

Summary
Significant advancements in understanding disease mechanisms can occur through combined analysis of next-generation sequencing datasets generated using purified cell populations. Here, we detail our optimized protocol for purification of mouse hepatic macrophages (or other liver non-parenchymal populations) suitable for use in various next-generation sequencing protocols. An alternative framework is described for sorting pre-fixed hepatic nuclei populations. This strategy has the advantage of rapidly preserving the nuclei and can facilitate success with ChIP-seq for more challenging molecules.
For complete details on the use and execution of these protocols, please refer to Muse et al. (2018), Sakai et al. (2019), and Seidman et al. (2020).
Subject areas: Cell isolation, Flow cytometry/mass cytometry, Sequencing, ChIP-seq, High-throughput screening, Immunology, Chromatin immunoprecipitation (ChIP)
Graphical abstract
Highlights
-
•
Isolation of high-purity hepatic non-parenchymal cells or hepatic nuclei using FACS
-
•
Recovered cells are suitable for (sc)RNA-seq, (sc)ATAC-seq, ChIP-seq, etc
-
•
Recovered nuclei are suitable for ChIP-seq, chromatin conformation assays, etc
Significant advancements in understanding disease mechanisms can occur through combined analysis of next-generation sequencing datasets generated using purified cell populations. Here, we detail our optimized protocol for purification of mouse hepatic macrophages (or other liver non-parenchymal populations) suitable for use in various next-generation sequencing protocols. An alternative framework is described for sorting pre-fixed hepatic nuclei populations. This strategy has the advantage of rapidly preserving the nuclei and can facilitate success with ChIP-seq for more challenging molecules.
Before you begin
Experimental considerations
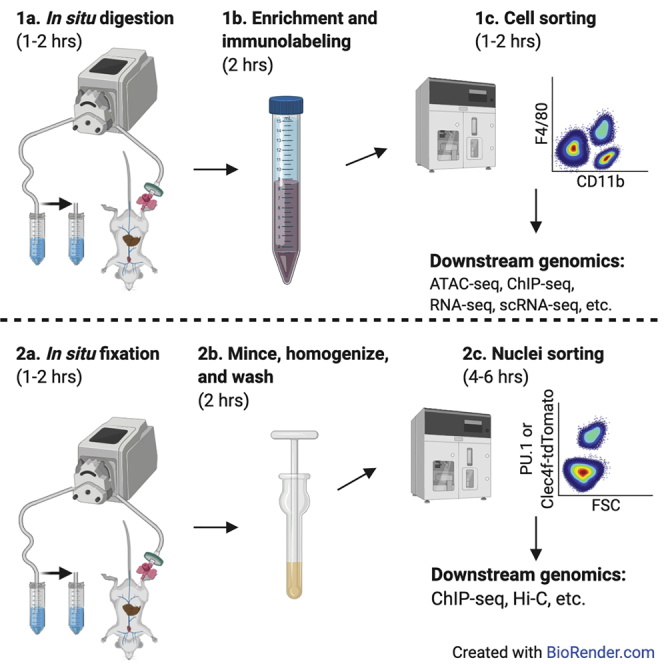
This protocol contains two modular strategies for preparing mouse liver cell populations for chromatin immunoprecipitation with sequencing (ChIP-seq), or other next-generation sequencing assays. In the first module, we describe an optimized protocol for preparing single-cell suspensions of mouse liver non-parenchymal cells (NPCs) for immunolabeling with fluorescent antibodies and population purification using fluorescence-activated cell sorting (FACS). We describe in detail a strategy for FACS purification of Kupffer cells and provide abbreviated examples of FACS strategies for purification of either liver sinusoidal endothelial cells or hepatic stellate cells. Additional cell populations can also be defined as suited. At the end of this module, the cells are ready for use in diverse next-generation sequencing assays. Examples are described in processing FACS-purified cells for RNA-seq, ATAC-seq, or ChIP-seq.
The second module describes an alternative strategy for cell population purification better suited for ChIP-seq of challenging transcriptional regulators. The rationale for this is that preparation of cells and purification by FACS can take many hours, in which time transcriptional regulators can dissociate from the genome. Instead, in this module mouse livers are perfused immediately after euthanasia with fixative and nuclei are prepared for sorting. This module contains a detailed strategy for purification of Kupffer cell nuclei using either a genetic nuclear reporter or immunolabeling of nuclei with fluorescent antibodies to the macrophage lineage determining transcription factor PU.1. This strategy can be extended to other liver populations of interest. Samples from either of these strategies are suitable for use in an extensively validated ChIP-seq library preparation protocol described by Texari et al.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse/human CD11b PE (clone M1/70) | BioLegend | 101208; RRID: AB_312791 |
| Anti-mouse CD146 PE/Cy7 (clone ME-9F1) | BioLegend | 134714; RRID: AB_2563109 |
| Anti-mouse CD16/32 (clone 93) | BioLegend | 101302; RRID: AB_312801 |
| Anti-mouse CD45 BB515 (clone 30-F11) | BD Biosciences | 564590; RRID: none |
| Anti-mouse F4/80 BV421 (clone BM8) | BioLegend | 123132; RRID: AB_11203717 |
| Anti-mouse Tim4 Alexa647 (clone RMT4-54) | BioLegend | 130008; RRID: AB_2271648 |
| Anti-PU.1 Alexa647 (7C2C34) | BioLegend | 681304; RRID:AB_2566591 |
| Chemicals, peptides, and recombinant proteins | ||
| 0.5 M EGTA, pH 8.0, DNase- and RNase-free (ethyleneglycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid) | bioWORLD | 40121266-3 |
| 0.5 M UltraPure EDTA, pH 8.0 (ethylenediaminetetraacetic acid) | Thermo Fisher | 15575020 |
| 10% Tween 20 | Teknova | T0027; CAS: 900564-5 |
| 1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) | Teknova | H1090 |
| 1 M magnesium chloride | Millipore Sigma | 63069 |
| 1 M UltraPure Tris-HCl pH 8.0 | Invitrogen | 15568-025 |
| 1× RBC lysis buffer | Invitrogen | 00-4333-57 |
| Dimethyl sulfoxide (DMSO) | Millipore Sigma | D8418 |
| Disuccinimidyl glutarate (DSG) | ProteoChem | c1104-100mg; CAS: 79642-50-5 |
| DNase I | Worthington | LS002139 |
| Fetal bovine serum (FBS) | NA | NA |
| Flavopiridol | Sigma-Aldrich | F3055; CAS: 131740-09-5 |
| Formaldehyde | Thermo Fisher Scientific | BP531-500; CAS: 7732-18-5, 50-00-0, 67-56-1 |
| Glycine | Fisher Scientific | BP381-5 |
| Hank’s balanced salt solution (no calcium, no magnesium, no phenol red) | Thermo Fisher | 14175095 |
| Hank’s balanced salt solution (with calcium, with magnesium, no phenol red) | Thermo Fisher | 14025092 |
| HBSS (10×), no calcium, no magnesium, no phenol red | Thermo Fisher | 14185052 |
| IGEPAL CA-630 | Millipore Sigma | I8896 |
| Invitrogen OneComp eBeads | Thermo Fisher | 01-1111-42 |
| Liberase™ | Sigma-Aldrich | 5401127001 |
| OptiPrep | Sigma-Aldrich | D1556; CAS: 92339-11-2 |
| Percoll | Sigma-Aldrich | P1644; CAS: 65455-52-9 |
