

Abstract
Black root rot (BRR) is an economically important disease of cotton and other crops, especially in cooler regions with short growing seasons. Symptoms include black discoloration of the roots, reduced number of lateral roots and stunted or slow plant growth. The cultivated tetraploid Gossypium species are susceptible to BRR. Resistance to BRR was identified in G. arboreum accession BM13H and is associated with reduced and restricted hyphal growth and less sporulation. Transcriptome analysis indicates that BM13H responds to infection at early time points 2- and 3-days post-inoculation, but by day 5, few differentially expressed genes are observed between infected and uninfected roots. Inheritance of BM13H resistance to BRR was evaluated in an F6 recombinant inbred population and shows a single semi-dominant locus conferring resistance that was fine mapped to a region on chromosome 1, containing ten genes including five putative resistance-like genes.
Keywords: Thielaviopsis basicola, Berkleyomyces rouxiae, black root rot, disease resistance, Gossypium arboreum, transcriptome analysis, asiatic cotton
1. Introduction
Black root rot (BRR) is a seedling disease of cotton caused by a soil-borne filamentous, hemibiotrophic fungus previously known as Thielaviopsis basicola, but recently reclassified into two species, Berkleyomyces basicola and B. rouxiae [1]. BRR is considered a significant threat to cotton and other crops in Australia, especially in the southern regions with shorter production seasons. The fungus survives in the soil primarily as chlamydospores [2], which upon germination invade the roots either directly or through wounds, root hairs or epidermal cells of young seedlings, and then spreads throughout the cortex. Invasion of the root is associated with cooler soil temperatures and occurs during the first 2–8 weeks of cotton growth, whereas older roots appear to be more resistant to infection [3]. The seedling disease characterized by dark brown or black discoloration of the roots as a result of the rotting of the cortex [4]. Diseased cotton plants show stunted or slow growth early in the season compared to uninfected plants, causing delayed flowering or maturity that can result in up to a 46% decrease in seed cotton yield [5]. In addition to the direct effect of BRR infection, lesions caused by the fungus may facilitate infection by other cotton seedling pathogens such as Rhizoctonia, Pythium, and Fusarium [6] and in North America significant interactions have been observed between T. basicola and the root-knot nematode Meloidogyne incognita [7].
BRR is widespread throughout Australian cotton-growing areas and through the production of chlamydospores BRR can survive in the soil for many years once established [2]. Management of this disease is currently accomplished by a range of measures that are designed to limit fungal inoculum levels from building up in the soil, and include crop rotation, flooding and fungicide seed treatments [6]. Improved host plant resistance to BRR is required for the long-term control of this disease in affected fields. However, large-scale screening of tetraploid cotton has not identified significant sources of resistance to BRR [8,9,10] but two diploid cotton accessions have been found with differing degrees of BRR resistance [11]. Gossypium arboreum accession PI 1415 had reduced root necrosis compared to susceptible checks at moderate levels of infection, but resistance was overcome at high inoculations levels. Whereas G. herbaceum accession ‘A20’ was found to possess high levels of BRR resistance even at high inoculation levels [11]. A quantitative trait loci (QTL) analysis of an interspecific F2-3 population derived from PI 1415 and A20 identified the presence of at least three loci that conferred increased resistance to BRR [12].
In the present study, BRR was shown to be caused by B. rouxiae and BRR resistance identified for G. arboreum accession BM13H [13] was found to be associated with reduced and restricted hyphal growth in root tissue and limited sporulation as compared to the susceptible G. arboreum accession Yuenanzhongmian (YZ). Transcriptome analysis of BM13H and YZ roots infected with B. rouxiae revealed very contrasting responses with BM13H responding early to infection but the number of transcriptional changes reduced over time, whereas in YZ, large changes were observed starting at day 3 and then increasing dramatically with the duration of infection. Inheritance of BM13H resistance to BRR was analyzed in an F6 BM13H x YZ recombinant inbred (RIL) population and was found to be conferred by a single semi-dominant locus that was fine mapped to a ~128.5 kbp region on chromosome 1, a region containing ten annotated genes including five putative resistance-like genes, including two; Ga01G2671 and Ga01G2672 that are differentially expressed between uninfected BM13H and YZ roots.
2. Results
2.1. Classification of BRR Fungal Species Infecting Australian Cotton
A molecular phylogenetic analysis by Nel et al. [1] of geographically isolated fungal isolates that cause BRR disease in a number of countries, showed that what was previously classified as T. basicola is in fact two species that were renamed; B. basicola and B. rouxiae. To determine which species is associated with BRR of cotton in Australia, DNA was isolated from a single spore culture of the fungal isolate WTB-001 used throughout this study. The isolate was recovered from a field-grown cotton plant displaying severe BRR symptoms. Amplification and sequencing of the internal transcribed spacer (ITS) region located between the 18S and 28S rRNA genes using the primers ITS1 and ITS4 [14] (Table S1) revealed that this isolate perfectly matched B. rouxiae (Figure S1). Analysis of previously deposited ITS sequences in GenBank from Australian BRR-infected cotton samples such as HM031125 [15] and an extensive collection of cotton BRR isolates from different cotton-growing regions in Australia carried out by Dr D. Le (NSW DPI, personal communication) also showed that all BRR-causing isolates from cotton were B. rouxiae, and not B. basicola, indicating that this species is most likely the only cause of BRR of cotton in Australia.
2.2. Microscopic Characterization of BRR Infection in BM13H and YZ
A systematic survey for BRR-resistant germplasm using a glasshouse bioassay with soil taken from fields containing the disease, revealed no resistance in a wide range of the tetraploid G. hirsutum and G. barbadense accessions (Dr W. Stiller personal communication), similar to that reported by Wheeler et al. [10] in the U.S.A. However, two G. arboreum accessions with contrasting levels of BRR resistance were identified in this screen; BM13H which displayed few obvious symptoms, and YZ which displayed symptoms similar to that observed in all G. hirsutum cultivars. Under controlled infection conditions in a glasshouse bioassay with the B. rouxiae isolate WTB-001 (250 colony forming units (CFU)/gram), BM13H plants were largely unaffected. Whereas YZ plants were stunted, had few lateral roots and almost complete blackening of the roots (Figure 1). Under sterile culture conditions, microscopic examination of the infection process in BM13H and YZ plants showed that fungal hyphae could be detected and associated with the taproots by 3 days post inoculation (dpi) and produced spores by 5 dpi on the susceptible YZ (Figure 2a–c). Hyphae were much less abundant on the surface of BM13H roots, infection sites were smaller, and spores were rarely seen until 7–10 dpi (Figure 2d–f and Figure 3c,d).
Figure 1.

Parental phenotypes of the black root rot (BRR)-susceptible YZ uninfected (a) and infected (b) and resistant BM13H uninfected (c) and infected (d) seedlings 25 days after germination in soil without (a,c) and with (b,d) BRR spores. Bars = 2 cm.
Figure 2.

