

Abstract
The N-methyl-d-aspartate receptor (NMDAR) is an ion channel that mediates the slow, Ca2+-permeable component of glutamatergic synaptic transmission in the central nervous system (CNS). NMDARs are known to play a significant role in basic neurological functions, and their dysfunction has been implicated in several CNS disorders. Herein, we report the discovery of second-generation GluN2C/D-selective NMDAR-positive allosteric modulators (PAMs) with a dihydropyrrolo[1,2-a]pyrazin-3(4H)-one core. The prototype, R-(+)-EU-1180–453, exhibits log unit improvements in the concentration needed to double receptor response, lipophilic efficiency, and aqueous solubility, and lowers cLogP by one log unit compared to the first-generation prototype CIQ. Additionally, R-(+)-EU-1180–453 was found to increase glutamate potency 2-fold, increase the response to maximally effective concentration of agonist 4-fold, and the racemate is brain-penetrant. These compounds are useful second-generation in vitro tools and a promising step toward in vivo tools for the study of positive modulation of GluN2C- and GluN2D-containing NMDA receptors.
Graphical Abstract
INTRODUCTION
The N-methyl-d-aspartate receptor (NMDAR) belongs to the family of mammalian ligand-gated excitatory ionotropic glutamate receptors (iGluRs) that includes 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid receptors (AMPARs) and (2S,3S,4S)-3-(carboxymethyl)-4-prop-1-en-2-ylpyrrolidine-2-carboxylic acid (kainate) receptors.1 NMDARs are known to play a role in learning,2,3 memory,4 brain development,5 and synaptic plasticity6,7 and have been implicated in numerous neurological conditions including schizophrenia,8,9 Alzheimer’s disease,10 Parkinson’s disease,11 Huntington’s chorea,12 epilepsy,13 neuropathic pain,14 ischemic brain injury,15–17 and depression.18,19 This has driven interest in the development of both positive and negative modulators of the NMDAR for potential therapeutic gain.20,21
Structurally, NMDARs are heterotetrameric assemblies of two GluN1 and two GluN2 protein subunits, which bind co-agonists glycine and glutamate, respectively. Each subunit contains four semiautonomous domains: an amino terminal domain (ATD), an agonist-binding domain (ABD), a transmembrane domain (TMD), and a carboxyl terminal domain (CTD).1 Upon co-agonist binding at the ABD, membrane depolarization occurs, which removes Mg2+ block22,23 from the pore. This allows Ca2+ and Na+ ions to flow into the cell, which contribute to postsynaptic signal transmission.24 The four isoforms of the GluN2 subunit (GluN2A-D) are expressed with different spatiotemporal patterns in the brain25–27 and endow the receptor with unique pharmacological properties including open probability,28–31 agonist potency,32 and deactivation time.33 For example, GluN2C- and GluN2D-containing NMDA receptors are expressed more prominently in the cerebellum, basal ganglia, thalamus, and cortical and hippocampal interneurons,25,26,34 compared to GluN2A- and GluN2B-containing receptors. Functionally, GluN2C/GluN2D-containing NMDA receptors show increased glutamate and glycine sensitivity32,35 and reduced open probability,29,30,36 Mg2+ sensitivity,25 Ca2+ permeability, and single-channel conductance compared to GluN2A/GluN2B-containing receptors.37
Because these subunit-specific expression patterns and GluN2-specific functional properties allow NMDARs to serve unique roles at different synapses, subunit-selective modulators provide an opportunity to target the aforementioned diseases through selective modification of specific circuits.38 However, FDA-approved drugs that target NMDARs nonselectively block the highly conserved NMDAR pore, irrespective of subunit composition. This results in the inhibition of all NMDAR subtypes, potentially leading to the neurological side effects that include altered cardiovascular activity, hallucinations, delusions, and impaired motor function.39
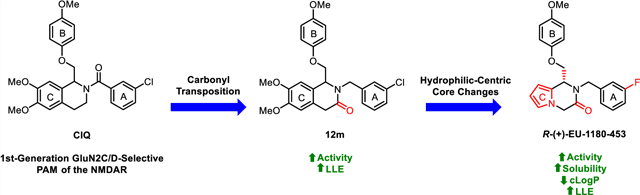
Subunit-selective NMDAR modulators have therefore become a focus of the research community, given their potential to reduce side effects by targeting one subunit in specific areas of the brain for therapeutic gain. Specifically, NMDAR-positive allosteric modulators (PAMs) could be useful in conditions associated with reduced NMDAR activity such as schizophrenia,40–42 autism spectrum disorders,43,44 anti-NMDAR encephalitis,45–47 and age-related memory loss.2,38,48,49 Further, there is evidence suggesting that positive therapeutic outcomes could be achieved with the development of GluN2C- and/or GluN2D-subtype-selective PAMs. One hypothesis is that the hypofunction of GluN2C-containing receptors plays a role in schizophrenic symptoms50 and, thus, GluN2C-selective PAMs could have therapeutically relevant actions. Additionally, postmortem examinations of schizophrenic patients with a specific NRG1 gene polymorphism revealed reduced GluN2C expression in the right cerebellum.51 GluN2D-containing receptors may also play a role in schizophrenia.51–54 For example, there is evidence to suggest that decreased inhibitory drive can lead to hyperactivity of principal cells and enhanced dopamine release.55 Expression of GluN2D in interneurons25,56,57 suggests that selective enhancement of GluN2D-containing receptor function could potentially rectify this hypothesized circuit imbalance. Finally, (3-chlorophenyl)(6,7-dimethoxy-1-((4-methoxyphenoxy)-methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (CIQ, 1), a GluN2C/D-selective PAM previously developed in our lab (Figure 1),58,59 has shown promising results in behavioral mouse models. This includes the ability to recover striatal synaptic plasticity deficit in a Parkinson’s model,60 facilitate the retention of fear and extinction learning,61 and prevent MK-801 (a noncompetitive antagonist of NMDARs)-induced prepulse inhibition deficit,62 suggesting the clinical potential of potent positive modulators of these subtypes.
Figure 1.
PAMs of the NMDAR.
There have been numerous other nonselective and subunit-selective NMDAR PAMs identified in addition to CIQ (Figure 1). For example, we have previously identified a GluN2B-preferring class of isopropoxy-substituted tetrahydroisoquinolines (TIQs) based on CIQ (prototype compound 2).63 Wang et al. published GNE-9278 (3), a nonselective benzenesulfonamide-based PAM that acts on each of the GluN1/GluN2 subtypes in the low micromolar (3–16 μM) range.64 Structural determinants suggest a possible binding site near the GluN1 pre-M1 helix, and it was shown to potentiate the receptor by increasing co-agonist affinity, increasing receptor response at saturating co-agonist concentrations, and slowing deactivation after glutamate removal. Other nonselective PAMs include the neurosteroid pregnenolone sulfate (PS, 4)65 and 24(S)-hydroxycholesterol (5).66 Genentech also published a GluN2A-selective series of thiazole derivatives that bind in a similar pocket to the N-(4-(2-benzoylhydrazine-1-carbonyl)-benzyl)-3-chloro-4-fluorobenzenesulfonamide (TCN) class of negative allosteric modulators (NAMs). These compounds potentiate via stabilization of the agonist-bound state.67,68 Significant optimization resulted in GNE-5729 (6), a PAM of GluN2A-containing receptors (EC50 = 37 nM) showing improved AMPAR selectivity and unbound clearance and an excellent secondary pharmacology profile.69 The only known series of compounds able to distinguish between GluN2C- and GluN2D-containing receptors are the pyrrolidinone-based (prototype PYD-106 (7)) GluN2C-selective PAMs.70,71 These compounds appear to increase mean open time and open probability via actions on the ATD and S1 domains.
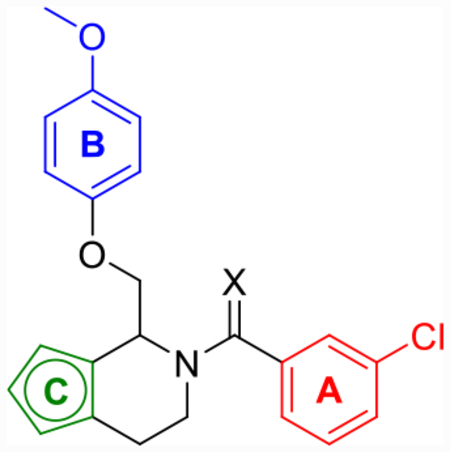
Although CIQ has made a significant impact as a first-in-class tool compound, it exhibits several liabilities limiting its use both in vitro and in vivo. These include modest potency, modest potentiation, low aqueous solubility, and high lipophilicity.58,59 Herein, we report the discovery of second-generation dihydropyrrolo[1,2-a]pyrazin-3(4H)-one-based GluN2C/D-selective PAMs that are the result of successive core changes from CIQ. First, we modified the TIQ scaffold to a 1,4-dihydroisoquinolin-3(2H)-one core (Figure 2, left), which resulted in improved concentrations needed to double receptor response (doubling concentration, 0.5 log unit improvement on average) and lipophilic ligand efficiency (LLE, defined here as LLE = −log10(doubling conc.) −cLogP, 0.4 log unit improvement on average). We have chosen to report the doubling concentration alongside the EC50 throughout this manuscript as it captures both the potency and potentiation contributions of a PAM’s biological activity in one directly comparable number. Since it considers both contributions, we believe it to be more useful than EC50 in describing the biological activity of these PAMs, as the EC50 describes only the potency without information on maximal achievable response. We have also chosen to define LLE using the doubling concentration (compared to the standard definition using EC50) due to its increased relevance. Finally, we have chosen 2-fold as the cutoff for several reasons: (1) it is currently unknown what percent potentiation of GluN2C/D-containing receptors will lead to pharmacologically relevant actions, (2) this percent potentiation was achieved by most compounds studied, (3) it is a convenient and memorable cutoff, and (4) to be consistent with previous reports.63 Building on this core modification resulted in a dihydropyrrolo[1,2-a]pyrazin-3(4H)-one series (Figure 2, right), with derivatives showing roughly log unit improvements in doubling concentration, aqueous solubility, and LLE, and a log unit decrease in cLogP compared to CIQ. The selectivity for GluN2C and GluN2D is maintained at subsaturating concentrations of co-agonists for both new cores. The enantiomers of promising derivatives were separated via chiral high-performance liquid chromatography (HPLC) and show stereo-dependent activity, with one enantiomer inactive against all examined NMDAR subtypes. The active enantiomers exhibit improved potency over the racemic mixture in each case. Finally, a crystal structure was solved for prototypical compound R-(+)-EU-1180–453 that allowed for assignment of the absolute configuration of the active enantiomers. Therefore, the improvements in activity and physicochemical properties observed in these two new structural modifications compared to the parent CIQ have created improved in vitro tool compounds for GluN2C- and GluN2D-containing NMDA receptors.
Figure 2.
Labeled 1,4-dihydroisoquinolin-3(2H)-one (left) and dihydropyrrolo[1,2-a]pyrazin-3(4H)-one (right) to distinguish between the A-, B-, and C-rings.
RESULTS AND DISCUSSION
Two-Electrode Voltage Clamp (TEVC) Recordings.
All compounds synthesized were evaluated via TEVC recordings of Xenopus laevis oocytes expressing four recombinant NMDAR subtypes: GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2C, and GluN1/GluN2D. Each compound was co-applied at 30 μM with saturating concentrations of glutamate (100 μM) and glycine (30 μM). When the mean potentiated response exceeded 130% of control, concentration–effect curves were generated by co-applying increasing concentrations of compound with saturating co-agonists. We determined the pEC50 value and maximal degree of potentiation by fitting the concentration–effect curves with eqs 1 and 2 (see Experimental Procedures). Each concentration–effect curve was generated with data from 6–19 oocytes from 2–3 frogs; all inactive compounds were tested in 4–10 oocytes from 1 to 2 frogs. Tables 1–7 summarize the mean pEC50 value with 95% confidence interval and average maximum potentiation (max) for each active compound at GluN1/GluN2C and GluN1/GluN2D receptors. The negative log of the doubling concentration (eqs 3 and 4; see Experimental Procedures), aqueous solubility (see the Supporting Information for Methods), and LLE (eq 5; see Experimental Procedures) are also reported when appropriate. Table 1 compares the doubling concentration of select compounds reported here to previously reported compounds. For compounds in which a pEC50 value was not determined (ND), the maximum potentiation is reported as the average potentiation at 30 μM drug as a percent of control. The average percent potentiation is defined as the mean ratio of current upon application of drug at the stated concentration to the current response in its absence. The doubling concentration is defined as the concentration of test compound at which the response to maximally effective concentration of co-applied glutamate and glycine is increased 2-fold over the response to glutamate and glycine alone. All compounds were also tested at GluN1/GluN2A and GluN1/GluN2B receptors at 10 or 30 μM and were typically found to have no effect (4–17 oocytes from 1 to 3 frogs). Only two compounds (12c–d) exhibited potentiation above 130% at GluN2A- or GluN2B-containing receptors and neither exceeded 2-fold potentiation (Supporting Information Table S1).
Table 1.
1,4-Dihydroisoquinolin-3(2H)-one Compounds and TIQ Counterparts
| # | R1 | R2 | R3 | X | Y | pEC50 [95% CI] (max) (%)a | p(doubling conc.)b | Δp(doubling conc.) | cLogP | LLE | ΔLLE | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GluN2C | GluN2D | GluN2C | GluN2D | GluN2C | GluN2D | GluN2C | GluN2D | GluN2C | GluN2D | |||||||
| CIQc | OMe | OMe | CI | O | - | 5.3 (230) | 5.3 (220) | 4.9 | 4.5 | 0.2 | 0.6 | 5.6 | −0.7 | −1.1 | 0.1 | 0.5 |
| 12m | - | O | 5.3 [5.1–5.4] (270) | 5.0 [4.9–5.1] (340) | 5.1 | 5.1 | 5.7 | −0.6 | −0.6 | |||||||
| 117c | OMe | H | CI | O | - | 5.9 (240) | 6.0 (220) | 5.5 | 5.3 | 0.1 | 0.4 | 5.9 | −0.4 | −0.6 | 0.1 | 0.4 |
| 12e | - | O | 5.7 [5.6–5.8] (270) | 5.6 [5.5–5.8] (330) | 5.6 | 5.7 | 5.9 | −0.3 | −0.2 | |||||||
| 2*c,d | OiPr | H | CI | O | - | 5.7 (240) | 5.5 (250) | 5.3 | 5.2 | 1.3 | 1.4 | 6.7 | −1.4 | −1.5 | 1.2 | 1.3 |
| 12a | - | O | 6.4 [6.3–6.5] (350) | 6.3 [6.1–6.6] (370) | 6.6 | 6.6 | 6.8 | −0.2 | −0.2 | |||||||
| 88c | OiBu | H | CI | O | - | 6.0 (450) | 5.9 (520) | 6.4 | 6.3 | −0.2 | 0.1 | 7.3 | −0.9 | −1.0 | −0.3 | 0 |
| 12d | - | O | 6.4 [6.3–6.4] (280) | 6.2 [6.1–6.3] (370) | 6.2 | 6.4 | 7.4 | −1.2 | −1.0 | |||||||
| 127c | OiPr | H | CF3 | O | - | 5.8 (380) | 5.6 (360) | 6.0 | 5.8 | 0.3 | 0.5 | 6.9 | −0.9 | −1.1 | 0.3 | 0.5 |
| 12b | - | O | 6.4 [6.2–6.6] (280) | 6.2 [6.2–6.3] (310) | 6.3 | 6.3 | 6.9 | −0.6 | −0.6 | |||||||
| 114c | OiPr | H | F | O | - | 5.2 (370) | 5.1 (430) | 5.4 | 5.5 | 0.6 | 0.3 | 6.1 | −0.7 | −0.6 | 0.5 | 0.2 |
| 12c | - | O | 6.0 [5.9–6.0] (320) | 5.7 [5.6–5.8] (330) | 6.0 | 5.8 | 6.2 | −0.2 | −0.4 | |||||||
Fitted pEC50 values are shown to two significant figures when potentiation at 30 μM exceeded 130% of control; values in brackets are the 95% confidence interval for the corresponding fitted pEC50 mean value; and values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. Data are between 8 and 19 oocytes from 2 to 3 frogs for each compound and receptor.
p(doubling conc.) is the negative log of the doubling concentration.
Previously published59,63 data for compounds CIQ, 117, 2*, 88, 127, and 114 were included for comparison. The numbering corresponds to the compound’s numbering in its respective publication.
Compound 2* is compound 2 from Santangelo et al.59
Table 7.
Stereodependence of Select Pyrrolopyrazine Corese
 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| pEC50 [95% CI] (max) (%)a | |||||||||
| # | X | Y | R | GluN2C | GluN2D | p(doubling conc.) (GluN2C/2D)b | sol. (μM)c | cLogP | LLE (GluN2C/2D) |
| CIQd (TIQ core) | Cl | 5.3 (230) | 5.3 (220) | 4.9/4.5 | 8 | 5.6 | −0.7/−1.1 | ||
| S-(−)-31a | O | CF3 | ND (84) | ND (88) | 57 | 5.2 | |||
| R-(+)-31a | O | CF3 | 6.1 [6.0–6.3] (280) | 6.1 [6.0–6.2] (330) | 6.0/6.2 | 57 | 5.2 | 0.8/1.0 | |
| S-(−)-25i | O | Cl | ND (98) | ND (99) | 36 | 4.9 | |||
| R-(+)-25i | O | Cl | 5.7 [5.7–5.8] (410) | 5.7 [5.7–5.8] (450) | 6.1/6.2 | 36 | 4.9 | 1.2/1.3 | |
| S-(−)-25j | O | CF3 | ND (99) | ND (98) | 58 | 5.1 | |||
| R-(+)-25j | O | CF3 | 5.9 [5.8–6.0] (460) | 5.9 [5.8–5.9] (540) | 6.3/6.4 | 58 | 5.1 | 1.2/1.3 | |
| S-(−)-EU-1180-453 | O | F | ND (100) | ND (88) | 74 | 4.5 | |||
| R-(+)-EU-1180-453 | O | F | 5.5 [5.4–5.6] (410) | 5.5 [5.4–5.6] (390) | 5.9/5.8 | 74 | 4.5 | 1.4/1.3 | |
Fitted pEC50 values are shown to have two significant figures when potentiation at 30 μM exceeded 130% of control; values in brackets are the 95% confidence interval for the corresponding log-fitted pEC50 value; and values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. For compounds in which a pEC50 value was not determined, maximum potentiation is reported as percent potentiation at 30 μM drug. Data are between 6 and 13 oocytes from 2 frogs for each compound and receptor.
p(doubling conc.) is the negative log of the doubling concentration.
Aqueous solubilities reported from racemic mixtures.
Previously published data for CIQ were included for comparison.
ND = not determined.
Design of NMDAR Potentiators.
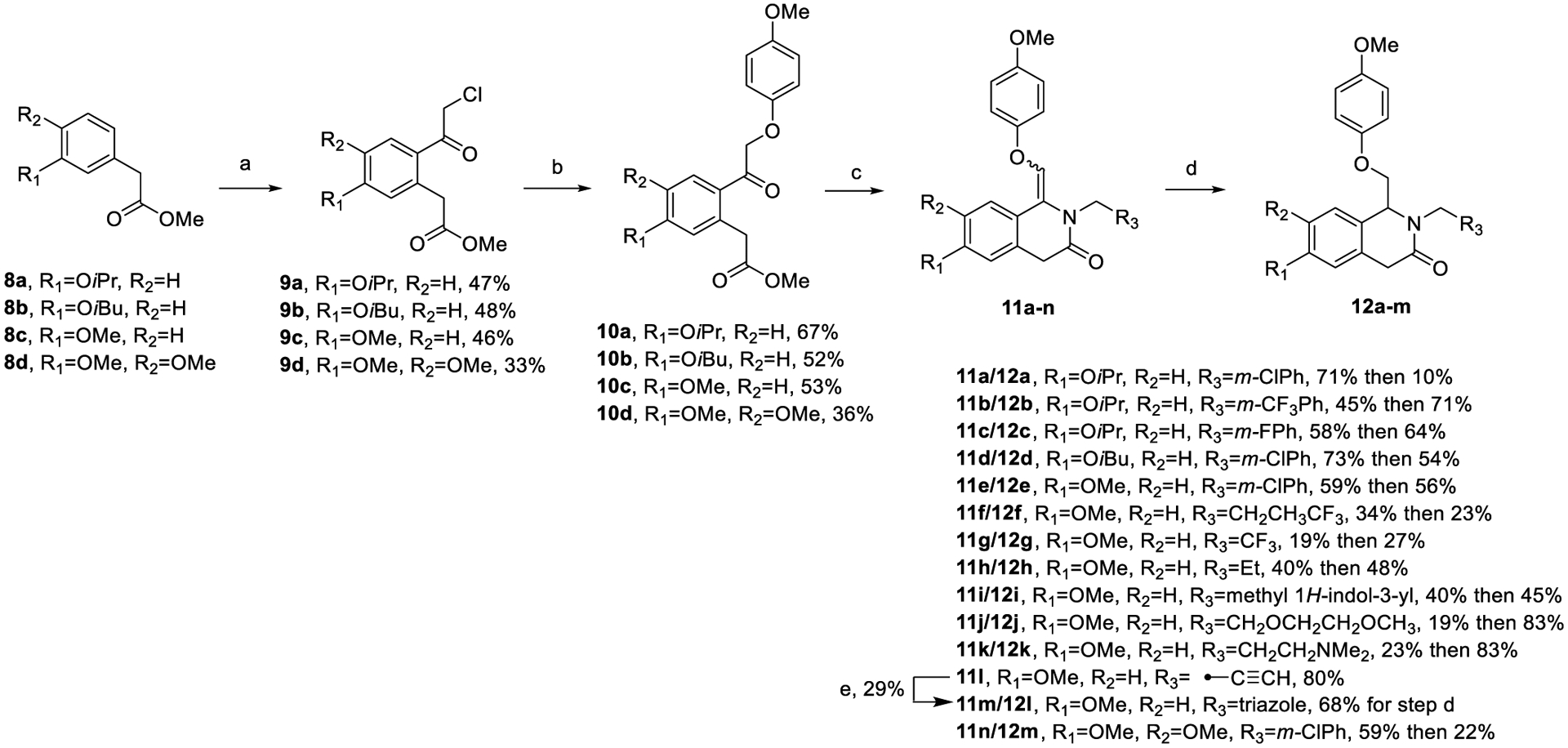
Previous computational studies on a CIQ analogue suggested that potency could be improved if the A-ring could be directed to adopt an s-trans conformation within the binding site, with s-cis/s-trans defined by the orientation of the amide carbonyl with respect to the stereocenter.63 To investigate this hypothesis, a novel 1,4-dihydroisoquinolin-3(2H)-one scaffold was designed and synthesized (Scheme 1). We predicted that translocation of the amide carbonyl of CIQ into the TIQ ring would provide enough steric influence to favor the s-trans conformation and therefore increase the fraction of drug in the predicted more active pose (Figure 3). This phenomenon can be visualized via a crystal structure of prototype R-(+)-EU-1180–453. The crystallographic data and methods can be viewed in Table S2 and the Crystal Structure Methods portion of the Supporting Information, respectively.
Scheme 1.
Synthesis of Dihydroisoquinolinonesa
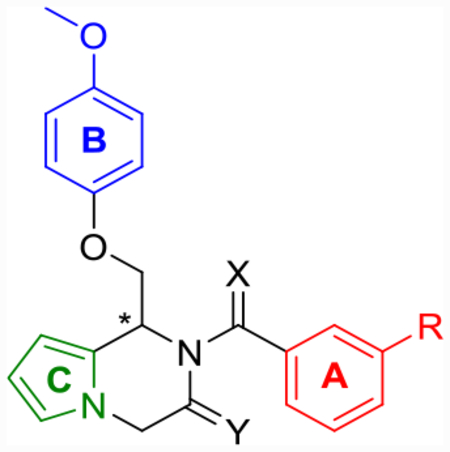
aReagents and conditions: (a) chloroacetyl chloride, SnCl4, 1,2-dichloroethane (DCE), reflux, 90 min; (b) 4-methoxyphenol, KI, K2CO3, reflux, 4 h; (c) substituted primary amines, Ti(OiPr)4, NaBH(OAc)3, DCE, 120 °C, mw, 20 min; (d) (i) PtO2, H2 (balloon), EtOH, rt, 10 h, (ii) Pd/C, H2 (50 psi), EtOH, rt, 24 h; (e) NaN3, CH2O (37% in H2O), AcOH, cat. sodium ascorbate, cat. CuSO4, tetrahydrofuran (THF), rt, 24 h.
