

Graphical abstract
Keywords: Cancer, SF3B1, Splicing, Missense mutations, Functional impact, Protein conformation, In-silico analysis
Highlights
-
•
Aberrant splicing patterns are key markers of the impact of splicing gene mutations.
-
•
SF3B1 Mut-induced aberrant splicing is due to the protein conformational change.
-
•
SF3B1 conformational change depends on the position and charge of AA substitution.
-
•
SF3B1 mutations present unequal pathogenic and prognostic potentials.
Abstract
The hotspot mutations of SF3B1, the most frequently mutated splicing gene in cancers, contribute to oncogenesis by corrupting the mRNA splicing. Further SF3B1 mutations have been reported in cancers but their consequences remain unclear. Here, we screened for SF3B1 mutations in the vicinity of the hotspot region in tumors. We then performed in-silico prediction of the functional outcome followed by in-cellulo modelling of different SF3B1 mutants. We show that cancer-associated SF3B1 mutations present varying functional consequences that are loosely predicted by the in-silico algorithms. Analysis of the tertiary structure of SF3B1 mutants revealed that the resulting splicing errors may be due to a conformational change in SF3B1 N-terminal region, which mediates binding with other splicing factors. Our study demonstrates a varying functional impact of SF3B1 mutations according to the mutated codon and the amino acid substitution, implying unequal pathogenic and prognostic potentials of SF3B1 mutations in cancers.
1. Introduction
Aberrant alternative splicing is emerging as a hallmark of cancer. Splice aberrations are mainly associated with mutations of genes encoding for splicing factors including SF3B1, U2AF1, SRSF2 and ZRSR2 as the most frequently mutated splicing genes in cancers. Mutations in these genes were initially discovered in myelodysplastic syndromes (MDSs) and chronic lymphocytic leukemia (CLL) [1], [2], [3]. Interestingly, these genes encode proteins that are all involved in the initial steps of RNA splicing, including 3′ splice site (3′ss) recognition during RNA splicing [4], [5]. SF3B1 has also been found mutated in a significant proportion (~20%) of uveal melanoma (UM) and in other solid tumors at lesser frequencies [6], [7], [8], [9], [10]. Intriguingly, mutations of SF3B1, U2AF1, ZRSR2 and SRSF2 are almost mutually exclusive and lead to distinct aberrant splice patterns. There is a growing interest in these splice aberrations, not only to understand the mechanisms and consequences of the causing mutations, but also to determine whether such aberrations are related to the oncogenesis or have a clinical significance. The analysis of these aberrant splice patterns has been shown to be of prognostic value for multiple cancer types, including non-small cell lung cancer, ovarian cancer, breast cancer, uveal melanoma and glioblastoma [11], [12], [13], [14], [15], [16], [17].
SF3B1 (Splicing Factor 3b Subunit 1) mediates U2 snRNP recruitment to the branchpoint (BP) by interacting with the intronic pre-mRNA (precursor messenger RNA) [18]. Cancer-associated missense mutations in SF3B1 are change-of-function mutations with three main hotspots targeting the HEAT (Huntingtin, Elongation factor 3, protein phosphatase 2A, Targets of rapamycin 1) repeat domains at codons R625, K666 and K700. Interestingly, K700 mutations are by far the most frequent in hematopoietic malignancies, whereas R625 mutations are prevailing in UM. These alterations affect residues that are predicted to be spatially close to one another [1]. The hotspot mutations are associated with a unique aberrant splice pattern characterized by recognition of alternative 3′ss in a subset of pre-mRNA. SF3B1 mutations targeting further codon positions have been reported at lesser frequencies, but little is known about their consequences. It has been reported that SF3B1 hotspot mutations disrupt interaction with the spliceosomal protein SUGP1 (SURP and G-patch domain containing 1) during BP recognition and that the loss of this interaction solely accounts for the splicing errors caused by mutant SF3B1 [19]. Very recently, we and others revealed that SUGP1 somatic mutations combined with its loss-of-heterozygosity induce the SF3B1-mutant splice pattern (we will denote by SF3B1Mut all SF3B1 mutations that lead to aberrant 3′ss usage) in tumors and cell models [20], [21].
Cretu et al. provided the first description of the crystal structure of SF3b core complex, revealing how the distinctive conformation of SF3B1 HEAT domains is maintained by multiple contacts with SF3b130, SF3b10, and SF3b14b [22]. Protein-protein crosslinking enabled the localization of the binding proteins p14 and U2AF65 within SF3B1 HEAT superhelix, which together with SF3b14b forms a composite RNA-binding platform. SF3B1 residues, targeted by cancer-related mutations, contribute to the tertiary structure of the HEAT domain and its surface properties in the proximity of p14 and U2AF65 [22]. Recently, descriptive studies of human cancers highlighted splicing variations between SF3B1 hotspots, and suggested correlation of these hotspots with clinical differences [23], [24], [25], [26]. Notably, RNA-sequencing data analysis of tumors and isogenic cell lines harboring SF3B1 hotspot mutations suggested that the distribution of distances from the canonical to the cryptic 3′ss varies among the different hotspot mutations with altered sequence motif associated with the aberrant 3′ss [25].
In the present study, we investigated a set of cancer-associated SF3B1 mutations in order to determine the functional and structural impact of these mutations. We performed in-silico prediction of the functional outcome followed by in-cellulo modelling of different SF3B1 mutants and validation of the induction of the aberrant splice pattern. We show that residues whose mutations are involved in cancer present varying functional consequences that are loosely predicted by the currently available in-silico algorithms. Analysis of the predictive tertiary structure of different SF3B1 mutants revealed that the splicing defects induced by SF3B1 mutations may be due to a conformational change in SF3B1 N-terminal region, which mediates binding with other splicing factors. Furthermore, our findings suggest that SF3B1 hotspot mutations induce different intensities of the aberrant splice pattern. We provide the first evidence that the location and the charge of amino acid substitution are determinant factors of the burden of aberrant splicing induced by SF3B1 mutants. Such findings imply unequal pathogenic and prognostic potentials of the different SF3B1 mutations in cancers.
