

Summary
Here, we present a protocol to analyze de novo genetic variants derived from the whole-exome sequencing (WES) of proband-parent trios. We provide stepwise instructions for using existing pipelines to call de novo mutations (DNMs) and determine whether the observed number of such mutations is enriched relative to the expected number. This protocol may be extended to any human disease trio-based cohort. Cohort size is a limiting determinant to the discovery of high-confidence pathogenic DNMs.
For complete details on the use and execution of this protocol, please refer to Dong et al. (2020).
Subject areas: Bioinformatics, Sequence analysis, Genetics, Genomics, Sequencing, High-throughput screening
Graphical Abstract
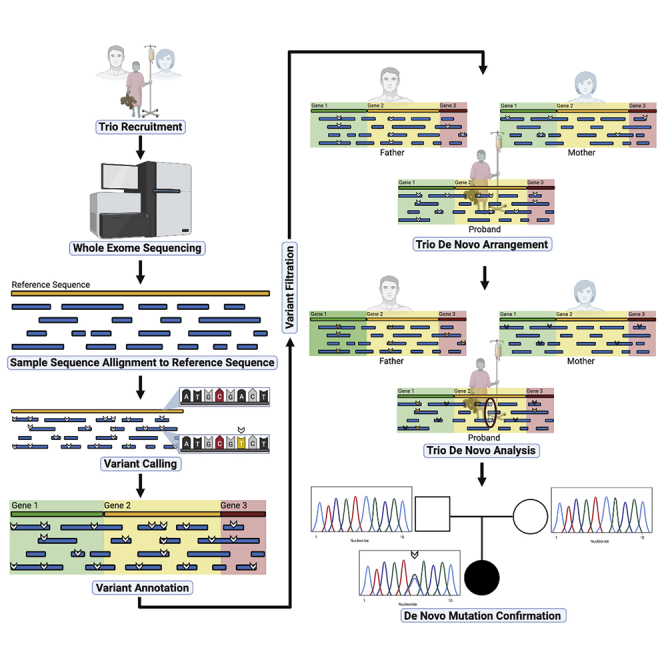
Highlights
-
•
We demonstrate the ability to call de novo mutations from whole-exome sequencing data
-
•
This protocol is applied to WES from cohorts composed of proband and both parents
-
•
We demonstrate how to perform enrichment analysis using denovolyzR
-
•
The size of the trio-based cohort is a limiting determinant of this protocol’s accuracy
Here, we present a protocol to analyze de novo genetic variants derived from the whole-exome sequencing (WES) of proband-parent trios. We provide stepwise instructions for using existing pipelines to call de novo mutations (DNMs) and determine whether the observed number of such mutations is enriched relative to the expected number. This protocol may be extended to any human disease trio-based cohort. Cohort size is a limiting determinant to the discovery of high-confidence pathogenic DNMs.
Before you begin
Timing: hours to days; factors that affect timing include the number of samples and available computing resources
Note: The user should already have recruited and performed WES on a cohort of trios (proband and both parents). Following this, the user will need to take unmapped sequence information and create a variant call format (VCF) file for downstream analysis. The genome analysis toolkit (GATK), developed by the Broad Institute, features a “best practices” workflow for data preprocessing and variant discovery (The 1000 genomes project consortium, 2015; McKenna et al., 2010; Van der Auwera et al., 2013).
-
1.Data preprocessing: data preprocessing generates binary alignment/map (BAM) files from unmapped sequencing reads. Preprocessing includes: alignment of unmapped reads to the human reference genome. The Broad Institute preprocessing workflow can be found at the location listed below:
- a.
-
2.Variant discovery:
-
a.The BAM files generated during preprocessing are fed into GATK’s HaplotypeCaller to perform per-sample variant calling, outputting a genomic VCF (gVCF) for a single sample. The Broad Institutes GATK preprocessing workflow can be found at the locations listed below.
- i.
- ii.
-
b.Joint-calling and variant quality score recalibration (VQSR) generates a final, multisample VCF that is the starting point for downstream analysis. The Broad Institute joint genotyping workflow can be found at the locations listed below.