| Phosphate buffered saline (PBS) | Millipore Sigma | 806552-500ML |
| Sucrose | MP Biomedicals | 194747 |
| Surgical glue (Loctite 4013) | All-Spec | 237041-30769 |
| Tris-EDTA (TE) buffer, pH 8.0 | Invitrogen | AM9849 |
| Triton X-100 | Millipore Sigma | T8787 |
| Trizol reagent (optional) | Thermo Fisher Scientific | 15596018; CAS: 108-95-2, 1762-95-4, 593-84-0 |
| UltraPure DNase/RNase-free distilled water | Invitrogen | 10977023 |
| Critical commercial assays | ||
| LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit | Invitrogen | L10119 |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6J-Clec4fem1(cre)Glass/J | Glass Lab | JAX stock #033296 |
| Software and algorithms | ||
| FlowJo | Becton, Dickinson & Company | https://www.flowjo.com/ |
| Summit | Beckman Coulter | NA |
| Sony Cell Sorter Software | Sony | NA |
| Other | ||
| 0.2 mL PCR Strip | NA | NA |
| 0.22 μm syringe filters | NA | NA |
| 1.5 inch 18-gauge needles | NA | NA |
| 1.5 mL DNA LoBind tubes | Eppendorf | 022431021 |
| 10 mL syringes | NA | NA |
| 15 mL conical tubes | NA | NA |
| 22-gauge needles | NA | NA |
| 24-gauge catheters | NA | NA |
| 50 mL conical tubes | NA | NA |
| Adjustable flow rate peristalsis pump | NA | NA |
| Adjustable temperature swinging bucket centrifuge | NA | NA |
| Cell sorter | NA | NA |
| Dissection tray | NA | NA |
| Falcon 40 μm cell strainer | Corning | 352340 |
| Falcon 100 μm cell strainer | Corning | 352360 |
| Falcon 5 mL round bottom polystyrene test tube, with cell strainer snap cap | Corning | 352235 |
| Heat lamp(s) | NA | NA |
| Incubator at 37°C | NA | NA |
| Liquid nitrogen or dry ice | NA | NA |
| MACSMix (or similar) | Miltenyi | 130-090-753 |
| Micropipettes and sterile DNase-/RNase-free filter tips | NA | NA |
| Nalgene Super Versi-dry surface protectors | Thermo Fisher Scientific | 74000-00 |
| Pasteur pipettes | NA | NA |
| Pipette aid | NA | NA |
| Primary IV solution set (Example: Baxter Healthcare Model: 2C8401) | NA | NA |
| Serological pipettes | NA | NA |
| Surgical glue (Loctite4013 or similar) | NA | NA |
| Sutures | NA | NA |
| Water bath at 37°C | NA | NA |
Materials and equipment
CRITICAL: Many chemicals used in this protocol are hazardous and/or toxic. Institutional safety guidelines should be followed for safe use and disposal.
Cell sorter
This equipment is necessary for Module 1: Preparation of hepatic Kupffer cells (or other NPCs) and Module 2, alternative strategy: in situ fixation and isolation of nuclei from Kupffer cells (or other liver cells). Cell sorters can be configured differently between institutes. Prior to beginning experiment, ensure the chosen fluorescent antibodies are suitable for the available equipment. It is recommended to use a cell sorter equipped with an ultraviolet laser to identify hepatic stellate cells using retinoid autofluorescence. Good results for FACS of cells prepared from Module 1 have been obtained using a BD FACSAria and a Beckman Coulter MoFlo Astrios. Good results for FACS of nuclei prepared from Module 2 have been obtained using a Sony SH800 and a Sony MA900.
DNase I (2 mg/mL)
This enzyme is used for Module 1: Preparation of hepatic Kupffer cells (or other NPCs). Prepare 100 mg lyophilized DNase I at 2 mg/mL in 50 mL ice cold phosphate buffered saline. Do not vortex or vigorously pipette! Store single use aliquots at −80°C. Aliquots of 10.5 mL are adequate for preparations of 8 healthy mice. Single use aliquots can be thawed in a 22°C–25°C water bath, then stored on ice immediately after thawing.
Flavopiridol (10 mM)
This reagent is used for Module 1: Preparation of hepatic Kupffer cells (or other NPCs). Prepare flavopiridol in dimethyl sulfoxide to 10 mM. Store single use aliquots at −80°C.
Glycine (2.625 M)
Place 9.85 g glycine in a 50 mL conical and add 45 mL 20°C–25°C UltraPure DNase/RNase-free distilled water. Mix (e.g., rocking, stir-bar) at 20°C–25°C until dissolved and adjust the final volume to 50 mL with 20°C–25°C UltraPure DNase/RNase-free distilled water. Store at 20°C–25°C.
Nuclei isolation buffer
This buffer is used for Module 2, alternative strategy: in situ fixation and isolation of nuclei from Kupffer cells (or other liver cells). Prepare nuclei isolation buffer at 20°C–25°C using the table below. Store at 4°C and discard after 2–3 weeks.
| Reagent | Final concentration | Amount |
|---|---|---|
| UltraPure DNase/RNase-free distilled water | 500 mL | |
| 1 M Tris (pH 8.0) | 0.5 mM | 500 μL |
| 0.5 M EDTA | 0.5 mM | 1 mL |
| 1 M magnesium chloride | 5 mM | 2.5 mL |
| Sucrose | 0.1 M | 17.11 g |
| 10% Triton X-100 | 0.05% | 2.5 mL |
| Total | NA | ~500 mL |
Nuclei sorting buffer
This buffer is used for Module 2, alternative strategy: in situ fixation and isolation of nuclei from Kupffer cells (or other liver cells). Prepare 500 mL nuclei sorting buffer by adding 5 mL 0.5 M EDTA to a new bottle of phosphate buffered saline. Store on ice or at 4°C. Discard after 1 week.
Peristalsis pump
Prepare necessary tubing for perfusion with the peristalsis pump. The tubing must permit transfer of liquids from a reservoir (example: media bottle, conical tubes) to a 22-gauge catheter. Primary IV solution sets are ideal for at the delivery end as they contain a bubble trap and a Luer lock for connecting to the catheter. Determine pump settings delivering 5 and 7 mL of fluid per minute.
Note: It is recommended to use tubing separately dedicated for each module to eliminate the risk of cross contamination. At the conclusion of module 1 experiments, clean the tubing by sequentially perfusing 500 mL of deionized or distilled water, 500 mL of 70% ethanol, and finally 500 mL of deionized or distilled water. At the conclusion of module 2 experiments, clean the tubing by sequentially perfusing 500 mL of deionized or distilled water, 500 mL of 0.1 M glycine, and finally 500 mL of deionized or distilled water.
