

Abstract
Purpose:
Preclinical data suggest that radiotherapy (RT) is beneficial in combination with immune checkpoint blockade. Clinical trials have explored RT with single-agent immune checkpoint blockade, but no trials have reported RT with the combination of nivolumab and ipilimumab.
Patients and Methods:
We conducted a phase 1 study of patients with stage IV melanoma receiving nivolumab and ipilimumab with two different dose-fractionation schemes of RT. Patients had at least one melanoma metastasis that would benefit from palliative RT and one metastasis that would not be irradiated. Nivolumab 1 mg/kg + ipilimumab 3 mg/kg and extracranial RT with a dose of 30 Gy in 10 fractions was administered in Cohort A, and then 27 Gy in 3 fractions was administered in Cohort B. The primary outcome was safety.
Results:
Twenty patients were treated (10 in each cohort). The rates of treatment-related grade 3–4 adverse events in Cohort A and B were 40% and 30%, respectively. There were no grade ≥3 adverse events attributed to RT. Patients responded to treatment outside of the irradiated volume (Cohort A 5/10; Cohort B 1/9). No evaluable patients had progression of irradiated metastases. Immunologic changes were seen in the peripheral blood with increases in T-cell receptor diversity in some responding patients.
Conclusions:
RT with nivolumab and ipilimumab was safe compared with historical data of nivolumab and ipilimumab alone. Immunologic effects were observed in the peripheral blood. Randomized studies are ongoing to assess whether RT increases the efficacy of nivolumab and ipilimumab.
Introduction
Combination immunotherapy with nivolumab (nivo) + ipilimumab (ipi) can be effective in patients with malignancies, including advanced melanoma (1), microsatellite-instability high colorectal cancer, renal cell carcinoma, and lung cancer (2), yet is associated with a high rate of immune-related adverse events. Radiotherapy (RT) is also commonly used to treat patients with these cancers. An important, practical question facing clinicians is whether RT can be safely administered to patients also receiving the nivo + ipi combination.
RT can result in favorable immunologic effects such as enhanced antigen expression and increased T-cell infiltration into tumors (3). Preclinical models also suggest that RT can improve efficacy of immune checkpoint blockade (4). The most promising results occur when RT is given together with combination immune checkpoint inhibition (5). However, the effects of RT are dose dependent, and the manner in which the total RT dose is fractionated may be of immunologic relevance (6). Palliative RT is typically given with conventional low-dose fractions of 2–4 gray (Gy), but preclinical data have suggested that RT with fraction sizes in the range of 8–10 Gy may be optimal to combine with immune checkpoint inhibition (7). Moreover, RT dose fraction sizes of 8–9 Gy are commonly used in metastatic melanoma, to a total equivalent dose that is higher than most palliative RT regimens because of the perceived resistance of melanoma to RT (8, 9).
Prior studies in patients suggest combining RT with either single-agent ipi or anti-programmed death 1 (PD-1) antibodies appears safe and is preliminarily promising (10–15). However, little is known about RT in combination with both nivo and ipi in patients. We conducted a phase 1, prospective clinical trial to investigate the safety and efficacy of combining two different doses and schedules of RT with the combination of nivo and ipi in patients with advanced melanoma, with the hypothesis that this could be given safely, with low-dose fractions and high-dose fractions.
In addition, we evaluated immunologic changes in the peripheral blood during treatment. We focused correlative analyses on cellular populations that previously have been described to be relevant to antitumor immune responses in patients receiving checkpoint inhibitors such as the absolute lymphocyte count (ALC; ref. 16), degrees of lymphocyte proliferation (17, 18), and the proportions of naïve and effector memory T cells (13, 19). We also examined the diversity of the T-cell receptor (TCR) repertoire based upon preclinical data suggesting that RT enhances antitumor immunity, at least in part, by diversifying the number of T-cell clones (5).
Patients and Methods
Patients and treatment
Patients were required to have stage IV melanoma and be candidates to receive the nivo + ipi combination. All patients also had at least one melanoma metastasis that was determined to potentially benefit from palliative RT per clinician assessment, to alleviate or prevent pain, luminal obstruction, bleeding, or other symptoms. More than one metastasis could be irradiated, but at least one metastasis evaluable by RECIST could not be irradiated to be eligible for inclusion. In both cohorts, immunotherapy was given at FDA-approved doses for melanoma: nivo 1 mg/kg with ipi 3 mg/kg for up to 4 doses every 3 weeks during the induction period, and then nivo 240 mg every 2 weeks or 480 mg every 4 weeks during maintenance. RT was required to start between the first and second doses of nivo + ipi immunotherapy as per schema in Supplementary Fig. S1. First, in Cohort A, patients received extracranial RT using a conventional dose and fractionation scheme of 30 Gy in 10 fractions of 3 Gy each. If ≤7 out of 9 evaluable patients had treatment-related grade 3–5 toxicity in Cohort A, this regimen would be deemed acceptable. If the treatment-related grade 3–5 adverse event (AE) rate was acceptable in Cohort A, Cohort B would subsequently open. Then, in Cohort B, patients received extracranial RT using a hypofractionated dose and fractionation scheme of 27 Gy in 3 fractions of 9 Gy each. Patients were treated until unacceptable toxicity, disease progression, or clinician discretion. As this was a safety study, patients could have any number of prior therapies, including prior anti–PD-1, prior anti-cytotoxic T lymphocyte antigen 4 (CTLA-4), and the combination of both as long as prior toxicities had resolved to grade 1 or less. After institutional review board (IRB) approval, all patients provided written informed consent before enrollment. The trial adhered to all ethical considerations governing research participants in accordance with the Declaration of Helsinki.
Safety
The primary endpoint of the study was safety as assessed by the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. To be more comprehensive with safety reporting, we report all 10 patients in each cohort who received any treatment. Because one patient in each cohort was not evaluable for safety specifically in the per protocol safety population (required at least 75% of scheduled doses of study drugs and RT), we enrolled one additional patient in each cohort (n = 10 total per cohort) over the initially planned n = 9. Separately for each cohort, treatment-related adverse events (TRAE) and treatment-emergent adverse events (TEAE; any AE arising during treatment) were summarized on a patient level for any AE by CTCAE grade. In the event of multiple instances of the same toxicity, the maximum grade per patient for the given category was taken.