- i.
- ii.
-
c.Additional best practices information for GATK germline pipelines can be found here: https://gatk.broadinstitute.org/hc/en-us/articles/360035535932-Germline-short-variant-discovery-SNPs-Indels-Note: Dong et al. used GATK best practices version 3 for all preprocessing steps, including mapping sequence reads at each site to the GRCh37 reference genome (Dong et al., 2020). Please note, however, that GATK version 3 is no longer supported by the developers. For this reason, the above discussion gives instructions specific to GATK4 and links user to GATK4 resources. We recognize that different users will be operating with access to different levels of computational resources and support, and so implementation of GATK4 best practices is likely to feature user-specific needs that are beyond the scope of this manuscript. For this reason, we recommend users visit the links posted above if they have questions about implementing GATK. In addition, we have provided two links below that for users interested in other pipelines for data preprocessing and germline variant discovery:
- i.
-
ii.https://bcbio-nextgen.readthedocs.io/en/latest/Note: Steps 3 and 4 pertain to kinship analysis and sample duplicate removal. Kinship analysis is a prerequisite to the discovery of de novo genetic variation. There are multiple established and accepted methods for verifying biological relationships among self-reported mother-father-proband trios. Dong et al. used identity-by-descent (IBD) (Purcell et al., 2007) and an analysis of high quality ultrarare single nucleotide polymorphisms (SNPs) absent from ExAC and gnomAD (Lek et al., 2016) for this purpose.
-
a.
-
3.Pedigree confirmation
-
a.Pairwise PLINK IBD calculation (Purcell et al., 2007)
-
a.
-
4.Sample duplicate removal
-
a.Remove individuals with ≥90% IBD (Purcell et al., 2007)
-
b.Remove individuals with shared ultrarare SNPs
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Published paper and data | Dong et al., 2020 | dbGap accession number: phs000744 |
| 1,000 genomes | The 1000 genomes project consortium, 2015 | https://www.internationalgenome.org/data |
| ExAC database | Lek et al., 2016 | https://gnomad.broadinstitute.org/ |
| Exome variant server (EVS) | Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA | https://evs.gs.washington.edu/EVS/ |
| BRAVO | Program, 2018 | https://bravo.sph.umich.edu/freeze8/hg38/ |
| Software and algorithms | ||
| TrioDeNovo | Wei et al., 2014 | https://genome.sph.umich.edu/wiki/Triodenovo |
| GATK | McKenna et al., 2010 | https://github.com/broadinstitute/gatk/ |
| DeNovo protocol code | Scripts, intermediate files, and data generated for this protocol | https://github.com/jinlab-washu/de-novo-wes-star-protocol |
Step-by-step method details
We have created a github that features all of the necessary files, scripts, and example outputs for the method presented herein. The github can be found at:
https://github.com/jinlab-washu/de-novo-wes-star-protocol
Users are advised to refer to github to download example trios (in VCF format) and pedigree (PED) files if they wish to replicate the step-by-step instructions reported here exactly.
Run DeNovo Analysis for each trio
In this step users will perform DeNovo analysis using TriodenovoRearrange_pythonAuto.R, which outputs a filtered tsv (tab-separated values) file of calls for each trio in your cohort.
-
1.Generate the commands for each trio, replacing TRIO_NAME with the name of each trio: Rscript TriodenovoRearrange_pythonAuto.R exome_calls_chr4_2trios.vcf Trios.ped OUTPUT_FOLDER TRIO_NAME. This should be done in the command line, and if this is sufficient then users should continue to step 2. Otherwise, users should execute the following sub-steps in R (recommended version 4.0.2).
-
a.Create a new directory for the cohort
-
i.system(paste("mkdir ",CohortName,sep = ''),intern = F)
-
i.
-
b.Read in the pedigree file
-
i.Fam = read.table(file=ped,header=TRUE,stringsAsFactors=FALSE)
-
ii.See Trios.ped for an example pedigree format
-
i.
-
c.Change your working directory to the cohort directory
-
i.setwd(CohortName)
-
i.
