

Significance
Insertion/deletion (InDel) mutations play key roles in genome and protein evolution. Despite their prominence in evolutionary history, the potential of InDels for changing function in protein engineering by directed evolution remains unexplored. Instead point mutagenesis is widely used. Here we create antibody libraries containing InDels and demonstrate that affinity maturation can be achieved in this way, establishing an alternative to the point mutation strategies employed in all previous in vitro selections. These InDels mirror the observation of considerable length variation in loops of natural antibodies originating from the same germline genes and be combined with point mutations, making both natural sources of functional innovation available for artificial evolution in the test tube.
Keywords: antibody, directed evolution, InDel, protein engineering
Abstract
We report a systematic combinatorial exploration of affinity enhancement of antibodies by insertions and deletions (InDels). Transposon-based introduction of InDels via the method TRIAD (transposition-based random insertion and deletion mutagenesis) was used to generate large libraries with random in-frame InDels across the entire single-chain variable fragment gene that were further recombined and screened by ribosome display. Knowledge of potential insertion points from TRIAD libraries formed the basis of exploration of length and sequence diversity of novel insertions by insertional-scanning mutagenesis (InScaM). An overall 256-fold affinity improvement of an anti–IL-13 antibody BAK1 as a result of InDel mutagenesis and combination with known point mutations validates this approach, and suggests that the results of this InDel mutagenesis and conventional exploration of point mutations can synergize to generate antibodies with higher affinity.
Powerful selection technologies have made in vitro evolution of protein binders more efficient and paved the way for the use of tailor-made antibodies in therapy. After initial selections of antibody candidates with desired specificity, lead antibodies are typically improved by affinity maturation in multiple rounds of randomization and selection (1) to reach the subnanomolar affinities ideally required for targeting soluble ligands (2–4). This is usually attempted by introduction of point substitutions, either at random positions across the entire V-gene (5, 6) or in the complementary-determining regions (CDRs; e.g., by CDR walking mutagenesis) (7).
In Nature, diversification of the primary antibody repertoire occurs by several mechanisms that generate variation in the regions forming the antigen-binding site, the CDRs, including considerable length variation (8–11) that is initially introduced by recombination of V(D)J gene segments. Length variations are concentrated in the CDR3 region (12), at the junctions of the joined segments, where additional diversity is produced by N- or P-nucleotide additions that can further extend the CDR3. The length of the CDRs considerably affects the topography of the combining site, as different shapes brought about by extension or shortening can form pockets, grooves, or fill space (13, 14).
Following B cell stimulation by the antigen, further diversification of the antigen-binding interface is generated through somatic hypermutation (SHM) (15), involving mainly point mutagenesis that preferentially targets hotspots in the CDRs (16, 17). This process is initiated through deamination of cytosine to uracil by activation-induced cytidine deaminase (AID), leading to uracil:guanine mismatches (16). Upon removal of these uracil bases by base excision-repair enzymes, error-prone DNA polymerases are then recruited to fill in the gaps and introduce mutations around the position of the deaminated cytosines. Interestingly, up to 6% of the mutations generated by SHM are insertions and deletions (InDels) (18), which occur due to misalignment of repeated DNA sequences (19, 20). Thus, insertions occur by duplication, while deletions are brought about by removal of repeated sequences (21, 22).
A small percentage of antibodies selected by in vivo SHM contain InDels in the CDRs 1 and 2 (1.6 to 6.5%) (21–24), while junctional diversity by N- or P-nucleotide additions in the CDR3 confounds the analysis of SHM-derived InDels, leading to an underestimation of the total percentage of affinity-improving InDels. In vitro-directed evolution has been unsuitable for introduction of InDels at random positions into an antibody gene, because of restrictions in the diversity of InDels that could be introduced (i.e., insertions by duplication in in vitro SHM) (22, 25). Rational (26) or computational (27) strategies have been successful at introducing InDels in a few, carefully chosen positions instead of random sampling. In contrast, an unusually high percentage of InDels with a functional role among in vivo affinity matured broadly neutralizing antibodies (bnAbs) to HIV-1 (28–30): ∼40% of the reported anti–HIV-1 bnAbs contain InDels that accumulate during in vivo SHM (28). Based on the frequent occurrence of InDels among multispecific, cross-reactive antibodies, one could infer that they provide a molecular solution for recognizing multiple targets by providing an altered interface (enlarged or tightened), possibly even involving conformational diversity (31). The accumulation of InDels in bnAbs has been attributed to extensive in vivo SHM, so that even positions that are rarely modified by SHM are also altered (17, 28).
Insertions in the V-genes occur only by duplication of adjacent sequences (21, 22), so that the actual sequence diversity of the resulting insertions is limited because they repeat existing modules. To introduce more diversity in the inserted sequences, point mutations are required in subsequent rounds of SHM. However, since the CDRs can tolerate considerable length variation, it is likely that the antibody fold can accommodate a larger number of affinity-enhancing InDels compared to those observed in antibodies affinity-matured by SHM.
Affinity gains by introduction of InDels have indeed been recognized (22, 25, 26, 32, 33) in in vitro-directed evolution, but often were by-products of campaigns focused on point mutations and not elicited systematically (32, 33). Only in mammalian cell surface display does the action of AID lead to InDels, just as AID brings about InDels in SHM in vivo (22, 25). In a seminal study by Bowers et al. (22), overexpression of AID enabled in vitro SHM of 53 antibodies against 21 antigens to identify InDels in multiple regions likely to improve binding, in particular to variable heavy domain (VH) and variable light domain (VL) CDR1, where 9 of 53 antibodies contained InDels. Despite the comprehensive nature of this study, AID-enabled insertions mirrored in vivo SHM and were therefore limited to direct duplication of adjacent sequences, not allowing the full exploration of length and sequence diversity in the insertions, and the low frequency of incorporation of in-frame InDels by AID (<0.1%) limited the combinatorial diversity explored. Finally, InDels have been introduced rationally based on structural analysis and natural length variation (26, 27). Taken together, only limited diversity of InDels in terms of length, position, and insert sequence across the variable domains has been explored thus far.
Here we address this omission and explore libraries with in-frame InDels of different lengths and high diversity of inserted sequences at random positions across the entire antibody variable regions (Fig. 1). We applied a new transposon-based mutagenesis approach, dubbed TRIAD (transposition-based random insertion and deletion mutagenesis) (34) that introduces short in-frame insertions and deletions randomly across a gene (in sequences of steps following transposition that excise the transposon, religate the plasmid, and insert designed cassettes) (SI Appendix, Figs. S1 and S2). TRIAD was used here to build libraries with InDels at random positions across an entire single-chain variable fragment (scFv) gene. The antibody chosen for this campaign was the anti–IL-13 antibody BAK1 (35), a derivative of which, tralokinumab, is under clinical investigation for asthma (36). In addition, we built libraries that explore diversity in the different lengths of insertions in a semirandom approach, insertional-scanning mutagenesis (InScaM). These InDel libraries were starting points for antibody affinity evolution in vitro, leading to insertions in two loops that, together with two previously known point mutations, brought about a 256-fold affinity improvement. The observation of alternative routes to affinity maturation validate our strategy and suggest that InDel mutagenesis can complement existing approaches.
Fig. 1.

Overview of the affinity maturation of the antibody BAK1 by transposon-based TRIAD and subsequent insertional scanning mutagenesis. TRIAD (Left) was applied to make libraries with deletions of one to three amino acids (step 1a) or single amino acid insertions (step 1b) at random positions across the scFv gene. These libraries were recombined (step 2) and four rounds of ribosome display selections for improved affinity to IL-13 were carried out by panning (step 3). The best binder was carrying an insertion in the VL FWR3 (BAK1-INS1). Scanning (Right) was used to guide the design of libraries with different lengths of insertions at targeted positions. A fraction of the insertion library generated in step 1b (5,632 variants) was screened by HTRF to identify variants with insertions that retained binding to IL-13 (step 4). Based on sequencing analysis, regions able to tolerate single amino acid insertions were identified (Fig. 4) and the VL CDR3 was chosen for targeted insertional mutagenesis. Libraries with zero to five amino acid insertions in targeted positions in the VL CDR3 were constructed (step 5), followed by four rounds of phage display selections for improved affinity to IL-13 (step 6).
