

Abstract
Approximately one third of all epilepsy patients are resistant to current therapeutic treatments. Some patients with focal forms of epilepsy benefit from invasive surgical approaches that can lead to large surgical resections of human epileptic neocortex. We have developed a systems biology approach to take full advantage of these resections and the brain tissues they generate as a means to understand underlying mechanisms of neocortical epilepsy and to identify novel biomarkers and therapeutic targets. In this review, we will describe our unique approach that has led to the development of a ‘NeuroRepository’ of electrically-mapped epileptic tissues and associated data. This ‘Big Data’ approach links quantitative measures of ictal and interictal activities corresponding to a specific intracranial electrode to clinical, imaging, histological, genomic, proteomic, and metabolomic measures. This highly characterized data and tissue bank has given us an extraordinary opportunity to explore the underlying electrical, cellular, and molecular mechanisms of the human epileptic brain. We describe specific examples of how an experimental design that compares multiple cortical regions with different electrical activities has led to discoveries of layer-specific pathways and how these can be ‘reverse translated’ from animal models back to humans in the form of new biomarkers and therapeutic targets.
Introduction
Epilepsy is a chronic neurological disorder characterized by recurrent seizures and abnormal, synchronized neuronal activity(England, Liverman, Schultz, & Strawbridge, 2012). Epilepsy is one of the most common neurological diseases, with around 70 million people of all ages, genders, and races currently diagnosed worldwide(England et al., 2012; Moshe, Perucca, Ryvlin, & Tomson, 2015; Singh & Trevick, 2016). The first line of treatment for epilepsy is anti-epileptic drugs (AED). There are currently over 30 FDA approved AEDs, with specific uses for both generalized and focal seizure treatment. Unfortunately, despite the numerous options available, approximately one third of all patients develop drug-resistant or refractory epilepsy(Brodie, Barry, Bamagous, Norrie, & Kwan, 2012; Loscher & Schmidt, 2011). This may be due to the fact that current drug treatments target seizure symptoms and do not directly affect or prevent the underlying cause contributing to epileptogenesis or to the epileptic state(Brodie et al., 2012; Crepeau & Sirven, 2017; Loscher & Schmidt, 2011). This highlights a need to better understand the electrical, cellular, and molecular pathways in human epilepsy so that we can develop medication and effectively target and treat these patients.
While a majority of mechanistic studies have been carried out in animal models, patients who undergo highly invasive surgical procedures to map and remove epileptic brain regions offer an unparalleled approach to understand the human epileptic condition in humans (Loeb, 2010; Loeb, 2011). Another important approach to discover epileptic mechanisms in humans comes from the identification of genetic mutations in familial forms of epilepsy (Chen, Giri, Xia, Subedi, & Li, 2017; Rhys H Thomas & Samuel F Berkovic, 2014; Steinlein, 2004; Steinlein, 2008), however, the vast majority of patients with epilepsy are sporadic with unknown genetic or molecular causes. Even those with genetic abnormalities, many patients with focal forms of epilepsy have associated developmental or acquired brain abnormalities that themselves may not directly produce epileptic activities, but likely trigger epileptic activities in surrounding brain regions (Cavalleri et al., 2005).
For the past 20 years, our group has been collecting tissues and performing multimodal analyses on human epileptic neocortex tissues to build highly integrated dataset that links clinical, electrical, imaging, histological, and molecular data from patients who underwent two-stage epilepsy surgery. This unbiased approach has helped us uncover common molecular and cellular differences in epileptic brain networks. As all of the results are from human tissues, the biomarkers and drug targets that emerge have a high probability of translating back into clinical practice. Here, we will summarize our NeuroRepository and ‘Big Data’ mining approaches and review some of the findings that we hope will help patients with intractable epilepsy.
Precise co-registration of quantitative epileptic activities with brain imaging
Patients who have drug resistant epilepsy can benefit from surgery that removes the areas of the brain that is epileptic or demonstrates high epileptic activity(Spencer & Huh, 2008). Effective surgeries often require the direct placement of depth electrodes or subdural grid electrodes to monitor and localize abnormal brain activity over several days. Intracranial recordings reveal and localize epileptic brain regions in ways that cannot be achieved with scalp EEG recordings due to the low sensitivity and poor spatial resolution of scalp EEG (Tao, Ray, Hawes-Ebersole, & Ebersole, 2005). Two of the most common electrical abnormalities seen are interictal spiking and seizures. While the relationship between ictal and interictal activity is still unclear(Gotman, 1991; Staley, K. J., White, & Dudek, 2011; Staley, Kevin, Hellier, & Dudek, 2005), removing both ictal and high-spiking interictal tissue significantly improves the surgical outcome for patients in a number of studies (Bautista, Cobbs, Spencer, & Spencer, 1999; Kanazawa, Blume, & Girvin, 1996). Additionally, animal studies have shown that interictal activity often proceed seizures after a brain insult and should be considered a predicator of seizure activity (White et al., 2010). In young children, the epileptic focus predominantly occurs in neocortical brain regions, while in older children and adults the epileptic focus in many cases also involves the hippocampus (Annegers, 1993).
Interestingly, intracranial recordings from patients with known structural lesions have shown that many epileptogenic brain regions are often nearby, and not always overlying the structural abnormality (Lee, Spencer, & Spencer, 2000; Palmini, Andermann, Olivier, Tampieri, & Robitaille, 1991; Palmini et al., 2004; Seo, J.H., et al., 2009). Lesions or abnormalities commonly associated with neocortical epilepsy include cortical dysplasias, stokes, trauma, tumors, and previous infections (Chu-Shore, Major, Camposano, Muzykewicz, & Thiele, 2010; Liu, S., Yu, & LÃ, 2016; Shain et al., 2013; Verloes, Elmaleh, Gonzales, Laquerriere, & Gressens, 2007).
Intracranial recordings are done as part of a 1-stage surgery, where the surgery and recordings are done during the same operation. However, seizures during the time of surgery are rarely seen with this approach in part because it is mostly done under general anesthesia, that suppresses seizures, and because the recording time is very brief. The gold standard for identification of seizure onset zone requires a 2-stage surgery where the first stage involves implantation of intracranial depth and/or surface grids of electrodes. Following electrode implantation, the patient leaves the operating room for 3-7 days of recordings to localize seizures, interictal spikes, as well as normal functioning brain regions that need to be preserved. A second surgery is then planned either immediately after the recording or at a later date to remove the epileptic brain regions, while maximizing the retention of normal brain structures.
Figure 1 displays our protocol that enables us to collect and subdivide cortical tissues at precise brain regions corresponding to specific electrical activities and imaging characteristics. The first step is 3-dimensional co-registration of MRI brain imaging with the precise electrode locations from a CAT scan. This enables the generation of 3D images that detail the patient’s epileptic activities. Next, the sheer volume of data produced and the variability between different EEG readers prompted us and others to develop automated spike and seizure detection algorithms to quantify abnormal electrical properties at each electrode location(Barkmeier, D.T., Shah, A.K., Flanagan, D., Atkinson, M.D., Agarwal, R., Fuerst D.R., Jafari-Khouzani, K., Loeb, J.A., 2011; Maharathi et al., 2019; Yadav, Shah, Loeb, Swamy, & Agarwal, 2012) . Quantitative measures can then be visualized on the co-registered brain surface as shown in Figure 1 where green indicates low spiking activity and red indicates high spiking activity. This approach can be used for both non-lesional as well as lesional cases, including those with various epileptic activities in patients with brain tumors (Mittal, Shah, Barkmeier, & Loeb, 2013).
Figure 1. Human epilepsy systems biology workflow.
