

Abstract
Single-cell RNA sequencing (scRNA-seq) has emerged as a powerful tool for investigating cell states and functions at the single-cell level. It has greatly revolutionized transcriptomic studies in many life science research fields, such as neurobiology, immunology, and developmental biology. With the fast development of both experimental platforms and bioinformatics approaches over the past decade, scRNA-seq is becoming economically feasible and experimentally practical for many biomedical laboratories. Drosophila has served as an excellent model organism for dissecting cellular and molecular mechanisms that underlie tissue development, adult cell function, disease, and aging. The recent application of scRNA-seq methods to Drosophila tissues has led to a number of exciting discoveries. In this review, I will provide a summary of recent scRNA-seq studies in Drosophila, focusing on technical approaches and biological applications. I will also discuss current challenges and future opportunities of making new discoveries using scRNA-seq in Drosophila.
This article is categorized under:
Technologies > Analysis of the Transcriptome
Keywords: drosophila, single-cell RNA sequencing
1 |. INTRODUCTION
Transcriptomic analysis has been widely used to study gene expression patterns for understanding the functions of a tissue or a population of cells. The advent of next-generation sequencing has transformed transcriptomic studies (Schuster, 2008; Soon, Hariharan, & Snyder, 2013). Most of what we have learned from transcriptomic research about fundamental principles underlying biological processes comes from bulk RNA-seq, which provides averaged gene expression from a whole tissue or from a large number of individual cells at scale of the entire transcriptome. However, the bulk approach can mask meaningful differences between molecularly similar cell types within a tissue. The analysis of single cells has the potential to overcome these limitations and can provide unprecedent molecular resolution for understanding cell functions at the single-cell level. Meanwhile, the existing high throughput datasets from bulk RNA-seq, such as modENCODE (Consortium, 2010), provide good resources for validating the sequencing data of scRNA-seq studies.
The past decade has witnessed the emergence and rapid development of a host of single-cell sequencing technologies (Gawad, Koh, & Quake, 2016; Labib & Kelley, 2020; Schwartzman & Tanay, 2015; Shapiro, Biezuner, & Linnarsson, 2013; Stuart & Satija, 2019; Tanay & Regev, 2017), including single-cell RNA sequencing (scRNA-seq), which allows the survey of transcriptomes of individual cells. scRNA-seq offers unique opportunities for both basic and clinical research, such as identifying new cell types, exploring cell heterogeneity, revealing developmental trajectories, studying drug resistance, and investigating cancer relapse (Haque, Engel, Teichmann, & Lönnberg, 2017; Hwang, Lee, & Bang, 2018; Liu & Trapnell, 2016; Potter, 2018). scRNA-seq has already significantly impacted our conceptual understanding of diverse biological processes.
The fruit fly, Drosophila melanogaster, is a premier model organism to study fundamental and evolutionarily conserved biological mechanisms ranging from development to aging, largely owing to the availability of sophisticated genetic tools. Combining scRNA-seq with powerful genetic tools holds a great potential for making new discoveries. Indeed, recent scRNA-seq studies in Drosophila have revealed novel biological findings, such as characterizing new cell types in different tissues including the whole embryo, whole brain, ventral nerve cord, gut, blood, abdominal cuticle, testis, and ovary (Allen et al., 2020; Brunet Avalos, Maier, Bruggmann, & Sprecher, 2019; Cattenoz et al., 2020; Cho et al., 2020; Croset, Treiber, & Waddell, 2018; Davie et al., 2018; Fu, Huang, Zhang, Leemput, & Han, 2020; Ghosh et al., 2019; Guo et al., 2019; Hung et al., 2020; Jevitt et al., 2020; Karaiskos et al., 2017; Rust et al., 2019; Shin, Jones, Petkau, Panteluk, & Foley, 2019; Slaidina, Banisch, Gupta, & Lehmann, 2020; Tattikota et al., 2020; Witt, Benjamin, Svetec, & Zhao, 2019), revealing unrealized mechanisms underlying neural development and brain aging (Davie et al., 2018; Konstantinides et al., 2018; Kurmangaliyev, Yoo, LoCascio, & Zipursky, 2019; Li et al., 2017, 2020), and uncovering transcriptional regulation or signaling pathways controlling development and tumorigenesis (Ariss, Islam, Critcher, Zappia, & Frolov, 2018; Deng et al., 2019; Genovese et al., 2019; Ji et al., 2019). So far, scRNA-seq profiling has been performed in various Drosophila tissues from multiple stages (Figure 1 and Table 1), providing valuable resources for future studies of those individual tissues or tissue-tissue interactions.
FIGURE 1.
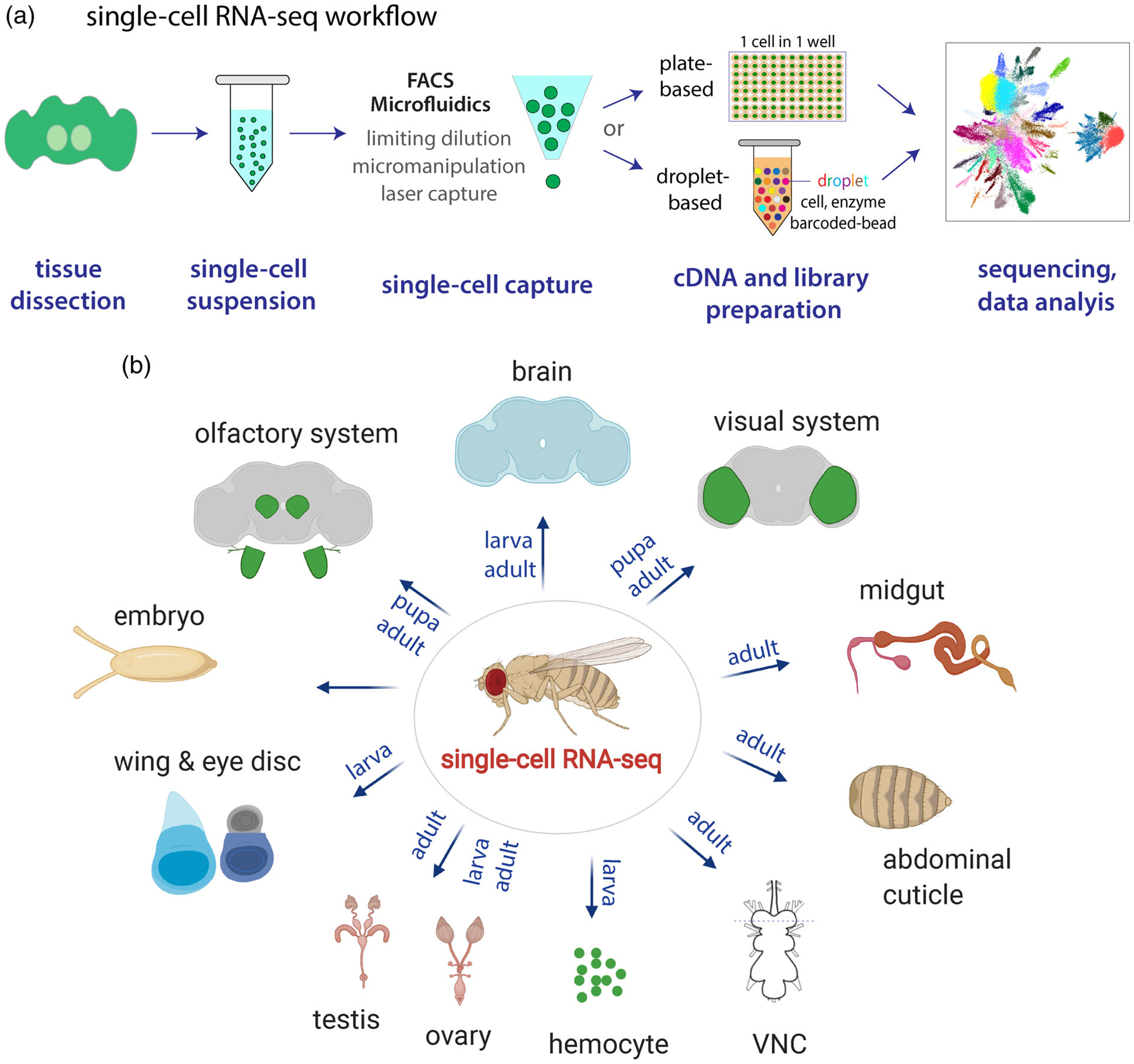
(a) Single-cell RNA-seq workflow. It contains five major steps: tissue dissection and dissociation, single-cell capture, cDNA and library preparation, sequencing, and data analysis. FACS- and microfluidics-based methods are two most commonly used methods for single-cell capture. In plate-based methods, each individual cell is captured in one well. In droplet-based methods, cells are captured in droplets with enzymes and barcoded-beads. (b) Summary of scRNA-seq studies in Drosophila (see Table 1 for details). Tissue stages are indicated. The abdominal cuticle is profiled through single-nucleus RNA-seq, and all other tissues are sequenced by single-cell RNA-seq. VNC, ventral nerve cord
TABLE 1.
