped with a differential refractometry (Shimadzu RID-10A), a light scattering detector (Wyatt Technology DAWN TREOS), a solvent pump (Shimadzu LC-20AD), and a column oven operating at 60 °C (Shimadzu CTO-20A). Polymer solutions were dissolved at 1 mg mL−1 DMF/0.05 M lithium chloride (LiCl) and analyzed using a 3 μm linear gel column (Tosoh TSK-Gel Super HM-N, 6.0 mm ID × 15 cm) in series with a 3 μm guard column (Tosoh Biosciences) with a mobile phase consisting of spectroscopic grade DMF/0.05 m LiCl and a flow rate of 0.35 mL min−1. Diblock copolymer molecular weights and polydispersity indices were calculated using ASTRA 6 software (Wyatt Technology) and a refractive index increment (dn/dc) of PSMA-b-PS polymers determined experimentally was 0.142 mL g−1 using a differential refractometer (Shimadzu RID-10A) and ASTRA 6 software (Wyatt Technology).[28,30,56]
NP formation was completed using self-assembly via previously documented approaches.[30] Briefly, ddH2O was added at a rate of 24 μL min−1[58] via syringe pump to amphiphilic diblock copolymers dissolved in 30 mL DMF (200 mg/30 mL). Upon completion of ddH2O addition, polymer NP solutions were dialyzed against water for 72 h (MWCO 6–8 kDa) and filtered using 0.2 μm sterile cellulose acetate filters. Gravimetric NP concentrations were determined using lyophilization and NPs were stored at 4 °C in ddH2O. Dynamic light scattering (Malvern Instrument, Worcestershire, UK) was used to measure size and zeta potential of NPs at 0.2 mg mL−1 concentrations in 0.1 M sodium phosphate buffered solution (PBS), pH 7.4. To determine the number of particles per mL, nanoparticle tracking analysis (NTA) was performed using a NS300 Nanosight (Malvern). Nanosight NTA software version 3.2 Dev Build 3.2.16 (Malvern Instruments Ltd., Worcestershire, UK) was used for data collection and processing was performed at a detection threshold of 3. Transmission electron microscopy was used to visualize PSMA-b-PS NP morphology and size. Untargeted and cCBP-NPs were prepared in 1:9 PBS:ddH2O at 0.2 mg mL−1 and incubated 1:1 with 2% (v/v in water) phosphotungstic acid negative stain on 150 mesh formvar/carbon coated grids for 5 min. Excess solution was removed prior to imaging. Samples were photographed at 80 000–200 000 magnification using a Hitachi 7650 transmission electron microscope operating at 80 with an attached Gatan 11-megapixel Erlangshen digital camera.
Preparation and Characterization of cCBP Functionalized NPs:
To produce a series of multivalent cCBP-NPs, peptide-NP conjugations were performed with 1:1, 10:1, 25:1, 50:1, 75:1, 100:1, and 200:1 [EDC]/[polymer], 5 mM sulfo-NHS, and peptide in 0.1 M sodium phosphate buffer, pH 7.4 (1:1, 10:1, 25:1, 50:1, 75:1, 100:1, and 200:1 [TR-peptide]/[polymer]). Peptide-NP reactions were stirred overnight and transferred into dialysis tubing (MWCO 6–8 kDa) and dialyzed against ddH2O for 72 h with ddH2O changes twice a day. Ligand densities were quantified via TR-cCBP fluorescence using a Biotek Cytation 5 (Ex/Em: 583/603 nm). Unlabeled NPs were used for background subtraction.