2. Materials and methods
2.1. In-silico quantification for SF3B1Mut impact in tumors
As previously described, Sequence Bloom Trees (SBTs) were generated based on the RNA-sequencing fastq files for all samples of the 33 tumor types of the TCGA, and the occurrence of 1443 aberrant splice junctions specific to SF3B1Mut tumors (40-nts sequences centered on the aberrant 3′ splice site) was screened using SBTs [20], [27], [28], [29], [30]. SBT scores corresponded to the number of junctions found for each tumor sample in the fastq file (0 ≤ SBT score ≤ 1443).
Linear model SBT scores ~ Million reads per mutated allele (total number of reads covering splice junctions multiplied by VAF of SF3B1 mutation) was built based on 34 cases with K700E and R625H/C mutations (R2 = 0.75, p < 10−10, Supplementary Fig. 1A). Mutation impact was estimated as deviation from the model value relative to the model value, i.e. the difference of the SBT score and the predicted value divided by the predicted value for all mutations (Supplementary Fig. 1B). Linear model and the boxplot were built using R functions.
2.2. Wild-type and mutated SF3B1 constructs
A pCMV-3tag-1A vector containing wild-type SF3B1 was synthesized by Genscript Corporation as previously described [8]. The full sequence of codon-optimized SF3B1 is available upon request. R625H, H662R, I665F, K666T, K700E, G740E, K741E, K741N, K741Q, G742D, D781E missense mutations and deletion of HEAT repeats included between amino acids 622 and 781 (Del (622–781)) were introduced using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies). All constructs were verified by DNA sequencing. The primer sequences used to generate SF3B1 constructs are provided in Supplementary Table 1.
2.3. Cell culture and transfection
HEK293T was cultured in DMEM supplemented with 10% fetal bovine serum and was tested to be Mycoplasma free. Authentication of the cell line was verified by Sanger sequencing for its mutational status and by RNA-Seq. Plasmid transfections were carried out in HEK293T cell line using 2 μg of plasmid construct and LipofectAMINE 2000 reagent (Invitrogen) according to the manufacturer’s instructions.
2.4. Reverse transcription and quantitative PCR amplification of splicing products
Total RNA was extracted with NucleoSpin RNA kit (Macherey-Nagel). The quantity and quality of RNA was determined by spectrophotometry (NanoDrop Technologies). Five hundred nanograms of RNA was used as a template for cDNA synthesis with the High Capacity cDNA ReverseTranscription Kit (Applied Biosystems). Twenty-five nanograms of the synthesized cDNA was used as a template for RT–PCR amplification with specific primers. The primer sequences are provided in Supplementary Table 2.
2.5. RNA-sequencing analysis
The total RNA was isolated from HEK293T cells using a NucleoSpin Kit (Macherey-Nagel). Cells were transfected with pCMV-3tag-1A expression vectors for wild-type SF3B1 (SF3B1WT), or for SF3B1 mutations (R625H, K666T, K700E) at comparable transfection levels as shown in Supplementary Fig. 2 (1 µg of expression vectors for R625H and K666T, and 2 µg for K700E). cDNA synthesis was conducted with MuLV Reverse Transcriptase in accordance with the manufacturer’s instructions (Invitrogen), with quality assessments conducted on an Agilent 2100 Bioanalyzer. Libraries were constructed using the TruSeq Stranded mRNA Sample Preparation Kit (Illumina) and sequenced on an Illumina NovaSeq platform using a 100-bp paired-end sequencing strategy. RNA-sequencing data analysis was performed as previously described [20]. Sequencing data are available as GSE167001.
2.6. Immunoblot analysis
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer, and proteins were quantified using a BCA Protein Assay (Pierce). Equal amounts were separated by SDS–polyacrylamide gel electrophoresis. Proteins were transferred to nitrocellulose membranes followed by immunoblotting with specific primary antibodies for SF3B1 (1:500; #170854; Abcam), Flag (1: 1,000, #3165; Sigma), and ß-actin (1: 5,000; #5313; Sigma). The membrane was then incubated at room temperature for one hour with either goat anti-rabbit or goat anti-mouse Odyssey secondary antibodies. Immunolabelled proteins were detected using the Odyssey Infrared Imaging System (Li-cor). ß-Actin immunoblotting was used to quantify and normalize results.
2.7. In-silico prediction of protein function and structure
Seven in-silico prediction tools (MAPP, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT, SNAP and PANTHER) were used to assess the effects of amino acid substitutions on protein function. Among these seven tools, the six best (MAPP, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT, SNAP) performing methods were combined into a consensus classifier PredictSNP, which represents a robust and accurate alternative to the predictions delivered by individual tools [31].
The visualization of SF3B1 structure was done using PyMOL [The PyMOL Molecular Graphics System, Delano Scientific, San Carlos, CA, USA, version 1.4]. The structure prediction of SF3B1 wild-type and SF3B1 mutants was done using the I-TASSER server [32], [33], [34], [35], [36]. I-TASSER provides the structural alignments between the target (in our case, SF3B1 wild-type and SF3B1 mutants) and the homologous structure templates from the PDB library that are ranked by TM-score [37]. The alignments between SF3B1 wild-type and SF3B1 mutants were performed using PyMOL and the RMSD (Root-Mean-Square Deviation) was used to calculate the quality of the alignment of two predicted protein structures [38]. Additionally, the RMSD per residue along the mutant backbone as compared to the wild-type backbone was analyzed by MultiSeq, an extension of the Multiple Alignement tool that is provided as part of VMD (Visual Molecular Dynamics) [39], [40], [41].