Figure 3.
Model of the steric encumbrance upon translocation of the carbonyl in the 1,4-dihydroisoquinolin-3(2H)-one series. The s-cis conformation was hypothesized to create steric clash between the carbonyl and A-ring, creating preference for the more active s-trans conformation (left). This is visualized on this scaffold through a crystal structure of R-(+)-EU-1180–453 (right).
Chemistry.
The synthesis of 1,4-dihydroisoquinolin-3(2H)-one analogues is shown in Scheme 1. Methyl esters 8a and 8b were prepared according to modified literature procedures,63 while all other similar intermediates were commercially available. α-Chloro ketones 9a–d were prepared via Friedel–Crafts acylation of the appropriate methyl esters with chloroacetyl chloride and tin(IV) chloride. The resulting α-chloroamide was then reacted with 4-methoxyphenol and potassium iodide to give ketones 10a–d, which were then cyclized in a microwave reactor using the appropriate amine, titanium(IV) isopropoxide, and sodium triacetoxyborohydride to afford the penultimate eneamide intermediates 11a–n as inconsequential mixtures of their E and Z isomers. Each eneamide could then either be reduced via hydrogenation at 50 psi using palladium on carbon or in cases with aryl chlorides, hydrogenated with platinum oxide at 1 atm to give the final 1,4-dihydroisoquinolin-3(2H)-one compounds (12a–m).
The procedure for the synthesis of B-ring derivatives is outlined in Scheme 2. The synthesis began with commercially available 3,4-dimethoxyphenethylamine (13), which was acylated with chloroacetyl chloride to give α-chloroamide 14. This was then reacted with sodium hydride and 4-methoxyphenol to give α-hydroxy amides 15a–d. Intermediates 15a and 15d were then cyclized to their respective imine intermediate using modified Bischler–Napieralski conditions and subsequently reduced via sodium borohydride or hydrogenation to yield their respective TIQ amines (16a in the case of 15a). These TIQ amines were then acylated using the appropriate benzoyl chloride, deprotected to their respective primary alcohols 17a–b, and further functionalized using coupling or SNAr conditions to produce the final compounds (18a–c). Alternatively, in the case where the final B-ring substitution was installed in step (b), such as with 15b–c, the respective secondary amine (16b–c) obtained via the Bischler–Napieralski cyclization was directly acylated with 3-(trifluoromethyl)benzoyl chloride to give the final compounds (18d–e).
Scheme 2.
Synthesis of B-Ring Derivativesa
aReagents and conditions: (a) chloroacetyl chloride, Et3N, dichloromethane (DCM), 0 °C, 30 min; (b) R1OH, NaH or Cs2CO3; (c) POCl3, ACN, reflux then NaBH4, MeOH, 0 °C, 10 min; (d) substituted benzoyl chloride, Et3N, DCM, rt, 1 h; (e) Pd/C, H2 (50 psi), MeOH, o/n; (f) methane sulfonic acid, DCM, rt, 6 h; (g) heteroaryl fluoride, NaH, THF, reflux, o/n, (ii) heteroaryl iodide, 1,10-Phen, CuI, Cs2CO3, PhMe, 80 °C, o/n.
Heterocyclic derivatives of the C-ring were synthesized utilizing a modified synthesis of CIQ59 described in Scheme 3. Ethylamine starting materials 21a–e were synthesized from the corresponding aldehyde via a Henry-type nitroalkenylation and subsequent reduction via in situ generated alane (Scheme 3, step a and step b). 2-Methoxy-5-formylthiophene was synthesized according to the literature.72 The remaining synthesis proceeded as described with only slight modifications.59
Scheme 3.
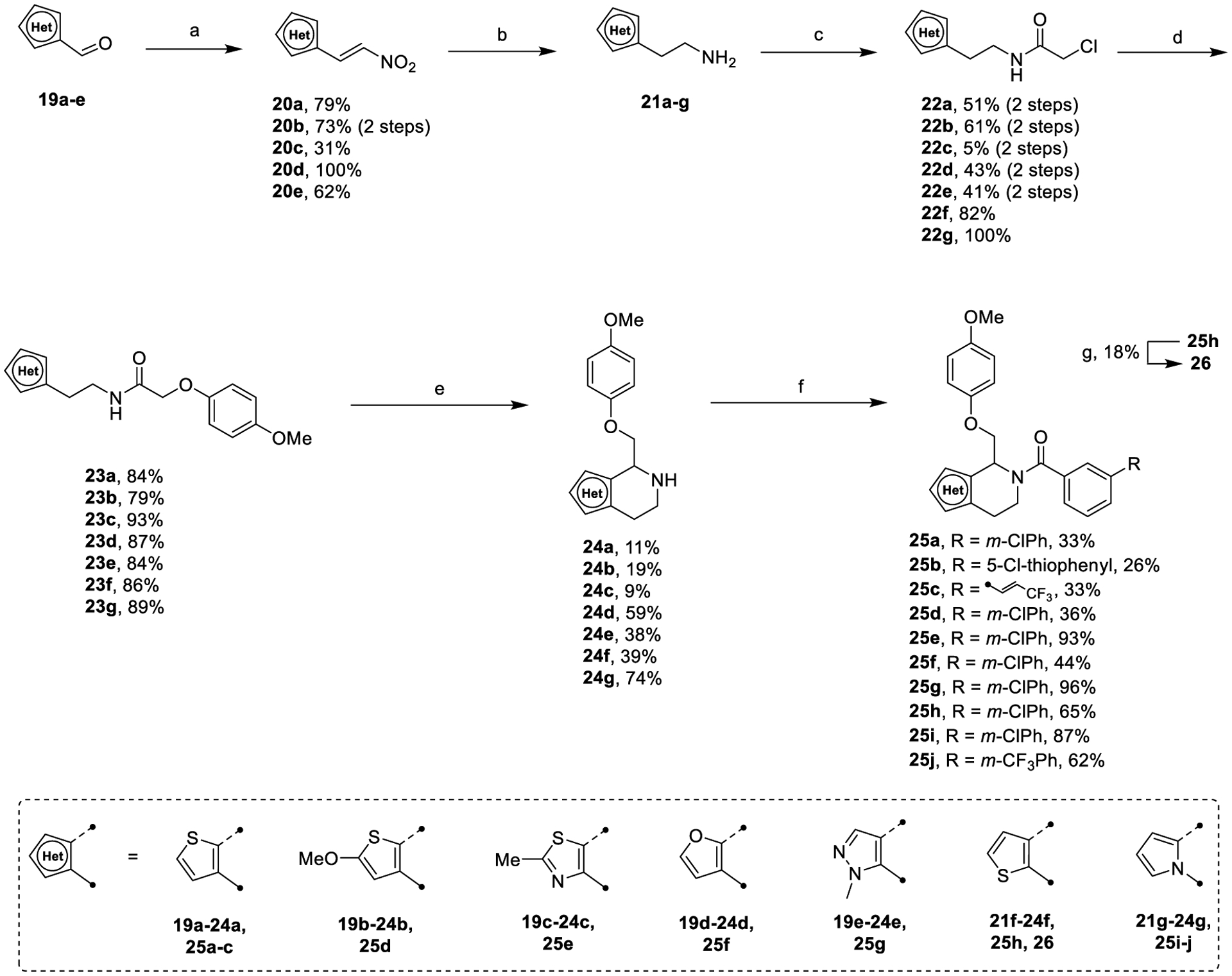
Synthesis of Heterocyclic C-Ringsa
aReagents and conditions: (a) cat. butylamine, cat. AcOH, 4 Å MS, MeNO2, reflux, 30 min; (b) LiAlH4/H2SO4, THF, 0 °C to reflux, 5 min; (c) chloroacetyl chloride, Et3N, DCM, 0 °C, 30 min; (d) 4-methoxyphenol, Cs2CO3, o/n, 79–93%; (e) POCl3, ACN, reflux, o/n then NaBH4, MeOH, 0 °C, 10 min; (f) (i) acid chloride, Et3N, DCM, rt, 1 h, (ii) carboxylic acid, EDCI, 4-dimethylaminopyridine (DMAP), rt, o/n; (g) Lawesson’s reagent, PhMe, 150 °C, mw, 2 h.
Finally, pyrrolopyrazinone derivatives were synthesized as described in Scheme 4. This synthesis is a modified version of the synthesis described in Scheme 1, where the pyrrole starting material was made through the Clauson–Kaas reaction starting with glycine methyl ester hydrochloride.
Scheme 4.
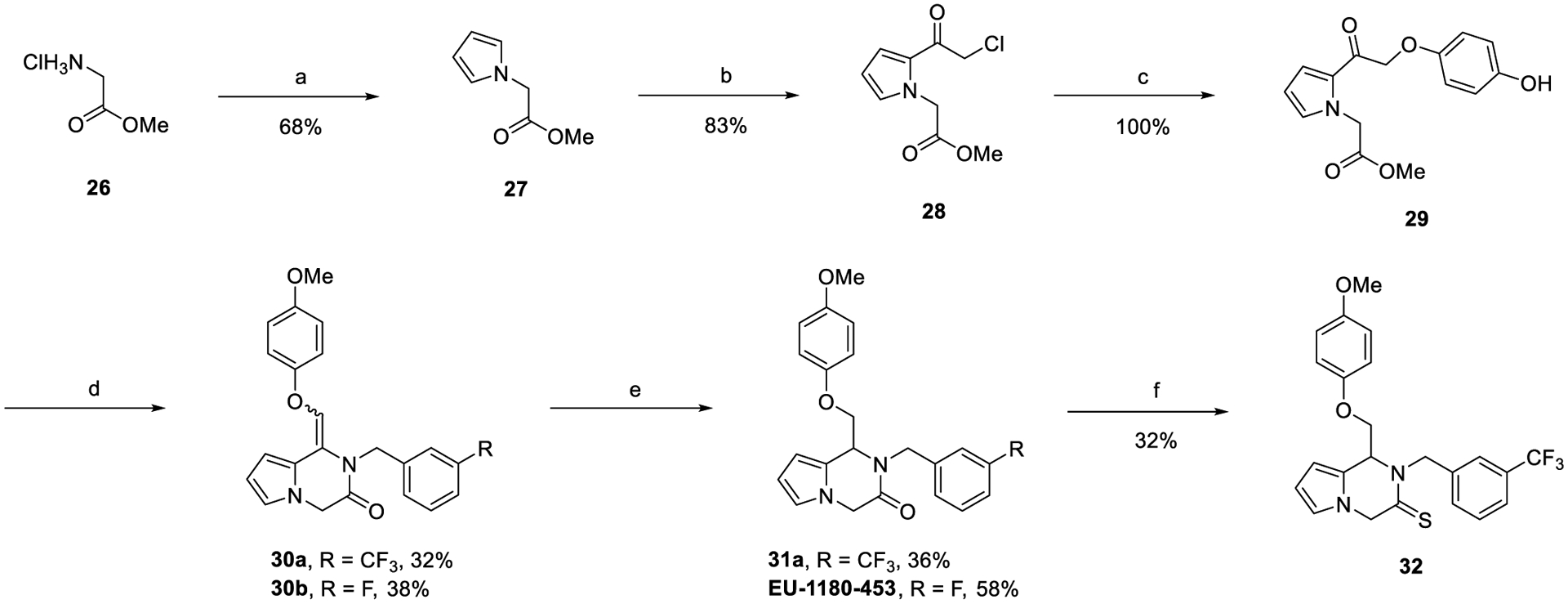
Synthesis of Pyrrolopyrazinonesa
aReagents and conditions: (a) 2,5-dimethoxytetrahydrofuran, NaOAc, 2:1 v/v AcOH:water, reflux, 4 h; (b) chloroacetyl chloride, DBN, PhMe, 115 °C, 4 h; (c) 4-methoxyphenol, KI, K2CO3, acetone, rt, o/n; (d) substituted primary amines, Ti(OiPr)4, sodium triacetoxyborohydride (STAB), DCE, 120 °C, mw, 2 h; (e) Pd/C, H2 (1 atm), EtOAc, rt, o/n; (f) Lawesson’s reagent, PhMe, 150 °C, mw, 2 h.
1,4-Dihydroisoquinolin-3(2H)-one Compounds Show Improved Doubling Concentrations Over TIQ Counterparts.
As predicted, compound 12m showed improved potentiation (270–340%) of GluN2C/D-containing NMDAR responses to saturating co-agonists compared to CIQ (220–230%) (Table 1). Compound 12m also exhibited similar potency (pEC50 = 5.0–5.3 at GluN2C/D) and subunit selectivity (no activity at GluN2A/B) to CIQ, resulting in improved doubling concentrations (p(doubling conc.) = 5.1). Selectivity for GluN2C and GluN2D was also maintained upon application of 12m and subsaturating levels of co-agonists glutamate and glycine (Table S3). Based on this promising result, we designed and synthesized a targeted library of dihydroisoquinolinones to demonstrate that this trend is maintained irrespective of substitution.
We calculated the doubling concentration using eq 3 for all synthesized 1,4-dihydroisoquinolin-3(2H)-ones with corresponding TIQs59,63 to demonstrate the trend of improved activity for 1,4-dihydroisoquinolin-3(2H)-one analogues (Table 1). The doubling concentration at GluN2C or GluN2D was improved in the 1,4-dihydroisoquinolin-3(2H)-one scaffold in 92% (11/12) of cases. The only case in which it decreased (compare 12d and 88 at GluN2C) resulted in only a 0.2 log unit drop. The average doubling concentration improvement was 0.5 log units, which led to a 0.4 log unit improvement in LLE due to the similar cLogP values of the isomeric scaffolds. These data demonstrate the ability of the 1,4-dihydroisoquinolin-3(2H)-one core to improve activity over the TIQ core.
The modification to a dihydroisoquinolinone core improved doubling concentrations substantially over the previous TIQ series; however, there were still opportunities to improve the physicochemical properties of the series (e.g., its high lipophilicity). Prototypical compounds (e.g., CIQ, 12m, etc.) exhibited cLogP values near 6 (and >8 for the most potent analogues) and aqueous solubilities in the single-digit micromolar range (<10 μM for CIQ). The highly lipophilic nature of CIQ is also likely the cause of its low free fraction that limits its concentration and receptor occupancy in vivo,63 making optimization of this property vital in medicinal chemistry efforts. Highly lipophilic compounds also typically exhibit liabilities including high clearance,73,74 low membrane permeability,75 and low oral bioavailability.76 There have been scattered previous attempts at decreasing the lipophilicity of the series,59 but in nearly each case, this led to a substantial or complete loss in potency. With this goal in mind, we developed a series of hydrophilic-focused changes to the three phenyl rings of CIQ, which resulted in the discovery of a brain-penetrant pyrrolopyrazinone-based scaffold with dramatically improved lipophilic efficiency for GluN2C- and GluN2D-containing NMDARs. Prototypical compounds also display an approximate order of magnitude improvement in aqueous solubility, a relatively clean off-target profile, and improved ease of analysis due to elimination of rotamers observed with CIQ. From this point forward, we assume that the structure–activity relationship (SAR) from the TIQ series is transferrable to the dihydroisoquinolinone series. A subset of analogues is provided to demonstrate the ability to extrapolate between scaffolds (Table S4).
Evaluation of A-Ring Replacements.
First, we explored replacements of the A-ring on the dihydroisoquinolinone scaffold aimed at lowering cLogP using the synthesis described in Scheme 1. Of the alternative substituents examined, only alkyl trifluoromethyl derivative 12f (p(doubling conc.) = 5.0–5.1) (Table 2) was tolerated. Despite lowering cLogP values by nearly two log units, 12f still displayed doubling concentrations in the single-digit micromolar range. Although this result is below desired potency ranges, this substitution may prove to be useful for future promising analogues to fine-tune absorption, distribution, metabolism, and excretion (ADME) properties due to the relatively favorable change in LLE (LLE = 0.9–1.0). Other A-ring substitutions such as aliphatic chains (12g, pEC50 < 4.5 or 12h, max = 94–97%), heterocycles (12l, max = 98–110% or 12i, max = 85%), and heterolinear derivatives (12j, max = 98–100% or 12k, max = 96–110%) were not tolerated. These results provide evidence that hydrophobic effects dominate receptor–ligand interactions in this portion of the molecule. Therefore, based on previous optimization,59,63 meta-Cl- or meta-CF3-substituted benzyl derivatives were synthesized from this point forward almost exclusively since they provided the most promising combination of activity and lipophilic efficiency.
Table 2.
Hydrophilic A-Ring Substitutionsd
| # | R1 | R2 | X | Y | pEC50 [95% CI] (max) (%)a | p(doubling conc.) (GluN2C/2D)b | cLogP | LLE (GluN2C/2D) | |
|---|---|---|---|---|---|---|---|---|---|
| GluN2C | GluN2D | ||||||||
| CIQc | OMe | 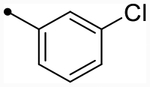 |
O | - | 5.3 (230) | 5.3 (220) | 4.9/4.5 | 5.6 | −0.7/−1.1 |
| 117c | H |  |
- | O | 5.9 (240) | 6.0 (220) | 5.5/5.3 | 5.9 | −0.4/−0.6 |
| 12f | H | - | O | 5.1 [4.9–5.3] (290) | 4.7 [4.5–4.9] (450) | 5.0/5.1 | 4.1 | 0.9/1.0 | |
| 12g | H | 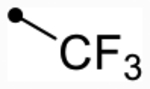 |
- | O | <4.5 (330) | <4.5 (160) | <4.5/ID | 4.1 | - |
| 12h | H | 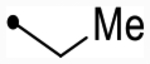 |
- | O | ND (97) | ND (94) | - | 4.0 | - |
| 12l | H | 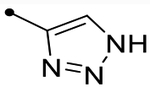 |
- | O | ND (110) | ND (98) | - | 2.9 | - |
| 12i | H | 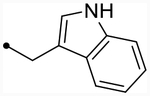 |
- | O | ND (85) | ND (85) | - | 5.1 | - |
| 12j | H | - | O | ND (98) | ND (100) | - | 3.1 | - | |
| 12k | H | 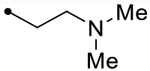 |
- | O | ND (110) | ND (96) | - | 3.6 | - |
Fitted pEC50 values are shown to have two significant figures when potentiation at 30 μM exceeded 130% of control; values in brackets are the 95% confidence interval for the corresponding fitted pEC50 mean value; and values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. For compounds in which a pEC50 value was not determined, maximum potentiation is reported as percent potentiation as a percent of control at 30 μM drug. Data are between 6 and 11 oocytes from 2 frogs for each compound and receptor.
p(doubling conc.) is the negative log of the doubling concentration.
Previously published data for CIQ and 117 were included for comparison. Single methoxy substitution on the C-ring maintains similar or better activity to dimethoxy substitution.59
ND = not determined.
Evaluation of Hydrophilic B-Ring Replacements.
Next, we examined hydrophilic replacements of the B-ring via the divergent synthesis described in Scheme 2. Upon TEVC evaluation, none of the derivatives synthesized were tolerated at any GluN2 subtype. Nitrogen heterocycles such as the 2-pyridinyl (18b), 3-pyridinyl (18e), and 2-pyrimidinyl (18c) derivatives all showed responses when co-applied with agonist that were 88–97% of control for GluN2C- and GluN2D-containing receptors. Incorporation of saturation (18d), bioisosteric replacement with thiophene (18a), and removal of the B-ring completely (17b) were also not tolerated. Para-methoxyphenyl substitution was therefore maintained for subsequent analogues based on previous optimization (Table 3).59,63
Table 3.
Hydrophilic B-Ring Substitutionsb
 |
Fitted pEC50 values are shown to have two significant figures when potentiation at 30 μM exceeded 130% of control; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. For compounds in which a pEC50 value was not determined, maximum potentiation is reported as percent potentiation as a percent of control at 30 μM drug. Data are from 4 oocytes from 1 frog.
Previously published data for compound 198 were included for comparison.59
Effect of Bioisosteric Replacement of the C-Ring.
Although derivatization of the three rings of CIQ has led to useful tool compounds,59,63 major changes to the C-ring and TIQ core itself have not been thoroughly explored. We viewed this as an underdeveloped area of the SAR in which opportunities for decreasing lipophilicity and improving affinity for the target protein could emerge, assuming appropriate choice of a hydrophilic core.
Due to thiophene’s known use as a classical phenyl isostere, tetrahydrothienopyridine 25a was synthesized (Scheme 3) and its activity against each of the GluN2 subunits (Table 4) was examined. Compound 25a showed improved maximum potentiation (280–350%) while maintaining similar potency (pEC50 = 5.4) and selectivity (no activity at GluN2A/B) to CIQ. Due to the improved potentiation, 25a showed improved doubling concentrations and LLE (p(doubling conc.) = 5.3–5.6 and LLE = −0.3 to 0.0) in comparison to CIQ (p(doubling conc.) = 4.5–4.9 and LLE = −0.7 to −1.1). A regioisomer of 25a, compound 25h, also showed improved LLE over CIQ (ΔLLE = 0.6–0.9) due to improved potentiation (max = 330–340%). Using these results as a starting point, several tetrahydrothienopyridine-based derivatives were subsequently explored (Table 4 and Scheme 3). First, bioisosteric 2-chlorothiophene replacement of the A-ring provided compound 25b, which was similarly tolerated (LLE = −0.1 to 0.2) to 25a (LLE = −0.3 to 0.0). The trifluorocrotonic acid derivative 25c suffered a loss in potency (pEC50 = 4.8–4.9) compared to CIQ, but resulted in a large improvement in LLE (LLE = 1.2–1.3) due to its notable decrease in cLogP (cLogP = 3.6). Similar to 12f, this may be an attractive substitution for fine-tuning the ADME properties of more potent future derivatives but rendered this compound outside the range of useful potency. Finally, methoxy substitution on the thiophene (25d) was examined due to previous promising SAR trends,59,63 but this compound showed no advantages in terms of LLE over CIQ. However, each of these derivatives exemplified that changes to the TIQ core were tolerated, and we hypothesized that other more hydrophilic substitution may follow a similar trend.
Table 4.
Optimization of A- and C-Ring Functionality for Thiophene Corec
| # | R1 | C-ring | R2 | pEC50 [95% CI] (max) (%)a | p(doubling conc.) (GluN2C/2D)b | sol. (μM) | cLogP | LLE (GluN2C/2D) | |
|---|---|---|---|---|---|---|---|---|---|
| GluN2C | GluN2D | ||||||||
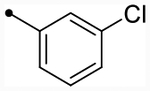
| CIQc (TIQ core) |  |
5.3 (230) | 5.3 (220) | 4.9/4.5 | 8 | 5.6 | −0.7/−1.1 | ||
| 25a | H |  |
 |
5.4 [5.3–5.5] (280) | 5.4 [5.3–5.5] (350) | 5.3/S.6 | 46 | 5.6 | −0.3/0.0 |
| 25h | H |  |
 |
5.4 [5.3–5.4] (330) | 5.3 [5.2–5.3] (340) | 5.5/5.4 | - | 5.6 | −0.1/−0.2 |
| 25b | H | 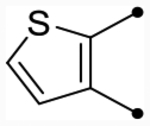 |
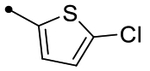 |
5.3 [5.3–5.3] (290) | 5.3 [5.2–5.4] (400) | 5.3/5.6 | - | 5.4 | −0.1/0.2 |
| 25c | H | 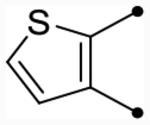 |
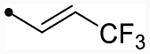 |
4.9 [4.8–5.0] (290) | 4.8 [4.6–4.9] (330) | 4.8/4.9 | - | 3.6 | 1.2/1.3 |
| 25d | OMe | 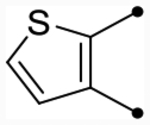 |
 |
5.3 [5.2–5.4] (220) | 5.2 [5.1–5.2] (220) | 4.6/4.5 | - | 5.7 | −1.1/−1.2 |
Fitted pEC50 values are shown to have two significant figures when potentiation at 30 μM exceeded 130% of control; values in brackets are the 95% confidence interval for the corresponding log-fitted pEC50 value; and values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. Data are between 10 and 12 oocytes from 2 frogs for each compound and receptor.
p(doubling conc.) is the negative log of the doubling concentration.
Previously published data for CIQ were included for comparison.
SAR of Additional TIQ Core Changes.