Efficacy
To assess responses outside of the irradiated volume, all patients had at least one measurable lesion, per Response Evaluation Criteria in Solid Tumors (RECIST) v. 1.1 that was not irradiated. Efficacy was assessed by both RECIST as well as the immune-related RECIST (irRECIST) criteria (20). Response rates with binomial 95% confidence intervals (CI) were determined at week 12, week 18, and best overall response during the study period. If patients clinically progressed without an available RECIST read, these patients were considered to have progressed outside of the irradiated volume. Disease control rate (DCR) was defined as patients with complete or partial responses or stable disease. Because many metastases intended to be irradiated were anticipated to be in bone, irradiated metastases were not required to be measurable per RECIST. When a RECIST measurable lesion was irradiated, the change in tumor size within the irradiated tumor burden was assessed.
Progression-free survival (PFS) was assessed from the start of treatment until documented progression or death from any cause. Patients alive and without progression were censored at next treatment date, end of study, or last off-study follow-up visit. Overall survival (OS) was assessed from start of treatment until death from any cause. Living patients were censored at last off-study follow-up visit, last scan date, or at the end of study if no follow-up was available. PFS and OS were estimated by Kaplan–Meier methodology. All analyses were performed with SAS 9.4 (The SAS Institute).
Immune correlates
Peripheral blood was collected at baseline before treatment initiation, 6–9 weeks following treatment initiation (after the completion of RT, at time of the third dose of immunotherapy), and 10–14 weeks following treatment start (time of the first efficacy assessment). Mass cytometry (CyTOF) and Luminex cytokine analyses were performed in the Human Immune Monitoring Center at Stanford University Medical Center. Flow cytometry was performed in the Immune Monitoring Facility at Memorial Sloan Kettering Cancer Center (MSKCC) to additionally examine T-cell phenotypic markers not tested by CyTOF. The ALC was obtained from routine clinical laboratory testing and additionally enabled assessment of absolute changes in specific lymphocyte populations identified by CyTOF and flow cytometry. TCR repertoire sequencing was performed using previously described methodology (Adaptive Biotechnologies as described in ref. 17) and analyzed at MSKCC. The Simpson index was used to estimate diversity of the TCR clone structures in each patient at each time point, with a high index representing a less diverse and more monoclonal population. See Supplementary Methods for details of the assays and analyses.
Results
Patients and treatment
Twenty patients were enrolled. Specific demographic details for each cohort are included in Table 1. Notably, five patients had prior anti–CTLA-4 therapy, 13 patients had prior anti–PD-1, and 2 patients had prior nivo + ipi. The duration of time between prior treatment and treatment within this protocol is shown in Supplementary Table S1. The median duration of follow-up for survivors in both cohorts was 19.4 months (range, 2.8–33.5); Cohort A was 24.0 months (range, 2.8–33.5) and Cohort B was 18.0 months (range, 5.6–19.6). The first patient started treatment on October 13, 2016, and the last patient started treatment on April 18, 2018. The median number of nivo + ipi doses was four. One patient had one dose, three had two doses, three had three doses, and 13 had four doses of combination immunotherapy. In the 7 patients who discontinued before four doses, 4 discontinued due to adverse events, 2 discontinued due to progression, and 1 patient discontinued due to death. RT was administered to various sites of metastatic melanoma, most commonly lymph node, bone, skin, and muscle/soft tissue metastases (Supplementary Table S2).
Table 1.
Clinical characteristics.
| All Patients | Cohort A | Cohort B | ||
|---|---|---|---|---|
| N (%) | N (%) | N (%) | ||
| Patients | 20 | 10 (50) | 10 (50) | |
| Age at consent, y | Median (range) | 60 (28–86) | 65 (28–86) | 51 (37–76) |
| Sex | Male | 12 (60) | 4 (40) | 8 (80) |
| Female | 8 (40) | 6 (60) | 2 (20) | |
| Race | White | 18 (90) | 8 (80) | 10 (100) |
| Asian | 2 (10) | 2 (20) | 0 (0) | |
| ECOG at Screening | 0 | 9 (45) | 4 (40) | 5 (50) |
| 1 | 11 (55) | 6 (60) | 5 (50) | |
| Stage at Enrollment | IV | 18 (90) | 9 (90) | 9 (90) |
| IVA | 2 (10) | 1 (10) | 1 (10) | |
| M Stage at Enrollment | M1a | 3 (15) | 2 (20) | 1 (10) |
| M1b | 1 (5) | 1 (10) | 0 (0) | |
| M1c | 13 (65) | 6 (60) | 7 (70) | |
| M1d | 3 (15) | 1 (10) | 2 (20) | |
| LDH at Enrollment | Median (range) | 265 (143–856) | 284 (148–856) | 234 (143–582) |
| Not elevated | 11 (55) | 3 (30) | 8 (80) | |
| Elevated | 9 (45) | 7 (70) | 2 (20) | |
| Prior treatment | ||||
| Anti-CTI_A4 | 5 (25) | 3 (30) | 2 (20) | |
| Anti-PD-1 | 13 (65) | 4 (40) | 9 (90) | |
| Combined anti-CTLA-4 and anti-PD-1 | 2 (10) | 0 (0) | 2 (20) | |
| BRAF/MEK Inhibitor | 1 (5) | 0 (0) | 1 (10) | |
| Radiotherapy | 10 (50) | 5 (50) | 5 (50) | |
| Doses of Nivo + Ipi | 1 | 1 (5) | 0 (0) | 1 (10) |
| 2 | 3 (15) | 3 (30) | 0 (0) | |
| 3 | 3 (15) | 0 (0) | 3 (30) | |
| 4 | 13 (65) | 7 (70) | 6 (60) | |
| Baseline tumor volume | ||||
| Baseline RECIST target sum outside irradiated volume (mm) | Median (range) | 33.0 (10.0–134.0) | 43.5 (10.0–134.0) | 32.8 (10.0–105.0) |
| N = | 20 | 10 | 10 | |
| Baseline RECIST target sum inside irradiated volume (mm) | Median (range) | 41.8 (14.0–116.0) | 43.0 (14.0–116.0) | 39.4 (25.0–63.9) |
| N = | 17 | 9 | 8 | |
Safety
Twenty patients (n = 10 Cohort A, n = 10 Cohort B) were evaluated for safety. In Cohort A, all 10 had at least 1 TRAE of any grade. The most common TRAE were fatigue (n = 6), decreased appetite (n = 5), and rash (n = 5). Four patients out of 10 (40%; 95% CI, 12–74) had grade 3–4 TRAE (Supplementary Table S3). Two patients had grade 4 TRAE in Cohort A: one elevated lipase value and one thrombocytopenia. Because ≤7 patients had grade 3–4 TRAE in Cohort A, Cohort B was opened. In Cohort B, all 10 patients had at least 1 TRAE of any grade. The most common TRAE were diarrhea (n = 5), rash (n = 5), pruritus (n = 5), and fatigue (n = 4; Supplementary of Table S3). Three of 10 patients (30%; 95% CI, 7–65) had grade 3–4 TRAE. No patients in Cohort B had grade 4 TRAE. Specific TEAE are detailed in Supplementary Table S4. Five patients had radiation-related dermatitis. There were no specific grade 3–5 toxicities attributed to RT. Nine patients died during the conduct of the study from progressive melanoma; there were no treatment-related deaths.
