

Abstract
Altered signaling through transforming growth factor-beta (TGFβ) increases the risk of aortic dissection in patients, which has been confirmed in mouse models. It is well known that altered TGFβ signaling affects matrix turnover, but there has not been a careful examination of associated changes in structure-function relations. In this paper, we present new findings on the rupture potential of the aortic wall following late postnatal smooth muscle cell (SMC)-specific disruption of type I and II TGFβ receptors in a mouse model with demonstrated dissection susceptibility. Using a combination of custom computer-controlled biaxial tests and quantitative histology and immunohistochemistry, we found that loss of TGFβ signaling in SMCs compromises medial properties but induces compensatory changes in the adventitia that preserve wall strength above that which is needed to resist in vivo values of wall stress. These findings emphasize the different structural defects that lead to aortic dissection and rupture – compromised medial integrity and insufficient adventitial strength, respectively. Relative differences in these two defects, in an individual subject at a particular time, likely reflects the considerable phenotypic diversity that is common in clinical presentations of thoracic aortic dissection and rupture. There is, therefore, a need to move beyond examinations of bulk biological assays and wall properties to cell- and layer-specific studies that delineate pathologic and compensatory changes in wall biology and composition, and thus the structural integrity of the aortic wall that can dictate differences between life and death.
Keywords: aorta, smooth muscle, TGFβ, collagen, dissection, rupture
Graphical Abstract
Introduction
Thoracic aortopathies – notably aneurysm, dissection, and rupture – are responsible for significant morbidity and mortality. Notwithstanding the large percentage of sporadic lesions, advances in medical genetics continue to reveal increasing numbers of mutations that predispose to familial and syndromic lesions (Brownstein et al., 2018; Isselbacher et al., 2016; Pinard et al., 2019). The confluence of many of the predisposing mutations about the mechanotransduction axis, with some mutations affecting actomyosin activity and others affecting extracellular matrix, increasingly suggests that dysfunctional mechanosensing and/or mechanoregulation of matrix contributes to many of these lesions (Humphrey et al., 2015; Jeremy et al., 2013; Karimi and Milewicz, 2016; Ramirez et al., 2018). Such dysfunction can lead to compromised biomechanical properties of the extracellular matrix and, thus, of the aortic wall as a structure, affecting its resilience, compliance, and strength. In particular, thoracic aortic aneurysms tend to be characterized by fragmented or degraded elastic fibers, remodeled collagen, and focal accumulation of glycosaminoglycans in addition to dysfunction or loss of smooth muscle cells (SMCs), all of which are often presumed to contribute to the risk of dissection and rupture (El-Hamamsy and Yacoub, 2009; Milewicz et al., 2008).
Genetically modified mouse models continue to provide significant insight into the molecular and cellular mechanisms underlying thoracic aortopathies, particularly via longitudinal information that is otherwise difficult to obtain (Cook et al., 2015; Li et al., 2014). Notwithstanding complementary information on the biomechanics in some of these mouse models (Bellini et al., 2017a; Bersi et al., 2019; Korneva et al., 2019), much less attention has been directed toward failure mechanisms associated with dissection or rupture. We previously showed that disruptions in thrombospondin-2 can affect collagen fiber integrity and thereby increase wall vulnerability to rupture (Bellini et al., 2017b) whereas increased smooth muscle cell contractility can stress-shield an otherwise vulnerable matrix and protect against intramural delamination (Ferruzzi et al., 2016), but there remains a pressing need for detailed information on wall strength in the many different thoracic aortopathies. In this paper, we present new findings on altered histological features and reduced strength of the aortic wall in a mouse model of late post-natal SMC-specific disruption of transforming growth factor-beta (TGFβ) signaling. Alterations to this pathway result in thoracic dissections in humans (Inamoto et al., 2010; Isselbacher et al., 2016), hence conferring potential clinical relevance to studies in this mouse model.
Materials and Methods
Animal Studies.
All animal protocols were approved by the Institutional Animal Care and Use Committee at Yale University. mT/mG.Myh11-CreERT2;Tgfbr1f/f;Tgfbr2f/f (denoted Tgfbr1r2 herein) mice backcrossed onto a C57BL/6J background for >10 generations were injected with tamoxifen (Tmx) at 1 mg per day for 5 consecutive days beginning at 11 weeks of age. Age- and sex-matched C57BL/6J mice served as wild-type controls; only male mice were used since the Myh11-CreERT2 inserts on the Y chromosome. All mice were euthanized at 14 weeks of age, after which the left ventricle was perfused with Hank’s Buffered Saline Solution (HBSS) to flush the central vasculature. The descending thoracic aorta was excised from the left subclavian artery to the third intercostal arteries and cleaned of adipose tissue; intercostal branches were ligated with suture to enable ex vivo pressurization.
Biaxial mechanical testing.
Vessels were cannulated on glass pipets and placed within a custom computer-controlled device (Gleason et al., 2004) for biaxial mechanical testing. The distance between the two glass pipets was controlled by two linear actuators, each mounted on a fine triaxial translational stage, and pressurization was controlled via a pump (see Appendix Figure A.1). The applied axial force was measured using an in-line load cell attached to one of the glass pipets and the pressure was measured with two in-line transducers; axial stretch was inferred from the actuator motions and outer diameter via video-microscopy. Details of the biaxial testing protocol are available elsewhere (Ferruzzi et al., 2013) but, briefly, vessels were submerged in HBSS at 37˚C and equilibrated within the device for 15 minutes at the estimated in vivo value of axial stretch with luminal pressures cycled between 80 and 120 mmHg. Next, the vessels were preconditioned via four cycles of pressurization from 10 to 140 mmHg, again at the in vivo axial stretch. Finally, vessels were subjected to seven different cyclic loading protocols: pressurization from 10 to 140 mmHg separately at 95%, 100%, and 105% of the in vivo value of axial stretch, then axial stretching at fixed luminal pressures of 10, 60, 100, and 140 mmHg. Pressure, axial force, outer diameter, and axial length were recorded on-line and used to construct pressure-diameter and axial force-length curves and associated Cauchy stress-stretch curves.
Failure Tests.
Next, the vessels were re-mounted on two concentric, coaxial metal cannulae of differing gauges, then adjusted to recover the in vivo axial stretch, after which failure tests were conducted by continuous perfusion of vessels with HBSS until failure. Luminal pressure and outer diameter were again measured on-line. The associated pressure-diameter curves were fit with an exponential function using a nonlinear least squares regression to estimate the outer diameter at failure. Radially averaged failure stresses were calculated assuming incompressibility and a thin wall relative to luminal radius.
Histochemistry and Immunofluorescence.
