

Abstract
Beckwith-Wiedemann syndrome (BWS) is the most common epigenetic overgrowth and cancer predisposition disorder. Due to both varying molecular defects involving chromosome 11p15 and tissue mosaicism, patients can present with a variety of clinical features, leading to the newly defined Beckwith-Wiedemann spectrum (BWSp). The BWSp can be further divided into three subsets of patients: those presenting with classic features, those presenting with isolated lateralized overgrowth (ILO) and those not fitting into the previous two categories, termed atypical BWSp. Previous reports of patients with BWS have focused on those with the more recognizable, classic features, and limited information is available on those who fit into the atypical and ILO categories. Here, we present the first cohort of patients recruited across the entire BWSp, describe clinical features and molecular diagnostic characteristics, and provide insight into practical diagnosis and management recommendations that we have gained from this cohort.
Keywords: Beckwith-Wiedemann spectrum, Beckwith-Wiedemann syndrome, cancer predisposition, lateralized overgrowth, macroglossia
1 |. INTRODUCTION
1.1 |. Overview
Beckwith-Wiedemann Syndrome (BWS, OMIM 130650) is the most common overgrowth and cancer predisposition disorder, affecting 1 in 10,340 patients (Mussa et al., 2013). First described in 1963 and 1964 by Drs. J. Bruce Beckwith (Beckwith, 1963) and Hans-Rudolf Wiedemann (Wiedemann, 1964), the disorder was initially characterized by macroglossia, omphalocele, and macrosomia. Since BWS was first described, it has been recognized that patients could be affected by a variety of clinical features, leading to designation of “complete” and “incomplete” forms of the syndrome (Gaston et al., 2001; Sotelo-Avila, Gonzalez-Crussi, & Fowler, 1980). In recognition of the variety of clinical features that can occur in patients with BWS, the syndrome was recently redefined as the Beckwith-Wiedemann Spectrum (BWSp) during an international BWS consensus meeting (Brioude et al., 2018).
1.2 |. Clinical features
The most common features of BWSp, designated as “cardinal features” (Figure 1a) by the BWS consensus, include macroglossia, omphalocele, lateralized overgrowth, multifocal, and/or bilateral Wilms tumor or nephroblastomatosis, and hyperinsulinism (Brioude et al., 2018). Additional “suggestive features” that can occur in BWSp include being large for gestational age (birth weight > 2 SDS above mean), facial nevus simplex, polyhydramnios, placentomegaly, ear creases/pits, transient hypoglycemia, nephromegaly, hepatomegaly, umbilical hernia, diastasis recti, and tumors including neuroblastoma, rhabdomyosarcoma, unilateral Wilms tumor, hepatoblastoma, adreno-cortical carcinoma, and pheochromocytoma (Brioude et al., 2018). The suggestive features can also occur in the general population and therefore have less weight in calculating the BWS clinical score (see below) (Figure 1b). Pathology findings that are considered cardinal features of BWSp include adrenal cortex cytomegaly, placental mesenchymal dysplasia, and pancreatic adenomatosis (Figure 1c).
FIGURE 1.
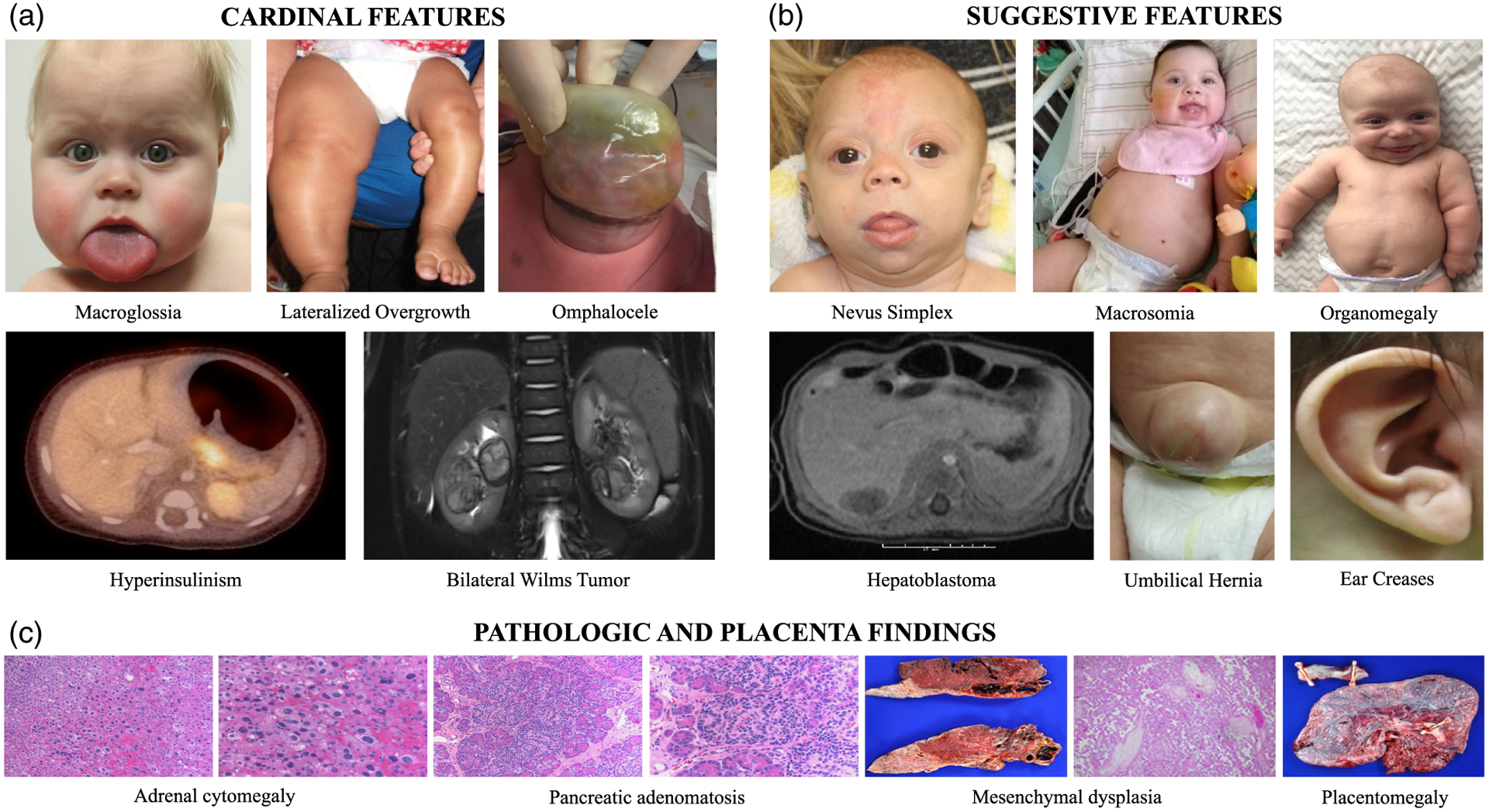
BWS diagnostic features. (a) Cardinal and (b) suggestive clinical features (not pictured: Hypoglycemia, ear pits, diastasis recti, polyhydramnios, and other embryonal tumors are also suggestive features of BWS). (c) Pathologic and placenta findings in patients with BWS. Pathologic findings including adrenal cytomegaly, pancreatic adenomatosis, and mesenchymal dysplasia are cardinal features and placentomegaly is a suggestive feature
1.3 |. Lateralized overgrowth
Lateralized overgrowth (LO; OMIM 235000), formerly referred to as hemihypertrophy or hemihyperplasia, is defined as asymmetric overgrowth of one or more regions of the body (Kalish et al., 2017a). The feature can be isolated (ILO) or accompany other major/minor findings suggestive of a syndrome. In patients with BWSp, the LO is characterized by increased muscle bulk. Skeletal asymmetry can also occur; however, skeletal asymmetry without associated muscle bulk differences is not typically seen in BWSp. Additional growth disorders that can cause asymmetry include neurofibromatosis (NF1; OMIM 162200), Proteus syndrome (OMIM 176920), PIK3CA-related segmental overgrowth (OMIM 612918), and Klippel-Trenaunay-Weber syndrome (OMIM 149000). These disorders are most often categorized by other abnormalities not typically seen in BWSp, such as skin pigmentation differences or asymmetry in adipose tissue rather than muscle (Hoyme et al., 1998; Mirzaa, Conway, Graham Jr., & Dobyns, 1993).
Asymmetry in muscle bulk differences can also occur in the under-growth disorder, Russell-Silver syndrome (RSS, OMIM 180860). Some molecular defects causing RSS occur in the same region of chromosome 11p15 as BWS; however, with the opposite methylation and gene dysregulation changes to those seen in BWS. A recent case series highlights patients referred for BWS/asymmetry and subsequently found to have RSS instead (Mackay et al., 2019).
1.4 |. Clinical diagnosis
The BWS consensus developed a clinical scoring system to aid in the categorization of BWSp (Brioude et al., 2018). Cardinal features were assigned two points each and suggestive features were assigned one point each. For a clinical diagnosis of BWSp, patients require a clinical score greater than or equal to four points. Patients with a clinical score greater than or equal to two points were recommended to warrant molecular analysis. The consensus also introduced three subtypes of BWS: patients presenting with “classic” features (i.e., macroglossia, omphalocele, overgrowth, etc.); patients presenting with isolated lateralized overgrowth (ILO); and patients with a BWS molecular defect that do not fit into the previous groups, termed “atypical” (Brioude et al., 2018). Patients with BWSp can fall anywhere on the “spectrum,” ranging from ILO to atypical to classic.
1.5 |. Mosaicism
The clinical spectrum in BWSp is likely due to the postzygotic nature of the epigenetic changes in most cases of BWSp. The earlier the epigenetic change occurs, the more cells with the change and the more parts of the body that demonstrate clinical features. The later the change, the fewer physical features observed. Mosaicism here is defined as a mixture of normal cells and cells with the genetic or epigenetic change causing BWS. This also occurs within multiple tissues in which normal and BWS cells can occur in differing ratios depending on the tissue tested in a given patient. We and others have demonstrated this mosaicism (Alders et al., 2014; Brioude et al., 2018; Eggermann et al., 2016; Kalish et al., 2016; Russo et al., 2016).
1.6 |. Molecular diagnosis and frequencies
Molecular testing in BWS includes methylation analysis at imprinting control regions 1 and 2 (IC1 and IC2) on chromosome 11p15, chromosome microarray analysis, and CDKN1C analysis (Brioude et al., 2018). In the case of negative blood analysis, testing of multiple affected tissues may increase the diagnostic yield due to mosaicism (Alders et al., 2014; Brioude et al., 2018; Eggermann et al., 2016; Kalish et al., 2016; MacFarland et al., 2018; Russo et al., 2016).