MV411 Cell Culture and Uptake:
Human bi-phenotypic B myelomonocytic leukemia MV411 cells (ATCC, CRL-9591) were maintained at 1 – 5×106 cells mL−1 in Iscove’s modified Dulbecco Media (IMDM) supplemented with 10% v/v heat inactivated fetal bovine serum (FBS) and 1% v/v penicillin-streptomycin at 37 °C in 5% CO2. For serum-free studies (Figure S4, Supporting Information), cells were maintained in serum-free media (IMDM supplemented with 0.005 mg mL−1 transferrin (MilliporeSigma, Cat # 61-639-7100MG), 0.005 mg mL−1 insulin (Gibco TM, Cat # 12585014), and 5 ng mL−1 granulocyte-macrophage colony-stimulating factor (GM-CSF, R&D systems, Cat # 215GM010).
To assess CLL1 expression on MV411 cells, Alexa Fluor 647-CLL1 monoclonal antibody, Clec12A Mouse anti-Human, Clone: 50C1 (BD Biosciences, Cat # BDB562568) was added at a volume of 1.25 μL per 1×106 cells in PBS for 30 min on ice. Cells were washed with PBS three times by centrifuging at 400 relative centrifugal forces (RCF) for 5 min followed by flow cytometry analysis. In a subset of experiments, human periodontal ligament cells, PC12, and mouse mesenchymal stem cells were assessed for CLL1 expression (Figure S1, Supporting Information) following the aforementioned protocol. Additionally, immunocytochemistry analysis of CLL1 was performed by incubating MV411 cells with 2.5 μL of CLL1 antibody in FACs buffer for 30 min on ice and washing with PBS. Cells were fixed with 4% paraformaldehyde (PFA) for 10 min and permeabilized with 0.3% Triton X-100 in PBS for 5 min. To stain for phalloidin actin, 5 μL of Alexa Fluor 546 Phalloidin (Thermo Fisher, Cat #: A22283) (6.6 μM in methanol) in 200 μL 1% bovine serum albumin in PBS was added to cell suspension for 15 min in 37 °C. Cells were washed and nuclei was stained with 4´,6-diamidino-2-phenylindole (DAPI) containing mounting media (Abcam, Cat #: ab104139). Cells were imaged using an Andor Dragonfly Spinning Disc Confocal system.
To test peptide binding and uptake of cCBP, peptides were labeled with Alexa Fluor 488 cadaverine via carbodiimide chemistry by dissolving cCBP in ddH2O along with 1:1 [EDC]/[peptide], 5 mM sulfo-NHS, and AF488 cadaverine in 0.1 M sodium phosphate buffer, pH 7.4 (1:1 [AF488]/[peptide]). Reactions were mixed overnight, transferred into dialysis tubing (MWCO 100–500 Da), and dialyzed against ddH2O for 72 h with ddH2O changes twice daily for three days. For binding studies, MV411 cells at 500 000 cells per well were incubated at 4 °C for 30 min with 25, 50, 100, 250, and 500 μg mL−1 fluorescently labeled cCBP. For uptake studies, MV411 cells at 500 000 cells per well were incubated at 4 or 37 °C for 30 min with 50 μg mL−1 fluorescently labeled cCBP. Cells were removed and washed twice with PBS and processed for flow cytometry.
To determine uptake of cCBP-NPs, untargeted NPs and peptide functionalized NPs were further labeled with Alexa Fluor 488 cadaverine via carbodiimide chemistry for flow cytometry analysis. NPs were dissolved in ddH2O with 10:1 [EDC]/[polymer], 5 mM sulfo-NHS, and AF488 cadaverine in 0.1 M sodium phosphate buffer, pH 7.4 (10:1 [AF488]/[polymer]). Reactions were mixed overnight, transferred into dialysis tubing (MWCO 6–8 kDa) and dialyzed against ddH2O for 72 h with ddH2O changes twice daily for three days. MV411 cells were plated at 500 000 cells per well in a 24-well plate, treated with 50 μg mL−1 fluorescently labeled cCBP-NPs or untargeted-NPs and incubated at 37 °C. At t = 2 and 24 h, cells were washed twice with PBS and processed for flow.