Alternatives: A good option is the Cole Palmer Masterflex L/S Precision Variable-Speed Console Drive with a 4 or 8 channel pump head (1 channel per parallel perfused liver). It is more expensive but more adaptable long-term, with less flow-rate variability observed. Alternatively, the Fisherbrand Variable-Flow Peristaltic Pumps (1 pump unit per parallel-perfused liver) is an inexpensive single channel option, but each unit requires frequent calibration of the flow rate. Many additional options are available.
Triton X-100 (10%)
This reagent is necessary for and Module 2, alternative strategy: in situ fixation and isolation of nuclei from Kupffer cells (or other liver cells). Prepare 40 mL 10% Triton X-100 by adding 10 mL (using a 10 mL syringe) of the detergent to 35 mL UltraPure water in a 50 mL conical. Seal the tube with parafilm and rock overnight at 20°C–25°C. Adjust to a final 40 mL volume using UltraPure water and store at 4°C for up to 1 year.
Step-by-step method details
This protocol was developed specifically for isolation of hepatic macrophages and downstream use in diverse next-generation sequencing technologies. Conceptually this framework can easily be extended for other liver non-parenchymal cells (NPCs) including sinusoidal endothelial cells, stellate cells, or alternative lineages of immune cells.
Module 1: preparation of hepatic Kupffer cells (or other NPCs)
Timing: >10 h
Liver tissue is enzymatically digested in situ to generate a single-cell suspension enriched for liver non-parenchymal cells that are then immunolabeled for fluorescence-activated cells sorting (FACS). The protocol for preparing non-parenchymal cells was optimized from several other available protocols (Mederacke et al., 2015; Movita et al., 2012; Seki et al., 2009). Excellent protocols with videos detailing the procedure for perfusion-based digestion of a mouse liver exist and can be referred to (Charni-Natan and Goldstein, 2020; Mederacke et al., 2015).
Note: The protocol benefits from two people working in parallel if processing more than four mice. Mouse dissection and digestion is time-consuming and delays at this stage can affect sample quality.
Prepare necessary buffers and reagents
In this section, necessary buffers for are made ready for non-parenchymal cell preparations.
-
1.Freshly prepare the following solutions on the same day:
-
a.1 mM flavopiridol: dilute 10 mM aliquot of flavopiridol stock to 1 mM using water. Do not use phosphate buffered saline as this can cause a precipitate to form. Store the 1 mM solution on ice and discard at the end of the day.CRITICAL: Tissue digestion leads to cell stress responses and the potential for confounding changes in measured analytes. To minimize the effect of tissue digestion on profiled transcriptional regulators, the RNA polymerase inhibitor flavopiridol is added to perfusion and digestion steps where the hepatic cells are at 37°C and active. Use of flavopiridol can affect the viability of purified cells in culture though and it can be excluded or titrated as appropriate for experimental purposes. For comparison of genomics data though, the digestion protocol used should not be altered.
-
b.Wash buffer 1: store and use at 4°C or on ice. The below table is adequate for 8 mice. Discard at the end of the day.
Reagent Final concentration Amount Hank’s Balanced Salt Solution (with calcium, with magnesium, no phenol red) 500 mL DNase I 20 μg/mL 5 mL Fetal bovine serum 2% volume/volume 10 mL 1 M HEPES 20 mM 10 mL 10× Hank’s Balanced Salt Solution (no calcium, no magnesium, no phenol red) 1× 1 mL Total 512.5 mL -
c.Wash buffer 2: store and use at 4°C or on ice. The below table is adequate for 16 mice. Discard within 1 week.
Reagent Final concentration Amount Hank’s Balanced Salt Solution (no calcium, no magnesium, no phenol red) 500 mL Fetal bovine serum 2% volume/volume 10 mL 1 M HEPES 20 mM 10 mL 0.5 M EDTA 5 mM 5 mL 10× Hank’s Balanced Salt Solution (no calcium, no magnesium, no phenol red) 1× 1 mL Total 512.5 mL -
d.Isotonic Percoll. Percoll must be made isotonic. Mixing 90% Percoll with 10% 10× Hank’s Balanced Salt Solution (no calcium, no magnesium, no phenol red) renders the solution safe for use with cells. Store the solution on ice and discard at the end of the day.
Reagent Final concentration Amount Percoll 9 mL 10× Hank’s Balanced Salt Solution (no calcium, no magnesium, no phenol red) 1× 1 mL Total NA 10 mL -
e.Liberase TM (5 mg/mL). Prepare and store on ice. Do not vortex or vigorously pipette! Immediately freeze unused enzyme at −80°C in single use aliquots (1.25 mL per healthy mouse liver).CRITICAL: The enzyme blend Liberase TM is preferable to sequential digestion with Pronase and Collagenase. This preference is due to improved reproducibility in tissue digestion quality and reduced loss of surface antigens used in subsequent FACS purification that are cleaved by Pronase (examples: CD31 and Tim4).
Reagent Final Concentration Amount 50 mg lyophilized Liberase TM Phosphate buffered saline 10 mL Total NA 10 mL -
f.Liver clearing buffer. Store and use at 37°C. The below table is adequate for 8 mice.
Reagent Final Concentration Amount Hank’s Balanced Salt Solution (no calcium, no magnesium, no phenol red) 500 mL 0.5 M EGTA 0.5 mM 1 mL 0.5 M EDTA 0.5 mM 1 mL 1 M HEPES 20 mM 10 mL 1 mM flavopiridol 1 μM 0.5 mL 10× Hank’s Balanced Salt Solution (no calcium, no magnesium, no phenol red) 1× 1 mL Total NA ~500 mL -
g.Liver digestion buffer A. Store and use at 37°C.CRITICAL: Collagenases require calcium to function. Do not use Hank’s Balanced Salt Solution not containing calcium or magnesium.
Reagent Final Concentration Amount Hank’s Balanced Salt Solution (with calcium, with magnesium, no phenol red) 500 mL 1 M HEPES 20 mM 10 mL 1 mM flavopiridol 1 μM 0.5 mL 10× Hank’s Balanced Salt Solution (no calcium, no magnesium, no phenol red) 1× 1 mL Total NA ~500 mL -
h.Liver digestion buffer B. Prepare immediately prior to euthanizing a group of mice. Prepare 60 mL per mouse, or 80 mL if the liver is expected to be enlarged or diseased.
Reagent Final Concentration Amount Liver digestion buffer A 60 mL Liberase TM (5 mg/mL) 0.1 mg/mL 1.25 mL DNase I (2 mg/mL) 1 μM 0.625 mL Total NA 61.88 mL -
i.OptiPrep (28%): OptiPrep is a commercially available iso-osmolar solution of iodixanol (60%) and water. With this solution, the final iodixanol concentration is 16.8%. Prepare 10 mL per liver and store at 4°C or on ice. Discard at the end of the day.CRITICAL: Mix the OptiPrep stock solution prior to use. It has a tendency to settle.
Reagent Final Concentration Amount OptiPrep 28% 2.8 mL Wash buffer 2 72% 7.2 mL Total NA 10 mL -
j.PBS + 0.1% Tween 20: Prepare 50 mL by adding 50 μL of 10% Tween 20 to 50 mL PBS. Store on ice or at 4°C for 1 week.
-
a.