Finally, vessels were fixed in 10% neutral buffered formaldehyde for 12 hours, then stored in 70% ethanol at 4˚C until sectioning at 5 μm. Sections were stained with Movat pentachrome to delineate layers, Verhoeff-Van Gieson to identify elastic laminae, and picro-sirius red to visualize fibrillar collagens. Immunofluorescence and immunohistochemistry were performed using primary antibodies against smooth muscle α-actin (SMαA), smooth muscle myosin heavy chain (SMMHC), type I collagen, macrophages (CD68), or T-cells (CD3), and counterstained by DAPI to identify cell nuclei (Table A.1). Images were acquired using an Olympus BX-51 microscope at 20x magnification using bright-field (Movat, Verhoeff-Van Gieson), polarized light (picro-sirius red), or fluorescent (immunofluorescence) settings. Constituent areas were calculated by pixel thresholding using a custom MATLAB code (Bersi et al., 2012) and ImageJ (NIH). CT-FIRE (Bredfeldt et al., 2014) was used to compute undulation of the elastic laminae of the media using pixels isolated from images of Verhoff-Van Gieson-stained sections.
Transmission Electron Microscopy.
Additional segments of aorta were fixed in 2.5% glutaraldehyde / 2% paraformaldehyde in sodium cacodylate buffer at room temperature for 30 minutes immediately after harvest, followed by an additional 2 hours at 4˚C. Samples were then rinsed in sodium cacodylate buffer before post-fixation in 1% osmium tetraoxide for 1 hour and subsequent staining using 2% uranyl acetate for 1 hour. Samples were washed and embedded in resin and viewed using a FEI Technai Biotwin electron microscope.
Biomechanical Properties.
Passive biomechanical material properties were quantified using a radially-averaged microstructurally motivated four-fiber family constitutive model, which expresses the stored elastic energy density due to deformation (Ferruzzi et al., 2013)
| (1) |
Where C = FTF is the right Cauchy-Green tensor, with F the deformation gradient tensor. is a unit vector denoting the orientation of the ith fiber family in axial , circumferential , or two symmetric diagonal directions in a cylindrical vessel in the intact traction-free reference configuration; the value of α0 is determined via nonlinear regression. Furthermore, IC = tr(C) and , with the eight model parameters , and determined by fitting simultaneously the unloading curves from the seven biaxial testing protocols using nonlinear regression. Using the unloading curves yields the value of W that would be available to work on the blood. Although this constitutive equation is motivated by the microstructure, it is yet designed to capture phenomenologically the complex anisotropic material properties, which depend on copious cross-links and constituent-constituent interactions that are not quantified by standard histological imaging. Assuming incompressibility, which can be enforced using a Lagrange multiplier p, the Cauchy stress is
| (2) |
which can be used to determine the radially averaged (mean) values that are used herein given that the thickness:radius ratio is low and the presence of residual stresses tend to homogenize the transmural distribution of wall stress. Material stiffness is calculated easily given appropriate differentiations of the stored energy function (Ferruzzi et al., 2013).
Statistical analysis.
Groups were compared using an analysis of variance (ANOVA), with Bonferroni post-hoc testing when normality of distribution was established by the Anderson-Darling test. Correlation was assessed via a two-tailed t-test after validating the linearity assumption by examination of residual plots. A p-value less than 0.05 was deemed statistically significant.
Results
Physiologic Properties.
Gross inspection revealed neither dissection nor rupture of the descending thoracic aorta of either the wild-type mice or mice having late postnatal SMC-specific disruption of Tgfbr1r2, as desired so as to focus this study on possible structural vulnerabilities, not prior failures. Standard pressure-diameter testing revealed, however, that ~50% of the Tgfbr1r2 aortas were dilated by ~16% relative to controls, which is nonetheless well less than that which constitutes an aneurysm (Table A.2). Separating the Tgfbr1r2 aortas into non-dilated versus dilated groups (Figure 1, Tables A.3-A.4), wall thickness and the in vivo value of axial stretch differed significantly between the control and non-dilated Tgfbr1r2 groups, and similarly for circumferential wall stress and elastic energy storage. That these metrics did not differ between controls and the dilated Tgfbr1r2 group suggested some biomechanical adaptation during the mild dilatation. Importantly, however, circumferential material stiffness increased significantly in the non-dilated and especially in the dilated Tgfbr1r2 groups relative to control, consistent with a compromised ability of intramural cells to mechano-sense or mechano-regulate this key intrinsic property of the matrix.
Figure 1.

Mechanical and geometric metrics that define physiological mechanical function. (A) Pressure-diameter data revealing structural stiffness, with values of (B-D) the inner radius a, wall thickness h, and the energetically preferred value of in vivo axial stretch λz at 100 mmHg compared by group: o – wild-type (WT, n=6), ■ –Tgfbr1r2 non-dilated (KO ND, n=4), and ▲ – Tgfbr1r2 dilated (KO D, n=4). (E) Circumferential Cauchy stress-stretch data revealing material stiffness, with (F-H) the circumferential Cauchy stress and material stiffness as well as energy storage at 100 mmHg compared by group. See Appendix Figure A.2 for similar results though based on comparisons of WT (n=6) versus all KOs (n=8). Figures A.3-A.5 contain complementary mechanical data. *p<0.05, **p<0.01, ***p<0.001
Failure Properties.
Pressurization tests to failure revealed a significantly lower luminal pressure at rupture in the dilated Tgfbr1r2 group relative to both the control and non-dilated Tgfbr1r2 groups (Figure 2, Table A.3). Gross inspection of the ruptured vessels revealed small, axially oriented linear tears, suggesting that failure was due to an excessive circumferential stress that resulted in the transmural loss of structural integrity. Because the diameter at rupture was similar across groups, while the thickness at rupture was higher in the non-dilated Tgfbr1r2 group, the mean circumferential Cauchy stress at failure was similar in both Tgfbr1r2 groups and significantly less than control (2.25 and 2.10 MPa relative to 3.67 MPa in control; panel G in Fig. 2).
Figure 2.

Mechanical and geometric metrics that define failure properties at supra-physiologic pressures. (A) Representative pressure-diameter data revealing the burst pressure, with (B-C) the associated outer diameter and circumferential stretch at failure. (D) Picture of a representative failed sample, revealing a linear axial tear. (E-G) Values of pressure, wall thickness, and circumferential wall stress at failure. (H) Histological image of a representative failed wall, noting the transmural break through the media and adventitia. See Figure A.6 for similar results though based on comparisons of WT (n=6) versus all KOs (n=8). *p<0.05, **p<0.01, ***p<0.001
Histological Evidence.