Several hydrophilic heterocycles were designed and synthesized to test this hypothesis (Table 5 and Scheme 3). The furan derivative, 25f, was the first compound in the series to show a simultaneous improvement in doubling concentration (p(doubling conc.) = 5.4–5.5) and decrease in cLogP (cLogP = 5.1) compared to CIQ. This led to a dramatic improvement in LLE (LLE = 0.3–0.4). Even more impressive was pyrrolopyrazine 25i, which exhibited excellent doubling concentrations (p(doubling conc.) = 5.8–5.9) while also lowering cLogP by 0.7 (cLogP = 4.9) compared to CIQ. This resulted in an approximate two log unit improvement in LLE (LLE = 1.0–1.1). It also maintained the aqueous solubility gains observed with 25a, displaying a 4.5-fold improvement over CIQ at 36 μM. However, other more hydrophilic derivatives such as 2-methylthiazole 25e (max = 100%) and N-methylpyrazole 25g (pEC50 < 4.5) were both not tolerated. Unsubstituted phenyl derivative 36 (see the Supporting Information for synthesis) was also only weakly active (p(doubling conc.) = 5.0, LLE = −1.0). Replacement of the A-ring amide linker with a thioamide linker has previously been shown to improve potency > 10-fold in certain cases.63 However, this modification was not tolerated in analogue 26 (max = 94–97%), which contains the tetrahydrothienopyridine scaffold. Nonetheless, compound 25i was an exciting discovery in identifying compounds with improved lipophilic efficiencies and other derivatives with this same pyrrolopyrazine core were subsequently developed.
Table 5.
Optimization of Heterocyclic C-Ring Derivativesd
| # | C-ring | X | pEC50 [95% CI] (max) (%)a | p(doubling conc.) (GluN2C/2D)b | sol. (μM) | cLogP | LLE (GluN2C/2D) | |
|---|---|---|---|---|---|---|---|---|
| GluN2C | GluN2D | |||||||
| CIQc | 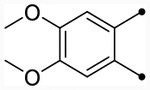 |
O | 5.3 (230) | 5.3 (220) | 4.9/4.5 | 8 | 5.6 | −0.7/−1.1 |
| 25f | 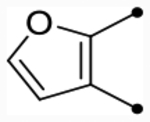 |
O | 5.2 [5.1–5.3] (340) | 5.1 [5.0–5.3] (470) | 5.4/5.5 | - | 5.1 | 0.3/0.4 |
| 25i | 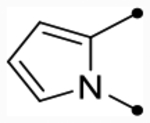 |
O | 5.6 [5.5–5.7] (370) | 5.5 [5.4–5.7] (370) | 5.9/5.8 | 36 | 4.9 | 1.0/1.1 |
| 25e | 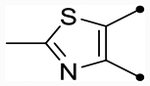 |
O | ND (100) | ND (100) | - | - | 4.8 | - |
| 25g | 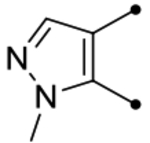 |
O | < 4.5 (210) | <4.5(220) | - | - | 4.2 | - |
| 36 |  |
O | 5.2 [5.1–5.3] (260) | 5.1 [4.9–5.2] (300) | 5.0/5.0 | - | 6.0 | −1.0/−1.0 |
| 26 |  |
S | ND (97) | ND (94) | - | - | 6.0 | - |
Fitted pEC50 values are shown to have two significant figures when potentiation at 30 μM exceeded 130% of control; values in brackets are the 95% confidence interval for the corresponding log-fitted pEC50 value; and values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. For compounds in which a pEC50 value was not determined, maximum potentiation is reported as percent potentiation at 30 μM drug. Data are between 7 and 15 oocytes from 2 frogs for each compound and receptor for which an EC50 value was determined and between 4 and 9 oocytes from 1 to 2 frogs for all other analogues.
p(doubling conc.) is the negative log of the doubling concentration.
Previously published data for CIQ were included for comparison.
ND = not determined.
Pyrrolopyrazine- and Pyrrolopyrazinone-Based Analogues.
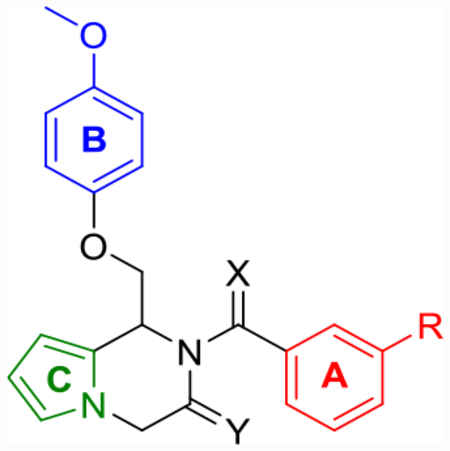
Based on previously established SAR within the series, a targeted sample of derivatives was designed and synthesized, keeping the pyrrole core consistent throughout (Table 6). First, based on previously described SAR,59 the chlorine of compound 25i was substituted for a trifluoromethyl substituent to give compound 25j (Scheme 3). This compound improved the doubling concentrations (p(doubling conc.) = 5.6–6.1) into the nanomolar range while also displaying the highest aqueous solubility (58 μM) of any derivative in the series tested thus far. To examine whether the LLE boosts seen individually with the pyrrolopyrazine core and dihydroisoquinolinone core would synergize, a hybridized pyrrolopyrazinone core was designed and synthesized as described in Scheme 4. The resulting pyrrolopyrazinone (31a) displayed similar doubling concentrations (p(doubling conc.) = 5.9–6.1) and aqueous solubility (57 μM) to 25j. Fluorine was also well tolerated (EU-1180–453) in place of the trifluoromethyl group. Although a slight potency drop was observed (pEC50 = 5.5), it maintained the improvements in LLE (LLE = 1.2) and doubling concentrations (p(doubling conc.) = 5.7) seen with 25i, while improving aqueous solubility nearly an order of magnitude (74 μM) compared to CIQ. EU-1180–453 is a free-flowing, white solid that is stable as a powder or a dimethyl sulfoxide (DMSO) stock for at least 30 days (longest time tested) at room temperature under regular atmosphere. Finally, the amide of 31a was converted to the thioamide to give 32, but this modification resulted in a complete loss of activity on the pyrrolopyrazinone core, similar to that observed for the tetrahydrothienopyridine core above.
Table 6.
Optimization of Potency and Aqueous Solubility for Pyrrolopyrazine- and Pyrrolopyrazinone-Based Derivativesd
 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| pEC50 [95% CI] (max) (%)a | |||||||||
| # | X | Y | R | GluN2C | GluN2D | p(doubling conc.) (GluN2C/2D)b | sol. (μM) | cLogP | LLE (GluN2C/2D) |
| CIQc (TIQcore) | Cl | 5.3 (230) | 5.3 (220) | 4.9/4.5 | 8 | 5.6 | −0.7/−1.1 | ||
| 25j | O | CF3 | 5.8 [5.7–5.9] (380) | 5.7 [5.5–5.9] (290) | 6.1/5.6 | 58 | 5.1 | 1.0/0.5 | |
| 31a | O | CF3 | 5.9 [5.7–6.0] (300) | 5.8 [5.6–6.0] (400) | 5.9/6.1 | 57 | 5.2 | 0.7/0.9 | |
| EU-1180–453 | O | F | 5.5 [5.4–5.6] (350) | 5.5 [5.4–5.6] (350) | 5.7/5.7 | 74 | 4.5 | 1.2/1.2 | |
| 32 | S | CF3 | ND (120) | ND (110) | 5.4 | ||||
Fitted pEC50 values are shown to have two significant figures when potentiation at 30 μM exceeded 130% of control; values in brackets are the 95% confidence interval for the corresponding log-fitted pEC50 value; and values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. For compounds in which a pEC50 value was not determined, maximum potentiation is reported as percent potentiation at 30 μM drug. Data are from between 7 and 15 oocytes from 2 frogs for each compound and receptor.
p(doubling conc.) is the negative log of the doubling concentration.
Previously published data for CIQ were included for comparison.
ND = not determined.
Enantiomers.
Four compounds with promising profiles (25i, 25j, 31a, and EU-1180–453) were selected for separation via chiral HPLC to examine the behavior of the individual enantiomers. Upon separation and isolation, X-ray crystallography studies on (+)-EU-1180–453 determined its absolute configuration to be the R-(+)-enantiomer and allowed for absolute stereochemical assignment of the enantiomers.
Consistent with CIQ and its derivatives, the R-(+)-enantiomers were active, while the S-(−)-enantiomers were not. We selected compound R-(+)-EU-1180–453 for further study because of its attractive doubling concentrations (p(doubling conc.) = 5.8–5.9) and improved aqueous solubility (74 μM), exhibiting a >50:1 aqueous solubility to doubling concentration ratio compared to <1:1 for CIQ. In addition, R-(+)-EU-1180–453 had virtually no effect on GluN1/GluN2A and GluN1/GluN2B, or a variety of other receptors (Tables S5 and S6). Together, these properties make this a more attractive and useful compound than CIQ (Table 7).
To better understand the scope of actions for EU-1180–453, we evaluated whether either enantiomer could alter glutamate potency, in addition to their potentiation of the response to maximally effective concentrations of glutamate plus glycine. We measured the ratio of a response to a subsaturating concentration of glutamate in vehicle or EU-1180–453 enantiomer to that of a maximally effective concentration of glutamate at GluN1/GluN2D receptors. We did not detect any meaningful effect of S-(−)-EU-1180–453 on the ratio of the current response at any receptor combination, suggesting that this stereoisomer has no effect on glutamate potency. This is consistent with the lack of a detectable effect on the maximal response to co-agonists (Table 8). A similar result was found for 30 μM R-(+)-EU-1180–453 for GluN2A and GluN2B, which had no effect on the ratio of submaximal to maximal response (Table 8) or response to maximally effective agonist for GluN1/GluN2A (108 ± 2% of control, n = 6) and GluN1/GluN2B (109 ± 2% of control, n = 7). By contrast, R-(+)-EU- 1180–453 had strong effects on the ratio of response of submaximal to maximal concentrations of glutamate for GluN1/GluN2C and GluN1/GluN2D (Table 8), suggesting that this enantiomer may shift the glutamate potency in addition to its ability to potentiate the response to maximally effective concentrations of agonist. We subsequently recorded full concentration–effect curves for glutamate activation (in saturating 30 μM glycine) at both GluN1/GluN2C and GluN1/GluN2D in the absence and presence of R-(+)-EU-1180–453. We obtained fitted pEC50 values (with non-overlapping 95% confidence intervals) of 6.1 [6.0–6.2] and 6.5 [6.4–6.6] for GluN1/GluN2C (Hill slopes 1.2–1.4, n = 15, 13), and 6.3 [6.2–6.3] and 6.6 [6.5–6.7] for GluN1/GluN2D (Hill slopes 1.1–1.5, n = 16–22) in the absence and presence of R-(+)-EU-1180–453, respectively. These data suggest that even larger potentiation will be observed for subsaturating concentrations of glutamate at GluN2C and GluN2D, as will occur at extrasynaptic receptors.77
Table 8.
Single Glutamate Concentration Screen for Changes in Glutamate Potency R-(+)-EU-1180–453a
| I30 μM/Icontrol (mean ± SEM, %) [low glutamate concentration/low glycine concentration, μM] | ||||
|---|---|---|---|---|
| # | GluN2A | GluN2B | GluN2C N = 13 | GluN2D N = 22 |
| vehicle | 40 ± 4 [3, 30] | 84 ± 1 [3, 30] | 54 ± 1 [0.5, 30] | 66 ± 2 [0.5, 30] |
| 30 μM S-(−)-EU-1180-453 | 45 ± 4 [3, 30] n = 11 | 85 ± 9 [3, 30] n = 6 | 57 ± 2 [0.5, 30] n = 13 | 72 ± 2* [0.5, 30] n = 7 |
| Vehicle | 46 ± 5 [3, 30–100] | 79 ± 1 [3, 30–100] | 19 ± 3 [0.3, 10] | 29 ± 2 [0.3, 10] |
| 30 μMR-(+)-EU-1180-453 | 53 ± 2 [3, 30–100] n = 28 | 77 ± 1 [3, 30–100] n = 22 | 48 ± 4* [0.3, 10] n = 13 | 55 ± 6* [0.3, 10] n = 16 |
Data mean ± standard error for the relative current response at 0.5–3 μM glutamate compared to maximally active (30–100 μM) glutamate. 30 μM glycine was present in all solutions.
p < 0.05, paired t-test between drug and vehicle.
We also found that R-(+)-EU-1180–453 only potentiates activated NMDARs that have bound both co-agonists and does not remove the requirement of either co-agonist. We observed that 30 μM R-(+)-EU-1180–453 had virtually no effect on GluN1/GluN2D receptors in 10 μM glycine plus 50 μM racemic 2-amino-5-phosphonopentanoic acid (DL-APV), a selective competitive antagonist of the NMDAR glutamate binding site. This produced 2.5% of the response to a maximally effective concentration of glutamate plus glycine (n = 8 oocytes), which we interpret to reflect an enhanced glutamate potency that increases the small response to contaminant glutamate. Similarly, 30 μM R-(+)-EU-1180–453 in 10 μM glutamate plus 10 μM 7-chlorokynurenic acid (7-CKA), a selective competitive antagonist of the NMDAR glycine binding site, produced 0.4% of the response to maximally effective concentration of glutamate and glycine (n = 8 oocytes).
Off-Target and Pharmacokinetic Profile.
Racemic compounds 25i, 31a, and EU-1180–453 were further examined for their behavior against common off-target receptors. First, their specificity against other ion channels in the brain were examined via two-electrode voltage clamp recordings of Xenopus oocytes expressing AMPA, kainate, nicotinic acetylcholine, serotonin, GABA, glycine, and ATP receptors.59 Current responses were tested with saturating concentrations of agonist in both the absence of drug and presence of 10 μM 25i, 31a, or EU-1180–453 (ca. 5–10 times the EC50). Only one receptor–ligand combination (α4β2-nACh/31a) exhibited >50% inhibition, and none exhibited >75% inhibition (Table S5).
Racemic compounds 25i, 31a, and EU-1180–453 were also submitted to the National Institute of Mental Health Psychoactive Drug Screening Program (PDSP, https://pdsp.unc.edu/ims/investigator/web/) to examine the activity of both the pyrrolopyrazine and pyrrolopyrazinone cores at additional off-target receptors (Table S6). The primary binding screen examined inhibition of ligand binding at 10 μM test compound, and if the drug exhibited >50% inhibition, a secondary binding screen was run to determine Ki. Compounds 25i, 31a, and EU-1180–453 showed >50% inhibition at an average of only 3 of the 45 off-target receptors examined, and all Ki values were in the micromolar range, suggesting reasonable selectivity for the GluN2C- and GluN2D-containing NMDA receptors.
Racemic78 EU-1180–453 was also dosed in mice to determine its pharmacokinetic behavior. EU-1180–453 was dosed i.p. in C57Bl/6 mice (n = 15, 3 per time point) at 10 mg/kg in vehicle consisting of 50% PEG400 in water, and samples were evaluated via LC/MS-MS at 0.25, 0.5, 1, 2, and 4 h (see Experimental Procedures). Concentrations in brain (962 ± 210 ng/mL, 2.5 ± 0.4 μM) and plasma (695 ± 111 ng/mL, 1.83 ± 0.29 μM) peaked at 0.25 h and steadily declined, resulting in excellent brain exposure (brain:plasma_AUCLAST = 1.4 ± 0.5) and an acceptable plasma half-life (t1/2 = 1.07 ± 0.04 h). CIQ has previously been reported as brain-penetrant but suffers from high plasma protein binding leading to low free fraction.63 We therefore evaluated the plasma protein binding (PPB) of EU-1180–453 using equilibrium dialysis and found it to be 98.7% bound to rat plasma protein at 1 μM of drug. This is a greater than 10-fold decrease compared to CIQ, for which we observed >99.9% bound. A comparison of pharmacokinetic data between EU-1180–453 and CIQ can be seen in Table 9.61
Table 9.
Plasma Pharmacokinetics Comparison Between EU-1180–453 and CIQ
| # | dose (mg/kg)a | Cmax (ng/mL) | Cmax (μM) | t1/2 (h) | PPB, fubb | [brain]: [plasma]c | cLogP | LLE (GluN2C/2D) |
|---|---|---|---|---|---|---|---|---|
| CIQ | 20 | 2090 ± 20 | 2.16 ± 0.03 | 1.35 ± 0.12 | <0.1 | 6.7 ± 2.2 | 5.6 | −0.7/−1.1 |
| EU-1180-453 | 10 | 695 ± 111 | 1.83 ± 0.29 | 1.07 ± 0.04 | 1.3 | 1.4 ± 0.5 | 4.5 | 1.2/1.2 |
Compounds were dosed i.p. in either 1:4:5 dimethylacetamide:PEG400:(5% dextrose in water) (CIQ, n = 3/group) or 1:1 PEG400:water (EU-1180–453, n = 3/group).
Rat plasma protein binding reported as percent unbound (fub ) at 1 μM.
Brain and plasma concentrations in ng/g and ng/mL, respectively, at Cmax.
CONCLUSIONS
Although CIQ is a useful in vitro tool compound for the study of GluN2C- and GluN2D-containing receptors, its in vivo and in vitro use is limited due to modest potency and potentiation, low aqueous solubility, and high lipophilicity. We have developed second-generation GluN2C- and GluN2D-selective PAMs that address each of these concerns. For example, racemic EU-1180–453 is a more potent potentiator (EC50 ~3 μM) that shows log unit improvements in lipophilic efficiency (ΔLLE = 1.9–2.3), doubling concentration (Δp(doubling conc.) = 0.8–1.2), aqueous solubility (8–74 μM), and a log unit decrease in cLogP (ΔcLogP = 1.1) compared to CIQ. It is brain-penetrant, exhibits a reasonable half-life, and shows minimal off-target activity. The active enantiomer, R-(+)-EU-1180–453, exhibits ca. 1–2 μM doubling concentrations due to 4-fold potentiation and results in a doubling concentration to aqueous solubility ratio of >50:1. Limitations at this time center around potency, as there is a question of whether the given combination of in vitro potency, free fraction, and Cmax,brain can drive measurable pharmacological activity in vivo. These in vivo experiments are currently ongoing in our lab, and the results will be reported in future publications. Overall, this work has resulted in second-generation in vitro tools to study positive modulation of GluN2C- and GluN2D-containing NMDARs and is an important step toward future in vivo work.
EXPERIMENTAL PROCEDURES
Biology.
Unfertilized Xenopus laevis oocytes were purchased from Ecocyte (Austin, TX). The oocytes were injected with mRNA to express recombinant rat GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2C, and GluN1/GluN2D, and two-electrode voltage clamp recordings were performed. Drs. S. Heinemann (Salk Institute), S. Nakanishi (Kyoto University), and P. Seeburg (University of Heidelberg) provided the cDNAs for rat GluN1–1a (GenBank accession numbers U11418 and U08261, referred to as GluN1 henceforth), GluN2A (D13211), GluN2B (U11419), GluN2C (M91563), and GluN2D (D13213). GluN2C and GluN2D were altered according to the literature.25 Isolation of oocytes, synthesis of cRNA, and injections of cRNA were each done according to the literature.79 Oocytes were placed in a perfusion chamber and continuously washed during TEVC recordings with a solution consisting of the following (in mM): 90 NaCl, 0.5 BaCl, 0.005 ethylenediaminetetraacetic acid (EDTA), 1.0 KCl, and 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) at pH 7.4 and 23 °C. Glass electrodes were pulled from thin-walled glass capillaries (tip resistance, 0.5–2.5 MΩ) and filled with 0.3–3.0 M KCl, while the oocyte membrane potential was held constant at −40 mV via an OC-725C amplifier (Warner Instruments Co.). Each compound was brought up in 20 mM DMSO and diluted with recording solution containing 30 μM glycine and 100 μM glutamate to the target concentration. To prevent the current increase typically seen during experiments with oocytes expressing GluN1/GluN2A receptors, some oocytes were either injected with 20 nL of 100 mM K-BAPTA (potassium 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid) or pretreated with 50 μM BAPTA-AM (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetraacetoxymethyl ester) for 10 min. Compounds that potentiated GluN2A- and GluN2B-containing receptors were not studied further.
Every test compound was recorded at 5–7 concentrations in 4–19 oocytes from 1 to 3 different frogs at GluN2C- and GluN2D-containing NMDA receptors and in 4 to 17 oocytes from 1 to 4 frogs at GluN2A- and GluN2B-containing receptors. The potentiation of certain test compounds at a concentration of 10 μM was averaged and reported as I10μM/Icontrol (mean ± SEM, %), where I is the current. For test compounds with potentiation that exceeded 125% at 30 μM, an average EC50 value (the half-maximal effective concentration of potentiator) was determined by fitting the following equation
| (1) |
to individual concentration–response curves normalized to the current in the absence of the potentiator (100%), where N is the Hill slope and max is the maximal response predicted for saturating concentration of potentiator. This is converted to the negative log of the value through the following
| (2) |
The concentration that gave a 2-fold increase in the current response was determined by rearranging eq 1 to yield
| (3) |
where N is the Hill slope. This is also converted to the negative log of the value through the following
| (4) |
The lipophilic ligand efficiency (LLE) equation used in this paper is a modified version of the metric that was recently reviewed.80 The negative log of the potency (pEC50) is replaced by the negative log of the doubling concentration to normalize for positive modulation, while Log D is replaced by cLogP to give
| (5) |
IACUC-approved protocols and animal welfare regulations outlined in the “Guide for the Care and Use of Laboratory Animals” were both followed during pharmacokinetic evaluation of compound EU-1180–453. In vivo analysis was performed by Pharmaron (Irvine, CA). Briefly, a group (n = 15) of fed, male C57BL/6 mice approximately 6–8 weeks of age were injected IP with 10 mg/kg (10 mL/kg, 1 mg/mL) of drug using 50% PEG400 in water as a vehicle. Samples were collected from the blood and brain at 0.25, 0.5, 1, 2, and 4 h after administration (3 mice per time point) following CO2 anesthesia. Collection from the brain was performed as follows: the mouse is terminally anesthetized via increasing concentration of CO2 and as much blood is removed as possible via cardiac puncture. The cardiac puncture is done by opening the chest cavity to expose the heart, cutting an incision in the right auricle using surgical scissors and finally injecting a saline solution (~10 mL) slowly into the left ventricle via syringe. The mouse is placed head down at a 45° angle to facilitate blood removal. After perfusion, the skull is opened and the brain is removed. The brain is washed with saline, dried with surgical gauze, placed in tared tubes, and stored at −75 °C before analysis. For plasma sample preparation at 0.25–2 h time points, 15 μL of blank solution, 30 μL of plasma sample, and 150 μL of acetonitrile were added sequentially for protein precipitation. After centrifugation, 20 μL of the supernatant and 80 μL of CB were combined, and thus the final compound concentration in plasma (ng/mL) was corrected by multiplying by 5. For plasma sample preparation at the 4 h time point, 15 μL of blank solution, 30 μL of plasma sample, and 150 μL of acetonitrile were added sequentially for protein precipitation. For all brain sample preparation, 15 μL of blank solution, 30 μL of brain samples, and 150 μL of acetonitrile were added sequentially for protein precipitation. Brain samples were prepared by adding brain (g) to deionized water (mL) in a 1:4 ratio for homogenization. The mixtures were then vortexed for 30 s and subsequently centrifuged (~4000 rpm) for 15 min. The supernatant was diluted 3-fold with water, and a 2 μL aliquot of the diluted supernatant was injected into a Shimadzu LC-30A LC-MS/MS system with Phenomenex 2.6 μ PFP 100A column (30 mm × 2.1 mm) using verapamil as an internal standard. A gradient from 95% water (0.1% formic acid) to 95% ACN (0.1% formic acid) was run over 2 min at a flow rate of 0.6 mL/min. Brain and blood samples were collected at 15, 30, 60, 120, and 240 min from three C57Bl/6 mice at each time point.
The maximum aqueous solubility for compounds CIQ, 25a, 25i, 25j, 31a, and EU-1180–453 was determined using a BMG Labtech Nephelostar nephelometer (Offenburg, Germany) according to manufacturer’s instructions. Compound powder (1–3 mg) was taken up in DMSO (Sigma-Aldrich) to a 20 mM stock solution and serially diluted (~10 μL) to 15.0, 10.0, 5.00, 2.50, 1.25, and 0.625 μM stock solutions. These, along with a DMSO blank, were distributed in triplicate to black, clear-bottom 96-well plates in 3 μL increments and subsequently diluted 100-fold using the aqueous oocyte recording solution (pH 7.4) described above. DMSO content was ≤1.0% (v/v). Each solution was shaken gently for 30 min before submission to the nephelometer.