-
d.Create a directory for the sample and change to id
-
i.command0=paste("mkdir -p ",Familyno,sep="")
-
ii.system(command0,intern=F)
-
iii.setwd(Familyno)
-
i.
-
e.Generate a pedigree file for just one trio
-
i.index <- which(Fam$FamID == Familyno)
-
ii.write.table(Fam[index,],file = paste("Trio_",Familyno,".ped",sep = ""),col.names=FALSE,row.names=FALSE,sep="∖t",quote=FALSE)
-
iii.Fam = Fam[index,]
-
iv.index = which(Fam$Father != 0)
-
v.Proband_ID = Fam[index,2]
-
vi.Father_ID = Fam[index,3]
-
vii.Mother_ID = Fam[index,4]
-
i.
-
f.Generate a VCF file for each trio, removing all records with missing genotype, only extracting lines where AC != 0
-
i.command1=paste("java -Xmx64g -jar GenomeAnalysisTK_3.5.jar -nt 16 -R /ref_data/h_sapiens/1000genomes/2.5/b37/human_g1k_v37_decoy.fasta -T SelectVariants --variant ",Input," -o Trio_",Familyno,".vcf -env -sn ",Proband_ID,' -sn ',Father_ID,' -sn ',Mother_ID,sep="")
-
•-nt 16: use 16 threads
-
•-R: path to reference fasta
-
•-T SelectVariants --variant: select a subset of variants from a VCF
-
•-o: output file path
-
•-env
-
•-sn: specify a sample name from which to include genotypes
-
•
-
ii.system(command1,intern=F)
-
i.
-
g.Regenotype
-
i.system(paste('java -Xmx64g -jar GenomeAnalysisTK_3.5.jar -nt 16 -R /ref_data/h_sapiens/1000genomes/2.5/b37/human_g1k_v37_decoy.fasta -T RegenotypeVariants --variant Trio_',Familyno,'.vcf -o Trio_',Familyno,'_reGT.vcf',sep = ''),intern = F)
-
•-nt 16: use 16 threads
-
•-R: path to reference fasta
-
•-T RegenotypeVariants --variant: Regenotypes the variants from a VCF
-
•-o: output file path
-
•
-
i.
-
h.Split multi-allelic sites
-
i.system(paste('bcftools norm -m-both -o Trio_',Familyno,'_reGT_step1.vcf Trio_',Familyno,'_reGT.vcf',sep = ''),intern = F)
-
•norm: normalize indels
-
•-m-both
-
•-o: output file path
-
•
-
i.
-
i.Left normalization
-
i.system(paste('bcftools norm -f /ref_data/h_sapiens/1000genomes/2.5/b37/human_g1k_v37.fasta -o Trio_',Familyno,'_reGT_step2.vcf Trio_',Familyno,'_reGT_step1.vcf',sep = ''),intern = F)
-
•norm: normalize indels
-
•-f: path to reference fasta
-
•-o: output file path
-
•
-
i.
-
j.Remove extra information (PID, PGT)
-
i.system(paste('vcfkeepgeno Trio_',Familyno,'_reGT_step2.vcf GT AD DP GQ PL > Trio_',Familyno,'_reGT_step2_modified.vcf',sep = ''),intern = F)
-
i.
-
k.Remove ./., unfavored PL, and AC != 0
-
i.system(paste('python ParseTrioVCF.py ',Familyno,sep = ''),intern = F)
-
ii.Requires ParseTrioVCF.py
-
i.
-
l.Annotate the updated VCF with annovar
-
i.command7=paste("perl table_annovar.pl --vcfinput Trio_",Familyno,"_updated.vcf /programs/annovar/humandb/ -buildver hg19 -out Trio_",Familyno," -remove -protocol refGene,genomicSuperDups,snp138,dbnsfp33a,esp6500siv2_all,1000g2015aug_all,exac03,gnomad_exome,gnomad_genome,bravo -operation g,r,f,f,f,f,f,f,f,f -nastring '.'",sep = "")
-
•--vcfinput: specify the input vcf file
-
•-buildver: genome build version
-
•-out: specify the output file
-
•-remove: remove all temporary files
-
•-protocol: comma-delimited string specifying database protocol
-
•In this case we are using these protocols: refGene,genomicSuperDups,snp138,dbnsfp33a,esp6500siv2_all,1000g2015aug_all,exac03,gnomad_exome,gnomad_genome
-
•-operation: comma-delimited string specifying type of operation
-
•-nastring: string to display when a score is not available
-
•
-
ii.system(command7,intern=F)
-
i.