Immunofluorescence, immunohistochemistry, and standard histology revealed multiple subtle differences in wall composition (Figure 3). Late SMC-specific postnatal disruption of Tgfbr1r2 resulted in significant reductions in smooth muscle α-actin (SMαA) and a trend toward a reduction in smooth muscle myosin heavy chain (SMMHC) when compared across the three groups (WT, KO ND, KO D), with both reductions significant when compared between WT and KO (Figure A.7), consistent with reduced cell- and vessel-level contractility reported in a related mouse model (Ferruzzi et al., 2016; Li et al., 2014). Importantly, however, this reduction in contractile markers coincided with an increase in medial cell density, consistent with a phenotypic modulation of the SMCs towards a more proliferative phenotype with increased matrix turnover. There was, for example, a significant reduction in medial collagen I. It is possible that the increase in SMC density combined with the mild dilatation decreased the undulation of the elastic laminae in the dilated Tgfbr1r2 group (Figure A.8). Regardless, the decrease in medial collagen appeared to be offset, in part, by an increase in adventitial collagen, which was characterized by an increase in thicker fibers (red in polarized light analysis of picro-sirius red stained sections) and a decrease in thinner fibers.
Figure 3.

Immunohistochemical and histological findings. (A-F) Sections showing SMαA (red), SM-MHC (brown), VVG (elastin in black), Col1a1 (brown), medial fibrillar collagen (yellow), and adventitial fibrillar collagen (red) for the wild-type (WT) control aortas. Scale bar: 100μm. (G-L) Similarly for the non-dilated Tgfbr1r2 aortas and (M-R) the dilated Tgfbr1r2 aortas. PSR: picro-sirius red, m: media, a: adventitia. Finally, quantification by study group for (S) SMαA, (T) cell density, (U) medial collagen, (V) thin collagen fibers in the adventitia, (W) smooth muscle myosin heavy chain, (X) elastic laminae undulation, (Y) adventitial collagen, and (Z) thick collagen fibers in the adventitia. See Figure A.7 for similar results though based on comparisons of WT (n=6) versus all KOs (n=8). *p<0.05, **p<0.01, ***p<0.001, †p=0.058
Transmission electron microscopy further revealed some fine defects in the elastic laminae in the Tgfbr1r2 groups, while confirming a general increase in thicker collagen fibers in the adventitia (Figure 4). Moreover, the thicker collagen fibers were less uniform in cross-sectional shape, often non-circular, hence giving rise to a broader spectrum of collagen fibers by both diameter and shape, confirming significant collagen remodeling during this brief (3-week) period in maturity.
Figure 4.

Transmission electron microscopic images of elastic laminae within the media for (A-B) wild-type and (D-E) Tgfbr1r2 mice and similarly for (C,F) collagen fiber cross-sections; (G) a histogram of collagen fiber diameters comparing wild-type and mutant.
Microstructural Correlates.
Given the marked changes in failure properties and histological architecture due to late SMC-specific Tgfbr1r2 disruption, we contrasted key histo-mechanical metrics in an attempt to identify possible correlations between macroscale mechanics and microscale features (Figure 5). Decreased undulation of the elastic laminae correlated well with increased SMC density. Noting that intrinsic circumferential stiffness relates linearly to circumferential stress in health (Humphrey and Tellides, 2019), we plotted this material stiffness versus Cauchy wall stress for all three groups. As noted above (Fig 1G), circumferential stiffness was elevated in both Tgfbr1r2 groups, but now seen to correlate linearly with the associated circumferential stress differently in wild-type and Tgfbr1r2 disrupted aortas. Plotting circumferential stiffness versus medial and adventitial collagen revealed a negative correlation for the former and a positive correlation for the latter. That is, the circumferential material stiffness increased with increases in adventitial collagen that otherwise appeared to compensate, in part, for the loss of medial collagen due to the SMC-specific Tgfbr1r2 disruption – see the direct correlation of adventitial collagen fiber thickening in response to reductions in medial collagen in panel E. Finally, a greater rupture stress, that is, strength of the wall, correlated with higher SMαA and medial collagen in combination with a smaller quantity of adventitial collagen consisting largely of fibers with thinner, uniform diameter. Hence, even though the normal adventitia serves as a protective sheath, the strength of the normal media appears critical in protecting the wall from an initiating failure event and cell-mediated mechao-regulation of matrix is critical for maintaining structural integrity.
Figure 5.

Histo-mechanical correlations: (A) elastin undulation vs. cell density, (B) circumferential material stiffness vs. circumferential stress, (C) circumferential stiffness vs. medial collagen, (D) circumferential stiffness vs. adventitial collagen, (E) adventitial thick fibers vs. medial collagen, (F) failure stress vs. SMαA, (G) failure stress vs. medial collagen, and (H) failure stress vs. adventitial collagen.
Discussion
Although it is widely appreciated that biomechanical factors play decisive roles in the natural history of thoracic aortopathies (El-Hamamsy and Yacoub, 2009; Elefteriades, 2008; Humphrey and Tellides, 2019; Milewicz et al., 2008), there has been significantly more attention to the genetics, molecular and cell biology, and histological characteristics than to functional readouts such as mechanical properties of the aortic wall, especially for the diverse mouse models now available. Yet, ultimate failure resulting in lethality, via dissection and/or rupture, only occurs when mechanical stress exceeds strength. Our study was motivated, first, by the need to quantify failure properties during pressurization tests that preserve native geometry and axial loading and, second, to identify histopathological correlates, both in mice susceptible to aortic dissection.
The composition and layered organization of the aorta reflects well its primary functions: to store elastic energy during systole that can be used during diastole to augment flow while maintaining both appropriate compliance, to control the propagation of the pulse pressure wave including its reflection back toward the heart, and sufficient strength, to prevent catastrophic rupture. Thus, the normal aortic wall consists of a thick elastin-rich medial layer, with embedded smooth muscle cells of a matrix phenotype along with intralamellar collagen and glycosaminoglycans, plus a thin collagen-rich adventitial layer. The media bears most of the hemodynamically induced load under normal conditions, allowing the elastin to store and make available the elastic energy arising from wall deformation while the adventitia serves largely as a strong protective sheath that can be engaged if needed to resist supra-physiological loads (Bellini et al., 2014; Roccabianca et al., 2014). These unique properties emerge during aortic development and maturation. Normal elastic fibers are produced and cross-linked during the perinatal period and possess a remarkable half-life of decades under healthy conditions (Wagenseil and Mecham, 2009). We recently showed that elastic fiber mechanical functionality matures by postnatal day P21 in the mouse while that of fibrillar collagen matures later, between P42 and P98 for adventitial fibers (Murtada et al., 2021).
