

Summary
Co-fractionation/mass spectrometry (CF/MS) is a flexible and powerful method to detect physical associations of proteins. CF/MS can be applied to any tissue or organism without the need for protein-specific antibodies or epitope tags. Here, we outline two alternate protocols for MS preparation of samples (containing low or high salt) and a computational pipeline (cfmsflow) that together allow the successful application of this approach. These protocols are based on CF/MS of over 16 diverse organisms including plants and animals.
For complete details on the use and execution of this protocol, please refer to McWhite et al. (2020).
Subject areas: Bioinformatics, Proteomics
Graphical abstract
Highlights
-
•
Co-fractionation/mass spectrometry (CF/MS) detects native protein associations
-
•
Experimental methods for mass spec preparation of 96-well native fractions
-
•
Computational pipeline to generate protein interaction maps from CF/MS data
Co-fractionation/mass spectrometry (CF/MS) is a flexible and powerful method to detect physical associations of proteins. CF/MS can be applied to any tissue or organism without the need for protein-specific antibodies or epitope tags. Here, we outline two alternate protocols for MS preparation of samples (containing low or high salt) and a computational pipeline (cfmsflow) that together allow the successful application of this approach. These protocols are based on CF/MS of over 16 diverse organisms including plants and animals.
Before you begin
-
1.
Prepare a native protein extract from your sample using any method optimized for the chosen starting material that avoids using organic solvents or other denaturing compounds. Low concentrations of non-ionic detergents (e.g., up to 1% NP40 or equivalent) can aid cell lysis, however higher concentrations of detergents can dissociate protein-protein interactions and interfere with the subsequent electrospray-based liquid chromatography-mass spectrometry (LC/MS) analysis. Detergents used for the initial extraction are typically omitted from subsequent chromatographic separation buffers in order to reduce any potential impact on mass spectrometry.
Note: if you include detergent and must use ultrafiltration to concentrate your lysate prior to fractionation, be mindful that micelles may also be concentrated and increase detergent concentration with consequent disruption of protein assemblies.
-
2.
Fractionate 1–4 mg of total native protein extract into 12–100 protein fractions using any convenient method such as size exclusion chromatography, ion exchange chromatography, isoelectric focusing, glycerol gradient separation, etc.
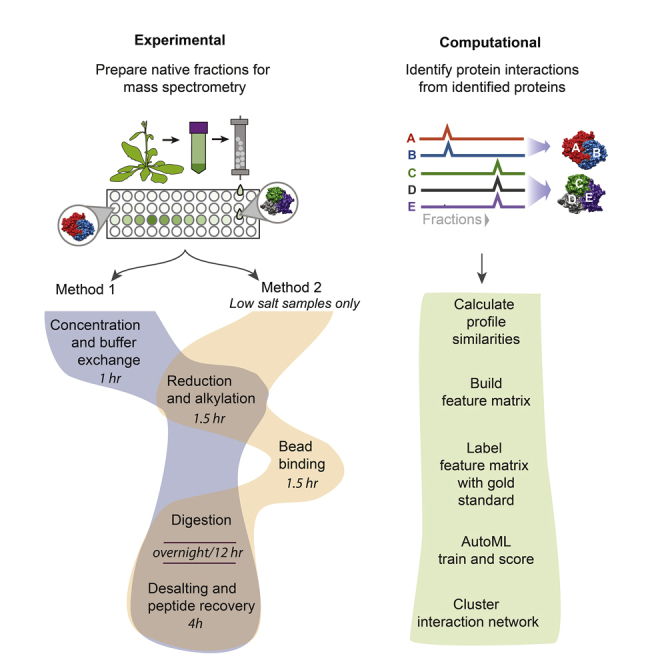
Note: The following MS preparation protocols assume you have collected your fractions in a 96-well format. There are two distinct MS preparation methods provided. If your samples contain >300 mM salt, then use Method 1. If your samples will be <300 mM salt, then use Method 2. Methods are compared in Figure 1.
Figure 1.

Workflow of the two alternative methods for preparation of LC/MS-ready samples from chromatographic fractions
Method 1 can be used in any salt condition but Method 2 will only work if salt is below 300 mM.
CRITICAL: If you plan to use MS sample preparation Method 2 then you must modify your 96-well fraction collection plate prior to collecting chromatographic fractions, as indicated in Figure 2.
Figure 2.

Modification of a 96-well fraction collection plate (shown here, Corning Axygen polypropylene #P-DW-20-C) for use with the 96-well magnetic plate separator (Thermo Fisher Scientific #A14179)
Four support ribs on the underside of the deep well plate (indicated by black arrows in lower left) interfere with prongs on the blue magnetic plate preventing close contact of the well bottoms with the magnet. The 4 ribs are cut and folded as follows to accommodate the prongs. For each of the 4 interfering support ribs use a straight-edge razor to make 2 vertical cuts of the approximate depth shown. Use metal forceps or other tool to bend the resultant plastic tabs over. After modification of the 4 ribs the deepwell plate should sit flush on the magnetic plate as shown in the upper right panel.