-
m.Run triodenovo
-
i.command6=paste("triodenovo --ped Trio_",Familyno,".ped --in_vcf Trio_",Familyno,"_updated.vcf --out Trio_",Familyno,".denovo.Bayfilter.vcf --mixed_vcf_records",sep = "")
-
•--ped: specifies the pedigree file to use
-
•--in_vcf: specifies the vcf file to use
-
•--out: specifies the output file
-
•--mixed_vcf_records
-
•
-
ii.system(command6,intern=F)
-
i.
-
n.Delete intermediate files
-
i.system(paste("rm Trio_",Familyno,".avinput",sep = ""),intern = F)
-
ii.system(paste("rm Trio_",Familyno,".hg19_multianno.txt",sep = ""),intern = F)
-
i.
-
o.Rearrangement
-
i.system(paste('python PrepareMerge.py ',Familyno,sep = ''),intern = F)
-
ii.Requires PrepareMerge.py
-
i.
-
p.Determine the order of members in the VCF
-
i.Order = unlist(strsplit(try(system(paste("grep -w '#CHROM' Trio_",Familyno,"_updated.vcf",sep = ""),intern = T)),'∖t'))
-
ii.col15 = Order[10]
-
iii.col16 = Order[11]
-
iv.col17 = Order[12]
-
i.
-
q.Process the triodenovo output
-
i.Bayfilter=readLines(paste("Trio_",Familyno,".denovo.Bayfilter.content.txt",sep = ""))
-
ii.Bayfilter=sapply(1:length(Bayfilter),function(i) unlist(strsplit(Bayfilter[i],"∖t")))
-
iii.Bayfilter=data.frame(t(Bayfilter),stringsAsFactors = F)
-
iv.colnames(Bayfilter)=c("CHROM","POS","ID","REF","ALT","QUAL","FILTER","INFO","FORMAT",Father_ID,Mother_ID,Proband_ID)
-
v.Bayfilter$POSITION=paste(Bayfilter$CHROM,Bayfilter$POS,Bayfilter$REF,Bayfilter$ALT,sep=":")
-
i.
-
r.Process the annovar output
-
i.Anno=readLines(paste("Trio_",Familyno,".hg19_multianno.content.txt",sep = ""))
-
ii.Anno=sapply(1:length(Anno),function(i) unlist(strsplit(Anno[i],"∖t")))
-
iii.Anno=data.frame(t(Anno),stringsAsFactors = F)
-
iv.colnames(Anno)=c("CHROM","POS","ID","REF","ALT","QUAL","FILTER","Info","Format",paste("Anno.",col15,sep = ""),paste("Anno.",col16,sep = ""),paste("Anno.",col17,sep = ""))
-
v.Anno$POSITION=paste(Anno$CHROM,Anno$POS,Anno$REF,Anno$ALT,sep=":")
-
i.
-
s.Delete intermediate files
-
i.system(paste("rm Trio_",Familyno,".hg19_multianno.content.txt",sep = ""),intern = F)
-
ii.system(paste("rm Trio_",Familyno,".denovo.Bayfilter.content.txt",sep = ""),intern = F)
-
i.
-
t.Merge based on the triodenovo file
-
i.AnnoBayfilter=merge(Bayfilter,Anno,by="POSITION",all.x=TRUE)
-
ii.AnnoBayfilter=AnnoBayfilter[,c(1:8,10:13,21:25)]
-
iii.colnames(AnnoBayfilter)=c("POSITION","CHROM","POS","ID","REF","ALT","QUAL","FILTER","FORMAT",Father_ID,Mother_ID,Proband_ID,"INFO","AnnoFormat",paste("Anno.",col15,sep = ""),paste("Anno.",col16,sep = ""),paste("Anno.",col17,sep = ""))
-
i.