Knowledge of developmental time-courses is important when studying disease progression, especially for diseases having early onset, and when seeking appropriately timed pharmacological treatments (Cook et al., 2015). Such knowledge is similarly important when designing “time of disruption” in conditional knock-outs. We previously showed that vulnerability to aortic dissection in the tamoxifen-inducible Myh11-CreERT2.Tgfbr2f/f mouse increases significantly when the disruption is introduced earlier in life: ~75% dissect when disrupted at P21, ~50% when disrupted at P28, and less than 25% when disrupted at P42 or after (Li et al., 2014). Given that the elastic fibers appear to mature largely by P21, decreased dissection potential with increased age of induction after P21 likely results from progressively reduced fragility of extracellular matrix constituents other than elastin, with fibrillar collagens the primary constituents that endow the wall with stiffness and strength. Indeed, vascular Ehlers-Danlos pathology often manifests as arterial dissection; it stems from mutations in genes that encode collagen III and collagen V, among others (Bowen et al., 2020; Cooper et al., 2010; Wenstrup et al., 2006), noting that collagens III and V also contribute to proper collagen I fibrillogenesis (Birk et al., 1990; Liu et al., 1997). Consequently, although defects in, damage to, or degradation of elastin is often thought of first in many thoracic aortopathies, collagen defects are yet critical (Powell and Länne, 2007), even in syndromic diseases such as Marfan that result from mutations that directly affect elastic fibers (Lindeman et al., 2010). We induced the SMC-specific disruption of Tgfbr1r2 at P77, well after P56 when the wall appears to be mature biomechanically (Murtada et al., 2021).
Our biaxial findings revealed that elastic energy storage was reduced slightly, though not dramatically, following late postnatal SMC-specific disruption of Tgfbr1r2, consistent with the integrity of the elastic laminae by standard histology and with the disruption in SMC-specific TGFβ signaling occurring well after elastic fiber maturation. Thus, notwithstanding transmission electron microscopy revealing some micro-defects in the elastic laminae, it appears that the reduced energy storage was due largely to the presence of additional matrix constituents that prevented otherwise competent elastic fibers from deforming and storing elastic energy under physiologic loading; this was particularly the case in the non-dilated mutant aortas, which had significantly increased wall thickness. In contrast to the long half-life of vascular elastin, that of vascular collagen is normally on the order of 90 days, though it can drop to as low as 7 days in disease (Nissen et al., 1978). Indeed, prior studies in our lab have shown dramatic collagen turnover within 7 to 14 days in angiotensin II induced hypertension, with circumferential material stiffness typically restored toward normal within ~21 days (Bersi et al., 2017).
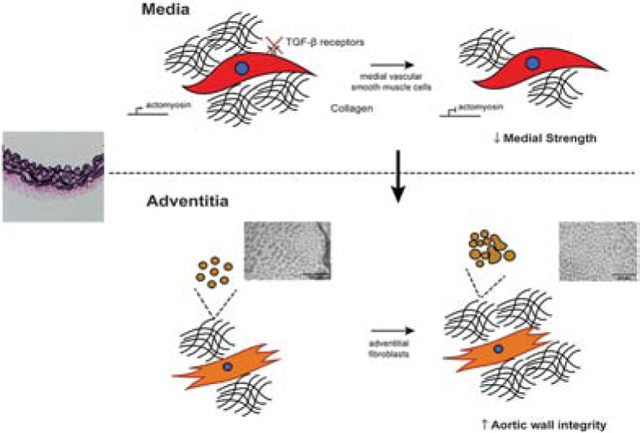
Consistent with findings in other mouse models of thoracic aortopathies and their propensity (Bellini et al., 2017a), there was a marked increase in circumferential material stiffness following late postnatal SMC-specific disruption of Tgfbr1r2 (for just 21 days), perhaps reflecting an altered mechano-regulation of medial matrix, with decreases in SMC-derived medial collagen compensated, in part, by increased adventitial collagen. Importantly, thoracic aortas from the current SMC-disrupted Tgfbr1r2 mice also had significantly reduced wall strength (i.e., lower failure stress) during pressurization tests, reductions that correlated with decreases in SMαA and medial collagen but also, perhaps surprisingly, with an increase in adventitial collagen that was characterized by an increased fraction of thick and decreased fraction of thin fibers. In a recent case study of a 73-year-old male patient with a chronic Type A dissected aorta, in vitro layer-specific mechanical testing of the descending thoracic aorta also revealed significant adventitial wall thickening as well as altered stress-strain responses compared to healthy descending thoracic aorta (Amabili et al., 2020). Although we did not assess cross-links, TGFβ signaling increases lysyl oxidase (Choudhary et al., 2009; Shanley et al., 1997), hence any newly deposited medial collagen may have not been well cross-linked. Regardless, transmission electron micrographs confirmed that the fibrillar collagen was not well formed due to heterogeneity in fiber diameters and cross-sectional shape; such heterogeneities inhibit effective linear and lateral fibrillogenesis (Wenstrup et al., 2006), likely leading to the reduced strength. Notwithstanding the decrease in failure stress, from ~3.67 to ~2.18 MPa within just three weeks, this reduced value is yet well above values of wall stress expected physiologically (<0.3 MPa). Our findings are thus consistent with a compensatory protection against catastrophic rupture despite increased dissection potential (noting the absence of death by aortic rupture in our colony), and similarly consistent with prior observations both that thoracic aortas from Myh11-CreERT2.Tgfbr2f/f mice often dissect but do not rupture in vivo over periods up to 26 weeks following tamoxifen-induction at 4 weeks of age (Li et al., 2014) and that these aortas experience medial delamination ex vivo but do not rupture under physiologic loading (Ferruzzi et al., 2016). It thus appears that, despite increased medial vulnerabilities to intramural delamination and dissection in this model of SMC-specific disruption of TGFβ signaling, rapid adventitial remodeling of collagen can be compensatory even late in maturity, thus protecting the wall from catastrophic failure though contributing to the compromised biomechanical functionality reflected by decreased energy storage and increased wall stiffness (Figure 6), which could have hemodynamic consequences (Humphrey and Tellides, 2019). Clearly, any pharmacological intervention should be mindful of this life-saving trade-off, striving to preserve the protective remodeling of adventitial collagen while targeting the pathology of the media.
Figure 6.

Schema illustrating biomechanical mechanisms associated with vessel remodeling and potential compensatory adaptations in response to late post-natal disruption of TGFβ signaling – the bottom row contrasts what appears to be a delicate balance between increased structural vulnerability with preserved physiological functionality and decreased mechanical functionality with preserved structural integrity. In either case, late compromised TGFβ signaling leads to adverse wall remodeling.