Digestion and removal of liver tissue
In this section, mouse livers are digested in situ, excised, then further digested in vitro. Following digestion, non-parenchymal cells from the crude cell preparation are enriched by successive washing and centrifugation, as well as density gradient centrifugation.
-
2.Flush blood from the liver of a mouse freshly euthanized following institutional approved guidelines.
-
a.Pin down the mouse and carefully open the abdominal cavity.
-
b.Gently move the intestines to the side exposing the inferior vena cava and the portal vein.
-
c.Insert a 24-gauge catheter into the inferior vena (Figure 1A). Attach the line of a peristalsis pump connected to a 0.22-μm filter to the catheter and pin into place. Nick the portal vein with scissors to release fluid pressure. Close the inferior vena cava above the diaphragm using a bulldog clamp. Pin the lines in place at the filter (Figure 1B).Note: All lobes of the liver should blanch (Figure 1B) as erythrocytes drain from the organ. If the liver is not blanching, carefully adjust the position of the catheter in the vessel.
-
d.Apply a small amount of surgical glue to hold the catheter securely in place. Alternatively, the catheter can be secured using a suture.
-
e.Turn on the heat lamp to maintain liver temperature at around 37°C, which is ideal for enzymatic digestion.
-
f.Perfuse with liver clearing buffer for 3 min at flow rate of 5 mL/min.
-
a.
-
3.Digest liver tissue in situ.Note: Enlarged fatty livers and fibrotic livers may require a higher flow rate and increased digestion volume (60 mL at 7 mL/min). This is described in the troubleshooting section.
-
a.Move the pump intake line to a tube with 40 mL of liver digestion buffer B and allow the contents to perfuse through the liver until the line is empty. The liver should begin to lose its shape during digestion, and the Glisson’s capsule may start to detach from the liver cells.
-
b.Remove the gallbladder using scissor and tweezers, the gallbladder contains digestive enzymes that can digest surface markers used for FACS.
-
a.
-
4.Continue digestion of liver tissue in vitro.
-
a.At this point the liver is now often “mush” and can be removed from the abdominal cavity by aspirating into a 1 mL syringe. Alternatively, remove the lobes with scissors and forceps and finely mince the tissue using a razor blade. Transfer the digested contents into a 50 mL conical filled with 20 mL of liver digestion buffer B. Refer to Troubleshooting for more details.
-
b.Mix digested liver tissue with digestion buffer by pipetting up and down 3–5 times or by gentle inversion of tubes.
-
c.Incubate digested liver tissue for 20 min at 37°C with Miltenyi MACSmix tube rotator at the highest speed.
-
a.
-
5.Remove hepatocytes from cell suspension by low-speed centrifugation followed by Percoll centrifugation.Note: If working with a fatty liver then perform the centrifugations described in this step at 20°C–25°C.Note: The Percoll wash (step 4) can be skipped if the liver is healthy and non-steatotic, instead simply centrifuge at 500 × g for 10 min at 4°C to pellet non-parenchymal cells. Furthermore, the Percoll wash depletes stellate cells. If stellate cells are desired, then skip the Percoll wash and simply centrifuge at 500 × g for 10 min to pellet non-parenchymal cells.
-
a.Filter 20 mL of cells suspended in liver digestion buffer through a 70 μM cell strainer into a 50 mL conical to remove undigested debris and connective tissue.
-
b.Add 20 mL of wash buffer 1 to dilute liver digestion buffer and slow enzymatic activity during hepatocyte removal.
-
c.Sediment the dense hepatocytes by low-speed centrifugation at 50 × g for 3 min at 4°C.
-
d.Transfer the supernatant to a new 50 mL conical and repeat spin at 50 × g for 3 min at 4°C and discard the pellet containing hepatocytes.
-
e.Transfer the supernatant enriched for NPCs to a new 50 mL conical.
-
f.Add 10 mL isotonic Percoll to 40 mL cell suspension. If the volume of cell suspension is less than 40 mL at this step adjust amount of isotonic Percoll that is added to maintain a 20% final concentration of Percoll.
-
g.Centrifuge cell suspension in 20% Percoll at 600 × g for 10 min 4°C.
-
h.Remove supernatant and resuspend NPCs in 20 mL of wash buffer 1.
-
a.
-
6.Isolate NPCs using OptiPrep gradient centrifugation (Figure 2).
-
a.Centrifuge the NPC cell suspension at 500 × g for 10 min at 4°C to pellet NPCs once more.
-
b.Resuspend pellet in 10 mL of OptiPrep (28%) using a 10 mL syringe with an 18-gauge needle attached (Figure 2). Take care to resuspend the cell pellet gently at this step.
-
c.Fill a 15 mL conical with 3 mL of wash buffer 1 and rest a glass Pasteur pipette at the bottom of the tube. Transfer the 10 mL cell suspension in OptiPrep buffer through the Pasteur pipette to create the cell layer underneath the less dense wash buffer 1 layer (Figures 2A and 2B). The narrow opening of the Pasteur pipette controls the flow rate and greatly simplifies the layering process.
-
d.Centrifuge gradient suspension at 1,400 × g for 25 min at 4°C with centrifuge acceleration set to very low and with brake mechanism turned off.
-
e.Following gradient centrifugation, non-parenchymal cells will be enriched at the interface between the upper 0% OptiPrep layer and the lower 28% OptiPrep layer (Figure 2C). Use a 1 mL pipette tip to extract all cells at this interface and transfer to a new 15 mL conical tube, then fill the tube to 12 mL with wash buffer 2.
-
a.
-
7.Lyse red blood cells remaining in non-parenchymal cell mixture.
-
a.Centrifuge cell suspension at 500 × g for 10 min at 4°C.
-
b.Remove supernatant and resuspend in 1 mL of 4°C RBC lysis buffer, incubate for 5 min on ice to lyse red blood cells.
-
c.Dilute the RBC lysis buffer cell suspension with 9 mL of 4°C phosphate buffered saline and pellet NPCs via centrifugation at 400 × g for 7 min at 4°C.
-
d.Remove the supernatant and wash the cells with 10 mL of 4°C phosphate buffered saline.
-
a.
Figure 1.

Catheter placement for fluid perfusion through the mouse inferior vena cava
(A) Example of successful insertion of a catheter into the inferior vena cava. Often the catheter hub will fill with blood. If it does not, it is sometimes helpful to apply a slight vacuum using a 1 mL syringe.
(B) Example of an ongoing successful perfusion. The catheter and peristalsis lines are secured by pinning the filter in place on the dissection mount (Styrofoam board). Note the blanched appearance of the liver in comparison to (A) as a result of blood evacuation through the portal vein.
Figure 2.

Setup of the OptiPrep density gradient
(A) Depiction of how to set up the gradient underlay.
(B) An example of a prepared gradient prior to centrifugation.
(C) Depiction of the resulting gradient layers after centrifugation. Collect the desired NPC layers by pipetting and discard the debris fraction. It can be helpful to assess a small portion of debris layer by flow cytometry if the yields for the desired cell population are consistently lower than expected. This could necessitate optimization of the gradient percentages.
Created with BioRender.com.
Staining of non-parenchymal cells
In this section, the enriched non-parenchymal cells are labeled with fluorescent antibodies in preparation for FACS.