Results
BAK1 Affinity Maturation Using Libraries with Random InDels across the Variable Domains.
Four libraries of the BAK1 antibody with InDels randomly distributed throughout the entire variable domains were constructed using TRIAD (34): Three deletion libraries (3nt-Del, 6nt-Del, 9nt-Del and 3nt-Ins, with deletions of 3, 6, and 9 nucleotides) and one single random nucleotide triplet (NNN) insertion library (3nt-Ins) (Fig. 2A and Table 1). The functional library sizes exceeded the theoretical library sizes at least 10-fold, suggesting that the percentages of variants with in-frame InDels were sufficient to cover the theoretical diversity of each library. Next-generation sequencing had been used previously to show the near absence of transposition bias in TRIAD, resulting in excellent coverage of InDels across a gene (34). Consistent with this assessment, the very high proportion of variants with insertions as well as deletions at unique positions (>90%) suggests that InDels were randomly distributed across the entire scFv coding sequence. In-frame InDel variants with additional point mutations across the scFv sequence were not observed. As a consequence, epistatic interactions between InDels and additional point substitutions should not affect the outcome of the selection, as deleterious or beneficial point substitutions would not mask the effect of beneficial indels. Overall, this quality control suggests that we successfully generated libraries with random InDels across the entire scFv gene, confirms the absence of point substitutions, and enables us to explore the vast custom-made sequence diversity relying on InDels alone in library selections.
Fig. 2.

Selections of BAK1 libraries with random InDels across the scFv gene for binding to IL-13. (A) Prior to selection (i.e., after step 2 in Fig. 1), insertions (red) were randomly distributed across the entire VH and VL regions. Insertions or substitutions are shown in blue or red, respectively, in the pop-out. (B) After selection (i.e., after step 3 in Fig. 1) a group of variants with two consecutive mutations was enriched: an Insertion (VL 67a) and a point substitution (VL 68), of different amino acid residues at the same position in VL FWR3 (BAK1-INS variants). The predominant variant in round four (40%), BAK1-INS1, had an insertion (E67a) and a substitution (G68W). Sequence numbering according to Wu and Kabat (75).
Table 1.
Sequence analysis of BAK1 libraries with random InDels
| BAK1 InDel library | Variants with in-frame InDel of desired length, % | Variants with frameshifts, % | Parent (or linker), % | Variants with InDel at unique position, % | Theoretical library size | Functional library size |
| 3 nt-Del* | 86 | 14 | 0 | 90 | 7.5 × 102 | 1.7 × 106 |
| 6 nt-Del* | 17 | 67 | 16 | 93 | 7.5 × 102 | 3.4 × 105 |
| 9 nt-Del* | 29 | 52 | 19 | 83 | 7.5 × 102 | 5.8 × 105 |
| 3 nt-Ins* | 36 | 47 | 17 | 100 | 4.8 × 104 | 7.1 × 105 |
| Rec library† | 36 | 33 | 31 | n/a | 1.3 × 109 | 1.9 × 1011 |
Based on sequencing of 88 variants from each library (3 nt-Del, 6 nt-Del, 9 nt-Del, and 3 nt-Ins containing 3, 6, 9 nucleotide [nt] deletions [Del], and 3 nucleotide insertions [Ins], respectively). The actual library size (∼2 × 106 variants) was calculated by measuring the colony number after each cloning step. For each library, the functional library size shows the number of variants with in-frame InDel of desired length.
Based on sequencing of 176 variants from the Rec library. The number of possible variants with a single InDel was the sum of possible variants of each InDel library (SI Appendix, Table S1). The actual library size was determined by the concentration of plasmid DNA that was added in the in vitro transcription reaction (1 μg of 1,200 bp DNA is 7.5 × 1011 molecules). The functional library size shows the number of in-frame variants (either parent or variants with single or double in-frame InDels per gene).
To identify variants with improved affinity from these libraries, panning selections were performed by ribosome display (37) in order to take advantage of recombination before selecting from very large library sizes (>5 × 1011 variants). Easier recombination and access to such large libraries (including oversampling) triggered the choice of this in vitro technology (instead of phage display). A shuffling library (dubbed “Rec”) was thus generated by a staggered extension process (38) using the four InDel libraries of BAK1 (3nt-Del, 6nt-Del, 9-nt Del and 3nt-Ins) as templates. Sequence analysis (Table 1 and SI Appendix, Table S1) suggested that all possible BAK1 variants with single and with two InDels per gene were present in the Rec library (with 146-fold oversampling). Thus, the library Rec (SI Appendix, Table S1) should be able to tap the potential of synergistic effects between InDels as a result of recombination of the BAK1 InDel libraries (3nt-Del, 6nt-Del, 9-nt Del and 3nt-Ins).
In order to enrich the variants with the highest affinities, four rounds of affinity-based selections of the Rec library were carried out by panning with recombinant biotinylated IL-13. Sequencing analysis of the selection outputs from rounds one to four revealed a group of variants with two consecutive mutations at the same position: A single amino acid insertion (VL 67a) and accidental point substitutions (VL G68X) in VL FWR3 (fourth loop in the third framework region) (BAK1-INS variants) (Fig. 2B) were enriched over the rounds, reaching 98% of the total InDel variants in round four (SI Appendix, Fig. S4). No deletion variants were enriched and selected, despite the quality of the deletion libraries and sampling well above their size. The other in-frame InDel variant that was found in round four (represented by the remaining 2%) contained a 52aG insertion in the VH CDR2, after the residue S52.* The amino acid types that were enriched were insertions 67aE, 67aV, 67aD, or 67aA combined with substitution G68W or G68R (Fig. 2B). One variant, BAK1-INS1, dominated the selection outcome in round four (40%): It has a 67aE insertion and a G68W substitution. The 67aE insertion is located within FWR3 (see, for example, Fig. 6A), sometimes referred to as CDR4 (39).
Fig. 6.

Sites of affinity-enhancing mutations modeled on the tralokinumab::IL-13 complex crystal structure (50). (A) Insertions with large beneficial effect on BAK1 affinity were found in two different loops, the VL FWR3 loop (G68) and the VL CDR3 (D92-S95), in close proximity to the antigen, IL-13 (brown cartoon/surface). The residues that were replaced by the insertion are shown as red sticks on the tralokinumab cartoon representation of the structure, while blue sticks show affinity-improving BAK1.1 substitutions (VL CDR1 N27Y and VH CDR3 N99S). (B) Both beneficial insertions are located on the periphery of the antigen binding site. The blue dotted line includes residues of the structural paratope of the BAK1.1 antibody. The colors indicate residues of the structural paratope, the positions of beneficial insertions and substitutions in yellow, red, and blue, respectively. The variable heavy domains (VH) is highlighted in gray, while the variable light domain (VL) is shown in cyan.
For quantitative characterization, the most enriched variant, BA1-INS1, was converted into an IgG1 format and binding measurements revealed a 14.3-fold affinity increase (KD = 140 pM) compared with the parent (KD = 2,300 pM) (Table 2). No gain in affinity was observed in variants bearing either substitution G68E or G68W (BAK1-E and BAK1-W). This observation suggests that the mutation G68W was beneficial only in the presence of the 67a insertion, leading to the conclusion that the insertion (67a) was the seminal affinity improving feature of BAK1-INS1.
Table 2.