Surface views of roots from susceptible (YZ—a–c) and resistant (BM13H—d–f) G. arboreum seedlings 5 days after inoculation with BRR spores in sterile culture. The roots became colonized in both accessions (black and brown patches, small arrows), but fungal growth is much more advanced on YZ roots, with abundant spores (arrowheads) already formed and more extended root blackening evident. Some BM13H roots support considerable fungal growth (e), but little sporulation. Bars = 0.5 mm.
Figure 3.

Cross-sections of roots from susceptible (YZ—a,b) and resistant (BM13H—c,d) G. arboreum seedlings 7 days after inoculation with BRR spores in sterile culture. The epidermis and cortex became colonized to varying degrees in both accessions, with dark fungal hyphae and cortical blackening generally more extensive in YZ roots. Abundant spores (arrowheads in a) are evident emerging from fungal mycelium on YZ roots. co = cortex, en = endodermis, xy = xylem poles (2 of 5 indicated in c) Bar in a for a–d = 0.2 mm.
Cross-sections taken through root segments showing dark surface discoloration at 7 dpi revealed considerable invasion into the root cortex by B. rouxiae hyphae (Figure 3). In sterile culture, more abundant hyphae were evident on YZ roots (Figure 4a) with abundant spores (arrowheads in Figure 4a). Colonization of the cortex was variable in both cultivars but was generally shallower in BM13H roots (e.g., Figure 3c) than in YZ roots (Figure 3b), although some sections of BM13H roots showed deep penetration of the black fungal hyphae (Figure 3d).
Figure 4.
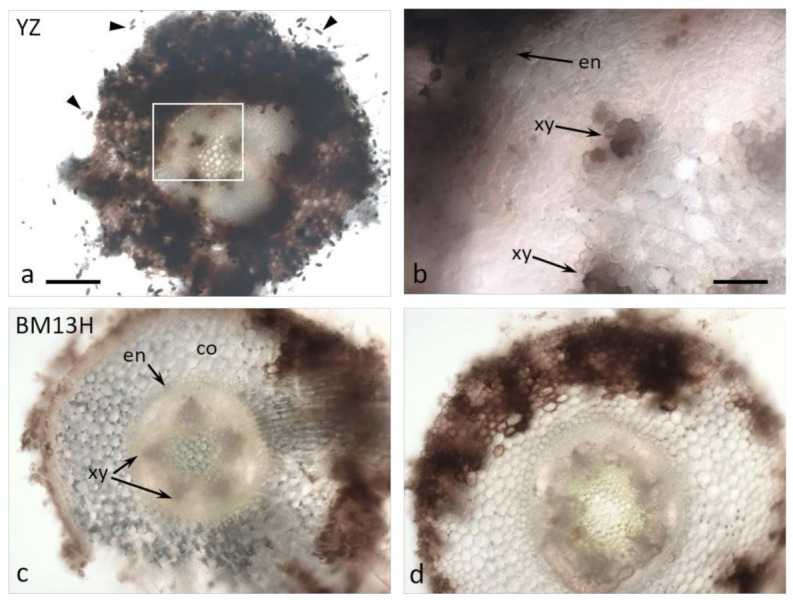
Cross-sections of roots from susceptible (YZ—a,b) and resistant (BM13H—c,d) G. arboreum seedlings 10 days after inoculation with BRR spores in sterile culture. (a,b) Abundant spores emerge from the surface of YZ roots (arrowheads in a), the cortex (co) is very heavily colonized, and black fungal hyphae commonly penetrate through the endodermis (en) and colonize the xylem (xy). b = enlarged area outlined in a. (c,d) There is less fungal colonization of BM13H roots and penetration through the endodermis is rare. co = cortex, en = endodermis, xy = xylem poles (2 of 5 indicated) Bar in a for a, c, d = 0.2 mm, bar in c = 50 mm.
By 10 dpi, the hyphae had penetrated the endodermis in some replicates of the susceptible YZ cultivar (Figure 4a,b), and either the hyphae themselves or the root response appeared to block the xylem (Figure 4b, arrows). None of the resistant BM13H roots appeared to contain fungal hyphae within the stele, although the outer cell layers of epidermis and cortex were often strongly colonized to the endodermis (Figure 3d and Figure 4c,d).
Both root hairs and epidermal cells were penetrated by hyphae in both cultivars (Figure 5), and infection generally occurred earliest in the zone of young, fully elongated root hairs.
Figure 5.

Cross-sections of roots from susceptible (YZ—a) and resistant (BM13H—b,c) G. arboreum seedlings 10 days after inoculation with BRR spores in sterile culture. Epidermal cells (arrowhead in a) and root hairs (arrows) are heavily colonized with dark fungal hyphae. (b,c) are different optical sections of the same root cross-section. Bar in a for a–c = 50 mm.
BM13H is therefore not immune to B. rouxiae but seems to reduce and restrict fungal growth such that by eye, infection is not visible on the roots, and growth is not significantly affected (Figure 1c,d).
2.3. Comparative Transcriptome Analysis in BM13H and YZ Accessions
To explore the gene expression associated with response to BRR infection in resistant BM13H and susceptible YZ, RNA-seq analysis was performed on root tissue taken from uninfected and infected plants at 2, 3, 5, 7, and 10 dpi. On average, each of the 60 samples sequenced had over 24 million clean paired-end reads, and the average Q30 percentage (sequences with sequencing error rates lower than 0.03%) was 93.3%, indicating the high quality of the RNA-seq data. Between 71% and 82% of clean reads from these samples could be aligned to the G. arboreum reference genome [16] (Table S2). When comparing infected to uninfected root tissue in each accession there was a difference in the number and type of differentially expressed genes (DEG) observed (Table 1, Tables S3 and S4). BM13H had 53 DEGs at 2 dpi (43 upregulated, 10 downregulated) that increased to 133 (59 upregulated, 74 downregulated) by 3 dpi, but this then decreased to 8 DEGs (5 upregulated, 3 downregulated) at 5 dpi and only two DEGs at 7 and 10 dpi (Table 1, Table S3). Meanwhile, YZ had 8 DEGs at 2 dpi (3 upregulated, 5 downregulated), but this number increased from 72 (57 upregulated, 15 downregulated) at 3 dpi to 1969 (1606 upregulated, 363 downregulated) at 5 dpi, 2321 (1660 upregulated, 661 downregulated) at 7 dpi and 6332 (3740 upregulated, 2592 downregulated) at 10 dpi (Table 1, Table S4). To look at accession-based (not infection dependent) gene expression differences, all uninfected samples were compared between BM13H and YZ, and 3197 DEGs were observed (Table S5). To identify biological processes and functions associated with the different transcriptional responses of BM13H and YZ, all BM13H infected and uninfected samples were compared directly to YZ infected and uninfected samples, respectively, and Gene Ontology (GO) enrichment analysis was performed on genes specific to infection and uninfected expression differences. Infection-specific DEGs associated with BM13H (Table S6) were enriched in 16 biological processes (FDR < 0.01), including carbohydrate metabolic processes, starch biosynthesis and plant cell wall organization (Table 2, Figure S2). The DEGs associated with YZ infection (Table S7) were enriched for 102 biological processes with major enriched terms for regulation of signaling and the immune system, defense and stress responses, and responses to the hormone salicyclic acid (Table 3, Figure S3). There were no enriched GO terms found for the DEGs between uninfected BM and YZ root samples.