-
8.Stain non-parenchymal cells with viability dye. Set aside some unstained cells which can be helpful for determining background fluorescence.
-
a.Centrifuge cell suspension at 500 × g for 10 min at 4°C.
-
b.Resuspend cell pellet in 250 to 500 μL of 4°C PBS containing anti-CD16/CD32 to block FC and Live/Dead NIR Viability Dye or similar to label dead cells.Note: A 250 μL stain volume per healthy liver is often appropriate for steps 8b and 9. Enlarged fatty livers may require a 500 μL stain volume.
Reagent Final concentration Amount Phosphate buffered saline anti-CD16/CD32 (0.5 mg/mL) 1 μg/mL 1:500 Live/Dead NIR 1 μM 1:300 -
c.Stain at 4°C for 10 min.
-
a.
-
9.
Freshly prepare 250 to 500 μL of the antibody staining cocktail per liver.
| Reagent | Final concentration | Amount |
|---|---|---|
| Wash buffer 2 | ||
| Anti-CD16/CD32 (0.5 mg/mL) | 1 μg/mL | 1:500 |
| Anti-F4/80 BV421 (0.2 mg/mL) | 2 μg/mL | 1:100 |
| Anti-CD45 BB515 (0.2 mg/mL) | 2 μg/mL | 1:100 |
| Anti-CD11b PE (0.2 mg/mL) | 2 μg/mL | 1:100 |
| Anti-CD146 PE/Cy7 (0.2 mg/mL) | 2 μg/mL | 1:100 |
| Anti-Tim4 Alexa 647 BV421 (0.5 mg/mL) | 2 μg/mL | 1:250 |
Note: Many excellent alternative antibody cocktails may be used. We refer the readers to the literature for examples (Devisscher et al., 2017; Ramachandran et al., 2012; Remmerie et al., 2020; Seidman et al., 2020).
-
10.
Add an equivalent volume used in step 8b of the 2× antibody staining cocktail to the cells. Mix together by inversion 20 times or using gentle flicking.
-
11.
Stain at 4°C for 20 min.
-
12.
Fill the tube with wash buffer 2 at the end of the staining process.
-
13.
Pellet the NPCs by centrifugation at 500 × g for 10 min at 4°C, remove the supernatant, then resuspend pellet in 2 mL wash buffer 2.
-
14.
Filter the cells through a 35- or 40-μm cell strainer to remove particulate matter that may clog the cell sorter. Flush the strainer with wash buffer 2, then fill the tube with wash buffer 2.
-
15.
Pellet the non-parenchymal cell suspension at 500 × g for 10 min at 4°C.
-
16.
Remove supernatant and resuspend in 500 μL wash buffer 2. Keep the cells on ice and in the dark (covered). Cells are now ready for FACS.
Sorting of non-parenchymal cells
In this section, the stained single-cell suspension is sorted into purified cell populations using FACS.
-
17.
Prepare single-color controls using compensation beads. We use OneComp eBeads Compensation Beads sourced from Invitrogen. Follow the manufacturer’s protocol. Typically, using 1–2 μg of antibody per compensation control provides necessary fluorescent signal higher than the signal of stained cells. Of note, we have found the popular UltraComp eBeads Compensation Beads are not easily detected by this lab’s MoFlo Astrios or Sony MA900. Yet, these beads are an excellent option for use on BD systems.
-
18.Set a gating strategy for Kupffer cells (Figure 3). We have found success with the following strategy:
-
a.Generate a dot plot of CD45 versus CD146 and set gates for CD146−CD45+ immune cells and CD146+CD45− endothelial cells.
-
b.Generate a dot plot CD11b versus F4/80 for the CD146−CD45+ population identified in step 20a. Set gate for CD11bloF4/80hi tissue macrophages.
-
c.Generate a dot plot of side scatter pulse height versus side scatter pulse area to identify single cells and exclude doublets. Doublet particles will have an increased ratio of area signal relative to height signal. It is also acceptable to identify singlets in an analogous manner using forward scatter.
-
d.Generate a dot plot of forward scatter versus Zombie NIR to identify live cells and exclude dead cells. Live cells will be Zombie NIR negative.Note: It is helpful to use a fluorescence minus one control for Zombie NIR to define your viable cell gate.
-
e.Generate a dot plot of Tim4-A647. Tim4+ cells are long-term resident or embryonic-derived Kupffer cells while Tim4− cells are monocyte-derived macrophages recruited during disease to the Kupffer cell niche.
-
a.
-
19.Optional: Set gating strategy for other hepatic macrophages which may arise during the course of a chosen disease model.
-
a.Select the CD11bhiF4/80lo population displayed in step 11b.
-
b.Select CX3CR1+Ly6G− cells to exclude neutrophils.
-
c.These cells can be further divided based on expression of Ly6C into Ly6Clo recruited macrophages and Ly6Chi recruited macrophages.
-
d.Identify singlets and live cells as in step 18c and 18d. Further clarification on identification of these cells is described in Ramachandran et al., 2012 and Seidman et al., 2020.
-
a.
-
20.Optional: Set gating strategy for liver sinusoidal endothelial cells.
-
a.Identify liver sinusoidal endothelial cells by gating on CD146+CD45− cells described in step 18a and shown in Figure 3 at left.
-
b.Identify singlets and live cells as in step 18c and 18d. Further clarification on identification of these cells is described in Sakai et al., 2019.
-
a.
-
21.Optional: Set gating strategy for stellate cells.
-
a.Generate a plot of retinoid autofluorescence versus side scatter to identify autofluorescent stellate cells.
-
b.Identify singlets and live cells as in step 18c and 18d. Further clarification on identification of these cells is described in Kisseleva et al., 2012 and Mederacke et al., 2015.
-
a.
Note: In our experience detection of retinoid autofluorescence is best when excited by an ultraviolet (355 nm) laser and detected at approximately 500 nm.
-
22.
Purity of sorted cells can be assessed by flow cytometry of a small number of sorted cells diluted in wash buffer 2.
Figure 3.

Gating strategy for purification of mouse hepatic macrophages and sinusoidal endothelial cells
Representative flow cytometry gating to isolate Kupffer cells. The cells displayed here are from a mouse fed a NASH-inducing diet and display two population of Kupffer cells that can be segregated based on expression of Tim4. High expression of Tim4 is a useful surrogate for determine cells of long-term residence or embryonic origin (Devisscher et al., 2017; Sakai et al., 2019; Scott et al., 2016; Seidman et al., 2020).
Storage of sorted non-parenchymal cells
FACS-purified cell populations are now suitable for many next-generation sequencing applications. We provide example storage strategies for RNA-seq, ATAC-seq, and ChIP-seq below. Our labs have also successfully used cells prepared in this manner for scRNA-seq and Hi-C as examples of other suitable applications.
Optional: To prepare cells for RNA-seq, we recommend using 50,000 - 100,000 cells for common RNA-seq library preparation protocols. Cells can be processed as recommended using a supplied lysis buffer found in many commercial RNA-seq kits or alternatively with Trizol.
Optional: To prepare cells for ATAC-seq, we recommend using 50,000 cells (Buenrostro et al., 2015; Corces et al., 2017). Of note, sample preparation and tagmentation reaction conditions may require optimization for each cell type to yield the best quality data.