Summary of scRNA-seq studies in Drosophila
| Tissue | Stage | Dissociation | Technology | Reference |
|---|---|---|---|---|
| Olfactory projection neuron | Pupa | Papain, liberase | Smart-seq2 | Li et al. (2017) |
| Olfactory receptor neuron | Pupa | Papain, liberase | Smart-seq2 | Li, Li, et al. (2020) |
| Embryo | Embryo | Dounce homogenizer | Drop-seq | Karaiskos et al. (2017) |
| Whole brain | Adult & aging | Dispase, collagenase | 10× | Davie et al. (2018) |
| Midbrain | Adult | Papain, collagenase | Drop-Seq | Croset et al. (2018) |
| Whole brain | Larva | Collagenase | 10× | Brunet Avalos et al. (2019) |
| Optical lobe | Adult | Dispase, collagenase | Drop-Seq | Konstantinides et al. (2018) |
| Optical lobe (T4/T5) | Pupa | Papain, liberase | 10× | Kurmangaliyev et al. (2019) |
| Abdominal cuticle | Adult | Dounce homogenizer | 10× | Ghosh et al. (2019) |
| Blood | Larva | NA | inDrop; 10× | Tattikota et al. (2020) |
| Blood | Larva | NA | 10× | Fu et al. (2020) |
| Blood | Larva | NA | 10× | Cattenoz et al. (2020) |
| Lymph gland | Larva | Papain, liberase | Drop-seq | Cho et al. (2020) |
| Eye disc | Larva | Typsin, collagenase | Drop-seq | Ariss et al. (2018) |
| Wing disc | Larva | TrypLE | Drop-seq | Bageritz et al. (2019) |
| Wing disc | Larva | Typsin | 10× | Ji et al. (2019) |
| Gut (EEs) | Adult | Elastase | 10× | Guo et al. (2019) |
| Gut | Adult | — | — | Shin et al. (2019) |
| Gut | Adult | Elastase | inDrop | Hung et al. (2020) |
| Ovary | Adult | Elastase, collagenase | 10× | Rust et al. (2019) |
| Ovary | Adult | Papain | 10× | Jevitt et al. (2020) |
| Ovary | Larva | Trypsin, collagenase | 10× | Slaidina et al. (2020) |
| Testis | Adult | Typsin, collagenase | 10× | Witt et al. (2019) |
| VNC (tumor model) | Adult | Papain, collagenase | 10× | Genovese et al. (2019) |
| VNC | Adult | Papain collagenase | 10× | Allen et al. (2020) |
In this review, I will summarize scRNA-seq technologies in general with an emphasis on those that have been used in Drosophila and discuss how scRNA-seq is employed as a tool for exciting biological discoveries in this model. For a broader view of single-cell analysis beyond scRNA-seq in Drosophila research, see (Gawad et al., 2016; Labib & Kelley, 2020; Packer & Trapnell, 2018; Schwartzman & Tanay, 2015; Tanay & Regev, 2017).
2 |. SINGLE-CELL RNA-SEQ IN DROSOPHILA: TECHNOLOGIES
The first scRNA-seq method was reported in 2009 (Tang et al., 2009). Since then many different scRNA-seq platforms have been developed (Chen, Ning, & Shi, 2019). scRNA-seq faces a number of challenges. The two primary challenges are the low cell capture efficiency and the low amount of input RNA material from individual cells. Drosophila cells are much smaller than mammalian cells with fewer RNA transcripts per cell, further magnifying the second challenge.
Depending on biological systems or study purposes, different scRNA-seq protocols have been used. Almost all of these protocols can be divided into five major steps (Figure 1a): (a) preparing single-cell suspension, (b) capturing individual cells, (c) making cDNA and barcoded libraries, (d) sequencing, and (e) analyzing data. With the maturation of scRNA-seq technologies and bioinformatics, commercially available kits and standardized data analysis platforms are rapidly increasing. Here, I will focus on the first three steps of these protocols and will not discuss sequencing, as it is relatively standardized. Data analysis methods will be discussed in the following section.
2.1 |. Tissue dissociation and single-cell suspension
For all large-scale scRNA-seq experiments, preparing the single-cell suspension is the first step. If starting materials are cultured cell lines or circulating blood cells (called hemocytes in Drosophila), making single-cell suspension is relatively easy (Cattenoz et al., 2020; Fu et al., 2020; Tattikota et al., 2020). In most other cases, dissected tissues need to be dissociated using either specific set of enzymes or mechanical force or both. Widely used dissociating enzymes in Drosophila include trypsin, collagenase, papain, liberase, and elastase, and in most cases these enzymes are used in combination to improve the dissociation efficiency (Table 1). When choosing a dissociation method, it is best to test multiple methods as their efficiency can vary significantly, depending on the cell type, tissue type, and developmental stage. Another important factor that needs to be considered is the cell viability. Since tissue dissociation is a harsh process, it can cause cellular stress and transcriptional changes. Thus, if two methods can both adequately dissociate the desired tissue, the less damaging one with higher cell viability should be used.
As stated above, cell dissociation can lead to endogenous transcriptional alterations, and minimizing these changes is important for downstream analyses. Tissue fixation can preserve transcriptome integrity, and recent studies have begun to explore the feasibility of adding a fixation step for scRNA-seq. For example, paraformaldehyde (PFA) was used to fix human radial glial cells for single-cell transcriptomic analysis (Thomsen et al., 2016). It is worth mentioning that PFA fixation-induced cross-linking prevents primer annealing in the reverse transcription step and reversal of cross-linking may cause RNA degradation. Alles et al. (2017) showed that methanol fixation could preserve dissociated cells for several weeks without compromising scRNA-seq data quality, and this method was validated in both mouse brain cells and Drosophila embryos. Attar et al. (2018) reported that the reversible cross-linker, dithiobis(succinimidyl propionate) (DSP), could be used to preserve cells for subsequent single-cell suspension and that the scRNA-seq data quality from these fixed cells is similar to fresh cells. Additionally, fixation steps increase the flexibility of sample handling, especially for samples that cannot be processed immediately. In addition to tissue fixation, another promising method is to add transcription inhibitor. For instance, the general transcription inhibitor actinomycin D (ActD) was recently used for mouse brain tissues to reduce transcriptional alterations resulting from dissociation (Hrvatin et al., 2018; Wu, Pan, Zuo, Li, & Hong, 2017). This method has not been applied to Drosophila but is an important consideration in future studies. In general, the cell dissociation should be done as quickly as possible to minimize the transcriptional changes introduced in this step. In addition, single-nucleus RNA-seq allows researchers to start with frozen tissues so that the in vivo transcriptional profiles are better preserved. This will be discussed in the later section.
2.2 |. Single cell capture
Next step is to capture individual cells. Currently, there are several approaches available for single cell capture: limiting dilution, micromanipulation or micropipetting, laser capture microdissection, fluorescence-activated cell sorting (FACS), and microfluidics. Limiting dilution, a traditional method that is commonly used for isolating monoclonal cell lines, employs statistical strategy to isolate single cells by diluting cells into a concentration of less than one cell per aliquot (Fuller, Takahashi, & Hurrell, 2001). Micromanipulation allows manual cell picking with micropipettes via microscope observation followed by transfer of cells to lysis buffer to preserve RNA molecules, which has been used for early embryos and cardiomyocytes in mice and Drosophila mushroom body neurons (Crocker, Guan, Murphy, & Murthy, 2016; Flynn, Santana, & Melov, 2011; Guo et al., 2019). Laser capture microdissection method combines a laser system with a computer system to isolate single cells from solid tissues (Nichterwitz et al., 2016). These three methods, although very useful in certain applications, are time-consuming or low-throughput and thus are not widely utilized in scRNA-seq studies.