Binding kinetics and thermodynamics of BAK1 variants (in IgG format) selected by ribosome display against IL-13. Substitutions are shown in blue, insertions in red
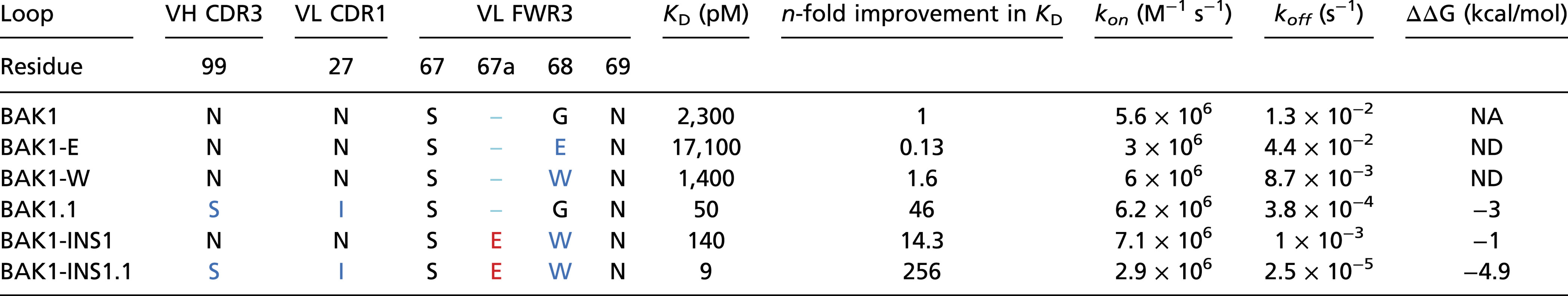 |
KD values of the human IgG1 variants were determined by surface plasmon resonance. The n-fold improvement in KD is the ratio KD Variant/KD Parent. ΔΔG was calculated from the equation ΔΔG = −RT In (KD Parent/KD Variant). NA, not applicable; ND, not determined.
Although the entire BAK1 scFv had previously been scanned for affinity-improving point mutations (35), no change in affinity could be attributed to point substitutions in the VL FWR3 in the present study. Furthermore, no affinity-enhancing substitutions in the VL FWR3 were found (as in ref. 35), despite enrichment of variants with other point mutations, namely VL CDR1 N27Y (BAK1.4) and VH CDR3 N99S (BAK1.55) (SI Appendix, Table S2). These are substitutions that make up the variant BAK1.1 [identified previously by ribosome display selections of BAK1 antibody libraries (35)], suggesting that our experiments tap similar effects as a past affinity maturation campaign. In our study, large affinity effects seem enabled by targeting VL FWR3 with an insertion (at position 67) and not through point substitutions (Table 2 and SI Appendix, Table S2). This is an example of a deliberate library strategy in which insertions are targeted as the main cause of antibody affinity maturation.
While the presence of the variant 67aE/G68 in the original insertion library was confirmed (SI Appendix, Table S4), only the 67aE/G68W variant (40%) was enriched, implying a positive synergistic effect between the two mutations. This idea is supported by the observation that similar variants combining G68W with a different residue inserted at position 67 were also enriched: 67aV/G68W and 67aA/G68W with 26% and <3% in round four, respectively. Taken together, these results underscore the importance of sampling insertions with high amino acid diversity and point substitutions in positions flanking the insertion to identify insertions with beneficial effect on affinity.
Synergistic Interaction between Beneficial Insertion and Point Substitutions.
To study epistatic interactions of insertions and point substitutions on antibody affinity, we determined the combined affinity effect of the beneficial insertion in FWR3 (BAK1-INS1) and two beneficial substitutions found in a previous study (35) (BAK1.1) (Fig. 3 and Table 2). Combination of both the insertion and substitutions resulted in an IgG variant (BAK1-INS1.1; KD = 9 pM) with higher affinity than either point substitutions (BAK1.1; KD = 50 pM) or the insertion alone (BAK1-INS1; KD = 142 pM). BAK1-INS1.1 possessed an overall 256-fold improvement in affinity compared to the parent, which is 6-fold higher than that of the substitution variant BAK1.1, with a 6.5-fold lower dissociation constant koff (2.45 × 10−5 s−1), consistent with an off-rate selection. The effect of combining insertion and point substitutions was synergistic, as the free energy change that resulted from combining insertion and point substitutions (BAK1-INS1.1; −4.9 kcal/mol) was higher than the sum (−4 kcal/mol) of the free energies of the variants with insertion (BAK1-INS1; −1 kcal/mol) and substitutions (BAK1.1; −3 kcal/mol), while for additive mutations the free energy change would be equal to the sum. These results demonstrate that beneficial insertions and point substitutions can have synergistic effects on affinity. This suggests a protein engineering strategy in which selections of libraries with insertions and substitutions could be performed separately and then combined to exploit synergistic effects on affinity.
Fig. 3.

Synergy between affinity-improving substitutions in the CDRs and an insertion in VL FWR3. (A) Mutations in the sequence of BAK1 variants: BAK1.1, a previously generated (35) variant with two beneficial substitutions (VH CDR3 N99S/VL CDR1 N27I); BAK1-INS1, combining two consecutive mutations, an insertion and a substitution in VL FWR3 (E67a/G68W; present study); BAK1-INS1.1, combining four mutations from both BAK1.1 and BAK1-INS1. (B) Mutational trajectories and improvements in affinity between parental BAK1 and variants BAK1.1, BAK1-INS1 and BAK1-INS1.1. Substitutions are shown in blue, insertions in red.
BAK1 Affinity Maturation by InScaM.
To identify further affinity-enhancing insertions in BAK1, insertions with diverse lengths and amino acid compositions were sampled in a second strategy. This semirandom approach, InScaM, consisted of two stages: Identification of sites in which insertions were tolerated, followed by exploration of diverse insertions in these sites. Practically, 5,632 variants of the 3nt-Ins TRIAD library (in scFv format) with single amino acid insertions (Fig. 2A and Table 1) were screened for variants with insertions that retained binding to IL-13 in a microplate-based homogeneous time-resolved fluorescence (HTRF) assay. Despite the relatively low throughput, multiple insertion mutants for each position exist, so that the question of insertional tolerance could be addressed by the library outcome. Positions tolerant to insertions across the variable regions were revealed by sequencing analysis of the hits (Fig. 4 and SI Appendix, Table S4), by determining the number of hits with insertion at a given position, even though different amino acid residues were inserted. Fig. 4 displays the sequence analysis of the IL-13 binders obtained in this experiment. After disregarding insertions in the N terminus of the VH (a possibly unfolded or structurally plastic terminus likely to be insertion-tolerant), 11 positions tolerating insertions in the loops VH FWR3, VH CDR2, VL CDR3, and VL FWR3 were found. Indeed, position G68 in the VL FWR3 loop (five hits) had previously been shown to tolerate an insertion in BAK1-INS1. A cluster of four positions with high frequency was found in the VL CDR3 before the residues D92, T93, G94, and S95 with 4, 10, 18, and 1 hit, respectively. Only one position with high frequency (before A53) was observed in the VH CDR2.
Fig. 4.

Tolerance for insertions across the variable regions of the BAK1 antibody: 5,632 variants from the TRIAD library with single amino acid insertions (Fig. 2A and Table 1) were randomly picked and screened for retention of IL-13 binding. The positions where insertions resulted in binding levels similar or better than BAK1 are indicated by the frequency observed for insertions before the residue of the (A) VH and (B) VL sequence of the BAK1 antibody, respectively. The frequency refers to the number of hits at a given position (even if the inserted amino acid differs). The loop showing the highest frequency of insertion tolerance (i.e., VL CDR3) was chosen as the target for insertional mutagenesis. The positions are color-matched to Fig. 6, where the yellow color indicates residues that constitute the paratope, positions shown in blue are those for which substitutions had been shown previously to improve affinity (in BAK1.1) (35) and positions in red correspond to the insertions identified in this work.