Efficacy
Outside irradiated volume
Among the 20 patients enrolled, one patient in Cohort B was not evaluable for efficacy outside of the irradiated volume as the patient experienced toxicity and withdrew consent before response determination. As the best overall response, in Cohort A, five patients (5/10; 50%; 95% CI, 18.7–81.3) had a response by RECIST outside of the irradiated volume. One of these responses was a complete response outside the irradiated volume (Fig. 1). In Cohort B, one patient had a partial response outside of the irradiated volume (1/9; 11.1%; 95% CI, 0.3–48.2). All patients with irRECIST responses also had responses by RECIST; the irRECIST response rates were 5/10 [50% (95% CI, 18.7–81.3)] and 1/9 [11.1% (95% CI, 0.3–48.2)] in Cohorts A and B, respectively. Among the six patients who had a response, three had no prior systemic therapy, two patients had prior anti–CTLA-4 and two patients had prior anti–PD-1.
Figure 1.
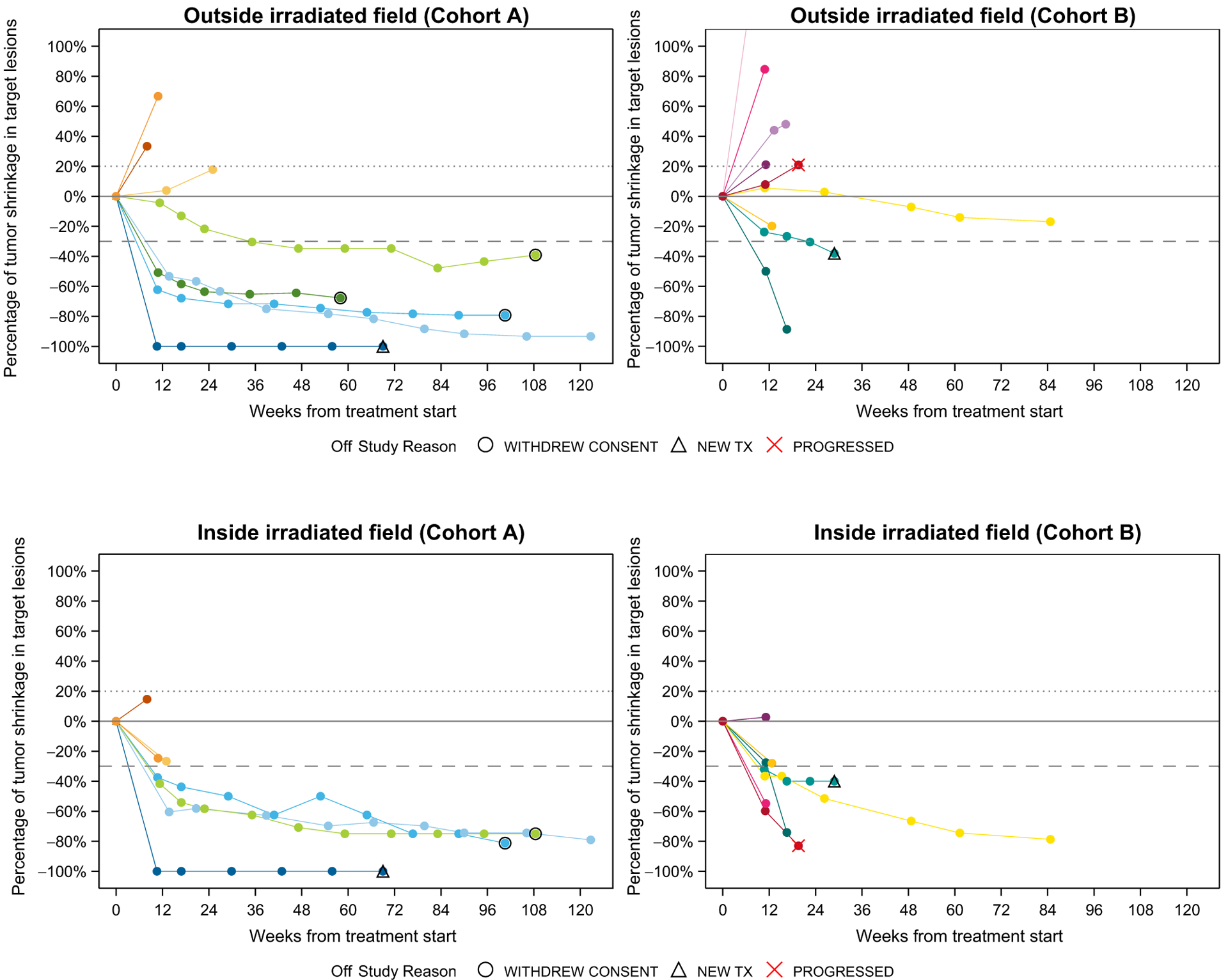
Spider plots of maximum target lesion change. In Cohort A, 10 patients were assessed for overall response rate. Only eight were included in the outside irradiated volume spider plot, as two patients experienced clinical progression and did not receive additional scans for RECIST assessment. Only seven patients were included within the irradiated volume as one additional patient did not have measurable lesions within the irradiated volume at baseline. In Cohort B, nine patients were evaluable for overall response rate outside the irradiated volume and included in the plot. Among these nine, seven patients were included in the plot of tumor changes inside the irradiated volume because one did not have a measurable lesion within the irradiated volume at baseline and another did not have a measurable lesion at a post-treatment initiation timepoint. Patients are matched by color.
At 6 months, estimated PFS was 50% (95% CI, 18.4–75.3) in Cohort A and 20% (95% CI, 3.1–47.5) in Cohort B. The median OS had not been reached in either cohort. At 12 months, estimated OS was 56.3% (95% CI, 20.9–80.9) in each cohort.