Pause Point: Collected fractions can be stored frozen at −80°C until you are ready to complete one of the mass spectrometry preparations outlined below.
-
3.
Before beginning to prepare your fractions for LC/MS analysis, prepare the required solutions from the Table 1 below using LC/MS-grade water and reagents as much as possible. If you are not using a commercially-prepared solution of TCEP, prepare a 0.5 M stock in water (this may require pH adjustment with potassium hydroxide for full solubility). If you will be using Method 2 additionally prepare and aliquot SpeedBeads (as described below).
Table 1.
Solutions and buffers
| Solutions stored 21°C–25°C | Needed for Method 1 | Needed for Method 2 |
|---|---|---|
| Buffer A (recipe below) | ✓ | ✓ |
| Buffer B (recipe below) | ✓ | ✓ |
| Buffer C (recipe below) | ✓ | ✓ |
| 60% Buffer B (recipe below) | ✓ | ✓ |
| Trypsin Digestion Buffer (recipe below) | ✓ | ✓ |
| 70% ethanol | N/A | ✓ |
| 2% DMSO | N/A | ✓ |
| 10% TFE in Trypsin Digestion Buffer | N/A | ✓ |
| 0.5 M TCEP | ✓ | ✓ |
| Aliquoted frozen stock solutions | ||
| 1 M DTT in LC/MS-grade water (store −20°C) | ✓ | ✓ |
| 550 mM 2-iodoacetamide in LC/MS-grade water (store −20°C, protect from light) | ✓ | ✓ |
| Trypsin 1 μg/10 μL in 10 mM acetic acid (store −80°C) | ✓ | ✓ |
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| 1 M Tris-HCl, pH 8.0 | No preference | N/A |
| CaCl2 | No preference | N/A |
| Tris(2-carboxyethyl)phosphine (TCEP) | No preference | N/A |
| 2-Iodoacetamide | No preference | N/A |
| 2,2,2-Trifluoroethanol (TFE) | No preference | N/A |
| Dimethyl sulfoxide (DMSO) | No preference | N/A |
| Ethanol | No preference | N/A |
| Formic acid | No preference | N/A |
| Acetonitrile (LC/MS-grade) | No preference | N/A |
| Water (LC/MS-grade) | No preference | N/A |
| Trypsin, MS-grade | No preference | N/A |
| Dithiothreitol (DTT) | No preference | N/A |
| Experimental models: organisms/strains | ||
| Arabidopsis thaliana | McWhite et al., 2020 | N/A |
| Brassica oleracea var. italica | McWhite et al., 2020 | N/A |
| Cannabis sativa | McWhite et al., 2020 | N/A |
| Ceratopteris richardii | McWhite et al., 2020 | N/A |
| Chenopodium quinoa | McWhite et al., 2020 | N/A |
| Chlamydomonas reinhardtii | McWhite et al., 2020 | N/A |
| Cocos nucifera | McWhite et al., 2020 | N/A |
| Glycine max | McWhite et al., 2020 | N/A |
| Homo sapiens | Mallam et al., 2019 | N/A |
| Mus Musculus | Mallam et al., 2019; Liebeskind et al., 2020 | N/A |
| Oryza sativa | McWhite et al., 2020 | N/A |
| Selaginella moellendorffii | McWhite et al., 2020 | N/A |
| Solanum lycopersicum | McWhite et al., 2020 | N/A |
| Triticum aestivum | McWhite et al., 2020 | N/A |
| Xenopus laevis | Drew et al., 2020 | N/A |
| Zea mays | McWhite et al., 2020 | N/A |
| Software and algorithms | ||
| Cfmsflow | This paper, https://github.com/marcottelab/cfmsflow | |
| TPOT | Olson and Moore, 2016 | Version >= 0.10.0 |
| Nextflow | Di Tommaso et al., 2017 | N/A |
| Docker | https://www.docker.com/ | N/A |
| Other | ||
| AcroPrep Advance 96-filter plate 3k MWCO | Pall | 8163 |
| QIAvac 96 or QIAvac Multiwell vacuum manifold | Qiagen | 19504 or 9014579 |
| 1-μm Sera-Mag SpeedBead magnetic carboxylate modified particles, hydrophilic | Cytiva | 45152105050250 |
| 1-μm Sera-Mag SpeedBead magnetic carboxylate modified particles, hydrophobic | Cytiva | 65152105050250 |
| Lab-in-a-plate Flow-thru plate, 5–7 μL C18 | Glygen | MFNSC18.10 |
| Magnetic plate, 96-well separator | Thermo Fisher Scientific | A14179 |
| Vacuum centrifuge | No preference | N/A |
| Deep 96-well plate for autosampler | Waters | 186005837 |
| Silicone cap-mat pre-slit for autosampler | Waters | 186006332 |
| 1.5 mL polypropylene microcentrifuge tubes | No preference | N/A |
| V-bottom, 0.45 mL, 96-well polypropylene microplates | No preference | N/A |
| Low-speed (1,000 × g) centrifuge with microplate adaptors | No preference | N/A |
| Organic-resistant plate sealing film | Eppendorf | 0030127870 |
| Plate sealing foil | Eppendorf | 0030127889 |
| Dionex UltiMate 3000 RSLCnano UHPLC system or other LC system | Thermo Scientific | ULTIM3000RSLCNANO |