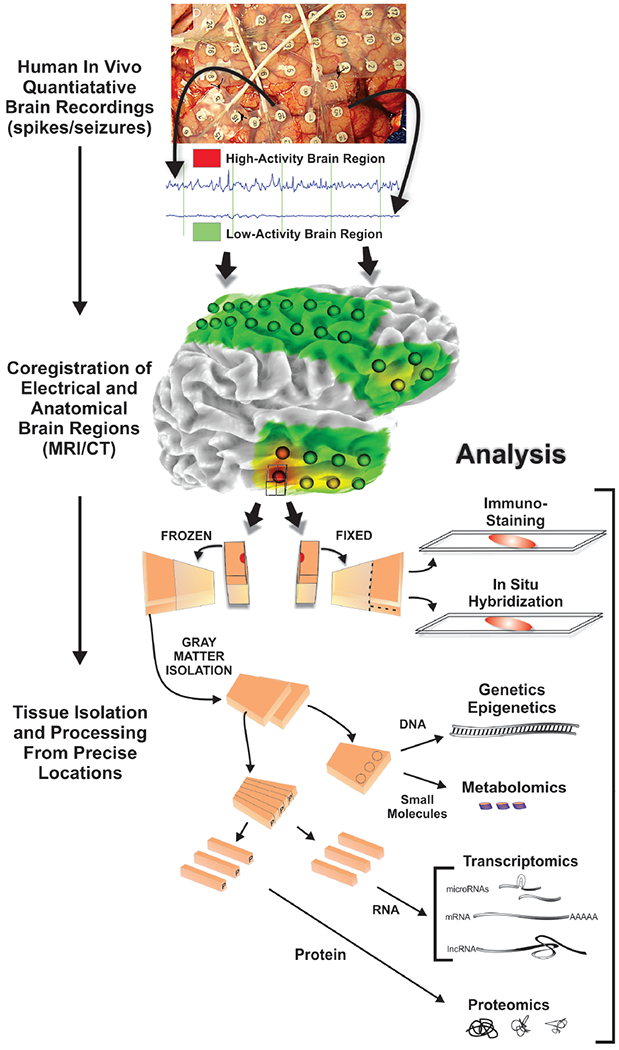
Patients who undergo long-term intracranial EEG monitoring followed by tissue removal to treat their epilepsy provide a unique opportunity to discover what is different about epileptic brain areas compared to non-epileptic brain areas. Precisely localized brain regions with high or low epileptic activities (spiking, non-spiking plus or minus seizures) quantified by a variety of algorithms are co-registered to MRI and CAT scans which aligns electrode locations with brain structures. Tissue blocks (1 cm3) from resected tissues at each electrode location are subdivided so that half is fixed for histology and the other half frozen immediately for molecular analyses. Using this approach, immunostaining and in situ hybridization are performed on the fixed tissue, whereas genomics, proteomics, and metabolomics are performed on alternating strips of even amounts of layers I-VI from the gray matter. In this way the molecular measures can be directly linked to histology, brain imaging, quantitative electrophysiology, and patient clinical information.
Resected tissues mapped to ictal and interictal brain activities can be subdivided for high-throughput ‘omic’ analyses
Immediately after tissue is removed, as part of the planned surgical procedure, photographs of the brain surface, with and without the grid electrodes, are used to identify their exact spatial locations. Only tissues that are not needed for diagnosis and treatment are used for research. As a quality control measure, using the digital photographs, the unique surface arterial pattern from each piece of resected tissue is used to confirm its location. The precise co-registration of resected tissues from specific electrode recording locations is critical for accurate downstream histological, cellular and molecular studies in order to be certain that they are linked to abnormal or to normal electrical activities. It is also important that following their removal, tissues reserved for ‘omic’ studies are immediately frozen on dry ice to prevent molecular degradation.
Figure 1 demonstrates the histological and molecular workflow that follows once the human epileptic neocortical tissues are removed and their locations are confirmed. As a first step, 1 cm3 blocks of tissue containing fairly equal amounts of grey and white matter are prepared at each electrode location and photographed. Each block is then subdivided into two facing pieces. One half is immediately fixed in 4% paraformaldehyde for 2 days for histological staining and the other half is rapidly frozen on dry ice and stored at −80°C for molecular analyses (genomic, proteomic, and metabolomic). For these molecular studies, alternating strips of full-thickness (layer I-VI) cortex are pooled from each frozen block for RNA and protein studies so that results are not biased by specific neuronal layers or white matter contamination. By dividing the tissue in this manner, tissue histology can be directly linked to molecular studies as well as to the electrical, imaging, and clinical data from the same patient. The tissue histology also allows for additional validation studies that can localize potential cell and layer-specific expression patterns in the same tissue block.
Following surgery and tissue collection, the tissue samples and their corresponding clinical and molecular data are stored and managed in the NeuroRepository. The NeuroRepository is more than a tissue repository because of the extensive ‘metadata’ not only for each patient that is further coregistered for each sampled brain region. Data is stored in a web-based, database that integrates all of the different types of data ranging from clinical data, imaging (MRI, CT, PET), EEG quantitative metrics, tissue inventory, and molecular results from each piece of tissue studied including histology and molecular data (genomics, proteomics, metabolomics).
Finding a needle in the haystack: Internally controlled, unbiased transcriptomic studies reveal constitutive activation of specific pathways in epileptic brain regions
A potential pitfall for any high throughput study on human epileptic tissue is that changes may or may not be directly related to the epileptic state of the tissue. When looking at thousands of genes, many gene expression changes can be due to differences not explicitly controlled for in the study. For example, the comparison of gene expression between fresh versus postmortem ‘control’ tissues are likely to show differential gene expression due to many other variabilities in addition to electrical differences including the rate of RNA degradation, genetic differences between the epileptic patient and the control, and the effects of medications the patient was taking. Equally important, even if freshly isolated, ‘control’ tissue from a patient with a brain tumor or head trauma may also have frequent epileptic potentials that are not being measured unless this is excluded by intracranial recordings. To avoid these pitfalls, our approach only compares epileptic brain regions with nearby cortical regions from the same patient with little to no epileptic activity, thus eliminating many potentially confounding variables. This approach has allowed us to identify highly significant, common ‘epileptic’ genes and pathways in relatively small numbers of patients with high statistical significance (Beaumont, Yao, Shah, Kapatos, & Loeb, 2012; Dachet et al., 2015; Lipovich et al., 2012; Rakhade et al., 2005; Rakhade et al., 2007). We further reduce variability by doing all of our microarray studies in quadruplicates with a flip-dye experimental design.
For example, using 5 paired patient cortical samples, we identified 137 differentially expressed genes at regions of seizure onset compared to nearby regions without seizure onset(Beaumont, Yao, Shah, Kapatos, & Loeb, 2012) (Figure 2). We next focused on interictal spiking and performed an expanded pairwise study of 15 patients that demonstrated 1700 differentially expressed genes at regions of high interictal spiking activity, many of which overlapped with genes in the seizure onset data set. Once highly significant populations of differentially expressed genes are linked to a specific variable, such as seizure onset, follow-up ontological analysis of these genes can be used to predict that the highest ranked pathways. Using this statistical approach, we found that one of the highest ranked pathway was the Mitogen Activated Protein Kinase Pathway (MAPK). We further discovered that within our 137 seizure onset genes, approximately 40% of these are known to be induced by a specific transcription factor called CREB (cyclic AMP response element binding protein) (Beaumont et al., 2012).
Figure 2. Internally controlled studies identify common molecular pathways linked to epileptic brain activity that can be confirmed and localized specific cell populations.