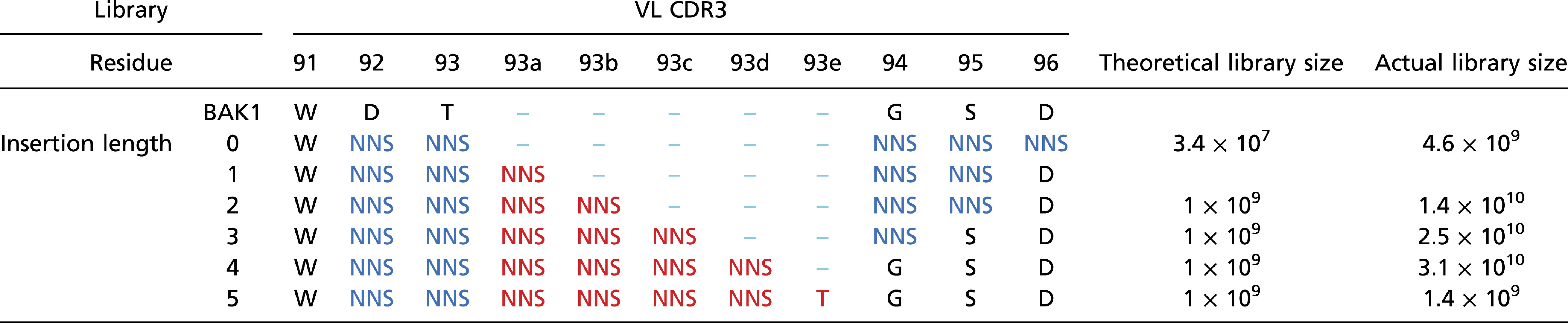
The positions tolerating insertions in the CDRs and FWR3 loop were considered possible sites to target by insertional mutagenesis, for which libraries with different lengths of insertions and with high diversity of amino acid replacements were designed and synthesized. Leaving aside 97 N-terminal insertions, 49 of the remaining 62 hits (79%) are located in the CDRs or FWR3 loop. Among these 49, 33 are located in VL CDR3 (in positions D92–S95 of the VL CDR3; 67%), which was thus chosen as the target for saturation mutagenesis. Five libraries with zero to five insertions as NNS codons between residues 93 and 94 and randomized flanking residues (92, 93, and 94 to 96) were constructed (Table 3). In total, residues 5 or 6 NNS positions lead to library sizes between 109 and 1010 variants.
Table 3.
Design of libraries with different lengths of insertions in the VL CDR3
 |
To enrich for the highest affinity variants, four rounds of KD-based phage display selections of the libraries with insertions in the VL CDR3 were performed. For each library, 384 variants of the round four selection output (SI Appendix, Fig. S5) were rescreened in a competition HTRF screen for binding to human IL-13, followed by sequencing of all of the hits and affinity determination in IgG format (Table 4 and SI Appendix, Table S3). Analysis of the KD values of IgG variants (Table 4) showed that large improvements (>5-fold), INS2.1, INS3.4, INS4.3, INS4.4, and INS5.3 (14-, 7.7-, 26-, 68-, and 48-fold improvements in KD, respectively), were associated with insertions longer than two amino acids (two to five amino acids) and a motif (93aW/93bD or 93aF/93bD for the insertions of two to four or five amino acids, respectively). These data (displayed in Fig. 5A) suggest that longer inserts make it more likely to find large effects on affinity, but length alone is not a sufficient criterion for high affinity. The lengths of the VL CDR3 for the parent BAK1 is 11, which is extended in insertion mutants (to 13 in INS2.1, and up to 16 in INS5.3). In contrast, the VL CDR3 loops of the λ-light chains observed in Nature range between 7 and 13 amino acids (11). The successful selection of affinity-enhancing insertions with high-diversity amino acid replacements stands in contrast to the selection from libraries with single or no insertion. Screening of these showed only one variant with a small improvement in affinity (INS1.6; 2.9-fold), indicating that variants without insertion in VL CDR3 were hard to improve, unless an insertion in VL CDR3, is present.
Table 4.
Binding affinity of improved BAK1 variants with insertions in the VL CDR3
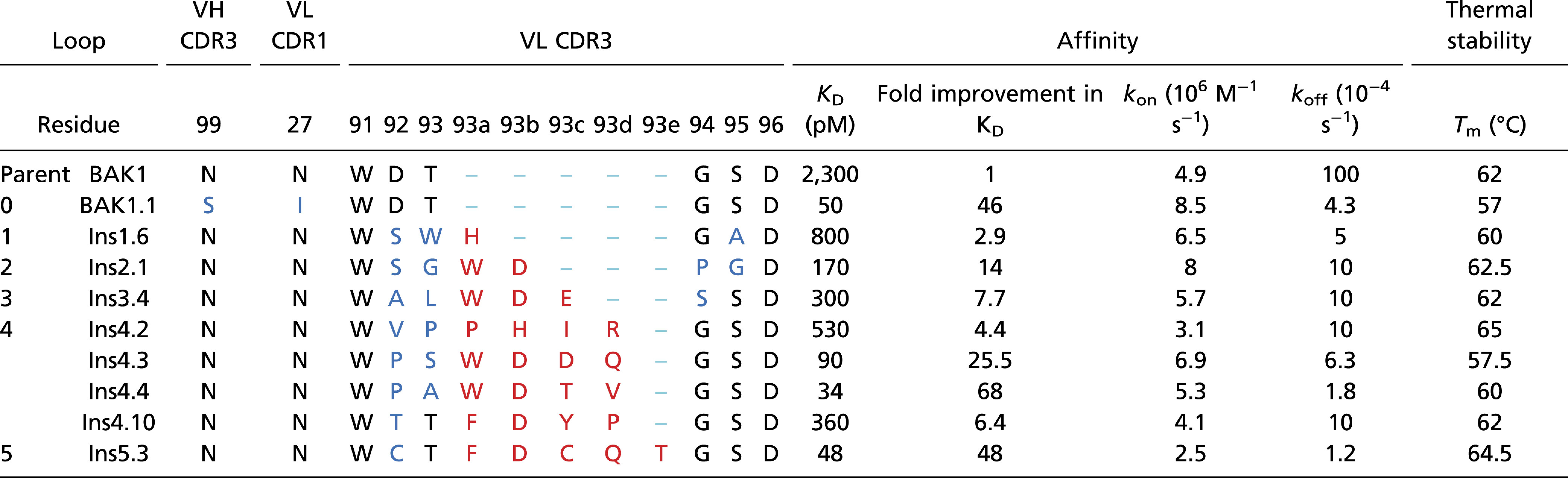 |
KD values of the human IgG1 variants were determined by surface plasmon resonance (T200, Biacore). The fold improvement in KD is the ratio KDVariant/KDParent. The errors of the kon and koff measurements were between 0.01 and 0.09 × 106 M−1s−1 and between 0.001 and 0.34 × 10−4 s−1, respectively.
Fig. 5.

Correlations of functional and structural features with the binding enhancements. (A) The length of the insertion plays a partial role in affinity maturation: Increasing insert sizes apparently provide more opportunity for interactions and antigen complementarity. While longer inserts generally tend to bind more tightly, the correlation also shows considerable scatter, suggesting that idiosyncrasies of specific interactions may or may not contribute. (B) The correlation between melting temperatures and fold improvement in KD shows a trade-off between affinity and stability for the variants with the largest improvements in affinity. There is one exception: INS 5.3 is a variant with a five-amino acid insertion, which possessed higher thermodynamic stability than the parent BAK1 (Tm = 64.5) and a large gain in affinity. Thermal stability measurements of improved BAK1 IgG variants with insertions in the VL CDR3 were carried out by differential scanning fluorimetry (49).