3. Results
3.1. In-silico predictions of the functional impact of missense mutations in SF3B1
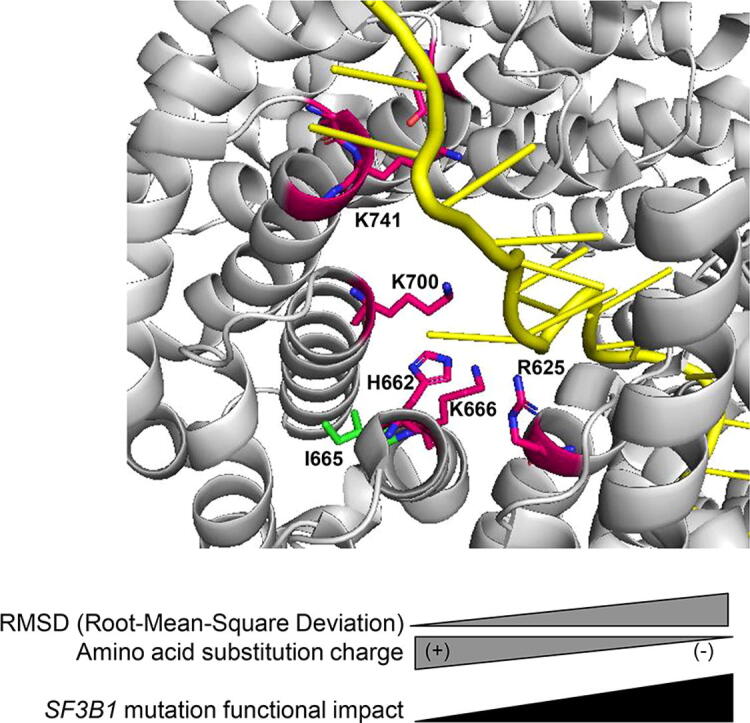
In order to gain insight into cancer-associated SF3B1 mutations, we interrogated COSMIC dataset to screen for SF3B1 mutations in 33 different types of tumors (Fig. 1A). In total, 8% of all samples (3,299/40,189) of this database harbored missense/nonsense mutations or insertions/deletions in SF3B1. Missense mutations are, by far, the most frequent event representing 81% of all events. Eighty-five percent (2,278/2,679) of the missense mutations are localized in the HEAT domain region 529 – 880 AA (amino acids) of SF3B1, including hotspots on codons R625, K666 and K700 (Fig. 1B). We focused on mutations with high recurrence or adjacent to hotspots. As shown in Fig. 2A, the mutated codons are all located at the helix of the HEAT domain encompassing the pre-mRNA. We then investigated the pathogenicity of SF3B1 amino acid substitutions: R625H, H662R, I665F, K666T, K700E, G740E, K741E, K741N, K741Q, G742D, D781E by using a consensus classifier PredictSNP, an in-silico tool combining the best performing prediction computational methods (MAPP, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT and SNAP). Based on various parameters such as physico-chemical properties of amino acids, the conservation of amino acids between species and structural characteristics provided by databases like PDB (Protein Data Bank) and Uniprot, this analysis predicts whether a substitution of a particular amino acid is neutral or deleterious with a confidence score reflecting the degree of veracity of its predictions. Fig. 2B shows that the tested SF3B1 mutations are all predicted to be deleterious according to computational prediction tools except for D781E, which was predicted to be neutral with a confidence score of 63%. Thus, the in-silico prediction analysis displays variations of the functional consequences of SF3B1 missense mutations.
Fig. 1.

Screening for SF3B1 mutations in COSMIC database. Graphical representation of the mutation spectrum across the SF3B1 gene on the amino acid scale (A) and a zoom on the hotspot mutational HEAT domain (blue regions) (B). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 2.

In-silico predictions of functional impact of SF3B1 missense mutations. A. Overview of residues mutated in cancers (magenta spheres) mapped to the humain SF3B1 structure (grey) in the activated spliceosome (Bact complex) (PDB ID: 5Z56). The figure was rendered using PyMOL (The PyMOL Molecular Graphics System; http://www.pymol.Org). B. Prediction of functional impact of SF3B1 missense mutations using PredictSNP. PredictSNP combined six best computational tools (MAPP, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT and SNAP) to predict the effect of a substitution of amino acid. The colors red and green indicate that the substitution is predicted respectivey deleterious and neutral. A confidence score estimated with a percentage is indicated for each mutation and prediction tool. The PredictSNP’s confidence score is assessed from the confidence scores of six individual tools. C. Relative impact of SF3B1 mutations measured by SBT scores obtained for the TCGA RNA-sequencing data. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We then opted to assess the occurrence of SF3B1 mutations based on the Sequence Bloom Tree (SBT) approach we used previously for scanning the TCGA for the functional SF3B1 mutations [20], [27], [28]. SBT is an indexing structure calculated based on the fastq files (thus avoiding alignment step), which allows fast and sensitive screening of the large datasets for a specific short sequences [20], [27], [28]. SBT score represents the number of pre-defined SF3B1Mut-specific aberrant junctions found at least once in raw RNA-sequencing data (Supplementary Table 3, [20], [27], [28]). SBT scores were validated by direct analysis of junction expression [20], [27], [28] and here we used these SBT scores to estimate the impact of SF3B1 mutations.
While exploring SBT scores for the set of SF3B1 mutations, we observed that the confounding effects of RNA-sequencing coverage and SF3B1 mutation VAF (variant allele frequency) on SBT scores could be efficiently modelled and the most frequent mutations K700E and R625H/C could be used as a baseline to estimate the impact of other mutations (Supplementary Fig. 1). Indeed, we found that correlation coefficient between SBT scores and million reads per SF3B1 mutated allele (an index characterizing the tumor sample and prevalence of SF3B1 mutation) equals 0.87 (Pearson correlation) and corresponding linear model had a perfect fit (R2 = 0.76, Fisher test p < 10−10). We estimated the relative impact of all mutations as the difference between mutation SBT score and the predicted value divided by the predicted value (Fig. 2C, Supplementary Table 3).