FACS is a powerful tool for purifying or enriching specific cells if they can be labeled by fluorescent markers. Two of the most frequently used labeling strategies are genetic labeling (e.g., Cre-loxP system in mice and GAL4-UAS system in Drosophila) (Brand & Perrimon, 1993; Schwenk, Baron, & Rajewsky, 1995) and antibody staining (e.g., CD cell-surface proteins for immune cells). In addition, multi-color and negative selection is also possible for desired cells. FACS-based method possesses several unique features for scRNA-seq studies. First, dead cells can be removed. Combining live/dead florescent dye staining with cell-type specific marker is sufficient to reduce cell damage effects that are introduced by the tissue dissociation procedure. This has been proven to be useful for isolating Drosophila neurons (Li et al., 2017). Second, single cells can be isolated from doublets or cell aggregates according to the cells size and fluorescence intensity, which can be visualized during cell sorting. Third, cell debris, usually generated in the single-cell suspension step and consisting of small pieces of membranes or other parts of a cell, will decrease the capture efficiency if collected as cells. They can be removed through FACS because they lack fluorescence labeling and show different forward scatter (FSC) and side scatter (SSC) signals from intact cells. The advantage of this feature is more profound for Drosophila, because most Drosophila cells are very small and cannot be easily distinguished from debris by size. Through FACS, individual cells can be either collected into single wells of 96- or 384-well plates for plate-based scRNA-seq or collected as a whole in one tube for droplet-based scRNA-seq (see below). The disadvantages of the FACS method include longer waiting time and additional stress introduced by the FACS procedure.
Microfluidics-based methods for capturing single cells are commonly used. Microfluidic technology was initially used in biochemical assays for quantifying DNA and protein molecules, and later was adopted for long-term monitoring of single bacteria and for gene expression profiling of single cells (Balagaddé, You, Hansen, Arnold, & Quake, 2005; Marcus, Anderson, & Quake, 2006). The first widely used commercial microfluidic system is Fluidigm C1, which provides automated single-cell capture, lysis, and reverse transcription and cDNA amplification (Pollen et al., 2014). For each run, Fuidigm C1 can capture and process 96 individual cells and the later upgraded version (high-throughput integrated fluidics circuits, HT IFCs) can process up to 800 cells. However, the average cost per cell is high and a large number of cells is required as input, limiting the adoption of Fluidigm C1. In 2015, two high-throughput droplet-based microfluidics methods were developed, called inDrop and Drop-seq (Klein et al., 2015; Macosko et al., 2015). These two methods, together with the commercialized 10× Genomics Chromium system (Zheng et al., 2017), have tremendously boosted the scRNA-seq field in recent years, owing to their high-throughput and low-cost features. inDrop, Drop-seq, and 10x Genomics share a similar workflow: individual cells are captured with uniquely barcoded beads in water-in-oil droplets; cells are lysed and cDNAs are generated; cDNAs are amplified and gene expression libraries are constructed. The pros and cons of plate-based and droplet-based scRNA-seq methods will be further discussed below.
How do we make sure captured cells are single cells, but not doublets or multiplets? This is a frequently asked question in the scRNA-seq field. There is no perfect method to completely solve this issue. However, here are several steps to help minimize the issue. First, after tissue dissociation, the single-cell suspension can be validated under microscope using cell counting slides. We find this step is very useful to determine if a tissue dissociation protocol should be further optimized. Next, if FACS is used, doublets can be distinguished by cell size and fluorescent intensity. However, it is worth mentioning that if cell sizes are largely varied in the single cell suspension, this can be challenging because large cells may show similar sizes or florescent intensity to doublets of small cells. Fuidigm C1 system has an imaging step to check if captured cells are single cells. Finally, multiple bioinformatic methods have been developed to detect and remove doublets, for example, DoubletFinder (McGinnis, Murrow, & Gartner, 2019), Scrublet (Wolock, Lopez, & Klein, 2019), and DoubletDecon (DePasquale et al., 2019), Solo (Bernstein et al., 2020).
2.3 |. scRNA-seq platforms: Smart-seq2 and 10× genomics chromium system
For most established scRNA-seq platforms, the cell capture process is integrated with downstream steps: reverse transcription, cDNA amplification, and sequencing library preparation. So far, numerous scRNA-seq platforms have been developed, such as CEL-seq (Hashimshony, Wagner, Sher, & Yanai, 2012), Smart-seq2 (Picelli et al., 2013), MARS-seq (Jaitin et al., 2014), inDrop (Klein et al., 2015), Drop-seq (Macosko et al., 2015), 10× Genomics (Zheng et al., 2017), Sci-RNA-seq (Cao et al., 2017), MATQ-seq (Sheng, Cao, Niu, Deng, & Zong, 2017), SPLIT-seq (Rosenberg et al., 2018), and SEQ-well (Aicher et al., 2019). Key differences between these approaches include cDNA coverage (full-length or 5′/3′ counting), the use of unique molecular identifier (UMI), handling platforms (plate- or droplet-based), targeted read depth, throughput, and cost. Detailed comparison has been discussed by other reviews (Haque et al., 2017; See, Lum, Chen, & Ginhoux, 2018).
Among those platforms, four have been used in Drosophila scRNA-seq studies: plate-based Smart-seq2 and droplet-based inDrop, Drop-seq, and 10× Genomics. The three droplet-based methods share many key features (Table 2), and 10× Genomics is becoming popular (15 out of 23 published Drosophila studies used this method) (Table 1), owing to its high accessibility. Here I will focus on Smart-seq2 and 10× Genomics system.
TABLE 2.
Comparison between plate-based Smart-seq2 and droplet-based scRNA-seq platforms
| Smart-seq2 | inDrop | Drop-seq | 10× genomics | |
|---|---|---|---|---|
| cDNA coverage | Full length | 3′ end | 3′ end | 3′ or 5′ end |
| Plate or droplet | 96- or 384-well plate | Droplet | Droplet | Droplet |
| UMI | None | Yes | Yes | Yes |
| Throughput (number of cells) | 96 or 384 | 1k–10k | 1k–10k | 1k–10k |
| Sequencing depth (read per cell) | 106 | 104–105 | 104–105 | 104–105 |
| Feature | FACS sorting, isoform analysis | Emulsion, low cost | Emulsion, low cost | Emulsion, low cost |
| Long-term storage | Yes, cells sorted into lysis buffer | No, must process immediately | No, must process immediately | No, must process immediately |
Smart-seq was developed to increase read coverage across transcripts for scRNA-seq studies (Ramsköld et al., 2012). Smart-seq2 is an improved version of Smart-seq, featuring the generation of full-length cDNAs using template switching in the reverse transcription step (Picelli et al., 2014). A Smart-seq2 based scRNA-seq protocol follows following steps: (a) After tissue dissociation, individual cells are FAC-sorted into single wells of 96- or 384-well plates, with lysis buffer preloaded. Sorted plates can be either stored in −80°C for long-term storage or processed immediately. (b) The first strand full-length cDNA is synthesized with the customized oligo-dT primer and the template switching oligo. (c) Full-length cDNAs are PCR-amplified for 18–25 cycles depending the amount of starting RNA materials. We have used 25 cycles for Drosophila neurons (Li et al., 2017; Li, Li, et al., 2020). (d) Sequencing libraries are made by Tn5 tagmentation according to standard procedure (Adey et al., 2010). (e) Libraries are pooled and sequenced.
The 10× Genomics Chromium system takes advantage of rapid droplet-based encapsulation of single cells with a gel bead in emulsion (Zheng et al., 2017). Each gel bead is tagged with millions of oligonucleotides containing a bead-specific barcode, different unique molecular identifiers (UMIs), and the oligo-dT with sequencing primer. The bead-specific barcode is used to index individual cells; UMIs can be used to index individual mRNA molecules, allowing transcripts to be directly counted to reduce PCR-introduced amplification bias; oligo-dT primers allow cDNA generation from poly(A) mRNAs. 10× Genomics provides different kits that allow to profile either 3′ or 5′ end of mRNAs. Up to eight different samples can be processed simultaneously, and about 10k cells can captured from each sample. The downstream processing for reverse transcription and library preparation is very simple because all cells from one sample are processed together in one tube.
As described above, both Smart-seq2 and 10× Genomics have advantages and disadvantages (Table 2). Compared to 10x Genomics, Smart-seq2 method allows full-length cDNA coverage thus enabling isoform analysis, provides higher sequencing depth allowing better detection of lowly-expressed transcripts, and offers a stable, long-term storage option after cell capture which increases experimental flexibility. Another advantage of Smart-seq2 is the capability to capture large polyploid cells, which is a challenge for 10× Genomics. This is a significant factor for Drosophila research because polyploid cells are very common for many Drosophila tissues. On the other hand, 10× Genomics provides much higher throughput, does not require cell sorting by FACS, and utilizes UMIs to remove PCR-amplification bias. Meanwhile, the cost per cell for 10× Genomics is much lower than that of Smart-seq2. As a result, all these parameters should be taken into account when choosing a scRNA-seq platform.