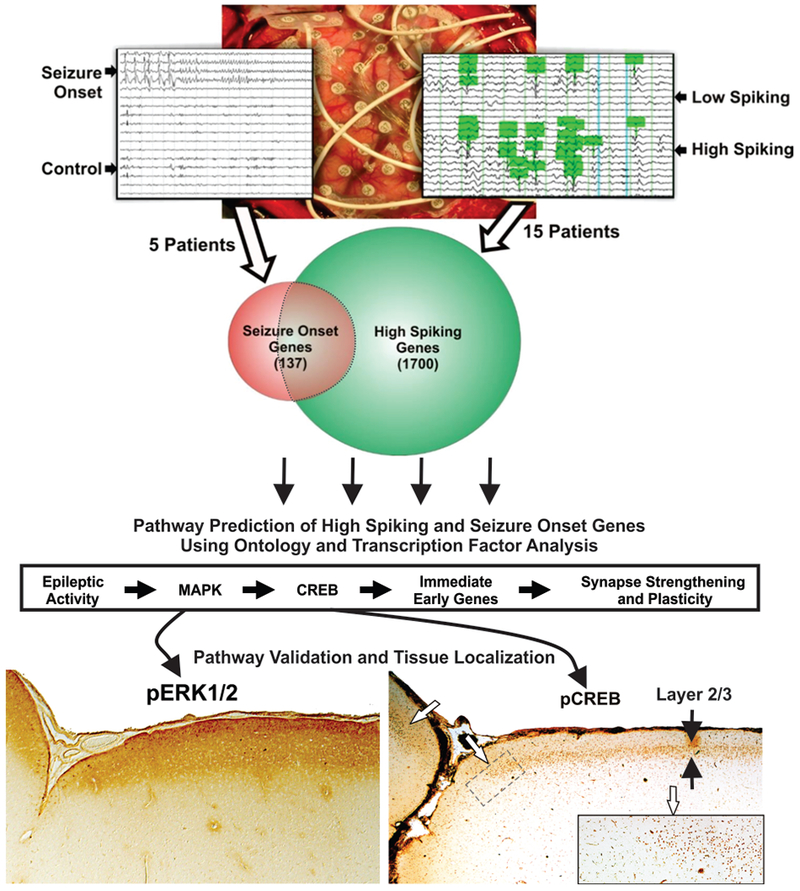
Common, genome-wide transcriptional differences were identified either between seizure onset versus non-seizure onset or high-versus low-spiking brain regions (n=5 patients for seizure onset; n=15 patients for interictal spiking). A Venn diagram shows considerable overlap between seizure and spiking genes, but also demonstrates genes that are unique to seizure onset and high spiking brain regions. Using ontological analysis and known patterns of transcription factor regulation, a highly significant pathway was predicted and linked epileptic activity to MAPK and CREB signaling, immediately early genes, and plasticity genes. Once implicated, validation studies confirmed these pathways, as shown on the bottom images with Western blots and immunostaining. This demonstrates that these predicted pathways are indeed overactive in epileptic, but not non-epileptic brain regions, and highly localized to specific neuronal lamina. Both MAPK (pERK1/2) and downstream nuclear CREB activation are restricted to layers 2 and 3 suggesting that epileptic spiking involves these specific layers.
Transcriptomic studies by other research groups have also observed MAPK upregulation in human epileptic tissues (Salman et al., 2017). The MAPK-CREB pathway is a ubiquitous pathway that is known for cell cycle progression and growth (Burotto, Chiou, Lee, & Kohn, 2014; Roux et al., 2007; Shaul & Seger, 2007; Thomas & Huganir, 2004) . In neurons, the MAPK-CREB pathway is activated by increased neuronal activity and is necessary for long-term potentiation and dendritic spine stabilization (Atkins, Selcher, Petraitis, Trzaskos, & Sweatt, 1998; English & Sweatt, 1996; English & Sweatt, 1997; Thomas & Huganir, 2004). The MAPK-CREB pathway can signal to the nucleus to increase the expression of known synaptic genes such as ARC, BDNF, and EGR1 (Herdegen & Leah, 1998; Kandel, 2001; Matynia, Kushner, & Silva, 2002; Montminy, Gonzalez, & Yamamoto, 1990; West, Griffith, & Greenberg, 2002). These MAPK-CREB target genes have also been shown to contribute to neuronal activity and synapse stability in epilepsy (Duclot & Kabbaj, 2017; Janz et al., 2018; Liu, X. et al., 2016; Parrish, R.R., Albertson, A.J., Buckingham, S.C., Hablitz, J.J., Mascia, K.L., Haselden D.W., Lubin, F.D., 2013) .
The fact that these genes were detected in almost all patients, most of whom had not had a seizure for at least 24 hours prior to tissue resection, suggests that their constitutive induction may more closely relate to interictal rather than ictal activity. In fact, when going back to the physiology on a larger number of patients, the MAPK-CREB genes are more closely correlated with interictal rather than ictal activity. Since these genes are well-known in pathways of LTP and synaptic plasticity, it seem likely that their sustained activation many be mediated by ongoing interictal spiking and in fact some of these correlate quite nicely with interictal spiking frequency(Rakhade et al., 2007)
Differentially expressed long non-coding RNAs from human epileptic brain define a new class of potential epileptic therapeutics
Not all genes are expressed in the brain encode for proteins. In fact, about half of the human genome produces RNA molecules that do not encode for proteins and are called long non-coding RNAs (lncRNAs) that are highly expressed in the brain(Clark et al., 2015; Harrow et al., 2012). Unlike micro-RNAs that are quite short, lncRNAs are transcripts larger than 200 base pairs (Dinger, Pang, Mercer, & Mattick, 2008; Guttman et al., 2009). LncRNAs are expressed at higher levels in the brain than in any other tissues (Harrow et al., 2012) and have been suggested to contribute to the complexity of the nervous system and neurological diseases such as Alzheimer’s. Parkinson’s, and Epilepsy(Quan, Zheng, & Qing, 2017). LncRNAs have been shown to carry out a variety of regulatory functions including transcriptional regulation, chromosomal imprinting, and gene silencing (Rinn & Chang, 2012; Wang, K. C. & Chang, 2011; Wu, Z. et al., 2014). Importantly, as lncRNAs are more rapidly evolving, they are species specific so in order to truly understand the potential role of lncRNA in human epilepsy, human tissue must be used(Guttman et al., 2009; Ponjavic & Ponting, 2007).
In parallel studies to our work on coding genes, we examined lncRNA differential expression in high- versus low-spiking human neocortical epileptic tissues. We utilized a custom lncRNA microarray that also included many known protein-coding differentially expressed genes from 8 patients (Lipovich et al., 2012). We found 1200 differentially expressed lncRNAs in the human epileptic brain. Interestingly, one of these differentially expressed lncRNA was BDNFOS. BDNFOS is an lncRNA located anti-sense to BDNF, a previously defined pro-epileptogenic MAPK signaling gene (Beaumont et al., 2012). Follow-up in vitro studies using the human SH-SY5Y cell line demonstrated that the loss of BDNFOS results in an increase in BDNF(Lipovich et al., 2012), suggesting that the lncRNA is an important regulator of BDNF in epileptic human brain regions. Taken together, these findings demonstrate that not only are lncRNAs differentially expressed in epilepsy, but they have the potential to regulate the same population of MAPK signaling genes and could offer an alternative therapeutic approach for human epilepsy.
Localization of the MAPK-CREB pathway to superficial neuronal layers as a “Tissue biomarker” of interictal spiking
Key to any high-throughput discovery-based study is an experimental plan for validating predictions made. Because we collect tissues and subdivide them for histology, genomics, proteomics, and metabolomics, we can rapidly validate predicted pathways directly on the same human tissue block where differential gene expression was previously identified. Specifically, Western blots of protein fractions from the same blocks of tissue that demonstrated sustain MAPK-CREB gene expression, showed sustained activation (phosphorylation) of both MAPK and CREB only in the high spiking tissues. As a means to localize where in the 6-layered cortex MAPK and CREB are activated, we next probed the fixed piece of tissue from the same block with these same antibodies. Surprisingly, as shown on the bottom of figure 2, both MAPK and CREB were activated selectively in the most superficial layers of the neocortex layers 1–3 (Beaumont et al., 2012). As layer 2/3 neurons project to the molecular layer 1, they are known to significantly contribute to lateral connectivity of the neocortex and therefore could play an important role in generating the hypersynchrony critical for generating epileptic discharges (Marin Padilla, 2001; Petersen & Crochet, 2013). Consistently we found that these same layers had a significant downstream induction of plasticity genes and a 3-fold increase in presynaptic nerve terminals compared to non-epileptic brain regions. Taken together, the laminar distribution of activated MAPK-CREB signaling may contribute to the epileptic state by promoting lateral connectivity and excitation that contributes to the synchronized neuronal activity necessary to produce epileptic discharges (Figure 2). Therefore, in addition to being a tissue biomarker, the MAPK-CREB pathway could also be a potential therapeutic target.