Selected Variants with Insertions Are Not Thermally Destabilized.
The perception that the stability cost of the mutational load that accompanies improvements in protein function has led to the model of trade-offs between affinity and stability in directed evolution (40, 41), which is supported by many studies (42–44). Indeed, the “one amino acid at a time” model (45, 46) proposes to proceed through relatively conservative mutation regimes, to allow cycles of structural destabilization and repair (47) and satisfy the stability requirement for clones with novel functions. It has been postulated (48) that, despite being potential sources of functional innovation, InDels are more disruptive than point mutations, implying their mutational damage to structure is higher and InDel strategies intrinsically less likely to succeed. To address this issue experimentally, we measured the thermal stability of the improved IgG variants by differential scanning fluorimetry (49) (Fig. 5B, Table 4, and SI Appendix, Fig. S6). Most variants with small improvements in affinity (INS2.1, INS3.4, INS4.2) were as stable as the parent IgG (BAK1; Tm = 62 °C), thus the insertions did not have a negative impact on antibody stability. Most variants with large improvements in affinity had a slight loss in stability (INS4.3; Tm = 58 °C, INS4.4; Tm = 60 °C), and the same loss was observed for the variant with two point substitutions (BAK1.1; Tm = 57 °C), indicating that the trade-off between affinity and stability was observed for both the insertion and the substitution variants with large improvements in affinity. The only exception was the variant with a five-amino acid insertion (INS5.3), which besides conferring large gain in affinity, also possessed higher stability than the parent (Tm = 65 °C). This improvement in stability could be attributed to the insertion of two cysteine residues that likely form a disulphide bond that could stabilize the loop. Intraloop disulphide bonds in the VL CDR3 have not been observed in natural antibodies. Thus, different effects were observed for insertions with different lengths and amino acid composition in the same region in the VL CDR3, suggesting that specific amino acid substitutions have idiosyncratic effects on function. By and large, however, in most cases a longer insert has an increased potential to exert a substantial effect on affinity.
Overall, these results show that despite the low tolerance for insertions, similar effects as point substitutions on antibody stability are observed: They can be neutral, deleterious, or beneficial, being subject to affinity/stability trade-off but, according to our present data, InDels are no worse than point mutants, leading to a similar conclusion as in the InDel mutagenesis of an enzyme (34).
Discussion
Insertion Hotspots in the CDRs and FWR3 Loops Emerge from InDel Evolution.
This work identified insertions that brought about large affinity improvements in two different loops of the same antibody, the VL CDR3 and the VL FWR3 loops: 1) Two to five amino acid residue insertions in the VL CDR3, replacing the residues D92–S95 and 2) a single amino acid insertion in the VL FWR3 loop, replacing the residue G68. In a previous study, two point substitutions that improved BAK1 affinity (BAK1.1 variant) were found in different loops, the VH CDR3 (N99S) and VL CDR1 (N27I) (35).
The crystal structure of tralokinumab (derived from the variant BAK1.1) in complex with human IL-13 (50) provides a basis for rationalization of the roles of affinity-enhancing mutations with respect to the antigen binding site. Fig. 6B highlights the structural paratope evident from this structure (50), which reveals that the positions of the insertions are on the periphery of the antigen binding site (Fig. 6), with residues G68 in VL FWR3 loop and T93 in VL CDR3 constituting part of the paratope. FWR3 and VL CDR3 loop insertions are likely to enable new interactions with the antigen, resulting in additional buried surface contacts in an enlarged antigen binding site.
The observation that loop extensions confer affinity enhancements is reminiscent of natural affinity maturation. CDR3 loop length variation can result from the V(D)J rearrangement, leading to some length variation (and as such complicates the estimation of InDel frequency during somatic hypermutation of these loops). The identification of beneficial insertions in the VL CDR3 of the BAK1 antibody in this study combined with the high tolerance in length variation of this loop in Nature suggests that length diversification of the VL CDR3 might also represent a mechanism of affinity maturation. The 67aE insertion is located within FWR3 (Fig. 6A), sometimes referred to as CDR4 (39). In contrast to the other hypervariable loops (CDR1 to -3), antibodies in nature show minimal variation in this loop in the germline (51, 52). The antibody PGT121, which originated from the same germline gene (L-Vλ3-21*02) as the BAK1 VL, has a three amino acid insertion in the same position in the FWR3 as BAK1-INS1 (53). The frequent presence of insertions in the FWR3 loop in extensively affinity matured antibodies (anti–HIV-1 bnAbs) has been noted previously. For example, 5 of 17 antibodies with broad neutralization activity (29.4%) have an FWR3 loop insertion (29). These insertions have been shown to confer broad neutralization activity of the 8ANC195 and 3BNC60 bnAbs (29, 30) and suggest that the framework region three is crucial for specificity determination.
The idea that insertions may exert their effects by making new contacts with the antigen is supported by studies of the role of insertions in naturally affinity-matured anti–HIV-1 bnAbs, which show that insertions extend loops to form new intermolecular contacts, either directly or by enabling other loop residues flanking the position of the insertion. For example, an FWR3 loop insertion in the anti–HIV-1 bnAb 8ANC195 (30) enabled new contacts of FWR3 loop residues flanking the insertion with residues of the gp120 loops D and V5. Another example is the four-residue insertion in the VH CDR3 of the bnAb NIH45-46, which belongs to the VRC01 clonal lineage (54, 55), facilitating new hydrogen bonds and electrostatic interactions of the insertion residues with the antigen from the CDR3 loop extension. Finally, in addition to static shape complementarity, insertions may change antibody affinity by changing the conformations of adjacent loops, as in the 2D1 anti-HA antibody insertion (56) at the junction of CDR2 and FWR3. This insertion altered the conformation of the CDR1 loop, resulting in 35-fold higher affinity.
Loop extensions have also resulted in introduction of additional specificities to create a multispecific “two-in-one antibody” (57). Such versatile reagents offer the opportunity of neutralizing multiple human targets for therapy (e.g., multiple, related cytokines). Similarly, making antibodies that hit both the human and rodent orthologs of a particular target would be desirable at the preclinical stage of drug development in order to understand the biology of the target in a different species. These approaches have been exemplified for point substitutions (57–61), but given the potential for introducing conformational changes via InDels, their potential in this respect may be explored in future work.
Epistatic Interactions and Synergy of Point Substitutions in Positions Flanking the Insertion Site and InDels.
Epistatic interactions between point substitutions are often governing the functional consequences of individual mutations, so that the combined effect of mutations is often not simply additive (7, 62). Even though epistasis was shown to be largely beneficial among CDR variants (63), mutations may still clash, requiring consensus design to avoid incompatible combinations (e.g., in libraries generated by error-prone PCR) (64). Whether the acquisition of beneficial insertions would depend on the presence of other point substitutions across the antibody variable region was not known before designing the insertion libraries. The discovery of insertions that improved BAK1 affinity in two different loops, independent of the presence of other point substitutions across the scFv gene, suggests that the effect of insertions does not necessarily depend on the presence of other mutations. This fact has important engineering implications, as it allows the identification of beneficial insertions without the need to sample large areas of sequence space with point substitutions, thus streamlining the search for beneficial insertions.
Although the effect of insertions did not depend on point substitutions far from the positions of the insertions, positive epistasis was observed with positions around the insertions. Insertion in the FWR3 (67aE) was beneficial only in the presence of a specific amino acid residue at position 68 (G68W), indicating epistatic interaction between those modifications, with a specific substitution at the position flanking the insertion and enabling the insertion to exert its function (i.e., permissive mutation). These results underline the importance of sampling high diversity of point substitutions: To identify an insertion variant with improved affinity, a combination of mutants flanking the position of the insertion with insertions of high diversity may be necessary to create a synergistic effect.