BWSp is caused by epigenetic or genetic defects on chromosome 11p15 (Figure 2). The most common cause is loss of methylation at KCNQ1OT1:TSS DMR (IC2 LOM), occurring in 50% of all patients (Brioude et al., 2018). Other causes include paternal uniparental isodisomy of chromosome 11 (pUPD11) (20% of patients), gain of methylation at H19/IGF2:IG DMR (IC1 GOM) (5% of patients), and mutations in the CDKN1C gene (5% of patients) (Brioude et al., 2018). More rarely, duplications, deletions, or chromosome translocations/inversions can occur which affect the 11p15 region (11p15 anomalies), affecting approximately 3–6% of patients (Brioude et al., 2018). Defects involving pUPD11 can occur beyond the 11p15 region and affect all chromosomes (genome-wide paternal uniparental isodisomy, GWpUPD) and it is estimated that up to 10% of patients with pUPD11 have GWpUPD (Brioude et al., 2018).
FIGURE 2.
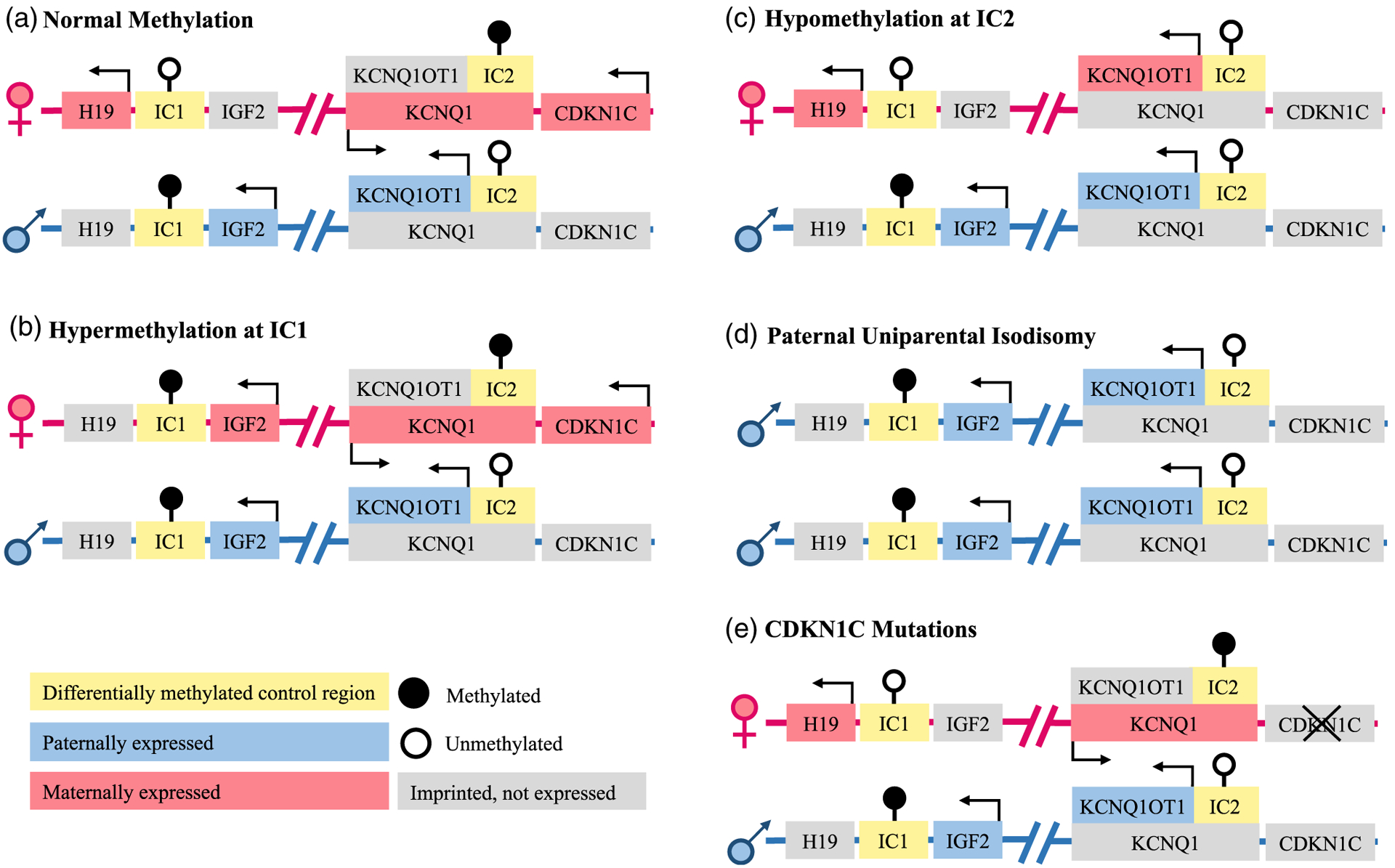
Schematic representations of the differential genetic and epigenetic causes of BWS on chromosome 11p15. (a) Normal methylation patterns demonstrate methylation at imprinting control region 1 (IC1) on the paternally inherited allele and methylation at imprinting control region 2 (IC2) on the maternally inherited allele. (b) Hypermethylation at IC1 results in the overexpression of IGF2. (c) Hypomethylation at IC2 results in the reduced expression of CDKN1C. (d) Paternal uniparental isodisomy results in the overexpression of IGF2 and the reduced expression of CDKN1C. (e) CDKN1C mutations of the maternal allele
1.7 |. Epigenotype-phenotype correlations
Patients with IC2 LOM have been reported to have higher frequencies of omphalocele, macroglossia, ear creases/pits, facial nevus simplex, and prematurity and lower frequencies of organomegaly/nephromegaly, large for gestational age, lateralized overgrowth, and tumors compared with patients with IC1 GOM and pUPD11 (Bliek et al., 2004; Brioude et al., 2013; Cooper et al., 2005; DeBaun et al., 2002; Engel et al., 2000; Gaston et al., 2001; Ibrahim et al., 2014; Maas et al., 2016; Mussa et al., 2016; Weksberg et al., 2001). Lateralized overgrowth appears most commonly in patients with pUPD11 (Brioude et al., 2013; Cooper et al., 2005; DeBaun et al., 2002; Ibrahim et al., 2014; Maas et al., 2016; Mussa et al., 2016). Other less frequent associations reported include diastasis recti and polyhydramnios in IC1 GOM patients (Mussa et al., 2016). Although some previous reports have indicated that hypoglycemia is more common in patients with pUPD11 (Brioude et al., 2013; DeBaun et al., 2002), others have not found an association with a specific molecular subtype (Cooper et al., 2005; Ibrahim et al., 2014; Maas et al., 2016; Mussa et al., 2016). In a previous case series of patients with severe hypoglycemia (hyperinsulinism) and Beckwith-Wiedemann syndrome, pUPD11 was the molecular defect identified in 26/28 of patients (Kalish et al., 2016). Patients with mutations in CDKN1C are less well characterized, but appear to be more affected by omphalocele and preterm birth (Mussa et al., 2016).
Here, we present a previously unreported cohort that represents the full BWSp and evaluate the BWS consensus guidelines in regards to clinical and molecular diagnosis of this cohort and provide some amendments to the consensus to aid in the practical application of those guidelines.
2 |. METHODS
A growth and genetic/epigenetic disorder registry was created at Children’s Hospital of Philadelphia (CHOP) in 2014 to systematically collect clinical information and samples from patients with a goal to understand more about these rare disorders. Consent was obtained from all participants/guardians and the study is approved by CHOP’s Institutional Review Board (IRB 13–010658). At time of enrollment in the study, patients are assigned a unique identification number and their associated diagnosis/diagnoses are recorded. Medical records are reviewed by the study team and information about prenatal, birth, and surgical histories in addition to hospital discharge summaries, specialist notes, and genetic testing results are collected and entered into the database by their unique ID numbers. Efforts are made to collect medical records from all institutions related to the patient. In the case of incomplete data, interviews with the patient, family members, and other physicians involved in their care are conducted as available. Records are reviewed at regular intervals and any updates are entered into the system.
2.1 |. Patient selection
The registry was queried to identify patients that fit criteria for the Beckwith-Wiedemann spectrum (BWSp). Patients enrolled and processed prior to March 2019 were eligible for the search. Search terms included cardinal features of BWSp: macroglossia, omphalocele, lateralized overgrowth/asymmetry/hemihypertrophy, hyperinsulinism, and Wilms tumor. Additional search terms included Beckwith-Wiedemann syndrome and overgrowth disorder.
The initial search yielded 462 potentially eligible patients. Due to insufficient data, 30 patients were initially excluded. The remaining 432 patients were then screened in a two-step process. Genetic testing results were reviewed and 25 patients were excluded due to a molecularly confirmed alternative diagnosis. The remaining 407 patients were then assessed for eligibility for a BWSp diagnosis: 33 were excluded due to the presence of clinical features suggestive of an alternative diagnosis and 17 were excluded due to the presence of an isolated feature (i.e., isolated omphalocele or hyperinsulinism without other features to meet criteria for BWSp). The less affected among monozygotic twins with concordant BWS molecular testing and discordant features were also excluded (n = 13), as the less affected twin of monozygotic pairs often presents with little to no BWS features (Cohen et al., 2019).
2.2 |. Data collection
Medical records were reviewed and data regarding demographic information, prenatal and birth history, BWS clinical features, and molecular testing results were abstracted. In some cases, interviews with patients and/or family members or outside hospital physicians were conducted to gather additional data. The BWSp clinical score was calculated according to the consensus criteria (Brioude et al., 2018): 2 points were assigned for the presence of each cardinal feature and 1 point was assigned for the presence of each suggestive feature. As pathology testing may not be routinely performed in patients, we excluded pathology findings from the clinical score calculations.
2.3 |. Clinical feature definitions
Lateralized overgrowth was defined as visible/palpable and/or measureable 5% difference in asymmetry in one or more limbs presumed to be related to muscle bulk differences. The presence of hypoglycemia overall was recorded and separated into two forms: transient hypoglycemia defined as low blood glucose levels lasting <1 week, and hyperinsulinism defined as prolonged hypoglycemia (lasting >1 week) requiring escalated treatment. The presence of tumor(s) overall was recorded and separated into two groups based on the consensus criteria: multifocal and/or bilateral Wilms tumor (WT) or nephroblastomatosis (NB), and typical BWSp tumor (unilateral WT, hepatoblastoma, neuroblastoma, pheochromocytoma, etc). Pathology results on the tumors were reviewed for classification when available.