Predicting novel cellular and histological changes in human epileptic brain through genomic and proteomic clustering
Understanding the underlying cellular networks that enable the neocortex to generate epileptic potentials has been a major challenge. Studies of human neocortical resections of epileptogenic zones have been less than revealing, sometimes showing no clear pathology or subtle observations of mild gliosis or dysplasias (Lee et al., 2000; Palmini et al., 2004; Seo, J.H., et al, 2009). We used an unbiased approach to predict abnormal histopathology by identifying gene clusters coordinately, differentially-expressed between high and low spiking brain areas using a combination of genomic (Dachet et al., 2015) and proteomic (Keren-Aviram et al., 2018) studies. From 30 paired cortical samples from 15 epileptic patients, we identified 11 gene clusters, where each cluster contains a group of genes that share an identical pattern of expression across the 30 samples. Each of the 11 individual clusters were assigned a putative cell-type using an automated script that performs repetitive, online PubMed searches using all pairs of genes and known brain cell types to see how many ‘hits’ occur with each combination. Based on this approach, four neuronal subtypes, three glial cell subtypes, three cell types associated with blood/blood products, and one “dividing” cell type were identified to be changed in the human epileptic neocortical tissue compared to the control regions (Figure 3A). Interestingly, the cell clustering results demonstrated an inverse expression pattern between an upregulated microglia cell type (Microglia 1) and a downregulated neuron (Neuron 1). Neuron 1 correlated with the gene RBFOX3 or NeuN, the most commonly used immunohistochemical marker for neurons (Dent, Segura-Anaya, Alva-Medina, & Aranda-Anzaldo, 2010) and one of the genes in this cluster was RORB, a layer 4 specific gene.
Figure 3. Gene clustering predicts a ‘Cellular Interactome’ composed of microlesions in deeper cortical layers.
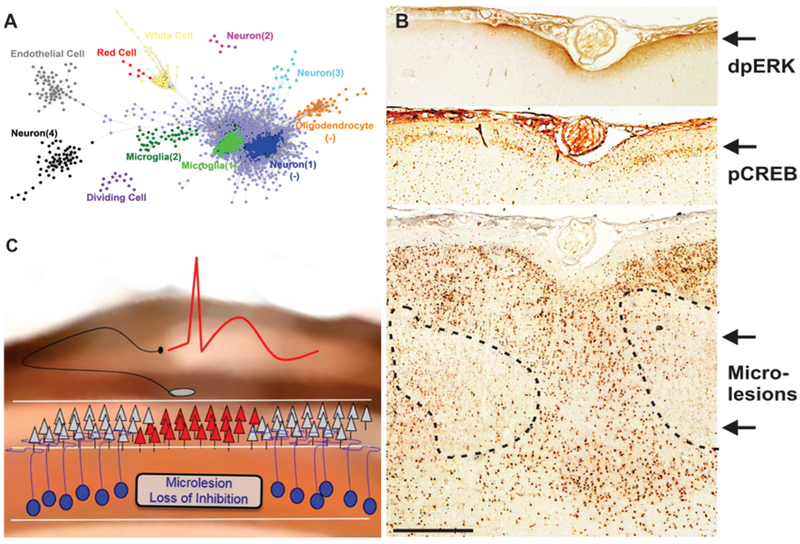
From 30 brain samples with varying amounts of interictal epileptic spiking, we developed a novel gene expression clustering approach to identify highly co-expressed genes clusters (A). Each dot in a cluster corresponds to a specific gene and the proximity of dots corresponds to the closeness of their correlation. Examination of how each predicted cell cluster changes relative to each other makes further predictions of histological differences in epileptic brain regions. Each prediction is then validated by tissue staining as shown in (B), where novel microlesions were discovered in deeper cortical layers adjacent to activation of MAPK and CREB in superficial layers. (C) A model of how these deep microlesions could contribute to generating synchronous epileptic spikes in superficial lamina due to a loss of inhibition from the microlesions.
These computational studies suggest that high spiking brain regions will have reduced NeuN staining in layer 4 of the cortex. Follow-up histological studies confirmed these predictions in high spiking tissue. Moreover, the remarkable inverse relationship between Microglia 1 and Neuron 1 could be demonstrated histologically by an increase in microglial numbers and size in the exact same brain regions where NeuN staining was reduced. We now call these focal cortical regions with reduced NeuN staining ‘microlesions.’ Microlesions were found in all patients we examined, including those with a variety of other associated lesions. Interestingly, microlesions that involved mostly deeper layers of the cortex were spatially opposed to superficial layer MAPK and CREB activation (Figure 3B). This suggests an abnormal circuitry caused by deeper microlesions leading to increased MAPK-CREB signaling in superficial layers and ongoing interictal spiking (Figure 3C). We hypothesize that microlesions within deeper layers of the cortex lack functional inhibitory neurons that lead to increased excitability and synchrony in superficial layers.
In addition to microlesions, our unbiased approach also predicted an increase of vascular structures in high-spiking brain regions (Dachet et al., 2015). This observation was supported also by a later proteomic study on the same samples (Keren-Aviram et al., 2018) other studies with both increases in vascularity and angiogenesis in animal models of epilepsy (Morin-Brureau et al., 2011; Pitkänen & Sutula, 2002). One explanation is that frequent ongoing spiking in these brain regions triggers an increase in vascular density due to increased oxygen need.
Parallel proteomic and genomic studies reveal both complementary and divergent changes
In other studies, proteomic and genomic analysis of the same samples often reveal changes in gene transcription that are not always seen through proteomics (Guoan Chen et al., 2002; Pascal et al., 2008). Using 2-D differential gel electrophoresis (2D-DIGE) on 6 patients, comparing epileptic (high interictal spiking) regions to nearby control regions, 8 proteins stood out as significantly upregulated, and 10 were downregulated (Keren-Aviram et al., 2018). While these proteins were consistently up- or down-regulated across all samples, a number of proteins had an exceptionally high variance compared to proteins that were not changing. We hypothesized that proteins that are highly variable in their expression in epileptic tissues may be mechanistically important compared to proteins whose expression levels are constant. This led to the identification of additional proteomic changes. Altogether, 146 unique proteins were considered to be differentially expressed from 397 highly variable spots on the 2D gels. Hierarchical clustering of these proteins revealed groups of co-regulated proteins, similar to what we found by clustering gene expression, and, similar to the genomic studies predicted increased blood proteins including carbonic anhydrase 1, fibrin and fibrinogen (Keren-Aviram et al., 2018).
The proteomic analysis also revealed changes not seen by transcriptomics (Keren-Aviram et al., 2018). One protein that was found to be significantly down-regulated in epileptic neocortical tissue was glial fibrillary acid protein (GFAP), specifically the 50kDa splice variant (Middeldorp & Hol, 2011). This was unexpected as GFAP, an intermediate filament protein, is commonly used as a marker for astrocytes (Middeldorp & Hol, 2011) that are often increased in epilepsy associated with hippocampal sclerosis (Oberheim et al., 2008). Results from other publisehd proteomic studies in human epilepsy have demonstrated GFAP to be increased in epileptic tissue(Yang, Czech, & Lubec, 2004). Yet, these other studies focused on lesional tissue without coregistration of electrical recordings, whereas here we focused our studies on non-lesional, human epileptic neocortical tissue precisely mapped to long term recordings. Studies on neocortical epilepsy, however, show a similar down-regulation in astrocytic proteins, such as the inwardly rectifying potassium channel Kir4.1 (Tessandra H. Stewart et al., 2010). Taken together, these results suggest that the expression or involvement of astrocytes in the epileptic process may be different for lesional versus non-lesional, neocortical epilepsy; however, additional studies are needed to fully understand the dynamic roles of astrocytes in neocortical epilepsy.
Metabolomics, combined with genomics, reveals cell specific metabolism associated will regions of high epileptic activity
Epilepsy is a disease of hypersynchronous neuronal activity, so it would not be surprising to find metabolomic changes consistent with heightened activity. Taking a similar approach to the previously described genomic and proteomic studies, we identified a highly significant metabolomic signature between high and low spiking human epileptic neocortex(Wu, H. C. et al., 2017). Using high resolution magic angle spinning (HRMAS) H MRS we found that the relative expression of 6 out of a total of 14 metabolites could be used to clearly differentiate epileptic versus non-epileptic cortical brain regions. These included choline, creatine + phospho-creatine (Cr+PCr), glycerophosphorylcholine (GPC), myo-inositol, lactate, and N-acetylaspartylglutamate (NAAG).