The idea that longer insertions generally lead to higher affinity because an additional structural feature provides additional interactions is addressed in Fig. 5A. Apparently, longer insertions can, but do not have to lead to affinity maturation, so the specific context that determines strength of newly formed interactions matters.
Variants with insertions in the VL FWR3 and the VL CDR3 loops were found to have higher affinity for IL-13 but no “substitution-only” variants in these loops were found to have conferred a beneficial effect on affinity. In contrast, variants with beneficial substitutions were found in different loops, namely the VH CDR3 and VL CDR1, in which no beneficial insertions were found. When these are combined, they act synergistically, creating BAK1-INS1.1 as the antibody with the highest affinity for IL-13 reported so far. This observation contrasts with the prior suggestion that most InDels share the same hotspots with point mutations because InDel duplications or deletions involve AGY motifs (21).
Strategy of InDel Mutagenesis Followed by Targeted Insertion (TRIAD/InScaM).
This work presents evidence of a deliberate strategy in which insertions and point substitutions are exploited to bring about synergistic effects on affinity. The implication of the observation of affinity enhancement is that libraries used in selections would benefit from exploiting the effects of InDels and substitutions to explore additive effects on affinity. For protein engineering purposes, the fact that both insertions and point substitutions can have complementary functions suggests that insertions can offer an alternative in cases where the identification of further beneficial point substitutions has proven difficult, thus increasing the chances of success in antibody evolution and offering additional pathways to affinity maturation.
Combining multiple mutagenesis approaches leads to libraries that are too large to be screened comprehensively. Therefore, a strategy for combining mutagenesis approaches is necessary. Generally, point mutations can be introduced at any stage of this process, as long as the epistatic properties of the protein fold allow synergy of mutations. For the introduction of InDels, we have explored in this work a two-step sequence that extends the simple introduction of InDels by TRIAD, where short (single) amino acid insertions are sampled entirely randomly. Given the experimental limits of screening, it is necessary to break down this procedure into two steps that answer the question where InDels are tolerated before the combinatorial diversity of the identity and length of potential insertions are addressed.
Step 1: Identification of InDel-tolerant positions. TRIAD is used to scan insertion points via single amino acid insertions by screening for binding or catalytic function. The outcome of this experiment will identify regions of the protein in which insertions are tolerated. Alternatively, insertion points can be deduced from previous knowledge of insertion tolerance (see discussion below).
Step 2: Insertional mutagenesis (InScaM). A library with insertion points based on the results of step 1 is made that contains high diversity insertions: That is, with different lengths (in our case up to six amino acids) and amino acid diversity in each position of the inserts, followed by screening or selection.
Step 1 can be achieved in an entirely combinatorial way: In contrast to previous methods of insertional mutagenesis (26, 65, 66), scanning by TRIAD identifies positions that tolerate insertions, even when no structural information is available or when analysis of natural length variation would indicate too many positions to choose from. It is conceivable that prior knowledge can also be used as a guide to help random exploration: A crystal structure of the complex (as in ref. 66), the comprehensive mapping of fitness effects in response to InDels (67), or comparative bioinformatic and phylogenetic analysis may suggest amenable regions as insertion points for combinatorial elaboration in step 2. However, a precise prediction enabling tight interactions with a new antigen will be difficult, and for antibodies there are many CDR positions to choose from, so it remains to be seen whether such predicted insertion points can rival combinatorially identified ones derived by TRIAD. For enzyme sequences toggling between known functions, the location and size of InDels has been shown to be indicative of functional switches (68), and such knowledge has, for example, guided active site remodeling to interconvert related promiscuous lactonases/phosphotriesterases (65).
Insertions will have different affinity-enhancing mechanism of action than substitutions: Extending loops and making new contacts with the antigen will result in much more profound changes (at least in structural terms) than those typically introduced by single amino acid substitution. This idea is underscored by the observation of mutations in loops where no beneficial substitutions had been found previously, suggesting different functional potential of InDels vs. point substitutions. In this work, insertions and substitutions seem to have complementary mechanisms of function, meaning that insertions offer alternative trajectories to affinity maturation. While it is too early to say whether a TRIAD/InScaM approach achieves better affinity enhancements than point mutagenesis, their complementarity is likely to rescue stalled evolution campaigns and synergistically lead to ultimately better affinity reagents.
Materials and Methods
Construction of BAK1 Libraries with Random InDels.
BAK1 libraries with random indels throughout the entire scFv coding sequence were constructed using TRIAD, as described previously (34, 69) (SI Appendix, Figs. S1 and S2). Briefly, the transposition reactions were performed starting from the vector pID-Tet (34) containing the BAK1 scFv coding sequence (pID-Tet-BAK1) using either the TransDel or TransIns engineered transposon, to create libraries with deletions or insertions, respectively, and the enzyme MuA Transposase (Finnzymes). The cassettes Ins1, Del1, Del2, and Del3 were extracted from pUC57 plasmids (34). For each step, the ligation reactions were concentrated using the Zymo Research, DNA Clean & ConcentratorTM-5 and then transformed into ∼30 μL of E. cloni 10G Electrocompetent Cells (Lucigen) (>5 × 109 cfu/μg) by electroporation. The transformation efficiency was ∼2 × 106 colonies in each step. The libraries were subcloned into the ribosome display vector (33, 70) and the quality of the libraries was assessed by sequencing (Table 1).
Construction of the BAK1 InDel Rec Library.
DNA of the BAK1 libraries 3nt-Del, 6nt-Del, 9nt-Del and 3nt-Ins was mixed and recombined following a Staggered Extension Process In-Vitro DNA Recombination (SteP) protocol (38), using a high-fidelity DNA polymerase (Phusion High-Fidelity DNA Polymerase, error rate 4.4 × 10−7) to keep the point mutagenesis as low as possible. The recombined BAK1 InDel library (Rec library) into the ribosome display vector was analyzed by sequencing (SI Appendix, Table S2). This library (∼5 × 1011 variants, quantified by determining the concentration of plasmid DNA that was added into the transcription reaction, using the correlation 1 µg of 1,200 bp DNA = 7.5 × 1011 molecules) was the input of the first round of ribosome display selection. In vitro transcription, translation, and selections were performed as described previously (33, 70), but using a high-fidelity DNA polymerase for the amplification steps between the rounds of selection.
Ribosome Display Selection from the Rec Library.
scFv–ribosome–mRNA complexes were subjected to selection in solution using recombinant biotinylated human IL-13 (Peprotech) to allow capture using streptavidin-coated paramagnetic beads (Dynabeads M-280), as described previously (35). Biotinylation of IL-13 surface lysine residues was done using EZ link NHS-LC-Biotin according to the manufacturer’s instructions (Pierce). Four rounds of KD-based selections were performed by panning with various biotinylated IL-13 concentrations below the KD (<2,300 pM). More specifically, the output of the selection with the lowest IL-13 concentration that gave a positive selection outcome (100 to 20 pM) was used in the following round of selection.
Construction of Libraries with Insertions in the VL CDR3.
Insertional mutagenesis in the VL CDR3 of the BAK1 scFv was achieved by generation of libraries in the phagemid vector pCANTAB6, as previously described (71), by oligonucleotide-directed mutagenesis. Five libraries were constructed using five different oligonucleotides designed to introduce randomized insertions of different lengths, from zero to five amino acid residues. Following the residue W91, blocks of five or six consecutive residues (NNS randomization) were randomized by Kunkel mutagenesis (72). Each library contained >109 variants, estimated from the number of transformants after electroporation of Escherichia coli TG1 cells with the DNA product of each mutagenesis reaction.
Phage Display Selections of VL CDR3 Insertion Libraries.