Receptor binding profiles, Ki determinations, and hERG activity of compounds 25i, 31a, and EU-1180–453 were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract #HHSN-271-2013-00017-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda, MD. Radioligand binding was measured in the presence of 10 μM test compound. A Ki value was determined for each receptor at which test compound showed >50% inhibition. For experimental details, refer to the PDSP website https://pdsp.unc.edu/ims/investigator/web/.
Compounds 25i, 31a, and EU-1180–453 were also tested at 10 μM for actions at AMPA (GluA1–4), kainate (GluK2), GluN3 (GluN3A-B), nicotinic acetylcholine (α1β2γδ, α4β2, α7), serotonin (5-HT3A), GABAA (α1β2γ2S), GABAC (ρ1), glycine (α1), and purinergic (P2X human) receptors expressed in Xenopus oocytes. cRNA encoding the receptor subunits was synthesized from linear template cDNA using the mMessage mMachine kit (Ambion). Oocytes were injected with 1–5 ng cRNA in a 50 nL volume and incubated in Barth’s solution at 15 °C for 2–5 days prior to recording. α1β2γδ nAChR subunits were injected at a 1:1:1:1 ratio, while α4β2 and α7 nAChR subunits were injected at a 1:1 ratio. The cDNAs encoding GABAC, glycine, and serotonin subunits were provided by Dr. D. Weiss (University of Texas Health Science Center at San Antonio). cDNAs encoding nicotinic acetylcholine receptor subunits were provided by Drs. R. Papke (University of Florida) and S. Heinemann (Salk Institute). cDNAs encoding the purinergic receptors were provided by Dr. R. Hume (University of Michigan). GABAA receptors were activated by 100 μM GABA, and GABAC receptors were activated by 100 μM GABA. Acetylcholine was used at the indicated concentrations (in μM) to activate the nicotinic acetylcholine receptors: α1β2γδ (1), α4β2 (10), α7(300). 100 μM glycine was used to activate the glycine receptor, 100 μM serotonin was used to activate 5-HT3A receptor, and 9 μM ATP was used to activate the purinergic receptors. Responses in the presence of test compound were expressed as a percent of control.
Plasma protein binding was determined by equilibrium dialysis using the Rapid Equilibrium Dialysis RED device (Pierce, Catalog #89810), according to manufacturer’s protocol. Compounds were prepared as 100 μM DMSO stocks and spiked into 1 mL of rat plasma (Bioreclamations) to make a final concentration of 1 μM. Plasma (300 μL) was dispensed into wells separated by an 8 KDa permeable cellulose membrane from wells containing 100 mM potassium phosphate, pH 7.4 (500 μL). The compound was tested in triplicate. The RED device was sealed, and the system was allowed to reach equilibrium for 6 h in a 37 °C incubator with gentle agitation at 100 rpm. After incubation, plasma samples were prepared by transferring 10 μL from plasma wells to 90 μL of fresh 100 mM potassium phosphate, pH 7.4, and buffer samples were prepared by transferring 90 μL from buffer wells to 10 μL of naïve plasma. In addition, a reference sample without equilibration was prepared in triplicate by mixing 10 μL of plasma containing 1 μM compound with 90 μL of buffer to determine compound recovery from the assay. Two volumes of 1:1 acetonitrile:methanol spiked with the internal standard phenytoin (0.2 μg/mL) were added to reference and samples. Precipitation of plasma protein binding was allowed for 15 min before the reference and samples were centrifuge clarified. Supernatant (10 μL) was used for LC/MS/MS analyses. Compounds were quantified on an API4000 MS/MS System (Applied Biosystems, Concord, Ontario, Canada) interfaced with an Agilent 1100 Series HPLC as previously described.81
Chemistry.
General Experimental.
All starting materials were purchased from commercial sources and used directly without further purification. Purification by flash column chromatography was done using a Teledyne ISCO Combiflash Companion instrument using Teledyne Redisep normal phase columns. 1H and 13C NMR spectra were recorded on a Mercury-300, VNMR-400, INOVA-400, INOVA-500, or INOVA-600 NMR spectrometer. Chemical shifts were reported in ppm and referenced to the residual deuterated solvent. Reactions were monitored by thin-layer chromatography on pre-coated aluminum plates (silica gel 60 F254, 0.25 mm) or liquid chromatography–mass spectrometry (LCMS) on an Agilent Technologies 1200 series instrument. High-resolution mass spectra were recorded on a VG 70-S Nier Johnson or JEOL instrument by the Emory University Mass Spectroscopy Center. Purity was established via LCMS (Varian) in at least two solvent systems (MeOH:water/ACN:water or MeOH:water/MeOH:water) unless otherwise noted. All compounds described were >95% pure by LCMS besides one compound (12k) that was >85% pure. Optical rotations were established using a PerkinElmer 314 instrument.
General Procedure for α-Chloro Ketones (Procedure I).
Methyl ester (1 equiv) was dissolved in DCE (1.0 M), and 1 M tin(IV) chloride in DCM (5 equiv) was added dropwise for 10 min at room temperature. 2-Chloroacetyl chloride (4 equiv) was then added at room temperature, and the resulting mixture was heated to reflux for 90 min. Upon completion, the reaction was cooled to room temperature and quenched carefully with 1 M HCl. The aqueous layer was extracted with DCM, washed with saturated NaHCO3 (2×) and brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for Biaryl Ketones (Procedure II).
To a suspension of potassium iodide (1.2 equiv) and potassium carbonate (2 equiv) in acetone (0.17 M) were added α-chloro ketone (1 equiv) and phenol (1.2 equiv). The resulting mixture was then heated open to atmosphere at reflux for 4 h. Upon completion, the reaction was cooled to room temperature and solvent was removed in vacuo. The resulting residue was then dissolved in EtOAc and washed with water. The organic layer was then extracted with EtOAc, dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for Eneamides (Procedure III).
An appropriate microwave vial was charged with ketone (1 equiv), primary amine (1.2 equiv), tetraisopropoxytitanium (3 equiv), and DCE (0.2 M). The solution was stirred for 10 min before sodium triacetoxyborohydride (3 equiv) was added, and the mixture was irradiated at 120 °C for 30 min. Upon completion, the reaction was cooled to room temperature, diluted with DCM, and washed with 1 M HCl. The organic layer was extracted with DCM, washed with NaHCO3, washed with brine (3×), dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound as a mixture of E/Z isomers, the mass was confirmed via LCMS, and carried forward without further characterization.
General Procedure for Final 1,4-Dihydroisoquinolin-3(2H)-one Compounds (Procedure IV-A).
A 10 mL microwave vial was charged with eneamide (1 equiv), platinum(IV) oxide (10 mol %), and ethanol. The vial was then purged with hydrogen, evacuated (3×) and reacted at room temperature under hydrogen for 6–24 h, and monitored via LCMS to ensure no dehalogenation occurred. Upon first sight of dehalogenation via LCMS, the reaction was diluted with MeOH, filtered through a plug of celite washing with MeOH, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for Final 1,4-Dihydroisoquinolin-3(2H)-one Compounds (Procedure IV-B).
Eneamide (1 equiv) was dissolved in ethanol, 10% Pd/C (0.15 equiv) was added, and the mixture was hydrogenated at 50 psi overnight. The reaction mixture was filtered through a pad of celite, washed with methanol, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for α-Chloro Amides (Procedure V).
Primary amine (1 equiv) was dissolved in dry DCM, and triethylamine (2 equiv) was added. The solution was brought to 0 °C, 2-chloroacetyl chloride (1.2 equiv) was added, and the mixture was reacted at room temperature for 1 h, when deemed complete by LCMS. The reaction was then quenched with 1 M HCl, extracted into DCM, washed with brine (3×), dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for Alkylation of Alkyl Halides Using Cesium Carbonate (Procedure VI).
Phenol (1.2 equiv) was dissolved in acetonitrile and cesium carbonate (4 equiv) was added. This mixture was stirred at room temperature for 2 h before α-chloroamide (1 equiv) in acetonitrile was added and allowed to react at room temperature overnight. The reaction was then quenched with saturated ammonium chloride, and the product extracted into EtOAc, washed with brine (3×), dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for Alkylation of Alkyl Halide Using Sodium Hydride (Procedure VII).
60% Sodium hydride dispersion in mineral oil (1.4–3 equiv) was added to a solution of alcohol (1.2 equiv) in dry THF or DMF (ca. 0.4–0.8 M) at 0 °C under argon. After 30 min, alkyl halide in dry THF (~0.4–0.8 M) was added dropwise, and the mixture was stirred at room temperature overnight. Upon reaction completion, excess sodium hydride was quenched with water, poured over ice, and 1 M HCl was added. The organic layer was extracted with ethyl acetate (3×), and the organic layers were combined, washed with brine (1×), dried over Na2SO4, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for Bischler–Napieralski Cyclization (Procedure VIII).
Acetamide (1 equiv) was dissolved in dry acetonitrile (~0.05 M), and POCl3 (3 equiv) was added. This was reacted at reflux overnight when the reaction was deemed complete via thin-layer chromatography (TLC). The reaction was cooled to room temperature and concentrated in vacuo to afford the crude compound. The crude imine was dissolved in dry MeOH and cooled to 0 °C before sodium borohydride (3 equiv) was added in ~100 mg portions. The mixture was stirred at room temperature for 10 min. The reaction was then concentrated and subsequently partitioned between EtOAc and H2O. The product was then extracted into EtOAc, washed with brine (3×), dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for Tetrahydroisoquinolines (Procedure IX).
Secondary amine (1 equiv) was dissolved in dry DCM, triethylamine (2 equiv) was added, and the solution was brought to 0 °C before acid chloride (1.2 equiv) was added dropwise. The mixture was stirred at room temperature for 1 h. The reaction was then quenched with 1 M HCl, and the organic layer was extracted into DCM, washed with brine (3×), dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for SNAr Reactions (Procedure X).
Primary alcohol (1 equiv) was dissolved in dry THF or DMF (~0.2 M), and 60% sodium hydride dispersion in mineral oil (2–3 equiv) was added. This mixture was stirred for 30 min at room temperature before heteroaryl halide (1.5 equiv) was added. The reaction was stirred at 70 °C overnight and then cooled to room temperature, quenched carefully with water, and diluted with both EtOAc and water. The aqueous layer was extracted with EtOAc, the organic layers were combined, washed with brine (3×), dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for Nitro Alkenes (Procedure XI).
Aldehyde (1 equiv) was dissolved in dry nitromethane (~1 M), and to this solution butylamine (0.12 equiv), acetic acid (0.2 equiv), and 4 Å molecular sieves (~10 wt %) were added. The solution was brought to reflux for 30 min before the mixture was concentrated in vacuo, and the crude material was purified via flash column chromatography to afford the title compound.
General Procedure for Ethylamines (Procedure XII).
Sulfuric acid (2.23 equiv) was added dropwise to a solution of lithium aluminum hydride (4.46 equiv) in THF (~0.15 M) at 0 °C and stirred for 20 min. Nitroalkene (1 equiv) dissolved in dry THF (~1.5 M) was then added dropwise at 0 °C and stirred for 10 min. The mixture was then heated to reflux for 5 min, cooled to 0 °C, and quenched carefully with iPrOH (6 equiv) and NaOH (9 equiv). The resulting suspension was then filtered and concentrated in vacuo to afford the crude title compound that was carried forward without further purification.
General Procedure for Carboxylic Acid Coupling to Final Compounds (Procedure XIII).
Carboxylic acid (1.1 equiv) was dissolved in dry DCM and cooled to 0 °C. EDCI (1.2 equiv) and DMAP (1.2 equiv) were then added and stirred for 2 h before the free amine (1 equiv) was added, and the mixture was stirred at room temperature overnight. The reaction was then quenched with deionized water, and the organic layer was extracted into DCM, washed with brine (3×), dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for Thioamides (Procedure XIV).
Amide (1 equiv) in a microwave vial was dissolved in toluene and Lawesson’s reagent (1.5 equiv) was added. This mixture was allowed to react in microwave at 150 °C for 20 min. The reaction was then diluted with DCM; washed with saturated NaHCO3, water, and brine; dried over MgSO4; and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for α-Chloro Ketones Using DBN (Procedure XV).
Methyl ester (1 equiv) was dissolved in PhMe (0.5 M) and DBN (0.15 equiv) was added followed by chloroacetyl chloride (1.2 equiv). The mixture was heated to 115 °C and reacted for 4 h. The reaction was then diluted with DCM, and the organic layer was washed with 1 M HCL followed by 1 M NaOH. The organic layer was then dried over MgSO4 and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for Biaryl Ketones (Procedure XVI).
To a suspension of potassium iodide (1.2 equiv) and potassium carbonate (2 equiv) in acetone (0.3 M) were added α-chloro ketone (1 equiv) and phenol (1.2 equiv). The resulting mixture was then stirred open to atmosphere at room temperature overnight. Upon completion, the solvent was removed in vacuo and the resulting residue was dissolved in EtOAc and washed with water. The aqueous layer was then extracted with EtOAc, and all organic layers were combined, dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
General Procedure for Tertiary Eneamides (Procedure XVII).
An appropriate microwave vial was charged with ketone (1 equiv), primary amine (1.2 equiv), tetraisopropoxytitanium (3 equiv), and DCE (0.2 M), and the solution was stirred for 10 min before sodium triacetoxyborohydride (3 equiv) was added. The mixture was irradiated at 120 °C for 2 h. Upon completion, the reaction was cooled to room temperature, diluted with DCM, and washed with 1 M HCl. The organic layer was extracted with DCM, washed with NaHCO3, washed with brine (3×), dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound as a mixture of E/Z isomers, mass was confirmed via LCMS, and it was carried forward without further characterization.
General Procedure for Final Pyrrolopyrazinone Compounds (Procedure XVIII).
Eneamide (1 equiv) was dissolved in EtOAc, and 10% Pd/C (0.15 equiv) was added. The flask was purged with hydrogen from a balloon (3×), and the mixture was hydrogenated (1 atm) overnight. The reaction mixture was filtered through a pad of celite, washed with methanol, and concentrated in vacuo. The crude product was purified via flash column chromatography to afford the title compound.
Methyl 2-(3-Isopropoxyphenyl)acetate (8a).
Methyl 2-(3-hydroxyphenyl)acetate (7.83 g, 47.1 mmol) was dissolved in THF (196 mL), and isopropanol (5.40 mL, 70.7 mmol), triphenylphosphine (18.5 g, 70.7 mmol), and 40% diethyl azodicarboxylate in toluene (32.2 mL, 70.7 mmol) were added and allowed to react overnight at room temperature. The crude product was purified via flash column chromatography (ISCO, Redisep 120 g column, 0–60% EtOAc/hexanes gradient) to afford the title compound as a clear oil (8.19 g, 83%). Rf (1:1 EtOAC/Hex): 0.88; 1H NMR (400 MHz, CDCl3) δ 7.22 (t, J = 8.1 Hz, 1H), 6.88–6.76 (m, 3H), 4.55 (hept, J = 6.1 Hz, 1H), 3.69 (s, 3H), 3.59 (s, 2H), 1.34 (d, J = 6.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 171.9, 158.0, 135.3, 129.5, 121.3, 116.8, 114.3, 69.7, 52.0, 41.2, 22.0. HRMS calcd for C12H17O3, 209.11722 [M + H]+; found 209.11720 [M + H]+.
Methyl 2-(3-Isobutoxyphenyl)acetate (8b).
Methyl 2-(3-hydroxyphenyl)acetate (2.50 g, 15.0 mmol) was dissolved in dry DMF (46 mL) and potassium carbonate (8.32 g, 60.2 mmol) was added. This mixture was stirred at room temperature for 2 h before 1-iodo-2-methylpropane (3.70 mL, 30.1 mmol) was added. The resulting mixture was allowed to react at 60 °C overnight, after which the reaction was deemed complete via TLC. The product was then extracted into EtOAc, washed with brine (3×), dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–20% EtOAc/hexanes gradient) to afford the title compound as a clear oil (1.64 g, 49%). Rf (1:1 EtOAC/Hex): 0.91; 1H NMR (400 MHz, CDCl3) δ 7.25–7.20 (m, 1H), 6.88–6.77 (m, 3H), 3.72 (d, J = 6.5 Hz, 2H), 3.70 (s, 3H), 3.60 (s, 2H), 2.16–2.01 (m, 1H), 1.03 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 172.0, 159.4, 135.3, 129.5, 121.3, 115.5, 113.2, 74.3, 52.1, 41.2, 28.3, 19.3. HRMS calcd for C13H19O3 , 223.13287 [M + H]+; found 223.13264 [M + H]+.
Methyl 2-(2-(2-Chloroacetyl)-5-isopropoxyphenyl)acetate (9a).
General procedure I was followed using 8a (8.10 g, 38.9 mmol), 1 M tin(IV) chloride in DCM (194 mL, 194 mmol), and 2-chloroacetyl chloride (12.5 mL, 156 mmol) in DCE (39 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 80 g column, 0–30% EtOAc/hexanes gradient) to afford the title compound as a white solid (5.20 g, 47%). Rf (1:1 EtOAC/Hex): 0.81; 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 8.7 Hz, 1H), 6.82 (dd, J = 8.7, 2.6 Hz, 1H), 6.75 (d, J = 2.6 Hz, 1H), 4.63 (hept, J = 5.8 Hz, 3H), 4.63 (s, 2H), 3.91 (s, 2H), 3.68 (s, 3H), 1.35 (d, J = 6.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 191.6, 171.6, 161.4, 138.6, 132.3, 125.9, 120.5, 113.1, 70.2, 51.95, 47.2, 40.7, 21.9. HRMS calcd for C14H18O4 Cl, 285.08881 [M + H]+; found 285.08867 [M + H]+.
Methyl 2-(2-(2-Chloroacetyl)-5-isobutoxyphenyl)acetate (9b).
General procedure I was followed using 8b (1.08 g, 4.86 mmol), 1 M tin(IV) chloride in DCM (24.3 mL, 24.3 mmol), and 2-chloroacetyl chloride (1.56 mL, 19.4 mmol) in DCE (4.9 mL). The crude product was purified via flash column chromatography (ISCO,Redisep 80 g column, 0–30% EtOAc/hexanes gradient) to afford the title compound as a white solid (0.70 g, 48%). Rf (1:1 EtOAC/Hex): 0.88; 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 8.7 Hz, 1H), 6.85 (dd, J = 8.7, 2.6 Hz, 1H), 6.79 (d, J = 2.6 Hz, 1H), 4.63 (s, 2H), 3.92 (s, 2H), 3.77 (d, J = 6.5 Hz, 2H), 3.69 (s, 3H), 2.08 (hept, J = 6.7 Hz, 1H), 1.01 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 191.7, 171.6, 162.6, 138.5, 132.2, 126.1, 119.6, 112.4, 74.5, 52.0, 47.2, 40.7, 28.1, 19.1. HRMS calcd for C15H20O4Cl, 299.10446 [M + H]+; found 299.10431 [M + H]+.
Methyl 2-(2-(2-Chloroacetyl)-5-methoxyphenyl)acetate (9c).
General procedure I was followed using methyl 2-(3-methoxyphenyl)-acetate (8c) (2.23 mL, 13.9 mmol), 1 M tin(IV) chloride in DCM (69.4 mL, 69.4 mmol), and 2-chloroacetyl chloride (4.45 mL, 55.5 mmol) in DCE (14 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–50% EtOAc/hexanes gradient) to afford the title compound as a white solid (1.65 g, 46%). Rf (1:1 EtOAC/Hex): 0.66; 1H NMR (399 MHz, CDCl3) δ 7.75 (d, J = 8.7 Hz, 1H), 6.86 (dd, J = 8.7, 2.6 Hz, 1H), 6.78 (d, J = 2.6 Hz, 1H), 4.63 (s, 2H), 3.92 (s, 2H), 3.85 (s, 3H), 3.69 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 191.8, 171.5, 162.8, 138.6, 132.1, 126.6, 119.1, 112.0, 55.5, 51.9, 47.1, 40.6. HRMS calcd for C12H14O4Cl, 257.05751 [M + H]+; found 257.05751 [M + H]+.
Methyl 2-(2-(2-Chloroacetyl)-4,5-dimethoxyphenyl)acetate (9d).
General procedure I was followed using methyl 2-(3,4-dimethoxyphenyl)-acetate (8d) (2.50 g, 11.9 mmol), 1 M tin(IV) chloride in DCM (59.5 mL, 59.5 mmol), and 2-chloroacetyl chloride (3.81 mL, 47.6 mmol) in DCE (12 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–55% EtOAc/hexanes gradient) to afford the title compound as a white solid (1.13 g, 33%). Rf (1:1 EtOAC/Hex): 0.45; 1H NMR (400 MHz, CDCl3) δ 7.23 (s, 1H), 6.74 (s, 1H), 4.63 (s, 2H), 3.92 (s, 3H), 3.91 (s, 3H), 3.88 (s, 2H), 3.68 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 191.9, 171.8, 152.4, 147.4, 130.5, 126.0, 115.6, 112.7, 56.2, 56.0, 52.0, 47.2, 40.0. HRMS calcd for C13H16O5Cl, 287.06808 [M + H]+; found 287.06795 [M + H]+.
Methyl 2-(5-Isopropoxy-2-(2-(4-methoxyphenoxy)acetyl)-phenyl)acetate (10a).
General procedure II was followed using 9a (332 mg, 1.17 mmol), potassium iodide (232 mg, 1.40 mmol), 4-methoxyphenol (174 mg, 1.40 mmol), and potassium carbonate (322 mg, 2.33 mmol) in acetone (7 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–50% EtOAc/hexanes gradient) to afford the title compound as a white solid (289 mg, 67%). Rf (1:1 EtOAC/Hex): 0.73; 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J = 8.7 Hz, 1H), 6.92–6.74 (m, 6H), 5.11 (s, 2H), 4.65 (hept, J = 6.0 Hz, 1H), 3.94 (s, 2H), 3.75 (s, 3H), 3.67 (s, 3H), 1.36 (d, J = 6.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 195.5, 171.8, 161.3, 154.3, 152.3, 138.32, 131.8, 126.3, 120.4, 115.9, 114.6, 113.1, 76.8, 72.1, 70.2, 55.7, 52.0, 40.5, 21.9. HRMS calcd for C21H25O6, 373.17043 [M + H]+; found 373.17143 [M + H]+.
Methyl 2-(5-Isobutoxy-2-(2-(4-methoxyphenoxy)acetyl)phenyl)-acetate (10b).
General procedure II was followed using 9b (700 mg, 2.34 mmol), potassium iodide (467 mg, 2.81 mmol), 4-methox-yphenol (349 mg, 2.81 mmol), and potassium carbonate (648 mg, 4.69 mmol) in acetone (14 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–30% EtOAc/hexanes gradient) to afford the title compound as a white solid (469 mg, 52%). Rf (1:1 EtOAC/Hex): 0.90; 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J = 8.7 Hz, 1H), 6.90–6.73 (m, 6H), 5.11 (s, 2H), 3.94 (s, 2H), 3.78 (d, J = 6.6 Hz, 2H), 3.75 (s, 3H), 3.67 (s, 3H), 2.10 (hept, J = 6.5 Hz, 1H), 1.03 (d, J = 6.7 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 195.6, 171.8, 162.5, 154.3, 152.3, 138.3, 131.8, 126.5, 119.5, 114.6, 112.4, 74.6, 72.1, 55.7, 52.0, 40.5, 28.2, 19.2. HRMS calcd for C22H27O6, 387.18022 [M + H]+; found 387.18013 [M + H]+.
Methyl 2-(5-Methoxy-2-(2-(4-methoxyphenoxy)acetyl)phenyl)-acetate (10c).
General procedure II was followed using 9c (10.2 g, 39.7 mmol), potassium iodide (7.92 g, 47.7 mmol), 4-methoxyphenol (5.92 g, 47.7 mmol), and potassium carbonate (11.0 g, 79.0 mmol) in acetone (238 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 120 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a white solid (7.23 g, 53%). Rf (1:1 EtOAC/Hex): 0.59; 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 8.7 Hz, 1H), 6.88–6.77 (m, 6H), 5.10 (s, 2H), 3.94 (s, 2H), 3.85 (s, 3H), 3.74 (s, 3H), 3.66 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 195.6, 171.7, 162.7, 154.3, 152.2, 138.2, 131.7, 126.8, 119.0, 115.8, 114.6, 112.0, 72.0, 55.6, 55.5, 51.9, 40.5. HRMS calcd for C19H21O6, 345.13326 [M + H]+; found 345.13321 [M + H]+.