3 |. SINGLE-CELL RNA-SEQ IN DROSOPHILA: APPLICATIONS
scRNA-seq enables direct comparison of transcriptomes among individual cells. Therefore, an immediate application of scRNA-seq is to characterize the cellular heterogeneity within a complex tissue, for example the fast-developing embryo or complex brain regions (Guo et al., 2017; Karaiskos et al., 2017; Tasic et al., 2016; Zeisel et al., 2018). By cluster analysis, single cells can be classified into different groups according to transcriptomic similarity. This allows the identification of rare cell populations, and also permits comparison of cell states in a number of biological contexts, such as development, aging, stem cell differentiation, and disease. In addition to revealing transcriptomic differences of individual cells, scRNA-seq can also provide critical information about fundamental features of gene regulation. For example, characterizing the gene co-expression patterns in single cells allows the identification co-regulated gene modules and gene-regulatory networks that may underline cellular heterogeneity (Wagner, Regev, & Yosef, 2016). Next, I will discuss those applications in general and then in Drosophila scRNA-seq studies (Figure 2).
FIGURE 2.

Summary of current applications of single-cell RNA-seq in Drosophila, including classifying cell types and identifying rare cells, constructing cellular developmental trajectories, deciphering gene regulatory networks, and discovering mechanisms that control development and aging and that contribute to diseases
3.1 |. Classifying cell types
Before the molecular biology era, cell types were defined and classified typically by their morphology, and later by their function and physiology (Fischbach & Dittrich, 1989; Kepecs & Fishell, 2014; Waddintong, 1957). These characteristics are largely determined by a cell’s molecular signature or gene expression pattern. Thus, the advent of scRNA-seq has pushed the cell type clarification to new heights in recent years (Brbić et al., 2020; Darmanis et al., 2015; Hung et al., 2020; Jaitin et al., 2014; Li et al., 2017; Zeisel et al., 2015).
Most scRNA-seq studies in Drosophila, as well as in other model organisms, start with classifying cells according to transcriptomic similarity. Numerous computational platforms have been developed for cluster analysis, for example Monocle (Trapnell et al., 2014), ASAP (Gardeux, David, Shajkofci, Schwalie, & Deplancke, 2017), Seurat (Butler, Hoffman, Smibert, Papalexi, & Satija, 2018), Scope (Davie et al., 2018), SCANPY (Wolf, Angerer, & Theis, 2018), MARS (Brbić et al., 2020), CellFindR (Yu et al., 2019), and many others. Cluster analysis is a common and efficient strategy for investigating complex tissues, for example the brain. Davie et al. (2018) profiled the entire adult Drosophila brain and revealed 87 primary cell clusters, many of which can be further divided when the clustering resolution is enhanced. Specific parts of the Drosophila nervous system from different stages have also been profiled, including pupal olfactory projection neurons and olfactory receptor neurons (Li et al., 2017; Li, Li, et al., 2020), pupal and adult optic lobes (Konstantinides et al., 2018; Kurmangaliyev et al., 2019), adult central brain (Croset et al., 2018), larval brain (Brunet Avalos et al., 2019), and adult ventral verve cord (Allen et al., 2020).
The Drosophila midgut is a great model system for studying adult stem cell biology and aging mechanisms (Jasper, 2020; Micchelli & Perrimon, 2006; Ohlstein & Spradling, 2006). Four major epithelial cell types in the midgut have been assumed from a traditional view, intestinal stem cells, enteroblasts, enterocytes, and enteroendocrine cells (Li & Jasper, 2016). Recently, Hung et al. (2020) provided the first cell atlas of the adult Drosophila midgut and revealed 22 clusters representing these four cell types, suggesting a high heterogeneity of these cells. Guo et al. (2019) performed scRNA-seq analysis of FAC-sorted enteroendocrine cells which formed 10 clusters, confirming the enteroendocrine cell heterogeneity. From the intestinal immunity view, scRNA-seq was performed in wild-type midgut and in midguts where IMD innate immune signaling was inactivated, and it was found that IMD inactivation resulted in appearance of a new enterocyte population and absence of one enteroendocrine cell population (Shin et al., 2019). These studies provide useful resources for understanding intestinal stem cell function and gut physiology.
How is a cell type’s identity encoded in the transcriptome? There are two possibilities: each cell type is defined by a unique marker, or each cell type is specified by a combinatorial code. It is clear that most major cell types from different tissues, such as neurons, glia, muscles, hemocytes, or distinct cell types from the same tissue, such as the four epithelial cells in the fly midgut discussed above, can be distinguished from one another using unique markers. However, often these cell types can be further divided into functionally distinct subtypes, which typically requires the use of multiple markers rather than a single unique marker. For example, the identity of fly olfactory projection neurons can be easily distinguished from astrocytes using one neuronal marker but differentiation of each of the 50 projection neuron subtypes requires the use of a combinatorial code (Li et al., 2017), and in the midgut each enteroendocrine cell is specified by 2–5 different classes of hormone peptides (Guo et al., 2019).
3.2 |. Characterizing rare cells
Following cluster analysis, one immediate question is whether these transcriptomic clusters represent meaningful cell types (or subtypes). There are three commonly used strategies to address this question: (a) using previously characterized marker genes, (b) sequencing specific subtypes of cells and re-clustering them with the large population to see if they form expected clusters, and (c) identifying novel markers for specific clusters and validating their expression pattern in vivo. These can be achieved in Drosophila without too much difficulty, because for most genes there are available GAL4 lines for direct validation and generating a new transgenic fly line is relatively simple, especially with combination of CRISPR technology and fly genetics (Diao et al., 2015; Jenett et al., 2012; Kanca et al., 2019; Lee et al., 2018).
During the validation, many clusters can be assigned to different known cell types and some uncharacterized cell clusters may reflect rare cell types. For example, in the adult Drosophila brain, analysis of dopaminergic neurons revealed a Fer2+ cluster of protocerebral anterior-medial dopaminergic cells, and analysis of peptidergic neurons, another rare cell type, revealed multiple specific subtypes of petidergic neurons (Davie et al., 2018). scRNA-seq of adult Drosophila testis and ovary have revealed transcriptomes of rare germline stem cells, allowing detailed characterization of spermatogenesis and oogenesis at the single cell level (Jevitt et al., 2020; Rust et al., 2019; Witt et al., 2019). scRNA-seq of the developing larval ovary enabled the identification of a new cell type corresponding to the elusive follicle stem cell precursors (Slaidina et al., 2020). Drosophila blood cells (hemocytes) from the larval stage have recently been profiled from four independent studies and they all reported the high heterogeneity of plasmatocytes, the major cell type of Drosophila hemocytes (Cattenoz et al., 2020; Cho et al., 2020; Fu et al., 2020; Tattikota et al., 2020). Interestingly, Fu et al. (2020) revealed two new blood cell types, which were named as thanacytes and primocytes. Tattikota et al. (2020) discovered rare subsets within crystal cells and lamellocytes, two other less frequent hemocytes. These studies provide a rich resource for understanding Drosophila blood cell types and physiologies.
3.3 |. Developmental trajectory
Another important application for scRNA-seq is to construct the cellular trajectory from one state to another (Packer & Trapnell, 2018). In tissues where cell fates are not fully terminated, individual cells undergo dynamic processes, such as stem cell proliferation and differentiation, in response to internal developmental clock or external environmental stimuli. This dynamic process is partially encoded in a cell’s transcriptome. Thus, scRNA-seq data can be utilized to map cell developmental trajectory from early states to terminal states along a pseudotime axis. Multiple methods have been invented to visualize the developmental trajectory, such as Monocle (Trapnell et al., 2014), SCUBA (Marco et al., 2014), Wanderlust (Bendall et al., 2014), Waterfall (Shin et al., 2015), and Wishbone (Setty et al., 2016). Detailed comparison of these methods has been discussed (Hwang et al., 2018). RNA velocity, which can be calculated by comparing unspliced and spliced mRNA ratios from scRNA-seq data, is a powerful indicator of future state of individual cells (La Manno et al., 2018). It is conceivable that combination of RNA velocity with single-cell trajectory analysis will provide complementary insights into cellular dynamics during tissue development and regeneration. It is worth mentioning that current RNA velocity analysis tools do not perform very well for 10x data from Drosophila cells (personal communication with the Reviewer).