Four rounds of KD-based selections of the BAK1 libraries with insertions in the VL CDR3 to enrich variants with improved affinity were performed in solution by capturing biotinylated human IL-13 with streptavidin-coated paramagnetic beads, following the principles described previously (73). Starting from an IL-13 concentration in the KD range of 10 nM in the first round, we decreased the antigen concentration by 10-fold in each round.
Construction of BAK1 IgG Variants by Site-Directed Mutagenesis.
Site-directed mutagenesis of BAK1 scFv coding sequence into pCANTAB6 was performed following the manufacturer’s instructions in the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies). The mutations introduced in the BAK1 or the BAK1-INS1 template sequences were: N99S in the VH CDR3, N27I in VL CDR1, G68W or G68E in the VL FWR3. The BAK1 VH and VL sequences were then subcloned into the heavy chain (pEU1.3) and the λ light-chain (pEU4.4) mammalian IgG1 expression vectors (74), respectively, which were designed as described previously (33).
Expression and Purification of scFv and IgG Variants.
E. coli periplasmic extracts containing His6-tagged scFv variants of either the 3nt-Ins library or the outputs of phage display selections were produced for screening by HTRF assays (SI Appendix), using the vector pCANTAB6 (71). scFv expression in E. coli was induced using isopropyl β-d-1-thiogalactopyranoside (IPTG). To induce cell lysis, the cell pellets were resuspended in osmotic shock buffer MES (containing 50 mM Mops, 0.5 mM EDTA and 0.5 M sorbitol; final pH 7.4).
His6-tagged scFv variants were purified for affinity-based screening using a competition HTRF assay. The expression of scFv variants in E. coli was induced using IPTG, while the cells were lysed using osmotic shock buffer TES (containing 200 mM Tris⋅HCl, 0.5 mM EDTA, 0.5 M sucrose; pH 8.0). The periplasmic fractions were purified on HisTrap HP (GE Healthcare) packed columns followed by buffer exchanged to PBS (NAP-10 columns; GE Healthcare).
IgGs were produced by cotransfection of the heavy-chain and light-chain pEU vectors (74) in mammalian cells and the IgG1 variants were purified by protein A affinity chromatography (MabSelect Sure columns, GE Healthcare) using the AKTA express system and eluted with 0.1 M sodium citrate (pH 3.0). PD10 columns (GE Healthcare) were used for buffer exchange of the antibodies in PBS and the concentrations were measured by absorption spectrophotometry based on calculation of the extinction coefficient (ε) of each IgG variant based on their amino acid content.
Supplementary Material
Acknowledgments
We thank Medimmune’s Biologics Expression Team for assistance with protein production, their DNA Chemistry Group for primer synthesis and sequencing, and Dr. Mariana Rangel for preliminary crystallization experiments. This research was funded by the Biological and Biotechnological Research Council (BBSRC) via Grant BB/L002469/1 and “sparking impact” funds, the European Union Programme H2020, and MedImmune/AstraZeneca. K.S. received a BBSRC Collaborative Awards in Science and Engineering studentship (BB/K012665/1) in collaboration with MedImmune/AstraZeneca. F.H. is a European Research Council Advanced Investigator (Grant 695669).
Footnotes
Competing interest statement: M.C., J.A., D.G.R., D.C., B.P., A.B., and R.R.M. are employees of AstraZeneca.
This article is a PNAS Direct Submission.
*Insertions are denoted by the letter a after the residue after which the insertion took place, followed by the one letter code of the inserted residue: e.g., a single residue G inserted after position 52 is referred to as '52aG.'
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2002954117/-/DCSupplemental.
Data Availability.
All study data are included in the main text and SI Appendix.
References
- 1.Dufner P., Jermutus L., Minter R. R., Harnessing phage and ribosome display for antibody optimisation. Trends Biotechnol. 24, 523–529 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Razai A.et al., Molecular evolution of antibody affinity for sensitive detection of botulinum neurotoxin type A. J. Mol. Biol. 351, 158–169 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Shim H., One target, different effects: A comparison of distinct therapeutic antibodies against the same targets. Exp. Mol. Med. 43, 539–549 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shealy D. J.et al., Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor α. MAbs 2, 428–439 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stemmer W. P., DNA shuffling by random fragmentation and reassembly: In vitro recombination for molecular evolution. Proc. Natl. Acad. Sci. U.S.A. 91, 10747–10751 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neylon C., Chemical and biochemical strategies for the randomization of protein encoding DNA sequences: Library construction methods for directed evolution. Nucleic Acids Res. 32, 1448–1459 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang W. P.et al., CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into the picomolar range. J. Mol. Biol. 254, 392–403 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Chothia C., Lesk A. M., Canonical structures for the hypervariable regions of immunoglobulins. J. Mol. Biol. 196, 901–917 (1987). [DOI] [PubMed] [Google Scholar]
- 9.Al-Lazikani B., Lesk A. M., Chothia C., Standard conformations for the canonical structures of immunoglobulins. J. Mol. Biol. 273, 927–948 (1997). [DOI] [PubMed] [Google Scholar]
- 10.North B., Lehmann A., Dunbrack R. L. Jr., A new clustering of antibody CDR loop conformations. J. Mol. Biol. 406, 228–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chailyan A., Marcatili P., Cirillo D., Tramontano A., Structural repertoire of immunoglobulin λ light chains. Proteins 79, 1513–1524 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Xu J. L., Davis M. M., Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity 13, 37–45 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Vargas-Madrazo E., Lara-Ochoa F., Almagro J. C., Canonical structure repertoire of the antigen-binding site of immunoglobulins suggests strong geometrical restrictions associated to the mechanism of immune recognition. J. Mol. Biol. 254, 497–504 (1995). [DOI] [PubMed] [Google Scholar]
- 14.Collis A. V. J., Brouwer A. P., Martin A. C. R., Analysis of the antigen combining site: Correlations between length and sequence composition of the hypervariable loops and the nature of the antigen. J. Mol. Biol. 325, 337–354 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Victora G. D., Nussenzweig M. C., Germinal centers. Annu. Rev. Immunol. 30, 429–457 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Di Noia J. M., Neuberger M. S., Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 76, 1–22 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Hwang J. K.et al., Sequence intrinsic somatic mutation mechanisms contribute to affinity maturation of VRC01-class HIV-1 broadly neutralizing antibodies. Proc. Natl. Acad. Sci. U.S.A. 114, 8614–8619 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goossens T., Klein U., Küppers R., Frequent occurrence of deletions and duplications during somatic hypermutation: Implications for oncogene translocations and heavy chain disease. Proc. Natl. Acad. Sci. U.S.A. 95, 2463–2468 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Diaz M., Kunkel T. A., Mechanism of a genetic glissando: Structural biology of indel mutations. Trends Biochem. Sci. 31, 206–214 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Streisinger G., Owen J., Mechanisms of spontaneous and induced frameshift mutation in bacteriophage T4. Genetics 109, 633–659 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Wildt R. M., van Venrooij W. J., Winter G., Hoet R. M., Tomlinson I. M., Somatic insertions and deletions shape the human antibody repertoire. J. Mol. Biol. 294, 701–710 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Bowers P. M.et al., Nucleotide insertions and deletions complement point mutations to massively expand the diversity created by somatic hypermutation of antibodies. J. Biol. Chem. 289, 33557–33567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Briney B. S., Willis J. R., Crowe J. E. Jr., Location and length distribution of somatic hypermutation-associated DNA insertions and deletions reveals regions of antibody structural plasticity. Genes Immun. 