Importantly, the aortic phenotype observed herein shares features with that in mice having other mutations that affect intramural collagen. Biglycan is a small leucine-rich proteoglycan that contributes to collagen fibrillogenesis. Its deficiency results in considerable mortality in male mice by P63, with aortic rupture often evidenced by thoracic hemorrhage, though with intramural blood also contained between the media and adventitia (Heegaard et al., 2007). Uniaxial mechanical tests on non-ruptured segments reveal reduced strength, with a two-step failure process – medial, then adventitial. Transmission electron microscopy reveals increased heterogeneity in collagen fibers by diameter and cross-sectional shape, comparable to that in Figure 4 herein. Similar observations have been reported for Col5a1+/− mice, namely reduced aortic strength in uniaxial tests and presumably an increased collagen fiber heterogeneity within the aorta similar to that which is evident in the dermis (Wenstrup et al., 2006). Results are also similar for Col3a1+/− mice, noting that normal collagen III plays an important structural role in the media but also as a key participant in collagen I fibrillogenesis in the adventitia. Again, overall wall strength is reduced and the collagen fibers exhibit more heterogeneity in diameter and cross-sectional shape (Cooper et al., 2010; Liu et al., 1997). It was noted that the aortic “rupture crossed the media, which led to a blood-filled channel between the media and adventitia…The adventitia eventually ruptured elsewhere, and blood leaked into the peritoneal cavity” (Liu et al., 1997). Importantly, Tgfbr2 expression is lower, by ~18%, in these haploinsufficient Col3a1+/− mice. Combined with the present results, appropriate TGFβ signaling is a critical contributor to aortic homeostasis in maturity, not just aortic morphogenesis. With regard to the latter, the homozygous Col5a1−/− mutation is embryonic lethal (~E10.5) while the homozygous Col3a1−/− mutation is perinatally lethal (~P0-P2), not unlike the case of embryonic lethality in germline and cell-specific Tgfbr1 and Tgfbr2 mutations (Langlois et al., 2010; Renard et al., 2014). Because haploinsufficient Tgfbr1 mice do not present with cardiovascular malformations (Renard et al., 2014), conditional postnatal disruption of the type I and II TGFβ receptors provide considerable insight (Li et al., 2014; Hu et al., 2015; Tellides, 2017), though there had not been any measurement of aortic failure previously in these mice.
In conclusion, mouse models can provide new insight into the evolving biology and mechanics of thoracic aortopathies, but there is clearly a need for similar studies based on human data. Although there are numerous studies on the mechanics of the human aorta (cf. García-Herrera et al., 2012; Iliopoulos et al., 2009; Pasta et al., 2013; Tong et al., 2016), mechanobiological correlates remain wanting. Moreover, it is essential to distinguish cell- and tissue-level mechanisms that either promote or protect against aortic dissection and rupture. We focused herein on a mild aortic phenotype, with dilatation but not aneurysmal enlargement and without prior dissection or rupture, to focus on histo-mechanical vulnerabilities rather than consequences of prior failures. We also focused on a period after which the physiological properties and function of the aorta are mature, but just before finalization of the protective functionality of the adventitia (Murtada et al., 2021). We found that a short-term (3 weeks), late postnatal (after aortic maturation at P56) disruption of type I and II TGFβ receptors had modest effects on thoracic aorta resilience, but marked increases (up to 2.3-fold) in circumferential stiffness and reductions (by 43%) in wall strength. Increased stiffness is consistent with compromised mechano-sensing and/or mechano-regulation of matrix, as suggested earlier based on molecular findings (Li et al., 2014), but herein related for the first time to decreased medial collagen and an apparently compensatory increase in adventitial collagen characterized by an increase in thicker, less uniform diameter versus thinner, uniform diameter fibers. Reduced wall strength is a particularly important finding, also correlating largely with histological changes characterized by decreases in medial and increases in adventitial collagen. Taken together, appropriate smooth muscle-specific TGFβ signaling plays a critical role in preserving material stiffness and sufficient strength, largely via effects on fibrillar collagens, though supported by a protective adventitial sheath that can be engaged to help protect against catastrophic rupture.
Highlights.
TGFβ pathway disruption in vascular SMC drives medial degeneration reducing strength
Compensatory changes in adventitia augment wall strength and prevent aortic rupture
Adventitial collagen density and fiber thickness underlie altered matrix properties
Medial defects drive aortic dissection, adventitial defects lead to aortic rupture
Acknowledgments
This work was supported, in part, by grants from the US National Institutes of Health (P01 HL134605, U01 HL142518, and R01 HL146723 – GT and JDH) and from the Masason Foundation (YK). The authors thank the Yale Histology Service for their expert assistance.
APPENDIX.
Figure A.1.

Schematic diagram of the biaxial testing device. Computer-controlled actuators (1) mounted onto triaxial translational stages (2) control axial stretch (λz). The force transducer (3) is attached to thin glass micropipettes (4) which allow luminal pressurization (P) via perfusion of physiologic solutions; pressure transducers not shown, but mounted equidistantly from the center of the sample and connected in-line via stiff tubing to the pump that drives fluid in- and out-flow (not shown). The sample (6), cannulated onto the micropipettes, was tested herein in a bath filled with a Hank’s buffered saline solution (7) at room temperature. See Gleason et al. (2004) for further details.
Figure A.2.

Additional comparisons of mechanical and geometric metrics that define physiological mechanical function. (A-F) Similar to Figure 1, but with results for Tgfbr1r2 aortas pooled, to include non-dilated and dilated. (G) Circumferential structural stiffness (material stiffness multiplied by wall thickness) and (H) loaded thickness at 100 mmHg for different values of axial stretch.
Figure A.3.

Circumferential stress (A), stiffness (B), energy (C), and thickness (D) in relation to pressure: o – wild-type (WT, n=6) ■ – non-dilated Tgfbr1r2 (KO ND, n=4) ▲ – dilated Tgfbr1r2 (KO D, n=4).
Figure A.4.

Analysis of axial mechanical parameters associated with physiological function. (A) Cauchy stress-stretch data revealing material stiffness: o – wild-type (“WT”, n=6) ■ – non-dilated Tgfbr1r2 (“KO ND”, n=4), and ▲ – dilated Tgfbr1r2 (“KO D”, n=4). (B-C) Axial Cauchy stress and material stiffness compared by group.
Figure A.5.

Altered elastic energy storage function of aortas with disrupted TGFβ signaling (KO). (A) Representative iso-energy contours for stored energy for each group; the small solid dot shows values at 120 mmHg and group specific in vivo axial stretch. The iso-energetic contours represent values of 0.1, 1, 5, 10, 20, 40, 60, 100, 250, 500 kPa, in order of increasing size.
Figure A.6.