| Acclaim PepMap 100 C18 Analytical Column, 75 μm × 25 cm NanoViper | Thermo Scientific | 164941 |
| Acclaim PepMap 100 C18 trap column, 75 μm × 2 cm NanoViper | Thermo Scientific | 164535 |
| Orbitrap Fusion Lumos or other mass spectrometer | Thermo Scientific | IQLAAEGAAPFADBMBHQ |
| Orbitrap Fusion or other mass spectrometer | Thermo Scientific | IQLAAEGAAPFADBMBCX |
| Orbitrap Elite or other mass spectrometer | Thermo Scientific | AAEGAAPFADBMAZQ |
Materials and equipment
The two alternate experimental methods below each begin with separated protein fractions in a deepwell 96-well plate and are described briefly in McWhite et al., 2020. See the key resources table for necessary specialty plates and equipment described in the protocols. Both methods require a 96-well plate-compatible vacuum manifold and a vacuum centrifuge (e.g., Eppendorf Vacufuge; #022820044) with a microplate adaptor. Method 2 additionally requires a magnetic slab (e.g., Thermo Fisher Scientific 96-well magnetic plate separator; A14179) and may require modification of the 96-well plate as in Figure 2 to allow the plate to adequately contact the magnet. Multichannel pipettes are helpful throughout. Solutions to make up in advance are listed in Table 1 with recipes below. Unless otherwise specified all solutions are made in LC/MS-grade water.
Solution preparations
| Reagent | Final concentration LC/MS-grade water | Final concentration LC/MS-grade Acetonitrile | Final concentration Formic Acid |
|---|---|---|---|
| Buffer A | 100% | 0% | 0.1% |
| Buffer B | 0% | 100% | 0.1% |
| Buffer C | 95% | 5% | 0.1% |
| 60% B | 40% | 60% | 0.1% |
| Trypsin digestion buffer | Final concentration |
|---|---|
| Tris-HCL pH 8.0 | 50 mM |
| CaCl2 | 2 mM |
Step-by-step method details
Prepare protein fractions for MS analysis
Method 1: Ultrafiltration and in-solution digest
Timing: 2 days
We use two different methods for reduction/alkylation and trypsin digestion of samples in 96-well plates for MS analysis (outlined in Figure 1). If your sample contains >300 mM salt, then use Method 1 directly below. If your sample contains <300 mM salt, then use Method 2.
-
1.Buffer exchange and sample concentration.
-
a.Use a vacuum manifold and vacuum of up to −24 Hg for all plate conditioning and sample concentration steps. Wash any preservatives or wetting agents from a 3k MWCO AcroPrep Advance 96 filter plate (see key resources table) by adding a 300-μL volume of LC/MS-grade H2O and pulling it through the membrane using vacuum. Then condition the filter plate by adding a 300-μL volume of Trypsin Digestion Buffer and pulling the solution through the membrane using vacuum.Note: If all 96 wells are not required place a sealing film over unused wells. The plate can be saved after use, and the unused wells uncovered for use at a later date.Note: Throughout all washes and subsequent steps do not let the wells dry out or proteins may stick irreversibly to the membrane.
-
b.Stop the vacuum and shake excess liquid from the washed plate.
-
c.Transfer your samples to the washed AcroPrep plate. Apply vacuum to the 96-well filter plate to reduce the sample volume to ~100 μL.Note: Samples will flow through the filter-plate wells at variable rates due to varied macromolecular content. Monitor the liquid volumes throughout filtration and add Trypsin Digestion Buffer to samples as necessary to prevent rapidly flowing wells from drying before all wells are adequately concentrated.
-
d.Add a 100-μL volume of Trypsin Digestion Buffer to all wells, and mix using several aspirate and dispense cycles of a pipette.
-
e.Continue vacuum filtration to reduce the sample volume to ~ 50 μL.
-
f.Transfer concentrated samples back into the original deep well plate for digestion.
-
a.
-
2.Reduction/Alkylation
-
a.To the 50-μL volume of sample, add a 50-μL volume of 100% TFE.Note: Even if your sample volume is >50-μL do NOT add more than a 50-μL volume of 100% TFE, or the final concentration at the digestion step could inhibit trypsin activity.
-
b.Add a sufficient volume of the 0.5 M TCEP stock for a final concentration of 5 mM in each sample well, seal the plate with clear film, and incubate for 30 min at 37°C.