Methyl 2-(4,5-Dimethoxy-2-(2-(4-methoxyphenoxy)acetyl)-phenyl)acetate (10d).
General procedure II was followed using 9d (1.08 g, 3.77 mmol), potassium iodide (750 mg, 4.52 mmol), 4-methoxyphenol (561 mg, 4.52 mmol), and potassium carbonate (1.04 g, 7.53 mmol) in acetone (23 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 20–90% EtOAc/hexanes gradient) to afford the title compound as a white solid (503 mg, 36%). Rf (1:1 EtOAC/Hex): 0.45; 1H NMR (500 MHz, CDCl3) δ 7.36 (s, 1H), 6.89–6.78 (m, 4H), 6.75 (s, 1H), 5.08 (s, 2H), 3.93 (s, 3H), 3.91 (s, 3H), 3.90 (s, 2H), 3.75 (s, 3H), 3.67 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 196.1, 172.0, 154.4, 152.3, 152.2, 147.4, 130.2, 126.4, 115.9, 115.5, 114.7, 112.6, 72.6, 56.3, 56.1, 55.7, 52.0, 39.9. HRMS calcd for C20H23O7, 375.14383 [M + H]+; found 375.14387 [M + H]+.
(E/Z)-2-(3-Chlorobenzyl)-6-isopropoxy-1-((4-methoxyphenoxy)-methylene)-1,4-dihydroisoquinolin-3(2H)-one (11a).
General procedure III was followed using compound 10a (148 mg, 0.397 mmol), 3-chlorobenzylamine (58 μL, 0.48 mmol), tetraisopropoxytitanium (362 μL, 1.19 mmol), and sodium triacetoxyborohydride (253 mg, 1.19 mmol) in DCE (2.0 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–35% EtOAc/hexanes gradient) to afford the title compound as a mixture of E/Z isomers and an orange oil (130 mg, 71%). HRMS calcd for C27H27O4NCl, 464.16231 [M + H]+; found 464.16224 [M + H]+.
(E/Z)-6-Isopropoxy-1-((4-methoxyphenoxy)methylene)-2-(3-(trifluoromethyl)benzyl)-1,4-dihydroisoquinolin-3(2H)-one (11b).
General procedure III was followed using compound 10a (75 mg, 0.22 mmol), 3-(trifluoromethyl)benzylamine (37 μL, 0.26 mmol), tetraisopropoxytitanium (198 μL, 0.653 mmol), and sodium triacetoxyborohydride (138 mg, 0.653 mmol) in DCE (1.1 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–35% EtOAc/hexanes gradient) to afford the title compound as a mixture of E/Z isomers and a yellow oil (45 mg, 45%). HRMS calcd for C28H29O4NF3, 500.20432 [M + H]+; found 500.20464 [M + H]+.
(E/Z)-2-(3-Fluorobenzyl)-6-isopropoxy-1-((4-methoxyphenoxy)-methylene)-1,4-dihydroisoquinolin-3(2H)-one (11c).
General procedure III was followed using compound 10a (50 mg, 0.13 mmol), 3-fluorobenzylamine (18 μL, 0.16 mmol), tetraisopropoxytitanium (122 μL, 0.403 mmol), and sodium triacetoxyborohydride (85 mg, 0.40 mmol) in DCE (0.7 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–35% EtOAc/hexanes gradient) to afford the title compound as a mixture of E/Z isomers and an orange oil (35 mg, 58%). HRMS calcd for C27H27O4NF, 448.19186 [M + H]+; found 448.19096 [M + H]+.
(E/Z)-2-(3-Chlorobenzyl)-6-isobutoxy-1-((4-methoxyphenoxy)-methylene)-1,4-dihydroisoquinolin-3(2H)-one (11d).
General procedure III was followed using compound 10b (500 mg, 1.29 mmol), 3-chlorobenzylamine (190 μL, 1.55 mmol), tetraisopropoxytitanium (1.18 mL, 3.88 mmol), and sodium triacetoxyborohydride (823 mg, 3.88 mmol) in DCE (6.5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–25% EtOAc/hexanes gradient) to afford the title compound as a mixture of E/Z isomers and an orange oil (450 mg, 73%). HRMS calcd for C28H29O4NCl, 478.17796 [M + H]+; found 478.17837 [M + H]+.
(E/Z)-2-(3-Chlorobenzyl)-6-methoxy-1-((4-methoxyphenoxy)-methylene)-1,4-dihydroisoquinolin-3(2H)-one (11e).
General procedure III was followed using compound 10c (100 mg, 0.290 mmol), 3-chlorobenzylamine (43 μL, 0.35 mmol), tetraisopropoxytitanium (260 μL, 0.87 mmol), and sodium triacetoxyborohydride (185 mg, 0.871 mmol) in DCE (1.5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–30% EtOAc/hexanes gradient) to afford the title compound as an ~1:2 mixture of E/Z isomers and an orange oil (74 mg, 59%). HRMS calcd for C25H23O4NCl, 436.13101 [M + H]+; found 436.13017 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 2.68 min). Method 2: 85–95% ACN in water over 6 min at 1 mL/min (retention time = 2.32 min).
(E/Z)-6-Methoxy-1-((4-methoxyphenoxy)methylene)-2-(4,4,4-trifluorobutyl)-1,2-dihydroisoquinolin-3(4H)-one (11f).
General procedure III was followed using compound 10c (100 mg, 0.290 mmol), 4,4,4-trifluorobutan-1-amine (44 mg, 0.35 mmol), tetraisopropoxytitanium (260 μL, 0.87 mmol), and sodium triacetoxyborohydride (185 mg, 0.871 mmol) in DCE (1.5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–30% EtOAc/hexanes gradient) to afford the title compound as a mixture of E/Z isomers and an orange oil (42 mg, 34%). HRMS calcd for C22H23O4NF3, 422.15737 [M + H]+; found 422.15750 [M + H]+.
(E/Z)-6-Methoxy-1-((4-methoxyphenoxy)methylene)-2-(2,2,2-trifluoroethyl)-1,2-dihydroisoquinolin-3(4H)-one (11g).
General procedure III was followed using compound 10c (150 mg, 0.436 mmol), 2,2,2-trifluoroethanamine (69 μL, 0.87 mmol, 2 equiv), tetraisopropoxytitanium (400 μL, 1.3 mmol), and sodium triacetoxyborohydride (277 mg, 1.31 mmol) in DCE (2 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–30% EtOAc/hexanes gradient) to afford the title compound as a mixture of E/Z isomers and a yellow oil (33 mg, 19%). HRMS calcd for C20H19O4NF3, 394.12607 [M + H]+; found 394.12617 [M + H]+.
E/Z)-6-Methoxy-1-((4-methoxyphenoxy)methylene)-2-propyl-1,4-dihydroisoquinolin-3(2H)-one (11h).
General procedure III was followed using compound 10c (75 mg, 0.22 mmol), propylamine (21 μL, 0.26 mmol), tetraisopropoxytitanium (198 μL, 0.653 mmol), and sodium triacetoxyborohydride (138 mg, 0.653 mmol) in DCE (1.1 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–40% EtOAc/hexanes gradient) to afford the title compound as a mixture of E/Z isomers and a brown oil (31 mg, 40%). HRMS calcd for C21H28O4N, 358.20128 [M + H]+; found 358.20118 [M + H]+.
(E/Z)-2-(2-(1H-Indol-3-yl)ethyl)-6-methoxy-1-((4-methoxyphenoxy)methylene)-1,4-dihydroisoquinolin-3(2H)-one (11i).
General procedure III was followed using compound 10c (75 mg, 0.22 mmol), tryptamine (42 mg, 0.26 mmol), tetraisopropoxytitanium (198 μL, 0.653 mmol), and sodium triacetoxyborohydride (138 mg, 0.653 mmol) in DCE (1.1 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–35% EtOAc/hexanes gradient) to afford the title compound as a mixture of E/Z isomers and a red oil (20 mg, 40%). HRMS calcd for C28H27O4N2, 455.19653 [M + H]+; found 455.19586 [M + H]+.
(E/Z)-6-Methoxy-2-(2-(2-methoxyethoxy)ethyl)-1-((4-methoxyphenoxy)methylene)-1,4-dihydroisoquinolin-3(2H)-one (11j).
General procedure III was followed using compound 10c (60 mg, 0.18 mmol), 2-(2-methoxyethoxy)ethanamine (25 mg, 0.21 mmol), tetraisopropoxytitanium (159 μL, 0.524 mmol), and sodium triacetoxyborohydride (111 mg, 0.524 mmol) in DCE (0.9 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a mixture of E/Z isomers and a red oil (14 mg, 19%). HRMS calcd for C23H28O6N, 414.19111 [M + H]+; found 414.19038 [M + H]+.
(E/Z)-2-(3-(Dimethylamino)propyl)-6-methoxy-1-((4-methoxyphenoxy)methylene)-1,4-dihydroisoquinolin-3(2H)-one (11k).
General procedure III was followed using compound 10c (50 mg, 0.15 mmol), 3-(dimethylamino)-1-propylamine (37 μL, 0.29 mmol, 2 equiv), tetraisopropoxytitanium (132 μL, 0.436 mmol), and sodium triacetoxyborohydride (92 mg, 0.44 mmol) in DCE (0.7 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–10% DCM/(1% NH4OH in MeOH) gradient) to afford the title compound as a mixture of E/Z isomers and a red oil (13 mg, 23%). HRMS calcd for C23H29O4N2, 397.21218 [M + H]+; found 397.21243 [M + H]+.
(E/Z)-6-Methoxy-1-((4-methoxyphenoxy)methylene)-2-(prop-2-yn-1-yl)-1,4-dihydroisoquinolin-3(2H)-one (11l).
General procedure III was followed using compound 10c (200 mg, 0.581 mmol), propargylamine (112 μL, 1.74 mmol), tetraisopropoxytitanium (495 μL, 1.74 mmol), and sodium triacetoxyborohydride (369 mg, 1.74 mmol) in DCE (3 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–30% EtOAc/hexanes gradient) to afford the title compound as a single E/Z isomer and a white solid (160 mg, 80%). HRMS calcd for C21H20O4N, 350.13868 [M + H]+; found 350.13837 [M + H]+.
2-((1H-1,2,3-Triazol-4-yl)methyl)-6-methoxy-1-((4-methoxyphenoxy)methylene)-1,4-dihydroisoquinolin-3(2H)-one (11m).
37% Formaldehyde in H2O (0.17 mL, 2.2 mmol, 10 equiv) and acetic acid (0.018 mL, 0.32 mmol, 1.5 equiv) were dissolved in THF (0.2 mL) and stirred for 15 min. Sodium azide (21 mg, 0.32 mmol, 1.5 equiv) and 11l (75 mg, 0.22 mmol, 1 equiv) dissolved in THF (0.2 mL) were then added in succession and stirred for 10 min. Then, sodium ascorbate (9.0 mg, 0.045 mmol, 0.2 equiv) was added, followed by an aqueous CuSO4 solution (200 mg/mL, 0.05 equiv), and the reaction was stirred at room temperature for 24 h. The reaction was then concentrated before being dissolved in EtOAc and washed with 1 M NaOH. The organic layer was extracted with EtOAc, washed with brine (3×), dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–10% DCM/(1% NH4OH in MeOH) gradient) to afford the title compound as a single E/Z isomer and a yellow oil (24 mg, 29%). Rf (90:10:0.1 DCM/MeOH/NH4OH): 0.46; 1H NMR (399 MHz, CDCl3) δ 7.51 (s, 1H), 7.20 (d, J = 8.5 Hz, 1H), 6.99–6.95 (m, 2H), 6.88–6.83 (m, 3H), 6.75 (dd, J = 8.5, 2.6 Hz, 1H), 6.70 (d, J = 2.4 Hz, 1H), 6.53 (s, 1H), 5.23 (s, 2H), 3.79 (s, 3H), 3.78 (s, 3H), 3.68 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.5, 159.5, 156.0, 150.7, 132.1, 130.8, 130.8, 123.8, 123.7, 121.9, 117.8, 114.9, 114.8, 113.2, 112.1, 55.7, 55.4, 40.0, 39.0. HRMS calcd for C21H21O4N4, 393.15573 [M + H]+; found 393.15487 [M + H]+.
(E/Z)-2-(3-Chlorobenzyl)-6,7-dimethoxy-1-((4-methoxyphenoxy)methylene)-1,4-dihydroisoquinolin-3(2H)-one (11n).
General procedure III was followed using compound 10d (200 mg, 0.534 mmol), 3-chlorobenzylamine (78 μL, 0.64 mmol), tetraisopropoxytitanium (486 μL, 1.60 mmol), and sodium triacetoxyborohydride (340 mg, 1.60 mmol) in DCE (2.7 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–35% EtOAc/hexanes gradient) to afford the title compound as a mixture of E/Z isomers and a white solid (150 mg, 59%). HRMS calcd for C26H25O5NCl, 466.14158 [M + H]+; found 466.14082 [M + H]+.
2-(3-Chlorobenzyl)-6-isopropoxy-1-((4-methoxyphenoxy)-methyl)-1,4-dihydroisoquinolin-3(2H)-one (12a).
General procedure IV-A was followed using compound 11a (150 mg, 0.323 mmol) and PtO2 (19 mg, 0.046 mmol) in ethanol (2.6 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–50% EtOAc/hexanes gradient) to afford the title compound as an orange oil (15 mg, 10%). Rf (1:1 EtOAc/Hex): 0.59; 1H NMR (500 MHz, CDCl3) δ 7.22–7.15 (m, 3H), 7.09–7.05 (m, 1H), 7.04 (d, J = 8.4 Hz, 1H), 6.81–6.69 (m, 6H), 5.46 (d, J = 15.4 Hz, 1H), 4.63–4.59 (m, 1H), 4.54 (hept, J = 6.1 Hz, 1H), 4.37 (d, J = 15.4 Hz, 1H), 4.08–3.99 (m, 2H), 3.91 (d, J = 19.3 Hz, 1H), 3.75 (s, 3H), 3.64 (d, J = 19.3 Hz, 1H), 1.33 (d, J = 6.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 169.7, 157.8, 154.3, 152.2, 139.2, 134.5, 134.1, 130.0, 128.0, 127.6, 127.1, 125.8, 124.3, 115.4, 114.7, 114.4, 114.3, 71.7, 70.0, 59.9, 55.7, 48.6, 37.7, 22.1. HRMS calcd for C27H29O4NCl, 466.17796 [M + H]+; found 466.17791 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 90–95% MeOH in water over 5 min at 1 mL/min (retention time = 2.56 min). Method 2: 85–95% ACN in water over 6 min at 1 mL/min (retention time = 3.00 min).
6-Isopropoxy-1-((4-methoxyphenoxy)methyl)-2-(3-(trifluoromethyl)benzyl)-1,4-dihydroisoquinolin-3(2H)-one (12b).
General procedure IV-B was followed using compound 11b (45 mg, 0.090 mmol) and 10% Pd/C (14 mg, 0.014 mmol) in ethanol (5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–50% EtOAc/hexanes gradient) to afford the title compound as a clear oil (32 mg, 71%). Rf (1:1 EtOAc/Hex): 0.65; 1H NMR (500 MHz, CDCl3) δ 7.51–7.44 (m, 2H), 7.39–7.34 (m, 2H), 7.04 (d, J = 8.4 Hz, 1H), 6.82–6.77 (m, 2H), 6.76–6.73 (m, 1H), 6.73–6.66 (m, 3H), 5.52 (d, J = 15.6 Hz, 1H), 4.65–4.59 (m, 1H), 4.58–4.51 (m, 1H), 4.48 (d, J = 15.5 Hz, 1H), 4.10–4.00 (m, 2H), 3.93 (d, J = 19.2 Hz, 1H), 3.75 (s, 3H), 3.66 (d, J = 19.3 Hz, 1H), 1.33 (d, J = 6.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 169.8, 157.9, 154.3, 152.1, 138.2, 134.0, 130.9, 130.9 (q, J = 32.0 Hz, 2J), 129.2, 127.1, 124.5 (q, J = 3.8 Hz, 3J), 124.3 (q, J = 3.9 Hz, 3J), 124.2, 124.0 (q, J = 272.5 Hz, 1J), 115.4, 114.7, 114.5, 114.3, 71.7, 70.0, 60.1, 55.7, 48.9, 37.7, 22.0. HRMS calcd for C28H29O4NF3, 500.20432 [M + H]+; found 500.20333 [M + H]+ Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 3.54 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 1.05 min).
2-(3-Fluorobenzyl)-6-isopropoxy-1-((4-methoxyphenoxy)-methyl)-1,4-dihydroisoquinolin-3(2H)-one (12c).
General procedure IV-B was followed using compound 11c (104 mg, 0.232 mmol) and 10% Pd/C (37 mg, 0.035 mmol) in ethanol (5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–50% EtOAc/hexanes gradient) to afford the title compound as a yellow oil (67 mg, 64%). Rf (1:1 EtOAc/Hex): 0.57; 1H NMR (400 MHz, CDCl3) δ 7.25–7.19 (m, 1H), 7.04 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 7.7 Hz, 1H), 6.94–6.88 (m, 2H), 6.81–6.69 (m, 6H), 5.49 (d, J = 15.5 Hz, 1H), 4.65–4.60 (m, 1H), 4.54 (hept, J = 6.0 Hz, 1H), 4.39 (d, J = 15.5 Hz, 1H), 4.10–3.99 (m, 2H), 3.92 (d, J = 19.2 Hz, 1H), 3.75 (s, 3H), 3.65 (d, J = 19.3 Hz, 1H), 1.34 (d, J = 6.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 169.7, 163.0 (d, J = 246.5 Hz, 1J), 157.8, 154.2, 152.2, 139.7 (d, J = 7.0 Hz, 3J), 134.1, 130.1 (d, J = 8.2 Hz, 3J), 127.1, 124.3, 123.2 (d, J = 2.8 Hz, 4J), 115.4, 114.6, 114.6 (d, J = 21.6 Hz, 2J), 114.4, 114.3 (d, J = 21.3 Hz, 2J), 114.2, 71.6, 69.9, 59.9, 55.7, 48.6, 37.6, 22.0. HRMS calcd for C27H29O4NF, 450.20751 [M + H]+; found 450.20731 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 90–95% MeOH in water over 5 min at 1 mL/min (retention time = 1.54 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 1.01 min).
2-(3-Chlorobenzyl)-6-isobutoxy-1-((4-methoxyphenoxy)methyl)-1,4-dihydroisoquinolin-3(2H)-one (12d).
General procedure IV-A was followed using compound 11d (200 mg, 0.418 mmol) and PtO2 (24 mg, 0.059 mmol) in ethanol (3.3 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–50% EtOAc/hexanes gradient) to afford the title compound as a red foam (82 mg, 54%). Rf (1:1 EtOAc/Hex): 0.76; 1H NMR (400 MHz, CDCl3) δ 7.22–7.14 (m, 3H), 7.09–7.03 (m, 2H), 6.81–6.69 (m, 6H), 5.46 (d, J = 15.5 Hz, 1H), 4.62 (t, J = 5.0 Hz, 1H), 4.37 (d, J = 15.5 Hz, 1H), 4.09–3.99 (m, 2H), 3.94 (d, J = 19.3 Hz, 1H), 3.75 (s, 3H), 3.71 (d, J = 6.5 Hz, 2H), 3.66 (d, J = 19.3 Hz, 1H), 2.08 (hept, J = 6.9 Hz, 1H), 1.02 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 169.7, 159.1, 154.2, 152.2, 139.2, 134.5, 134.0, 129.9, 127.9, 127.6, 127.0, 125.7, 124.3, 115.4, 114.7, 113.3, 113.0, 74.5, 71.6, 59.9, 55.7, 48.6, 37.6, 28.2, 19.2. HRMS calcd for C28H31O4NCl, 480.19361 [M + H]+; found 480.19404 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 2.05 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 0.86 min).
2-(3-Chlorobenzyl)-6-methoxy-1-((4-methoxyphenoxy)methyl)-1,2-dihydroisoquinolin-3(4H)-one (12e).
General procedure IV-A was followed using compound 11e (113 mg, 0.259 mmol) and PtO2 (8.4 mg, 0.037 mmol) in ethanol (2 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–30% EtOAc/hexanes gradient) to afford the title compound as a clear oil (63 mg, 56%). Rf (1:1 EtOAc/Hex): 0.59; 1H NMR (400 MHz, CDCl3) δ 7.22–7.11 (m, 3H), 7.09–7.02 (m, 2H), 6.82–6.67 (m, 6H), 5.44 (d, J = 15.5 Hz, 1H), 4.62 (t, J = 5.0 Hz, 1H), 4.37 (d, J = 15.5 Hz, 1H), 4.08–3.99 (m, 2H), 3.94 (d, J = 19.1 Hz, 1H), 3.78 (s, 3H), 3.73 (s, 3H), 3.66 (d, J = 19.2 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 169.6, 159.4, 154.2, 152.1, 139.2, 134.4, 134.1, 129.9, 127.8, 127.6, 127.1, 125.7, 124.6, 115.4, 114.6, 112.8, 112.3, 71.5, 59.9, 55.7, 55.3, 48.6, 37.6. HRMS calcd for C25H25O4NCl, 438.14666 [M + H]+; found 438.14644 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 1.83 min).
6-Methoxy-1-((4-methoxyphenoxy)methyl)-2-(4,4,4-trifluorobutyl)-1,2-dihydroisoquinolin-3(4H)-one (12f).
General procedure IV-B was followed using compound 11f (79 mg, 0.19 mmol) and 10% Pd/C (30 mg, 0.028 mmol) in ethanol (5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–30% EtOAc/hexanes gradient) to afford the title compound as a clear oil (18 mg, 23%). Rf (1:1 EtOAc/Hex): 0.47; 1H NMR (400 MHz, CDCl3) δ 7.19 (d, J = 8.4 Hz, 1H), 6.83 (d, J = 2.6 Hz, 1H), 6.82–6.69 (m, 5H), 4.68 (t, J = 5.4 Hz, 1H), 4.09–3.98 (m, 2H), 3.81 (d, J = 19.3 Hz, 1H), 3.81 (s, 3H), 3.74 (s, 3H), 3.56 (d, J = 19.3 Hz, 1H), 3.37–3.26 (m, 2H), 2.18–2.05 (m, 2H), 1.94–1.83 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 169.5, 159.6, 154.3, 152.2, 134.2, 127.1, 127.0 (q, J = 276.2 Hz, 1J), 124.6, 115.4, 114.7, 112.9, 112.4, 71.7, 61.2, 55.7, 55.4, 45.7, 37.7, 31.3 (q, J = 29.1 Hz, 2J), 20.5 (q, J = 2.6 Hz, 3J). HRMS calcd for C22H25O4NF3, 424.17302 [M + H]+; found 424.17256 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 6 min at 1 mL/min (retention time = 4.69 min). Method 2: 85–95% ACN in water over 6 min at 1 mL/min (retention time = 1.48 min).
6-Methoxy-1-((4-methoxyphenoxy)methyl)-2-(2,2,2-trifluoroethyl)-1,2-dihydroisoquinolin-3(4H)-one (12g).
General procedure IV-B was followed using compound 11g (33 mg, 0.084 mmol) and 10% Pd/C (13 mg, 0.013 mmol) in ethanol (5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–30% EtOAc/hexanes gradient) to afford the title compound as a clear oil (8.8 mg, 27%). Rf (1:1 EtOAc/Hex): 0.60; 1H NMR (400 MHz, CDCl3) δ 7.19 (d, J = 8.4 Hz, 1H), 6.84 (dd, J = 8.6, 2.8 Hz, 1H), 6.81–6.72 (m, 4H), 6.72 (d, J = 2.3 Hz, 1H), 5.07–4.96 (m, 1H), 4.94 (dd, J = 7.6, 4.0 Hz, 1H), 4.09 (dd, J = 9.7, 3.8 Hz, 1H), 3.99 (dd, J = 9.7, 7.8 Hz, 1H), 3.96–3.82 (m, 2H), 3.81 (s, 3H), 3.75 (s, 3H), 3.68 (d, J = 19.6 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 169.9, 159.7, 154.4, 151.9, 133.6, 127.3, 126.7 (q, J = 273.5 Hz, 1J), 123.6, 115.3, 114.7, 113.1, 112.6, 72.1, 61.3, 55.7, 55.4, 46.3 (q, J = 33.6 Hz, 2J), 37.4. HRMS calcd for C20H21O4NF3, 396.14172 [M + H]+; found 396.14143 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 85–95% MeOH in water over 10 min at 1 mL/min (retention time = 1.21 min). Method 2: 75–95% MeOH in water over 6 min at 1 mL/min (retention time = 3.78 min).