Additional analyses of mechanical and geometric metrics that define failure properties at supra-physiologic pressures. (A) Similar to Figure 2A, but plotted separately by group with inset showing close fit between measurement on biaxial device (curve) and burst device (circles) in the physiological range. Data shown for representative wild-type (WT), Tgfbr1r2 non-dilated (KO ND), Tgfbr1r2 dilated (KO D) samples. (B-F) Similar to Figure 2, but with pooled Tgfbr1r2 results. *p<0.05, **p<0.01
Figure A.7.

Additional analyses of immuno-histochemical findings. (A) Immunofluorescence images suggest absence of inflammatory cells (CD3: T-Cells, CD68: Macrophages). Quantification of immuno-histochemical findings for (B) SMαA, (C) cell density, (D) medial collagen, (E) adventitial thin collagen fibers, (F) SMMHC, (G) elastic laminae undulation, (H) adventitial collagen, and (I) adventitial thick collagen fibers. *p<0.05, **p<0.01, ***p<0.001
Figure A.8.
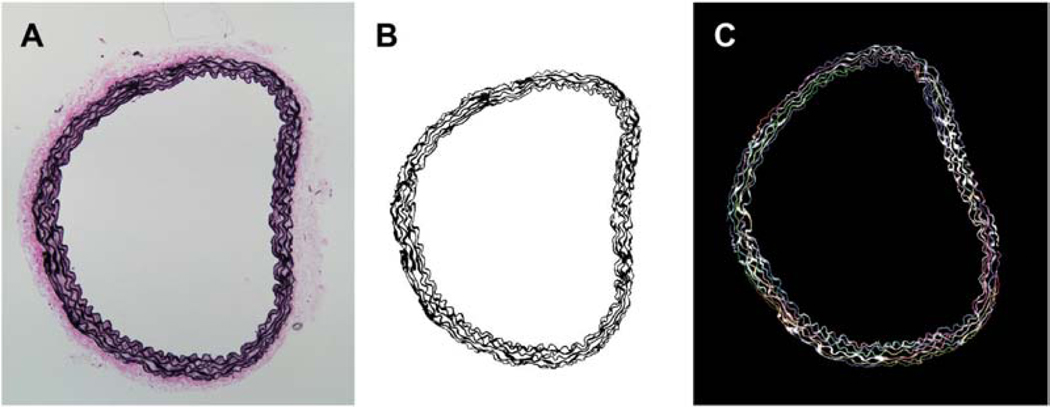
Analysis of elastin undulation using CT-Fire. (A) Original microscopy image stained with Verhoff-Van Gieson stain. (B) Extracted elastic fibers using a custom MATLAB code. (C) Fiber tracking using CT-Fire.
Table A.1.
Antibodies and associated dilutions used in the study. CD68 and CD3 stain macrophages and T-cells, respectively.
| Target | Dilution | Manufacturer |
|---|---|---|
| Collagen 1 | 1:150 | Abcam; ab34710 |
| Myosin heavy chain (SMMHC) | 1:250 | Abcam; ab53219 |
| Smooth muscle α-actin (SMαA) | 1:100 | Invitrogen; PA1–37024 |
| CD68 | 1:100 | NSJ Bioreagents; R30592 |
| CD3 | 1:150 | AbCam; ab166694 |
Table A.2.
Overview of the phenotypic classification of dilated or not dilated based on passive pressure-diameter data collected ex vivo from Tgfbr1r2 aortas.
| Phenotype categorization | Outer diameter at 120mmHg (μm) | % Difference from average WT diameter (1481.77μm) |
|---|---|---|
| Dilated | 1655.37 | 11.7% |
| Dilated | 1721.07 | 16.1% |
| Dilated | 1745.59 | 17.8% |
| Dilated | 1736.88 | 17.2% |
| Non-dilated | 1446.41 | −2.39% |
| Non-dilated | 1476.69 | −3.42% |
| Non-dilated | 1485.24 | 0.234% |
| Non-dilated | 1472.62 | −0.6% |
Table A.3.
Mechanical properties of aortas by group, with values of material stiffness and elastic energy storage calculated using best-fit constitutive parameters in Table A.4. WT – wild-type and KO is Tgfbr1r2 disrupted, with ND (non-dilated) and D (dilated).
| WT (n=6) | KO ND (n=4) | KO D (n=4) | ||
|---|---|---|---|---|
| Unloaded and Traction-Free | Outer Diameter (μm) | 940±20 | 950±21 | 994±22 |
| Wall Thickness (μm) | 112±3 | 136±4*** | 122±1* | |
| Loaded at 100mmHg | Outer Diameter (μm) | 1429±21 | 1445±7 | 1692±16*** |
| Wall Thickness (μm) | 44±2 | 63±4*** | 46±2 | |
| In vivo axial stretch | 1.52±0.03 | 1.27±0.05** | 1.42±0.03 | |
| Cauchy Stresses | Circumferential (kPa) | 204±8 | 143±9 | 235±12 |
| Axial (kPa) | 165±6 | 91±8 | 153±7 | |
| Material Stiffness | Circumferential (MPa) | 1.32±0.05 | 2.36±0.14*** | 3.01±0.11*** |
| Axial (MPa) | 1.35±0.12 | 1.08±0.12* | 1.52±0.08 | |
| Loaded at 120mmHg | Stored Energy (kPa) | 61.1±3.5 | 26.7±2.4*** | 53.2±3.1 |
| At Failure | Rupture Pressure (mmHg) | 943±39 | 1088±32** | 598±80*** |
| Wall Thickness (μm) | 33±2 | 54±3*** | 38±2 | |
| Failure Stress (MPa) | 3.67±0.34 | 2.25±0.19* | 2.10±0.25* |
p<0.05,
p<0.01,
p<0.001 with respect to WT using Student’s t-test.
Table A.4.
Best-fit values of the material parameters for the four-fiber family constitutive model, describing passive behaviors under physiologic conditions by group.
| c (kPa) | c11 (kPa) | c21 (−) | c12 (kPa) | c22 (−) | c13,4 (kPa) | c23,4 (−) | α(°) | |
|---|---|---|---|---|---|---|---|---|
| WT | 25.616 | 10.185 | 0.149 | 11.365 | 0.090 | 1.579 | 1.182 | 39.0 |
| KO ND | 22.129 | 4.692 | 3.139 | 0.156 | 2.370 | 0.737 | 2.977 | 37.8 |
| KO D | 30.666 | 1.262 | 3.842 | 0.040 | 1.291 | 1.308 | 2.113 | 37.1 |
Table A.5.