To inhibit the CLL1 receptor, MV411 cells were plated at 500 000 cells per well in 24-well plates and treated with 50 μg mL−1 unlabeled cCBP or 5 μL of CLL1 antibody in 2% FBS in PBS for 30 min on ice. Cells were washed twice with PBS and treated with complete media containing 50 μg mL−1 fluorescently labeled cCBP-NPs, or untargeted NPs and incubated at 37 °C. After 1 h, cells were washed twice with PBS and processed for flow cytometry.
To assess NP uptake pathways, MV411 cells were plated at 500 000 cells per well in 24-well plates and treated with 10 μg mL−1 chlorpromazine hydrochloride to inhibit clathrin-mediated endocytosis, 250 μM genistein to inhibit caveolae-mediated endocytosis, or 5 μg mL−1 rottlerin to inhibit micropinocytosis for 30 min prior to adding NPs. Cells were then treated with 50 μg mL−1 NPs in the presence of the inhibitors for an additional hour, followed by a PBS wash and analyzed via flow cytometry. Cytocompatibility of inhibitors was tested via flow cytometry after incubating for 1.5 and 24 h (Figure S5, Supporting Information).
Flow cytometry was performed on an Accuri C6 (BD Biosciences). 10 000 events were collected on cells suspended in 2% FBS in PBS (FACS buffer) supplemented with 0.01% trypan blue (to quench extracellular signal from bound fluorescent NPs) for uptake studies, FACs buffer in the absence of trypan blue (for binding studies) and FACS buffer supplemented with 0.01% trypan blue and 50 μg mL−1 propidium iodine (for cytocompatibility studies, Figures S3 and S5, Supporting Information). Uptake and cytocompatible experiments were repeated three times and had three replicates per plate and endocytic pathways and blocking experiments were repeated three times and had two replicates per plate. Unstained cells were used to identify NP positive populations and untargeted NPs were used as a positive control for uptake experiments as previous studies show that untargeted PSMA-b-PS NPs were uptaken by MV411 cells. [29] Fluorescence from cCBP-NPs and untargeted NPs were detected using the FL1 channel (488 nm excitation, 530/30 detector). Fluorescent signal from CLL1-antibody was detected using the FL4 channel (640 nm excitation, 675/25 detector). FCS express software V.6 was used for analysis. All flow cytometry data was gated on singlet cells identified through double discrimination by graphing FSC-A versus FSH-H.
Statistical Analysis:
Statistical testing was completed using Prism software (GraphPad Version 6.0). Multiple group comparisons were performed using one-way or two-way ANOVA with Tukey’s post-hoc analysis or Dunnett’s post-hoc analysis to determine statistical significance (p-values are indicated in figure legends).
Supplementary Material
Acknowledgements
The authors gratefully thank Karen Bentley, M.S. (University of Rochester Medical Center Electron Microscopy Shared Laboratory) for EM image acquisition, Dr. James McGrath (University of Rochester) for equipment use, Dr. Sayantani Basu, Dr. Jared Mereness, Clyde Overby, and Dr. David Fraser for manuscript review, Dr. Jim Miller (University of Rochester) for discussions regarding the uptake data and the University of Rochester, Center for Professional Development, and the University of Rochester 3-Day Writing Boot Camp for providing writing assistance. Funding for this study was provided by the National Science Foundation Career Award (CBET1450987 (D.S.W.B)), National Institutes of Health (NIH) P30 AR069655, R01 AR064200, R01 AR056696 (D.S.W.B), F31 CA228391 (M.A.A.-F), University of Rochester Clinical and Translational Science Awards (CTSA) award number UL1 TR002001 (D.S.W.B), University Research Award (D.S.W.B), and Drug Discovery Grant (D.S.W.B).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Marian A. Ackun-Farmmer, University of Rochester, Department of Biomedical Engineering, Rochester, NY, USA University of Rochester Medical Center, Department of Orthopaedics and Center for Musculoskeletal Research, Rochester, NY, USA.