Inside irradiated volume
Among the 7 patients (Cohort A) and 8 patients (Cohort B) with disease evaluable for response within the irradiated volume, 4/7 (57.1%; 95% CI, 18.4–90.1) and 5/8 (62.5%; 95% CI, 24.5–91.5) responded, respectively (Fig. 1). There were 3 complete responses within the irradiated volume in Cohort A and no complete responses in Cohort B. One patient had some enlargement of lesions in the irradiated volume not meeting criteria for progression. The DCRs, therefore, within the irradiated volume were 7/7 (100%; 95% CI, 59–100) and 8/8 (100%; 95% CI, 63.1–100), respectively.
Immune correlates
Eighteen patients had peripheral blood available for analyses by CyTOF, flow cytometry, and Luminex analyses. 15 patients had blood available for TCR sequencing analysis. At the first timepoint after initiation of therapy (6–9 weeks after pretreatment baseline), there were several immunologic changes observed in peripheral blood. The median ALC decreased from pretreatment more in Cohort A (median, 1.20–0.95) than in Cohort B (median, 1.22–1.20; Fig. 2A). CyTOF data suggested that the decrease in lymphocytes was primarily in the proportion of B cells because proportions of T and natural killer (NK) cells did not obviously change at this first timepoint. Not all changes were sustained by the second on-treatment timepoint (10–14 weeks after treatment initiation). The proportion of naïve CD8 T cells (CCR7+CD45RA+) was lower at baseline in Cohort A compared with Cohort B and remained lower at later timepoints. The proportion of T effector memory (CCR7−CD45RA−) cells was similar in both cohorts at baseline but increased in Cohort A at both post-treatment timepoints. However, in Cohort B, the T effector memory cell population did not change much from baseline. There was no obvious change in the proportion of T regulatory cells (Treg). Absolute changes in these populations are shown in Supplementary Fig. S2.
Figure 2.
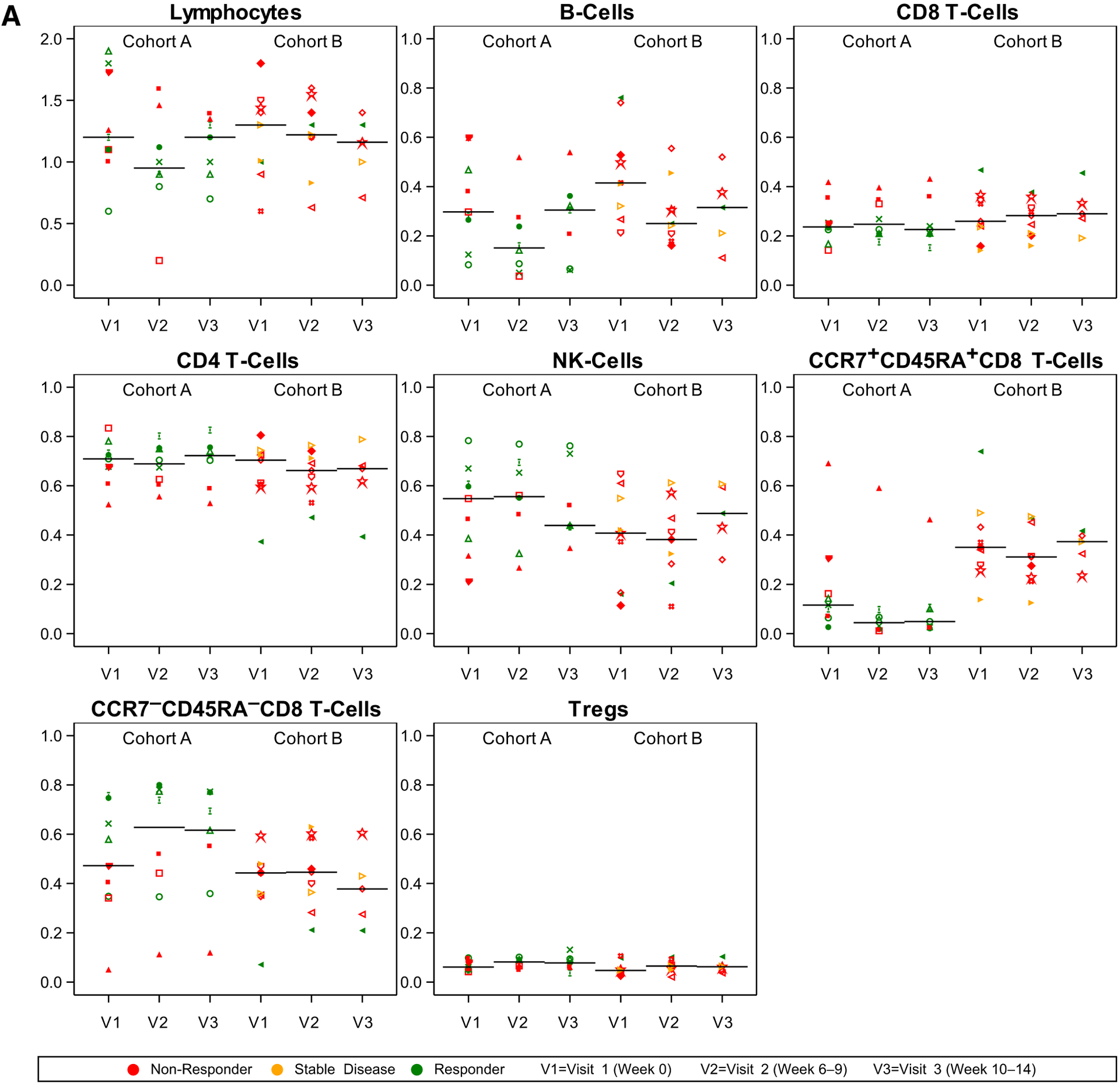
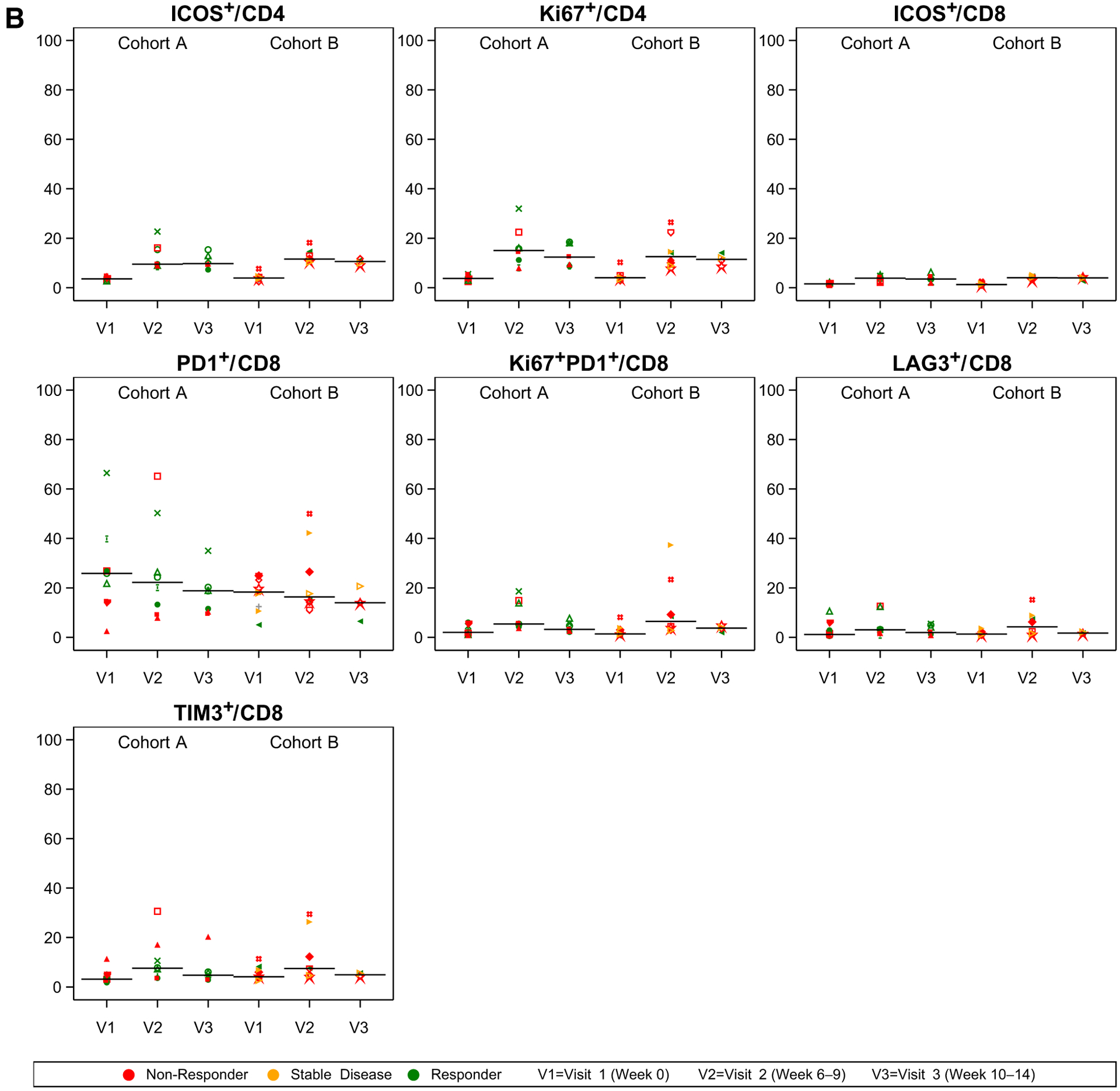