2.4 |. Diagnostic indication definitions
Patients were assigned to five groups based on the feature(s) that led to the clinical/molecular diagnosis of BWSp: Patients presenting with a constellation of BWS features (BWS features group); those presenting with asymmetry as their main indication (asymmetry group); those first presenting with hyperinsulinism (hyperinsulinism group) or tumor (tumor group) before BWSp was suspected; and those who were not suspected to have BWS but had genetic testing for another reason and had results consistent with BWS (incidental group). Patients were also grouped according to the age at diagnosis: prenatally confirmed; neonatal (<30 days); and postneonatal, which was assigned when the patient was diagnosed past 30 days old. The age in months at diagnosis in the postneonatal group was recorded when available.
2.5 |. BWSp group definitions
The BWS consensus established three groups of patients within the spectrum: classic, atypical, and isolated lateralized overgrowth (ILO). Although these groups were established, no set criteria for designation to these groups were defined. For the purposes of this study, we arbitrarily defined these groups for analysis. In an effort to properly categorize patients in their respective groups prior to presenting with a tumor (as ideally patients would be recognized clinically prior to the development of a tumor), tumors were excluded from the clinical score when classifying. Patients presenting with hyperinsulinism or tumors automatically were grouped into atypical, as many of these patients had subtle lateralized overgrowth and the combination of these features may artificially inflate a score and cause a patient to fit “classic” criteria when they are not initially presenting as “classic.”
Group definitions were as follows:
2.5.1 |. Classic BWSp
Patients with a clinical score ≥ 6 with two or more cardinal features (except if hyperinsulinism and lateralized overgrowth were the two features).
2.5.2 |. Atypical BWSp
Patients with a clinical score < 6 with at least one cardinal feature; or Patients with a clinical score ≥ 6 with hyperinsulinism and lateralized overgrowth as the two cardinal features; or
Patients diagnosed with BWS after presenting with hyperinsulinism; or
Patients diagnosed with BWS after presenting with tumor (except if LO was the only cardinal feature).
2.5.3 |. Isolated lateralized overgrowth
Patients with a clinical score < 4 with LO as the only cardinal feature.
2.6 |. Statistical analysis
Data were analyzed using SPSS Statistics (version 25.0). Continuous variables were summarized by means/SDs and nominal/categorical variables were summarized by frequencies. Differences between the groups were evaluated by independent t tests or one-way ANOVA for continuous variables and Fisher’s Exact and Pearson chi square analyses for nominal/categorical variables as applicable. For groups with significant differences overall, Tukey post hoc analysis was performed for continuous variables and column proportion testing (z test) with adjusted p values using the Bonferroni method was used to evaluate for differences within groups that were found to be overall significantly different by Pearson chi square. Significance was set at p < .05.
3 |. RESULTS
A total of 344 patients were included in the final cohort. Characteristics of the cohort are summarized in Table 1. There were slightly more females than males, more than half were Caucasian, and the majority lived in the United States. Patients were followed from birth for an average of 70.0 ± 111.22 months.
TABLE 1.
Characteristics of the Beckwith-Wiedemann spectrum (BWSp) cohort
| Characteristic | Total, N (%) |
|---|---|
| Sex, male: female | 154:190/344 (44.8%:55.2%) |
| From the United States | 302/344 (87.8%) |
| Race/ethnicity group | |
| Caucasian only | 227/344 (66.0%) |
| Mixed race/ethnicity | 49/344 (14.2%) |
| Non-Caucasian or Hispanic only | 49/344 (14.2%) |
| Unknown/not reported | 19/344 (5.5%) |
| BWSp subgroup | |
| Classic | 207/322 (64.3%) |
| Atypical | 55/322 (17.1%) |
| ILOa | 60/322 (18.6%) |
| Diagnosis group | |
| IC1 GOMb | 32/344 (9.3%) |
| IC2 LOMc | 118/344 (34.3%) |
| pUPD11d | 78/344 (22.7%) |
| CDKN1C mutation | 7/344 (2.0%) |
| GWpUPDe | 8/344 (2.3%) |
| 11p15 anomaly | 19/344 (5.5%) |
| Negative testing | 57/344 (16.6%) |
| No testing | 19/344 (5.5%) |
| Unknown | 6/344 (1.7%) |
ILO: isolated lateralized overgrowth.
IC1 GOM: gain of methylation at H19/IGF2:IG DMR.
IC2 LOM: loss of methylation at KCNQ1OT1:TSS DMR.
pUPD11: paternal uniparental isodisomy of chromosome 11p15.
GWpUPD: genome wide paternal uniparental isodisomy.
3.1 |. Clinical features
The most common clinical feature present in the group was lateralized overgrowth (Table 2). Differences in degree of severity of LO were observed between patients (Figure 3), as well as differences in degree over time were observed for some (Figure 4). Macroglossia was also common with approximately 2/3 of the cohort having some degree of the feature. Among those with macroglossia, about half required a tongue reduction. Slightly more than half of patients had some degree of hypoglycemia and ear creases/pits. Slightly less than half of patients were large for gestational age at birth and had facial nevus simplex. Overall tumor incidence was 14.5%.
TABLE 2.
Clinical features of the Beckwith-Wiedemann spectrum (BWSp) cohort
| Feature | Total n (%) or mean (SD) |
|---|---|
| Pregnancy/birth | |
| Assisted reproductive technologya | |
| None | 257/309 (83.2%) |
| IUIb and/or hormone stimulation | 6/309 (1.9%) |
| IVFc and/or ICSId | 46/309 (14.9%) |
| Multiple gestation | 44/315 (14.0%) |
| Polyhydramnios | 79/301 (26.2%) |
| Placentomegaly | 36/299 (12.0%) |
| Gestational age | 35.81 wks (3.96) |
| Premature (<37 weeks) | 139/319 (43.6%) |
| LGAe | 200/320 (62.5%) |
| Typical features | |
| Macroglossia | 226/340 (66.5%) |
| Tongue reduction | 96/205 (46.8%) |
| Facial nevus simplex | 138/313 (44.1%) |
| Ear creases/pits | 180/309 (58.3%) |
| Abdominal wall defect | 211/320 (65.9%) |
| Omphalocele | 65/332 (19.6%) |
| Umbilical hernia | 108/318 (34.0%) |
| Diastasis recti | 53/312 (17.0%) |
| Lateralized overgrowth | 244/329 (74.2%) |
| Hypoglycemia | 171/321 (53.3%) |
| Transient | 99/321 (30.8%) |
| Hyperinsulinism | 72/329 (21.9%) |
| Pancreatectomy | 26/67 (38.8%) |
| Organomegaly | 118/316 (37.3%) |
| Hepatomegaly | 64/302 (21.2%) |
| Nephromegaly | 63/305 (20.7%) |
| Splenomegaly | 37/294 (12.6%) |
| Tumor | 48/332 (14.5%) |
| Bilateral/multifocal WT/NBf | 16/331 (4.8%) |
| Typical BWSp tumor | 32/344 (9.6%) |
| Other features | |
| Cleft palate | 7/304 (2.3%) |
| Undescended testes | 37/123 (30.1%) |
| Family history of BWS | 8/344 (2.3%) |
ART: assisted reproductive technology.
IUI: intrauterine insemination.
IVF: in vitro fertilization.
ICSI: intracytoplasmic sperm injection.
LGA: large for gestational age (>2 SDs above the mean).
WT/NB: Wilms tumor or nephroblastomatosis.
FIGURE 3.
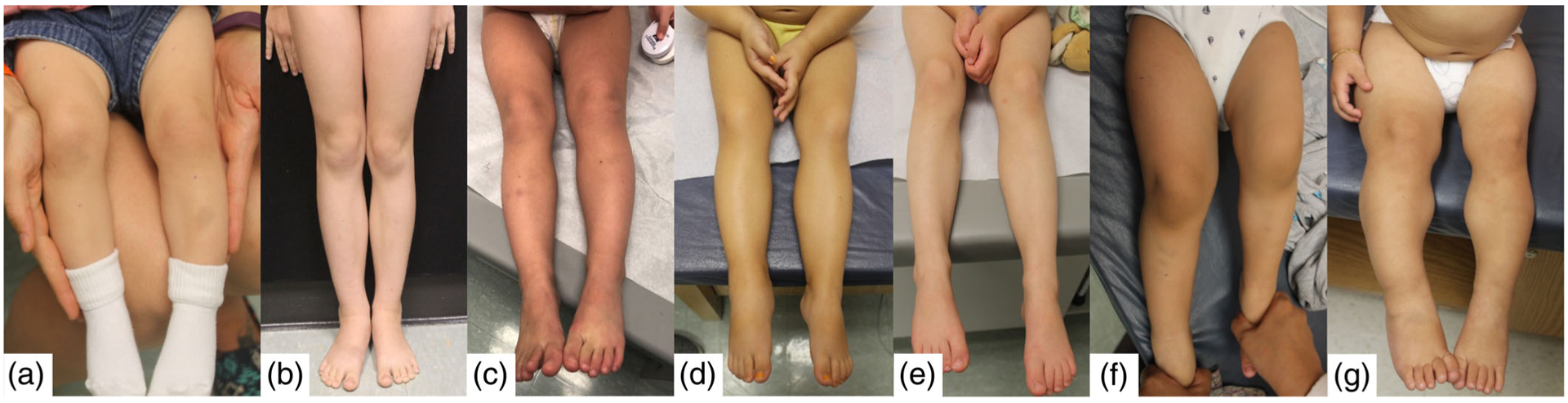
Spectrum of lateralized overgrowth in patients with BWS or isolated lateralized overgrowth. Abbreviations: IC1 GOM = imprinting center 1 gain of methylation; pUPD11 = paternal uniparental isodisomy of chromosome 11; and ILO = isolated lateralized overgrowth. (a) Clinical BWS (no molecular diagnosis), 11 months old; (b) IC1, 8 years old (c) pUPD11, 32 months old (d) ILO, 32 months old (e) pUPD11, 4 years old; (f) pUPD11, 22 months old; and (g) ILO, 14 months old
FIGURE 4.