Kharimat L. Alatise, University of Rochester, Department of Biomedical Engineering, Rochester, NY, USA
Griffin Cross, Washington University in St. Louis, Biomedical/Medical Engineering, St. Louis, MO, USA.
Danielle S. W. Benoit, University of Rochester, Department of Biomedical Engineering, Rochester, NY, USA University of Rochester Medical Center, Department of Orthopaedics and Center for Musculoskeletal Research, Rochester, NY, USA; University of Rochester, Materials Science Program, Rochester, NY, USA; University of Rochester, Department of Chemical Engineering, Rochester, NY, USA.
References
- [1].Anselmo AC, Mitragotri S, Bioeng. Transl. Med 2016, 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Anselmo AC, Mitragotri S, Bioeng. Transl. Med 2019, 4, e10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC, Adv. Drug Delivery Rev 2014, 66, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chittasupho C, Ther. Delivery 2012, 3, 1171. [DOI] [PubMed] [Google Scholar]
- [5].Alkilany AM, Zhu L, Weller H, Mews A, Parak WJ, Barz M, Feliu N, Adv. Drug Delivery Rev 2019, 143, 22. [DOI] [PubMed] [Google Scholar]
- [6].Treuel L, Jiang X, Nienhaus GU, Soc JR., Interface 2013, 10, 20120939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Oh N, Park JH, Int. J. Nanomed 2014, 9, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Compeer EB, Kraus F, Ecker M, Redpath G, Amiezer M, Rother N, Nicovich PR, Kapoor-Kaushik N, Deng Q, Samson GPB, Yang Z, Lou J, Carnell M, Vartoukian H, Gaus K, Rossy J, Nat. Commun 2018, 9, 1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Montealegre S, van Endert PM, Front. Immunol 2018, 9, 3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sandvig K, Pust S, Skotland T, van Deurs B, Curr. Opin. Cell Biol 2011, 23, 413. [DOI] [PubMed] [Google Scholar]
- [11].Bakker ABH, van den Oudenrijn S, Bakker AQ, Feller N, van Meijer M, Bia JA, Jongeneelen MAC, Visser TJ, Bijl N, Geuijen CAW, Marissen WE, Radosevic K, Throsby M, Schuurhuis GJ, Ossenkoppele GJ, de Kruif J, Goudsmit J, Kiuisbeek AM, Cancer Res. 2004, 64, 8443. [DOI] [PubMed] [Google Scholar]
- [12].Wang YY, Chen WL, Weng XQ, Sheng Y, Wu J, Hao J, Liu ZY, Zhu YM, Chen B, Xiong SM, Chen Y, Chen QS, Sun HP, Li JM, Wang J, Stem Cells Dev. 2017, 26, 1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, Moshaver B, Stigter-van Walsum M, Zweegman S, Ossenkoppele GJ, Jan Schuurhuis G, Blood 2007, 110, 2659. [DOI] [PubMed] [Google Scholar]
- [14].Ma H, Padmanabhan IS, Parmar S, Gong Y, J. Hematol. Oncol 2019, 12, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hutten TJ, Thordardottir S, Fredrix H, Janssen L, Woestenenk R, Tel J, Joosten B, Cambi A, Heemskerk MH, Franssen GM, Boerman OC, Bakker LB, Jansen JH, Schaap N, Dolstra H, Hobo W, J. Immunol 2016, 197, 2715. [DOI] [PubMed] [Google Scholar]
- [16].Jiang YP, Liu BY, Zheng Q, Panuganti S, Chen R, Zhu J, Mishra M, Huang J, Dao-Pick T, Roy S, Zhao X, Lin J, Banik G, Hsi ED, Mandalam R, Junutula JR, Blood Adv. 2018, 2, 1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhao X, Singh S, Pardoux C, Zhao J, Hsi ED, Abo A, Korver W, Haematologica 2010, 95, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wu H, Windmiller DA, Wang L, Backer JM, J. Biol. Chem 2003, 278, 40425. [DOI] [PubMed] [Google Scholar]