6-Methoxy-1-((4-methoxyphenoxy)methyl)-2-propyl-1,4-dihydroisoquinolin-3(2H)-one (12h).
General procedure IV-B was followed using compound 11h (31 mg, 0.088 mmol) and 10% Pd/C (14 mg, 0.013 mmol) in ethanol (5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–50% EtOAc/hexanes gradient) to afford the title compound as a clear oil (15 mg, 48%). Rf (1:1 EtOAc/Hex): 0.28; 1H NMR (500 MHz, CDCl3) δ 7.19 (d, J = 8.4 Hz, 1H), 6.83–6.68 (m, 6H), 4.68 (t, J = 5.3 Hz, 1H), 4.16–4.00 (m, 3H), 3.84 (d, J = 19.2 Hz, 1H), 3.80 (s, 3H), 3.74 (s, 3H), 3.56 (d, J = 19.2 Hz, 1H), 3.14–3.06 (m, 1H), 1.70–1.55 (m, 2H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 169.2, 159.4, 154.2, 152.4, 134.5, 127.4, 125.1, 115.5, 114.7, 112.7, 112.3, 71.4, 60.9, 55.7, 55.4, 48.3, 37.8, 21.0, 11.4.NMR (126 MHz, CDCl3) δ 169.3, 159.4, 154.2, 152.3, 134.3, 127.1, 124.6, 115.4, 114.7, 112.8, 112.2, 71.5, 63.1, 55.7, 55.4, 37.3, 34.7. HRMS calcd for C21H26O4N, 356.18563 [M + H]+; found 356.18553 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 1.35 min). Method 2: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 0.81 min).
2-(2-(1H-Indol-3-yl)ethyl)-6-methoxy-1-((4-methoxyphenoxy)-methyl)-1,4-dihydroisoquinolin-3(2H)-one (12i).
A modified version of general procedure IV-B was followed using compound 11i (21 mg, 0.046 mmol), 10% Pd/C (7.3 mg, 0.0069 mmol), and AcOH (catalytic) in ethanol (5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–70% EtOAc/hexanes gradient) to afford the title compound as a clear oil (10 mg, 45%). Rf (1:1 EtOAc/Hex): 0.17; 1H NMR (500 MHz, CDCl3) δ 7.88 (s, 1H), 7.65 (d, J = 7.9 Hz, 1H), 7.34 (d, J = 7.3 Hz, 1H), 7.19 (t, J = 7.6 Hz, 1H), 7.10 (t, J = 7.5 Hz, 1H), 6.89–6.83 (m, 2H), 6.78–6.62 (m, 6H), 4.42–4.33 (m, 2H), 3.94–3.89 (m, 2H), 3.84 (d, J = 19.2 Hz, 1H), 3.81 (s, 3H), 3.73 (s, 3H), 3.58 (d, J = 19.0 Hz, 1H), 3.53–3.43 (m, 1H), 3.08 (t, J = 7.2 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 169.3, 159.3, 154.1, 152.3, 136.2, 134.4, 127.3, 127.0, 125.2, 122.2, 122.0, 119.4, 118.8, 115.4, 114.6, 113.1, 112.6, 112.1, 111.1, 71.4, 61.9, 55.7, 55.4, 48.0, 37.9, 23.7. HRMS calcd for C28H29O4N2, 457.21218 [M + H]+; found 457.21230 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 2.29 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 0.94 min).
6-Methoxy-2-(2-(2-methoxyethoxy)ethyl)-1-((4-methoxyphenoxy)methyl)-1,4-dihydroisoquinolin-3(2H)-one (12j).
General procedure IV-B was followed using compound 11j (16 mg, 0.039 mmol) and 10% Pd/C (6.2 mg, 0.0059 mmol) in ethanol (5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a red oil (10 mg, 83%). Rf (1:1 EtOAc/Hex): 0.44; 1H NMR (500 MHz, CDCl3) δ 7.16 (d, J = 8.4 Hz, 1H), 6.83–6.66 (m, 6H), 4.93 (t, J = 5.0 Hz, 1H), 4.18–4.01 (m, 3H), 3.84 (d, J = 18.2 Hz, 1H), 3.80 (s, 3H), 3.74 (s, 3H), 3.73–3.68 (m, 1H), 3.65–3.59 (m, 1H), 3.59–3.51 (m, 3H), 3.51–3.45 (m, 1H), 3.44–3.40 (m, 2H), 3.31 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 169.6, 159.3, 154.1, 152.5, 134.4, 127.2, 125.4, 115.4, 114.6, 112.6, 112.2, 71.8, 71.4, 70.4, 70.0, 62.3, 59.0, 55.7, 55.4, 46.9, 37.8. HRMS calcd for C23H30O6N, 416.20676 [M + H]+; found 416.20700 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 6 min at 1 mL/min (retention time = 3.58 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 0.86 min).
2-(3-(Dimethylamino)propyl)-6-methoxy-1-((4-methoxyphenoxy)methyl)-1,4-dihydroisoquinolin-3(2H)-one (12k).
A modified version of general procedure IV-B was followed using compound 11k (16 mg, 0.039 mmol), 10% Pd/C (6.2 mg, 0.0059 mmol), and AcOH (catalytic) in ethanol (5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a red oil (10 mg, 83%). Rf (1:1 EtOAc/Hex): 0.40; 1H NMR (500 MHz, CDCl3) δ 7.16 (d, J = 8.4 Hz, 1H), 6.83–6.66 (m, 6H), 4.93 (t, J = 5.0 Hz, 1H), 4.18–4.01 (m, 3H), 3.84 (d, J = 18.2 Hz, 1H), 3.80 (s, 3H), 3.74 (s, 3H), 3.73–3.68 (m, 1H), 3.65–3.59 (m, 1H), 3.59–3.51 (m, 3H), 3.51–3.45 (m, 1H), 3.44–3.40 (m, 2H), 3.31 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 169.6, 159.3, 154.1, 152.5, 134.4, 127.2, 125.4, 115.4, 114.6, 112.6, 112.2, 71.8, 71.4, 70.4, 70.0, 62.3, 59.0, 55.7, 55.4, 46.9, 37.8. HRMS calcd for C23H30O4N2, 399.22783 [M + H]+; found 399.22743 [M + H]+. Purity was established to be 85% pure using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 50–95% MeOH in water over 6 min at 1 mL/min (retention time = 5.56 min). Method 2: 75–95% MeOH in water over 6 min at 1 mL/min (retention time = 1.18 min).
2-((1H-1,2,3-Triazol-4-yl)methyl)-6-methoxy-1-((4-methoxyphenoxy)methyl)-1,4-dihydroisoquinolin-3(2H)-one (12l).
A modified version of general procedure IV-B was followed using compound 11m (22 mg, 0.056 mmol), 10% Pd/C (8.9 mg, 0.0083 mmol), and AcOH (catalytic) in ethanol (5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–50% EtOAc/hexanes gradient) to afford the title compound as a yellow oil (15 mg, 68%). Rf (9:1 DCM/MeOH): 0.43; 1H NMR (400 MHz, CDCl3) δ 7.66 (s, 1H), 7.10 (d, J = 8.5 Hz, 1H), 6.80–6.65 (m, 6H), 5.27 (d, J = 15.3 Hz, 1H), 4.87 (t, J = 4.8 Hz, 1H), 4.68 (d, J = 15.2 Hz, 1H), 4.16–4.00 (m, 2H), 3.91 (d, J = 19.3 Hz, 1H), 3.77 (s, 3H), 3.73 (s, 3H), 3.60 (d, J = 19.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 170.1, 159.4, 154.2, 152.2, 133.7, 127.1, 124.3, 115.4, 114.6, 112.9, 112.2, 71.6, 61.1, 55.7, 55.4, 41.1, 37.5. HRMS calcd for C21H23O4N4, 395.17138 [M + H]+; found 395.17077 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 6 min at 1 mL/min (retention time = 1.78 min). Method 2: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 1.53 min).
2-(3-Chlorobenzyl)-6,7-dimethoxy-1-((4-methoxyphenoxy)-methyl)-1,4-dihydroisoquinolin-3(2H)-one (12m).
General procedure IV-A was followed using compound 11n (136 mg, 0.292 mmol) and PtO2 (17 mg, 0.041 mmol) in ethanol (2.3 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–50% EtOAc/hexanes gradient) to afford the title compound as a clear oil (30 mg, 22%). Rf (1:1 EtOAc/Hex): 0.28; 1H NMR (300 MHz, CDCl3) δ 7.24–7.16 (m, 3H), 7.12–7.06 (m, 1H), 6.82–6.63 (m, 6H), 5.49 (d, J = 15.5 Hz, 1H), 4.59 (t, J = 5.1 Hz, 1H), 4.37 (d, J = 15.5 Hz, 1H), 4.08–4.03 (m, 2H), 3.97–3.92 (m, 1H), 3.90 (s, 1H), 3.89 (s, 3H), 3.84 (s, 3H), 3.75 (s, 3H), 3.73–3.59 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 169.8, 154.3, 152.2, 149.0, 147.8, 139.2, 134.5, 130.0, 127.9, 127.6, 125.8, 124.7, 124.2, 115.4, 114.7, 110.2, 109.0, 71.5, 60.1, 56.1, 56.0, 55.7, 48.5, 36.9. HRMS calcd for C26H27O5NCl, 468.15723 [M + H]+; found 468.15734 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 1.45 min). Method 2: 85–95% ACN in water over 6 min at 1 mL/min (retention time = 1.34 min).
2-(3,4-Dimethoxyphenyl)ethanamine (14).
General procedure V was followed using compound 3,4-dimethoxyphenethylamine (13) (5.59 mL, 33.1 mmol), triethylamine (9.23 mL, 66.2 mmol), and 2-chloroacetyl chloride (3.16 mL, 39.7 mmol) in dry DCM (120 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 10–80% EtOAc/hexanes gradient) to afford the title compound as a white solid (6.32 g, 74%). Rf (1:1 EtOAc/Hex): 0.56; 1H NMR (300 MHz, CDCl3) δ 6.82 (d, J = 8.0 Hz, 1H), 6.77–6.69 (m, 2H), 6.64 (s, 0H), 4.02 (s, 2H), 3.87 (s, 3H), 3.86 (s, 3H), 3.53 (q, J = 6.9 Hz, 2H), 2.78 (t, J = 7.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 165.7, 149.0, 147.8, 130.8, 120.6, 111.8, 111.3, 55.9, 42.7, 41.1, 35.1. HRMS calcd for C12H17O3NCl, 258.08915 [M + H]+; found 258.08876 [M + H]+.
2-(Benzyloxy)-N-(3,4-dimethoxyphenethyl)acetamide (15a).
General procedure VII was followed using compound 14 (2.97 g, 11.5 mmol), benzyl alcohol (1.41 g, 13.0 mmol), and 60% sodium hydride dispersion in mineral oil (0.61 g, 15 mmol) in THF (28 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 30–100% EtOAc/hexanes gradient) to afford the title compound as a yellow oil (2.7 g, 70%). Rf (1:1 EtOAC/Hex): 0.19; 1H NMR (400 MHz, CDCl3) δ 7.40–7.20 (m, 4H), 6.85–6.65 (m, 4H), 4.49 (s, 2H), 3.97 (s, 2H), 3.84 (s, 3H), 3.83 (s, 3H), 3.54 (q, J = 6.8 Hz, 2H), 2.78 (t, J = 7.0 Hz, 2H).13C NMR (101 MHz, CDCl3) δ 171.1, 169.4, 149.0, 147.6, 136.8, 131.1, 128.5, 128.1, 127.8, 120.6, 111.7, 111.2, 73.4, 69.5, 55.8, 40.0, 35.2. HRMS calcd for C19H24O4N, 330.16998 [M + H]+; found 330.16945 [M + H]+.
N-(3,4-Dimethoxyphenethyl)-2-((tetrahydro-2H-pyran-4-yl)oxy)-acetamide (15b).
General procedure VII was followed using compound 14 (0.20 g, 0.78 mmol), 4-hydroxytetrahydropyran (95 mg, 0.93 mmol), and 60% sodium hydride dispersion in mineral oil (43 mg, 1.1 mmol) in THF (3.9 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–10% MeOH/DCM gradient) to afford the title compound as a white solid (0.19 g, 75%). Rf (9:1 DCM/MeOH): 0.62; 1H NMR (500 MHz, chloroform-d) δ 6.79 (d, J = 7.9 Hz, 1H), 6.75–6.70 (m, 2H), 6.62 (s, 1H), 3.93 (s, 2H), 3.90–3.83 (m, 8H), 3.53 (q, J = 6.9 Hz, 2H), 3.47 (tt, J = 8.7, 4.0 Hz, 1H), 3.39 (ddd, J = 12.0, 9.7, 2.7 Hz, 2H), 2.78 (t, J = 7.1 Hz, 2H), 1.85–1.78 (m, 2H), 1.56–1.45 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 169.7, 149.1, 147.8, 131.1, 120.6, 111.8, 111.3, 75.0, 67.3, 65.3, 55.9, 55.9, 39.9, 35.2, 32.1. HRMS calcd for C17H26O5N, 324.18055 [M + H]+; found 324.18056 [M + H]+.
N-(3,4-Dimethoxyphenethyl)-2-(pyridine-3-yloxy)acetamide (15c).
General procedure VI was followed using compound 14 (0.20 g, 0.78 mmol), 3-hydroxypyridine (89 mg, 0.93 mmol), and cesium carbonate (1.0 g, 3.1 mmol) in acetonitrile (4.6 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–10% MeOH/DCM gradient) to afford the title compound as a white solid (0.15 g, 60%). Rf (9:1 DCM/MeOH): 0.49; 1H NMR (500 MHz, CDCl3) δ 8.28–8.23 (m, 2H), 7.24–7.18 (m, 1H), 7.14–7.08 (m, 1H), 6.75 (d, J = 8.1 Hz, 1H), 6.70–6.63 (m, 3H), 4.47 (s, 2H), 3.83 (s, 3H), 3.81 (s, 3H), 3.57 (q, J = 6.9 Hz, 2H), 2.78 (t, J = 7.0 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 167.2, 153.4, 149.1, 147.8, 143.4, 138.3, 130.9, 124.0, 120.9, 120.7, 111.8, 111.4, 67.5, 55.9, 55.8, 40.2, 35.2. HRMS calcd for C17H21O4N2, 317.14958 [M + H]+; found 317.14920 [M + H]+.
N-(3,4-Dimethoxyphenethyl)-2-((4-methoxybenzyl)oxy)-acetamide (15d).
General procedure VII was followed using compound 14 (18.8 g, 73.0 mmol), 4-methoxybenzyl alcohol (12.1 g, 87.5 mmol), and 60% sodium hydride dispersion in mineral oil (4.38 g, 110. Mmol) in THF (196 mL). The crude material was purified via flash column chromatography (ISCO, Redisep 320 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as an orange solid (20.1 g, 74%). Rf (1:1 EtOAc/Hex): 0.67; 1H NMR (500 MHz, CDCl3) δ 7.22 (t, J = 7.8 Hz, 1H), 7.19–7.13 (m, 2H), 6.91–6.84 (m, 2H), 6.81–6.73 (m, 3H), 6.65 (s, 1H), 4.43 (s, 2H), 3.94 (s, 2H), 3.81 (s, 3H), 3.78 (s, 3H), 3.54 (q, J = 7.0 Hz, 2H), 2.80 (t, J = 7.1 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 169.6, 159.9, 159.6, 140.3, 129.6, 129.5, 128.9, 121.1, 114.4, 114.0, 112.0, 73.2, 69.3, 55.3, 55.2, 39.8, 35.7. HRMS calcd for C20H26O5N, 360.18055 [M + H]+; found 360.18023 [M + H]+.
1-((Benzyloxy)methyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (16a).
General procedure VIII was followed using compound 15a (2.67 g, 8.11 mmol), phosphorous oxychloride (2.27 mL, 24.3 mmol), and sodium borohydride (0.64 g, 24 mmol) in acetonitrile (~40 mL) and MeOH (~10 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 10–90% EtOAc/hexanes gradient) to afford the title compound as a white foam (1.6 g, 62% over two steps). Rf (1:1 EtOAC/Hex): 0.29; 1H NMR (300 MHz, CDCl3) δ 7.38–7.26 (m, 5H), 6.58 (s, 2H), 4.68–4.50 (m, 2H), 4.17 (dd, J = 8.2, 3.9 Hz, 1H), 3.83 (s, 3H), 3.79 (s, 3H), 3.75–3.62 (m, 2H), 3.25–3.10 (m, 1H), 3.04–2.90 (m, 1H), 2.74 (t, J = 5.8 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 147.6, 147.1, 138.0, 128.4, 127.8, 127.7, 127.7, 126.6, 111.8, 109.2, 73.3, 72.9, 55.9, 55.8, 55.0, 40.0, 29.0. HRMS calcd for C19H24O3N, 314.17507 [M + H]+; found 314.17453 [M + H]+.
6,7-Dimethoxy-1-(((tetrahydro-2H-pyran-4-yl)oxy)methyl)-1,2,3,4-tetrahydroisoquinoline (16b).
General procedure VIII was followed using compound 15b (187 mg, 0.578 mmol), phosphorous oxychloride (0.27 mL, 2.9 mmol), and sodium borohydride (66 mg, 1.7 mmol) in acetonitrile (9.2 mL) and MeOH (2.4 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–10% MeOH/DCM gradient) to afford the title compound as a clear oil (29 mg, 16%). Rf (9:1 DCM/MeOH): 0.44; 1H NMR (399 MHz, chloroform-d) δ 6.62 (s, 1H), 6.58 (s, 1H), 4.08 (dd, J = 8.8, 3.9 Hz, 1H), 3.98–3.87 (m, 2H), 3.83 (s, 3H), 3.82 (s, 3H), 3.72 (dd, J = 9.2, 4.0 Hz, 1H), 3.63–3.49 (m, 2H), 3.48–3.37 (m, 2H), 3.18 (dt, J = 11.7, 5.7 Hz, 1H), 2.96 (dt, J = 11.8, 5.9 Hz, 1H), 2.73 (t, J = 5.8 Hz, 2H), 2.40 (s, 1H), 1.97–1.85 (m, 2H), 1.69–1.52 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 147.6, 147.0, 127.9, 127.2, 111.9, 109.3, 74.4, 70.9, 65.6, 56.0(0), 55.9(9), 55.8, 55.2, 40.2, 32.6, 32.3, 29.3. HRMS calcd for C17H26O4N, 308.18563 [M + H]+; found 308.18552 [M + H]+.
6,7-Dimethoxy-1-((pyridin-3-yloxy)methyl)-1,2,3,4-tetrahydroisoquinoline (16c).
General procedure VIII was followed using compound 15c (0.20 g, 0.64 mmol), phosphorous oxychloride (0.30 mL, 3.2 mmol), and sodium borohydride (72 mg, 1.9 mmol) in acetonitrile (7.4 mL) and MeOH (1.9 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–20% MeOH/DCM gradient) to afford the title compound as a white solid (0.12 g, 60%). Rf (8:2 DCM/MeOH): 0.66; 1H NMR (500 MHz, CDCl3) δ 8.34 (dd, J = 2.6, 0.9 Hz, 1H), 8.21 (dd, J = 4.1, 1.9 Hz, 1H), 7.25–7.16 (m, 2H), 6.65 (s, 1H), 6.61 (s, 1H), 4.36 (dd, J = 7.9, 4.6 Hz, 1H), 4.21–4.12 (m, 2H), 3.84 (s, 4H), 3.82 (s, 3H), 3.19 (ddd, J = 12.2, 6.8, 5.4 Hz, 1H), 3.03 (dt, J = 12.1, 5.5 Hz, 1H), 2.75 (q, J = 5.3 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 148.0, 147.3, 142.4, 138.1, 128.3, 126.0, 123.9, 121.2, 112.1, 109.5, 71.2, 56.1, 55.8, 54.6, 39.8, 29.2. HRMS calcd for C17H21O3N2, 301.15467 [M + H]+; found 301.15442 [M + H]+.
(3-Chlorophenyl)(1-(hydroxymethyl)-6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanone (17a).
General procedure IX was followed using compound 16a (1.56 g, 4.98 mmol), triethylamine (1.39 mL, 9.96 mmol), and 3-chlorobenzoyl chloride (0.77 mL, 6.0 mmol) in DCM (80 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 10–90% EtOAc/hexanes gradient) to afford the title compound as a white foam (1.4 g) that was carried forward without further characterization. This material (1.4 g, 3.0 mmol) was dissolved in a 2:3 mixture of chloroform (40 mL) and methane sulfonic acid (60 mL) and allowed to react at room temperature for 6 h. The reaction was then poured over ice, dilute with EtOAc, and quenched with saturated NaHCO3. The organic layer was then washed with brine (3×), dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 10–90% EtOAc/hexanes gradient) to afford the title compound as a white foam (0.48 g, 44% over two steps). Rf (1:1 EtOAC/Hex): 0.07; 1H NMR (400 MHz, CDCl3) δ 7.60–6.97 (m, 4H), 6.78–6.38 (m, 2H), 5.75 (dd, J = 8.6, 3.7 Hz, 0.5H), 4.83–4.74 (m, 0.5H), 4.08 (dd, J = 11.6, 3.9 Hz, 1H), 3.93 (dd, J = 11.5, 8.9 Hz, 1H), 3.89–3.70 (m, 6H), 3.67 (dd, J = 11.8, 4.2 Hz, 0.5H), 3.57 (td, J = 13.5, 12.9, 4.0 Hz, 1H), 3.23 (td, J = 13.0, 4.2 Hz, 0.5H), 3.06 (td, J = 14.1, 12.2, 6.0 Hz, 0.5H), 2.92 (ddd, J = 17.1, 11.9, 5.8 Hz, 1H), 2.69 (t, J = 15.6 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 171.1, 170.4, 148.3, 148.2, 147.8, 147.4, 138.0, 137.6, 134.6, 134.3, 130.1, 130.0, 129.7, 129.6, 129.6, 128.4, 128.0, 127.8, 127.0, 126.6, 125.7, 125.7, 124.9, 124.4, 124.3, 111.7, 111.3, 110.0, 109.5, 66.5, 65.8, 64.6, 64.6, 59.6, 56.0, 56.0, 55.9, 55.2, 42.1, 35.5, 28.8, 27.6, 15.3. HRMS calcd for C19H21O4NCl, 362.11536 [M + H]+; found 362.11479 [M + H]+.
(1-(Hydroxymethyl)-6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)(3-(trifluoromethyl)phenyl)methanone (17b).
In step 1, a modified version of general procedure VIII was followed, in which the material was refluxed for only 1 h instead of overnight and only one equivalent of POCl3 was used due to the loss of the PMB-protecting group and conversion to the alkyl chloride upon continued heating. This proceeded using compound 15d (11.1 g, 30.8 mmol), phosphorous oxychloride (2.87 mL, 30.8 mmol), and sodium borohydride (3.50 g, 92.5 mmol) in acetonitrile (493 mL) and MeOH (123 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–15% MeOH/DCM gradient) to afford crude 6,7-dimethoxy-1-(((4-methoxybenzyl)oxy)-methyl)-1,2,3,4-tetrahydroisoquinoline as a clear oil (4.71 g) that was carried forward without further characterization. General procedure IX was followed for step 2 using the above compound (4.71 g, 13.7 mmol), triethylamine (3.82 mL, 27.4 mmol), and 3-(trifluoromethyl)-benzoyl chloride (2.48 mL, 16.4 mmol) in DCM (214 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 80 g column, 0–100% EtOAc/hexanes gradient) to afford impure (6,7-dimethoxy-1-(((4-methoxybenzyl)oxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(3-(trifluoromethyl)phenyl)methanone as a clear oil (2.77 g) that was carried forward without further characterization. Finally, this compound (2.77 g, 5.37 mmol) was hydrogenated at 50 psi using 10% Pd/C (0.57 g, 0.54 mmol) in MeOH (32 mL) overnight. The reaction mixture was filtered through a pad of celite, washed with methanol, and concentrated in vacuo. The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a white foam (1.01 g, 8% over three steps). 1H NMR (500 MHz, CDCl3) δ 7.84–7.54 (m, 4H), 6.76–6.37 (m, 2H), 5.77 (dd, J = 8.8, 3.6 Hz, 0.8H), 4.88–4.71 (m, 0.2H), 4.14–4.07 (m, 1H), 4.02–3.93 (m, 1H), 3.88 (s, 2.5H), 3.86 (s, 3.5H), 3.79–3.70 (m, 2H), 3.64–3.55 (m, 0.8H), 3.31–3.21 (m, 0.2H), 3.09 (ddd, J = 17.7, 12.5, 6.5 Hz, 0.2H), 2.94 (ddd, J = 17.0, 11.9, 5.7 Hz, 0.8H), 2.74–2.66 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 171.4, 148.4, 148.1, 136.7, 130.2, 129.3, 126.8, 125.8, 124.3, 123.9, 111.5, 110.1, 66.9, 56.1, 55.9, 55.5, 42.3, 28.7. (Unable to discern between fluorine splitting and minor peaks; only major peaks reported; major CF3 peak not reported.) HRMS calcd for C20H21O4NF3, 396.14172 [M + H]+; found 369.14133 [M + H]+.