In Drosophila, scRNA-seq analysis of the adult ovary and testis allowed the reconstruction of developmental trajectories of germ cells during oogenesis and spermatogenesis, respectively (Jevitt et al., 2020; Rust et al., 2019; Witt et al., 2019). In the ovary, pseudotime analysis revealed germ cell trajectory with three branches, one representing early stages of differentiation and other two representing the paths to oocytes and nurse cells (Jevitt et al., 2020; Rust et al., 2019). In the testis, such analysis revealed de novo gene expression bias during spermatogenesis (de novo genes represent new genes that evolve from DNA sequences that were ancestrally nongenic). For example, the top five most differentially expressed de novo genes tend to be biased toward early and middle pseudotime (Witt et al., 2019). Developmental trajectory analysis of neuroblast tumors in Drosophila larvae identified a subset of genes, responsible for temporal patterning of normal neuroblasts, that are redeployed in tumors to generate a differentiation trajectory leading to tumor cell heterogeneity (Genovese et al., 2019). scRNA-seq in adult Drosophila midgut allowed the lineage analysis to reveal the differentiation trajectory of intestinal stem cells, allowing future characterization of stem cell functions in homeostasis and in response to injuries (Hung et al., 2020).
Another interesting question is how transcriptomic differences of two cell types evolve during development. We (Li et al., 2017) performed scRNA-seq analysis of two types of Drosophila olfactory projection neurons from five stages covering middle pupa to adulthood and found that these two transcriptomes are quite distinct in developmental stages, but become indistinguishable in adulthood. Similar findings of transcriptomic convergence from development to adulthood were reported in the mouse lateral geniculate nucleus (Kalish et al., 2018), as well as in mouse retinal ganglion cells (Tran et al., 2019). These findings suggest that using scRNA-seq to characterize cell types from just one stage, such as adulthood, may be not sufficient to reveal the differences between two functionally distinct cell types. Although this principle may not be applicable to other cell types, it is advised to take it into account when performing scRNA-seq studies for classifying cells and characterizing cell types.
3.4 |. Mechanisms of development, aging, and disease
Besides trajectory analysis-related applications, scRNA-seq can also provide valuable insights into additional mechanisms controlling development. For example, single-cell transcriptomic profiling of the Drosophila embryo, consisting of about 6,000 cells, revealed a new mechanism underlying the embryo pattern formation (Karaiskos et al., 2017). The Drosophila embryo is an excellent model for studying pattering principles that specify cellular identities. By combining scRNA-seq and a computational mapping strategy to predict spatial gene expression, Karaiskos et al. (2017) obtained a 3D virtual in situ hybridization map of the embryo. This 3D in situ map enabled the researchers to identify the expression of multiple Hippo pathway components in an anterior region of the embryo and to reveal a new role of Hippo signaling in embryo patterning.
How does aging affect cell-identity at the transcriptomic level? By comparing scRNA-seq data from young and old Drosophila brains, Davie et al (Davie et al., 2018) obtained several interesting observations. First, it was found that mRNA abundance of almost all brain cell types, including neurons and glial cells, displayed a decline with age. Second, the decline of mRNA abundance did not affect cell identity, because most cell type clusters that were characterized in young brains remained in old flies. Third, genes involved in oxidative phosphorylation and mitochondrial turnover showed most significant decline. These characterizations provide a valuable resource to study brain aging. It will be of interest for future studies to characterize how aging impact other cell types at the single-cell level beyond the brain.
scRNA-seq has also greatly contributed to our understanding of numerous disease processes, such as tumorigenesis (Potter, 2018). Tumors usually consist of a heterogenous mix of multiple cell types, including cancer, vascular, immune, and fibroblast cells, each of which can be further divided into subtypes. Thus, scRNA-seq can be used to dissect tumor heterogeneity to understand tumor development and to devise treatment approaches. Drosophila wing disc has served as a model system for studying conserved mechanisms underlying tumorigenesis (Morimoto & Tamori, 2017). Ji et al. (2019) performed scRNA-seq on scrib mutant-induced wing disc tumors and found that dynamic MAPK signaling activity control the transition from growth arrest to cell proliferation during tumorigenesis.
3.5 |. Gene regulatory network
Deciphering the gene regulatory network is another nice feature of scRNA-seq (Shalek et al., 2013; Xue et al., 2013). At the transcriptomic level, two cell types may carry expression differences of tens or hundreds of genes. However, at the transcriptional regulation level, these differences can be attributed to a limited number of transcription factors (TFs) or cofactors. In other words, genes can be grouped into co-regulated modules based on their shared upstream regulators. Inferring gene regulatory network from scRNA-seq data is not only a strategy to refine cluster analysis, but also a powerful tool to discover regulatory mechanisms driving cellular heterogeneity. Recently, multiple methods have been developed to reconstruct the gene expression network from scRNA-seq data (Aibar et al., 2017; Chan, Stumpf, & Babtie, 2019; Matsumoto et al., 2017). For example, Aibar et al. (2017) developed SCENIC, which utilizes a computational strategy combining gene co-expression and cis-regulatory motif information to infer gene regulatory network, and they showed that SCENIC can accurately predict the interactions between TFs and their targets.
In the Drosophila nervous system, gene regulatory network analysis revealed that different modular transcriptional programs regulate distinct neural wiring features during development (Kurmangaliyev et al., 2019). In this study, scRNA-seq profiling was performed on developing T4 and T5 neurons, two cell types in the Drosophila visual system involved in motion detection, and modular analysis identified eight transcriptional programs that represent eight T4/T5 subtypes defined by a combination of dendrite and axon wiring patterns. Importantly, analysis-instructed gain- and loss-of-function experiments revealed a new role of the TF, grn, in controlling T4/T5 axon targeting. In the Drosophila immune system, gene regulatory network analysis identified hemocyte cluster-specific modular signatures that are associated with either a unique TF or a combination of TFs, which could be validated in vivo (Cattenoz et al., 2020). These data provide useful resources for generating more targeted genetic tools to study immune cell functions during homeostasis and upon infection.
4 |. CHALLENGES, OPPORTUNITIES, AND PERSPECTIVES
Currently, Drosophila researchers still face a number of challenges for conducting specific scRNA-seq studies. However, challenges usually lead to new opportunities in scientific fields. Here, I will focus on three challenges and discuss potential opportunities (Figure 3).
FIGURE 3.

Challenges and future opportunities to extend the applications of scRNA-seq by combining scRNA-seq with other technologies, including single-nucleus RNA-seq, single-cell genomics, and epigenomics, nonpoly(A) RNA profiling, single-cell proteomics, and single-cell spatial transcriptomics
How to perform scRNA-seq when intact cells cannot be isolated?
Performing scRNA-seq in some adult Drosophila tissues has proven to be difficult, as many cell types are strongly associated with surrounding cuticles, for example sensory neurons in the Drosophila antenna, wing, and body. It is extremely difficult to isolate these intact cells, because mild dissociation methods cannot break the tough cuticles while harsh dissociation methods will destroy cuticle as well as attached cells. Single-nucleus RNA-seq (snRNA-seq) methods provide a great opportunity to overcome this issue. Recently, snRNA-seq has been successfully applied to profile adult mouse and human brain cells and proven to be sensitive, efficient and unbiased for classifying cells (Habib et al., 2016, 2017; Lake et al., 2016). Importantly and encouragingly, direct comparison between snRNA-seq and scRNA-seq of mouse visual cortex cells suggest although the nuclear content and proportion varies among cell types, nuclear transcripts carry adequate information to identify highly related neuronal cell types with a resolution similar to whole cells (Bakken et al., 2018). snRNA-seq has other advantages: (a) tissues can be stored, long-term, at −80°C before nucleus extraction, which is very helpful when sample collection and downstream sequencing preparation are not in the same location, such as for clinical samples; (b) snRNA-seq will reduce sampling bias introduced in the tissue dissociation step. For scRNA-seq, specific cell types may be susceptible to damage-induced cell death and will be removed before cell capture, while for snRNA-seq, all nuclei will be applied; (c) large and fragile cells, such as adipocytes, may not easily flow into microfluidics-based channels, including 10× Genomics, but their nuclei should be easily captured. Ghosh et al. (2019) has recently reported the application of snRNA-seq for profiling adult fly abdominal cuticles. We have recently developed a reliable snRNA-seq protocol in Drosophila through both Smart-seq2 and 10× Genomics for profiling adult olfactory neurons (Li, McLaughlin, Luo, unpublished) and expect more snRNA-seq to be performed in other adult Drosophila tissues.
How to profile nonpolyadenylated transcripts?