Prepare sorted viable cells for ChIP-seq. We recommend starting with 0.5 to 1 million cells for ChIP-seq library preparation. For Kupffer cells, this can often be attained with 1–3 livers. For sinusoidal endothelial cells, this can be often be attained with 1 liver. Other populations of non-parenchymal cells would require testing to determine necessary sample size. This approach works well for assessment of histone modifications and genomic PU.1 occupancy. Other ChIP targets may require optimization.
-
23.
Wash the desired number of cells with wash buffer 2 and centrifuge at 500 × g for 5 min at 4°C.
-
24.
Remove the supernatant and resuspend pellet in 500 μL wash buffer 2 and transfer to 1.5 mL DNA LoBind tube. Wash the starting tube with an additional 500 μL wash buffer 2 and combine into the 1.5 mL DNA LoBind tube.
-
25.
Centrifuge the cells in the 1.5 mL DNA LoBind tube at 500 × g for 5 min at 4°C.
-
26.
Resuspend the pellet in 20°C–25°C 1% formaldehyde using approximately 1 million cells per mL. Incubate for 10 min 20°C–25°C with gentle rocking or rotation to cross link proteins and DNA.
-
27.
Quench the cross-linking reaction by adding 20°C–25°C glycine to a final 0.125 M concentration from the 2.625 M glycine stock solution. Additionally, add Tween 20 to a final 0.1% concentration from a 10% Tween 20 stock. Tween 20 aids in pelleting the cross-linked cells during centrifugation.
-
28.
Centrifuge cross-linked cells at 800 × g for 10 min at 4°C. Carefully confirm the presence of a cell pellet. If a pellet is not apparent, centrifuge again at 1,200 × g for 10 min at 4°C. Aspirate the supernatant taking care to avoid disturbing the cell pellet.
-
29.
Suspend the cross-linked cells in 1 mL PBS + 0.1% Tween 20 at 4°C and centrifuge at 800–1,200 × g for 10 min at 4°C, as in step 28. Aspirate the supernatant taking care to avoid disturbing the cell pellet.
-
30.
Wash the cells once more by repeating step 29.
-
31.
Quickly centrifuge the sample for a few seconds at 800–1,200 × g collect the residual fluid to the tube bottom. Aspirate as much residual supernatant as possible using a p10 pipette tip. Be very careful to avoid disturbing the cell pellet. Left over liquid in the sample at this step will crystallize at −80°C and can interfere with ChIP-seq library quality.
-
32.
Snap-freeze the pellet using liquid nitrogen and store the cross-linked cells at −80°C until ready for ChIP-seq library preparation described by Texari et al. (link to “An optimized protocol for rapid, sensitive, and robust on-bead ChIP-seq”). Alternatively, the samples can be snap-frozen using an alcohol and dry ice bath. Take care to protect the tube labeling!
Note: It can be beneficial for challenging ChIPs to double crosslink the sample at 20°C–25°C with gentle rocking for 30 min using 1 mL of a 2 mM solution of disuccinimidyl glutarate (DSG) per million cells. Then add formaldehyde to a final concentration of 1% and incubate for an additional 10 min of 20°C–25°C incubation. More details on this approach can be found in the linked protocol described by Texari et al. (link to “An optimized protocol for sensitive and robust on-bead ChIP-seq”). However, in our experience much higher quality ChIP-seq data for more challenging ChIP factors can be obtained through use of nuclei fixed in situ prior to fluorescence-activated nuclei sorting. This approach is described in the next section.
Module 2, alternative strategy: in situ fixation and isolation of nuclei from Kupffer cells (or other liver cells)
This section describes an approach for preparing nuclei from fixed livers for sorting that was optimized from other protocols (Nott et al., 2019). We have found that using around 1 million Kupffer cell nuclei prepared in this manner yield higher quality ChIP-seq data than material from Module 1. This is particularly true for transcription factors that are challenging to ChIP with limited material. Examples of the data quality can be found in Sakai et al., 2019 and Seidman et al., 2020.
Prepare buffers and reagents necessary for module 2
In this section, the buffers needed for in situ fixation are prepared.
-
33.
Freshly prepare 25 mL liver clearing buffer 2 per mouse liver and store at 20°C–25°C.
| Reagent | Final Concentration | Amount |
|---|---|---|
| PBS | 24.95 mL | |
| 0.5 M EDTA | 0.5 mM | 25 μL |
| 0.5 M EGTA | 0.5 mM | 25 μL |
| Total | NA | 25 mL |
-
34.Freshly prepare 180 mL 1 mg/mL DSG per mouse liver.
-
a.Dissolve 100 mg vials of DSG using 4 mL of DMSO.CRITICAL: DSG is toxic. Wear appropriate personal protective equipment (lab coat, safety glasses, and disposable gloves), refer to the safety data sheet and follow institutional guidelines for safe handling and disposal!
-
b.Dilute the DSG/DMSO solution to 1 mg/mL by rapidly dispensing it into 176 mL 20°C–25°C PBS followed by immediate mixing by inverting 5–6 times and keep at 20°C–25°C until use.Note: Occasionally the solution will form an immediate precipitate. In this event discard the solution and prepare fresh.
-
a.
-
35.Freshly prepare 60–65 mL of 1% formaldehyde per mouse liver.
-
a.Dilute stock formaldehyde solution to 1% volume/volume using 20°C–25°C PBS. Keep at 20°C–25°C until use.
-
a.
-
36.
Freshly prepare 20 mL 0.25 M glycine per mouse liver by diluting 1.9 mL 2.625 M stock glycine into 18.1 mL 20°C–25°C PBS. Store at 20°C–25°C.
-
37.
Chill and use nuclei isolation buffer and nuclei sorting buffer on ice.
In situ fixation and nuclei preparation
In this section, mouse livers are quickly fixed by in situ perfusion. Then nuclei are liberated and prepared for FACS.
-
38.Flush blood from the liver of a mouse freshly euthanized following institutional approved guidelines.
-
a.Pin down the mouse and carefully open the abdominal cavity.
-
b.Gently move the intestines to the side exposing the inferior vena cava and the portal vein.
-
c.Carefully insert a 24-gauge catheter into the inferior vena cava. Attach the line of a peristalsis pump connected to a 0.22-μm filter to the catheter and pin into place. Nick the portal vein with scissors to release fluid pressure. Close the inferior vena cava above the diaphragm using a bulldog clamp.
-
d.Insert a 24-gauge catheter into the inferior vena (Figure 1A). Attach the line of a peristalsis pump connected to a 0.22-μm filter to the catheter and pin into place. Nick the portal vein with scissors to release fluid pressure. Close the inferior vena cava above the diaphragm using a bulldog clamp (Figure 1B).Note: All lobes of the liver should blanch as erythrocytes drain from the organ. If the liver is not blanching, carefully adjust the position of the catheter in the vessel.
-
e.Apply a small amount of surgical glue to hold the catheter securely in place. Alternatively, the catheter can be secured using a suture.
-
f.Perfuse 20°C–25°C Liver clearing buffer 2 at 5 mL/min for 3 min.
-
a.