CyTOF and Flow Cytometry immune correlates. A, CyTOF showing the proportion (of 1.0, y axis) and flow cytometry. B, Flow Cytometry showing the percentage (of 100, y axis) of cells positive for respective markers at pretreatment baseline (V1), after the completion of RT/before the third dose of immunotherapy (week 6–9, V2), and at the timepoint of first radiographic assessment (Week 10–14, V3). Panel headers refer to numerator and denominator of respective cellular populations (i.e., “ICOS+/CD4” is the percentage of ICOS+CD4+ among all CD4+ cells). Patients with response (green), stable disease (yellow), and progressive disease (red) outside of the irradiated volume are shown. Symbols reflect unique patients.
Flow cytometry (Fig. 2B) demonstrated increases in the proportion of T cells expressing the activation marker inducible co-stimulator (ICOS), most notably in the CD4 T-cell populations at both on-treatment timepoints. Within CD4 T cells, the proportion of proliferating (Ki67+) cells also appeared to increase after treatment initiation in both cohorts. Although the proportion of CD8 T cells expressing PD1 decreased slightly from baseline to both on-treatment timepoints, the proportion of proliferating (Ki67+) PD1+CD8+ T cells appeared to increase slightly and transiently at the first on-treatment timepoint, similar to other markers of T-cell activation such as LAG-3 and TIM-3.
Using the Simpson index to assess TCR diversity, all patients at baseline seem to have lower diversity (higher clonality/higher Simpson index, above 0.0001) compared with a healthy donor reference level (Fig. 3; ref. 21). The diversity did not seem to change dramatically during treatment for most patients, but generally responding patients tended to display decreased clonality (increased diversity) during treatment whereas clonality increased (decreased diversity) for non-responding patients. Two of three patients who had very high baseline clonality (Simpson indices above 0.005) were nonresponders whereas one responding patient from Cohort B with very high baseline clonality had the greatest decrease in clonality (increase in diversity) during the study.
Figure 3.