- [19].Walter RB, Raden BW, Kamikura DM, Cooper JA, Bernstein ID, Blood 2005, 105, 1295. [DOI] [PubMed] [Google Scholar]
- [20].Marshall ASJ, Willment JA, Lin HH, Williams DL, Gordon S, Brown GD, J. Biol. Chem 2004, 279, 14792. [DOI] [PubMed] [Google Scholar]
- [21].Wiersma VR, de Bruyn M, Shi C, Gooden MJM, Wouters MCA, Samplonius DF, Hendriks D, Nijman HW, Wei YW, Zhou J, Helfrich W, Bremer E, Mabs-Austin 2015, 7, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang JH, Chen SY, Xiao W, Li WD, Wang L, Yang S, Wang WD, Xu LP, Liao SY, Liu WJ, Wang Y, Liu NW, Zhang JN, Xia XJ, Kang TB, Chen G, Cai XY, Yang H, Zhang X, Lu Y, Zhou PH, J. Hematol. Oncol 2018, 11, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang HY, Luo JT, Li YP, Henderson PT, Wang YC, Wachsmann-Hogiu S, Zhao WX, Lam KS, Pan CX, Nanomedicine 2012, 8, 1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lin TY, Zhu Y, Li Y, Zhang H, Ma AH, Long Q, Keck J, Lam KS, Pan CX, Jonas BA, Nanomedicine 2019, 20, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Laborda E, Mazagova M, Shao S, Wang X, Quirino H, Woods AK, Hampton EN, Rodgers DT, Kim CH, Schultz PG, Young TS, Int. J. Mol. Sci 2017, 18, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lu H, Zhou Q, Deshmukh V, Phull H, Ma J, Tardif V, Naik RR, Bouvard C, Zhang Y, Choi S, Lawson BR, Zhu S, Kim CH, Schultz PG, Angew. Chem. Int. Ed. Engl 2014, 53, 9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].van Loo PF, Hangalapura BN, Thordardottir S, Gibbins JD, Veninga H, Hendriks LJA, Kramer A, Roovers RC, Leenders M, de Kruif J, Doornbos RP, Sirulnik A, Throsby M, Logtenberg T, Dolstra H, Bakker ABH, Expert Opin. Biol. Ther 2019, 19, 721. [DOI] [PubMed] [Google Scholar]
- [28].Wang Y, Newman MR, Ackun-Farmmer M, Baranello MP, Sheu TJ, Puzas JE, Benoit DSW, ACS Nano 2017, 11, 9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Baranello MP, Bauer L, Jordan CT, Benoit DSW, Cell. Mol. Bioeng 2015, 8, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Baranello MP, Bauer L, Benoit DS, Biomacromolecules 2014, 15, 2629. [DOI] [PubMed] [Google Scholar]
- [31].Lahoud MH, Proietto AI, Ahmet F, Kitsoulis S, Eidsmo L, Wu L, Sathe P, Pietersz S, Chang HW, Walker ID, Maraskovsky E, Braley H, Lew AM, Wright MD, Heath WR, Shortman K, Caminschi I, J. Immunol 2009, 182, 7587. [DOI] [PubMed] [Google Scholar]
- [32].Marshall AS, Willment JA, Pyz E, Dennehy KM, Reid DM, Dri P, Gordon S, Wong SY, Brown GD, Eur. J. Immunol 2006, 36, 2159. [DOI] [PubMed] [Google Scholar]
- [33].Toft-Petersen M, Stidsholt Roug A, Plesner T, Ebbesen L, Brown GD, Nederby L, Cytometry, Part B 2018, 94, 520. [DOI] [PubMed] [Google Scholar]
- [34].Matutes E, Morilla R, Farahat N, Carbonell F, Swansbury J, Dyer M, Catovsky D, Haematologica 1997, 82, 64. [PubMed] [Google Scholar]
- [35].Chen CH, Floyd H, Olson NE, Magaletti D, Li C, Draves K, Clark EA, Blood 2006, 107, 1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].ISO 10993–5:2009, Biological Evaluation of Medical Devices: Tests for In Vitro Cytotoxicity 2009.