-
39.In situ double-fixation of the liver.
-
a.Switch the perfusion line to a solution of 1 mg/mL DSG prepared in 20°C–25°C phosphate buffered saline. Perfuse for 30 min, which requires approximately 180 mL/mouse.Note: Some ChIP targets, (for example PU.1, H3K27ac, and RNA Polymerase II), do not require double cross-linking with DSG and step 39a should be excluded. This determination should be made empirically. Further, optimal fragmentation conditions for single and double-cross-linked samples will not be the same.
-
b.Switch the perfusion line to a solution containing 1% formaldehyde prepared in 20°C–25°C phosphate buffered saline. Perfuse for 10 min, which requires approximately 60 mL per mouse.
-
a.
-
40.Quenching fixation.
-
a.Switch the perfusion line to a vessel containing 20 mL of 20°C–25°C 0.25 M glycine and allow the perfusion line to fully empty.
-
a.
-
41.Homogenize the liver.
-
a.Remove the fixed liver and dispose of the hazardous mouse carcass following institutional guidelines.
-
b.Finely mince the liver with a razor in a 5 cm plastic dish, then transfer the minced tissue to a 50 mL conical.
-
c.Wash the liver twice by suspending in 20 mL nuclei isolation buffer and centrifuging for 1,200 × g for 7 min at 4°C. Discard the supernatant after each wash.
-
d.Suspend the washed liver tissue in 15 mL nuclei isolation buffer then transfer to a 15 mL Dounce homogenizer.
-
e.Using the loose pestle, homogenize slowly 10 times with ¼ to ½ rotation on the downward stroke and no rotation on the upward stroke.
-
f.Incubate on ice for 30 min.
-
g.Using the tight pestle, homogenize slowly 10 times with ¼ to ½ rotation on the downward stroke and no rotation on the upward stroke.
-
h.Transfer the homogenate to a 15 mL conical tube and centrifuge at 1,200 × g for 7 min at 4°C.
-
i.Repeat this step by resuspending the pellet in 10 mL fresh nuclei isolation buffer each time for a total of 2–4 times. The goal by this step is to have sufficiently cleaned the pellet of debris yielding a clear supernatant. Removing this debris is beneficial to reduce the frequency of clogging the cell sorter and the overall time spent sorting. A cleaner starting sample will also result in increased sort purity and yield.
-
j.Examine nuclei under a microscope using trypan blue and a hemocytometer. If the cells are intact, consider repeating steps 41g–i. Then suspend at ∼20 million nuclei per mL.
-
a.
Pause point: At this point, nuclei can be suspended in 10 mL 90% FBS and 10% DMSO (10 mL per liver) and aliquots stored at −80°C for downstream processing at a later time. We have not tested other nuclei preservation methods. To recover the cryopreserved sample, warm the frozen suspension at 37°C in a water bath, then wash the nuclei twice with nuclei isolation buffer.
Note: At this time nuclei can be stained for nuclear antigens specific for the population of interest. For the purpose of this protocol, we use fluorescence signal provided genetically via nuclear-localized tdTomato of the Clec4f-Cre-Reporter (Sakai et al., 2019) which is available at Jackson labs under Stock No: 033296. As shown in Figure 4, Kupffer cell nuclei can also be enriched at the steady state by staining the nuclei with anti-PU.1. We describe this procedure in the optional step below. This concept can be extended to labeling of other populations with lineage-specific antibodies.
Figure 4.

Gating strategy for purification of mouse hepatic Kupffer cell nuclei
Representative flow cytometry gating for isolation of Kupffer cell nuclei. The use of a Clec4f nuclear reporter is necessary to ensure separation of Kupffer cell nuclei from nuclei of other PU1+ recruited macrophages that can arise during disease. At the steady state, there are very few recruited macrophages in the liver isolation of Kupffer cell nuclei using PU1 staining alone is adequate.
Note: Liver disease can promote population expansion of many cell lineages, especially myeloid cells. In these instances, purification of nuclei by labeling with a single antibody may not result in a consistent nuclei population. During disease, we suggest considering genetically encoded drivers of nuclear-localized reporters.
-
42.Optional: Stain nuclei population(s) of interest using antibodies for lineage-specific transcription factors. We have found success labeling hepatic macrophage nuclei using anti-PU.1. This process is described below as a template for user specific applications. Save a small portion of the sample for use as an unstained control.
-
a.Add 1 μg/mL anti-CD16/CD32 to prevent nonspecific binding of antibodies to FC receptors and incubate at 4°C for 20 min.
-
b.Add 1 μg/mL Alexa Fluor 647 conjugated anti-PU.1 and stain at 4°C for 30 min or overnight as optimized empirically.
-
c.Fill the tube with nuclei isolation buffer and centrifuge at 1,200 × g for 7 min at 4°C.
-
d.Aspirate the supernatant and refill the tube with nuclei isolation buffer, then repeat the previous centrifugation.
-
e.Aspirate the supernatant.
-
a.
Nuclei sorting
In this section, the prepared nuclei are filtered and the desired populations are purified using FACS.
-
43.
Prepare the isolated nuclei for sorting by suspending the washed nuclei in nuclei sorting buffer, then slowly pass the suspension through a 22-gauge needle and a 40-μm cell strainer. Suspending nuclei at 10–20 million/mL is a good starting point. Store on ice until sorting.
-
44.Purify desired nuclei population using fluorescence-activated nuclei sorting.
-
a.During sorter setup, pay careful attention to instrument detector thresholds and voltages/gains for light scatter.
-
a.
-
45.
After purification, verify yield by microscopy and purity by flow cytometry.
-
46.
Centrifuge nuclei at 800 to 1,200 × g for 10 min at 4°C in PBS + 0.01% Tween 20
-
47.
Carefully aspirate all buffer then quick spin the nuclei once more at 800 to 1,200 × g.
-
48.
Carefully remove all buffer without disturbing the nuclei pellet.
-
49.
Snap-freeze the pellet using liquid nitrogen and store at −80°C until use for ChIP.
Expected outcomes
At the end of Module 1, yields of 0.3 to 0.75 million Kupffer cells, a million or more sinusoidal endothelial cells, and 50,000 to 250,000 stellate cells. Yields of other recruited macrophage populations are usually less than 50,000 in mice with disease and are not present in healthy mice. Variables contributing to cell yields include genetic background, the presence of disease, digestion quality, and cell sorter abort rates. Yields of 1–2 million Kupffer cell nuclei are typical of Module 2.
Limitations
Acquiring sufficient material for downstream assays can require extensive time investment. Further, yields can be variable, especially from livers with fatty liver disease or fibrosis. The procedure described in module 1 uses a Percoll centrifugation as a way to eliminate debris and fatty hepatocytes. This can also lead to loss of stellate cells, especially in fatty livers. If the primary experimental goal is to yield hepatic stellate cells, consider instead using the excellent protocol described elsewhere (Mederacke et al., 2015). Further, autofluorescence of liver NPC preparations can make robust population discrimination challenging during FACS.
Troubleshooting
Problem 1
Liver tissue is poorly digested following in situ digestion.