(3-Chlorophenyl)(6,7-dimethoxy-1-(((5-methoxythiophen-2-yl)-oxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (18a).
Compound 17a (100 mg, 0.276 mmol), 2-iodo-5-methoxythiophene (73 mg, 0.30 mmol), 1,10-phenanthroline (10 mg, 0.055 mmol), copper(I) iodide (11 mg, 0.055 mmol), and cesium carbonate (0.18 g, 0.55 mmol) were charged to a 10 mL microwave vial. The vial was capped, purged with argon, evacuated three times, and toluene (0.5 mL) was added. The vial was heated to 80 °C and allowed to react for 24 h. Upon completion, the reaction was cooled to room temperature, filtered through a pad of celite, washed with EtOAc, and concentrated in vacuo. The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 10–90% EtOAc/hexanes gradient) to afford the title compound as a white foam (28 mg, 21%). Rf (1:1 EtOAC/Hex): 0.47; 1H NMR (400 MHz, CDCl3) δ 7.58–7.28 (m, 4H), 6.79–6.41 (m, 2H), 5.92 (dd, J = 13.0, 4.5 Hz, 1H), 5.77 (s, 1H), 5.05 (dd, J = 9.0, 3.2 Hz, 0.5H), 4.88 (dd, J = 13.1, 5.4 Hz, 0.5H), 4.38 (d, J = 5.1 Hz, 1H), 4.18 (t, J = 10.0 Hz, 0.5H) 4.01 (dd, J = 10.5, 4.0 Hz, 0.5H), 3.95–3.72 (m, 9H), 3.62 (td, J = 13.1, 3.6 Hz, 1H), 3.24 (td, J = 12.9, 3.7 Hz, 0.5H), 3.09 (td, J = 16.8, 5.9 Hz, 0.5H), 2.86 (ddd, J = 15.3, 13.1, 5.9 Hz, 0.5H), 2.76 (dd, J = 16.2, 3.0 Hz, 0.5H), 2.68 (d, J = 15.4 Hz, 0.5H). 13C NMR (101 MHz, CDCl3) δ 170.1, 169.4, 168.4, 155.1, 155.0, 152.7, 152.0, 148.7, 148.5, 148.3, 147.9, 147.9, 147.6, 139.9, 137.9, 134.7, 134.6, 134.4, 130.0, 130.0, 129.9, 129.8, 129.8, 129.6, 127.7, 127.1, 126.8, 126.7, 126.1, 125.5, 124.7, 124.6, 124.3, 123.1, 116.4, 111.8, 111.4, 111.3, 109.9, 109.8, 109.5, 103.4, 102.8, 100.7, 77.2, 76.2, 74.8, 68.8, 65.9, 60.5, 60.4, 57.0, 56.0, 55.9, 51.8, 51.5, 51.1, 42.7, 42.2, 35.5, 28.9, 28.8, 27.6, 15.3. HRMS calcd for C24H25O5NClS, 474.11365 [M + H]+; found 474.11362 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 1.74 min). Method 2: 75–95% ACN in water over 5 min at 1 mL/min (retention time = 3.56 min).
(6,7-Dimethoxy-1-((pyridin-2-yloxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(3-(trifluoromethyl)phenyl)methanone (18b).
General procedure X was followed using compound 17b (25 mg, 0.063 mmol), 2-fluoropyrimidine (8.2 μL, 0.095 mmol), and 60% sodium hydride dispersion in mineral oil (4.6 mg, 0.19 mmol) in THF (0.3 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a white foam (8.0 mg, 27%). Rf (1:1 EtOAC/Hex): 0.40; 1H NMR (600 MHz, CDCl3) δ 8.08 (d, J = 3.7 Hz, 0.5H), 7.91 (d, J = 4.1 Hz, 0.5H), 7.67 (s, 0.5H), 7.59 (d, J = 7.6 Hz, 0.5H), 7.52 (t, J = 7.1 Hz, 1.5H), 7.45 (t, J = 8.4 Hz, 1H), 7.40–7.35 (m, 1H), 7.33 (t, J = 7.6 Hz, 0.5H), 6.86–6.76 (m, 1.5H), 6.71 (d, J = 8.3 Hz, 0.5H), 6.66 (d, J = 8.3 Hz, 0.5H), 6.61 (s, 0.5H), 6.54 (s, 1H), 6.06–6.01 (m, 0.5H), 5.14–5.09 (m, 0.5H), 4.89–4.78 (m, 1H), 4.63 (dd, J = 11.2, 4.2 Hz, 0.5H), 4.50 (t, J = 10.8 Hz, 0.5H), 4.39 (dd, J = 11.8, 3.4 Hz, 0.5H), 3.83 (s, 1.5H), 3.81 (s, 1.5H), 3.79 (s, 1.5H), 3.75 (s, 1.5H), 3.71–3.63 (m, 0.5H), 3.63–3.57 (m, 0.5H), 3.29 (td, J = 12.9, 4.2 Hz, 0.5H), 3.06 (ddd, J = 17.7, 12.6, 6.2 Hz, 0.5H), 2.83–2.70 (m, 1H), 2.60 (d, J = 15.2 Hz, 0.5H). 13C NMR (151 MHz, CDCl3) δ 170.0, 169.3, 163.4, 162.9, 148.7, 148.3, 147.9, 147.6, 146.6, 146.5, 139.0, 137.3, 137.0, 131.2, 130.9, 130.9, 130.6, 130.5, 129.8, 129.2, 128.7, 127.0, 126.2, 126.1, 126.0, 125.8, 124.7, 124.7, 123.9, 123.5, 117.3, 117.2, 111.8, 111.4, 111.4, 111.1, 110.4, 109.8, 66.8, 66.6, 57.0, 56.1, 55.9, 51.7, 42.0, 35.5, 28.9, 27.7. (Unable to discern between fluorine splitting and minor peaks, all peaks reported as seen.) HRMS calcd for C25H24O4N2F3, 473.16827 [M + H]+; found 473.16871 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 50–95% MeOH in water over 6 min at 1 mL/min (retention time = 4.27 min). Method 2: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 1.10 min).
(6,7-Dimethoxy-1-((pyrimidin-2-yloxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(3-(trifluoromethyl)phenyl)methanone (18c).
General procedure X was followed using compound 17b (25 mg, 0.063 mmol), 2-chloropyrimidine (11 mg, 0.095 mmol), and 60% sodium hydride dispersion in mineral oil (3.1 mg, 0.13 mmol) in DMF (0.3 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a clear oil (14 mg, 47%). Rf (9:1 DCM/MeOH): 0.67; 1H NMR (600 MHz, CDCl3) δ 8.45 (d, J = 4.5 Hz, 1H), 8.37 (d, J = 4.6 Hz, 1H), 7.66–7.29 (m, 4H), 6.90–6.82 (m, 1.5H), 6.62 (s, 0.5H), 6.54 (d, J = 7.0 Hz, 1H), 6.07–6.02 (m, 0.5H), 5.11 (dd, J = 9.2, 3.3 Hz, 0.5H), 4.87 (dd, J = 11.4, 4.2 Hz, 0.5H), 4.81 (dd, J = 13.1, 6.0 Hz, 0.5H), 4.71 (dd, J = 11.3, 6.8 Hz, 0.5H), 4.61–4.54 (m, 0.5H), 4.45 (dd, J = 11.8, 3.8 Hz, 0.5H), 3.84 (s, 1.5H), 3.82 (s, 1.5H), 3.79 (s, 1.5H), 3.76 (s, 1.5H), 3.72–3.67 (m, 1H), 3.34–3.27 (m, 0.5H), 3.06 (ddd, J = 17.5, 12.5, 6.3 Hz, 0.5H), 2.81 (ddd, J = 17.4, 10.8, 6.9 Hz, 0.5H), 2.77–2.71 (m, 0.5H), 2.65 (d, J = 16.1 Hz, 0.5H). 13C NMR (151 MHz, CDCl3) δ 170.0, 169.4, 165.0, 164.6, 159.3, 148.8, 148.3, 147.9, 147.7, 137.2, 136.9, 131.2, 131.0, 130.9, 130.8, 130.7, 123.0, 129.2, 128.8, 127.2, 126.4, 126.3, 126.0, 125.9, 124.6, 124.5, 123.7, 123.5, 122.8, 121.0, 115.5, 115.3, 111.8, 111.4, 110.2, 109.8, 68.7, 67.9, 56.6, 56.1, 55.9, 51.2, 42.5, 35.6, 28.9, 27.7 (Unable to discern between fluorine splitting and minor peaks, all peaks reported as seen.) HRMS calcd for C24H23O4N3F3, 474.16325 [M + H]+; found 474.16477 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 50–95% MeOH in water over 3 min at 1 mL/min (retention time = 2.84 min). Method 2: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 0.82 min).
(6,7-Dimethoxy-1-(((tetrahydro-2H-pyran-4-yl)oxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(3-(trifluoromethyl)phenyl)methanone (18d).
General procedure IX was followed using compound 16b (29 mg, 0.094 mmol), triethylamine (26 μL, 0.19 mmol), and 3-(trifluoromethyl)benzoyl chloride (17 μL, 0.11 mmol) in DCM (1.5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a white foam (45 mg, quant.). Rf (1:1 EtOAC/Hex): 0.62; 1H NMR (500 MHz, chloroform-d) δ 7.85 (s, 0.5H), 7.71–7.45 (m, 3.5H), 6.79 (s, 0.5H), 6.64 (s, 0.5H), 6.60 (s, 0.5H), 6.41 (s, 0.5H), 5.77 (t, J = 5.5 Hz, 0.5H), 4.83 (ddd, J = 13.8, 11.3, 5.1 Hz, 1H), 3.95–3.83 (m, 7.5H), 3.77 (s, 1.5H), 3.74–3.53 (m, 2.5H), 3.51–3.36 (m, 2.5H), 3.22 (td, J = 12.7, 4.2 Hz, 0.5H), 3.09 (td, J = 14.4, 12.3, 6.2 Hz, 0.5H), 2.85 (ddd, J = 17.0, 11.7, 6.0 Hz, 0.5H), 2.75 (dd, J = 16.2, 3.4 Hz, 0.5H), 2.65 (d, J = 15.7 Hz, 0.5H), 1.93–1.82 (m, 2H), 1.65–1.49 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 170.2, 169.1, 148.6, 148.2, 147.8, 147.5, 137.4, 137.3, 131.3, 131.0, 130.8, 130.5, 129.9, 129.3, 129.0, 128.8, 126.8, 126.3, 126.2, 126.1, 125.8, 125.7, 125.0(8), 125.0(6), 124.4, 123.6, 111.8, 111.4, 110.3, 109.8, 75.0, 74.0, 70.0, 69.6, 65.5, 65.4, 65.3, 57.9, 56.0, 55.9, 52.0, 42.6, 35.4, 32.4, 32.3, 32.1, 32.0, 29.0, 27.6. (Unable to discern between fluorine splitting and minor peaks, all peaks reported as seen.) HRMS calcd for C25H29O5NF3, 480.19923 [M + H]+; found 480.19912 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 50–95% MeOH in water over 6 min at 1 mL/min (retention time = 3.62 min). Method 2: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 0.88 min).
(6,7-Dimethoxy-1-((pyridin-3-yloxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(3-(trifluoromethyl)phenyl)methanone (18e).
General procedure IX was followed using compound 16c (25 mg, 0.082 mmol), triethylamine (23 μL, 0.16 mmol), and 3-(trifluoromethyl)-benzoyl chloride (15 μL, 0.10 mmol) in DCM (1.3 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a white foam (17 mg, 44%). Rf (9:1 DCM/MeOH): 0.64; 1H NMR (600 MHz, CDCl3) δ 8.08 (d, J = 3.7 Hz, 0.5H), 7.91 (d, J = 4.1 Hz, 0.5H), 7.67 (s, 0.5H), 7.59 (d, J = 7.6 Hz, 0.5H), 7.52 (t, J = 7.1 Hz, 1.5H), 7.45 (t, J = 8.4 Hz, 1H), 7.40–7.35 (m, 1H), 7.33 (t, J = 7.6 Hz, 0.5H), 6.86–6.76 (m, 1.5H), 6.71 (d, J = 8.3 Hz, 0.5H), 6.66 (d, J = 8.3 Hz, 0.5H), 6.61 (s, 0.5H), 6.54 (s, 1H), 6.06–6.01 (m, 0.5H), 5.14–5.09 (m, 0.5H), 4.89–4.78 (m, 1H), 4.63 (dd, J = 11.2, 4.2 Hz, 0.5H), 4.50 (t, J = 10.8 Hz, 0.5H), 4.39 (dd, J = 11.8, 3.4 Hz, 0.5H), 3.83 (s, 1.5H), 3.81 (s, 1.5H), 3.79 (s, 1.5H), 3.75 (s, 1.5H), 3.71–3.63 (m, 0.5H), 3.63–3.57 (m, 0.5H), 3.29 (td, J = 12.9, 4.2 Hz, 0.5H), 3.06 (ddd, J = 17.7, 12.6, 6.2 Hz, 0.5H), 2.83–2.70 (m, 1H), 2.60 (d, J = 15.2 Hz, 0.5H). 13C NMR (151 MHz, CDCl3) δ 170.0, 169.3, 163.4, 162.9, 148.7, 148.3, 147.9, 147.6, 146.6, 146.5, 139.0, 137.3, 137.0, 131.2, 130.9, 130.9, 130.6, 130.5, 129.8, 129.2, 128.7, 127.0, 126.2, 126.1, 126.0, 125.8, 124.7, 124.7, 123.9, 123.5, 117.3, 117.2, 111.8, 111.4, 111.4, 111.1, 110.4, 109.8, 66.8, 66.6, 57.0, 56.7, 55.9, 51.7, 42.0, 35.5, 28.9, 27.7. (Unable to discern between fluorine splitting and minor peaks, all peaks reported as seen.) HRMS calcd for C26H20O4N2F3, 473.16827 [M + H]+; found 473.16919 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 50–95% MeOH in water over 3 min at 1 mL/min (retention time = 2.59 min). Method 2: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 0.77 min).
3-(2-Nitrovinyl)thiophene (20a).
General procedure XI was followed using thiophene-3-carbaldehyde (19a) (3.00 g, 26.7 mmol), butylamine (0.32 mL, 3.2 mmol), acetic acid (0.31 mL, 5.4 mmol), and 4 Å molecular sieves (300 mg) in nitromethane (27 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a yellow solid (3.3 g, 79%). Rf (1:1 EtOAc/Hex): 0.58; 1H NMR (500 MHz, CDCl3) δ 8.01 (dd, J = 13.5, 0.5 Hz, 1H), 7.73 (ddd, J = 2.5, 1.3, 0.6 Hz, 1H), 7.49 (d, J = 13.5 Hz, 1H), 7.44 (ddd, J = 5.1, 2.9, 0.7 Hz, 1H), 7.28 (ddd, J = 5.1, 1.3, 0.5 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 136.7, 132.6, 132.5, 132.2, 128.2, 125.1. HRMS calcd for C6H6O2NS, 156.01138 [M + H]+; found 156.01151 [M + H]+.
2-Methoxy-5-(2-nitrovinyl)thiophene (20b).
2-Methoxythiophene (1.06 g, 9.28 mmol) was dissolved in Et2O (19 mL, 0.5 M), and n-butyllithium (5.57 mL, 13.9 mmol, 2.5 M in hexanes) was added dropwise under nitrogen at 0 °C. The mixture was then heated to reflux and allowed to react for 1 h. The reaction was then cooled to 0 °C and quenched carefully via dropwise addition of water before the organic layer was extracted with Et2O, washed with brine (3×), dried over MgSO4, and concentrated in vacuo to give the crude 5-methoxythiophene-2-carbaldehyde (19b) as a yellow oil (1.32 g, 100%) that was carried forward without further purification. General procedure XI was then followed using the crude material (1.32 g, 9.28 mmol), butylamine (0.110 mL, 1.11 mmol), acetic acid (0.106 mL, 1.86 mmol), and 4 Å molecular sieves (132 mg) in nitromethane (19 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a red solid (1.25 g over two steps, 73%). Rf (1:1 EtOAc/Hex): 0.66; 1H NMR (399 MHz, CDCl3) δ 8.02 (d, J = 13.1 Hz, 1H), 7.24 (d, J = 13.1 Hz, 2H), 7.18 (d, J = 4.2 Hz, 1H), 6.25 (d, J = 4.2 Hz, 1H), 3.97 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 172.2, 136.2, 133.7, 131.9, 120.4, 106.4, 60.6. HRMS calcd for C7H8O3NS, 186.02194 [M + H]+; found 186.02186 [M + H]+.
2-Methyl-4-(2-nitrovinyl)thiazole (20c).
General procedure XI was followed using 2-methylthiazole-4-carbaldehyde (19c) (1.00 g, 7.86 mmol), butylamine (93 μL, 0.94 mmol), acetic acid (90 μL, 1.6 mmol), and 4 Å molecular sieves (100 mg) in nitromethane (8 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–80% EtOAc/hexanes gradient) to afford the title compound as a yellow solid (0.41 g, 31%). Rf (1:1 EtOAc/Hex): 0.70; 1H NMR (400 MHz, CDCl3) δ 7.85 (s, 1H), 7.85 (s, 1H), 7.54 (s, 1H), 2.75 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.9, 146.9, 139.1, 130.5, 126.2, 19.4. HRMS calcd for C6H7O2N2S, 171.02227 [M + H]+; found 171.02191 [M + H]+.
3-(2-Nitrovinyl)furan (20d).
General procedure XI was followed using furan-3-carbaldehyde (19d) (1.80 mL, 20.8 mmol), butylamine (0.25 mL, 2.5 mmol), acetic acid (0.24 mL, 4.2 mmol), and 4 Å molecular sieves (200 mg) in nitromethane (21 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a yellow solid (2.9 g, quant.). Rf (1:1 EtOAc/Hex): 0.83; 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 13.5 Hz, 1H), 7.86–7.81 (m, 1H), 7.56–7.48 (m, 1H), 7.39 (d, J = 13.5 Hz, 1H), 6.59–6.55 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 147.3, 145.3, 136.7, 129.6, 118.2, 107.2. No ionization observed by HRMS.
1-Methyl-5-(2-nitrovinyl)-1H-pyrazole (20e).
General procedure XI was followed using 1-methyl-1H-pyrazole-5-carbaldehyde (19e) (2.00 g, 18.2 mmol), butylamine (0.22 mL, 2.2 mmol), acetic acid (0.21 mL, 3.6 mmol), and 4 Å molecular sieves (200 mg) in nitromethane (18 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a yellow solid (1.7 g, 62%). Rf (1:1 EtOAc/Hex): 0.65; 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 13.4 Hz, 1H), 7.54 (d, J = 2.2 Hz, 1H), 7.51 (d, J = 13.4 Hz, 1H), 6.66 (d, J = 2.2 Hz, 1H), 4.02 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 139.4, 137.6, 133.2, 124.4, 107.5, 37.3. HRMS calcd for C6H8O2N3, 154.06110 [M + H]+; found 154.06109 [M +H]+.
2-(Thiophen-3-yl)ethan-1-amine (21a).
General procedure XII was followed using compound 20a (3.27 g, 21.1 mmol), sulfuric acid (2.51 mL, 47.0 mmol), and 2 M lithium aluminum hydride in THF (47.0 mL, 94.0 mmol) in THF (144 mL) to afford the title compound as a red oil (2.10 g). The crude material was confirmed via LCMS and carried forward without further purification.
2-(5-Methoxythiophen-2-yl)ethan-1-amine (21b).
General procedure XII was followed using compound 20b (1.19 g, 6.43 mmol), sulfuric acid (0.76 mL, 14 mmol), and 2 M lithium aluminum hydride in THF (14.3 mL, 28.7 mmol) in THF (39 mL) to afford the crude title compound as a pale yellow oil (1.01 g). The crude material was confirmed via LCMS and carried forward without further purification.
2-(2-Methylthiazol-4-yl)ethan-1-amine (21c).
General procedure XII was followed using compound 20c (411 mg, 2.42 mmol), sulfuric acid (0.29 mL, 5.4 mmol), and 1 M lithium aluminum hydride in THF (10.8 mL, 10.8 mmol) in THF (17 mL) to afford the title compound as a red oil (60 mg). The crude material was confirmed via LCMS and carried forward without further purification.
2-(Furan-3-yl)ethan-1-amine (21d).
General procedure XII was followed using compound 20d (1.45 g, 10.4 mmol), sulfuric acid (1.24 mL, 23.2 mmol), and 2 M lithium aluminum hydride in THF (23.2 mL, 46.5 mmol) in THF (71 mL) to afford the title compound as a yellow oil (0.89 g). The crude material was confirmed via LCMS and carried forward without further purification.
2-(1-Methyl-1H-pyrazol-5-yl)ethan-1-amine (21e).
General procedure XII was followed using compound 20e (1.60 g, 10.5 mmol), sulfuric acid (1.24 mL, 23.3 mmol), and 2 M lithium aluminum hydride in THF (23.3 mL, 46.6 mmol) in THF (71 mL) to afford the title compound as a pale yellow oil (0.80 g). The crude material was confirmed via LCMS and carried forward without further purification.
2-Chloro-N-(2-(thiophen-3-yl)ethyl)acetamide (22a).
General procedure V was followed using crude compound 21a (2.10 g, 16.5 mmol), triethylamine (3.34 mL, 33.0 mmol), and 2-chloroacetyl chloride (1.59 mL, 19.8 mmol) in DCM (60 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a white solid (2.20 g, 51% over two steps). Rf (1:1 EtOAc/Hex): 0.47; 1H NMR (500 MHz, CDCl3) δ 7.30 (dd, J = 4.9, 2.9 Hz, 1H), 7.05–7.01 (m, 1H), 6.96 (dd, J = 4.9, 1.3 Hz, 1H), 6.65 (s, 1H), 4.02 (s, 2H), 3.56 (d, J = 6.1 Hz, 2H), 2.89 (t, J = 6.9 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 165.8, 138.6, 127.9, 126.2, 121.6, 42.7, 40.2, 23.0. HRMS calcd for C8H11ONClS, 204.02444 [M + H]+; found 204.02490 [M + H]+.
2-Chloro-N-(2-(5-methoxythiophen-2-yl)ethyl)acetamide (22b).
General procedure V was followed using crude compound 21b (1.01 g, 6.43 mmol), triethylamine (1.79 mL, 12.9 mmol), and 2-chloroacetyl chloride (0.62 mL, 7.7 mmol) in DCM (23 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as an orange solid (0.92 g, 61% over two steps). Rf (1:1 EtOAc/Hex): 0.43; 1H NMR (400 MHz, CDCl3) δ 6.75 (s, 1H), 6.47–6.41 (m, 1H), 6.01 (d, J = 3.7 Hz, 1H), 4.04 (s, 2H), 3.85 (s, 3H), 3.51 (q, J = 6.5 Hz, 2H), 2.89 (t, J = 7.0 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 166.0, 165.0, 126.6, 122.8, 103.2, 60.2, 42.6, 40.9, 30.1. HRMS calcd for C9H13O2NClS, 234.03500 [M + H]+; found 234.03492 [M + H]+.
2-Chloro-N-(2-(2-methylthiazol-4-yl)ethyl)acetamide (22c).