Based on this analysis, R625H/C and K700E mutations seem to induce high aberrant splice burden together with other mutations including D781E. The lowest aberrant splice burden was obtained for I665F and A633V. Furthermore, different substitutions on K741 (E, N, Q) are intriguingly associated with different intensities of the aberrant splicing. Thus, SBT analysis does not reflect in-silico predictions. Of note, SBT scores were obtained based on the splicing pattern defined in datasets enriched in SF3B1R625H and SF3B1K700E tumors [8], [29], however, all further unsupervised analysis of the splice junctions were consistent with these SBT scores.
3.2. Alternative 3′ss usage is a marker of functional impact of SF3B1 mutations
We then evaluated in cellulo the functional impact of SF3B1 amino acid substitutions (R625H, H662R, I665F, K666T, K700E, G740E, K741E, K741N, K741Q, G742D, D781E) by assessing the relative expression of aberrantly-spliced transcripts of DPH5, DLST, ENOSF1 and ARMC9 as markers of the aberrant splice pattern detected in SF3B1Mut tumors [8]. HEK293T (SF3B1WT) cell line was transiently transfected with pCMV-3tag-1A expression vectors for wild-type SF3B1 (SF3B1WT), for SF3B1 missense mutations mentioned above and for SF3B1 in which the HEAT repeats included between amino acids 622 and 781 are deleted (SF3B1Del (622−781)). Transfection levels were controlled by immunoblotting (Fig. 3A). We evaluated the aberrant splice index (AG’/AG) based on the mRNA expression ratio of the aberrantly-spliced transcript (AG’) to the canonical transcript (AG) as determined by quantitative reverse transcription (RT)-PCR (Fig. 3B). While the overexpression of SF3B1WT did not have any significant effect on AG’/AG index, the overexpression of hotspot mutants (SF3B1R625H, SF3B1K666T, SF3B1K700E) significantly increased the AG’/AG index in HEK293T cell line. Of note, SF3B1R625H induced a considerably high AG’/AG index as compared to SF3B1K666T and SF3B1K700E mutants. The observed differences in AG’/AG index between the SF3B1 hotspot mutants imply variable intensities of the aberrant splice pattern induced by the different mutants. To validate this finding, we introduced the hotspot mutants at increasing ranges of expression levels in HEK293T cells (Supplementary Fig. 2A). Our findings show that the intensity of the aberrant splice pattern significantly varies between the hotspot mutations with R625H presenting the highest intensity of aberrant splice pattern (Supplementary Fig. 2B). Of note, the experimentally determined AG’/AG index correlated with SBT scores suggesting that low expression of aberrant transcripts may limit their detection, and vice versa. RNA sequencing of HEK293T cells transfected with SF3B1WT or for SF3B1R625H, K666T, K700E allowed a further validation on a panel of previously validated SF3B1Mut-aberrant splice events [8], [42], [43], [44] (Fig. 3C, Supplementary Fig. 3).
Fig. 3.

In-cellulo evaluation of the aberrant splice pattern of SF3B1 mutants. A. Protein expression analysis of exogenous flagged-SF3B1 and endogenous SF3B1 in transfected HEK293T cells by immunoblotting with anti-Flag and anti-SF3B1, respectively. ß-actin antibody was used as a loading control. B. Effect of the various mutations of SF3B1 on the AG’/AG transcript expression ratio of DPH5, DLST, ENOSF1 and ARMC9 in HEK293T cells. Ratios of expression levels of the alternative AG’ and canonical AG forms (AG’/ AG) were determined by RT-qPCR. The results are average of three replicates and are represented as mean ± sd. Paired t-test was used to generate the p-values comparing each condition to the SF3B1 wild-type vector transfection (SF3B1-WT): *p < 0.05; **p < 0.005; ***p < 0.0005. C. Heatmap of ratios of aberrant to canonical 3′ss junction expression (AG’/AG index) for a panel of previously validated SF3B1Mut-aberrant splice events in HEK293T cells transfected with the SF3B1 wild-type or hotspot mutants (left panel), and examples of IGV visualizations of SF3B1Mut-aberrant splice events (right panel).
Strikingly, our results show that the different amino acid substitutions on the codon 741 (K741E, K741N, K741Q) lead to varying aberrant splice index. As shown in Fig. 3B, SF3B1K741Q induces a considerably low AG’/AG index as compared to SF3B1K741N and SF3B1K741E. Such finding implies that the intensity of the aberrant splice pattern of SF3B1 mutants depends on the type of the substituting amino acid, in agreement with SBT analysis.
Interestingly, the I665F mutation does not induce the aberrant splice pattern despite its vicinity to the K666T hotspot mutation. Similarly, the aberrant splice pattern was not induced by SF3B1Del (622−781) mutant, which confirms that the pattern-inducing mutations lead to a change of function rather than a loss of function of the mutated SF3B1 region.
Altogether, these results provide evidence that SF3B1 mutants present different consequences on splicing and suggest that the burden and intensity of aberrant splice pattern are dependent on SF3B1 amino acid substitutions.
3.3. Structural impact of SF3B1 amino acid substitutions reflects the functional impact of SF3B1 mutations
In order to better understand the differential intensities of aberrant splice pattern induced by SF3B1 mutants, we analysed the structural impact of SF3B1 hotspot mutations (R625H, K666T, K700E) and that of the different amino acid substitutions on the codon 741 (K741Q, K741N, K741E). The I665F mutation, which does not induce the aberrant splice pattern, was also analysed. As shown in Fig. 4A and B, the side chains of the hotspot amino acids (R625, K666, K700) are all oriented towards the pre-mRNA, whereas the side chain of isoleucine (I665) is oriented toward the protein surface. Considering the structural and biochemical characteristics of amino acids at physiological pH (pH ~ 7), the side chain of arginine at position 625 (R625) contains three carbons followed by a guanidinium group, making arginine a positively charged aliphatic amino acid. When a hotspot mutation replaces arginine by histidine (R625H), its side chain becomes shorter with an imidazole ring (Fig. 4C). Therefore, the substitution R625H loses its proximity to pre-mRNA and its polarity as compared to R625 (Fig. 4B and C), which may explain the high AG’/AG index induced by R625H.
Fig. 4.