-
u.Write the final output to a file
-
a.
-
2.
Run the list of commands generated in step 1
-
3.Concatenate the output files into one final file, in the command line
-
a.head -1 TRIO_NAME/∗DenovoM > Trios.BayesianFilter.DenovoM
-
b.for file in TRIO_PREFIX∗/∗DenovoM;do sed '1d' $file >> Trios.BayesianFilter.DenovoM;done
-
c.Output: Trios.BayesianFilter.DenovoM
-
a.
-
4.Filter the concatenated file using Python 3.7.3 These commands should be executed in the command line with paths to the Python scripts.
-
a.python ParseBayOutput_SciencePaper.py Trios.BayesianFilter.DenovoM Trios.BayesianFilter.DenovoM.filtered
-
b.Requires ParseBayOutput_SciencePaper.py
-
c.Output: Trios.BayesianFilter.DenovoM.filtered
-
a.
Denovolyze preparation and visualization of candidate DNMs
The DeNovo Analysis output will now be manually filtered and annotated to prepare for the Denovolyze step.
-
5.After compiling a list of candidate DNMs, all calls must be verified manually using the integrative genomics viewer (IGV) (Robinson et al., 2011). IGV can be accessed at igv.org and manual visualization of DNM calls can be accomplished as follows:
-
a.Open IGV and load in a BAM, CRAM (and any associated index file), or other supported file format from the first subject you wish to analyze.
-
b.Select the appropriate reference sequence for your analysis and enter the chromosome and position of the called variant to the search bar at the top.
-
c.Zoom in to the user-determined threshold window to visualize all recorded reads.
-
d.Export the image by clicking "Save Image" under the "File" menu and saving to your desired folder.
-
e.Repeat steps for all candidate DNMs in the call list for the specific subject file loaded into IGV.
-
f.End the current session and create a new session loading the BAM or CRAM file of the next subject you wish to analyze.
-
g.Repeat the above steps until all candidate DNM calls have been manually analyzed in IGV.
- h.
-
a.
-
6.Open the output in your preferred spreadsheet software and classify the mutations into different categories in the column denovo_metasvm_cadd30
-
a.Use the ExonicFunc.refGene column to aid in this process
-
b.The MetaSVM and CADD columns may also be helpful
-
c.Example: A nonsynonymous_SNV with a CADD (v1.3) score ≥ 30 or a MetaSVM prediction of “D” would be classified as misD (Kircher et al., 2014)
-
d.Output: Trios.BayesianFilter.DenovoM.filtered.PlotReads.xlsx
-
a.
Enrichment analysis
The enrichment analysis relies on the denovolyzeR library and will output mutability or enrichment tables depending on the script. The sample data was generated by the DeNovo Analysis step and prepared for denovolyzeR in the previous step. The gene lists used by Dong et al. are available in the github. Users will need curate their own gene lists based upon published literature for the disease they are studying.
-
7.Run the scripts
-
a.Rscript denovolyzR.script.R
-
b.Requires: TN_n70_Input.txt, 0819_hs37d5_coding_idt_med_v2_spikein_padded_Mar2018_adj_modified.txt
-
c.Output: Trios_ObserveExpect_metasvm_cadd30.txt
-
a.
Expected outcomes
Steps 3 and 4 from the step-by-step method details generate tab-separated files. Example outputs are provided in the github.
Step 5 requires the user to generate IGV plots. An example IGV plot is given in the troubleshooting section of this protocol.
Table 1 shows the expected outcome of the enrichment analysis for DNMs for all genes (Table 1). The results match that of supplemental table 2b in our companion paper by Dong et al. The analysis can and should be extended to controls in order to interpret the results; the full findings can be found in Dong et al., 2020.
Table 1.