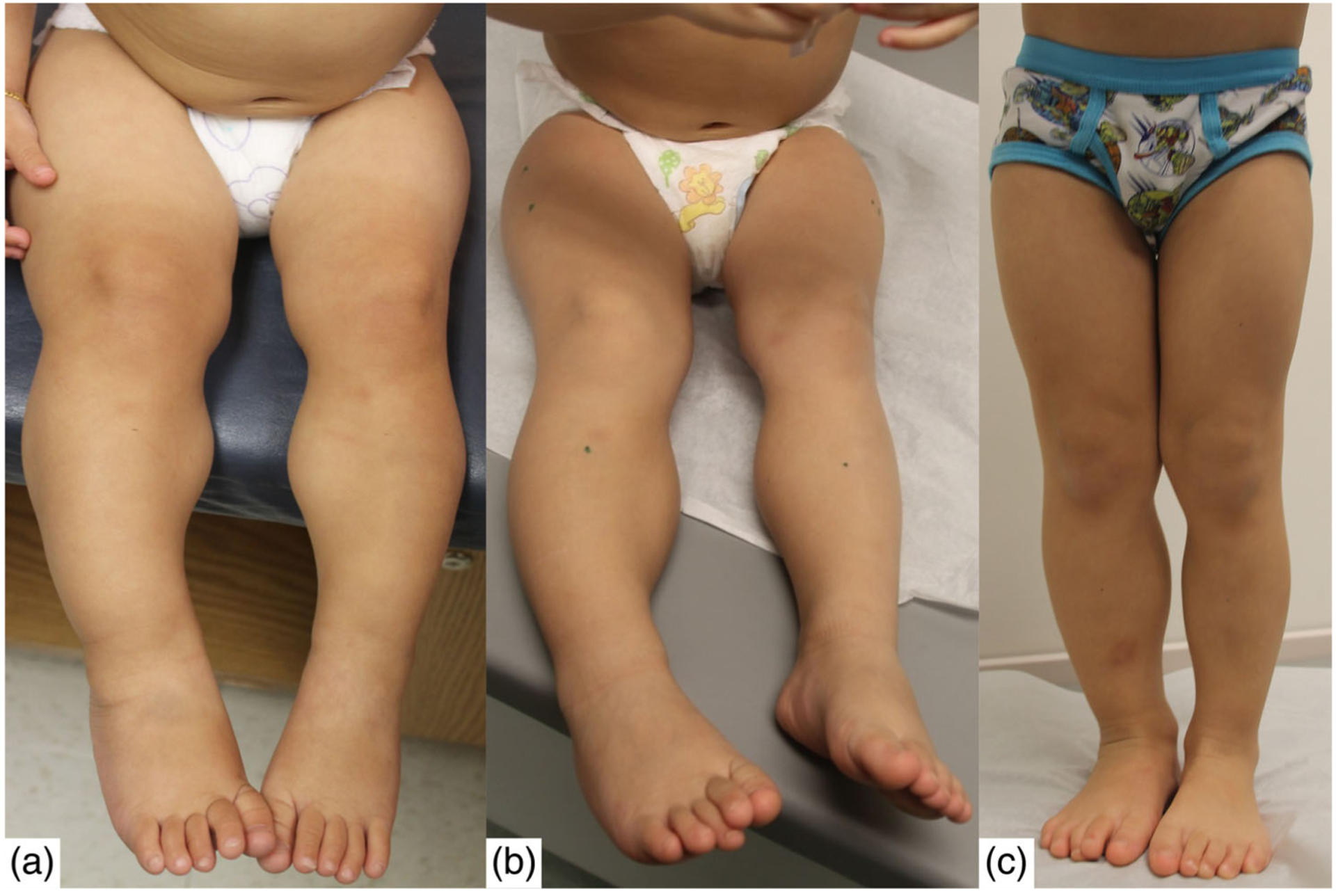
Isolated lateralized overgrowth in one patient overtime. (a) 14 months old, (b) 32 months old, and (c) 45 months old
3.2 |. Molecular testing
The recommended molecular testing for BWS includes methylation testing at IC1 and IC2 followed by copy number testing using a variety of techniques (Brioude et al., 2018). Single nucleotide polymorphism (SNP) array analysis is also recommended to distinguish pUPD11 from GWpUPD. The molecular breakdown of this BWSp cohort is demonstrated through the molecular diagnostic algorithm proposed in the consensus (Figure 5). The most common molecular subtype in the cohort was IC2 LOM. The majority of patients were molecularly confirmed through blood analysis (n = 224) and an additional 38 patients were molecularly confirmed through tissue testing (Figure 6). Among patients molecularly tested, tissue analysis increased the diagnostic yield from 70.2% (224/319) to 82.1% (262/319). The most common molecular subtype among those confirmed through tissue testing was pUPD11. Among patients with pUPD11, 9.3% were found to have GWpUPD.
FIGURE 5.

Testing algorithm and the molecular breakdown of the BWSp cohort based on testing. Testing at H19/IGF2:IG DMR and KCNQ1OT1: TSS DMR by methylation, copy number variant (CNV), and single nucleotide polymorphism (SNP) array analysis
FIGURE 6.

Patients with BWS with (1) IC1 gain of methylation, (2) IC2 loss of methylation, (3) paternal uniparental isodisomy (pUPD11), (4) genome-wide paternal uniparental isodisomy (GWpUPD), (5) CDKN1C mutations, (6) an 11p15 anomaly (deletions or duplications), (7) an unknown subtype, (8) no genetic testing, and (9) no defect identified. (a) Patients with BWS reported by Brioude et al., 2018. (b) Patients with BWS in the BWSp cohort at the Children’s Hospital of Philadelphia. (c) Patients in the cohort with a molecularly confirmed diagnosis. (d) Patients in the cohort with positive testing in blood. (e) Patients in the cohort with positive testing in tissue
3.3 |. Diagnostic characteristics of group
Among the 321 patients with information regarding reason for referral, the majority were referred due to the presence of BWS features (64.2%) or asymmetry (17.8%). Other indications for diagnosis included presenting with hyperinsulinism (10.3%) or tumor (5.9%) and a few patients were incidentally diagnosed with BWS by genome-wide microarray analysis performed for another reason (1.9%). Age at diagnosis was available for 308 patients and were divided into three groups: prenatally confirmed (7.5%), neonatal (<30 days; 47.4%), and postneonatal (45.1%). The average age at diagnosis for the postneonatal group was 14.06 ± 23.51 months.
3.4 |. BWSp clinical score
The average clinical score of the group was 6.72 ± 2.58 points. The average number of cardinal features was 1.85 ± 0.81 points and the average number of suggestive features was 3.04 ± 1.70 points. A wide variation in BWSp clinical scores among the molecular subgroups was observed.
3.5 |. EpiGenotype/phenotype correlations
3.5.1 |. Clinical features
Clinical data was available for 228 patients with IC2 LOM, IC1 GOM, and pUPD11 molecular defects (Table 3). Patients with IC2 LOM were found to have significantly increased incidence of macroglossia, abdominal wall defects overall, omphalocele, facial nevus simplex, and ear creases/pits compared with patients with IC1 GOM and pUPD11 subtypes. Polyhydramnios, prematurity (<37 weeks gestation), and undescended testes were also more common in patients with IC2 LOM compared with patients with pUPD11, but no significant difference was observed between IC2 LOM and IC1 GOM for these characteristics. Patients with pUPD11 were found to have a significantly increased incidence of lateralized overgrowth, hyperinsulinism, and pancreatectomy and patients with IC1 GOM were found to have a significantly increased incidence of diastasis recti compared with the other subtypes. Tumor incidence was significantly increased in patients with IC1 GOM and pUPD11 compared with patients with IC2 LOM. A significant increase in multifocal/bilateral Wilms tumor was observed in patients with IC1 GOM compared with pUPD11 patients; however, no difference was observed for incidence of other typical BWSp tumors.
TABLE 3.
