

Abstract
This editorial refers to ‘C-type natriuretic peptide co-ordinates cardiac structure and function’†, by A.J. Moyes et al., on page 1006.
Heart failure (HF) is a growing pandemic and remains one of the leading causes of morbidity and mortality in the USA and worldwide.1 A hallmark in the development and progression of HF is cardiac remodelling which includes cardiomyocyte hypertrophy and apoptosis, vascular rarefaction and dysfunction, and the formation of interstitial fibrosis.2 Despite improvements in therapies that target cardiac remodelling, such as renin–angiotensin–aldosterone system (RAAS) blockade and dual RAAS and neprilysin inhibition (via sacubitril–valsartan), in many instances, pathological cardiac remodelling and HF progression remain unabated. This suggests the unmet clinical need to understand the mechanisms of cardiac remodelling and to discover novel cardiac anti-remodelling therapies to alleviate the growing burden of HF.
Numerous studies have clearly demonstrated that the heart functions beyond a pump, and is also a remarkable endocrine organ that fine-tunes the body’s ability to regulate blood pressure, fluid volume, and water and salt balance, and to maintain cardiac structure and function.3 This cardiovascular homeostasis is achieved, in part, through the natriuretic peptide (NP) family, which is comprised of three structurally conserved peptides, atrial, B-type, and C-type NP (ANP, BNP, and CNP, respectively).4 Traditionally, ANP and BNP have been thought to mediate the balance in cardiovascular homeostasis and cardioprotection via the activation of the particulate guanylyl cyclase receptor A (GC-A, also known as NPR-A) and the generation of its second messenger, cGMP. However, it is now recognized that CNP is also a key hormone that possesses anti-fibrotic, anti-hypertrophic, anti-atherogenic, anti-inflammatory, and vasorelaxing properties.5,6 The majority of these cardiovascular protective properties assigned to CNP have been reported to be mediated by the particulate guanylyl cyclase receptor B (GC-B, also known as NPR-B) and cGMP signalling. Intriguingly, some of the favourable actions of CNP, particularly in the vasculature, involve a non-cGMP pathway via the NP clearance receptor, NPRC,7–9 which generally has been thought to be the receptor responsible for clearing and breaking down NPs.
In the current issue of the European Heart Journal, Moyes and colleagues present provocative new data that broaden our understanding of the invaluable role of CNP in regulating cardiac structure and function, with a special focus on NPRC (see Take home figure).10 These investigators performed a comprehensive series of experiments that span in vitro cell-based systems, in vivo models of HF (i.e pressure overload and sympathetic hyperactivation), ex vivo investigations of ischaemia/reperfusion (I/R) injury, and coronary reactivity, as well as gene and protein expression studies in human HF tissue, to provide novel evidence of the cardioprotective actions of CNP. Using various unique transgenic mouse lines that either expressed cell-specific (i.e. endothelial, cardiomyocyte, or fibroblast) deletion of CNP or global knockout of CNP’s cognate receptors, NPR-B or NPRC, the investigators report that under basal, non-stress conditions, none of the three cell-specific deletions of CNP nor global NPRC or NPR-B deletion led to significant changes in cardiac structure or systolic function. However, under pathophysiological conditions induced by abdominal aortic constriction (AAC) pressure overload, both cardiomyocyte- and fibroblast-specific deletion of CNP had a deleterious effect, with greater reduction in ejection fraction (EF), greater left ventricular (LV) dilatation, and increased cardiac fibrosis and hypertrophy. Further, these detrimental cardiac structural and functional indices were mimicked in a second pre-clinical model of HF by isoprelanine-induced sympathetic hyperactivation in mice with cardiomyocyte-specific deletion of CNP. It is tempting to speculate that the mice with fibroblast-specific deletion of CNP would have exhibited a similar outcome due to sympathetic hyperactivation; however, this was not