De novo enrichment analysis among cases
|
De novo enrichment analysis in 70 TN cases | ||||||
|---|---|---|---|---|---|---|
| Class | Observed | rate_obs_cases | Expected | rate_exp_cases | Enrichment | p value |
| all | 74 | 1.057142857 | 79.5 | 1.135714286 | 0.931 | 0.747 |
| syn | 22 | 0.314285714 | 22.5 | 0.321428571 | 0.977 | 0.571 |
| misT | 27 | 0.385714286 | 37.4 | 0.534799393 | 0.721 | 0.968 |
| misD | 17 | 0.242857143 | 12.7 | 0.181428571 | 1.34 | 0.141 |
| lof | 8 | 0.114285714 | 6.9 | 0.098571429 | 1.16 | 0.387 |
| protD | 25 | 0.357142857 | 19.6 | 0.28 | 1.28 | 0.134 |
| prot | 52 | 0.742857143 | 57 | 0.814285714 | 0.912 | 0.764 |
Results of the enrichment analysis for DNMs across all genes in cases. Corresponds to supplemental table 2b in Dong et al. (2020).
The rate_obs_cases is the number of DNMs divided by the number of individuals in the cohort. The enrichment is the ratio of observed to expected numbers of mutations. misD is damaging missense mutations as predicted by MetaSVM and CADD algorithms; misT is the non-damaging missense mutations; protD is the damaging category (misD +lof); prot is the protein altering category (misT + misD + lof). All genes from the genome were considered for the analysis.
Quantification and statistical analysis
Table 2 details the filtering criteria used by Dong et al. for putative DNMs.
Table 2.
Filtering criteria for putative DNMs
| Cases | Controls |
|---|---|
|
|
| |
Example filtering criteria to analyze DNMs in cases and controls. The criteria are the same as those used by Dong et al. (2020).
Dong et al. used a previously developed mutability model (Samocha et al., 2014) to compute a mutability table containing the expected number of DNMs per gene per variant class. We have provided a copy of the mutability table generated by Dong et al. on the github for this protocol for users wishing to replicate their analysis.
The observed versus expected number of DNMs for each variant class were compared using a one-tailed Poisson test. The statistical analyses for DNMs were implemented as part of “denovolyzeR” (Ware et al., 2015) and we included the instructions for replicating these analyses in the step-by-step method details.
Limitations
While WES can identify genetic variation that may predispose an individual to certain disease states, bioinformatic analyses and statistical genetic approaches do not provide functional insight into the role of candidate genes/variants in disease pathogenesis. Statistically significant genetic variation can sometimes be found to be functionally irrelevant when the candidate mutations are reproduced in animal models. Functional validation is a valuable and necessary counterpart to unbiased sequencing approaches if one hopes to obtain mechanistic insight into their disease of interest.
Dong et al., reconstituted the human DNM p.Cys188Trp in the GABAA receptor Cl− channel γ-1 subunit (GABRG1) using mice, and found that mice with these mutations showed signs of trigeminal neuralgia (Dong et al., 2020). However, the resource and time costs of creating and testing such mouse models preclude the possibility of reconstituting each candidate hit. As such, our mechanistic understanding of how genetic variation shapes disease pathogenesis often lags behind our knowledge of what genetic variation exists between patients and healthy controls.
Our protocol is limited by the small number of trios available to Dong et al (70 trios). Small cohorts limit the power to identify candidate genes with more than one damaging DNM. Other studies that used the method illustrated here have featured larger cohorts (Homsy et al., 2015; Jin et al., 2017). For example, studies by Homsy et al. and Jin et al. were able to identify 21 and 66 (respectively) genes with greater than one DNM in cohorts of congenital heart disease (CHD) patients. Informatively, the cohort size in Homsy et al. was 1,213 probands with CHD and this increased to 2,645 CHD probands for the study by Jin et al., reflecting that as the number of cases increases so too does the ability to detect genes with more than one DNM. Despite not being able to directly compare results in a trigeminal neuralgia cohort to a CHD cohort, one appreciates that regardless of disease indication there is a benefit to having larger numbers of cases. Still, even with only 70 trios Dong et al was able to discover pathogenic genetic lesions that arose from DNMs.