EpiGenotype/phenotype correlations
| Feature | Totala n (%) or mean (SD) | IC1 GOMb | IC2 LOMc | pUPD11d | p valuee |
|---|---|---|---|---|---|
| Sex, male | 99/228 (43.4%) | 12/32 (37.5%) | 54/118 (45.8%) | 33/78 (42.3%) | .684 |
| Diagnosis source, blood | 192/228 (84.2%) | 25/32 (78.1%) | 116/118 (98.3%) | 51/78 (65.4%) | <.001*** |
| Clinical score | 7.14 (2.53) | 6.09 (2.60) | 7.74 (2.22) | 6.71 (2.71) | .001** |
| Suggestive features | 3.25 (1.65) | 2.78 (1.70) | 3.59 (1.42) | 2.93 (1.84) | .006** |
| Cardinal features | 1.95 (0.82) | 1.66 (0.83) | 2.07 (0.81) | 1.89 (0.81) | .031* |
| Diagnosis indication | <.001*** | ||||
| BWS features | 151/213 (70.9%) | 15/29 (51.7%) | 106/113 (93.8%) | 30/71 (42.3%) | |
| Asymmetry | 21/213 (9.9%) | 4/29 (13.8%) | 3/113 (2.7%) | 14/71 (19.7%) | |
| Hyperinsulinism | 19/213 (8.9%) | 0/29 (0%) | 2/113 (1.8%) | 17/71 (23.9%) | |
| Tumor | 19/213 (8.9%) | 10/29 (34.5%) | 2/113 (1.8%) | 7/71 (9.9%) | |
| Incidental | 3/213 (1.4%) | 0/29 (0%) | 0/113 (0%) | 3/71 (4.2%) | |
| Diagnosis age group | .003** | ||||
| Prenatal (confirmed) | 15/209 (7.2%) | 0/29 (0%) | 14/109 (12.8%) | 1/71 (1.4%) | |
| Neonatal (<30 days) | 108/209 (51.7%) | 12/29 (41.1%) | 60/109 (55.0%) | 36/71 (50.7%) | |
| Postneonatal | 86/209 (41.1%) | 17/29 (58.6%) | 35/109 (32.1%) | 34/71 (47.9%) | |
| Postneonatal age at diagnosis (m) | 16.36 (27.60) | 27.80 (28.92) | 12.78 (24.60) | 14.81 (29.39) | .198 |
| Race/ethnicity group | .125 | ||||
| Caucasian | 149/221 (67.4%) | 22/31 (71.0%) | 80/112 (71.4%) | 47/78 (60.3%) | |
| Mixed | 39/221 (17.6%) | 7/31 (22.6%) | 19/112 (17.0%) | 13/78 (16.7%) | |
| Non-Caucasian | 33/221 (14.9%) | 2/31 (6.5%) | 13/112 (11.6%) | 18/78 (23.1%) | |
| Pregnancy/birth | |||||
| ARTf use | <.001*** | ||||
| Natural | 168/208 (80.8%) | 27/30 (90.9%) | 75/109 (68.8%) | 66/69 (95.7%) | |
| IUIg and/or hormone stim | 2/208 (1.0%) | 2/30 (6.7%) | 0/109 (0%) | 0/69 (0%) | |
| IVFh and/or ICSIi | 38/208 (18.3%) | 1/30 (3.3%) | 34/109 (31.2%) | 3/69 (4.3%) | |
| Multiple gestation | 31/213 (14.6%) | 3/30 (10.0%) | 22/111 (19.8%) | 6/72 (8.3%) | .074 |
| Polyhydramnios | 52/196 (26.5%) | 6/29 (20.7%) | 34/98 (34.7%) | 12/69 (17.4%) | .033* |
| Placentomegaly | 27/194 (13.9%) | 3/29 (10.3%) | 19/97 (19.6%) | 5/68 (7.4%) | .069 |
| Gestational age | 36.01 (3.74) | 35.92 (3.73) | 35.50 (3.76) | 36.81 (3.63) | .079 |
| Premature (<37 weeks) | 88/213 (41.3%) | 12/30 (40.0%) | 55/111 (49.5%) | 21/72 (29.2%) | .023* |
| LGAj | 135/212 (63.7%) | 18/28 (64.3%) | 70/112 (62.5%) | 47/72 (65.3%) | .927 |
| Typical features | |||||
| Macroglossia | 162/226 (71.7%) | 17/32 (53.1%) | 112/118 (94.9%) | 33/76 (43.4%) | <.001*** |
| Tongue reduction | 78/145 (53.8%) | 8/16 (50.0%) | 57/101 (56.4%) | 13/28 (46.4%) | .610 |
| Facial nevus simplex | 103/205 (50.0%) | 6/27 (22.2%) | 77/108 (71.3%) | 20/71 (28.2%) | <.001*** |
| Ear creases/pits | 125/201 (62.2%) | 9/27 (33.3%) | 81/104 (77.9%) | 35/70 (50.0%) | <.001*** |
| Abdominal Wall defect | 149/213 (70.0%) | 17/30 (56.7%) | 92/110 (83.6%) | 40/73 (54.8%) | <.001*** |
| Omphalocele | 52/220 (23.6%) | 0/32 (0%) | 47/115 (40.9%) | 5/73 (6.8%) | <.001*** |
| Umbilical hernia | 73/211 (34.6%) | 8/30 (26.7%) | 40/109 (36.7%) | 25/72 (34.7%) | .593 |
| Diastasis recti | 35/205 (17.1%) | 12/27 (44.4%) | 9/107 (8.4%) | 14/71 (19.7%) | <.001*** |
| Lateralized overgrowth | 158/215 (73.5%) | 23/30 (76.7%) | 64/110 (58.2%) | 71/75 (94.7%) | <.001*** |
| Hypoglycemia | 129/212 (60.8%) | 14/31 (45.2%) | 63/109 (57.8%) | 52/72 (72.2%) | .023* |
| Transient | 81/212 (38.2%) | 11/31 (35.5%) | 49/109 (45.0%) | 21/72 (29.2%) | .096 |
| Hyperinsulinism | 48/218 (22.0%) | 3/32 (9.4%) | 14/112 (12.5%) | 31/74 (41.9%) | <.001*** |
| Pancreatectomy | 22/47 (46.8%) | 0/3 (0%) | 2/14 (14.3%) | 20/30 (66.7%) | .001** |
| Organomegaly | 80/206 (38.8%) | 13/29 (44.8%) | 37/106 (34.9%) | 30/71 (42.3%) | .478 |
| Hepatomegaly | 42/197 (21.3%) | 9/28 (32.1%) | 15/100 (15.0%) | 18/69 (26.1%) | .072 |
| Nephromegaly | 42/198 (21.2%) | 8/28 (28.6%) | 17/100 (17.0%) | 17/70 (24.3%) | .307 |
| Splenomegaly | 29/193 (15.0%) | 5/28 (17.9%) | 12/97 (12.4%) | 12/68 (17.6%) | .584 |
| Tumor | 43/219 (19.6%) | 16/31 (51.6%) | 5/114 (4.4%) | 22/74 (29.7%) | <.001*** |
| Bilateral/multifocal WT/NBk | 14/219 (6.4%) | 10/31 (32.3%) | 1/114 (0.9%) | 3/74 (4.1%) | <.001*** |
| Typical BWSp | 29/220 (13.2%) | 6/31 (19.4%) | 4/115 (3.5%) | 19/74 (25.7%) | <.001*** |
| Other features | |||||
| Cleft palate | 3/202 (1.5%) | 0/28 (0%) | 2/104 (1.9%) | 1/70 (1.4%) | .756 |
| Undescended testes | 29/81 (35.8%) | 3/9 (33.3%) | 24/46 (52.2%) | 2/26 (7.7%) | .001s |
Total refers to totals of the three subgroups compared (IC1 GOM, IC2 LOM, pUPD11).
IC1 GOM: gain of methylation at H19/IGF2:IG DMR.
IC2 LOM: loss of methylation at KCNQ1OT1:TSS DMR.
pUPD11: paternal uniparental isodisomy of chromosome 11p15.
p values refer to the frequency of each feature between the three subgroups (IC1 GOM, IC2 LOM, pUPD11), so all subgroups are compared at the same time.
ART: assisted reproductive technology.
IUI: intrauterine insemination.
IVF: in vitro fertilization.
ICSI: intracytoplasmic sperm injection.
LGA: large for gestational age (>2 SDs above the mean).
WT/NB: Wilms tumor or nephroblastomatosis.
Significant at p < .05;
Significant at p < .01;
Significant at p < .001.
3.5.2 |. Diagnostic characteristics
Patients with IC2 LOM were more likely to be diagnosed prenatally or in the neonatal period while patients with IC1 GOM and pUPD11 were more likely to be diagnosed in the neonatal period or post-neonatally. Indications for diagnosis varied between the groups, with the majority of IC2 LOM patients diagnosed based on presenting with BWS features, while HI indication was significantly increased in patients with pUPD11 and tumor indication was significantly increased in patients with IC1 GOM. Asymmetry indication was significantly greater in patients with IC1 GOM and pUPD11 compared with patients with IC2 LOM. A significant difference in source of positive molecular testing was found with positive blood analysis increased in patients with IC2 LOM, while approximately three-quarters of patients with IC1 GOM and pUPD11 were diagnosed through positive tissue analysis after negative blood analysis. The BWSp groups were also found to significantly differ between the molecular subtypes. Patients with IC2 LOM were more likely to be “classic” and less likely to be “ILO.”
3.5.3 |. BWSp clinical score
Patients with IC2 LOM were found to have increased clinical scores compared with IC1 GOM (p = .001) and patients with pUPD11 (p = .008). In comparison with patients with IC1 GOM, patients with IC2 LOM had an increased number of cardinal features (p = .012) and suggestive features (p = .008). An increased number of suggestive features was found in patients with IC2 LOM compared with pUPD11 (p = .011); however, no significant difference was found in the number of cardinal features (p = .137). No significant differences between patients with IC1 GOM and pUPD11 were found for BWSp clinical score or the number of cardinal or suggestive features.
3.6 |. BWSp group correlations
The most common BWSp group among the cohort was classic (Table 4). A variation of facial features was observed among the molecular subtypes and BWSp groups (Figure 7). A significant decrease in molecular confirmation was found in the ILO group compared with the classic and atypical groups. Patients in the classic group were significantly more often diagnosed through blood while patients in the atypical and ILO groups were more often diagnosed through tissue. Molecular subtype was found to differ significantly between the three groups. A significant difference in IC2 LOM diagnosis was observed across all three groups with IC2 LOM most common in the classic group and least common in the ILO group. The incidence of pUPD11 was greater in the atypical group compared with the classic group.
TABLE 4.
BWSp group correlations
| Feature | Totala n (%) or mean (SD) | Classic | Atypical | ILOb | p valuec |
|---|---|---|---|---|---|
| Molecular confirmation, yes | 248/305 (81.3%) | 178/195 (91.3%) | 45/53 (84.9%) | 25/57 (43.9%) | <.001*** |
| Molecular subtype | <.001*** | ||||
| IC1 GOMd | 32/322 (9.9%) | 15/207 (7.2%) | 7/55 (12.7%) | 10/60 (16.7%) | |
| IC2 LOMe | 110/322 (34.2%) | 97/207 (46.9%) | 12/55 (21.8%) | 1/60 (1.7%) | |
| pUPD11f | 73/322 (22.7%) | 38/207 (18.4%) | 21/55 (38.2%) | 14/60 (23.3%) | |
| CDKN1C mutation | 7/322 (2.2%) | 5/207 (2.4%) | 2/55 (3.6%) | 0/60 (0%) | |
| GWpUPDg | 7/322 (2.2%) | 4/207 (1.9%) | 3/55 (5.5%) | 0/60 (0%) | |
| 11p15 anomaly | 19/322 (5.9%) | 19/207 (9.2%) | 0/55 (0%) | 0/60 (0%) | |
| Negative testing | 57/322 (17.7%) | 17/207 (8.2%) | 8/55 (14.5%) | 32/55 (53.3%) | |
| No testing | 15/322 (4.7%) | 10/207 (4.8%) | 2/55 (3.6%) | 3/55 (5.0%) | |
| Unknown | 2/322 (0.6%) | 2/207 (1.0%) | 0/55 (0%) | 0/60 (0%) | |
| Diagnosis source, blood | 210/248 (84.7%) | 174/178 (97.8%) | 26/45 (57.8%) | 10/25 (40.0%) | <.001*** |
| Diagnosis indication | <.001*** | ||||
| BWS features | 195/308 (63.3%) | 179/199 (89.9%) | 15/53 (28.3%) | 1/56 (1.8%) | |
| Asymmetry | 56/308 (18.2%) | 8/199 (4.0%) | 5/53 (9.4%) | 43/56 (76.8%) | |
| Hyperinsulinism | 32/308 (10.4%) | 7/199 (3.5%) | 25/53 (47.2%) | 0/56 (0%) | |
| Tumor | 19/308 (6.2%) | 2/199 (1.0%) | 6/53 (11.3%) | 11/56 (19.6%) | |
| Incidental | 6/308 (1.9%) | 3/199 (1.5%) | 2/53 (3.8%) | 1/56 (1.8%) | |
| Diagnosis age group | <.001*** | ||||
| Prenatal (confirmed) | 22/295 (7.5%) | 22/190 (11.6%) | 0/53 (0%) | 0/52 (0%) | |
| Neonatal (<30 days) | 140/295 (47.5%) | 115/190 (60.5%) | 21/53 (39.6%) | 4/52 (7.7%) | |
| Postneonatal | 133/295 (45.1%) | 53/190 (27.9%) | 32/53 (60.4%) | 48/52 (92.3%) | |
| Postneonatal age at diagnosis (m) | 12.3 (1.9) | 6.3 (8.2) | 14.6 (22.7) | 19.9 (28.1) | .006** |
| Race/ethnicity group | .098 | ||||
| Caucasian | 216/307 (70.4%) | 136/194 (70.1%) | 33/54 (61.1%) | 47/59 (79.7%) | |
| Mixed | 48/307 (15.6%) | 32/194 (16.5%) | 8/54 (14.8%) | 8/59 (13.6%) | |
| Non-Caucasian | 43/307 (14.0%) | 26/194 (13.4%) | 13/54 (24.1%) | 4/59 (6.8%) |
Total refers to totals of the three subgroups compared (Classic, Atypical, ILO).