Table 1.
Number of differentially expressed genes (DEGs) between infected and uninfected root tissue during BRR infection of BM13H and YZ.
| YZ | BM13H | |||
|---|---|---|---|---|
| Days Post Infection | Upregulated | Downregulated | Upregulated | Downregulated |
| 2 | 3 | 5 | 43 | 10 |
| 3 | 57 | 15 | 59 | 74 |
| 5 | 1606 | 363 | 5 | 3 |
| 7 | 1660 | 661 | 0 | 2 |
| 10 | 3740 | 2592 | 2 | 0 |
Table 2.
Gene Ontology (GO) terms enriched in BM13H during infection by B. rouxiae.
| Description | Ontology | Number of Genes | False Discovery Rate |
|---|---|---|---|
| glucan metabolic process | Biological process | 35 | 1.3 × 10−5 |
| cellular glucan metabolic process | Biological process | 35 | 1.3 × 10−5 |
| glucan biosynthetic process | Biological process | 29 | 1.6 × 10−5 |
| carbohydrate metabolic process | Biological process | 84 | 2.6 × 10−5 |
| polysaccharide biosynthetic process | Biological process | 34 | 3.3 × 10−5 |
| cellular carbohydrate metabolic process | Biological process | 43 | 3.3 × 10−5 |
| cellular polysaccharide metabolic process | Biological process | 37 | 3.3 × 10−5 |
| cellular polysaccharide biosynthetic process | Biological process | 31 | 6.4 × 10−5 |
| polysaccharide metabolic process | Biological process | 41 | 6.4 × 10−5 |
| cellular carbohydrate biosynthetic process | Biological process | 32 | 0.000 |
| starch metabolic process | Biological process | 21 | 0.000 |
| starch biosynthetic process | Biological process | 18 | 0.000 |
| cellular component organization or biogenesis | Biological process | 113 | 0.002 |
| carbohydrate biosynthetic process | Biological process | 37 | 0.003 |
| cell wall organization or biogenesis | Biological process | 35 | 0.004 |
| cellular component organization | Biological process | 103 | 0.005 |
| external encapsulating structure organization | Biological process | 26 | 0.012 |
| single-organism carbohydrate metabolic process | Biological process | 53 | 0.012 |
| maltose metabolic process | Biological process | 12 | 0.012 |
| plant-type cell wall organization or biogenesis | Biological process | 22 | 0.012 |
| cell wall organization | Biological process | 25 | 0.014 |
| plastid organization | Biological process | 21 | 0.019 |
| plant-type cell wall organization | Biological process | 17 | 0.033 |
| extracellular region | Cellular component | 39 | 0.001 |
Table 3.
GO terms enriched in YZ during infection by B. rouxiae.
| Description | Ontology | Number of Genes | False Discovery Rate |
|---|---|---|---|
| defense response | Biological process | 68 | 1.1 × 10−6 |
| response to organonitrogen compound | Biological process | 26 | 5.0 × 10−6 |
| response to biotic stimulus | Biological process | 61 | 2.7 × 10−5 |
| regulation of defense response | Biological process | 32 | 2.7 × 10−5 |
| response to chitin | Biological process | 23 | 2.7 × 10−5 |
| regulation of response to stress | Biological process | 32 | 2.7 × 10−5 |
| response to nitrogen compound | Biological process | 34 | 2.8 × 10−5 |
| immune system process | Biological process | 44 | 2.9 × 10−5 |
| response to stress | Biological process | 138 | 4.7 × 10−5 |
| cell death | Biological process | 32 | 0.000 |
| regulation of innate immune response | Biological process | 25 | 0.000 |
| innate immune response | Biological process | 40 | 0.000 |
| regulation of immune response | Biological process | 25 | 0.000 |
| response to external biotic stimulus | Biological process | 54 | 0.000 |
| regulation of immune system process | Biological process | 25 | 0.000 |
| immune response | Biological process | 40 | 0.000 |
| response to other organism | Biological process | 54 | 0.000 |
| regulation of cell death | Biological process | 24 | 0.000 |
| response to external stimulus | Biological process | 70 | 0.000 |
| regulation of response to stimulus | Biological process | 42 | 0.000 |
| response to stimulus | Biological process | 218 | 0.000 |
| response to oxygen-containing compound | Biological process | 78 | 0.000 |
| response to chemical | Biological process | 125 | 0.000 |
| programmed cell death | Biological process | 27 | 0.001 |
| regulation of programmed cell death | Biological process | 22 | 0.001 |
| negative regulation of cell death | Biological process | 15 | 0.001 |
| negative regulation of programmed cell death | Biological process | 15 | 0.001 |
| host programmed cell death induced by symbiont | Biological process | 23 | 0.002 |
| plant-type hypersensitive response | Biological process | 23 | 0.002 |
| cell communication | Biological process | 88 | 0.002 |
| salicylic acid mediated signaling pathway | Biological process | 17 | 0.002 |
| response to endogenous stimulus | Biological process | 63 | 0.002 |
| cellular response to salicylic acid stimulus | Biological process | 17 | 0.003 |
| response to salicylic acid | Biological process | 19 | 0.003 |
| response to acid chemical | Biological process | 48 | 0.005 |
| cellular response to stress | Biological process | 51 | 0.005 |
| response to fungus | Biological process | 24 | 0.005 |
| regulation of plant-type hypersensitive response | Biological process | 18 | 0.006 |
| regulation of cellular response to stress | Biological process | 18 | 0.006 |
| response to organic substance | Biological process | 82 | 0.007 |
| cellular response to acid chemical | Biological process | 26 | 0.008 |
| respiratory burst involved in defense response | Biological process | 10 | 0.009 |
| endoplasmic reticulum unfolded protein response | Biological process | 12 | 0.009 |
| cellular response to unfolded protein | Biological process | 12 | 0.009 |
| cellular response to topologically incorrect protein | Biological process | 12 | 0.009 |
| response to unfolded protein | Biological process | 12 | 0.009 |
| multi-organism process | Biological process | 61 | 0.009 |
| signal transduction | Biological process | 75 | 0.010 |
| respiratory burst | Biological process | 10 | 0.010 |
| protein targeting to membrane | Biological process | 18 | 0.010 |
| single organism signaling | Biological process | 75 | 0.015 |
| signaling | Biological process | 75 | 0.016 |
| response to organic cyclic compound | Biological process | 27 | 0.025 |
| protein localization to membrane | Biological process | 18 | 0.026 |
| establishment of protein localization to membrane | Biological process | 18 | 0.026 |
| cellular response to stimulus | Biological process | 98 | 0.036 |
| defense response to other organism | Biological process | 33 | 0.046 |
| phosphotransferase activity, alcohol group as acceptor | Molecular function | 93 | 0.001 |
| kinase activity | Molecular function | 103 | 0.003 |
| protein kinase activity | Molecular function | 81 | 0.003 |
| protein serine/threonine kinase activity | Molecular function | 60 | 0.017 |
| catalytic activity | Molecular function | 471 | 0.017 |
2.4. Inheritance of BBR Resistance
To understand the genetics of the resistance observed in BM13H, crosses were made between BM13H and YZ. In a glasshouse based BRR infection assay of 51 F1 seedlings in soil containing 250 CFUs of BRR spores per gram of soil, the average BRR disease rating (0–100%) based on the percentage of necrotic root tissue [12] of the roots was 39% for the F1 plants compared to 0% for BM13H (17 plants) and 90% for YZ (18 plants) indicating an intermediate level of resistance. An F1 plant was self-pollinated and 263 F2 progeny generated and infected with WTB-001. The infection results at 25 dpi indicate a bimodal infection level with approximately 25% of plants with a root infection percentage of less than 30% (Figure S4). To aid the genetic characterization of the resistance an F6 recombinant inbred line (RIL) population from a BM13H x YZ cross was developed by single seed decent from the first 158 individuals from the F2 population. Each F6 line was subjected to at least two independent assays (two pots per assay, ~eight plants per pot) and the final disease score for each line was the average of the disease rating from ~32 plants. Each assay included four pots of parental plants (~eight plants per pot) and over the course of the assays the disease scores were 1% for BM13H and 88% for YZ lines. The distribution of infection percentages in the RIL population had a clearer bimodal infection distribution than in the F2 population, with most lines falling either into the ≤30% or ≥80% necrotic root ranges (Figure S5).