Recruitment of larger cohorts would certainly lend greater power to the study by Dong et al., but it is not always possible to recruit cohorts of the necessary size. Importantly, the method outlined in this manuscript does not require a minimum number of case trios. While the method benefits from increased case trios (like any study would), the lack of a minimum number of required case count allows this method to be applied to even very rare diseases where it may be impossible to recruit more than a few dozen patients. Regardless of how many patients one recruits, it is advisable to sequence subjects to coverage of at least 40x if trying to identify DNMs, as this level of coverage has been successful in prior studies (Homsy et al., 2015; Jin et al., 2017). Low coverage makes it more likely to count false positives as true hits, even with visualization in IGV.
Troubleshooting
Problem 1
Choice of reference genome: GRCh37 versus GRCh38
The cohort analyzed in our companion paper was mapped to GRCh37. Alignment against a more complete build of the human reference genome (GRCh38) could offer more accurate read-mapping and result in a more comprehensive variant call set.
Potential solution 1
Choice of reference genome
There is not a correct or incorrect choice when choosing between alignment to GRCh37 or GRCh38. The choice of reference genome used for alignment can have a substantial impact on variant discovery and the choice should be made in the context of what a user hopes to accomplish with their dataset. Increasingly, we would advise users to align to GRCh38 because it will offer greater compatibility with resources like version three of gnomAD (natively aligned to GRCh38). Nevertheless, for users who already have data aligned to GRCh37, it may not be worthwhile or feasible to remap to GRCh38. In these cases, one may consider a liftover of GRCh37 coordinates to GRCh38 if GRCh38 coordinates are required. Liftover can be accomplished by using any number of publicly available tools including:
-
•
The UCSC genome browser (http://www.genome.ucsc.edu/cgi-bin/hgLiftOver)
-
•
NCBI Remap (https://www.ncbi.nlm.nih.gov/genome/tools/remap)
While an in-depth discussion of these tools is beyond the scope of this manuscript, an informative introduction to these resources can be found online at https://genviz.org/module-01-intro/0001/06/02/liftoverTools/
Problem 2
Integrative Genomics Viewer (IGV)
During manual validation of potential DNMs using IGV, it is possible different users will have a different judgment about whether a particular call is a true DNM or a false-positive. This can influence the final set of candidate DNMs.
Potential solution
We have included example IGV images for a true DNM (Figure 1A) and a false-positive DNM (Figure 1B) for the user to contrast. The sample reads and coverage depth are visualized with the putative DNM site demarcated between two dashed lines. The example in Figure 1B is a false-positive call because the putative DNM site is mutated in several reads in parent 177-3. We recognize that the in-text versions of these visualizations may be low resolution for some users. Therefore, we have provided high resolution versions of the IGV plot for each individual from Figure 1 as part of the Data S1.
Figure 1.

Example IGV plots for two DNM calls
(A) A true-positive DNM in the gene GABRG1.
(B) A false-positive DNM in the gene HTT. Color scheme: A is green, C is blue, G is brown, and T is red.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the co-corresponding authors (kristopher.kahle@yale.edu and jin810@wustl.edu).
Materials availability
No biological reagents were used as part of this protocol.
Data and code availability
The WES data analyzed with this protocol and generated by the companion paper Dong et al. are available at dbGap with accession number phs000744. We have provided our Python and R scripts for the de novo variant analysis demonstrated in Dong et al. and this protocol. These can be found at the github repository for this protocol (https://github.com/jinlab-washu/de-novo-wes-star-protocol).
Acknowledgments
This work was supported by NIH Medical Scientist Training Program Training Grant T32GM136651 (N.S.D.) and by R01 NS111029-01A1, R01 NS109358, and K12 228168, as well as by the Rudi Schulte Research Institute (K.T.K.). W.D. is supported by American Heart Association Predoctoral Fellowship (19PRE34380842). S.C.J. is supported by a K99/R00 Pathway to Independence Award (K99HL143036 and R00HL143036-02).