13, 523–529 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reason D. C., Zhou J., Codon insertion and deletion functions as a somatic diversification mechanism in human antibody repertoires. Biol. Direct 1, 24 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowers P. M.et al., Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proc. Natl. Acad. Sci. U.S.A. 108, 20455–20460 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamminmäki U.et al., Expanding the conformational diversity by random insertions to CDRH2 results in improved anti-estradiol antibodies. J. Mol. Biol. 291, 589–602 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Mou Y.et al., Engineering improved antiphosphotyrosine antibodies based on an immunoconvergent binding motif. J. Am. Chem. Soc. 140, 16615–16624 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Kepler T. B.et al., Immunoglobulin gene insertions and deletions in the affinity maturation of HIV-1 broadly reactive neutralizing antibodies. Cell Host Microbe 16, 304–313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein F.et al., Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 153, 126–138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scharf L.et al., Antibody 8ANC195 reveals a site of broad vulnerability on the HIV-1 envelope spike. Cell Rep. 7, 785–795 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.James L. C., Roversi P., Tawfik D. S., Antibody multispecificity mediated by conformational diversity. Science 299, 1362–1367 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Main S.et al., A potent human anti-eotaxin1 antibody, CAT-213: Isolation by phage display and in vitro and in vivo efficacy. J. Pharmacol. Exp. Ther. 319, 1395–1404 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Finch D. K.et al., Whole-molecule antibody engineering: Generation of a high-affinity anti-IL-6 antibody with extended pharmacokinetics. J. Mol. Biol. 411, 791–807 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Emond S.et al., Accessing unexplored regions of sequence space in directed enzyme evolution via insertion/deletion mutagenesis. Nat. Commun. 11, 3469 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thom G.et al., Probing a protein-protein interaction by in vitro evolution. Proc. Natl. Acad. Sci. U.S.A. 103, 7619–7624 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.May R. D.et al., Preclinical development of CAT-354, an IL-13 neutralizing antibody, for the treatment of severe uncontrolled asthma. Br. J. Pharmacol. 166, 177–193 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanes J., Plückthun A., In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. U.S.A. 94, 4937–4942 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao H., Giver L., Shao Z., Affholter J. A., Arnold F. H., Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat. Biotechnol. 16, 258–261 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Fanning S. W., Horn J. R., An anti-hapten camelid antibody reveals a cryptic binding site with significant energetic contributions from a nonhypervariable loop. Protein Sci. 20, 1196–1207 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tokuriki N., Stricher F., Serrano L., Tawfik D. S., How protein stability and new functions trade off. PLoS Comput. Biol. 4, e1000002 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas V. L., McReynolds A. C., Shoichet B. K., Structural bases for stability-function tradeoffs in antibiotic resistance. J. Mol. Biol. 396, 47–59 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Julian M. C.et al., Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng. Des. Sel. 28, 339–350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Julian M. C., Li L., Garde S., Wilen R., Tessier P. M., Efficient affinity maturation of antibody variable domains requires co-selection of compensatory mutations to maintain thermodynamic stability. Sci. Rep. 7, 45259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Houlihan G., Gatti-Lafranconi P., Lowe D., Hollfelder F., Directed evolution of anti-HER2 DARPins by SNAP display reveals stability/function trade-offs in the selection process. Protein Eng. Des. Sel. 28, 269–279 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tracewell C. A., Arnold F. H., Directed enzyme evolution: Climbing fitness peaks one amino acid at a time. Curr. Opin. Chem. Biol. 13, 3–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tokuriki N., Tawfik D. S., Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 19, 596–604 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Campbell E.et al., The role of protein dynamics in the evolution of new enzyme function. Nat. Chem. Biol. 12, 944–950 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Studer R. A., Dessailly B. H., Orengo C. A., Residue mutations and their impact on protein structure and function: Detecting beneficial and pathogenic changes. Biochem. J. 449, 581–594 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Niesen F. H., Berglund H., Vedadi M., The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Popovic B.et al., Structural characterisation reveals mechanism of IL-13-neutralising monoclonal antibody tralokinumab as inhibition of binding to IL-13Rα1 and IL-13Rα2. J. Mol. Biol. 429, 208–219 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Williams S. C.et al., Sequence and evolution of the human germline V lambda repertoire. J. Mol. Biol. 264, 220–232 (1996). [DOI] [PubMed] [Google Scholar]
- 52.Solomon A., Weiss D. T., Structural and functional properties of human lambda-light-chain variable-region subgroups. Clin. Diagn. Lab. Immunol. 2, 387–394 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mouquet H.et al., Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc. Natl. Acad. Sci. U.S.A. 109, E3268–E3277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diskin R.et al., Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science 334, 1289–1293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bjorkman P., Structures of broadly neutralizing anti-HIV antibodies that target the CD4 binding site on the HIV envelope. Sci. Highlight 3, 4–6 (2012). [Google Scholar]
- 56.Krause J. C.et al., An insertion mutation that distorts antibody binding site architecture enhances function of a human antibody. mBio 2, e00345-10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bostrom J.et al., Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science 323, 1610–1614 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Koenig P., Sanowar S., Lee C. V., Fuh G., Tuning the specificity of a two-in-one Fab against three angiogenic antigens by fully utilizing the information of deep mutational scanning. MAbs 9, 959–967 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koenig P.et al., Deep sequencing-guided design of a high affinity dual specificity antibody to target two angiogenic factors in neovascular age-related macular degeneration. J. Biol. Chem. 290, 21773–21786 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee C. V., Koenig P., Fuh G., A two-in-one antibody engineered from a humanized interleukin 4 antibody through mutation in heavy chain complementarity-determining regions. MAbs 6, 622–627 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eigenbrot C., Fuh G., Two-in-one antibodies with dual action Fabs. Curr. Opin. Chem. Biol. 17, 400–405 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Barbas C. F. 3rdet al., In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity. Proc. Natl. Acad. Sci. U.S.A. 91, 3809–3813 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adams R. M., Kinney J. B., Walczak A. M., Mora T., Epistasis in a fitness landscape defined by antibody-antigen binding free energy. Cell Syst. 8, 86–93.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mankowska S. A.et al., A shorter route to antibody binders via quantitative in vitro bead-display screening and consensus analysis. Sci. Rep. 6, 36391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoque M. A.et al., Stepwise loop insertion strategy for active site remodeling to generate novel enzyme functions. ACS Chem. Biol. 12, 1188–1193 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Krykbaev R. A., Tsantili P., Jeffrey P. D., Margolies M. N., Modifying specificity of antidigoxin antibodies using insertional mutagenesis. Protein Sci. 11, 2899–2908 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gonzalez C. E., Roberts P., Ostermeier M., Fitness effects of single amino acid insertions and deletions in TEM-1 β-lactamase. J. Mol. Biol. 431, 2320–2330 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Loo B.et al., Balancing specificity and promiscuity in enzyme evolution: Multidimensional activity transitions in the alkaline phosphatase superfamily. J. Am. Chem. Soc. 141, 370–387 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Jones D. D., Triplet nucleotide removal at random positions in a target gene: The tolerance of TEM-1 β-lactamase to an amino acid deletion. Nucleic Acids Res. 33, e80 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hanes J., Jermutus L., Plückthun A., Selecting and evolving functional proteins in vitro by ribosome display. Methods Enzymol. 328, 404–430 (2000). [DOI] [PubMed] [Google Scholar]
- 71.McCafferty J.et al., Selection and rapid purification of murine antibody fragments that bind a transition-state analog by phage display. Appl. Biochem. Biotechnol. 47, 157–171, NaN–173 (1994). [DOI] [PubMed] [Google Scholar]
- 72.Kunkel T. A., Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. U.S.A. 82, 488–492 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hawkins R. E., Russell S. J., Winter G., Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J. Mol. Biol. 226, 889–896 (1992). [DOI] [PubMed] [Google Scholar]
- 74.Persic L.et al., An integrated vector system for the eukaryotic expression of antibodies or their fragments after selection from phage display libraries. Gene 187, 9–18 (1997). [DOI] [PubMed] [Google Scholar]
- 75.Wu T. T., Kabat E. A., An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J. Exp. Med. 132, 211–250 (1970). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All study data are included in the main text and SI Appendix.