investigated in the current study. Furthermore, mice with endothelial cell-restricted deletion of CNP or wild-type mice treated with a nitric oxide synthase (NOS) inhibitor and CNP (vs. NOS inhibitor alone) did not exhibit a worse cardiac phenotype when stressed with pressure overload. These data are highly supportive of the importance of cardiomyocyte- and fibroblast-derived, and not endothelial cell-derived, CNP in maintaining cardiac structural integrity and function which are NOS independent. Such observations markedly extend our understanding of the biology of CNP beyond the endothelial cell in the heart. Interestingly, the adverse cardiac structural and functional effects observed due to the cardiomyocyte- and fibroblast-specific deletion of CNP following pressure overload were also seen in global NPRC knockout mice. Importantly, in the NPRC knockout mice, the adverse phenotype related to fibrosis and myocardial function could not be rescued with chronic subcutaneous (via osmotic mini-pump) therapy of CNP. With these data to hand, the investigators make a compelling argument that CNP/NPRC signalling mediates the cardioprotection seen in the current studies. This is supported, in part, by the intriguing findings that isoprelanine-induced sympathetic hyperactivation in the global NPR-B (the classical functional receptor for CNP signalling) knock out mice did not have an adverse effect on systolic function or cardiac remodelling as seen in mice with cardiomyocyte-specific deletion of CNP. It is worthwhile to note, due to the characteristics of the global NPR-B deletion, that include early death, long-term investigations of cardiac structure and function are challenging, and in-depth interrogation of the cardiac phenotype under pathophysiological conditions in cardiomyocyte- and fibroblast-specific deletion of NPR-B are worthy of future studies. Indeed, an elegant study by Langenickel and co-workers11 demonstrated exaggerated LV hypertrophic response in dominant-negative NPR-B mutant transgenic rats with HF, thus providing evidence that the anti-hypertrophic actions of CNP may involve redundancy with the involvement of both NPRC and NPR-B. Regardless, the fact that up-regulation of well-established hypertrophic markers and pro-fibrotic genes, such as ANP, β-MHC, Col1A1, TGF-β, and fibronectin, and down-regulation of SERCA-2 in cardiomyocyte- and fibroblast-specific deletion of CNP and global NPRC knockout mice provide additional validation of molecular mechanisms that may mediate the favourable CNP/NPRC anti-remodelling effects on the heart.
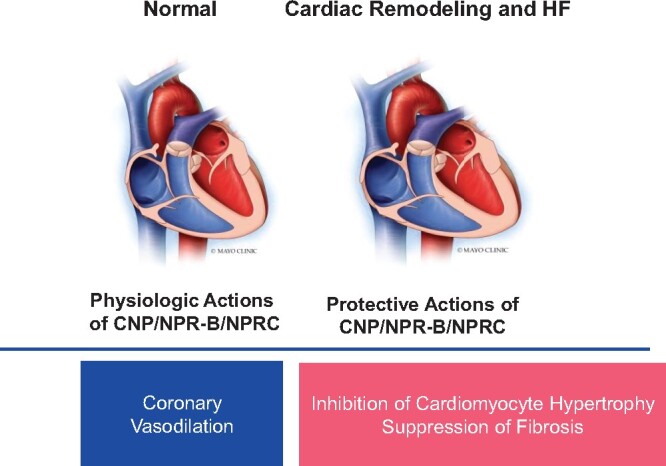
Take home figure.
The protective role of CNP/NPR-B/NPRC.
To support the role of the CNP/NPRC pathway in HF further, the investigators performed an elegant analysis of CNP, NPR-B, and NPRC expression in human HF ventricular tissue as well as in pre-clinical HF-induced AAC, and determined that CNP is down-regulated and NPRC is up-regulated in both pre-clinical and clinical HF, with no significant difference in NPR-B expression. Their findings with regards to CNP and NPRC in human HF are consistent with studies by Ichiki and colleagues;12 however, they differ in regards to NPR-B expression which was increased in the latter study. Moreover, while endothelium-derived CNP deletion did not develop pressure overload-induced cardiac structural and functional abnormalities, it did impair responsiveness to endothelium-dependent vasodilators and shear stress in the coronary vasculature in an ex vivo Langendorff setting. These data thus support an important role for endothelial cell-derived CNP in maintaining coronary vascular homeostasis, which appears to be regulated via NPRC given that coronary endothelium-dependent vasoreactivity and responsiveness were dampened with exogenous treatment of CNP in hearts from global NPRC knockout mice. To add to the complexity of the CNP system in anti-remodelling effects within the heart, mice with cardiomyocyte-specific deletion of CNP or global NPRC knockout had greater infarct size and extended LV functional impairment following I/R injury ex vivo, which was unexpectedly not observed in endothelial cell-specific CNP-disrupted mice.