Structural impact of amino acid substitutions of SF3B1 in the activated spliceosome (Bact complex) (PDB ID: 5Z56). A. Overview of wild-type SF3B1 amino acids (R625, H662, I665, K666, K700, G740, K741, G742, D781) with a zoom (black box) on the hotspot region (R625, H662, I665, K666 and K700E). The amino acids susceptible to damaging substitutions are R625, H662, K666, K700, G740, K741, G742, D781 and their carbon chains are represented in pink. The carbon chain of the amino acid I665 susceptible to substitution with no functional impact is represented in green. B-F. A zoom on the amino acids R625, H662, I665, K666 and K700 (B) and the corresponding mutations R625H (C), K666T (D), K700E (E) and I665F (F). The carbon chains of substitutions are represented in light blue. G. Representation of wild-type SF3B1 amino acids (R625, H662, I665, K666, K700, G740, K741, G742, D781) and an overview (red box) of the residue K741. The amino acids susceptible to damaging substitutions are R625, H662, K666, K700, G740, K741, G742, D781 and their carbon chains are represented in pink. The carbon chain of the amino acid I665 susceptible to substitution with no functional impact is represented in green. A zoom on the amino acid K741 and its substitutions K741Q, K741N and K741E. The carbon chains of substitutions are represented in light blue. (+): positively charged amino acid; (-): negatively charged amino acid. Oxygen (O), nitrogen (N) and pre-mRNA are shown respectively in red, dark blue and yellow. The residues are shown as stick in elemental colors and the figure was rendered using PyMOL. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
On the other hand, lysine (K) at positions 666 and 700 is positively charged and very polar (Fig. 4B). SF3B1 hotspot mutations K666T and K700E radically alter the residue charge as threonine (T) is uncharged and glutamate (E) is negatively charged. Of note, the two substitutions K666T and K700E maintain their polarity compared to that of lysine (Fig. 4B, D and E), possibly resulting in a lower impact of K666T and K700E as compared to R625H.
Regarding the isoleucine (I) at position 665 (I665), the hydrocarbon side chain makes isoleucine a hydrophobic amino acid (Fig. 4B). A substitution of isoleucine by a phenylalanine (I665F) results in a side chain with an aromatic ring while preserving its hydrophobicity (Fig. 4F), which may explain the absence of functional impact of this particular mutation.
At the codon K741, we investigated the three amino acid substitutions (K741Q, K741N, K741E). As shown in Fig. 4G, lysine (K) is a positively-charged amino acid with 4 carbons in its side chain while glutamine (Q) is uncharged and has 3 carbons. Aspargine (N) is also uncharged but its side chain has only 2 carbons. This observation implies closer length of the side chain of K741Q to the wild-type codon as compared to that of K741N, which explains the low AG’/AG index of K741Q as compared to K741N. On the other hand, the carboxyl group of glutamate (E) confers a negative charge to its side chain which makes it radically different from lysine (K), explaining the high AG’/AG index induced by K741E.
Overall, these findings suggest that the codon position and its proximity to the pre-mRNA, as well as the charge and polarity change caused by the residue substitution are all determinant factors of the resulting splice pattern intensity. The length of the side chain is also to be considered as a minor determinant of the impact.
3.4. Consequences of SF3B1 mutations on its tertiary structure
In order to assess the consequences of SF3B1 mutations on the protein conformation, we used the in-silico tool I-TASSER to predict the tertiary structure of SF3B1 mutants [32], [33], [34], [35], [36]. We analyzed SF3B1 hotspot mutants (R625H, K666T, K700E), the different amino acid substitutions at codon K471 (Q, N or E), and I665F which does not induce the aberrant splice pattern. Local accuracy and confidence scores estimating the quality of the predicted models by I-TASSER are reported in Supplementary Tables 4 and 5. We compared the tertiary structure of each SF3B1 mutant to that of SF3B1 wild-type protein. RMSD (Root-Mean-Square Deviation) value assesses the quality of the superposition of two protein 3D (three-dimensional) structures [38]. The lower the RMSD value, the better the superposition/alignment of two 3D protein structures and the more similar the conformation of two proteins will be. Fig. 5A and Supplementary Fig. 4 show that all of the tested SF3B1 mutants induce a conformational change at the N-terminal domain of the protein. RMSD values of the superpositions between SF3B1 mutants and wild-type varied from 0.235 Å to 0.335 Å. Strikingly, the intensity of aberrant splice pattern induced by the different amino acid substitutions at the codon 741 (K741Q, K741N, K741E) correlated positively with RMSD (Pearson correlation coefficient r = 0.99, Fig. 5B), indicating that the functional impact of mutations correlates positively with the extent of the associated change of conformation of the protein. Furthermore, SF3B1I665F, which does not induce the aberrant splice pattern, results in a less significant change in the 3D structure with a low RMSD (0.235 Å) as compared to the other mutants inducing the aberrant splice signature. Accordingly, the predicted conformation of SF3B1I665F is closer to that of SF3B1WT than those of other mutants. We further analyzed the hypothetical mutations K700R (a variant with physico-chemical properties similar to wild-type), K700G and I665C (two variants that are radically different from the wild-type). Interestingly, we found the RMSD value of K700R (0.270 Å) to be lower than that of K700E (0.274 Å), in contrast to the RMSD of K700G (0.293 Å). Additionally, the RMSD of I665C (0.322 Å) is higher than that of I665F (0.235 Å), which is in line with our physico-chemical model.
Fig. 5.