2.5. Genetic Mapping of Black Root Rot Resistance
A total of 178 genetic markers (Tables S8–S10) comprising simple sequence repeat (SSR), cleaved amplified polymorphic sequences (CAPS), and single nucleotide polymorphism (SNP)-based markers (Kompetitive Allele Specific PCR (KASP) or Fluidgm) were analyzed on the 158 RIL F6 lines. After linkage analysis, the markers were assembled into 17 linkage groups with a total length of 1294 cM, and linkage groups were assigned to chromosomes using the DNA marker sequences matches to the genome assembly of the G. arboreum cultivar Shixiya1 (SXY1) [16] (Figure S6).
Single marker analysis, using a nonparametric mapping test equivalent to a one-way analysis of variance [17], found nine markers on chromosome 1 (Chr01_11 to Chr01_19) associated with BRR resistance (p ≤ 0.001) (Figure 6A). A single QTL (BRR01) above the permutation threshold, was detected for BRR resistance by multiple QTL model analysis [17] between markers Chr01_17 and Chr01_18 which explained 98% of the phenotypic variation (Figure 6a). The QTL analysis, therefore, indicated that resistance was conferred by a single locus that we call ‘Resistance to Berkleyomyces rouxiae 1′ (RBR01). The RIL disease data were then re-analyzed such that plants with roots with disease ratings of ≤30% were designated as resistant, roots having a disease rating of ≥70% were designated as susceptible, and families that possessed plants with infection scores that included at least one resistant and one susceptible plant were designated as heterozygous. Using this simplified scoring system, RBR01 was mapped between the markers Chr01_16 and Chr01_18 (Figure 6a), which is 0.9 cM in genetic distance, and physically covers 587,500 bp (110,831,711-111,419,245) based on the SXY1 genome [16].
Figure 6.

Mapping of the BRR resistance locus RBR01. (a) Single marker analysis associated resistance with markers Chr01_11 to Chr01_19 (line with closed circles at end), and a single QTL was detected between markers Chr01_17 to Chr01_18 (dotted box). Scoring the phenotype as a simple trait indicated the interval was between Chr01_16 and Chr01_18 (open box). (b) Fine mapping of the region with key recombinants reduced the interval to between GaBRR01 and GaBRR04 markers (bold), S = susceptible (Blue), H = Heterozygous (green) and R- resistant (red).
2.6. Fine Mapping of BRR01
To reduce the size of the genetically defined interval, two new populations were developed from RILF6#59 and RILF6#141 families that were found to be segregating for BRR resistance and were scored as heterozygous for the Chr01_16, Chr01_17 and Chr01_18 markers (Table S11). Eight hundred RILF6-7 plants from each family were planted and genotyped for the Chr01_16 and Chr01_18 markers, and eleven plants with recombination between the two markers were identified (Table S11). These plants were self-pollinated and their progeny tested for BRR resistance. A further eight KASP markers (designated GaBRR01 to 08) were developed in the interval between the Chr01_16 and Chr01_18 markers (Table S8) and used to genotype the original F6 recombinants and the eleven fine mapping recombinants (Table S11). The results indicate that the BRR01 locus was located between GaBRR01 and GaBRR04, a distance of 128,488 bp (Figure 6b). This interval has 10 annotated genes Ga01g2664 to Ga01g2673 that includes five putative disease resistance like genes (Ga01G2668, Ga01G2670, Ga01G2671, Ga01G2672, Ga01G2673) (Figure 7).
Figure 7.

Gene expression ratios of 10 annotated genes in the BRR01 mapping interval, gene expression values are fold-change ratio values (infected/uninfected), statistically significant DEG values are outlined.
Analysis of the expression profiles of the ten genes in the mapping interval based on the RNA-seq dataset in Section 2.3 ‘Comparative Transcriptome Analysis in BM13H and YZ accessions’, revealed that only three of those genes were differentially expressed between infected and uninfected roots, during the period of infection analyzed. Ga01G2665, Ga01G2666 and Ga01G2671 were differentially expressed in YZ at 10 dpi (Figure 7). However, three genes (Ga01G2669, Ga01G2671, Ga01G2672) were differentially expressed between BM13H and YZ when only uninfected roots samples were compared (Figure 7, Table S5). Ga01G2669 was expressed 3.4-fold higher in BM13H, Ga01G2671 was expressed 2.3-fold higher in BM13H, and Ga01G2672 was expressed 3-fold higher in YZ.
3. Discussion
BRR has been found in a wide range of economically important plant species, both indigenous and exotic in Australia [8], but the BRR pathogen is thought to be introduced as it has not been identified from undisturbed Australian soils [6]. Comparisons of the ITS sequence from the fungal isolate WTB-001 and other Australian cotton samples infected with BRR [15] indicate that the newly recognized species B. rouxiae is the causative agent of BRR in Australian cotton. In addition, B. rouxiae has been identified as the cause of BRR on lettuce in Japan [18], implying that the species has a wide host range, although it may show some degree of host specificity. Nevertheless, this wide host range could be useful for the evaluation of diverse mechanisms of resistance that could benefit cotton improvement.
In the absence of any known resistant germplasm in cultivated G. hirsutum and G. barbadense [12], the impact of the disease on the Australian cotton industry is currently limited solely by a range of cultural practices such as rotation of non-host crops and fungicide application that attempt to limit the build-up of inoculum levels in the soil [6]. Therefore, improved host plant resistance to BRR is needed for the long-term control. Niu et al. [12] identified strong resistance to BRR in the G. herbaceum accession ‘A20’ to an T. basicola isolate, and QTL analysis revealed that resistance was genetically complex involving at least three loci (BRR5.1, BRR9.1 and BRR13.1) that together explained approximately 40% of the total resistance observed. We identified strong resistance in the G. arboreum accession BM13H to a B. rouxiae isolate. BM13H is also a diploid cotton with an A-genome, but in this accession, resistance was due to a single locus called RBR01 on Chromosome 1. As none of the QTL identified in G. herbaceum [12] are located on Chromosome 1, RBR01 represents a new BRR resistance locus in cotton.