Potential solution
Fibrotic, fatty, or hypertrophied liver tissue may not enzymatically digest as well as healthy liver tissue. If this is expected, we have found success by increasing the perfusion rate to 7 mL/min and perfusing with 60 mL of liver digestion buffer. Further, perfusion quality will impact liver digestion efficiency. Thorough descriptions of strategies to improve liver digestions have already been described elsewhere. (Charni-Natan and Goldstein, 2020; Mederacke et al., 2015).
Problem 2
Cell populations are difficult to discriminate in setup for FACS.
Potential solution
Residual hepatocytes and stellate cells have significant autofluorescence that interferes with detection of true signals of interest. It is helpful during setup of the cell sorter to crudely identify your population(s) of interest using fluorescent signals then determine their SSC profiles. This information can then be used to apply a first gate on SSC versus autofluorescence of retinoids to exclude the undesired residual hepatocytes and stellate cells from other downstream gates. Additionally, Kupffer cells also have significant autofluorescence and many antigen:fluorophor combinations do have adequate signal to noise profiles. It is valuable to create and carefully assess fluorescence minus one controls, especially when testing new antibodies. We have often found success with Kupffer cells using the following fluorophores: BUV395, BUV737, BV421, BV786, Alexa Fluor 488 or BB515, PE or TdTomato, PE-Cy7 or PE-eFluor-770, Alexa Fluor 647 or APC, and Live/dead NIR. We have found use of detector channels for PE-Dazzle, BV510, BV650, PerCP-Cy5.5, and Alexa Fluor 700 often have inadequate signal to noise ratio for reliable protein detection on mouse Kupffer cells without very careful attention to proper controls and antibody titrations.
Problem 3
Detection and/or yields for nuclei sorts are lower than expected.
Potential solution
Detecting nuclei of some cell populations using flow cytometry can be trickier than intact cells due to the smaller particle size. It can be helpful to co-stain the nuclei with titrated DAPI. Nuclei will be very brightly labeled with DAPI and the resulting signal can be used to assist setup of detector voltages and gains for proper detection of the desired nuclei population. It is also important to check the trigger threshold setting to ensure the desired population is not simply being excluded as noise by the controlling software.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Christopher K. Glass (ckg@health.ucsd.edu).
Materials availability
This study did not generate new materials.
Data and code availability
This study did not generate new data or code.
Acknowledgments
These studies were supported by NIH grants DK091183, HL088083, DK063491, and GM085764 and Fondation Leducq grant 16CVD01. T.D.T. was supported by P30 DK063491, T32DK007044, and NRSA T32CA009523. H.B. was supported by the NIH grants T32DK007202 and F30DK124980. M.S. was supported by the Manpei Suzuki Diabetes Foundation of Tokyo, Japan, and the Osamu Hayaishi Memorial Scholarship for Study Abroad, Japan. J.S.S. was supported by a American Heart Association Fellowship 16PRE30980030 and NIH Predoctoral Training grant T32DK007541. S.H. received additional support from NIH grants R01GM129523, P30DK063491 and P30DK120515.
Author contributions
T.D.T., protocol development and optimization, manuscript conceptualization and writing, experimentation and data analysis, and manuscript final approval; H.R.B., protocol optimization, manuscript writing, experimentation; M.S., protocol development and optimization, manuscript editing, experimentation; J.S.S., protocol development and optimization, draft writing; S.H., protocol development and draft writing; C.K.G, manuscript conceptualization, manuscript editing, and manuscript final approval.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Ty D. Troutman, Email: ttroutman@health.ucsd.edu.
Christopher K. Glass, Email: ckg@health.ucsd.edu.
References
- Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charni-Natan M., Goldstein I. Protocol for primary mouse hepatocyte isolation. STAR Protoc. 2020;1:100086. doi: 10.1016/j.xpro.2020.100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devisscher L., Scott C.L., Lefere S., Raevens S., Bogaerts E., Paridaens A., Verhelst X., Geerts A., Guilliams M., Van Vlierberghe H. Non-alcoholic steatohepatitis induces transient changes within the liver macrophage pool. Cell. Immunol. 2017;322:74–83. doi: 10.1016/j.cellimm.2017.10.006. [DOI] [PubMed] [Google Scholar]
- Kisseleva T., Cong M., Paik Y., Scholten D., Jiang C., Benner C., Iwaisako K., Moore-Morris T., Scott B., Tsukamoto H. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. U S A. 2012;109:9448–9453. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mederacke I., Dapito D.H., Affò S., Uchinami H., Schwabe R.F. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015;10:305–315. doi: 10.1038/nprot.2015.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movita D., Kreefft K., Biesta P., Oudenaren A. van, Leenen P.J.M., Janssen H.L.A., Boonstra A. Kupffer cells express a unique combination of phenotypic and functional characteristics compared with splenic and peritoneal macrophages. J. Leukoc. Biol. 2012;92:723–733. doi: 10.1189/jlb.1111566. [DOI] [PubMed] [Google Scholar]
- Muse E.D., Yu S., Edillor C.R., Tao J., Spann N.J., Troutman T.D., Seidman J.S., Henke A., Roland J.T., Ozeki K.A. Cell-specific discrimination of desmosterol and desmosterol mimetics confers selective regulation of LXR and SREBP in macrophages. Proc. Natl. Acad. Sci. U S A. 2018;115:E4680–E4689. doi: 10.1073/pnas.1714518115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A., Holtman I.R., Coufal N.G., Schlachetzki J.C.M., Yu M., Hu R., Han C.Z., Pena M., Xiao J., Wu Y. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science. 2019;366:1134–1139. doi: 10.1126/science.aay0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran P., Pellicoro A., Vernon M.A., Boulter L., Aucott R.L., Ali A., Hartland S.N., Snowdon V.K., Cappon A., Gordon-Walker T.T. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. U S A. 2012;109:E3186–E3195. doi: 10.1073/pnas.1119964109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmerie A., Martens L., Thoné T., Castoldi A., Seurinck R., Pavie B., Roels J., Vanneste B., De Prijck S., Vanhockerhout M. Osteopontin expression identifies a subset of recruited macrophages distinct from Kupffer cells in the fatty liver. Immunity. 2020;53:641–657. doi: 10.1016/j.immuni.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai M., Troutman T.D., Seidman J.S., Ouyang Z., Spann N.J., Abe Y., Ego K.M., Bruni C.M., Deng Z., Schlachetzki J.C.M. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain Kupffer cell identity. Immunity. 2019;51:655–670.e8. doi: 10.1016/j.immuni.2019.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott C.L., Zheng F., De Baetselier P., Martens L., Saeys Y., De Prijck S., Lippens S., Abels C., Schoonooghe S., Raes G. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016;7:10321. doi: 10.1038/ncomms10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman J.S., Troutman T.D., Sakai M., Gola A., Spann N.J., Bennett H., Bruni C.M., Ouyang Z., Li R.Z., Sun X. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity. 2020;52:1057–1074.e7. doi: 10.1016/j.immuni.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E., de Minicis S., Inokuchi S., Taura K., Miyai K., van Rooijen N., Schwabe R.F., Brenner D.A. CCR2 promotes hepatic fibrosis in mice. Hepatology. 2009;50:185–197. doi: 10.1002/hep.22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate new data or code.