Functional impact of SF3B1 mutations on SF3B1 protein (PDB ID: 6AHD1). A. Representation of the alignment of wild-type SF3B1 structure (green) with that of SF3B1 mutant predicted proteins: R625H (purple), K666T (pink), K700E (magenta), K741E (red), K741N (blue), K741Q (orange), I665F (yellow). On the right panels: representation of the N-terminal domain of wild-type SF3B1 (green) and SF3B1 mutants: R625H (purple), K666T (pink), K700E (magenta), K741E (red), K741N (blue), K741Q (orange), I665F (yellow). The structures were visualized using PyMOL. For each superposition between SF3B1 mutant and the wild-type, an RMSD value was indicated for evaluating the quality of alignment between the two proteins. B. Correlation between the AG’/AG transcript expression ratio of DPH5 and the RMSD of SF3B1 mutations (I665F, K741E, K741N, K741Q). Ratios of expression levels of the alternative AG’ and canonical AG forms (AG’/ AG) were determined by RT-qPCR. A linear correlation between AG’/AG index and RMSD is represented with an orange line. Orange spheres represent the RMSD value for each mutant. RMSD value is obtained from the alignment between wild-type SF3B1 and the corresponding SF3B1 mutant using PyMOL. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Collectively, our results reveal that the studied SF3B1 mutations have a conformational impact on the N-terminal domain of SF3B1 although they are localized in the C-terminal HEAT domain of SF3B1. Our findings suggest also that the aberrant splice pattern correlates with the extent of the conformational change.
4. Discussion
SF3B1 is the most frequently mutated splicing gene in cancers. SF3B1 hotspot mutations affect codons R625, K666 and K700 inducing an aberrant splice pattern characterized by the usage of alternative 3′ss (AG’) upstream of the canonical 3′ss (AG) in a subset of pre-mRNA [8], [29], [30]. Previous studies addressed the aberrant splice pattern induced by SF3B1 hotspot mutations as compared to the canonical splice pattern associated with wild-type SF3B1. Mutations targeting other codons of SF3B1 have also been described, but little is known about their consequences on splicing and their biological contribution to oncogenesis [45]. Here, we addressed the functional and structural impact of SF3B1 recurrent mutations in the hotspot region of the HEAT domain of SF3B1 to determine whether these mutations equally impact the splicing factor function, and thereby comparably contribute to tumorigenesis. This work is the first to compare the consequences of SF3B1 mutations on splicing in a homogeneous experimental context.
Our results indicate that all studied SF3B1 missense mutations induce the expression of the aberrant form AG’, except for SF3B1I665F. This finding could not be predicted by the in-silico algorithms based on the structural and physico-chemical characteristics, but rather by the SBT scores highlighting the aberrant splice pattern as a reliable marker of functional impact of SF3B1 mutations. Our results also show variations in the intensity of the induced aberrant splice pattern between the SF3B1 mutations, suggesting a variable functional impact of the mutants. These data are in line with recent findings suggesting a differential usage of cryptic 3′ss between SF3B1K700E and SF3B1R625H mutations in different tumor samples [25].
Our molecular visualization analysis suggests that R625H and K666T induce a loss of proximity to the pre-mRNA. Indeed, a substitution of an arginine by a histidine at position 625 may result in steric clash that would prevent intramolecular hydrogen bonds in which R625 is involved [22]. Similarly, regarding K666T, the hydroxyl group of threonine could be a target of post-translational modifications, therefore may prevent intramolecular hydrogen bonds in which K666 is also involved. Substitutions of these residues may lead to a conformational change in the U2 snRNP complex, which would decrease or alter the interaction of SF3B1 with pre-mRNA and other components such as Prp5, p14 and splicing factors like U2AF65. This could lead to alternative BP recognition and subsequent cryptic 3′ss selection giving rise to aberrant transcript expression [8], [29], [30], [46]. It has been shown that K700E has no impact on the stability of SF3b-U2AF65 complex, and does not decrease its affinity towards pre-mRNA, suggesting that other spliceosomal proteins may be involved in the mechanism [22]. Recently, the 3D cryo-electron microscopy structure of the human 17S U2 snRNP showed that human PRP5 (DDX46) encompasses the entire HEAT domain of SF3B1 in U2 snRNP to facilitate the formation of a stable U2–BP interaction [46]. SF3B1 mutations may induce a change in the curvature of the HEAT domain, resulting in the destabilization of the SF3B1–PRP5 interaction [46]. Such destabilization may lead to the selection of aberrant BP with higher base-pairing affinity to U2 snRNP and subsequently the recognition of aberrant 3′ ss. Zhang et al. demonstrated that K700E disrupts SF3B1 interaction with another splicing factor called SUGP1, resulting in splicing defects and production of aberrant mRNA [19]. Moreover, residues (529–880 AA) within HEAT repeats of SF3B1 are exposed to solvent supporting that hotspot mutations may disrupt SF3B1 interactions with other splicing factors altering its selectivity for BP [22]. In contrast, I665F mutation does not induce the aberrant splice pattern even though it is adjacent to K666T hotspot mutation. Actually, isoleucine and phenylalanine at position 665 are both oriented towards the protein surface, implying that isoleucine at position 665 would not have a key role in the interaction of U2 snRNP complex with BP. Furthermore, the hydrophobic side chain of isoleucine and phenylalanine may have limited impact on the conformation of SF3B1 and would not prevent its interaction with other spliceosomal proteins or SF3b subunits. In addition to the key role of proximity of SF3B1 mutants towards pre-mRNA, our findings indicate that the charge of amino acid substitution is also determinant for the aberrant splice pattern. Hence, we demonstrate that the codon position as well as the type of substitution are both determinant for the burden of the aberrant splice pattern induced by SF3B1 mutations.
Based on crystallography analysis, Cretu et al. reported that many residues mutated in cancer cells (mostly clustered in HEAT repeats H4-H7) are involved in the tertiary structure of SF3B1 [22]. Interestingly, our results suggest that all the studied SF3B1 mutations induce a conformational change at the N-terminal domain of SF3B1, known to be the binding domain with other splicing factors. A recent work demonstrated that TAT-SF1, which is involved in the earliest stage of spliceosome assembly, interacts with the N-terminal domain of SF3B1 to stabilize the SF3B1 open conformation and must be displaced for a stable interaction between U2 snRNP complex and BP sequences [46]. A conformational change at the N-terminal region of SF3B1 may destabilize the removal of TAT-SF1, diminishing the interaction of SF3B1 with pre-mRNA and leading to the selection of alternative BP sequences that causes expression of aberrant transcripts [46]. Moreover, our data imply that the extent of the conformational change in the N-terminal domain correlates positively with the intensity of aberrant splice pattern.