While these metabolomic changes are highly consistent with and may help develop non-invasive ways to someday ‘visualize’ human epileptic brain regions by MRI spectroscopic imaging, they also may reveal important new aspects about the metabolic state of the epileptic neocortex. Two of the differentially expressed metabolites share similar expression patterns with cell types predicted by transcriptomics. Specifically, an increase in Cr+PCr correlates with increased microglia and the decrease in lactate correlates with a decrease in type I neurons in epileptic tissues. This suggests that some metabolomic changes may be due to changes in the cellular architecture in the epileptic tissue, including changes in cell number or cell phenotype. While many think that lactate should be elevated, particularly after an acute seizure, a similar, unbiased study examining resected tissue samples from patients with epilepsy found a similar decrease in lactate expression in epileptic tissue (Detour et al., 2018). This highly consistent finding in all of our samples suggests that the cell populations in epileptic brain regions rely on more heavily on aerobic than on anaerobic respiration and may somehow drive the increase seen in total brain vascularity seen both by genomics and proteomics (Dachet et al., 2015; Keren-Aviram et al., 2018) .
Validation
While the results from human tissue studies are highly relevant to the human condition, they remain correlative in nature. To determine whether a specific change seen in human tissues is in fact causative or simply results from ongoing epileptic activity requires studies in animals where changes in the epileptic state can be quantified after interventions that increase or decrease a specific gene or pathway. Because most of the gene expression changes correlated better to interictal spiking than seizures, we optimized a chronic rat model of epilepsy that produces predominantly interictal spiking after injection of tetanus toxin into the rat somatosensory cortex(Barkmeier, D. T. & Loeb, 2009). We found that the generation of interictal spiking using this approach was sufficient to induce the exact same layer 2/3 specific pattern of CREB activation and MAPK gene expression as we discovered in human epileptic neocortex (Barkmeier, D. T. et al., 2012) (Figure 4). We further showed “cause and effect” in that a one-week treatment using a drug that blocks MAPK signaling following tetanus toxin injection prevented both downstream CREB activation and the development of interictal spiking, suggesting that MAPK signaling is required in the epileptogenic process and could therefore be a potential therapeutic target to prevent epilepsy after a brain insult or injury (Barkmeier et al., 2012).
Figure 4. Layer-specific activation of CREB and downstream plasticity genes in a rat model of cortical interictal spiking closely parallels human epileptic cortex.
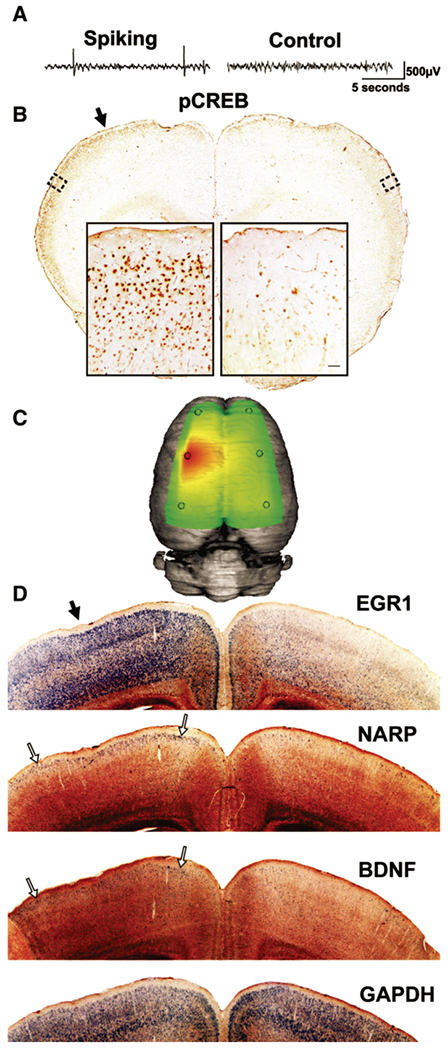
As a mean to test for cause and effect of genes and pathways predicted from human cortex, we optimized a rat model of interictal spiking using a single injection of tetanus toxin into somatosensory cortex followed by long-term video EEG and histological analysis. (A) A sample recording at one week after tetanus toxin injection shows interictal spikes confined to the left hemisphere. (B) At this time selective CREB phosphorylation is seen in layer 2/3 of the spiking hemisphere relative to the contralateral, non-spiking hemisphere. This phosphorylation extends far beyond the injection site (solid arrow; slice approximately 1 mm anterior to injection site). (C) A three-dimensional heatmap shows the interictal spike field size two weeks after tetanus toxin (D) within 1 mm of the injection site (solid arrow). At two weeks, layer specific gene expression of genes induced in human cortex are seen to spreads laterally in this model away from the injection site. Using radioactive in situ hybridizations, EGR1 is diffusely induced, but especially strong in the superficial part of layer 2/3. NARP and BNDF induction are highly restricted to layer 2/3 of the spiking hemisphere (open arrows), while the non-spiking hemi-sphere shows comparatively sparse signal. The housekeeping gene GAPDH was used as a control and shows an even signal across both hemispheres in layers 2–6.
In addition to in vivo studies, we have utilized an in vitro approach to model repetitive spiking seen in epilepsy by repeated depolarization of the human neuronal like SY5Y cells as described above for BDNFOS (Lipovich et al.,2012). This model uses repeated pulses of 100mM Potassium Chloride (KCl), which results in an influx of calcium ions into the neuronal like cells activating multiple cellular pathways, one of which is MAPK-CREB signaling (Wang, W. et al., 2018). Compared to a single depolarization, repeated depolarizations over an 8 hour period leads to sustained MAPK and CREB activation similar to what we have seen in human epileptic neocortical tissues(Lipovich et al., 2012) (Beaumont et al., 2012; Rakhade et al., 2005; Rakhade et al., 2007). The ability to create an in vitro model with human cells that mimics the MAPK activation and gene expression of the human epileptic brain allows us to perform mechanistic studies, both on coding and non-coding transcripts, and will allow for future compound testing for drug development studies.
Reverse Translation
Generating new knowledge from human tissues is often referred to as a ‘reverse translation.’ Once targets and biomarkers are identified, the major goal will be to ‘forwardly’ translate findings in animal models back into human treatments. As described above, administration of a highly specific MAPK inhibitor was sufficient to prevent the development of epileptic discharges from tetanus toxin injection into the neocortex without any other identified side effect on the brain activity. This finding confirms a critical role for MAPK signaling pathway in the development of epileptic activity and highlights MAPK inhibition as a possible therapeutic treatment (Barkmeier et al., 2012). At present time, there are three FDA approved MEK inhibitors available for use in patients (Cohen & Sullivan, 2019). These inhibitors are currently used only for the treatment metastatic melanoma and rare childhood brain tumors as they carry significant side effects, particularly in the GI tract. In addition to these or other drugs that could be repurposed, our detailed systems biology approach is a virtual pipeline of additional targets both within and outside the MAPK signaling pathway that could be tested in both in vivo and in vitro models to identify novel regulators of epileptic activities. As described above, some of these novel approaches could include manipulation of lncRNAs that have the ability to alter the expression of pro-epileptogenic signaling genes. LncRNA manipulation as a potential therapy has been suggested in other research fields, most prominently in oncology(Arun, Diermeier, & Spector, 2018). However, technological advancements for targeting lncRNAs, including molecular delivery and stability, need to be made in order for this to occur(Leti & DiStefano, 2017; Ozcan, Ozpolat, Coleman, Sood, & Lopez-Berestein, 2015) .
Conclusions and Future Directions
Through an unbiased, big data mining approach, highly consistent patterns of differentially expressed genes, proteins, and metabolites, together with their associated signaling pathways can be discovered from carefully localized human neocortical tissues from patients undergoing respective epilepsy surgery. This experimental approach generates large, unbiased data sets that can readily be organized into a multivariate interactome, that also includes clinical data such as medications and seizure characteristics. Using a variety of correlation analyses, statistically significant clusters containing a number of these variables can be visualized as shown in Figure 5. This interactome is thus a discovery machine that can be used through reverse-translation back into animal and in vitro models to validate novel biomarkers as well as therapeutic targets that can then be forward translated back to patients with epilepsy.