So far, almost all published scRNA-seq studies have focused on profiling poly(A) mRNAs because all current scRNA-seq protocols get first-stand cDNAs using the oligo-dT based primers. However, many regulatory noncoding RNAs, such as microRNAs, long noncoding RNAs (lncRNAs), circular RNAs, are not polyadenylated, but have essential functions in lots of biological processes, including development, aging, and disease (Batista & Chang, 2013; He & Hannon, 2004; Memczak et al., 2013; Yang, Duff, Graveley, Carmichael, & Chen, 2011). Note that in some cases nonpolyadenylated RNAs can be detected in oligo-dT primer-based profiling, and this is presumably due to internal poly(A) priming (Nam et al., 2002). Systematic investigation of those nonpolyadenylated RNAs in single cells is still greatly needed. Random hexamer priming is a potential strategy to capture all RNAs with or without poly(A) tails (Fan et al., 2015; Kang et al., 2011). Incorporating such a strategy into existing scRNA-seq technologies will allow us to explore the functions of those critical RNA species at the single-cell level. Encouragingly, the recently developed MATQ-seq method nicely demonstrates that nonpolyadenylated RNAs can be profiled by utilizing primers based on multiple annealing and looping-based amplification cycles in a small number of single cells (Sheng et al., 2017; Zong, Lu, Chapman, & Xie, 2012). It is anticipated that further development of MATQ-seq will allow high-throughput profiling of nonpolyadenylated RNAs in a large scale.
How to integrate other complementary single-cell technologies with scRNA-seq?
Although scRNA-seq is a powerful tool for studying cell states and functions, it has some limitations. For example, scRNA-seq does not carry the spatial information of profiled cells, it cannot infer epigenetic landscapes, and it does not directly reflect the protein level of genes whose post-transcriptional modifications alter their translation. Recently, several methods have been developed to detect spatial transcriptomes at single-cell resolution, including FISSEQ (Lee et al., 2014), MERFISH (Chen, Boettiger, Moffitt, Wang, & Zhuang, 2015), seqFISH (Shah, Lubeck, Zhou, & Cai, 2016), and STARmap (Wang et al., 2018). The development of single-cell ATAC-seq allows researchers to measure chromatin accessibility in the genomic level of single cells (Buenrostro et al., 2015; Cusanovich et al., 2015). Peterson et al (Peterson et al., 2017) developed REAP-seq which allows simultaneous measurement of mRNAs and certain proteins with barcoded antibodies in single cells. in vivo tissue-specific proteomic profiling methods have been recently applied to Drosophila, providing valuable information that cannot be revealed by transcriptomic analysis (Droujinine et al., 2020; Li et al., 2020). High throughput single-cell proteomic profiling is still a dream, but not far from being achieved (Aebersold & Mann, 2016; Labib & Kelley, 2020). Several integration algorithms have been developed to either integrate different scRNA-seq datasets or integrate scRNA-seq data with data from scATAC and spatial transcriptomes (Korsunsky et al., 2019; Stuart et al., 2019). Combining scRNA-seq with these complementary single-cell technologies will help us to draw a complete picture of Drosophila cells, as well as in other systems.
5 |. CONCLUSION
Biological findings are largely driven by technology development. In the past decade, scRNA-seq emerged as one of the most important techniques in biomedical fields and has profoundly changed our comprehension of many biological phenomena. Due to the smaller size of Drosophila cells, the application of scRNA-seq to Drosophila fell slightly behind compared to mammals. However, recent scRNA-seq studies in Drosophila, researchers have gained numerous insights into mechanisms underlying embryo cell patterning, neural development, germ cell development, intestinal stem cell differentiation, brain aging, tumorigenesis, immune cell specification and many others to come. Combining scRNA-seq with other single-cell technologies hold a high potential for making new exciting discoveries in the next decade.
ACKNOWLEDGMENTS
I thank my mentor Liqun Luo for his strong support and valuable comments on this manuscript and my colleagues Justus Kebschull, Jiefu Li, Tongchao Li, Zhuoran Li and Colleen McLaughlin, for their constructive comments on this manuscript. I also thank my family, my wife Yanyan Qi and my daughter Jieni Li, for their support when I prepare the manuscript during the work-from-home time caused by Covid-19. I acknowledge the support from Stanford Wu Tsai Neurosciences Institute, Stanford University Interdisciplinary Scholar Awards. This work was supported by NIH grant 1K99AG062746-01.
Funding information
National Institutes of Health, Grant/Award Number: 1K99AG062746-01
Footnotes
CONFLICT OF INTEREST
The author declares no competing interests.
RELATED WIREs ARTICLE
Single cell transcriptomics of noncoding RNAs and their cell-specificity
REFERENCES
- Adey A, Morrison HG, Asan Xun X., Kitzman JO, Turner EH, et al. (2010). Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol, 11, R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R, & Mann M (2016). Mass-spectrometric exploration of proteome structure and function. Nature, 537, 347–355. [DOI] [PubMed] [Google Scholar]
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, … Aerts S (2017). SCENIC: Single-cell regulatory network inference and clustering. Nature Methods, 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aicher TP, Carroll S, Raddi G, Gierahn T, Wadsworth MH, Hughes TK, … Shalek AK (2019). Seq-well: A sample-efficient portable picowell platform for massively parallel single-cell RNA sequencing. Methods in Molecular Biology, 1979, 111–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen AM, Neville MC, Birtles S, Croset V, Treiber CD, Waddell S, & Goodwin SF (2020). A single-cell transcriptomic atlas of the adult drosophila ventral nerve cord. eLife, 9, e54074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alles J, Karaiskos N, Praktiknjo SD, Grosswendt S, Wahle P, Ruffault P-L, … Rajewsky N (2017). Cell fixation and preservation for droplet-based single-cell transcriptomics. BMC Biology, 15, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariss MM, Islam ABMMK, Critcher M, Zappia MP, & Frolov MV (2018). Single cell RNA-sequencing identifies a metabolic aspect of apoptosis in Rbf mutant. Nature Communications, 9, 5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar M, Sharma E, Li S, Bryer C, Cubitt L, Broxholme J, … Bowden R (2018). A practical solution for preserving single cells for RNA sequencing. Scientific Reports, 8, 2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bageritz J, Willnow P, Valentini E, Leible S, Boutros M, & Teleman AA (2019). Gene expression atlas of a developing tissue by single cell expression correlation analysis. Nat. Methods 16, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken TE, Hodge RD, Miller JA, Yao Z, Nguyen TN, Aevermann B, … Tasic B (2018). Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One, 13, e0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balagaddé FK, You L, Hansen CL, Arnold FH, & Quake SR (2005). Long-term monitoring of bacteria undergoing programmed population control in a microchemostat. Science, 309, 137–140. [DOI] [PubMed] [Google Scholar]
- Batista PJ, & Chang HY (2013). Long noncoding RNAs: Cellular address codes in development and disease. Cell, 152, 1298–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir E-AD, Tadmor MD, Simonds EF, Chen TJ, … Pe’er D (2014). Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell, 157, 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein NJ, Fong NL, Lam I, Roy MA, Hendrickson DG, & Kelley DR (2020). Solo: doublet identification in single-cell RNASeq via semi-supervised deep learning. Cell Systems.11, 95–101.e5. [DOI] [PubMed] [Google Scholar]
- Brand AH, & Perrimon N (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development, 118, 401–415. [DOI] [PubMed] [Google Scholar]
- Brbi[notdef]c M, Zitnik M, Wang S, Pisco AO, Altman RB, Darmanis S, & Leskovec J (2020). Discovering novel cell types across heterogeneous single-cell experiments. BioRxiv. 10.1101/2020.02.25.960302. [DOI] [PubMed] [Google Scholar]
- Brunet Avalos C, Maier GL, Bruggmann R, & Sprecher SG (2019). Single cell transcriptome atlas of the drosophila larval brain. eLife, 8, e50354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, … Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature, 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, & Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology, 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, … Shendure J (2017). Comprehensive single-cell transcriptional profiling of a multicellular organism. Science, 357, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattenoz PB, Sakr R, Pavlidaki A, Delaporte C, Riba A, Molina N, … Giangrande A (2020). Temporal specificity and heterogeneity of drosophila immune cells. EMBO Journal, 39, e104486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TE, Stumpf MPH, & Babtie AC (2019). Gene regulatory networks from single cell data for exploring cell fate decisions. Methods in Molecular Biology, 1975, 211–238. [DOI] [PubMed] [Google Scholar]