ILO: isolated lateralized overgrowth.
p values refer to the frequency of each feature between the three subgroups (Classic, Atypical, ILO), so all subgroups are compared at the same time.
IC1 GOM: gain of methylation at H19/IGF2:IG DMR.
IC2 LOM: loss of methylation at KCNQ1OT1:TSS DMR.
pUPD11: paternal uniparental isodisomy of chromosome 11p15.
GWpUPD: genome wide paternal uniparental isodisomy.
Significant at p < .05;
Significant at p < .01;
Significant at p < .001.
FIGURE 7.

Facial photographs of patients with BWS demonstrating the variation of facial gestalt within each molecular subtype. Abbreviations: IC2 = imprinting center 2 loss of methylation; IC1 = imprinting center 1 gain of methylation; pUPD11 = paternal uniparental isodisomy of chromosome 11; CDKN1C = CDKN1C mutation; ILO = isolated lateralized overgrowth. Top row, classic BWS patients: (a) IC2, 1 month old (b) IC1, 12 months old (c) pUPD11, 13 months old (d) CDKN1C, 12 months old; middle row, atypical BWS patients: (e) IC2, 24 months old (f) IC1, 12 months old (g) pUPD11, 12 months old (h) CDKN1C, 8 months old; bottom row, ILO patients: (i) IC2, 7 years old (j) IC1, 26 months old (k) pUPD11, 30 months old (l) there are no patients with CDKN1C and ILO in our cohort
The indication for diagnosis significantly differed between groups. The presenting feature of typical BWS features was most common in the classic group, hyperinsulinism as the presenting feature was most common in the atypical group, and asymmetry was most common in the ILO group. Tumor as the presenting feature was more common in the atypical and ILO group compared with the classic group.
A significant difference in age at diagnosis was also observed between the groups. Patients in the classic group were more often prenatally confirmed compared with patients in the atypical and ILO groups. All groups were different for neonatal diagnosis, with the highest incidence in the classic group and lowest incidence in the ILO groups. Postneonatal diagnosis was most common in the ILO group and least common in the classic group. The age at diagnosis in the ILO group significantly differed from the classic group by one-way ANOVA testing with Tukey post hoc testing (mean difference 13.58 ± 4.19 months, p = .004).
4 |. DISCUSSION
In evaluating a cohort of patients with BWSp that represent the complete spectrum, we observed that IC2 LOM patients were most commonly recognized as indicated by prenatal and early postnatal diagnoses and their composition of features had the highest clinical score. These patients looked most like the classic textbook cases of BWS. Patients with IC1 GOM and pUPD11 demonstrated much more variability in diagnostic features. Based on our further designation of classic, atypical, and ILO versions of BWS in the methods, we have made distinct recommended updates to the current BWS consensus guidelines to aid in diagnosis of patients in the atypical category as described below.
4.1 |. Epigenotype–phenotype correlations
Our results are consistent with previous epigenotype–phenotype correlations (Bliek et al., 2004; Brioude et al., 2013; Cooper et al., 2005; DeBaun et al., 2002; Engel et al., 2000; Gaston et al., 2001; Ibrahim et al., 2014; Maas et al., 2016; Mussa et al., 2016; Weksberg et al., 2001). An unreported phenotype we found was the increased incidence of undescended testes in patients with IC1 GOM and IC2 LOM compared with patients with pUPD11. This finding is most likely caused by the increased incidence of prematurity observed in these groups, as in the general population undescended testes are more common in premature patients (Niedzielski, Oszukowska, & Slowikowska-Hilczer, 2016). We also found an increased incidence of hyperinsulinism in the pUPD11 group, consistent with some previous reports (Brioude et al., 2013; DeBaun et al., 2002). As these reports evaluated hypoglycemia overall, further characterization of hyperinsulinism is necessary as we previously described (Kalish et al., 2016).
Sex distribution is variably reported in the large case series of patients with BWS. Some series have reported higher frequencies of female patients (Brioude et al., 2013; Maas et al., 2016), while others have shown slightly higher frequencies of males (DeBaun et al., 2002; Ibrahim et al., 2014; Zarate et al., 2009). To our knowledge, only one series has reported the frequency of sexes among the most common molecular subtypes (Maas et al., 2016). In this series, males represented 45.9% of the cohort, with similar frequencies observed within the subtypes evaluated. We observed similar frequencies to Maas et al. (2016) in our cohort, with the exception of IC1 GOM, in which females were overrepresented (62.5%). In the general pediatric population, patients with bilateral WT are more often female and the reasoning for this is unknown (Charlton, Irtan, Bergeron, & Pritchard-Jones, 2017). In our analysis, we demonstrated that IC1 GOM patients have bilateral/multifocal WT significantly more often than patients with IC2 LOM or pUPD11. The increased prevalence of IC1 GOM in female patients may be explained by the established connection between females and bilateral WT.
4.2 |. Cardinal features and limitations
In looking at the cardinal features present in patients with a confirmed molecular diagnosis, the most useful features were macroglossia, omphalocele, LO, and hyperinsulinism.
Patients with IC2 LOM most often fit into the classic group while an increased incidence of pUPD11 and IC1 GOM was observed in the atypical and ILO groups. These results indicate that the current BWS criteria are focused on diagnosing IC2 LOM patients and may not be useful in diagnosing patients with other molecular defects and those at the highest risk for tumors.
In the consensus, multifocal/bilateral WT/NB is considered a cardinal feature while unifocal/unilateral WT and other embryonal tumors are considered a suggestive feature. In our cohort, multifocal/bilateral WT/NB were not common however unifocal/unilateral WT or other embryonal tumors were, suggesting that perhaps identification of one of these other tumor types and/or not limiting WT to bilateral or multifocal should be considered in this analysis. In our cohort, 6% of patients were diagnosed with BWSp after presenting with a tumor and more than half (55.6%) had a unilateral/unifocal WT or other typical BWSp tumor. Interestingly, one patient was molecularly diagnosed with BWS in blood after presenting with a presumed WT but final pathology revealed unilateral nephroblastomatosis, which would not have warranted BWS molecular testing at all.
4.3 |. Clinical diagnosis
The consensus developed the clinical scoring system to recognize BWS as a spectrum and acknowledge that not all patients with BWSp will look like the textbook cases (classic patients) with BWS. The scoring system was developed to aid in diagnosis and to prevent a potential diagnosis being dismissed due to the absence of classic BWS features. In this study, we have also noted that requiring 4 points on the clinical scoring scale for a clinical diagnosis may not be sufficient without further caveats. In practical application of this system in a cohort that spans the BWSp, we recommend several modifications:
4.3.1 |. Isolated lateralized overgrowth
Patients with ILO and no other cardinal or suggestive features warrant consideration for inclusion in the spectrum, assuming the asymmetry is caused by proportionate growth and muscle bulk differences. Tissue analysis through skin biopsies of the larger limb is likely to aid in molecular characterization.
4.3.2 |. Hyperinsulinism and tumor as presenting features
Patients presenting with hyperinsulinism or a tumor typically occurring in patients with BWS may present with ILO as their only cardinal feature, or may be affected by a constellation of suggestive features that would not provide a high suspicion for BWS diagnosis without the occurrence of the tumor or hyperinsulinism. Despite this, approximately 16% of our cohort was diagnosed with BWS after presenting with hyperinsulinism or a tumor. Patients referred for hyperinsulinism were most often grouped in the atypical group and patients referred for tumors were most often characterized in the atypical and ILO groups. These results support the notion that these patients most often do not fit into the classic BWS presentation, however can still be affected by the disorder thus representing the spectrum. As a result, we suggest that all patients presenting with hyperinsulinism and typical BWSp tumors be evaluated for subtle asymmetry and other BWSp suggestive features. These patients also warrant molecular testing in available tissues in addition to molecular blood analysis.
In the consensus scoring system, the presence of multifocal and/or bilateral Wilms tumor/nephroblastomatosis receives two points to warrant genetic testing, while unilateral Wilms tumor and hepatoblastoma receive one point. We suggest that all patients presenting with Wilms tumor and hepatoblastoma receive genetic testing on both blood and affected tissue.
4.3.3 |. Assisted reproductive technology
The increased risk of BWS and use of assisted reproductive technology (ART) (~four–sixfold) is well documented (DeBaun, Niemitz, & Feinberg, 2003; Gicquel et al., 2003; Maher et al., 2003), with an increased risk as high as 1 in 1,126 reported (Mussa et al., 2017). In industrialized countries, ART accounts for 1–3% of all live births (Brioude et al., 2018). In this study, ART was used for conception for 14.9% of patients. Furthermore, approximately a quarter of patients conceived via IVF and/or ICSI were characterized in the atypical or ILO groups (data not shown). This observation clearly shows that patients conceived by IVF or ICSI may not have classic BWS features and supports a proposal that conception by IVF or ICSI could be viewed as one of the diagnostic criteria for BWSp.
The majority of patients with BWS born after ART (BWS-ART) are affected by IC2 LOM (Brioude et al., 2018). To this date, three BWS-ART patients with pUPD11 have been reported (Johnson et al., 2018; Mussa et al., 2017). Additionally, a BWS-ART patient was found to have isolated IC2 LOM and IC1 GOM after pUPD11 was excluded (DeBaun et al., 2003). To our knowledge no patient with isolated IC1 GOM after ART has been previously reported. In this study, three patients had pUPD11 and one patient had IC1 GOM. These patients displayed classic features, besides one pUPD11 patient who was diagnosed after presenting with hyperinsulinism and the pUPD11 was identified in pancreatic tissue. These findings support the previous suggestion that ART may be implicated in genomic events beyond methylation defects (Mussa et al., 2017).
4.4 |. Molecular diagnosis
Based on the analysis of this cohort, the consensus recommendations to test multiple tissues are quite useful in expanding the molecular diagnosis in these patients. Among our cohort, we found that testing multiple tissues increased the diagnostic yield from 70% to 82% (Figure 5). As a result, we recommend molecular testing on affected tissue whenever possible, especially in atypical and ILO presentations of BWSp.