Figure 5. Multivariate Interactome.
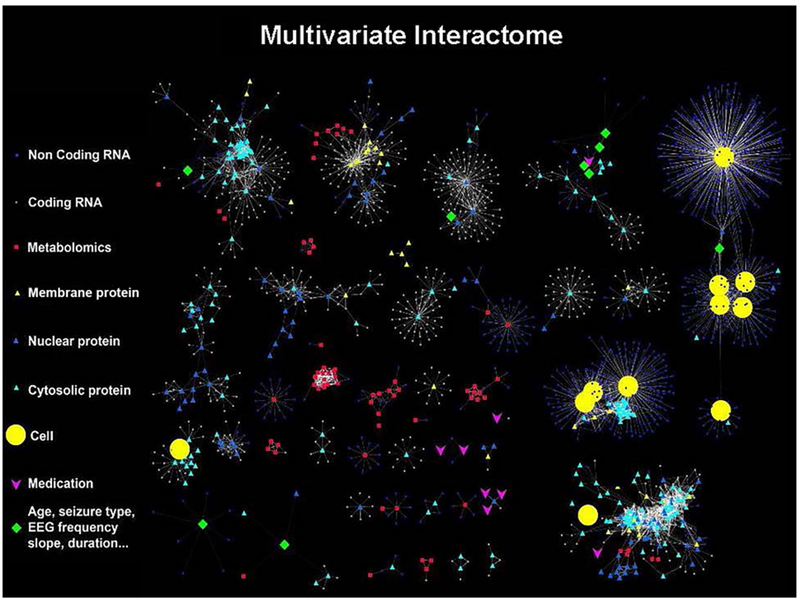
Integration of data from our systems biology approach allows all data to be linked together to identify significant correlations between all variables. These relationships were visualized using Cytoscape software (https://cytoscape.org/) that shows correlations between genes, proteins, metabolites, predicted cell populations, electrical and clinical variables. Each type of data has a different shape and the closer two objects are two each other, the stronger the correlation. Many of these independent variables now fall into clusters that integrate many types of variables related to different aspects of epilepsy. Continued expansion of this discovery-based approach will provides an unbiased approach to explore the molecular, cellular, electrical and clinical underpinnings of human epilepsy.
Moving forward, new database structures that have the ability to automatically draw in, integrate, and analyze data from electronic health records could make this highly laborious approach more tenable for most institutions and could be applied to other disorders as well. Some of the major challenges will include the development of digital tools that automatically place a mark on imaging studies where tissue is removed, improved algorithms for quantifying EEG studies, and programs that link these together, both spatially and longitudinally, over each patient’s life. Single cell RNA-sequencing is an additional new tool that will be complementary to our mathematical approaches and can identify distinct cell populations that could be therapeutically targeted.
Novel approach to combine quantitative electrical, brain imaging, and genomics for refractory epilepsy
Internally controlled study designs yield unbiased clues for why some brain regions are epileptic
A multivariate interactome is presented as a discovery machine for human epilepsy
Acknowledgements
This work was funded by NIH grants UL1TR002003 (JAL), NS083527 (JAL), and NS109515 (JAL)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Annegers J (1993). The& treatment of epilepsy. Philadelphia [u.a.]: Lea & Febiger. [Google Scholar]
- Arun G, Diermeier SD, & Spector DL (2018). Therapeutic targeting of long non-coding RNAs in cancer. Trends in Molecular Medicine, 24(3), 257–277. doi: 10.1016/j.molmed.2018.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, & Sweatt JD (1998). The MAPK cascade is required for mammalian associative learning. Nature Neuroscience, 1(7), 602–609. doi: 10.1038/2836 [doi] [DOI] [PubMed] [Google Scholar]
- Barkmeier DT, & Loeb JA (2009). An animal model to study the clinical significance of interictal spiking. Clinical EEG and Neuroscience, 40(4), 234–238. doi: 10.1177/155005940904000405 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkmeier DT, Senador D, Leclercq K, Pai D, Hua J, Boutros NN, … Loeb JA (2012). Electrical, molecular and behavioral effects of interictal spiking in the rat. Neurobiology of Disease, 47(1), 92–101. doi: 10.1016/j.nbd.2012.03.026 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkmeier DT, Shah AK, Flanagan D, Atkinson MD, Agarwal R, Fuerst DR, Jafari-Khouzani K, Loeb JA (2011). High inter-reviewer variability of spike detection on intracranial EEG addressed by an automated multi-channel algorithm. Clinical Neurophysiology, 123(6), 1088–1095. doi: 10.1016/j.clinph.2011.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista RE, Cobbs MA, Spencer DD, & Spencer SS (1999). Prediction of surgical outcome by interictal epileptiform abnormalities during intracranial EEG monitoring in patients with extrahippocampal seizures. Epilepsia, 40(7), 880–890. doi: 10.1111/j.1528-1157.1999.tb00794.x [DOI] [PubMed] [Google Scholar]
- Beaumont TL, Yao B, Shah A, Kapatos G, & Loeb JA (2012). Layer-specific CREB target gene induction in human neocortical epilepsy. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 32(41), 14389–14401. doi: 10.1523/JNEUROSCI.3408-12.2012 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie MJ, Barry SJ, Bamagous GA, Norrie JD, & Kwan P (2012). Patterns of treatment response in newly diagnosed epilepsy. Neurology, 78(20), 1548–1554. doi: 10.1212/WNL.0b013e3182563b19 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burotto M, Chiou VL, Lee JM, & Kohn EC (2014). The MAPK pathway across different malignancies: A new perspective. Cancer, 120(22), 3446–3456. doi: 10.1002/cncr.28864 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalleri GL, Lynch JM, Depondt C, Burley MW, Wood NW, Sisodiya SM, & Goldstein DB (2005). Failure to replicate previously reported genetic associations with sporadic temporal lobe epilepsy: Where to from here? Brain : A Journal of Neurology, 128(Pt 8), 1832–1840. doi:awh524 [pii] [DOI] [PubMed] [Google Scholar]
- Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, & Thiele EA (2010). The natural history of epilepsy in tuberous sclerosis complex. Epilepsia, 51(7), 1236–1241. doi: 10.1111/j.1528-1167.2009.02474.x [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MB, Mercer TR, Bussotti G, Leonardi T, Haynes KR, Crawford J, … Dinger ME (2015). Quantitative gene profiling of long noncoding RNAs with targeted RNA sequencing. Nature Methods, 12(4), 339–342. doi: 10.1038/nmeth.3321 [doi] [DOI] [PubMed] [Google Scholar]
- Cohen JV, & Sullivan RJ (2019). Developments in the space of new MAPK pathway inhibitors for BRAF-mutant melanoma. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, doi:clincanres.0836.2018 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepeau AZ, & Sirven JI (2017). Management of adult onset seizures. Mayo Clinic Proceedings, 92(2), 306–318. doi:S0025-6196(16)30771-6 [pii] [DOI] [PubMed] [Google Scholar]
- Dachet F, Bagla S, Keren-Aviram G, Morton A, Balan K, Saadat L, … Loeb JA (2015). Predicting novel histopathological microlesions in human epileptic brain through transcriptional clustering. Brain : A Journal of Neurology, 138(Pt 2), 356–370. doi: 10.1093/brain/awu350 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detour J, Bund C, Behr C, Cebula H, Cicek EA, Valenti-Hirsch MP, … Namer IJ (2018). Metabolomic characterization of human hippocampus from drug-resistant epilepsy with mesial temporal seizure. Epilepsia, 59(3), 607–616. doi: 10.1111/epi.14000 [doi] [DOI] [PubMed] [Google Scholar]
- Dinger ME, Pang KC, Mercer TR, & Mattick JS (2008). Differentiating protein-coding and noncoding RNA: Challenges and ambiguities. PLoS Computational Biology, 4(11), e1000176. doi: 10.1371/journal.pcbi.1000176 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duclot F, & Kabbaj M (2017). The role of early growth response 1 (EGR1) in brain plasticity and neuropsychiatric disorders. Frontiers in Behavioral Neuroscience, 11, 35. doi: 10.3389/fnbeh.2017.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- England MJ, Liverman CT, Schultz AM, & Strawbridge LM (2012). Epilepsy across the spectrum: Promoting health and understanding. A summary of the institute of medicine report. Epilepsy & Behavior : E&B, 25(2), 266–276. doi: 10.1016/j.yebeh.2012.06.016 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, & Sweatt JD (1996). Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. The Journal of Biological Chemistry, 271(40), 24329–24332. [DOI] [PubMed] [Google Scholar]