-
c.Place the plate at room temperature (20°C–25°C) and add a sufficient volume of the 550 mM iodoacetamide stock for a final concentration of 15 mM in each sample well.
-
d.Seal the plate and incubate for 30 min in the dark, and at room temperature (20°C–25°C).Note: Iodoacetamide is light sensitive and reactive in aqueous solution. We generally store small aliquots of the 550 mM stock solution of iodoacetamide in water at −20°C. These are thawed directly prior to use in this protocol. Alternatively, a fresh stock can be made from powder directly before use.
-
e.Quench the unreacted iodoacetamide by the addition of a sufficient volume of the 1 M DTT stock for a final concentration of 7.5 mM in each sample well.
-
a.
-
3.Digestion
-
a.Add ~880-μL volume of the Trypsin Digestion Buffer to bring each sample to a final volume of 1 mL, this dilution reduces the TFE concentration to less than 5%.
-
b.Add a 10 μL volume (1 μg of enzyme) of a proteomics-grade Trypsin stock solution in 10 mM acetic acid to each well.
-
c.Seal the plate with clear film and digest overnight (12–16 h) at 37°C.
-
d.Stop the protein digestion by the addition of a sufficient volume of formic acid for a final concentration of 0.1% (v/v) per sample well.
-
a.
-
4.Desalting and Peptide recoveryNote: This step uses the solid-phase C18 Lab-in-a-Plate device indicated in the key resources table. As with the ultrafiltration plates, unused wells should be covered with sealing film during plate use so that they may be saved for future use.
-
a.Condition each utilized well of the C18 Lab-in-a-Plate using vacuum filtration as follows:
-
i.Add a 100-μL volume of the 60% Buffer B per well and vacuum through. Repeat a second time.
-
ii.Add a 100-μL volume of Buffer A per well and vacuum through. Repeat 3–4 times.
-
i.
-
b.Add the 1-mL volume of each sample to an individual conditioned well on the plate, and apply vacuum to filter the samples through.
-
c.Wash the wells by adding a 100-μL volume of Buffer A, and apply vacuum to filter. Repeat 2 more times.Note: If your sample initially had very high salt add some additional Buffer A washes here to thoroughly de-salt the sample prior to peptide elution for LC/MS analysis.
-
d.Place a 96-well collection plate into the appropriate vacuum manifold adaptor to collect the eluate.CRITICAL: Strong vacuum can lead to bubbling/foaming of the eluting liquid causing cross-contamination of samples so pay attention at this step and adjust the vacuum accordingly. Elution at <−15 Hg is recommended. Alternatively, samples can be eluted by using gentle centrifugation (<1,000 × g) in a swinging bucket rotor equipped with microplate adaptors. During centrifugation, the C18 plate is directly seated on top of the collection plate.
-
e.Elute the digested peptides into the collection plate using a 50-μL volume of 60% Buffer B per well. Repeat the elution step a second time for a total eluate volume of 100 μL.
-
f.Evaporate the eluate solvent in a vacuum centrifuge to dryness. This step takes approximately 3 h.Pause Point: If you will not be able to load samples onto a MS system within ~24 h these samples can be stored dry, sealed with adhesive foil at −80°C.
-
g.For LC/MS analysis, resuspend peptides in a 20- to 35-μL volume of Buffer C per well, pipette up and down several times, and transfer the reconstituted sample to the appropriate sample vial or plate for your autosampler system.Note: We use Waters deep 96-well plates appropriate to our autosampler with a pre-slit silicone cap-mat as listed in the key resources table. A 5-μL volume of each sample is sufficient for LC/MS analysis using the mass spectrometry parameters described below. Unused sample can be stored for a period of weeks to months at −80°C in sealed plates, but solvent may evaporate.
-
a.
Prepare protein fractions for MS analysis
Method 2: Bead binding and on-bead digest
Method 2 for reduction/alkylation and trypsin digestion can be used if your sample contains < 300 mM salt. Otherwise you must use Method 1 (see step 1 above).
Prepare SpeedBead slurry for Method 2
-
5.
Completely resuspend each of the 2 types of commercial beads (hydrophilic and hydrophobic, see key resources table) provided as a 50 mg/mL slurry containing preservative (0.05% sodium azide). Be thorough with vortex-mixing and pipetting to ensure homogeneity.
-
6.
Combine 50 μL of each commercial bead suspension in a single 1.5 mL microcentrifuge tube and mix well.
-
7.
Collect the beads by low-speed pulse centrifugation in a microfuge and remove and discard the supernatant.
-
8.
Wash the beads by thorough resuspension in 1 mL LC/MS-grade H2O.
-
9.
Repeat steps 7 and 8.
-
10.
Collect the beads as in step 7 and resuspend in a 500-μL volume of LC/MS-grade H2O for a working microparticle dispersion density of 10 μg/μL total (5 μg/μL of each bead type). This slurry can be aliquoted for ease of resuspension, and stored at 4°C (do not freeze) for at least 6 months. Resuspend beads thoroughly before each use.