Almost all patients in the classic group with molecular confirmation were diagnosed by blood, compared with approximately half of the patients in the atypical and ILO groups. The patients in these groups were significantly more often diagnosed through tissue compared with the classic group and the most common molecular subtype among those diagnosed through tissue was pUPD11. Patients in the atypical or ILO groups also had fewer BWS features, indicating the (epi)genetic changes occurred later in development and are more mosaic compared with patients with classic BWS. As a result, testing additional tissues in these groups increases the diagnostic yield of detecting mosaicism and is important to help characterize patients in the BWS spectrum. This is especially important in patients presenting with hyperinsulinism or a tumor.
4.5 |. Tumor risk
Although all patients with BWS have an increased tumor risk, epigenotype–phenotype correlations have identified tumor risk differs in regard to molecular subtypes. Patients with IC1 GOM and pUPD11 defects are at the greatest risk, while tumor risk is lower in patients with IC2 LOM and mutations in CDKN1C (Brioude et al., 2018). Tumor incidence in our group overall (14.5%) was higher than previously published (8%; Maas et al., 2016). Similarly, we observed higher tumor incidences within the three most common molecular subtypes compared with the previously published data (Maas et al., 2016): IC1 GOM with 51.6% compared with 28%; pUPD11 with 29.7% compared with 16%; and IC2 LOM with 4.4% compared with 2.6%. Tumor incidence observed in our population may be higher compared with previous published groups due to increased recognition of subtle features, as well as molecular diagnosis through tissue testing. As a number of patients in our cohort were diagnosed with BWS due to presenting with a tumor or hyperinsulinism, it is likely these patients would not have been included in previous reports. Patients at the highest risk for tumors (IC1 GOM and pUPD11) were also observed more often to be diagnosed through tissue testing. This observation highlights the importance of recognizing patients along the spectrum, as all are at an increased risk of developing tumors. In this analysis, we did not explore specific tumor types or clinical features that may increase the risk of tumors, and additional analysis is warranted in order to better characterize BWSp patients at the greatest risk.
4.6 |. Management recommendations
For the most part, management recommendations are inline with the consensus recommendations (Brioude et al., 2018). There are two areas in which these differ based on our experience with this BWSp cohort. These include hyperinsulinism and tumor screening.
4.6.1 |. Hyperinsulinism
Hypoglycemia occurred in half of the patients in our cohort with 20% of patients having hyperinsulinism. Among those with hyperinsulinism, more than a third required a pancreatectomy. Patients with pUPD11 molecular defects were most often affected by hyperinsulinism; however, hyperinsulinism occurs in other BWSp molecular defects as well. Management of hyperinsulinism is further discussed in the hyperinsulinism article in this issue.
4.6.2 |. Tumor screening
Until recently, there were uniform recommendations that all patients with BWS undergo routine tumor surveillance for detection of the two most common tumors (Wilms tumor and hepatoblastoma), with serial alpha-fetoprotein measurements and abdominal/renal ultrasounds. Based on differing approaches to acceptable risk in different regional and practice environments in combination with epigenotype–phenotype correlations, some groups have used a 5% tumor risk cutoff and have begun stratifying tumor surveillance to those subtypes at the greatest risk (Brioude et al., 2018), while others maintain a more conservative approach using a 1% tumor risk cutoff, similar to that used by the American Association for Cancer Research (AACR) and continue to screen all patients (Kalish et al., 2017b). The largely European BWS consensus advocated for tumor screening in just those patients with the highest risk: IC1 GOM, pUPD11, and those without a molecular defect identified, as well as patients with CDKN1C mutations and did not advocate surveillance in patients with IC2 LOM (Brioude et al., 2018). Tumor surveillance strategy recommended by the consensus includes screening by abdominal ultrasound every 3 months from time of diagnosis until age 7 years for detection of embryonal tumors (Brioude et al., 2018). The consensus did not recommend using serial alpha-fetoprotein (AFP) measurements for detection of hepatoblastoma due to low risk and difficultly in interpretation.
Based on an acceptable risk threshold of >1%, our tumor screening recommendations are in line with recommendations from the AACR (Kalish, Doros, et al., 2017b) for all patients with BWSp:
abdominal ultrasounds and AFP measurements every 3 months until the 4th birthday;
renal ultrasounds every 3 months until the 7th birthday.
Although the utility of AFP screening has been debated (Duffy, Deardorff, & Kalish, 2017; Kalish & Deardorff, 2016; Maas et al., 2016; Mussa & Ferrero, 2015, 2017), previous reports highlight the usefulness of this measure in detecting hepatoblastoma before detection by ultrasonography (Clericuzio et al., 2003; Mussa et al., 2011; Zarate et al., 2009) thus downgrading tumor stage at diagnosis (Clericuzio et al., 2003; Trobaugh-Lotrario, Venkatramani, & Feusner, 2014), which is why we advocate for AFP in our tumor surveillance recommendations. Recent evidence shows that hepatoblastoma in patients with BWSp occurs before the age of 30 months (Mussa, Duffy, Carli, Ferrero, & Kalish, 2019), so guidelines may be further amended to reduce the length of AFP screening and full abdominal ultrasounds. AFP norms in BWS were recently published to aid in interpretation of values when screening (Duffy, Cohen, Elci, & Kalish, 2019). There are also additional recommendations for patients with CDKN1C mutations due to a risk for development of neuroblastoma (urine HVA/VMA and chest X-rays; Kalish, Doros, et al., 2017b), however further data are needed for this patient population.
4.6.3 |. Multidisciplinary team approach
Patients with BWSp can be affected by a variety of clinical features requiring a multidisciplinary team approach. At time of suspected diagnosis, patients should be evaluated for which clinical features are present and referred to other specialists as appropriate. In patients with macroglossia, referral to a plastic surgeon for consideration for tongue reduction is warranted, as approximately half of patients with macroglossia in our cohort required a tongue reduction. Referral to a pulmonologist for polysomnography is also warranted in patients with macroglossia, as a high prevalence of obstructive sleep apnea (OSA) in children with BWSp and macroglossia has been observed (Cielo, Duffy, Taylor, Marcus, & Kalish, 2019). Patients with LO should be referred to an orthopedist to evaluate for an associated leg length (Brioude et al., 2018). Referrals to nephrology and/or urology may be warranted in some patients, as renal and genitourinary issues have been reported in patients with BWS (Goldman et al., 2002; Mussa et al., 2012; Tong et al., 2017; Wong, Cuda, & Kirsch, 2011). Other referrals to specialists should be made according to clinical features present. Cardiac and renal evaluation is recommended to screen for congenital lesions (Brioude et al., 2018). While there are no specific developmental associations with BWS, hyperinsulinism and prematurity can both cause delays so developmental evaluation is recommended if there are any concerns. Adult management is largely focused on specific clinical concerns and genetic counseling regarding family planning, especially if the genetic etiology is not known (Brioude et al., 2018). A recent study indicates that early surgical management of BWS features including tongue reduction, treatment of leg length difference, and orchiopexy are important to prevent later complications (Gazzin et al., 2019). Further study is warranted to improve recommendations for adult management.
Another challenging aspect of BWS is the best strategy to manage partially discordant twins. Current recommendations include evaluating both twins for clinical features of BWS and testing the patients with a clinical score of greater than 4 including at least one cardinal feature (Cohen et al., 2019). Screening is currently recommended for clinically affected twins but not for a twin without clinical features unless molecular testing is positive in a tissue other than blood (Cohen et al., 2019).
5 |. CONCLUSION
Although the BWS consensus scoring system was designed with the goal to recognize BWS as a spectrum and aid in diagnosis, our results suggest that this scoring system may benefit may from modifications to improve its utility. Further delineation of the full spectrum suggests the need for updating the scoring methodology especially for patients presenting with hyperinsulinism or a tumor as more of these patients may fall into the spectrum than previously considered.
ACKNOWLEDGMENTS
The authors thank the patients and their families for participating in this study. Funding was provided by K08 CA193915, St Baldrick’s Foundation Scholar award and Alex’s Lemonade Stand Foundation.
Alex’s Lemonade Stand Foundation for Childhood Cancer; National Cancer Institute, Grant/Award Number: K08 CA193915; St. Baldrick’s Foundation, Grant/Award Number: Scholar Award
Footnotes
CONFLICT OF INTERESTS
The authors have no conflicts of interest relevant to this article to disclose. Author J.L.C. was a one-time consultant for Sobi, Inc.