5. Conclusions
In conclusion, cancer-associated SF3B1 mutations may present different functional consequences, and thereby varying pathogenic impact and prognosis potentials, depending on the position and the type of the amino acid substitution. Our study highlights also the aberrant splice pattern as a reporter of the functional impact of mutations of SF3B1 or other splicing genes and exposes the limits of in-silico prediction tools. Further work is required to understand the molecular mechanism by which SF3B1 HEAT domain tolerates conformational changes induced by missense mutations to develop clinical applications in UM and other diseases harboring SF3B1 mutations.
CRediT authorship contribution statement
Christine Canbezdi: Data curation, formal analysis, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing - original draft, Writing - review & editing. Malcy Tarin: Validation, Writing - review & editing. Alexandre Houy: Data curation, formal analysis, Methodology, Resources, Software, Visualization. Dorine Bellanger: Methodology, Validation, Writing - review & editing. Tatiana Popova: Data curation, formal analysis, Methodology, Resources, Software, Visualization, Writing - review & editing. Marc-Henri Stern: Methodology, Project administration, Resources, Writing - review & editing. Sergio Roman-Roman: Data curation, formal analysis, Funding acquisition, Project administration, Resources, Writing - review & editing. Samar Alsafadi: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing - original draft, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The translational group of UM at Institut Curie is supported by SIRIC Curie (Grant INCa-DGOSInserm_12554) and the European Union’s Horizon 2020 project “UM Cure 2020” (grant no. 667787). This work benefits also from financial support of the Ligue Nationale Contre le Cancer (PhD funding for CC, grant no. 20117). This work benefits also from a collaboration with The Seven Bridges Cancer Genomics Cloud which has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, Contract No. HHSN261201400008C and ID/IQ Agreement No. 17X146 under Contract No. HHSN261201500003I.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.02.012.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Quesada V., Conde L., Villamor N., Ordóñez G.R., Jares P., Bassaganyas L. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44(1):47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida K., Sanada M., Shiraishi Y., Nowak D., Nagata Y., Yamamoto R. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 3.Wang L., Lawrence M.S., Wan Y., Stojanov P., Sougnez C., Stevenson K. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497–2506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J., Manley J.L. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013;3(11):1228–1237. doi: 10.1158/2159-8290.CD-13-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida K., Ogawa S. Splicing factor mutations and cancer. Wiley Interdiscip Rev RNA. 2014;5(4):445–459. doi: 10.1002/wrna.1222. [DOI] [PubMed] [Google Scholar]
- 6.Furney S.J., Pedersen M., Gentien D., Dumont A.G., Rapinat A., Desjardins L. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013;3(10):1122–1129. doi: 10.1158/2159-8290.CD-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harbour J.W., Roberson E.D.O., Anbunathan H., Onken M.D., Worley L.A., Bowcock A.M. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013;45(2):133–135. doi: 10.1038/ng.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alsafadi S., Houy A., Battistella A., Popova T., Wassef M., Henry E. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016;7(1) doi: 10.1038/ncomms10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maguire S.L., Leonidou A., Wai P., Marchiò C., Ng C.K.Y., Sapino A. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol. 2015 doi: 10.1002/path.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong Y, Krauthammer M, Halaban R. Rare SF3B1 R625 mutations in cutaneous melanoma. Melanoma Res 2014. https://doi.org/10.1097/CMR.0000000000000071. [DOI] [PMC free article] [PubMed]
- 11.Liu Y., Zhang Y.u., Feng G., Niu Q., Xu S., Yan Y. Comparison of effectiveness and adverse effects of gefitinib, erlotinib and icotinib among patients with non-small cell lung cancer: a network meta-analysis. Exp Ther Med. 2017 doi: 10.3892/etm10.3892/etm.2017.5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu J., Chen Z., Yong L. Systematic profiling of alternative splicing signature reveals prognostic predictor for ovarian cancer. Gynecol Oncol. 2018;148(2):368–374. doi: 10.1016/j.ygyno.2017.11.028. [DOI] [PubMed] [Google Scholar]
- 13.Bjørklund S.S., Panda A., Kumar S., Seiler M., Robinson D., Gheeya J. Widespread alternative exon usage in clinically distinct subtypes of Invasive Ductal Carcinoma. Sci Rep. 2017;7(1) doi: 10.1038/s41598-017-05537-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robertson A.G., Shih J., Yau C., Gibb E.A., Oba J., Mungall K.L. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell. 2017;32(2):204–220.e15. doi: 10.1016/j.ccell.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marcelino Meliso F., Hubert C.G., Favoretto Galante P.A., Penalva L.O. RNA processing as an alternative route to attack glioblastoma. Hum Genet. 2017;136(9):1129–1141. doi: 10.1007/s00439-017-1819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kahles A., Lehmann K.-V., Toussaint N.C., Hüser M., Stark S.G., Sachsenberg T. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. 2018;34(2):211–224.e6. doi: 10.1016/j.ccell.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seiler M., Yoshimi A., Darman R., Chan B., Keaney G., Thomas M. H3B–8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med. 2018;24(4):497–504. doi: 10.1038/nm.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gozani O.r., Potashkin J., Reed R. A potential role for U2AF-SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol Cell Biol. 1998;18(8):4752–4760. doi: 10.1128/MCB.18.8.4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J., Ali A.M., Lieu Y.K., Liu Z., Gao J., Rabadan R. Disease-causing mutations in SF3B1 alter splicing by disrupting interaction with SUGP1. Mol Cell. 2019;76(1):82–95.e7. doi: 10.1016/j.molcel.2019.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alsafadi S., Dayot S., Tarin M., Houy A., Bellanger D., Cornella M. Genetic alterations of SUGP1 mimic mutant-SF3B1 splice pattern in lung adenocarcinoma and other cancers. Oncogene. 2021;40(1):85–96. doi: 10.1038/s41388-020-01507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z., Zhang J., Sun Y., Perea-Chamblee T.E., Manley J.L., Rabadan R. Pan-cancer analysis identifies mutations in SUGP1 that recapitulate mutant SF3B1 splicing dysregulation. Proc Natl Acad Sci U S A. 2020;117(19):10305–10312. doi: 10.1073/pnas.1922622117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cretu C., Schmitzová J., Ponce-Salvatierra A., Dybkov O., De Laurentiis E., Sharma K. Molecular architecture of SF3b and structural consequences of its cancer-related mutations. Mol Cell. 2016;64(2):307–319. doi: 10.1016/j.molcel.2016.08.036. [DOI] [PubMed] [Google Scholar]