Comparison of additional geometric features; WT is wild-type control, KO is postnatal disruption of Tgfbr1r2, and ND and D denote not dilated and dilated, respectively.
| WT | KO ND | KO D | |
|---|---|---|---|
| Adventitia:Media Thickness Ratio | 0.273748 | 0.265018 | 0.217255 |
| Unloaded cross-sectional area (pm2) | 243184 | 299826* | 298947* |
| Unloaded medial area (pm2) | 174337 | 219190* | 221155* |
| Unloaded adventitial area (pm2) | 68846 | 80637* | 77791* |
p<0.05,
p<0.01,
p<0.001 using Student’s t-test.
Table A.6.
Summary of altered vessel properties in aortas with disrupted TGFβ signaling at physiological conditions and rupture conditions.
| WT | KO Non-Dilated | KO Dilated | ||
|---|---|---|---|---|
| Physiological conditions | Outer Diameter | -- | -- | ↑ |
| Thickness | -- | ↑ | -- | |
| Circ. Stress | -- | ↓ | -- | |
| Axial stretch | -- | ↓ | -- | |
| Circ. Stiffness | -- | ↑ | ↑↑ | |
| Energy storage | -- | ↓ | -- | |
| Structural Stiffness | -- | ↑ | ↑ | |
| At rupture | Outer diameter | -- | -- | ↑ |
| Thickness | -- | ↑ | -- | |
| Failure stress | -- | ↓ | ↓ | |
| Rupture Pressure | -- | -- | ↓ | |
Footnotes
Conflict of interest statement
None of the authors declare any conflict, financial or otherwise.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amabili M, Arena GO, Balasubramanian P, Breslavsky ID, Cartier R, Ferrari G, Holzapfel GA, Kassab A, Mongrain R, 2020. Biomechanical characterization of a chronic type a dissected human aorta. J Biomech. 110:109978. [DOI] [PubMed] [Google Scholar]
- Bellini C, Bersi MR, Caulk AW, Ferruzzi J, Milewicz DM, Ramirez F, Rifkin DB, Tellides G, Yanagisawa H, Humphrey JD, 2017a. Comparison of 10 murine models reveals a distinct biomechanical phenotype in thoracic aortic aneurysms. Journal of The Royal Society Interface 14, 20161036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellini C, Ferruzzi J, Roccabianca S, Di Martino ES, Humphrey JD, 2014. A microstructurally motivated model of arterial wall mechanics with mechanobiological implications. Annals of Biomedical Engineering 42, 488–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellini C, Kristofik NJ, Bersi MR, Kyriakides TR, Humphrey JD, 2017b. A hidden structural vulnerability in the thrombospondin-2 deficient aorta increases the propensity to intramural delamination. Journal of the mechanical behavior of biomedical materials 71, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersi MR, Bellini C, Humphrey JD, Avril S, 2019. Local variations in material and structural properties characterize murine thoracic aortic aneurysm mechanics. Biomechanics and Modeling in Mechanobiology 18, 203–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersi MR, Collins M, Wilson E, Humphrey JD, 2012. Disparate changes in the mechanical properties of murine carotid arteries and aorta in response to chronic infusion of angiotensin-II. Int J Adv Eng Sci Appl Math. 4, 228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersi MR, Khosravi R, Wujciak AJ, Harrison DG, Humphrey JD, 2017. Differential cell-matrix mechanoadaptations and inflammation drive regional propensities to aortic fibrosis, aneurysm or dissection in hypertension. Journal of The Royal Society Interface 14, 20170327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk DE, Fitch JM, Babiarz JP, Doane KJ, Linsenmayer TF, 1990. Collagen fibrillogenesis in vitro: interaction of types I and V collagen regulates fibril diameter. Journal of Cell Science 95, 649. [DOI] [PubMed] [Google Scholar]
- Bowen CJ, Calderón Giadrosic JF, Burger Z, Rykiel G, Davis EC, Helmers MR, Benke K, Gallo MacFarlane E, Dietz HC, 2020. Targetable cellular signaling events mediate vascular pathology in vascular Ehlers-Danlos syndrome. The Journal of Clinical Investigation 130, 686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredfeldt JS, Liu Y, Pehlke CA, Conklin MW, Szulczewski JM, Inman DR, Keely PJ, Nowak RD, Mackie TR, Eliceiri KW, 2014. Computational segmentation of collagen fibers from second-harmonic generation images of breast cancer. J Biomed Opt 19, 16007–16007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownstein AJ, Kostiuk V, Ziganshin BA, Zafar MA, Kuivaniemi H, Body SC, Bale AE, Elefteriades JA, 2018. Genes associated with thoracic aortic aneurysm and dissection: 2018 update and clinical implications. Aorta (Stamford) 6, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary B, Zhou J, Li P, Thomas S, Kaartinen V, Sucov HM, 2009. Absence of TGFβ signaling in embryonic vascular smooth muscle leads to reduced lysyl oxidase expression, impaired elastogenesis, and aneurysm. genesis 47, 115–121. [DOI] [PubMed] [Google Scholar]
- Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, Nelson CA, Cheng SH, Wentworth BM, Ramirez F, 2015. Dimorphic effects of transforming growth factor-β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan Syndrome. Arteriosclerosis, Thrombosis, and Vascular Biology 35, 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TK, Zhong Q, Krawczyk M, Tae HJ, Müller GA, Schubert R, Myers LA, Dietz HC, Talan MI, Briest W, 2010. The haploinsufficient Col3a1 mouse as a model for vascular Ehlers-Danlos syndrome. Vet Pathol 47, 1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hamamsy I, Yacoub MH, 2009. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nature Reviews Cardiology 6, 771–786. [DOI] [PubMed] [Google Scholar]
- Elefteriades JA, 2008. Thoracic aortic aneurysm: reading the enemy’s playbook. Current Problems in Cardiology 33, 203–277. [DOI] [PubMed] [Google Scholar]
- Ferruzzi J, Bersi MR, Humphrey JD, 2013. Biomechanical phenotyping of central arteries in health and disease: advantages of and methods for murine models. Annals of Biomedical Engineering 41, 1311–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferruzzi J, Murtada SI, Li G, Jiao Y, Uman S, Ting MY, Tellides G, Humphrey JD, 2016. Pharmacologically improved contractility protects against aortic dissection in mice with disrupted transforming growth factor-beta signaling despite compromised extracellular matrix properties. Arterioscler Thromb Vasc Biol 36, 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Herrera CM, Celentano DJ, Cruchaga MA, Rojo FJ, Atienza JM, Guinea GV, Goicolea JM, 2012. Mechanical characterisation of the human thoracic descending aorta: experiments and modelling. Computer Methods in Biomechanics and Biomedical Engineering 15, 185–193. [DOI] [PubMed] [Google Scholar]
- Gleason RG, Gray SP, Wilson E, Humphrey JD, 2004. A multiaxial computer-controlled organ culture and biomechanical device for mouse carotid arteries. Journal of Biomechanical Engineering 126, 787–795. [DOI] [PubMed] [Google Scholar]