- Chen G, Ning B, & Shi T (2019). Single-cell RNA-Seq technologies and related computational data analysis. Frontiers in Genetics, 10, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KH, Boettiger AN, Moffitt JR, Wang S, & Zhuang X (2015). RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science, 348, aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho B, Yoon S-H, Lee D, Koranteng F, Tattikota SG, Cha N, et al. (2020). Single-cell transcriptome maps of myeloid blood cell line-ages in drosophila. BioRxiv. 10.1101/2020.01.15.908350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium. (2010). Identification of functional elements and regulatory circuits by drosophila modENCODE. Science. 330, 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker A, Guan X-J, Murphy CT, & Murthy M (2016). Cell-type-specific transcriptome analysis in the drosophila mushroom body reveals memory-related changes in gene expression. Cell Reports, 15, 1580–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croset V, Treiber CD, & Waddell S (2018). Cellular diversity in the drosophila midbrain revealed by single-cell transcriptomics. eLife, 7. e34550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, … Shendure J (2015). Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science, 348, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, … Quake SR (2015). A survey of human brain transcriptome diversity at the single cell level. Proceedings of the National Academy of Sciences of the United States of America, 112, 7285–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie K, Janssens J, Koldere D, De Waegeneer M, Pech U, Kreft Ł, et al. (2018). A single-cell transcriptome atlas of the aging drosophila brain. Cell, 174, 982–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M, Wang Y, Zhang L, Yang Y, Huang S, Wang J, … Yan Y (2019). Single cell transcriptomic landscapes of pattern formation, proliferation and growth in drosophila wing imaginal discs. Development, 146, dev179754. [DOI] [PubMed] [Google Scholar]
- DePasquale EAK, Schnell DJ, Van Camp P-J, Valiente-Alandí Í, Blaxall BC, Grimes HL, … Salomonis N (2019). DoubletDecon: Deconvoluting doublets from single-cell RNA-sequencing data. Cell Reports, 29, 1718–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao F, Ironfield H, Luan H, Diao F, Shropshire WC, Ewer J, … White BH (2015). Plug-and-play genetic access to drosophila cell types using exchangeable exon cassettes. Cell Reports, 10, 1410–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droujinine IA, Wang D, Hu Y, Udeshi ND, Mu L, Svinkina T, et al. (2020). Proteomics of protein trafficking by in vivo tissue-specific labeling. BioRxiv. 10.1101/2020.04.15.039933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Zhang X, Wu X, Guo H, Hu Y, Tang F, & Huang Y (2015). Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biology, 16, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach KF, & Dittrich APM (1989). The optic lobe of Drosophila melanogaster. I. A Golgi analysis of wild-type structure. Cell and Tissue Research, 258, 441–475. [Google Scholar]
- Flynn JM, Santana LF, & Melov S (2011). Single cell transcriptional profiling of adult mouse cardiomyocytes. Journal of Visualized Experiments, 58, e3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Huang X, Zhang P, van de Leemput J, & Han Z (2020). Single-cell RNA sequencing identifies novel cell types in drosophila blood. Journal of Genetics and Genomics, 47, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller SA, Takahashi M, & Hurrell JG (2001). Cloning of hybridoma cell lines by limiting dilution. Current Protocols in Molecular Biology 10.1002/0471142727.mb1108s01 [DOI] [PubMed] [Google Scholar]
- Gardeux V, David FPA, Shajkofci A, Schwalie PC, & Deplancke B (2017). ASAP: A web-based platform for the analysis and interactive visualization of single-cell RNA-seq data. Bioinformatics, 33, 3123–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawad C, Koh W, & Quake SR (2016). Single-cell genome sequencing: Current state of the science. Nature Reviews. Genetics, 17, 175–188. [DOI] [PubMed] [Google Scholar]
- Genovese S, Clément R, Gaultier C, Besse F, Narbonne-Reveau K, Daian F, … Maurange C (2019). Coopted temporal patterning governs cellular hierarchy, heterogeneity and metabolism in drosophila neuroblast tumors. eLife, 8, e50375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AC, Tattikota SG, Liu Y, Comjean A, Hu Y, Barrera V, … Perrimon N (2019). Drosophila PDGF/VEGF signaling from muscles to hepatocyte-like cells protects against obesity. BioRxiv. 10.1101/2019.12.23.887059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Li L, Li J, Wu X, Hu B, Zhu P, … Tang F (2017). Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Research, 27, 967–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Yin C, Yang F, Zhang Y, Huang H, Wang J, … Xi R (2019). The cellular diversity and transcription factor code of drosophila enteroendocrine cells. Cell Reports, 29, 4172–4185. [DOI] [PubMed] [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, … Regev A (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature Methods, 14, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N, Li Y, Heidenreich M, Swiech L, Avraham-Davidi I, Trombetta JJ, … Regev A (2016). Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science, 353, 925–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque A, Engel J, Teichmann SA, & Lönnberg T (2017). A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Medicine, 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T, Wagner F, Sher N, & Yanai I (2012). CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Reports, 2, 666–673. [DOI] [PubMed] [Google Scholar]
- He L, & Hannon GJ (2004). MicroRNAs: Small RNAs with a big role in gene regulation. Nature Reviews. Genetics, 5, 522–531. [DOI] [PubMed] [Google Scholar]
- Hrvatin S, Hochbaum DR, Nagy MA, Cicconet M, Robertson K, Cheadle L, … Greenberg ME (2018). Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nature Neuroscience, 21, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung R-J, Hu Y, Kirchner R, Liu Y, Xu C, Comjean A, … Perrimon N (2020). A cell atlas of the adult drosophila midgut. Proceedings of the National Academy of Sciences of the United States of America, 117, 1514–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang B, Lee JH, & Bang D (2018). Single-cell RNA sequencing technologies and bioinformatics pipelines. Experimental & Molecular Medicine, 50, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, … Amit I (2014). Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science, 343, 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasper H (2020). Intestinal stem cell aging: Origins and interventions. Annual Review of Physiology, 82, 203–226. [DOI] [PubMed] [Google Scholar]
- Jenett A, Rubin GM, Ngo T-TB, Shepherd D, Murphy C, Dionne H, … Zugates CT (2012). A GAL4-driver line resource for Drosophila neurobiology. Cell Reports, 2, 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevitt A, Chatterjee D, Xie G, Wang X-F, Otwell T, Huang Y-C, & Deng W-M (2020). A single-cell atlas of adult drosophila ovary identifies transcriptional programs and somatic cell lineage regulating oogenesis. PLoS Biology, 18, e3000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji T, Zhang L, Deng M, Huang S, Wang Y, Pham TT, … Yan Y (2019). Dynamic MAPK signaling activity underlies a transition from growth arrest to proliferation in drosophila scribble mutant tumors. Disease Models & Mechanisms, 12, dmm040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalish BT, Cheadle L, Hrvatin S, Nagy MA, Rivera S, Crow M, … Greenberg ME (2018). Single-cell transcriptomics of the developing lateral geniculate nucleus reveals insights into circuit assembly and refinement. Proceedings of the National Academy of Sciences of the United States of America, 115, E1051–E1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanca O, Zirin J, Garcia-Marques J, Knight SM, Yang-Zhou D, Amador G, … Bellen HJ (2019). An efficient CRISPR-based strategy to insert small and large fragments of DNA using short homology arms. eLife, 8, e51539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Norris MH, Zarzycki-Siek J, Nierman WC, Donachie SP, & Hoang TT (2011). Transcript amplification from single bacterium for transcriptome analysis. Genome Research, 21, 925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaiskos N, Wahle P, Alles J, Boltengagen A, Ayoub S, Kipar C, … Zinzen RP (2017). The Drosophila embryo at single-cell transcriptome resolution. Science, 358, 194–199. [DOI] [PubMed] [Google Scholar]
- Kepecs A, & Fishell G (2014). Interneuron cell types are fit to function. Nature, 505, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, … Kirschner MW (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell, 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinides N, Kapuralin K, Fadil C, Barboza L, Satija R, & Desplan C (2018). Phenotypic convergence: Distinct transcription factors regulate common terminal features. Cell, 174, 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, … Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with harmony. Nature Methods, 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurmangaliyev YZ, Yoo J, LoCascio SA, & Zipursky SL (2019). Modular transcriptional programs separately define axon and dendrite connectivity. eLife, 8.e50822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. (2018). RNA velocity of single cells. Nature, 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib M, & Kelley SO (2020). Single-cell analysis targeting the proteome. Nature Reviews Chemistry, 4, 143–158. [DOI] [PubMed] [Google Scholar]
- Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, … Zhang K (2016). Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science, 352, 1586–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, … Church GM (2014). Highly multiplexed subcellular RNA sequencing in situ. Science, 343, 1360–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P-T, Zirin J, Kanca O, Lin W-W, Schulze KL, Li-Kroeger D, … Bellen HJ (2018). A gene-specific T2A-GAL4 library for drosophila. eLife, 7, e35574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Horns F, Wu B, Xie Q, Li J, Li T, … Luo L (2017). Classifying drosophila olfactory projection neuron subtypes by single-cell RNA sequencing. Cell, 171, 1206–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, & Jasper H (2016). Gastrointestinal stem cells in health and disease: From flies to humans. Disease Models & Mechanisms, 9, 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li T, Horns F, Li J, Xie Q, Xu C, … Luo L (2020). Single-cell transcriptomes reveal diverse regulatory strategies for olfactory receptor expression and axon targeting. Current Biology, 30, 1189–1198.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Han S, Li H, Udeshi ND, Svinkina T, Mani DR, et al. (2020). Cell-surface proteomic profiling in the Fly brain uncovers wiring regulators. Cell, 180, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, & Trapnell C (2016). Single-cell transcriptome sequencing: recent advances and remaining challenges. F1000Res. 5:F1000 Faculty Rev-182. 10.12688/f1000research.7223.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, … McCarroll SA (2015). Highly parallel genome-wide expression profiling of individual cells using Nanoliter droplets. Cell, 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco E, Karp RL, Guo G, Robson P, Hart AH, Trippa L, & Yuan G-C (2014). Bifurcation analysis of single-cell gene expression data reveals epigenetic landscape. Proceedings of the National Academy of Sciences of the United States of America, 111, E5643–E5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus JS, Anderson WF, & Quake SR (2006). Microfluidic single-cell mRNA isolation and analysis. Analytical Chemistry, 78, 3084–3089. [DOI] [PubMed] [Google Scholar]