The surface colonization of cotton roots by BRR [19,20] and the physiological consequences associated with seed cotton yield reductions from heavy infestations [5] have been described several times. Our analysis shows that the progress of hyphal invasion into susceptible roots of G. arboreum is very similar to that in tobacco, outlined in several earlier reports [21,22,23]. Indeed, the invasion process and sequence of disease progression appear similar in several other species including bean, pansy and lupin [24,25,26]. When comparing the infection process between YZ and BM13H, the initial stages of invasion into the root epidermis and root hair cells appeared similar (Figure 5), with both cell types harboring abundant hyphae. In the resistant BM13H roots, the fungus penetrated no further than the endodermis of the main root (the taproot), although it could colonize almost all of the enclosing cortex and epidermal tissues (e.g., Figure 4d). However, in the susceptible YZ roots, fungal hyphae were often detected within the stele, and in the xylem vessels (e.g., Figure 4b). The infection process and single-locus nature of BM13H resistance resembles the single-gene resistance in tobacco identified from Nicotiana deneyi Domin [27] called resistance to BRR 1 (RBRR1), that was transferred to burley tobacco production in the 1950s and has remained a durable source of BRR resistance ever since [28]. Hood and Shew [22] compared BRR infection in a BRR-resistant tobacco cultivar ‘Tennessee 90′ (TN90) containing RBRR1, to a susceptible tobacco cultivar ‘Burley 21 x Kentucky 10′ (B2IxI0). They found that for TN90 spore germination, germ tube growth and penetration of root tissue were similar to B2IxI0 but that hyphal invasion was more rapid and spores formed earlier on B2IxI0 roots, and that invasive hyphae penetrated the stele of B2IxI0 roots but not TN90 roots. Hyphal growth and lesion development were limited in TN90 with sporulation rarely being observed. Due to the limited fungal growth and absence of secondary infections, the root system of TN90 gradually outgrew the effects of the initial inoculation [22]. Our results for BM13H appear to be similar to that observed in tobacco although sporulation may be more common in BM13H than observed in TN90, it appears that BM13H can limit the primary infection and reduce secondary infection such that root growth is not adversely affected, and as older roots appear to be more resistant [3], this allows BM13H to outgrow the disease.
Transcriptome analysis reflected the microscopic observations on the infection process, as BM13H had early gene expression responses at 2 and 3 dpi with infection-specific DEG GO terms being associated with cell wall development, and carbohydrate biosynthesis and metabolic processes, but few DEGs were identified at later time points (Table 2, Figure S2). This lack of DEGs after 5 dpi possibly reflects that BM13H restricts fungal growth such that root growth and development is relatively unaffected at later time points leaving the transcriptomic landscape relatively unchanged. Whereas, the susceptible accession YZ had a very different transcriptional response with a small number of expression changes observed at 2 dpi, but then very large numbers of gene expression changes were observed for evaluations after 5 dpi with YZ infection specific GO terms associated with the regulation of signaling and the immune system, and defense and stress responses (Table 3, Figure S3). The increasing DEG changes probably reflect the increasing level of fungal infection on the roots and cell death occurring in YZ that results in blackened, stunted roots and are similar to the protein expression changes observed during a compatible interaction between B. basicola and G. hirsutum (Sicot 189) in which 29% and 24% of identified protein expression changes were associated with defense and stress, respectively [29].
Genetic analysis located the RBR01 locus to a 128,488 bp region on chromosome 1, a region annotated for ten genes including five putative resistance-like genes. Of these genes only Ga01G2665, Ga01G2666 and Ga01G2671 were differentially expressed between infected and uninfected YZ roots at 10 dpi (Figure 7). There were a large number of accession-based gene expression changes observed between uninfected BM13H and YZ roots that may be associated with the resistance phenotype. Three genes in the mapped interval were consistently differentially expressed between uninfected BM13H and YZ roots; Ga01G2669 and Ga01G2671, 3.4- and 2.3-fold higher in BM13H, respectively, and Ga01G2672 3-fold higher in YZ (Figure 7). Ga01G2669 is an uncharacterized protein, whereas Ga01G2671 and Ga01G2672 share homology to putative disease resistance proteins. Therefore, resistance may be associated with constitutive expression differences between YZ and BM13H and not infection associated DEGs between the two accessions. As RBR01 resistance is partially dominant the most likely BRR resistance candidate gene based on available information is Ga01G2671 that is overexpressed in the resistant accession. Although, it must be noted that the genome and transcriptome mapping analyses are dependent on the genome sequence information derived from the G arboreum accession SXY1 [16] as de novo genomes assemblies of BM13H and YZ are not available, and it is known in G. hirsutum that there can be extensive intraspecific gene order and gene structural variations between cultivars in some regions [30]. In the future, functional confirmation of the candidate genes using a transgenic approach must be performed before the gene/s responsible for resistance can be conclusively determined. However, G. arboreum is very difficult to stably transform; thus, this analysis may need to be performed in G. hirsutum that is more easily transformed.
Additionally G. arboreum is sexually incompatible with G. hirsutum due to genome size and structural differences so to transfer this new BRR resistance locus to commercial G. hirsutum cultivars will require either a molecular genetics approach via transgenic introgression of the resistance gene or a breeding approach via the formation of a synthetic tetraploid or synthetic hexaploid bridging species that enable sexual crossing to G. hirsutum [31,32]. The molecular approaches require knowledge of the causal gene and are potentially quicker, but also assumes that the resistance gene works independently or that the downstream molecular pathways required for the resistance mechanism are also present in G. hirsutum; although such disease resistance pathways are likely conserved due to the common origin of the A-genome for this species [33]. The breeding approach has been successful in the introgression of Rotylenchus reniformis resistance from the F-genome type G. longicalyx to G. hirsutum [34]. However, it is a very long process, even with modern high throughput genotyping and genome information, and has proven to be difficult for the introgression of the BRR resistance from G. herbaceum ‘A20’ [10]. Nonetheless, the simpler nature of the inheritance of the resistance present in BM13H may make this process easier.
4. Materials and Methods
4.1. Plant Materials
A population segregating for BRR resistance was created by crossing the highly resistant G. arboreum accession BM13H [13], with the susceptible G. arboreum accession YZ [35]. An F2 population of 263 individuals was developed from a single F1 plant. A RIL population was developed from 158 F2 lines advanced to the F6 generation by single seed descent.