Author contributions
Protocol Design and Conceptualization, N.S.D., S.K., W.D., K.T.K., and S.C.J. Pipeline Execution and Documentation, N.S.D., S.K., W.D., A.S., S.P., G.A., and S.C.J. Writing – Review & Editing, N.S.D., S.K., W.D., G.A., A.S., S.P., K.T.K., and S.C.J. Funding Acquisition and Supervision, K.T.K. and S.C.J.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100383.
Contributor Information
Kristopher T. Kahle, Email: kristopher.kahle@yale.edu.
Sheng Chih Jin, Email: jin810@wustl.edu.
Supplemental information
The file names within the zip correspond to the labeled individual-IDs from the figure.
Supp-File1_Sample-124-1 corresponds to the plot for individual TGRN124-1 in Figure 1A.
Supp-File1_Sample-124-3 corresponds to the plot for individual TGRN124-3 in Figure 1A.
Supp-File1_Sample-124-4 corresponds to the plot for individual TGRN124-4 in Figure 1A.
Supp-File1_Sample-177-1 corresponds to the plot for individual TGRN177-1 in Figure 1B.
Supp-File1_Sample-177-2 corresponds to the plot for individual TGRN177-2 in Figure 1B.
Supp-File1_Sample-177-3 corresponds to the plot for individual TGRN177-3 in Figure 1B.
References
- Dong W., Jin S.C., Allocco A., Zeng X., Sheth A.H., Panchagnula S., Castonguay A., Lorenzo L.É., Islam B., Brindle G. Exome sequencing implicates impaired GABA signaling and neuronal ion transport in trigeminal neuralgia. IScience. 2020;23:101552. doi: 10.1016/j.isci.2020.101552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homsy J., Zaidi S., Shen Y., Ware J.S., Samocha K.E., Karczewski K.J., DePalma S.R., McKean D., Wakimoto H., Gorham J. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–1266. doi: 10.1126/science.aac9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.C., Homsy J., Zaidi S., Lu Q., Morton S., DePalma S.R., Zeng X., Qi H., Chang W., Sierant M.C. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 2017;49:1593–1601. doi: 10.1038/ng.3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M., Witten D.M., Jain P., O'Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O'DonnellLuria A.H., Ware J.S., Hill A.J., Cummings B.B. Analysis of proteincoding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Program T.N.T. BRAVO variant browser: University of Michigan and NHLBI. 2018. https://bravosphumichedu/freeze5/hg38/
- Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha K.E., Robinson E.B., Sanders S.J., Stevens C., Sabo A., McGrath L.M., Kosmicki J.A., Rehnstrom K., Mallick S., Kirby A. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014;46:944–950. doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 genomes project consortium A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware J.S., Samocha K.E., Homsy J., Daly M.J. Interpreting de novo variation in human disease using denovolyzeR. Curr. Protoc. Hum. Genet. 2015;87:7.25.1–7.25.15. doi: 10.1002/0471142905.hg0725s87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q., Zhan X., Zhong X., Liu Y., Han Y., Chen W., Li B. A Bayesian framework for de novo mutation calling in parents-offspring trios. Bioinformatics. 2014;31:1375–1381. doi: 10.1093/bioinformatics/btu839. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The file names within the zip correspond to the labeled individual-IDs from the figure.
Supp-File1_Sample-124-1 corresponds to the plot for individual TGRN124-1 in Figure 1A.
Supp-File1_Sample-124-3 corresponds to the plot for individual TGRN124-3 in Figure 1A.
Supp-File1_Sample-124-4 corresponds to the plot for individual TGRN124-4 in Figure 1A.
Supp-File1_Sample-177-1 corresponds to the plot for individual TGRN177-1 in Figure 1B.
Supp-File1_Sample-177-2 corresponds to the plot for individual TGRN177-2 in Figure 1B.
Supp-File1_Sample-177-3 corresponds to the plot for individual TGRN177-3 in Figure 1B.
Data Availability Statement
The WES data analyzed with this protocol and generated by the companion paper Dong et al. are available at dbGap with accession number phs000744. We have provided our Python and R scripts for the de novo variant analysis demonstrated in Dong et al. and this protocol. These can be found at the github repository for this protocol (https://github.com/jinlab-washu/de-novo-wes-star-protocol).