- [37].Loike JD, Silverstein SC, J. Immunol. Methods 1983, 57, 373. [DOI] [PubMed] [Google Scholar]
- [38].Ronzani C, Safar R, Diab R, Chevrier J, Paoli J, Abdel-Wahhab MA, Le Faou A, Rihn BH, Joubert O, Cell Biol. Toxicol 2014, 30, 137. [DOI] [PubMed] [Google Scholar]
- [39].Leipold DD, Figueroa I, Masih S, Latifi B, Yip V, Shen BQ, Dere RC, Carrasco-Triguero M, Lee MV, Saad OM, Liu L, He J, Su D, Xu K, Vuillemenot BR, Laing ST, Schutten M, Kozak KR, Zheng B, Polson AG, Kamath AV, Mabs-Austin. 2018, 10, 1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Macri C, Dumont C, Panozza S, Lahoud MH, Caminschi I, Villadangos JA, Johnston AP, Mintern JD, Mol. Immunol 2017, 81, 143. [DOI] [PubMed] [Google Scholar]
- [41].Kasahara S, Clark EA, J. Leukocyte Biol 2012, 91, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Darwish NH, Sudha T, Godugu K, Elbaz O, Abdelghaffar HA, Hassan EE, Mousa SA, Oncotarget 2016, 7, 57811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Moradi E, Vllasaliu D, Garnett M, Falcone F, Stolnik S, RSC Adv. 2012, 2, 3025. [Google Scholar]
- [44].Cao J, Zhang Y, Wu Y, Wu J, Wang W, Wu Q, Yuan Z, Colloids Surf., B 2018, 161, 508. [DOI] [PubMed] [Google Scholar]
- [45].Roug AS, Larsen HO, Nederby L, Just T, Brown G, Nyvold CG, Ommen HB, Hokland P, Br. J. Haematol 2014, 164, 212. [DOI] [PubMed] [Google Scholar]
- [46].Behzadi S, Serpooshan V, Tao W, Hamaly MA, Alkawareek MY, Dreaden EC, Brown D, Alkilany AM, Farokhzad OC, Mahmoudi M, Chem. Soc. Rev 2017, 46, 4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Frohlich E, Int. J. Nanomed 2012, 7, 5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Foroozandeh P, Aziz AA, Nanoscale Res. Lett 2018, 13, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, Agarwal RG, Lam KS, Biomaterials 2011, 32, 3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Li Y, Gu N, J. Phys. Chem. B 2010, 114, 2749. [DOI] [PubMed] [Google Scholar]
- [51].Dalal C, Saha A, Jana NR, J. Phys. Chem. C 2016, 120, 6778. [Google Scholar]
- [52].Dalal C, Jana NR, J. Phys. Chem. B 2017, 121, 2942. [DOI] [PubMed] [Google Scholar]
- [53].Hillaireau H, Couvreur P, Cell. Mol. Life Sci 2009, 66, 2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Shen Y, Maupetit J, Derreumaux P, Tuffery P, Chem J. Theory Comput. 2014, 10, 4745. [DOI] [PubMed] [Google Scholar]
- [55].Thevenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tuffery P, Nucleic. Acids. Res 2012, 40, W288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Baranello MP, Ph.D. Thesis, University of Rochester 2015. [Google Scholar]
- [57].Moad G, Rizzardo E, Thang SH, Aust. J. Chem 2005, 58, 379. [Google Scholar]
- [58].Rodriguez VB, Henry SM, Hoffman AS, Stayton PS, Li XD, Pun SH, J. Biomed. Opt 2008, 13, 014025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.