4.2. Identification of BRR Isolate, Inoculum Preparation and Inoculation System
The fungal isolate WTB-001 was recovered by Dr S. Allen from a cotton plant displaying severe BRR symptoms in a field at the Australian Cotton Research Institute (Narrabri, NSW, Australia). A single spore culture derived from WTB-001 was isolated and grown on carrot agar [36] for 5–6 weeks at 25 °C on Petri plates (145 × 20 mm). The spores were then washed from the plates with sterile water and mixed with glycerol to a final concentration of 15% and stored at −80 °C. To classify the fungal isolate at the species level, a pure culture of WTB-001 was grown on half-strength potato dextrose (PDA) broth (19.5 g potato dextrose (Oxoid Microbiology Products, Hampshire, UK), distilled water 1 L) incubated at 25 °C for 3 days and DNA was isolated using the technique described by de Beer et al. [37]. The ITS region of the rRNA genes was amplified using the primers ITS1 and ITS4 [14] (Table S1) and compared to the published sequences [1].
For plant disease assays, spores from the single spore culture derived from WTB-001 were washed from the Petri plates with 10 mL of sterile water and blended with a Waring blender (Model HGBTAWTS3, Torrington, CT, USA) on high speed for 30 s. The mixture was calibrated for CFU/mL on half-strength PDA agar (19.5 g potato dextrose (Sigma-Aldrich, St Louis, MI, USA), 14.5 g agar course powder (Ajax FineChem, Wollongong, NSW, Australia), distilled water 1 L) incubated at 25 °C for 3 days. CFU counts represent the number of viable propagules and can be composed of chlamydospores, endoconidia, and mycelia.
BRR disease phenotypes for genetic mapping were assessed based on the following method. Initially, 51 F1 plants and the F2 population were evaluated to assess the genetics of resistance with more extensive evaluation conducted for the RIL population. Fungal inoculum was added such that there were 250 CFU of spores per gram of soil (60:40 mix of compost and perlite) and made in 20 kg batches mixed in a concrete mixer for 30 s. The inoculated soil was added to polypropylene pots (10 cm diameter, 10 cm length) and 8 seeds per F6 line were planted per pot. The lines were grown in a temperature-controlled glasshouse watered daily for 4–5 weeks, with a daytime (16 h) temperature of 21–23 °C and night (8 h) temperature of 18–20 °C. A single assay consisted of at least two replicates of each line (~16 plants per replicate), resistant (BM13H) and susceptible (YZ) parents in both infected and uninfected soil (as a control for soil quality). Pots were randomized with respect to their position the glasshouse for each experiment. After ~25 days the soil was carefully washed from the roots. Disease rating (0–100%) was assessed visually as per Niu et al. [12] based on the percentage of necrotic root tissue. A 50% disease rating denotes that necrosis was observed on half the total root area. Each line was subjected to at least two independent assays and the final disease score for each line was the average of the disease rating across all plants and all assays.
For transcriptome experiments, three 2-week-old plants for each parent that were grown in polypropylene pots (10 cm diameter, 10 cm length) and drenched with 100 mL of solution containing 1 × 106 CFU of fungal inoculum suspension. At designated time points (2, 3, 5, 7 and 10 dpi) the soil was carefully washed from the roots of the plants, and plants were rapidly dissected into roots and hypocotyl tissue. Whole roots tissue was collected and placed in liquid nitrogen, and then stored at −80 °C. Three independent root samples were collected for each time point.
For microscopic examination of infection, seeds were surface sterilized in a solution containing 1% NaOCl and 10% ethanol for 15 min and then rinsed several times in sterile distilled water. Seeds were incubated in Petri dishes (145 × 20 mm) with Murashige and Skoog (MS) medium [38] with 0.8% agar for 2 days at 25 °C. Germinated seedlings free from bacterial and fungal contamination were then removed and root dip-inoculated in either a WTB-001 spore suspension (1 × 106 CFU/mL) or sterile water for 5 min. Excess liquid was removed and the seedlings were transferred to sterile, sealed transparent plastic tissue culture pots (65 mm diameter, 100 mm length) containing semi-solid MS media (0.5% agar). The seedlings were incubated at 25 °C with a 16h photoperiod (30 μmolm−2/s−1).
At one day intervals after inoculation, 4–8 seedlings were gently removed from the agar and transferred into 70% ethanol or lactic acid before staining and microscopic observation. Root surfaces were examined under a dissecting microscope either fresh or after storage in 70% ethanol. Hand sections from fresh roots, or roots preserved in 70% ethanol, were examined under a compound microscope either unstained or stained with 1% trypan blue.
4.3. Plant DNA Sample Preparation and Genotyping
The genomic DNA of the parental G. arboreum accessions and their derived F6 individuals was extracted from young leaves according to the method described by Ellis et al. [39]. DNA quantity was measured using the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and adjusted to a working concentration of 20 ng/µL. DNA integrity of each sample was checked on a 0.8% agarose gel against Lambda DNA/HindIII size ladder.
Genotyping of the F6 RIL population was performed using multiple methods. KASP markers (Supplementary Table S8) were designed based on SNPs between parental accessions and performed as previously described [39]. SSR and CAPS markers (Supplementary Table S9) were performed as previously described by Lopez-Lavalle et al. [40]. Fluidigm SNPtype™ assays were directed to specific sequences (Table S10) and were performed by Millennium Science Pty Ltd. (Mulgrave, Victoria, Australia) according to the manufacturer’s instructions.
4.4. Linkage and QTL Analysis
A linkage map was constructed from 178 polymorphic loci using Joinmap 4.0 software [41]. A LOD score of 3.0 and recombination fraction of 0.4 were used as the threshold criteria for linkage and the Kosambi map function was used to convert recombination frequency to genetic map distance cM [42]. The collinearity of each linkage group was compared to the G. arboreum (SXY1) genome sequence from Du et al. [16] to assist in the assignment of each linkage group.
QTL analysis was performed using MapQTL5 [17]. Significance thresholds were established by permutation tests (1000 iterations) [43]. The percentage of phenotypic variance explained by the QTL, was calculated at the likelihood peak.
4.5. Total RNA Extraction and Transcriptome Sequencing
Total RNA of whole root samples was extracted by Maxwell RSC Plant RNA Kit with the Maxwell RSC Instrument (Promega, Madison, WI, USA). Each sample included three biological replicates. The quality of the RNA samples was checked using the Agilent 2100 Bioanalyzer system (Agilent Technologies, Palo Alto, CA, USA). RNA samples from each time point with an RNA integrity number (RIN) above 7.0 were used for RNA-seq library construction. Libraries were constructed and sequenced by GENEWIZ (Suzhou, China) using the paired-end configuration on an Illumina HiSeq instrument according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Raw reads were first processed using Trimmomatic v0.39 [44] to remove low-quality sequences and adaptors. The quality of trimmed FASTQ files was evaluated using FastQC v0.11.8. Reads were mapped to the G. arboreum reference genome from the cultivar SXY1 [16] using Biokanga version 3.9.8 (https://github.com/csiro-crop-informatics/biokanga, accessed date 17 February 2021) and transcript per million mapped reads (TPM) was calculated for estimating gene expression levels with a custom Python script. HTSeqcount [45] was used to obtain the raw read counts of each gene. The R package DESeq2 was used to identify DEGs [46]. An adjusted p-value < 0.05 was used as criterion for the determination of DEGs. The raw RNA-Seq data are available from CSIRO data portal (https://data.csiro.au/dap/landingpage?pid=csiro:47747, accessed date 1 March 2021). Gene ontology (GO) enrichment analysis was performed using agriGO v2.0 based on the default settings [47]. The gene lists for GO analysis were obtained by finding DEGs between all pooled infected BM13H samples versus all YZ infected samples and removing all DEGs between all pooled uninfected BM13H samples versus all YZ uninfected samples.