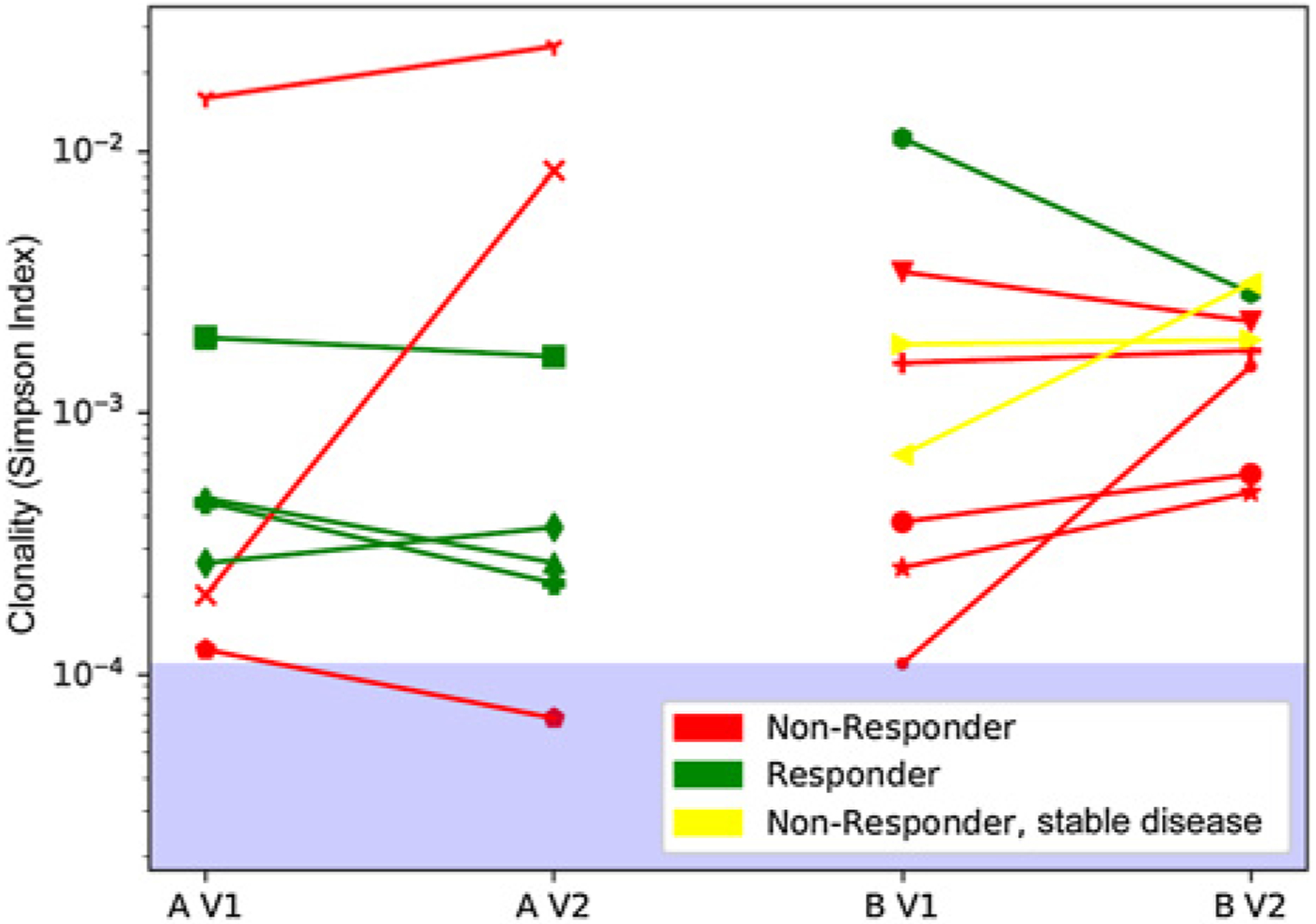
T-cell receptor (TCR) clonal diversity. Change in TCR clonal diversity during treatment. TCR-beta sequencing was assessed using the Adaptive Biotechnologies immunoSEQ (version 4) assay using DNA from peripheral blood mono-nuclear cells gathered before and at the first post-baseline timepoint (6–9 weeks after treatment initiation). The Simpson index (a measure of TCR clonal diversity) remained mostly stable indicating no change in the TCR diversity. All patients had lower diversity than healthy individuals (range for healthy indicated by blue area in the log scale plot; ref. 21). Patients who had extremely low diversity (Simpson above 0.005) and did not increase diversity upon treatment, were consistently nonresponders.
Among all cytokines analyzed by Luminex (see Supplementary Methods), eotaxin levels appeared higher in Cohort A. Patients with response in Cohort A had particularly high levels of eotaxin (Supplementary Fig. S3).
Discussion
This is the first prospective study testing the safety of nivo + ipi + RT. Although the number of patients was small, the rate of grade 3–4 TRAE (40% in Cohort A and 30% in Cohort B) did not appear higher than published literature for nivo 1mg/kg + ipi 3 mg/kg without RT (22). RT did not compromise the ability of patients to receive intended combination immunotherapy as the majority (65%) were able to complete the intended four doses of nivo + ipi, similar to prior literature without RT where 57% of patients received four doses of nivo + ipi (1).
As expected for patients with melanoma, most irradiated sites were lymph node, soft tissue, or bone metastases. However, a few patients received RT to visceral sites, including the lung, liver, and rectum. Our data do not indicate untoward adverse events with RT to visceral sites, but we nevertheless would advise careful management in these cases.
We did not intend to determine whether RT improved the efficacy of nivo + ipi, as this would have required a much larger, randomized study. Nonetheless, RT did not obviously enhance nivo + ipi efficacy outside the irradiated volume. Only one patient among nine evaluable patients responded in Cohort B. Whether this is due to a potentially detrimental effect of hypofractionated RT or to the fact that nine of the patients in Cohort B had been exposed to prior immunotherapy, remains unclear.
Because RT may be immunosuppressive in certain contexts (23, reviewed in ref. 24), we sought to explain the lower response rates outside of irradiated areas by examining immunologic changes in the peripheral blood, which may be relevant to antitumor immune responses. Lymphocytes have been positively associated with outcomes following checkpoint inhibition (16), and RT is known to deplete lymphocytes (25), especially B cells. In our previous retrospective study of ipi and RT, we noted a decrease in the ALC after 82% of the administered courses of RT (median dose 30 Gy in 5 fractions; ref. 26). In this trial, the ALC decreased during treatment with conventional RT (Cohort A), but to a lesser extent with hypofractionated RT (Cohort B). It is possible that RT-induced lymphocyte depletion was counter-balanced, in part, by an increase in lymphocyte proliferation, which we observed (slight increase in proportion of Ki67+PD1+CD8 T cells and increase in Ki67+CD4+ T cells) as has been previously described to occur with checkpoint inhibition induced T-cell reinvigoration in prior studies of anti–PD-1 and anti–PD-1 + anti–CTLA-4 without RT (17, 18, 27, 28). Although of interest to compare the pharmacodynamic effects of nivo + ipi + RT in our study to prior studies of nivo + ipi alone (28), low numbers of patients in these previously published datasets prevent a more conclusive comparison. Prior reports suggest that naïve T cells are negatively associated with response following checkpoint blockade and were lower at baseline in Cohort A than Cohort B (13, 19). Whether the greater number of responders in Cohort A was related to this difference or increase in T effector memory cells is not clear. In addition, RT destroys tumor cells and releases tumor antigens, which preclinical data suggest may be related to diversification of the TCR repertoire following RT (5). Responding patients in our study generally seemed to have increases in TCR diversity, possibly from increased T-cell responses to a wider variety of antigens, but low numbers of patients precluded an ability to formally test these associations with clinical outcome. Eotaxin is a chemokine involved in eosinophil trafficking, and eosinophils have been implicated in outcomes and toxicity following checkpoint inhibitors (29, 30). The high level of eotaxin in responding patients in Cohort A provides preliminary justification for further studying the role eosinophils may play in immune checkpoint blockade and RT combinations.