- Heegaard AM, Corsi A, Danielsen CC, Nielsen KL, Jorgensen HL, Riminucci M, Young MF, Bianco P, 2007. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation 115, 2731–2738. [DOI] [PubMed] [Google Scholar]
- Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN, Yan J, Allen JK, Kang I, Wight TN, Fox K, Smith A, Enstrom R, Dichek DA, 2015. Postnatal deletion of the type ii transforming growth factor-β receptor in smooth muscle cells causes severe aortopathy in mice. Arteriosclerosis, Thrombosis, and Vascular Biology 35, 2647–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey JD, Schwartz MA, Tellides G, Milewicz DM, 2015. Role of Mechanotransduction in Vascular Biology. Circulation Research 116, 1448–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey JD, Tellides G, 2019. Central artery stiffness and thoracic aortopathy. American Journal of Physiology-Heart and Circulatory Physiology 316, H169–H182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos DC, Deveja RP, Kritharis EP, Perrea D, Sionis GD, Toutouzas K, Stefanadis C, Sokolis DP, 2009. Regional and directional variations in the mechanical properties of ascending thoracic aortic aneurysms. Med Eng Phys 31, 1–9. [DOI] [PubMed] [Google Scholar]
- Inamoto S, Kwartler CS, Lafont AL, Liang YY, Fadulu VT, Duraisamy S, Willing M, Estrera A, Safi H, Hannibal MC, Carey J, Wiktorowicz J, Tan FK, Feng X-H, Pannu H, Milewicz DM, 2010. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovascular Research 88, 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isselbacher EM, Lino Cardenas CL, Lindsay ME, 2016. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation 133, 2516–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeremy RW, Robertson E, Lu Y, Hambly BD, 2013. Perturbations of mechanotransduction and aneurysm formation in heritable aortopathies. International Journal of Cardiology 169, 7–16. [DOI] [PubMed] [Google Scholar]
- Karimi A, Milewicz DM, 2016. Structure of the elastin-contractile units in the thoracic aorta and how genes that cause thoracic aortic aneurysms and dissections disrupt this structure. Canadian Journal of Cardiology 32, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneva A, Zilberberg L, Rifkin DB, Humphrey JD, Bellini C, 2019. Absence of LTBBP-3 attenuates the aneurysmal phenotype but not spinal effects on the aorta in Marfan syndrome. Biomechanics and Modeling in Mechanobiology. 18, 261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois D, Hneino M, Bouazza L, Parlakian A, Sasaki T, Bricca G, Li JY, 2010. Conditional inactivation of TGF-β type II receptor in smooth muscle cells and epicardium causes lethal aortic and cardiac defects. Transgenic Research 19, 1069–1082. [DOI] [PubMed] [Google Scholar]
- Li W, Li Q, Jiao Y, Qion L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G, 2014. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest 124, 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman JHN, Ashcroft BA, Beenakker J-WM, van Es M, Koekkoek NBR, Prins FA, Tielemans JF, Abdul-Hussien H, Bank RA, Oosterkamp TH, 2010. Distinct defects in collagen microarchitecture underlie vessel-wall failure in advanced abdominal aneurysms and aneurysms in Marfan syndrome. Proceedings of the National Academy of Sciences 107, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu H, Byrne M, Krane S, Jaenisch R, 1997. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proceedings of the National Academy of Sciences 94, 1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milewicz DM, Guo D-C, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, Kwartler CS, Pannu H, 2008. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annual Review of Genomics and Human Genetics 9, 283–302. [DOI] [PubMed] [Google Scholar]
- Murtada S-I, Kawamura Y, Li G, Schwartz MA, Tellides G, Humphrey JD, 2020. Developmental origins of mechanical homeostasis in the aorta. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen R, Cardinale GJ, Udenfriend S, 1978. Increased turnover of arterial collagen in hypertensive rats. Proceedings of the National Academy of Sciences 75, 451–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasta S, Rinaudo A, Luca A, Pilato M, Scardulla C, Gleason TG, Vorp DA, 2013. Difference in hemodynamic and wall stress of ascending thoracic aortic aneurysms with bicuspid and tricuspid aortic valve. Journal of biomechanics 46, 1729–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinard A, Jones GT, Milewicz DM, 2019. Genetics of thoracic and abdominal aortic diseases. Circulation Research 124, 588–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JT, Länne T, 2007. Through thick and thin collagen fibrils, stress, and aortic rupture: another piece in the jigsaw. Circulation 115, 2687–2688. [DOI] [PubMed] [Google Scholar]
- Ramirez F, Caescu C, Wondimu E, Galatioto J, 2018. Marfan syndrome: A connective tissue disease at the crossroads of mechanotransduction, TGFβ signaling and cell stemness. Matrix Biology 71-72, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard M, Trachet B, Casteleyn C, Campens L, Cornillie P, Callewaert B, Deleye S, Vandeghinste B, van Heijningen PM, Dietz H, De Vos F, Essers J, Staelens S, Segers P, Loeys B, Coucke P, De Paepe A, De Backer J, 2014. Absence of cardiovascular manifestations in a haploinsufficient Tgfbr1 mouse model. PLoS One 9, e89749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roccabianca S, Bellini C, Humphrey JD, 2014. Computational modelling suggests good, bad and ugly roles of glycosaminoglycans in arterial wall mechanics and mechanobiology. Journal of The Royal Society Interface 11, 20140397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanley CJ, Gharaee-Kermani M, Sarkar R, Welling TH, Kriegel A, Ford JW, Stanley JC, Phan SH, 1997. Transforming growth factor-beta 1 increases lysyl oxidase enzyme activity and mRNA in rat aortic smooth muscle cells. J Vasc Surg 25, 446–452. [DOI] [PubMed] [Google Scholar]
- Tellides G, 2017. Further evidence supporting a protective role of transforming growth factor-β (tgfβ) in aortic aneurysm and dissection. Arteriosclerosis, Thrombosis, and Vascular Biology 37, 1983–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Cheng Y, Holzapfel GA, 2016. Mechanical assessment of arterial dissection in health and disease: Advancements and challenges. Journal of biomechanics 49, 2366–2373. [DOI] [PubMed] [Google Scholar]
- Wagenseil JE, Mecham RP, 2009. Vascular extracellular matrix and arterial mechanics. Physiological Reviews 89, 957–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenstrup RJ, Florer JB, Davidson JM, Phillips CL, Pfeiffer BJ, Menezes DW, Chervoneva I, Birk DE, 2006. Murine model of the Ehlers-Danlos syndrome. The Journal of Biological Chemistry 281, 12888–12895. [DOI] [PubMed] [Google Scholar]