Process sample using Method 2
-
11.Reduction and alkylation
-
a.To each well containing your samples add 100% TFE to achieve 20% TFE final concentration and mix by pipetting.
-
b.Add a sufficient volume of the 0.5 M TCEP stock for a final concentration of 5 mM in each sample well.
-
c.Seal the plate with adhesive film and incubate 45 min at 37°C.
-
d.Place the plate at room temperature (20°C–25°C).
-
e.Add a sufficient volume of the 550 mM iodoacetamide stock for a final concentration of 25 mM in each sample well (see note at step 2d above).
-
f.Incubate for 30 min in the dark at room temperature (20°C–25°C).
-
i.Quench reactivity by the addition of a sufficient volume of the 1 M DTT stock for a final concentration of 12 mM in each sample well.
-
i.
-
a.
-
12.Bead binding of proteins
-
a.To each fraction add a 4-μL volume of bead suspension. The bead suspension is a stock slurry comprising a 1:1 mix of 5 μg/mL each of the two bead types (hydrophobic and hydrophilic) listed in the key resources table.
-
b.Add a sufficient volume of formic acid for a final concentration of 2% (v/v), and a sufficient volume of acetonitrile for final solvent proportion of 50% (v/v) and mix well by pipetting. These two ingredients can be made into a premix for easier dispensing.
-
c.Mix gently but thoroughly by pipetting.
-
d.Incubate for 30 min with gentle shaking or rocking to keep beads from settling.CRITICAL: incubation for prolonged periods of time does not help and may reduce recovery.
-
e.Collect beads from this large volume by brief centrifugation (e.g., 5 min at 1,000 × g) in a swinging bucket rotor equipped with deep well plate adaptors.
-
f.Place the sample plate on the magnetic plate. Remove and discard the bulk of the liquid leaving behind the beads and approximately a 250-μL volume of liquid.
-
g.Take the sample plate off the magnetic plate. Resuspend the beads in the remaining liquid and transfer them to a fresh 450 μL conical bottom, 96-well plate to facilitate the remaining steps.
-
h.Place this shallow plate on the magnetic plate. Once beads have collected to the bottom, remove and discard the remaining liquid.Note: beads will vary in appearance from rust color when diffuse to darker brown when compact, and appear more aggregated or dispersed depending on protein content and other unknown parameters. None of these appearances indicates failure to bind proteins.
-
i.Keep the sample plate on the magnetic plate for all the following wash steps and work rapidly. Pipette carefully to avoid losing beads. The washes need not resuspend the bead pellet.
-
i.Wash with a 100-μL volume of 70% ethanol per well. Repeat this wash step a second time.
-
ii.Wash with a 100 μL volume of acetonitrile. Repeat this wash step a second time.
-
iii.Air dry the beads briefly.
-
i.
-
a.
-
13.On-bead digestion of proteins
-
a.Remove the sample plate from the 96-well magnetic separator plate, and resuspend the beads in a 25-μL volume of the 10% TFE prepared in Trypsin Digestion Buffer solution.Note: Do not be concerned if beads appear “chunky” or aggregated at this stage.
-
b.Dilute enough trypsin stock solution with trypsin digestion buffer for the next step.
-
c.To each sample add a 25-μL volume of Trypsin Digestion Buffer that contains 0.25 μg of proteomics-grade Trypsin.
-
d.Seal the plate with adhesive film and incubate overnight (12–16 h) at 37°C.
-
a.
-
14.Peptide recovery
-
a.Place the sample plate on the magnet. Once the beads have collected transfer the bead-free supernatant to a fresh 96-well plate.
-
b.To the transferred volume add a sufficient volume of formic acid for a final concentration of 1% (v/v) to stop protein digestion.Note: Formic acid is very volatile and corrosive to metal parts of pipettors (e.g., plungers) at concentrations >10%. If you are pipetting undiluted formic acid we recommend using positive displacement pipettors.
-
c.Remove the plate from the 96-well magnetic plate separator, and elute peptides from the beads with the addition of a 50-μL volume of a 2% DMSO solution.Note: If possible, cover samples with adhesive film and sonicate 5 min in a bath sonicator to assist elution at this point.
-
d.Replace the plate on the 96-well magnetic plate separator to collect the beads. Remove each sample eluate and add it to the corresponding supernatant from step 14a.
-
e.Remove the plate from the magnet and elute the beads with another 50-μL volume of a 2% DMSO solution (no sonication necessary).
-
f.Replace the bead plate on the magnetic plate separator. Remove this second volume of eluate and pool it with the first corresponding matched eluate for a total volume of 150 μL for each fraction.
-
g.Desalting of the eluted peptides is identical to that in Method 1 (step 4) with the only difference being the sample volumes loaded on the conditioned solid-phase C18 Lab-in-a-Plate are 150 μL instead of 1 mL.
-
a.