Despite no obvious improvement in the efficacy of nivo + ipi outside the irradiated volume with the addition of RT, the efficacy of RT appeared high within irradiated tumors. Among patients evaluable for responses within irradiated volumes, no patients progressed within irradiated volumes during the time of observation. This degree of efficacy with relatively low doses of radiation (in Cohort A) appears favorable compared with prior studies of RT in melanoma, which used higher doses (such as Cohort B; ref. 8). Whether RT possibly enhanced the efficacy of checkpoint inhibition in irradiated tumors or checkpoint inhibition enhanced the efficacy of RT within irradiated tumors remains unknown. Unfortunately, we were unable to collect biopsies of irradiated sites to elucidate potential mechanisms.
We acknowledge several study limitations. First, our study was small and non-randomized, which precluded an ability to determine whether RT improves the efficacy of immunotherapy. A larger, randomized study in patients with melanoma brain metastases receiving nivo + ipi ± stereotactic radiosurgery is planned (NCT03340129) to formally address the question of whether intracranial RT improves efficacy of immunotherapy (31). The small size also did not offer the possibility to statistically test associations between immune correlates and clinical outcomes; correlative results remain preliminary and descriptive. Second, before enrolling in this study, many patients had progressed on prior treatment with anti–PD-1 or anti–CTLA-4 agents, which may have affected the efficacy of RT + nivo + ipi outside the irradiated volume. Nonetheless, some responders had received prior anti–PD-1 and/or anti–CTLA-4 therapy. We also cannot exclude the possibility that the safety of nivo + ipi + RT in patients naïve to anti–PD-1 and/or anti–CTLA-4 differs from the safety that we observed in our study. Yet, we believe our results are relevant to clinical practice because many patients with melanoma receive anti–PD-1 agents as monotherapy in the front-line setting for metastatic disease, or as adjuvant therapy, and then receive the nivo + ipi combination subsequently upon metastatic progression. We chose to give RT concurrently, starting after the first dose of nivo + ipi primarily out of practical considerations as patients with multiple sites of metastases most often start immunotherapy first and then receive RT. Although there could be clinical differences depending on whether RT is administered concurrently or sequentially with immunotherapy, prior preclinical data suggest no difference when treatment is given concurrently or sequentially (5). Finally, because diverse anatomic locations were irradiated, it is possible that immunologic effects of RT may depend upon the specific anatomic site irradiated as others have shown (14).
In summary, we completed the first prospective study of RT + nivo + ipi. RT did not obviously increase the rate of nivo + ipi side effects, and there were no high-grade RT toxicities. Efficacy of RT within irradiated volumes with concurrent combination checkpoint blockade appears promising. The optimal radiation dose-fractionation schema to combine with immunotherapy was not identified in this study; however, there were trends suggesting a difference in immune response between the two cohorts. Moreover, we found no evidence of RT increasing the efficacy of immunotherapy, which requires separate determination in other ongoing, randomized studies.
Supplementary Material
Translational Relevance.
Radiotherapy (RT) causes immunologic changes that may be favorable in combination with immune checkpoint blockade. Preclinical models suggest that the efficacy of immune checkpoint blockade can be increased when RT is added and that the immunologic effects of radiotherapy depend upon its dose-fractionation. The most effective combination in preclinical models is the triple combination of radiotherapy with blockade of both the programmed cell death 1 (PD-1) and cytotoxic T lymphocyte antigen-4 (CTLA-4) pathways. Prior clinical trials have explored radiotherapy with single-agent immune checkpoint blockade (either CTLA-4 or PD-1 blockade), but this is the first trial to report the safety, efficacy, and immunologic correlates of radiotherapy with the combination of PD-1 and CTLA-4 blockade.
Acknowledgments
The study was financially supported by Bristol-Myers Squibb, the Conquer Cancer Foundation, and Ludwig Institute for Cancer Research. Ludwig Institute for Cancer Research was involved in design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, and approval of the article; and decision to submit the article for publication. Conquer Cancer Foundation provided funding in support of the trial but was not involved directly in the study. Bristol-Myers Squibb was not involved in the design and conduct of the study, data collection, management, analysis, interpretation of the data, or preparation of the article. Bristol-Myers Squibb reviewed and approved the article before submission. We appreciate the assistance of Yael Rosenberg-Hasson in the Human Immune Monitoring Center (HIMC) at Stanford for completing the Luminex assays and Stacy Leanne Stamps-DeAnda for assistance with data collection and trial conduct. This research was funded in part through the Conquer Cancer Foundation, NIH/NCI Cancer Center Support Grant P30 CA008748, and support from NIH grants S10RR027582 and 5P30CA124435.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Disclosure of Potential Conflicts of Interest
M.A. Postow is an employee/paid consultant for BMS, Merck, Array BioPharma, Novartis, Incyte, NewLink Genetics, and Aduro; reports receiving other commercial research support from RGenix, Infinity, BMS, Merck, Array BioPharma, Novartis and AstraZeneca; and reports receiving speakers bureau honoraria from BMS and Merck. D.A. Goldman holds ownership interest (including patents) in Johnson and Johnson. A.N. Shoushtari is an employee/paid consultant for Bristol-Myers Squibb, Immunocore, and Castle Bioscience. M.K. Callahan is an employee/paid consultant for Bristol-Myers Squibb, AstraZeneca, Incyte, Moderna, and Merck, and reports receiving commercial research grants from Bristol-Myers Squibb. P.B. Chapman is an employee/paid consultant for Innunocore, Merck, and Takdea Millenium; reports receiving commercial research grants from Pfizer; and holds ownership interest (including patents) in Rgenix. J.D. Wolchok is an employee/paid consultant for Adaptive Biotech, Amgen, Apricity, Ascentage Pharma, Astellas, AstraZeneca, Bayer, Beigene, Bristol-Myers Squibb, Eli Lilly, Celgene, Chugai, Elucida, F Star, Janssen, Kyowa Hakko Kirin, Merck, Neon Therapeutics, Novartis, Polynoma, Psioxua, Syntalogic, and Surface; reports receiving commercial research grants from Bristol-Myers Squibb, AstraZeneca, Genentech, and Sephora; holds ownership interest (including patents) in Tizona Pharma, Adaptive Biotechnologies, Imvaq, Beigene, Linneaus, Arsenal, Potenza Therapeutics, and Serametrix; and is an advisory board member/unpaid consultant for Takara, Trieza, Truvax, and Syndax. K.S. Panageas reports receiving other remuneration from Pfizer. C.A. Barker reports receiving commercial research grants from Alpha Tau Medical, Merck, Amgen, and Elekta; reports receiving speakers bureau honoraria from Regeneron and Pfizer; is an advisory board member/unpaid consultant for Regeneron; and reports receiving other remuneration from Elsevier and Patient Resource. No potential conflicts of interest were disclosed by the other authors.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2019;381:1535–46. [DOI] [PubMed] [Google Scholar]