The investigators are to be commended on this important work, which extends the mounting evidence of CNP’s cardioprotective role within the heart, which has historically been underappreciated. As with all studies, this fascinating report raises new questions for the future. It would also be of interest to further tease apart commonalities or uniqueness of NPR-B and NPRC in the heart under physiological and pathophysiological conditions in various species, to confirm and extend the current findings. Such studies should be performed in vivo, with and without exogenous CNP treatment as well as with an NPR-A-activating comparator such as ANP, in cell-specific deletion of NPR-B which would hopefully overcome the limitation of early death and dwarfism seen with global NPR-B knockout. In addition, measuring plasma and tissue cGMP (the second messenger of NPR-B/-A activation) is needed to help understand the role of NPRC, or lack of NPR-B/-A activation, in mediating the beneficial effects of CNP on cardiac structure and function. It is also important to examine the CNP/NPRC and CNP/NPRB/cGMP systems in HF (large and small animal) models of reduced and preserved EF and/or in human cell lines using gene silencing techniques (as an example) to support these outstanding studies. Further, it would be of benefit to establish the downstream mechanisms of CNP/NPRC signalling to enhance our understanding of novel pathways mediating cardiac protection with CNP. Lastly, if NPRC functions beyond a clearance receptor, it would be helpful to define the physiological and pathophysiological triggers or mechanisms that switch the balance of NPRC from being a clearing receptor to a signalling receptor.
The biology of the CNP system in the cardiovascular arena is complex; however, it is becoming increasingly evident that CNP is a vital hormone that mediates favourable cardioprotective actions. What is unequivocally clear, and highly supported by this sophisticated study by Moyes and co-workers, is that a CNP deficiency has deleterious effects on cardiac structure and function in pathophysiological settings such as pressure overload as seen with HF. The clinical therapeutic implications of the current study are most important for the development of innovative CNP-based/NPR-B/NPRC therapies, which is also supported by other pre-clinical and translational studies in myocardial infarction, HF, atrial fibrosis/fibrillation, and hypertension.9,13–15
There is no doubt that we are only at the beginning of a new era regarding CNP biology. Certainly, unravelling the intricate mechanisms of CNP signalling is of utmost importance to facilitate the development of new therapies targeting cardiac remodelling and HF. With future prospective studies, innovative CNP-based drugs may offer a unique therapeutic and efficacious strategy to curb the growing challenge of preventing adverse pathological cardiac remodelling and HF.
Funding
This work was supported by grants from the National Heart, Lung and Blood Institute (R01 HL132854 to S.J.S. and R01 HL136340 to J.C.B) and the National Institute on Aging (R01 AG056315 to S.J.S. and J.C.B.).
Conflict of interest: J.C.B is a named inventor on patents related to CNP-based peptides. The other authors have no conflicts to declare.
Footnotes
doi:10.1093/eurheartj/ehz093.
The opinions expressed in this article are not necessarily those of the Editors of the European Heart Journal or of the European Society of Cardiology.