5. Conclusions
A new resistance locus RBR01 to BRR (B. rouxiae) was identified in the G. arboreum accession BM13H and mapped to chromosome 1. Analysis of the root infection process revealed that resistance was associated with the slowing down of fungal infection and reduction in sporulation, which enable the infected roots to outgrow the disease. Fine mapping reduced the locus region to 128.5 kbp containing ten annotated genes with the most likely candidate being a putative resistance-like gene Ga01G2671 that is constitutively more highly expressed in resistant accession BM13H compared to the susceptible accession YZ.
Acknowledgments
The authors would like to thank S. Allen for supplying the B. rouxiae isolate WTB-001. Tanya Phongkham for her contribution to the development of the BRR disease and transcriptome assays and genetic mapping. Melanie Soliveres, Vanessa Gillespie and Walter Tate for their excellent technical assistance, and Rachel Robbins, Bec Edwards and Patrick Moody for their assistance with glasshouse work. Tan Jiafu and Lucy Egan for their helpful reviews.
Supplementary Materials
Supplementary Materials can be found at https://www.mdpi.com/1422-0067/22/5/2642/s1.
Author Contributions
I.W.W. and M.E. conceived the study; M.E., I.W.W., and R.G.W. performed the experiments and D.L. supervised the study. Q.-H.Z. and P.M. analyzed the data; I.W.W. and R.G.W. wrote the manuscript; and W.S., D.L., P.M., M.E. and Q.-H.Z. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Cotton Breeding Australia, a joint venture between Cotton Seed Distributors Ltd. and CSIRO.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw paired-end Illumina RNA sequencing reads generated in the current study are available from the CSIRO data portal https://data.csiro.au/dap/landingpage?pid=csiro:47747 accessed date 1 March 2021.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nel W.J., Duong T.A., Wingfield B.D., Wingfield M.J., de Beer Z.W. A new genus and species for the globally important, multihost root pathogen Thielaviopsis basicola. Plant Pathol. 2018;67:871–882. doi: 10.1111/ppa.12803. [DOI] [Google Scholar]
- 2.Tsao P.H., Bricker J.L. Chlamydospores of Thielaviopsis basicola as surviving propagules in natural soils. Phytopathology. 1966;56:1012–1014. [Google Scholar]
- 3.Hillocks R.J. Seedling diseases. In: Hillocks R.J., editor. Cotton Diseases. CAB International; Wallingford, UK: 1992. pp. 1–39. [Google Scholar]
- 4.Allen S.J. Black Root Rot. In: Kirkpatrick T., Rothrock C., editors. Compendium of Cotton Diseases. 2nd ed. APS Press; St Paul, MN, USA: 2001. pp. 16–17. [Google Scholar]
- 5.Nehl D.B., Allen S.J., Mondal A.H., Lonergan P.A. Black root rot: A pandemic in Australian cotton. Australas. Plant Pathol. 2004;33:87–95. doi: 10.1071/AP03085. [DOI] [Google Scholar]
- 6.Pereg L.L. Black root rot of cotton in Australia: The host, the pathogen and disease management. Crop Pasture Sci. 2013;64:1112–1126. doi: 10.1071/CP13231. [DOI] [Google Scholar]
- 7.Walker N.R., Kirkpatrick T.L., Rothrock C.S. Interaction between Meloidogyne incognita and Thielaviopsis basicola on cotton (Gossypium hirsutum) J. Nematol. 1998;30:415–422. [PMC free article] [PubMed] [Google Scholar]
- 8.Honess T. Ph.D. Thesis. University of New England; Armidale, Australia: 1994. Atiological Aspects of Black Root Rot of Cotton. [Google Scholar]
- 9.Wang H.P., Davis R.M. Susceptibility of selected cotton cultivars to seedling disease pathogens and benefits of chemical seed treatments. Plant Dis. 1997;81:1085–1088. doi: 10.1094/PDIS.1997.81.9.1085. [DOI] [PubMed] [Google Scholar]
- 10.Wheeler T., Gannaway J. 1644 Identification of germplasm resistant to Thielaviopsis basicola in the USDA cotton germplasm collection; Proceedings of the 4th World Cotton Research Conference; Lubbock, TX, USA. 10–14 September 2007; Washington, DC, USA; Lubbock, TX, USA: International Cotton Advisory Committee; 2007. [Google Scholar]
- 11.Wheeler T.A., Gannaway J.R., Keating K. Identification of resistance to Thielaviopsis basicola in diploid cotton. Plant Dis. 1999;83:831–833. doi: 10.1094/PDIS.1999.83.9.831. [DOI] [PubMed] [Google Scholar]
- 12.Niu C., Lister H.E., Nguyen B., Wheeler T.A., Wright R.J. Resistance to Thielaviopsis basicola in the cultivated A genome cotton. Theor. Appl. Genet. 2008;117:1313–1323. doi: 10.1007/s00122-008-0865-5. [DOI] [PubMed] [Google Scholar]
- 13.Miyazaki J., Stiller W.N., Wilson L.J. Novel cotton germplasm with host plant resistance to twospotted spider mite. Field Crop. Res. 2012;134:114–121. doi: 10.1016/j.fcr.2012.05.006. [DOI] [Google Scholar]
- 14.White T., Bruns T., Lee S., Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis M., Gelfand D., Sninsky J., White T., editors. PCR Protocols: A Guide to Methods and Applications—A Laboratory Manual. Academic Press, Inc.; San Diego, CA, USA: 1990. pp. 315–322. [Google Scholar]
- 15.Coumans J.V.F., Harvey J., Backhouse D., Poljak A., Raftery M.J., Nehl D., Katz M.E., Pereg L. Proteomic assessment of host-associated microevolution in the fungus Thielaviopsis basicola. Environ. Microbiol. 2011;13:576–588. doi: 10.1111/j.1462-2920.2010.02358.x. [DOI] [PubMed] [Google Scholar]
- 16.Du X., Huang G., He S., Yang Z., Sun G., Ma X., Li N., Zhang X., Sun J., Liu M., et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 2018;50:796–802. doi: 10.1038/s41588-018-0116-x. [DOI] [PubMed] [Google Scholar]
- 17.Van Ooijen J.W. MapQTL® 5, Software for Mapping of Quantitative Triat Loci in Experimental Populations. 5th ed. Kyazma, B.V.; Wageningen, The Netherlands: 2004. [Google Scholar]
- 18.Nakane R., Miki S., Ikeda K., Sakai H., Hayashi K., Usami T. First report of black root rot of lettuce in Japan caused by Berkeleyomyces rouxiae. J. Gen. Plant Pathol. 2019;85:436–439. doi: 10.1007/s10327-019-00860-y. [DOI] [Google Scholar]