- English JD, & Sweatt JD (1997). A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. The Journal of Biological Chemistry, 272(31), 19103–19106. [DOI] [PubMed] [Google Scholar]
- Gotman J (1991). Relationships between interictal spiking and seizures: Human and experimental evidence. The Canadian Journal of Neurological Sciences. Le Journal Canadien Des Sciences Neurologiques, 18(4 Suppl), 573–576. doi: 10.1017/S031716710003273X [DOI] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, … Lander ES (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature, 458(7235), 223–227. doi: 10.1038/nature07672 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, … Hubbard TJ (2012). GENCODE: The reference human genome annotation for the ENCODE project. Genome Research, 22(9), 1760–1774. doi: 10.1101/gr.135350.111 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herdegen T, & Leah JD (1998). Inducible and constitutive transcription factors in the mammalian nervous system: Control of gene expression by jun, fos and krox, and CREB/ATF proteins. Brain Research.Brain Research Reviews, 28(3), 370–490. doi:S0165017398000186 [pii] [DOI] [PubMed] [Google Scholar]
- Janz P, Hauser P, Heining K, Nestel S, Kirsch M, Egert U, & Haas CA (2018). Position- and time-dependent arc expression links neuronal activity to synaptic plasticity during epileptogenesis. Frontiers in Cellular Neuroscience, 12, 244. doi: 10.3389/fncel.2018.00244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa O, Blume WT, & Girvin JP (1996). Significance of spikes at temporal lobe electrocorticography. Epilepsia, 37(1), 50–55. doi: 10.1111/j.1528-1157.1996.tb00511.x [DOI] [PubMed] [Google Scholar]
- Kandel ER (2001). The molecular biology of memory storage: A dialogue between genes and synapses. Science (New York, N.Y.), 294(5544), 1030–1038. doi: 10.1126/science.1067020 [doi] [DOI] [PubMed] [Google Scholar]
- Keren-Aviram G, Dachet F, Bagla S, Balan K, Loeb JA, & Dratz EA (2018). Proteomic analysis of human epileptic neocortex predicts vascular and glial changes in epileptic regions. PloS One, 13(4), e0195639. doi: 10.1371/journal.pone.0195639 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Spencer DD, & Spencer SS (2000). Intracranial EEG Seizure-Onset patterns in neocortical epilepsy. Epilepsia, 41(3), 297–307. doi: 10.1111/j.1528-1157.2000.tb00159.x [DOI] [PubMed] [Google Scholar]
- Leti F, & DiStefano JK (2017). Long noncoding RNAs as diagnostic and therapeutic targets in type 2 diabetes and related complications. Genes, 8(8), 207. doi: 10.3390/genes8080207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipovich L, Dachet F, Cai J, Bagla S, Balan K, Jia H, & Loeb JA (2012). Activity-dependent human brain coding/noncoding gene regulatory networks. Genetics, 192(3), 1133–1148. doi: 10.1534/genetics.112.145128 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Yu W, & LÃ Y (2016). The causes of new-onset epilepsy and seizures in the elderly. Neuropsychiatric Disease and Treatment, 12, 1425–1434. doi: 10.2147/NDT.S107905 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ou S, Xu T, Liu S, Yuan J, Huang H, … Chen Y, (2016). New differentially expressed genes and differential DNA methylation underlying refractory epilepsy. Oncotarget, 7(52), 87402–87416. doi: 10.18632/oncotarget.13642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JA (2010). A human systems biology approach to discover new drug targets in epilepsy. Epilepsia, 51 Suppl 3, 171–177. doi: 10.1111/j.1528-1167.2010.02635.x [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JA (2011). Identifying targets for preventing epilepsy using systems biology. Neuroscience Letters, 497(3), 205–212. doi: 10.1016/j.neulet.2011.02.041 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher W, & Schmidt D (2011). Modern antiepileptic drug development has failed to deliver: Ways out of the current dilemma. Epilepsia, 52(4), 657–678. doi: 10.1111/j.1528-1167.2011.03024.x [doi] [DOI] [PubMed] [Google Scholar]
- Maharathi B, Wlodarski R, Bagla S, Asano E, Hua J, Patton J, & Loeb JA (2019). Interictal spike connectivity in human epileptic neocortex. Clinical Neurophysiology, 130(2), 270–279. doi: 10.1016/j.clinph.2018.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin Padilla M (2001). The evolution of the structure of the neocortex in mammals: A new theory of cytoarchitecture. [Evolucion de la estructura de la neocorteza del mamifero: nueva teoria citoarquitectonica] Revista De Neurologia, 33(9), 843–853. [PubMed] [Google Scholar]
- Matynia A, Kushner SA, & Silva AJ (2002). Genetic approaches to molecular and cellular cognition: A focus on LTP and learning and memory. Annual Review of Genetics, 36, 687–720. doi: 10.1146/annurev.genet.36.062802.091007 [doi] [DOI] [PubMed] [Google Scholar]
- Middeldorp J, & Hol EM (2011). GFAP in health and disease. Progress in Neurobiology, 93(3), 421–443. doi: 10.1016/j.pneurobio.2011.01.005 [DOI] [PubMed] [Google Scholar]
- Mittal S, Shah AK, Barkmeier DT, & Loeb JA (2013). Systems biology of human epilepsy applied to patients with brain tumors. Epilepsia, 54(s9), 35–39. doi: 10.1111/epi.12441 [DOI] [PubMed] [Google Scholar]
- Montminy MR, Gonzalez GA, & Yamamoto KK (1990). Regulation of cAMP-inducible genes by CREB. Trends in Neurosciences, 13(5), 184–188. doi:0166-2236(90)90045-C [pii] [DOI] [PubMed] [Google Scholar]
- Morin-Brureau M, Lebrun A, Rousset M, Fagni L, Bockaert J, de Bock F, & Lerner-Natoli M (2011). Epileptiform activity induces vascular remodeling and zonula occludens 1 downregulation in organotypic hippocampal cultures: Role of VEGF signaling pathways. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 31(29), 10677–10688. doi: 10.1523/JNEUROSCI.5692-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moshe SL, Perucca E, Ryvlin P, & Tomson T (2015). Epilepsy: New advances. Lancet (London, England), 385(9971), 884–898. doi: 10.1016/S0140-6736(14)60456-6 [doi] [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Tian G, Han X, Peng W, Takano T, Ransom B, & Nedergaard M (2008). Loss of astrocytic domain organization in the epileptic brain. Journal of Neuroscience, 28(13), 3264–3276. doi: 10.1523/JNEUROSCI.4980-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan G, Ozpolat B, Coleman RL, Sood AK, & Lopez-Berestein G (2015). Preclinical and clinical development of siRNA-based therapeutics. Advanced Drug Delivery Reviews, 87, 108–119. doi: 10.1016/j.addr.2015.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmini A, Andermann F, Olivier A, Tampieri D, & Robitaille Y (1991). Focal neuronal migration disorders and intractable partial epilepsy: Results of surgical treatment. Annals of Neurology, 30(6), 750–757. doi: 10.1002/ana.410300603 [DOI] [PubMed] [Google Scholar]
- Palmini A, Najm I, Avanzini G, Babb T, Guerrini R, Foldvary-Schaefer N, … Vinters HV (2004). Terminology and classification of the cortical dysplasias. Neurology, 62(6 Suppl 3), S8. doi: 10.1212/01.WNL.0000114507.30388.7E [DOI] [PubMed] [Google Scholar]