References
- 1. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O’Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS, American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019;doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2. Burchfield JS, Xie M, Hill JA.. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation 2013;128:388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clerico A, Giannoni A, Vittorini S, Passino C.. Thirty years of the heart as an endocrine organ: physiological role and clinical utility of cardiac natriuretic hormones. Am J Physiol Heart Circ Physiol 2011;301:H12–H20. [DOI] [PubMed] [Google Scholar]
- 4. Matsuo A, Nagai-Okatani C, Nishigori M, Kangawa K, Minamino N.. Natriuretic peptides in human heart: novel insight into their molecular forms, functions, and diagnostic use. Peptides 2019;111:3–17. [DOI] [PubMed] [Google Scholar]
- 5. Kuhn M. Molecular physiology of membrane guanylyl cyclase receptors. Physiol Rev 2016;96:751–804. [DOI] [PubMed] [Google Scholar]
- 6. Spiranec K, Chen W, Werner F, Nikolaev VO, Naruke T, Koch F, Werner A, Eder-Negrin P, Dieguez-Hurtado R, Adams RH, Baba HA, Schmidt H, Schuh K, Skryabin BV, Movahedi K, Schweda F, Kuhn M.. Endothelial C-type natriuretic peptide acts on pericytes to regulate microcirculatory flow and blood pressure. Circulation 2018;138:494–508. [DOI] [PubMed] [Google Scholar]
- 7. Moyes AJ, Khambata RS, Villar I, Bubb KJ, Baliga RS, Lumsden NG, Xiao F, Gane PJ, Rebstock AS, Worthington RJ, Simone MI, Mota F, Rivilla F, Vallejo S, Peiro C, Sanchez Ferrer CF, Djordjevic S, Caulfield MJ, MacAllister RJ, Selwood DL, Ahluwalia A, Hobbs AJ.. Endothelial C-type natriuretic peptide maintains vascular homeostasis. J Clin Invest 2014;124:4039–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bubb KJ, Aubdool AA, Moyes AJ, Lewis S, Drayton JP, Tang O, Mehta V, Zachary IC, Abraham DJ, Tsui J, Hobbs AJ.. Endothelial C-type natriuretic peptide is a critical regulator of angiogenesis and vascular remodeling. Circulation 2018;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y, Sarkar O, Brochu M, Anand-Srivastava MB.. Natriuretic peptide receptor-C attenuates hypertension in spontaneously hypertensive rats: role of nitroxidative stress and Gi proteins. Hypertension 2014;63:846–855. [DOI] [PubMed] [Google Scholar]
- 10. Moyes AJ, Chu SM, Aubdool AA, Dukinfield MS, Margulies KB, Bedi Jr KC, Hodivala-Dilke K, Baliga RS, Hobbs AJ.. C-type natriuretic peptide co-ordinates cardiac structure and function. Eur Heart J 2020;41:1006–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Langenickel TH, Buttgereit J, Pagel-Langenickel I, Lindner M, Monti J, Beuerlein K, Al-Saadi N, Plehm R, Popova E, Tank J, Dietz R, Willenbrock R, Bader M.. Cardiac hypertrophy in transgenic rats expressing a dominant-negative mutant of the natriuretic peptide receptor B. Proc Natl Acad Sci USA 2006;103:4735–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ichiki T, Schirger JA, Huntley BK, Brozovich FV, Maleszewski JJ, Sandberg SM, Sangaralingham SJ, Park SJ, Burnett JC Jr.. Cardiac fibrosis in end-stage human heart failure and the cardiac natriuretic peptide guanylyl cyclase system: regulation and therapeutic implications. J Mol Cell Cardiol 2014;75:199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Soeki T, Kishimoto I, Okumura H, Tokudome T, Horio T, Mori K, Kangawa K.. C-type natriuretic peptide, a novel antifibrotic and antihypertrophic agent, prevents cardiac remodeling after myocardial infarction. J Am Coll Cardiol 2005;45:608–616. [DOI] [PubMed] [Google Scholar]
- 14. Kawakami R, Lee CYW, Scott C, Bailey KR, Schirger JA, Chen HH, Benike SL, Cannone V, Martin FL, Sangaralingham SJ, Ichiki T, Burnett JC Jr.. A human study to evaluate safety, tolerability, and cyclic GMP activating properties of cenderitide in subjects with stable chronic heart failure. Clin Pharmacol Ther 2018;104:546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jansen HJ, Mackasey M, Moghtadaei M, Liu Y, Kaur J, Egom EE, Tuomi JM, Rafferty SA, Kirkby AW, Rose RA.. NPR-C (natriuretic peptide receptor-C) modulates the progression of angiotensin II-mediated atrial fibrillation and atrial remodeling in mice. Circ Arrhythm Electrophysiol 2019;12:e006863. [DOI] [PubMed] [Google Scholar]