LC/MS system and acquisition conditions
To maximize its generality, this protocol was explicitly designed to work with CF/MS data collected using different LC/MS equipment and analysis protocols, including both label and label-free protein quantification. In practice, we frequently use the following LC/MS setup:
Peptides are separated using reverse phase chromatography on a Dionex UltiMate 3000 RSLCnano UHPLC system with a C18 trap to Acclaim C18 PepMap RSLC column configuration. We typically employ a data-dependent acquisition strategy on either a Thermo Orbitrap Elite, Orbitrap Fusion, or Orbitrap Fusion Lumos mass spectrometer. If using an Orbitrap Fusion or Lumos, we elute peptides with a 3%–45% acetonitrile gradient in 0.1% formic acid over 60 min, and if using a less sensitive machine (Orbitrap Elite) we employ a longer gradient separation, e.g., 5%–40% acetonitrile gradient in 0.1% formic acid over 120 min. Additional details are provided in McWhite et al., 2020.
Expected outcomes
Plates prepared as above are ready for protein detection and quantification by LC/MS. We typically exclude fractions corresponding to baseline UV signal in a chromatographic separation. In cases where a particular separation experiment is repeated we will also exclude those particular fractions that previously contained insufficient material to confidently identify proteins.
In our experience, starting with samples that contain too little protein is the biggest cause of failure. Fractionation experiments beginning with less than 1 mg total protein generally produce poor results. We recommend aiming for 1–4 mg total protein at a concentration of ≥ 5 mg/mL. Failure to reach this amount can occur when beginning with too little starting material. If grinding the material on liquid nitrogen and prior to protein extraction, aim for at least a 1 mL volume of frozen powder, ideally 5 mL. Likewise, failure to extract sufficient protein can occur when the sample inherently has a very low proportion of protein per overall mass (e.g., starch-filled seeds) or a high amount of confounding non-proteinaceous substances (e.g., abundant mucus). In these cases it is important to use extraction methods specialized for the particular organism or tissue.
Quality control
After fractionation, proteins in each fraction are identified and quantified by LC/MS. This information is used to create elution profiles across all fractions for each protein observed. After proteins have been identified, we typically spot-check the elution profile in Excel for intact protein complexes, typically highly stable and abundant protein complexes such as the proteasome, CCT chaperonin, and ribosome. Table S1 shows an example of one of these elution profiles, annotated with protein names and a selection of the typically highly stable and abundant complexes we use as internal controls. In practical terms, if a fractionation experiment shows, e.g., core proteasome subunits with different elution patterns, it means that the experimental preparation may have failed to preserve intact protein complexes. To simplify this quality control step, we add a sparkline column showing the elution profile of each protein, and an annotation column containing, e.g., gene names or protein names. As a quick-and-dirty sanity check, proteins that are members of the same complex are often named similarly (i.e., CCT1, CCT2, CCT3), etc, so sorting the Excel sheet by the annotation column will group members of some complexes together, allowing their intactness to be quickly visually assessed by the similarity of subunit sparklines.
Determine protein interactions from protein elution profiles
The computational protocol identifies sets of proteins which have related elution profiles, signifying that these sets are physically associating. For determining protein interactions, we always use a set of separations represented by at least two different fractionation techniques (e.g., size exclusion and ion exchange). Many complexes elute in the same size range, or the same charge range, however few elute at both the same size and charge. The orthogonality of separations of different biochemical properties highly increases the discovery power of CF/MS.
To simplify the computational process of detecting protein interactions from CF/MS data, we provide cfmsflow, a Nextflow pipeline for Linux systems. This pipeline takes as inputs identified protein profiles (elution profiles) and uses known protein interactions (provided by the user) to train a model of elution profile similarity.
The pipeline is divided into 5 main steps (Figure 3). To summarize, these steps are to 1) Calculate similarities between protein elution profiles, 2) Combine similarity scores into a feature matrix, 3) Label the feature matrix with gold standard protein-protein interactions, 4) Train a model to detect and score pairwise protein interactions, 5) Cluster the resulting protein interaction network into complexes.
Figure 3.

Overview of computational pipeline to detect protein-protein interactions and protein complexes
In step 1, a set of similarity scores between all proteins are calculated for each fractionation experiment. In step 2, these similarity scores are combined into one large table. In step 3, pairs of proteins that are known from prior literature to interact are labeled with a 1 (positive training label), and a set of random pairs of proteins are labeled with a −1 (negative training label). In step 4, a model is trained to distinguish these positive and negatively labeled pairs of proteins, giving a score to each pair, where a higher score indicates higher probability of interaction. In step 5, this interaction network is clustered to protein complexes.
By default, the pipeline begins at step 1 (calculating protein profile similarities) and continues through step 5 (detect complexes), however, a subset of steps or individual steps may be run as set in the user parameter file.