- 23.Taylor J, Mi X, North K, Binder M, Penson A, Lasho T, et al. Single-cell genomics reveals the genetic and molecular bases for escape from mutational epistasis in myeloid neoplasms. Blood 2020. https://doi.org/10.1182/blood.2020006868. [DOI] [PMC free article] [PubMed]
- 24.Seiler M., Peng S., Agrawal A.A., Palacino J., Teng T., Zhu P. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 2018;23(1):282–296.e4. doi: 10.1016/j.celrep.2018.01.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Z., Yoshimi A., Wang J., Cho H., Chun-Wei Lee S., Ki M. Mutations in the RNA splicing factor SF3B1 promote tumorigenesis through MYC stabilization. Cancer Discov. 2020;10(6):806–821. doi: 10.1158/2159-8290.CD-19-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brian Dalton W, Helmenstine E, Pieterse L, Li B, Gocke CD, Donaldson J, et al. The K666N mutation in SF3B1 is associated with increased progression of MDS and distinct RNA splicing. Blood Adv 2020. https://doi.org/10.1182/bloodadvances.2019001127. [DOI] [PMC free article] [PubMed]
- 27.Lau J.W., Lehnert E., Sethi A., Malhotra R., Kaushik G., Onder Z. The cancer genomics cloud: Collaborative, reproducible, and democratized - a new paradigm in large-scale computational research. Cancer Res. 2017;77(21):e3–e6. doi: 10.1158/0008-5472.CAN-17-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dehghannasiri R., Freeman D.E., Jordanski M., Hsieh G.L., Damljanovic A., Lehnert E. Improved detection of gene fusions by applying statistical methods reveals oncogenic RNA cancer drivers. Proc Natl Acad Sci U S A. 2019;116(31):15524–15533. doi: 10.1073/pnas.1900391116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darman R., Seiler M., Agrawal A., Lim K., Peng S., Aird D. Cancer-associated SF3B1 hotspot mutations induce Cryptic 3’ splice site selection through use of a different branch point. Cell Rep. 2015;13(5):1033–1045. doi: 10.1016/j.celrep.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 30.DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, et al. Transcriptome sequencing reveals potential mechanism of cryptic 3’ splice site selection in SF3B1-mutated cancers. PLoS Comput Biol 2015. https://doi.org/10.1371/journal.pcbi.1004105. [DOI] [PMC free article] [PubMed]
- 31.Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, et al. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol 2014;10:e1003440. https://doi.org/10.1371/journal.pcbi.1003440. [DOI] [PMC free article] [PubMed]
- 32.Yang J., Zhang Y. Protein structure and function prediction using I-TASSER. Curr Protoc Bioinforma. 2015;52(1) doi: 10.1002/0471250953.2015.52.issue-110.1002/0471250953.bi0508s52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y. The I-TASSER suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J., Wang Y., Zhang Y. ResQ: an approach to unified estimation of B-factor and residue-specific error in protein structure prediction. J Mol Biol. 2016;428(4):693–701. doi: 10.1016/j.jmb.2015.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinf. 2008;9(1) doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roy A., Kucukural A., Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y., Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins Struct Funct Genet. 2004;57(4):702–710. doi: 10.1002/(ISSN)1097-013410.1002/prot.v57:410.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 38.Kufareva I., Abagyan R. Methods of protein structure comparison. Methods Mol Biol. 2012 doi: 10.1007/978-1-61779-588-6_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(27–28):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 40.Roberts E., Eargle J., Wright D., Luthey-Schulten Z. MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinf. 2006;7:382. doi: 10.1186/1471-2105-7-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eargle J., Wright D., Luthey-Schulten Z. Multiple alignment of protein structures and sequences for VMD. Bioinformatics. 2006;22:504–506. doi: 10.1093/bioinformatics/bti825. [DOI] [PubMed] [Google Scholar]
- 42.Wang L., Brooks A., Fan J., Wan Y., Gambe R., Li S. Transcriptomic characterization of SF3B1 mutation reveals its pleiotropic effects in chronic lymphocytic leukemia. Cancer Cell. 2016;30(5):750–763. doi: 10.1016/j.ccell.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pozzo F, Bittolo T, Tissino E, Vit F, Vendramini E, Laurenti L, et al. SF3B1-mutated chronic lymphocytic leukemia shows evidence of NOTCH1 pathway activation including CD20 downregulation. Haematologica 2020;Online ahe. https://doi.org/10.3324/haematol.2020.261891. [DOI] [PMC free article] [PubMed]
- 44.Dolatshad H., Pellagatti A., Fernandez-Mercado M., Yip B.H., Malcovati L., Attwood M. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia. 2015;29(5):1092–1103. doi: 10.1038/leu.2014.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Damm F., Nguyen-Khac F., Fontenay M., Bernard O.A. Spliceosome and other novel mutations in chronic lymphocytic leukemia and myeloid malignancies. Leukemia. 2012;26(9):2027–2031. doi: 10.1038/leu.2012.86. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Z., Will C.L., Bertram K., Dybkov O., Hartmuth K., Agafonov D.E. Molecular architecture of the human 17S U2 snRNP. Nature. 2020;583(7815):310–313. doi: 10.1038/s41586-020-2344-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.