- 19.King C.J., Presley J.T. A root rot of cotton caused by Thielaviopsis basicola. Phytopathology. 1942;32:752–761. [Google Scholar]
- 20.Mathre D.E., Ravenscroft A.V., Garber R.H. The role of Thielaviopsis basicola as a primary cause of yield reduction in cotton in California. Phytopathology. 1966;56:1213–1216. [Google Scholar]
- 21.Conant G. Histological studies of resistance in tobacco to Thielaviopsis basicola. Am. J. Bot. 1927;14:457–480. doi: 10.1002/j.1537-2197.1927.tb04857.x. [DOI] [Google Scholar]
- 22.Hood M.E., Shew H.D. Pathogenesis of Thielaviopsis basicola on a susceptible and a resistant cultivar of burley tobacco. Phytopathology. 1996;86:38–44. doi: 10.1094/Phyto-86-38. [DOI] [Google Scholar]
- 23.Hood M.E., Shew H.D. Initial cellular interactions between Thielaviopsis basicola and tobacco root hairs. Phytopathology. 1997;87:228–235. doi: 10.1094/PHYTO.1997.87.3.228. [DOI] [PubMed] [Google Scholar]
- 24.Christou T. Penetration and host-parasite relationship of Thielaviopsis basicola in the bean plant. Phytopathology. 1962;52:194–198. [Google Scholar]
- 25.Mims C.W., Copes W.E., Richardson E.A. Ultrastructure of the penetration and infection of pansy roots by Thielaviopsis basicola. Phytopathology. 2000;90:843–850. doi: 10.1094/PHYTO.2000.90.8.843. [DOI] [PubMed] [Google Scholar]
- 26.Mosma S., Struck C. Studies on penetration, infection and colonization of lupin roots infected by Thielaviopsis basicola. Sci. Res. Rep. 2013;3:97–101. [Google Scholar]
- 27.Clayton E.E. The study of resistance to the black root rot disease of tobacco. Tob. Sci. 1969;13:30–37. [Google Scholar]
- 28.Haji H.M., Brammall R.A., Van Hooren D.L. Effect of the Nicotiana debneyi black root rot resistance gene on the yield and quality characteristics of flue-cured tobacco in Ontario. Can. J. Plant Sci. 2003;83:939–942. doi: 10.4141/P02-134. [DOI] [Google Scholar]
- 29.Coumans J.V.F., Polijak A., Raftery M.J., Backhouse D., Pereg-Gerk L. Analysis of cotton (Gossypium hirsutum) root proteomes during a compatible interaction with the black root rot fungus Thielaviopsis basicola. Proteomics. 2009;9:335–349. doi: 10.1002/pmic.200800251. [DOI] [PubMed] [Google Scholar]
- 30.Yang Z.E., Ge X.Y., Yang Z.R., Qin W.Q., Sun G.F., Wang Z., Li Z., Liu J., Wu J., Wang Y., et al. Extensive intraspecific gene order and gene structural variations in upland cotton cultivars. Nat. Commun. 2019;10:1–13. doi: 10.1038/s41467-019-10820-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brubaker C.L., Brown A.H.D., Stewart J.M., Kilby M.J., Grace J.P. Production of fertile hybrid germplasm with diploid Australian Gossypium species for cotton improvement. Euphytica. 1999;108:199–213. doi: 10.1023/A:1003641217653. [DOI] [Google Scholar]
- 32.Sacks E.J., Robinson A.F. Introgression of resistance to reniform nematode (Rotylenchulus reniformis) into upland cotton (Gossypium hirsutum) from Gossypium arboreum and a G. hirsutum/Gossypium aridum bridging line. Field Crop. Res. 2009;112:1–6. doi: 10.1016/j.fcr.2009.01.006. [DOI] [Google Scholar]
- 33.Wendel J.F., Cronn R.C. Polyploidy and the evolutionary history of cotton. Adv. Agron. 2003;78:139–186. [Google Scholar]
- 34.Robinson A.F., Bell A.A., Dighe N.D., Menz M.A., Nichols P.L., Stelly D.M. Introgression of resistance to nematode Rotylenchulus reniformis into upland cotton (Gossypium hirsutum) from Gossypium longicalyx. Crop Sci. 2007;47:1865–1877. doi: 10.2135/cropsci2006.12.0776. [DOI] [Google Scholar]
- 35.Zhu Q.H., Fan L., Liu Y., Xu H., Llewellyn D., Wilson I. miR482 regulation of NBS-LRR defense genes during fungal pathogen infection in cotton. PLoS ONE. 2013;8:e84390. doi: 10.1371/journal.pone.0084390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stelfox D., Herbut M. Growth of a Phytophthora Sp on Carrot Agar. Can. Plant Dis. Surv. 1979;59:61–62. [Google Scholar]
- 37.de Beer Z.W., Duong T.A., Barnes I., Wingfield B.D., Wingfield M.J. Redefining Ceratocystis and allied genera. Stud. Mycol. 2014;79:187–219. doi: 10.1016/j.simyco.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murashige T., Skoog F. A revised medium for rapid growth and bioassays with tobacco tissue culture. Physiol. Plant. 1962;15:473–497. doi: 10.1111/j.1399-3054.1962.tb08052.x. [DOI] [Google Scholar]
- 39.Ellis M.H., Stiller W.N., Phongkham T., Tate W.A., Gillespie V.J., Gapare W.J., Zhu Q.H., Llewellyn D.J., Wilson I.W. Molecular mapping of bunchy top disease resistance in Gossypium hirsutum L. Euphytica. 2016;210:135–142. doi: 10.1007/s10681-016-1713-3. [DOI] [Google Scholar]
- 40.Lopez-Lavalle L.A.B., Gillespie V.J., Tate W.A., Ellis M.H., Stiller W.N., Llewellyn D.L., Wilson I.W. Molecular mapping of a new source of Fusarium wilt resistance in tetraploid cotton (Gossypium hirsutum L.) Mol. Breed. 2012;30:1181–1191. doi: 10.1007/s11032-012-9705-z. [DOI] [Google Scholar]
- 41.Stam P. Construction of integrated genetic-linkage maps by means of a new computer package—Joinmap. Plant J. 1993;3:739–744. doi: 10.1111/j.1365-313X.1993.00739.x. [DOI] [Google Scholar]
- 42.Kosambi D. The estimation of map distance from recombination value. Ann. Hum. Genet. 1944;12:172–175. [Google Scholar]
- 43.Churchill G.A., Doerge R.W. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolger A.M., Lohse M., Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anders S., Pyl P.T., Huber W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:1–21. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian T., Liu Y., Yan H., You Q., Yi X., Du Z., Xu W., Su Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017;45:W122–W129. doi: 10.1093/nar/gkx382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw paired-end Illumina RNA sequencing reads generated in the current study are available from the CSIRO data portal https://data.csiro.au/dap/landingpage?pid=csiro:47747 accessed date 1 March 2021.