Quantification and statistical analysis
The pipeline requires two main data inputs 1) A set of known gold standard protein complexes, and 2) One table of protein identifications for each fraction per each separation experiment. Examples of all input files can be found in the example_input/ directory. For maximal power in deriving protein-protein interactions, it is ideal to use multiple separations using different separation techniques, as CF/MS gains substantial statistical power from reproducible co-elution behavior of interacting proteins across otherwise distinct separations. Each table of protein identifications for each fractionation plate should be formatted as a comma separated table with header, where the first column (named ID) contains protein identifiers, and the following columns contain protein quantification in each fraction. Values can be any abundance metric, such as peptide spectral matches (PSMs), peptide peak area or precursor ion intensity. We typically use gold standard protein complexes from the CORUM database. This file should be formatted with one protein complex per line, and each protein in the complex separated by a space.
Check the notes and CRITICAL points below carefully before proceeding to running the pipeline.
nextflow main.nf –params-file user_parameters.json
nextflow main.nf –params-file example_params/example_wholepipeline.json
Note: Examples of all input file formats are provided in the example_input directory of the github repository.
Note: For discrete count-based measures such as peptide spectral matches (PSMs), correlations may be run for N repetitions with Poisson noise, and scores averaged, controlled by the parameter `added_poisson_reps = N`. Intensity-based measures should not be run with added Poisson noise.
Note: To resume running a pipeline after fixing an error or changing a parameter value, run the same command as above with additionally –resume to run all steps downstream of the parameter change.
Computational pipeline steps
-
1.Calculate features
-
a.Correlation and distance metrics are calculated between all possible pairs of proteins in each elution file.
-
b.Input: File containing paths to elution files, or glob pattern matching paths to elution files.
-
a.
Note: Reduce feature length and run time by pre-filtering to only include well observed proteins, or proteins that are observed in at least n experiments.
-
2.Build a feature matrix
-
a.Features are joined into a single table
-
b.Input: Output of previous step, or file containing paths to feature files, or glob pattern matching paths to feature files.
-
a.
-
3.Label the feature matrix with gold standard interactions
-
a.A column “label” is added to the feature matrix, with a value of 1 if a positive training interaction and −1 if a negative interaction
-
b.Input: Output of previous step, or path to a feature matrix file
-
c.Input: Path to gold standard interactions
-
a.
Note: Training labels may be either provided by the user or generated from input gold standard complexes. Negative interactions can either be drawn from observed interactions (as in McWhite et al., 2020) or from faux interactions between different gold standard complexes (as in (Drew et al., 2017), as controlled by the parameter `negatives_from_observed = true/false`.
-
4.Train a model to score protein-protein interactions
-
a.The TPOT AutoML software scans machine learning pipelines and parameters to determine optimal model parameter settings based on positive and negative labeled training interactions.Note: Available algorithms are listed in accessory_files/all_classifiers.txt
-
b.A model is trained on the same training interactions using the parameter settings determined by TPOT
-
c.This model is applied to the full feature matrix to give a CF-MS score to all pairs of proteins in the feature matrix.Note: Interactions from the same gold standard complex are prevented from being split between different cross validation portionsNote: To reduce overtraining effects, lower the value of the max_features_to_select parameter. The model will then select no more than this number of features. After modifying this parameter, rerun the nextflow command with an additional –resume flag to avoid rerunning already complete portions of the pipeline.
-
a.
-
5.Cluster interactions
-
a.Scored interactions are thresholded at a parameter-input false discovery rate threshold (default 0.1), then clustered into protein complexes with diffusion clustering (see Supplemental methods section of McWhite et al., 2020).
-
a.
Limitations
No single extraction method will be able to comprehensively identify all protein complexes because of their diverse biochemical characteristics. For example, extraction conditions that readily solubilize cytoplasmic proteins may not be optimal for transmembrane proteins or cytoskeletal proteins. Therefore the original extraction conditions and chromatography method will most directly affect the range of detectable proteins identified by LC/MS. Failure to identify a specific expected protein complex is typically attributable to insufficient abundance of complex members or insufficient stability of the complex. If a protein is detected with less than approximately 10 PSMs per fractionation experiment, it is unlikely that this experimental approach will be able to confidently detect its interactors.
Troubleshooting
Problem 1
Pipeline fails to run
Potential solution
First, confirm that the example parameter json (example_params/example_wholepipeline.json) runs to completion. If it does not, use provided example parameter jsons to test and troubleshoot individual steps of the pipeline. Next, confirm that the file format of your input files exactly matches the corresponding example input files, including delimiters and column names when specified. Confirm at the command line that file paths in your parameter json exist. If missing parameters are warned, add those parameters to your parameter json.
Problem 2
Overfit model
Potential solution
When a model is overfit to the training data, it fails to generalize to data not used to train the model. Overfitting can be visually diagnosed by comparison of the precision-recall curves of training and test of interactions (Figure 4). While models will generally perform better on training labels, a large gap in performance between training and test precision-recall curves is diagnostic of an overfit model. Overfitting can often be reduced by reducing the number of features that are used to construct the model. The initial run of the pipeline will produce a .featureimportances file after step 4, which contains feature importances determined by Random Feature Elimination. To rerun the TPOT pipeline limiting the maximum number of features, modify the max_features_to_select value in your user parameters json to a smaller number than the total number of features and resume the pipeline with nextflow main.nf –params-file user_parameters.json –resume or create a new parameters file that begins at step 4.