- Matsumoto H, Kiryu H, Furusawa C, Ko MSH, Ko SBH, Gouda N, … Nikaido I (2017). SCODE: An efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics, 33, 2314–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, & Gartner ZJ (2019). DoubletFinder: Doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Systems, 8, 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, … Rajewsky N (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature, 495, 333–338. [DOI] [PubMed] [Google Scholar]
- Micchelli CA, & Perrimon N (2006). Evidence that stem cells reside in the adult drosophila midgut epithelium. Nature, 439, 475–479. [DOI] [PubMed] [Google Scholar]
- Morimoto K, & Tamori Y (2017). Induction and diagnosis of tumors in drosophila imaginal disc epithelia. Journal of Visualized Experiments, 125, 55901. 10.3791/55901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam DK, Lee S, Zhou G, Cao X, Wang C, Clark T, … Wang SM (2002). Oligo(dT) primer generates a high frequency of truncated cDNAs through internal poly(a) priming during reverse transcription. Proceedings of the National Academy of Sciences of the United States of America, 99, 6152–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichterwitz S, Chen G, Aguila Benitez J, Yilmaz M, Storvall H, Cao M, … Hedlund E (2016). Laser capture microscopy coupled with smart-seq2 for precise spatial transcriptomic profiling. Nature Communications, 7, 12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlstein B, & Spradling A (2006). The adult drosophila posterior midgut is maintained by pluripotent stem cells. Nature, 439, 470–474. [DOI] [PubMed] [Google Scholar]
- Packer J, & Trapnell C (2018). Single-cell multi-omics: An engine for new quantitative models of gene regulation. Trends in Genetics, 34, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, … Klappenbach JA (2017). Multiplexed quantification of proteins and transcripts in single cells. Nature Biotechnology, 35, 936–939. [DOI] [PubMed] [Google Scholar]
- Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, & Sandberg R (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature Methods, 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, & Sandberg R (2014). Full-length RNA-seq from single cells using smart-seq2. Nature Protocols, 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Shuga J, Wang X, Leyrat AA, Lui JH, … West JAA (2014). Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nature Biotechnology, 32, 1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter SS (2018). Single-cell RNA sequencing for the study of development, physiology and disease. Nature Reviews. Nephrology, 14, 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsköld D, Luo S, Wang Y-C, Li R, Deng Q, Faridani OR, … Sandberg R (2012). Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nature Biotechnology, 30, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, … Seelig G (2018). Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science, 360, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust K, Byrnes L, Shengyang Yu K, Park JS, Sneddon JB, Tward AD, & Nystul TG (2019). A single-cell atlas and lineage analysis of the adult drosophila ovary. BioRxiv . 10.1101/798223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SC (2008). Next-generation sequencing transforms today’s biology. Nature Methods, 5, 16–18. [DOI] [PubMed] [Google Scholar]
- Schwartzman O, & Tanay A (2015). Single-cell epigenomics: Techniques and emerging applications. Nature Reviews. Genetics, 16, 716–726. [DOI] [PubMed] [Google Scholar]
- Schwenk F, Baron U, & Rajewsky K (1995). A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Research, 23, 5080–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See P, Lum J, Chen J, & Ginhoux F (2018). A single-cell sequencing guide for immunologists. Frontiers in Immunology, 9, 2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setty M, Tadmor MD, Reich-Zeliger S, Angel O, Salame TM, Kathail P, … Pe’er D (2016). Wishbone identifies bifurcating developmental trajectories from single-cell data. Nature Biotechnology, 34, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Lubeck E, Zhou W, & Cai L (2016). In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron, 92, 342–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, … Regev A (2013). Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature, 498, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro E, Biezuner T, & Linnarsson S (2013). Single-cell sequencing-based technologies will revolutionize whole-organism science. Nature Reviews. Genetics, 14, 618–630. [DOI] [PubMed] [Google Scholar]
- Sheng K, Cao W, Niu Y, Deng Q, & Zong C (2017). Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nature Methods, 14, 267–270. [DOI] [PubMed] [Google Scholar]
- Shin J, Berg DA, Zhu Y, Shin JY, Song J, Bonaguidi MA, … Song H (2015). Single-cell RNA-Seq with waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell, 17, 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Jones LO, Petkau K, Panteluk A, & Foley E (2019). Cell-specific regulation of intestinal immunity in Drosophila. BioRxiv. 10.1101/721662. [DOI] [Google Scholar]
- Slaidina M, Banisch TU, Gupta S, & Lehmann R (2020). A single-cell atlas of the developing drosophila ovary identifies follicle stem cell progenitors. Genes & Development, 34, 239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soon WW, Hariharan M, & Snyder MP (2013). High-throughput sequencing for biology and medicine. Molecular Systems Biology, 9, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, … Satija R (2019). Comprehensive integration of single-cell data. Cell, 177, 1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, & Satija R (2019). Integrative single-cell analysis. Nature Reviews. Genetics, 20, 257–272. [DOI] [PubMed] [Google Scholar]
- Tanay A, & Regev A (2017). Scaling single-cell genomics from phenomenology to mechanism. Nature, 541, 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, … Surani MA (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods, 6, 377–382. [DOI] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, … Zeng H (2016). Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nature Neuroscience, 19, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattikota SG, Cho B, Liu Y, Hu Y, Barrera V, Steinbaugh MJ, … Perrimon N (2020). A single-cell survey of drosophila blood. eLife, 9, e54818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen ER, Mich JK, Yao Z, Hodge RD, Doyle AM, Jang S, … Ramanathan S (2016). Fixed single-cell transcriptomic characterization of human radial glial diversity. Nature Methods, 13, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NM, Shekhar K, Whitney IE, Jacobi A, Benhar I, Hong G, … Sanes JR (2019). Single-cell profiles of retinal ganglion cells differing in resilience to injury reveal neuroprotective genes. Neuron, 104, 1039–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, … Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology, 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddintong CH (1957). The strategy of the genes, London: George Allen & Unwin. [Google Scholar]
- Wagner A, Regev A, & Yosef N (2016). Revealing the vectors of cellular identity with single-cell genomics. Nature Biotechnology, 34, 1145–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, … Deisseroth K (2018). Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science, 361, eaat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt E, Benjamin S, Svetec N, & Zhao L (2019). Testis single-cell RNA-seq reveals the dynamics of de novo gene transcription and germline mutational bias in drosophila. eLife, 8, e47138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, & Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biology, 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, & Klein AM (2019). Scrublet: Computational identification of cell doublets in single-cell transcriptomic data. Cell Systems, 8, 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YE, Pan L, Zuo Y, Li X, & Hong W (2017). Detecting activated cell populations using single-cell RNA-Seq. Neuron, 96, 313–329. [DOI] [PubMed] [Google Scholar]
- Xue Z, Huang K, Cai C, Cai L, Jiang C, Feng Y, … Fan G (2013). Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature, 500, 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Duff MO, Graveley BR, Carmichael GG, & Chen L-L (2011). Genomewide characterization of non-polyadenylated RNAs. Genome Biology, 12, R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu KS, Frumm SM, Park JS, Lee K, Wong DM, Byrnes L, … Tward AD (2019). Development of the mouse and human cochlea at single cell resolution. BioRxiv, 10.1101/739680. [DOI] [Google Scholar]
- Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, et al. (2018). Molecular architecture of the mouse nervous system. Cell, 174, 999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, et al. (2015). Brain structure cell types in the mouse cortex and hippocampus revealed by single-cell RNA-Seq. Science, 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, … Bielas JH (2017). Massively parallel digital transcriptional profiling of single cells. Nature Communications, 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C, Lu S, Chapman AR, & Xie XS (2012). Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science, 338, 1622–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]