- 2.Ready N, Hellmann MD, Awad MM, Otterson GA, Gutierrez M, Gainor JF, et al. First-line nivolumab plus ipilimumab in advanced non–small cell lung cancer (Checkmate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J Clin Oncol 2019;37:992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol 2005;174: 7516–23. [DOI] [PubMed] [Google Scholar]
- 4.Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res 2005; 11:728–34. [PubMed] [Google Scholar]
- 5.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015;520:373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ko EC, Benjamin KT, Formenti SC. Generating antitumor immunity by targeted radiation therapy: role of dose and fractionation. Adv Radiat Oncol 2018;3: 486–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 2017;8:15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sause WT, Cooper JS, Rush S, Ago CT, Cosmatos D, Coughlin CT, et al. Fraction size in external beam radiation therapy in the treatment of melanoma. Int J Radiat Oncol Biol Phys 1991;20:429–32. [DOI] [PubMed] [Google Scholar]
- 9.Overgaard J, von der Maase H, Overgaard M. A randomized study comparing two high-dose per fraction radiation schedules in recurrent or metastatic malignant melanoma. Int J Radiat Oncol Biol Phys 1985;11:1837–9. [DOI] [PubMed] [Google Scholar]
- 10.Maity A, Mick R, Huang AC, George SM, Farwell MD, Lukens JN, et al. A phase I trial of pembrolizumab with hypofractionated radiotherapy in patients with metastatic solid tumours. Br J Cancer 2018;119:1200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luke JJ, Lemons JM, Karrison TG, Pitroda SP, Melotek JM, Zha Y, et al. Safety and clinical activity of pembrolizumab and multisite stereotactic body radiotherapy in patients with advanced solid tumors. J Clin Oncol 2018;36: 1611–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sundahl N, Vandekerkhove G, Decaestecker K, Meireson A, De Visschere P, Fonteyne V, et al. Randomized phase 1 trial of pembrolizumab with sequential versus concomitant stereotactic body radiotherapy in metastatic urothelial carcinoma. Eur Urol 2019;75:707–11. [DOI] [PubMed] [Google Scholar]
- 13.Hiniker SM, Reddy SA, Maecker HT, Subrahmanyam PB, Rosenberg-Hasson Y, Swetter SM, et al. A prospective clinical trial combining radiation therapy with systemic immunotherapy in metastatic melanoma. Int J Radiat Oncol Biol Phys 2016;96:578–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang C, Welsh JW, de Groot P, Massarelli E, Chang JY, Hess KR, et al. Ipilimumab with stereotactic ablative radiation therapy: phase I results and immunologic correlates from peripheral T cells. Clin Cancer Res 2017;23: 1388–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Theelen WSME, Peulen HMU, Lalezari F, van der Noort V, de Vries JF, Aerts JGJV, et al. Effect of pembrolizumab after stereotactic body radiotherapy vs. pembrolizumab alone on tumor response in patients with advanced non–small cell lung cancer: results of the PEMBRO-RT phase 2 randomized clinical trial. JAMA Oncol 2019;5:1276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Postow MA, Chasalow SD, Kuk D, Panageas KS, Cheng ML, Yuan J, et al. Absolute lymphocyte count as a prognostic biomarker for overall survival in patients with advanced melanoma treated with ipilimumab. Melanoma Res 2019;30:71–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti–PD-1 response. Nature 2017;545:60–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med 2019;25:454–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subrahmanyam PB, Dong Z, Gusenleitner D, Giobbie-Hurder A, Severgnini M, Zhou J, et al. Distinct predictive biomarker candidates for response to anti–CTLA-4 and anti–PD-1 immunotherapy in melanoma patients. J Immunother Cancer 2018;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishino M, Giobbie-Hurder A, Gargano M, Suda M, Ramaiya NH, Hodi FS. Developing a common language for tumor response to immunotherapy: immune-related response criteria using unidimensional measurements. Clin Cancer Res 2013;19:3936–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emerson RO, DeWitt WS, Vignali M, Gravley J, Hu JK, Osborne EJ, et al. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T-cell repertoire. Nat Genet 2017;49:659–65. [DOI] [PubMed] [Google Scholar]
- 22.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arina A, Beckett M, Fernandez C, Zheng W, Pitroda S, Chmura SJ, et al. Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat Commun 2019;10:3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kleinberg L, Sloan L, Grossman S, Lim M. Radiotherapy, lymphopenia, and host immune capacity in glioblastoma: a potentially actionable toxicity associated with reduced efficacy of radiotherapy. Neurosurgery 2019;85:441–53. [DOI] [PubMed] [Google Scholar]
- 25.Wild AT, Herman JM, Dholakia AS, Moningi S, Lu Y, Rosati LM, et al. Lymphocyte-sparing effect of stereotactic body radiation therapy in patients with unresectable pancreatic cancer. Int J Radiat Oncol Biol Phys 2016;94:571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barker CA, Postow MA, Khan SA, Beal K, Parhar PK, Yamada Y, et al. Concurrent radiotherapy and ipilimumab immunotherapy for patients with melanoma. Cancer Immunol Res 2013;1:92–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, et al. Proliferation of PD-1 CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A 2017;114:4993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, et al. Combination therapy with anti–CTLA-4 and anti–PD-1 leads to distinct immunologic changes in vivo. J Immunol 2015;194:950–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosner S, Kwong E, Shoushtari AN, Friedman CF, Betof AS, Brady MS, et al. Peripheral blood clinical laboratory variables associated with outcomes following combination nivolumab and ipilimumab immunotherapy in melanoma. Cancer Med 2018;7:690–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phillips GS, Hellmann MD, Postow MA, Rizvi NA, Freites-Martinez A, Chan D, et al. Treatment outcomes of immune-related cutaneous adverse events. J Clin Oncol 2019;37:2746–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez M, Hong AM, Carlino MS, Atkinson V, Wang W, Lo S, et al. A phase II, open label, randomized controlled trial of nivolumab plus ipilimumab with stereotactic radiotherapy versus ipilimumab plus nivolumab alone in patients with melanoma brain metastases (ABC-X Trial). J Clin Oncol 2019;37:TPS9600. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.