Figure 4.

Using precision-recall curves to evaluate overfitting
Precision-recall curves illustrating overfit, better fit, and underfit models. A substantial difference between Test and Train precision-recall curves suggests overfitting, and that the max number of features should likely be lowered to improve model performance. When a model is overfit, as features are removed, the Test precision-recall curve will shift right, while the training curve is minimally affected. Once too many features have been removed, performance in both test and training will decline.
Problem 3
Too few or too many proteins in the output clustering file. Large complexes broken apart.
Potential solution
The number of interactions forwarded into clustering is controlled by the fdr_cutoff value in your user parameters json, which thresholds interactions with CF-MS scores above a particular false discovery rate. False discovery rate (FDR) is calibrated from test positive and negative labeled interactions.
A high FDR cutoff that uses more interactions can cause an overly dense network that, in our experience, can lead to less cleanly distinguished complexes after clustering. However, an overly stringent FDR cutoff reduces recall of complexes.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Edward Marcotte (marcotte@utexas.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The cfmsflow pipeline is available at https://github.com/marcottelab/cfmsflow. Example data are provided along with the pipeline.
Acknowledgments
Research was funded by grants from the Welch Foundation (F-1515 to E.M.M.); NSF (1237975 to E.M.M.); Army Research Office (W911NF-12-1-0390); and NIH (GM123683 to C.D.M., K99 HD092613 to K.D., and R35 GM122480 and R01 HD085901 to E.M.M.).
Author contributions
Writing – Experimental Methodology, O.P.; Writing – Computational Methodology and Pipeline, C.D.M.; Editing, C.D.M., O.P., and E.M.M.; Scripts, C.D.M. and K.D.; Pipeline Testing, V.D., W.S., and J.C.L.; Funding Acquisition, C.D.M., K.D., and E.M.M.; Supervision, E.M.M.
Declaration of interests
The authors have no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100370.
Contributor Information
Claire D. McWhite, Email: cmcwhite@princeton.edu.
Ophelia Papoulas, Email: papoulas@austin.utexas.edu.
Edward M. Marcotte, Email: marcotte@utexas.edu.
Supplemental information
Data are a Chlamydomonas reinhardtii ion exchange fractionation experiment from McWhite et al., 2020 (ProteomeXchange: PXD013369).
References
- Di Tommaso P., Chatzou M., Floden E.W., Barja P.P., Palumbo E., Notredame C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017;35:316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- Drew K., Lee C., Cox R.M., Dang V., Devitt C.C., Papoulas O., Huizar R.L., Marcotte E.M., Wallingford J.B. A systematic, label-free method for identifying RNA-associated proteins in vivo provides insights into vertebrate ciliary beating machinery. Dev. Biol. 2020;467:108–117. doi: 10.1016/j.ydbio.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew K., Lee C., Huizar R.L., Tu F., Borgeson B., McWhite C.D., Ma Y., Wallingford J.B., Marcotte E.M. Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes. Mol. Syst. Biol. 2017;13(6):932. doi: 10.15252/msb.20167490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebeskind B.J., Young R.L., Halling D.B., Aldrich R.W., Marcotte E.M. Mapping functional protein neighborhoods in the mouse brain. bioRxiv. 2020 doi: 10.1101/2020.01.26.920447. [DOI] [Google Scholar]
- Mallam A.L., Sae-Lee W., Schaub J.M., Tu F., Battenhouse A., Jang Y.J., Kim J., Finkelstein I.J., Marcotte E.M., Drew K. Systematic discovery of endogenous human ribonucleoprotein complexes. Cell Rep. 2019;29:1351–1368.e5. doi: 10.1016/j.celrep.2019.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhite C.D., Papoulas O., Drew K., Cox R.M., June V., Dong O.X., Kwon T., Wan C., Salmi M.L., Roux S.J., Jr., Browning K.S., Chen Z.J., Ronald P.C., Marcotte E.M. A Pan-plant protein complex map reveals deep conservation and novel assemblies. Cell. 2020;181:460–474.e14. doi: 10.1016/j.cell.2020.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson R.S., Moore J.H. TPOT: A tree-based pipeline optimization tool for automating machine learning. In: Hutter F., Kotthoff L., Vanschoren J., editors. Automated Machine Learning. Springer; 2016. pp. 66–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data are a Chlamydomonas reinhardtii ion exchange fractionation experiment from McWhite et al., 2020 (ProteomeXchange: PXD013369).
Data Availability Statement
The cfmsflow pipeline is available at https://github.com/marcottelab/cfmsflow. Example data are provided along with the pipeline.