- Parrish RR, Albertson AJ, Buckingham SC, Hablitz JJ, Mascia KL, Haselden DW, Lubin FD (2013). Status epilepticus triggers early and late alterations in brain-derived neurotrophic factor and NMDA glutamate receptor Grin2b DNA methylation levels in the hippocampus. Neuroscience, 248, 602–619. doi: 10.1016/j.neuroscience.2013.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CC, & Crochet S (2013). Synaptic computation and sensory processing in neocortical layer 2/3. Neuron, 78(1), 28–48. doi: 10.1016/j.neuron.2013.03.020 [doi] [DOI] [PubMed] [Google Scholar]
- Pitkänen A, & Sutula TP (2002). Is epilepsy a progressive disorder? prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurology, 1(3), 173–181. doi: 10.1016/S1474-4422(02)00073-X [DOI] [PubMed] [Google Scholar]
- Ponjavic J, & Ponting CP (2007). The long and the short of RNA maps. BioEssays : News and Reviews in Molecular, Cellular and Developmental Biology, 29(11), 1077–1080. doi: 10.1002/bies.20669 [doi] [DOI] [PubMed] [Google Scholar]
- Quan Z, Zheng D, & Qing H (2017). Regulatory roles of long non-coding RNAs in the central nervous system and associated neurodegenerative diseases. Frontiers in Cellular Neuroscience, 11, 175. doi: 10.3389/fncel.2017.00175 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhade SN, Shah AK, Agarwal R, Yao B, Asano E, & Loeb JA (2007). Activity-dependent gene expression correlates with interictal spiking in human neocortical epilepsy. Epilepsia, 48 Suppl 5, 86–95. doi:EPI1294 [pii] [DOI] [PubMed] [Google Scholar]
- Rakhade SN, Yao B, Ahmed S, Asano E, Beaumont TL, Shah AK, … Loeb JA (2005). A common pattern of persistent gene activation in human neocortical epileptic foci. Annals of Neurology, 58(5), 736–747. doi: 10.1002/ana.20633 [doi] [DOI] [PubMed] [Google Scholar]
- Rinn JL, & Chang HY (2012). Genome regulation by long noncoding RNAs. Annual Review of Biochemistry, 81, 145–166. doi: 10.1146/annurev-biochem-051410-092902 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, … Blenis J (2007). RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. The Journal of Biological Chemistry, 282(19), 14056–14064. doi:M700906200 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salman MM, Sheilabi MA, Bhattacharyya D, Kitchen P, Conner AC, Bill RM, … Princivalle AP (2017). Transcriptome analysis suggests a role for the differential expression of cerebral aquaporins and the MAPK signalling pathway in human temporal lobe epilepsy. The European Journal of Neuroscience, 46(5), 2121–2132. doi: 10.1111/ejn.13652 [doi] [DOI] [PubMed] [Google Scholar]
- Seo JH, Noh BH, Lee JS, Kim DS, Lee SK, Kim TS, Kim SH, Kang HC, & Kim HD (2009). Outcome of surgical treatment in non-lesional intractable childhood epilepsy. Seizure: European Journal of Epilepsy, 18(9), 625–629. doi: 10.1016/j.seizure.2009.07.007 [DOI] [PubMed] [Google Scholar]
- Shain C, Ramgopal S, Fallil Z, Parulkar I, Alongi R, Knowlton R, … the EPGP Investigators. (2013). Polymicrogyria-associated epilepsy: A multi-center phenotypic study from the epilepsy phenome/genome project. Epilepsia, 54(8), 10.1111/epi.12238. Epub 2013 Jun 10 doi:10.1111/epi.12238. doi:10.1111/epi.12238 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul YD, & Seger R (2007). The MEK/ERK cascade: From signaling specificity to diverse functions. Biochimica Et Biophysica Acta, 1773(8), 1213–1226. doi:S0167-4889(06)00319-3 [pii] [DOI] [PubMed] [Google Scholar]
- Singh A, & Trevick S (2016). The epidemiology of global epilepsy. Neurologic Clinics, 34(4), 837–847. doi:S0733-8619(16)30037-8 [pii] [DOI] [PubMed] [Google Scholar]
- Spencer S, & Huh L (2008). Outcomes of epilepsy surgery in adults and children. The Lancet.Neurology, 7(6), 525–537. doi: 10.1016/S1474-4422(08)70109-1 [doi] [DOI] [PubMed] [Google Scholar]
- Staley KJ, White A, & Dudek FE (2011). Interictal spikes: Harbingers or causes of epilepsy? Neuroscience Letters, 497(3), 247–250. doi: 10.1016/j.neulet.2011.03.070 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley K, Hellier JL, & Dudek FE (2005). Do interictal spikes drive epileptogenesis? The Neuroscientist, 11(4), 272–276. doi: 10.1177/1073858405278239 [DOI] [PubMed] [Google Scholar]
- Tao JX, Ray A, Hawes-Ebersole S, & Ebersole JS (2005). Intracranial EEG substrates of scalp EEG interictal spikes. Epilepsia, 46(5), 669–676. doi: 10.1111/j.1528-1167.2005.11404.x [DOI] [PubMed] [Google Scholar]
- Stewart Tessandra H., Eastman Clifford L., Groblewski Peter A., Fender Jason S., Verley Derek R., Cook David G., & D’Ambrosio Raimondo. (2010). Chronic dysfunction of astrocytic inwardly rectifying K+ channels specific to the neocortical epileptic focus after fluid percussion injury in the rat. Journal of Neurophysiology, 104(6), 3345–3360. doi: 10.1152/jn.00398.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, & Huganir RL (2004). MAPK cascade signalling and synaptic plasticity. Nature Reviews.Neuroscience, 5(3), 173–183. doi: 10.1038/nrn1346 [doi] [DOI] [PubMed] [Google Scholar]
- Verloes A, Elmaleh M, Gonzales M, Laquerriere A, & Gressens P (2007). Genetic and clinical aspects of lissencephaly. [Lissencephalies: aspects cliniques et genetiques] Revue Neurologique, 163(5), 533–547. doi:MDOI-RN-05-2007-163-5-0035-3787-101019-200702448 [pii] [DOI] [PubMed] [Google Scholar]
- Wang KC, & Chang HY (2011). Molecular mechanisms of long noncoding RNAs. Molecular Cell, 43(6), 904–914. doi: 10.1016/j.molcel.2011.08.018 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zhong D, Lin Y, Fan R, Hou Z, Cao X, & Ren Y (2018). Responsiveness of voltage-gated calcium channels in SH-SY5Y human neuroblastoma cells on micropillar substrates. Journal of Biomaterials Science, Polymer Edition, 29(2), 125–144. doi: 10.1080/09205063.2017.1403714 [DOI] [PubMed] [Google Scholar]
- West AE, Griffith EC, & Greenberg ME (2002). Regulation of transcription factors by neuronal activity. Nature Reviews.Neuroscience, 3(12), 921–931. doi: 10.1038/nrn987 [doi] [DOI] [PubMed] [Google Scholar]
- White A, Williams PA, Hellier JL, Clark S, Dudek FE, & Staley KJ (2010). EEG spike activity precedes epilepsy after kainate-induced status epilepticus. Epilepsia, 51(3), 371–383. doi: 10.1111/j.1528-1167.2009.02339.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HC, Dachet F, Ghoddoussi F, Bagla S, Fuerst D, Stanley JA, … Loeb JA (2017). Altered metabolomic-genomic signature: A potential noninvasive biomarker of epilepsy. Epilepsia, 58(9), 1626–1636. doi: 10.1111/epi.13848.[doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Liu X, Liu L, Deng H, Zhang J, Xu Q, … Ji A (2014). Regulation of lncRNA expression. Cellular & Molecular Biology Letters, 19(4), 561–575. doi: 10.2478/s11658-014-0212-6 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav R, Shah AK, Loeb JA, Swamy MNS, & Agarwal R (2012). Morphology-based automatic seizure detector for intracerebral EEG recordings. IEEE Transactions on Biomedical Engineering, 59(7), 1871–1881. doi: 10.1109/TBME.2012.2190601 [DOI] [PubMed] [Google Scholar]
- Yang JW, Czech T, & Lubec G (2004). Proteomic profiling of human hippocampus. Electrophoresis, 25(7-8), 1169–1174. doi: 10.1002/elps.200305809 [doi] [DOI] [PubMed] [Google Scholar]