REFERENCES
- Alders M, Maas SM, Kadouch DJ, van der Lip K, Bliek J, van der Horst CM, & Mannens MM (2014). Methylation analysis in tongue tissue of BWS patients identifies the (EPI)genetic cause in 3 patients with normal methylation levels in blood. European Journal of Medical Genetics, 57, 293–297. [DOI] [PubMed] [Google Scholar]
- Beckwith JB. 1963. Extreme cytomegaly of the adrenal fetal cortex, omphalocele, hyperplasia of kidneys and pancreas, and Leydig-cell hyperplasia: another syndrome in Annual Meeting of Western Society of Pediatric Research Los Angeles, CA. [Google Scholar]
- Bliek J, Gicquel C, Maas S, Gaston V, Le Bouc Y, & Mannens M (2004). Epigenotyping as a tool for the prediction of tumor risk and tumor type in patients with Beckwith-Wiedemann syndrome (BWS). The Journal of Pediatrics, 145, 796–799. [DOI] [PubMed] [Google Scholar]
- Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, … Maher ER (2018). Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: An international consensus statement. Nature Reviews. Endocrinology, 14, 229–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brioude F, Lacoste A, Netchine I, Vazquez MP, Auber F, Audry G, … Rossignol S (2013). Beckwith-Wiedemann syndrome: Growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Hormone Research in Pædiatrics, 80, 457–465. [DOI] [PubMed] [Google Scholar]
- Charlton J, Irtan S, Bergeron C, & Pritchard-Jones K (2017). Bilateral Wilms tumour: A review of clinical and molecular features. Expert Reviews in Molecular Medicine, 19, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cielo CM, Duffy KA, Taylor JA, Marcus CL, & Kalish JM (2019). Obstructive sleep apnea in children with Beckwith-Wiedemann syndrome. Journal of Clinical Sleep Medicine, 15, 375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clericuzio CL, Chen E, McNeil DE, O’Connor T, Zackai EH, Medne L, … DeBaun M (2003). Serum alpha-fetoprotein screening for hepatoblastoma in children with Beckwith-Wiedemann syndrome or isolated hemihyperplasia. The Journal of Pediatrics, 143, 270–272. [DOI] [PubMed] [Google Scholar]
- Cohen JL, Duffy KA, Sajorda BJ, Hathaway ER, Gonzalez-Gandolfi CX, Richards-Yutz J, … Kalish JM (2019). Diagnosis and management of the phenotypic spectrum of twins with Beckwith-Wiedemann syndrome. American Journal of Medical Genetics. Part A, 179, 1139–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper WN, Luharia A, Evans GA, Raza H, Haire AC, Grundy R, … Maher ER (2005). Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. European Journal of Human Genetics, 13, 1025–1032. [DOI] [PubMed] [Google Scholar]
- DeBaun MR, Niemitz EL, & Feinberg AP (2003). Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. American Journal of Human Genetics, 72, 156–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBaun MR, Niemitz EL, McNeil DE, Brandenburg SA, Lee MP, & Feinberg AP (2002). Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. American Journal of Human Genetics, 70, 604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy KA, Cohen JL, Elci OU, & Kalish JM (2019). Development of the serum alpha-fetoprotein reference range in patients with Beckwith-Wiedemann Spectrum. The Journal of Pediatrics, 212, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy KA, Deardorff MA, & Kalish JM (2017). The utility of alpha-fetoprotein screening in Beckwith-Wiedemann syndrome. American Journal of Medical Genetics. Part A, 173, 581–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann K, Bliek J, Brioude F, Algar E, Buiting K, Russo S, … Eggermann T (2016). EMQN best practice guidelines for the molecular genetic testing and reporting of chromosome 11p15 imprinting disorders: Silver-Russell and Beckwith-Wiedemann syndrome. European Journal of Human Genetics, 24, 1377–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel JR, Smallwood A, Harper A, Higgins MJ, Oshimura M, Reik W, … Maher ER (2000). Epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. Journal of Medical Genetics, 37, 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston V, Le Bouc Y, Soupre V, Burglen L, Donadieu J, Oro H, … Gicquel C (2001). Analysis of the methylation status of the KCNQ1OT and H19 genes in leukocyte DNA for the diagnosis and prognosis of Beckwith-Wiedemann syndrome. European Journal of Human Genetics, 9, 409–418. [DOI] [PubMed] [Google Scholar]
- Gazzin A, Carli D, Sirchia F, Molinatto C, Cardaropoli S, Palumbo G, … Mussa A (2019). Phenotype evolution and health issues of adults with Beckwith-Wiedemann syndrome. American Journal of Medical Genetics. Part A, 179:1691–1702. [DOI] [PubMed] [Google Scholar]
- Gicquel C, Gaston V, Mandelbaum J, Siffroi JP, Flahault A, & Le Bouc Y (2003). In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. American Journal of Human Genetics, 72, 1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman M, Smith A, Shuman C, Caluseriu O, Wei C, Steele L, … Rosenblum ND (2002). Renal abnormalities in beckwith-wiedemann syndrome are associated with 11p15.5 uniparental disomy. Journal of the American Society of Nephrology, 13, 2077–2084. [DOI] [PubMed] [Google Scholar]
- Hoyme HE, Seaver LH, Jones KL, Procopio F, Crooks W, & Feingold M (1998). Isolated hemihyperplasia (hemihypertrophy): Report of a prospective multicenter study of the incidence of neoplasia and review. American Journal of Medical Genetics, 79, 274–278. [PubMed] [Google Scholar]
- Ibrahim A, Kirby G, Hardy C, Dias RP, Tee L, Lim D, … Maher ER (2014). Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clinical Epigenetics, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JP, Beischel L, Schwanke C, Styren K, Crunk A, Schoof J, & Elias AF (2018). Overrepresentation of pregnancies conceived by artificial reproductive technology in prenatally identified fetuses with Beckwith-Wiedemann syndrome. Journal of Assisted Reproduction and Genetics, 35, 985–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalish JM, Biesecker LG, Brioude F, Deardorff MA, Di Cesare-Merlone A, Druley T, … Hennekam RC (2017a). Nomenclature and definition in asymmetric regional body overgrowth. American Journal of Medical Genetics. Part A, 173, 1735–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalish JM, Boodhansingh KE, Bhatti TR, Ganguly A, Conlin LK, Becker SA, … Deardorff MA (2016). Congenital hyperinsulinism in children with paternal 11p uniparental isodisomy and Beckwith-Wiedemann syndrome. Journal of Medical Genetics, 53, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalish JM, & Deardorff MA (2016). Tumor screening in Beckwith-Wiedemann syndrome-to screen or not to screen? American Journal of Medical Genetics. Part A, 170, 2261–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalish JM, Doros L, Helman LJ, Hennekam RC, Kuiper RP, Maas SM, … Druley TE (2017b). Surveillance recommendations for children with overgrowth syndromes and predisposition to Wilms tumors and Hepatoblastoma. Clinical Cancer Research, 23, e115–e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas SM, Vansenne F, Kadouch DJ, Ibrahim A, Bliek J, Hopman S, … Hennekam RC (2016). Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. American Journal of Medical Genetics. Part A, 170, 2248–2260. [DOI] [PubMed] [Google Scholar]
- MacFarland SP, Duffy KA, Bhatti TR, Bagatell R, Balamuth NJ, Brodeur GM, … Kalish JM (2018). Diagnosis of Beckwith-Wiedemann syndrome in children presenting with Wilms tumor. Pediatric Blood & Cancer, 65, e27296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DJG, Bliek J, Lombardi MP, Russo S, Calzari L, Guzzetti S, … Eggermann T (2019). Discrepant molecular and clinical diagnoses in Beckwith-Wiedemann and silver-Russell syndromes. Genetic Research, 101, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher ER, Brueton LA, Bowdin SC, Luharia A, Cooper W, Cole TR, … Hawkins MM (2003). Beckwith-Wiedemann syndrome and assisted reproduction technology (ART). Journal of Medical Genetics, 40, 62–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaa G, Conway R, & Graham JM Jr. et al. PIK3CA-Related Segmental Overgrowth. 2013. August 15. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK153722/. [Google Scholar]
- Mussa A, Duffy KA, Carli D, Ferrero GB, & Kalish JM (2019). Defining an optimal time window to screen for hepatoblastoma in children with Beckwith-Wiedemann syndrome. Pediatric Blood & Cancer, 66, e27492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussa A, & Ferrero GB (2015). Screening Hepatoblastoma in Beckwith-Wiedemann syndrome: A complex issue. Journal of Pediatric Hematology/Oncology, 37, 627. [DOI] [PubMed] [Google Scholar]
- Mussa A, & Ferrero GB (2017). Serum alpha-fetoprotein screening for hepatoblastoma in Beckwith-Wiedemann syndrome. American Journal of Medical Genetics. Part A, 173, 585–587. [DOI] [PubMed] [Google Scholar]
- Mussa A, Ferrero GB, Ceoloni B, Basso E, Chiesa N, De Crescenzo A, … de Sanctis L (2011). Neonatal hepatoblastoma in a newborn with severe phenotype of Beckwith-Wiedemann syndrome. European Journal of Pediatrics, 170, 1407–1411. [DOI] [PubMed] [Google Scholar]
- Mussa A, Molinatto C, Cerrato F, Palumbo O, Carella M, Baldassarre G, … Ferrero GB (2017). Assisted reproductive techniques and risk of Beckwith-Wiedemann syndrome. Pediatrics, 140, e20164311. [DOI] [PubMed] [Google Scholar]
- Mussa A, Peruzzi L, Chiesa N, De Crescenzo A, Russo S, Melis D, … Ferrero GB (2012). Nephrological findings and genotype-phenotype correlation in Beckwith-Wiedemann syndrome. Pediatric Nephrology, 27, 397–406. [DOI] [PubMed] [Google Scholar]
- Mussa A, Russo S, De Crescenzo A, Chiesa N, Molinatto C, Selicorni A, … Ferrero GB (2013). Prevalence of Beckwith-Wiedemann syndrome in north west of Italy. American Journal of Medical Genetics. Part A, 161A, 2481–2486. [DOI] [PubMed] [Google Scholar]
- Mussa A, Russo S, De Crescenzo A, Freschi A, Calzari L, Maitz S, … Ferrero GB (2016). (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome. European Journal of Human Genetics, 24, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedzielski JK, Oszukowska E, & Slowikowska-Hilczer J (2016). Undescended testis - current trends and guidelines: A review of the literature. Archives of Medical Science, 12, 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo S, Calzari L, Mussa A, Mainini E, Cassina M, Di Candia S, … Larizza L (2016). A multi-method approach to the molecular diagnosis of overt and borderline 11p15.5 defects underlying silver-Russell and Beckwith-Wiedemann syndromes. Clinical Epigenetics, 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo-Avila C, Gonzalez-Crussi F, & Fowler JW (1980). Complete and incomplete forms of Beckwith-Wiedemann syndrome: Their oncogenic potential. The Journal of Pediatrics, 96, 47–50. [DOI] [PubMed] [Google Scholar]
- Tong CC, Duffy KA, Chu DI, Weiss DA, Srinivasan AK, Canning DA, & Kalish JM (2017). Urological findings in Beckwith-Wiedemann syndrome with chromosomal duplications of 11p15.5: Evaluation and management. Urology, 100, 224–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobaugh-Lotrario AD, Venkatramani R, & Feusner JH (2014). Hepatoblastoma in children with Beckwith-Wiedemann syndrome: Does it warrant different treatment. Journal of Pediatric Hematology/Oncology, 36, 369–373. [DOI] [PubMed] [Google Scholar]
- Weksberg R, Nishikawa J, Caluseriu O, Fei YL, Shuman C, Wei C, … Squire J (2001). Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Human Molecular Genetics, 10, 2989–3000. [DOI] [PubMed] [Google Scholar]
- Wiedemann HR (1964). Familial malformation complex with umbilical hernia and Macroglossia–A “new syndrome”. Journal de Génétique Humaine, 13, 223–232. [PubMed] [Google Scholar]
- Wong CA, Cuda S, & Kirsch A (2011). A review of the urologic manifestations of Beckwith-Wiedemann syndrome. Journal of Pediatric Urology, 7, 140–144. [DOI] [PubMed] [Google Scholar]
- Zarate YA, Mena R, Martin LJ, Steele P, Tinkle BT, & Hopkin RJ (2009). Experience with hemihyperplasia and Beckwith-Wiedemann syndrome surveillance protocol. American Journal of Medical Genetics. Part A, 149A, 1691–1697. [DOI] [PubMed] [Google Scholar]