General procedure V was followed using crude compound 21c (60 mg, 0.42 mmol), triethylamine (0.12 mL, 0.84 mmol), and 2-chloroacetyl chloride (34 μL, 0.42 mmol) in DCM (1.5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a yellow oil (27 mg, 5% over two steps). Rf (1:1 EtOAc/Hex): 0.39; 1H NMR (500 MHz, CDCl3) δ 7.59 (s, 0H), 6.81 (s, 1H), 4.03 (s, 2H), 3.62 (q, J = 6.5 Hz, 2H), 2.93 (t, J = 6.3 Hz, 2H), 2.68 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 166.3, 165.9, 153.5, 114.0, 42.8, 39.3, 30.4. HRMS calcd for C8H11ON2ClS, 219.03534 [M + H]+; found 219.03522 [M + H]+.
2-Chloro-N-(2-(furan-3-yl)ethyl)acetamide (22d).
General procedure V was followed using crude compound 21d (0.860 g, 7.74 mmol), triethylamine (2.16 mL, 15.5 mmol), and 2-chloroacetyl chloride (0.74 mL, 9.3 mmol) in DCM (28 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–70% EtOAc/hexanes gradient) to afford the title compound as a red oil (0.84 g, 43% over two steps). Rf (1:1 EtOAc/Hex): 0.61; 1H NMR (500 MHz, CDCl3) δ 7.39 (t, J = 1.7 Hz, 1H), 7.31–7.26 (m, 1H), 6.66 (s, 1H), 6.30 (dd, J = 1.8, 0.9 Hz, 1H), 4.03 (s, 2H), 3.50 (q, J = 6.2 Hz, 2H), 2.68 (t, J = 6.9 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 165.8, 143.4, 139.7, 121.3, 110.7, 42.7, 39.8, 24.8. HRMS calcd for C8H10O2NClNa, 210.02923 [M + H]+; found 210.02875 [M + H]+.
2-Chloro-N-(2-(1-methyl-1H-pyrazol-5-yl)ethyl)acetamide (22e).
General procedure V was followed using crude compound 21e (0.78 g, 6.2 mmol), triethylamine (1.74 mL, 12.5 mmol), and 2-chloroacetyl chloride (0.60 mL, 7.5 mmol) in DCM (23 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–10% (1% NH4OH in MeOH)/DCM gradient) to afford the title compound as a yellow oil (0.86 g, 41% over two steps). Rf (90:10:0.1 DCM/MeOH/NH4OH): 0.43; 1H NMR (500 MHz, CDCl3) δ 7.40 (d, J = 1.9 Hz, 1H), 6.79 (bs, 1H), 6.07 (dt, J = 1.9, 0.6 Hz, 1H), 4.03 (s, 2H), 3.81 (s, 3H), 3.57 (dt, J = 7.1, 6.0 Hz, 3H), 2.88 (t, J = 7.1 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 166.1, 139.0, 138.5, 104.8, 42.6, 38.5, 36.2, 25.5. HRMS calcd for C8H13ON3Cl, 202.07417 [M + H]+; found 202.07392 [M + H]+.
2-Chloro-N-(2-(thiophen-2-yl)ethyl)acetamide (22f).
General procedure V was followed using 2-(thiophen-2-yl)ethan-1-amine (21f) (2.00 g, 15.7 mmol), triethylamine (3.18 mL, 31.4 mmol), and 2-chloroacetyl chloride (1.51 mL, 18.9 mmol) in DCM (57 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–60% EtOAc/hexanes gradient) to afford the title compound as a red oil (2.64 g, 82%). Rf (1:1 EtOAc/Hex): 0.51; 1H NMR (500 MHz, CDCl3) δ 7.18 (dd, J = 5.2, 1.2 Hz, 1H), 6.95 (dd, J = 5.1, 3.4 Hz, 1H), 6.88–6.83 (m, 1H), 6.75 (s, 1H), 4.03 (s, 2H), 3.58 (q, J = 6.7 Hz, 2H), 3.07 (t, J = 6.7 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 165.9, 140.6, 127.2, 125.6, 124.2, 42.7, 41.1, 29.7. HRMS calcd for C8H10ONClNaS, 226.00638 [M + Na]+; found 226.00658 [M + Na]+.
N-(2-(1H-Pyrrol-1-yl)ethyl)-2-chloroacetamide (22g).
General procedure V was followed using 2-(1H-pyrrol-1-yl)ethanamine (21g) (1.00 g, 9.08 mmol), triethylamine (2.53 mL, 18.2 mmol), and 2-chloroacetyl chloride (0.87 mL, 11 mmol) in DCM (33 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as an orange solid (1.7 g, quant.). Rf (90:10:0.1 DCM/MeOH/NH4OH): 0.69; 1H NMR (500 MHz, CDCl3) δ 6.66 (t, J = 2.0 Hz, 2H), 6.19 (t, J = 2.0 Hz, 2H), 4.05 (t, J = 5.9 Hz, 2H), 4.03 (s, 2H), 3.62 (q, J = 5.9 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 166.3, 120.6, 109.0, 48.6, 42.5, 41.3. HRMS calcd for C8H12ON2Cl, 187.06327 [M + H]+; found 187.06360 [M + H]+.
2-(4-Methoxyphenoxy)-N-(2-(thiophen-3-yl)ethyl)acetamide (23a).
General procedure VI was followed using compound 22a (1.00 g, 4.91 mmol), 4-methoxyphenol (0.731 g, 5.89 mmol), and cesium carbonate (6.40 g, 19.6 mmol) in acetonitrile (15 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–70% EtOAc/hexanes gradient) to afford the title compound as a white solid (1.20 g, 84%). Rf (1:1 EtOAc/Hex): 0.40; 1H NMR (500 MHz, CDCl3) δ 7.28 (dd, J = 4.9, 3.0 Hz, 1H), 6.97–6.94 (m, 1H), 6.93 (dd, J = 4.9, 1.3 Hz, 1H), 6.87–6.77 (m, 4H), 6.66 (s, 1H), 4.42 (s, 2H), 3.77 (s, 3H), 3.60 (d, J = 6.1 Hz, 2H), 2.87 (t, J = 6.9 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 168.4, 154.7, 151.3, 138.8, 128.0, 126.1, 121.6, 115.6, 114.9, 68.1, 55.7, 39.3, 30.2. HRMS calcd for C15H18O3NS, 292.10019 [M + H]+; found 292.10028 [M + H]+.
2-(4-Methoxyphenoxy)-N-(2-(5-methoxythiophen-2-yl)ethyl)-acetamide (23b).
General procedure VI was followed using compound 22b (0.89 g, 3.8 mmol), 4-methoxyphenol (0.57 g, 4.6 mmol) and cesium carbonate (4.96 g, 15.2 mmol) in acetonitrile (38 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a white solid (0.97 g, 79%). Rf (1:1 EtOAc/Hex): 0.41; 1H NMR (400 MHz, CDCl3) δ 6.88–6.79 (m, 4H), 6.75 (bs, 1H), 6.37 (dt, J = 3.7, 0.8 Hz, 1H), 5.98 (d, J = 3.7 Hz, 1H), 4.43 (s, 2H), 3.84 (s, 3H), 3.77 (s, 3H), 3.55 (q, J = 6.6 Hz, 2H), 2.88 (t, J = 7.0 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 168.4, 164.9, 154.7, 151.3, 126.9, 122.7, 115.6, 114.8, 103.2, 68.1, 60.2, 55.7, 40.1, 30.3. HRMS calcd for C16H20O4NS, 322.11076 [M + H]+; found 322.11032 [M + H]+.
2-(4-Methoxyphenoxy)-N-(2-(2-methylthiazol-4-yl)ethyl)-acetamide (23c).
General procedure VI was followed using compound 22c (27 mg, 0.12 mmol), 4-methoxyphenol (18 mg, 0.15 mmol), and cesium carbonate (161 mg, 0.494 mmol) in acetonitrile (0.4 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a clear oil (35 mg, 93%). Rf (1:1 EtOAc/Hex): 0.08; 1H NMR (500 MHz, CDCl3) δ 7.31 (s, 1H), 6.82 (s, 4H), 6.74 (s, 1H), 4.43 (s, 2H), 3.76 (s, 3H), 3.67 (q, J = 6.1 Hz, 2H), 2.93 (t, J = 6.8 Hz, 2H), 2.65 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 168.5, 166.1, 154.6, 153.6, 151.5, 115.6, 114.8, 113.9, 68.2, 55.7, 38.3, 30.9, 19.2. HRMS calcd for C15H19O3N2S, 307.11109 [M + H]+; found 307.11127 [M + H]+.
N-(2-(Furan-3-yl)ethyl)-2-(4-methoxyphenoxy)acetamide (23d).
General procedure VI was followed using compound 22d (0.837 g, 4.46 mmol), 4-methoxyphenol (0.665 g, 5.35 mmol), and cesium carbonate (5.81 g, 17.8 mmol) in acetonitrile (13 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–70% EtOAc/hexanes gradient) to afford the title compound as a clear oil (1.07 g, 87%). Rf (1:1 EtOAc/Hex): 0.18; 1H NMR (500 MHz, CDCl3) δ 7.37 (t, J = 1.7 Hz, 1H), 7.23–7.18 (m, 1H), 6.87–6.76 (m, 4H), 6.67 (bs, 0H), 6.27 (dd, J = 1.8, 0.9 Hz, 1H), 4.42 (s, 2H), 3.77 (s, 3H), 3.54 (q, J = 6.9 Hz, 2H), 2.66 (t, J = 6.9 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 168.4, 154.7, 151.3, 143.3, 139.6, 121.5, 115.6, 114.8, 110.7, 68.1, 55.7, 38.9, 25.0. HRMS calcd for C15H18O4N, 276.12303 [M + H]+; found 276.12303 [M + H]+.
2-(4-Methoxyphenoxy)-N-(2-(1-methyl-1H-pyrazol-5-yl)ethyl)-acetamide (23e).
General procedure VI was followed using compound 22e (0.83 g, 4.1 mmol), 4-methoxyphenol (0.613 g, 4.94 mmol), and cesium carbonate (5.36 g, 16.5 mmol) in acetonitrile (24 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–10% EtOAc/hexanes gradient) to afford the title compound as a white solid (1.0 g, 84%). Rf (1:1 EtOAc/Hex): 0.76; 1H NMR (399 MHz, CDCl3) δ 7.39 (d, J = 1.8 Hz, 1H), 6.87–6.76 (m, 5H), 6.03 (d, J = 1.9 Hz, 1H), 4.43 (s, 2H), 3.81 (s, 3H), 3.77 (s, 3H), 3.61 (q, J = 6.9 Hz, 2H), 2.88 (t, J = 7.1 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 168.7, 154.8, 151.2, 139.1, 138.3, 115.6, 114.8, 104.7, 68.0, 55.7, 37.7, 36.2, 25.8. HRMS calcd for C15H20O3N3, 290.14992 [M + H]+; found 290.14994 [M + H]+.
2-(4-Methoxyphenoxy)-N-(2-(thiophen-2-yl)ethyl)acetamide (23f).
General procedure VI was followed using compound 22f (2.64 g, 13.0 mmol), 4-methoxyphenol (1.93 g, 15.6 mmol), and cesium carbonate (16.9 g, 51.8 mmol) in acetonitrile (39 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–70% EtOAc/hexanes gradient) to afford the title compound as an orange solid (3.26 g, 86%). Rf (1:1 EtOAc/Hex): 0.40; 1H NMR (500 MHz, CDCl3) δ 7.16 (dd, J = 5.1, 1.2 Hz, 1H), 6.93 (dd, J = 5.1, 3.4 Hz, 1H), 6.87–6.77 (m, 6H), 4.43 (s, 2H), 3.77 (s, 3H), 3.61 (q, J = 6.7 Hz, 2H), 3.07 (t, J = 6.8 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 168.5, 154.7, 151.3, 140.9, 127.1, 125.5, 124.1, 115.7, 114.8, 68.1, 55.7, 40.3, 29.9. HRMS calcd for C30H35O6N2S2, 583.19361 [2M + H]+; found 583.19343 [2M + H]+.
N-(2-(1H-Pyrrol-1-yl)ethyl)-2-(4-methoxyphenoxy)acetamide (23g).
General procedure VI was followed using compound 22g (0.87 g, 4.7 mmol), 4-methoxyphenol (0.694 g, 5.59 mmol), and cesium carbonate (6.08 g, 18.7 mmol) in acetonitrile (28 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a yellow oil (1.1 g, 89%). Rf (1:1 EtOAc/Hex): 0.30; 1H NMR (500 MHz, CDCl3) δ 6.88–6.76 (m, 4H), 6.69 (s, 1H), 6.61 (t, J = 2.1 Hz, 2H), 6.17 (t, J = 2.1 Hz, 2H), 4.43 (s, 2H), 4.08–4.01 (m, 2H), 3.78 (s, 3H), 3.65 (q, J = 6.1 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 168.8, 154.8, 151.2, 120.6, 115.6, 114.8, 108.9, 68.0, 55.7, 48.9, 40.4. HRMS calcd for C15H19O3N2, 275.13902 [M + H]+; found 275.13913 [M + H]+.
7-((4-Methoxyphenoxy)methyl)-4,5,6,7-tetrahydrothieno[2,3-c]-pyridine (24a).
General procedure VIII was followed using compound 23a (1.17 g, 4.02 mmol), POCl3 (1.87 mL, 20.1 mmol), and sodium borohydride (0.456 g, 12.1 mmol) in acetonitrile (60 mL) and then methanol (17 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–10% (1% NH4OH in MeOH)/DCM gradient) to afford the title compound as an orange oil (0.120 g, 11%). Rf (90:10:0.1 DCM/MeOH/NH4OH): 0.63; 1H NMR (500 MHz, CDCl3) δ 7.16 (d, J = 5.1 Hz, 1H), 6.91–6.88 (m, 2H), 6.85–6.82 (m, 3H), 4.49–4.43 (m, 1H), 4.17–4.06 (m, 2H), 3.77 (s, 3H), 3.70 (dd, J = 20.3, 11.2 Hz, 1H), 3.31 (dt, J = 12.5, 4.8 Hz, 1H), 3.06 (ddd, J = 13.0, 8.4, 4.9 Hz, 1H), 2.80–2.63 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 154.1, 152.7, 135.5, 133.9, 127.5, 123.0, 115.7, 114.6, 72.2, 55.7, 54.3, 41.7, 26.7. HRMS calcd for C15H18O2NS, 276.10528 [M + H]+; found 276.10526 [M + H]+.
2-Methoxy-4-((4-methoxyphenoxy)methyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine (24b).
General procedure VII was followed using compound 23b (0.94 g, 2.9 mmol), POCl3 (1.36 mL, 14.6 mmol), and sodium borohydride (0.33 g, 8.8 mmol) in acetonitrile (58 mL) and then methanol (15 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a red oil (0.17 g, 19%). Rf (EtOAc): 0.17; 1H NMR (500 MHz, CDCl3) δ 6.88–6.82 (m, 4H), 5.95 (s, 1H), 4.22–4.16 (m, 1H), 4.10 (dd, J = 9.2, 3.7 Hz, 1H), 4.02–3.94 (m, 1H), 3.83 (s, 3H), 3.76 (s, 3H), 3.28 (dt, J = 12.1, 5.2 Hz, 1H), 3.07 (ddd, J = 12.2, 7.4, 4.9 Hz, 1H), 2.76–2.59 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 163.9, 154.0, 152.9, 130.7, 122.2, 115.6, 114.7, 101.3, 71.2, 60.3, 55.7, 54.1, 41.4, 25.4. HRMS calcd for C16H20O3NS, 306.11584 [M + H]+; found 306.11537 [M + H]+.
4-((4-Methoxyphenoxy)methyl)-2-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridine (24c).
General procedure VIII was followed using compound 23c (35 mg, 0.11 mmol), POCl3 (53 μL, 0.57 mmol), and sodium borohydride (13 mg, 0.34 mmol) in acetonitrile (1.8 mL) and then methanol (0.5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–10% (1% NH4OH in MeOH)/DCM gradient) to afford the title compound as a clear oil (3.6 mg, 9%). Rf (90:10:0.1 DCM/MeOH/NH4OH): 0.40; 1H NMR (500 MHz, CDCl3) δ 6.79 (s, 4H), 4.03 (t, J = 5.2 Hz, 2H), 3.73 (s, 3H), 3.12–2.92 (m, 6H), 2.65 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 165.8, 154.3, 153.9, 152.8, 115.5, 114.6, 113.4, 67.5, 55.7, 48.7, 48.5, 31.4, 19.1. HRMS calcd for C15H19O2N2S, 291.11618 [M + H]+; found 291.11615 [M + H]+.
7-((4-Methoxyphenoxy)methyl)-4,5,6,7-tetrahydrofuro[2,3-c]-pyridine (24d).
General procedure VIII was followed using compound 23d (1.02 g, 3.71 mmol), POCl3 (1.73 mL, 18.5 mmol), and sodium borohydride (0.421 g, 11.1 mmol) in acetonitrile (59 mL) and then methanol (15 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–20% (1% NH4OH in MeOH)/DCM gradient) to afford the title compound as an orange oil (0.570 g, 59%). Rf (90:10:0.1 DCM/MeOH/NH4OH): 0.59; 1H NMR (500 MHz, CDCl3) δ 7.31–7.26 (m, 1H), 6.91–6.79 (m, 4H), 6.26 (d, J = 1.9 Hz, 1H), 4.33–4.25 (m, 2H), 4.11–4.03 (m, 1H), 3.76 (s, 3H), 3.21 (dt, J = 12.7, 5.0 Hz, 1H), 2.98 (ddd, J = 12.6, 7.7, 4.9 Hz, 1H), 2.62–2.44 (m, 2H), 2.21 (bs, 1H). 13C NMR (126 MHz, CDCl3) δ 154.0, 152.9, 148.3, 141.1, 117.4, 115.7, 114.6, 110.4, 69.5, 55.7, 52.8, 41.6, 23.9. HRMS calcd for C15H18O3N, 260.12812 [M + H]+; found 260.12799 [M + H]+.
4-((4-Methoxyphenoxy)methyl)-1-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine (24e).
General procedure VIII was followed using compound 23e (0.70 g, 2.4 mmol), POCl3 (1.13 mL, 12.1 mmol), and sodium borohydride (0.275 g, 7.26 mmol) in acetonitrile (39 mL) and then methanol (10 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–10% (1% NH4OH in MeOH)/DCM gradient) to afford the title compound as a clear oil (0.25 g, 38%). Rf (90:10:0.1 DCM/MeOH/NH4OH): 0.47; 1H NMR (399 MHz, CDCl3) δ 7.33 (s, 1H), 6.92–6.78 (m, 4H), 4.28 (dd, J = 8.5, 4.0 Hz, 1H), 4.11 (dd, J = 9.1, 4.0 Hz, 1H), 3.92 (t, J = 8.8 Hz, 1H), 3.76 (s, 3H), 3.76 (s, 3H), 3.32 (dt, J = 12.3, 5.0 Hz, 1H), 3.07–2.98 (m, 1H), 2.74–2.55 (m, 2H), 2.16 (bs, 1H). 13C NMR (100 MHz, CDCl3) δ 154.0, 152.9, 137.5, 134.3, 115.6, 115.2, 114.6, 71.6, 55.7, 51.2, 40.9, 35.6, 22.7. HRMS calcd. HRMS calcd for C15H20O2N3, 274.15500 [M + H]+; found 274.15504 [M + H]+.
4-((4-Methoxyphenoxy)methyl)-4,5,6,7-tetrahydrothieno[3,2-c]-pyridine (24f).
General procedure VIII was followed using compound 23f (1.00 g, 3.43 mmol), POCl3 (1.60 mL, 17.2 mmol), and sodium borohydride (0.390 g, 10.3 mmol) in acetonitrile (60 mL) and then methanol (17 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–10% (1% NH4OH in MeOH)/DCM gradient) to afford the title compound as an orange oil (371 mg, 39%). Rf (90:10:0.1 DCM/MeOH/NH4OH): 0.86; 1H NMR (500 MHz, CDCl3) δ 7.11 (d, J = 5.2 Hz, 1H), 6.91–6.80 (m, 5H), 4.39–4.33 (m, 1H), 4.19 (dd, J = 9.2, 3.8 Hz, 1H), 4.06–3.99 (m, 1H), 3.77 (s, 3H), 3.70 (dd, J = 21.8, 11.2 Hz, 1H), 3.37–3.28 (m, 1H), 3.14–3.05 (m, 1H), 2.97–2.80 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 154.0, 152.9, 135.9, 133.7, 124.6, 122.4, 115.6, 114.7, 71.2, 55.7, 54.2, 41.3, 25.9. HRMS calcd for C15H18O2NS, 276.10528 [M + H]+; found 276.10513 [M + H]+.
1-((4-Methoxyphenoxy)methyl)-1,2,3,4-tetrahydropyrrolo[1,2-a]-pyrazine (24g).
General procedure VIII was followed using compound 23g (1.12 g, 4.08 mmol), POCl3 (1.90 mL, 20.4 mmol), and sodium borohydride (0.463 g, 12.3 mmol) in acetonitrile (65 mL) and then methanol (16 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–10% (1% NH4OH in MeOH)/DCM gradient) to afford the title compound as a white solid (0.777 g, 74%). Rf (90:10:0.1 DCM/MeOH/NH4OH): 0.58; 1H NMR (500 MHz, CDCl3) δ 6.93–6.81 (m, 4H), 6.61 (t, J = 2.0 Hz, 1H), 6.18 (t, J = 3.0 Hz, 1H), 5.96 (dt, J = 3.3, 1.3 Hz, 1H), 4.46 (dd, J = 8.9, 3.3 Hz, 1H), 4.25 (dd, J = 9.2, 3.4 Hz, 1H), 4.06–3.93 (m, 3H), 3.78 (s, 3H), 3.37 (dt, J = 12.4, 4.0 Hz, 1H), 3.24–3.15 (m, 1H), 2.28 (s, 1H). 13C NMR (126 MHz, CDCl3) δ 154.0, 152.8, 126.8, 119.5, 115.5, 114.7, 107.8, 102.8, 71.3, 55.7, 53.0, 45.6, 42.0. HRMS calcd for C15H19O2N2, 259.14410 [M + H]+; found 259.14418 [M + H]+.
(3-Chlorophenyl)(7-((4-methoxyphenoxy)methyl)-4,7-dihydrothieno[2,3-c]pyridin-6(5H)-yl)methanone (25a).
General procedure IX was followed using compound 24a (100 mg, 0.363 mmol), triethylamine (101 μL, 0.726 mmol), and 3-chlorobenzoyl chloride (56 μL, 0.44 mmol) in DCM (5.7 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–50% EtOAc/hexanes gradient) to afford the title compound as rotamers in solution and a white foam (50 mg, 33%). Rf (1:1 EtOAc/Hex): 0.69; 1H NMR (600 MHz, CDCl3, probe temp: −25 °C) δ 8.05–8.01 (m, 0.05H), 7.96–7.92 (m, 0.05H), 7.63 (bs, 0.3H), 7.57–7.52 (m, 0.5H), 7.45–7.32 (m, 3.3H), 7.32–7.23 (m, 2H), 6.95–6.78 (m, 5.2H), 6.09 (t, J = 5.4 Hz, 0.6H), 5.31 (dd, J = 9.1, 4.3 Hz, 0.4H), 4.92 (dd, J = 13.3, 5.6 Hz, 0.4H), 4.36 (dd, J = 9.3, 5.2 Hz, 0.6H), 4.28 (dd, J = 9.3, 6.1 Hz, 0.6H), 4.19 (t, J = 9.6 Hz, 0.4H), 3.98 (dd, J = 9.9, 4.4 Hz, 0.4H), 3.89 (dd, J = 13.7, 4.8 Hz, 0.6H), 3.77 (s, 3H), 3.63–3.54 (m, 0.6H), 3.26–3.15 (m, 0.4H), 2.99 (ddd, J = 17.1, 12.4, 5.7 Hz, 0.4H), 2.83–2.74 (m, 1H), 2.71 (dd, J = 15.9, 3.1 Hz, 0.6H). 13C NMR (151 MHz, CDCl3, probe temp: −25 °C) δ 170.5, 169.6, 153.9, 153.8, 152.3, 151.8, 137.6, 137.4, 136.0, 134.7, 134.4, 134.1, 132.1, 130.3, 130.1, 130.0, 130.0, 128.2, 127.8, 127.3, 126.8, 126.7, 125.5, 124.8, 124.7, 124.6, 124.3, 69.9, 69.6, 55.7, 55.6, 50.7, 43.2, 35.9, 26.5, 25.4. HRMS calcd for C22H21O3NClS, 414.09252 [M + H]+; found 414.09298 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 2.42 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 0.702 min).
(5-Chlorothiophen-2-yl)(7-((4-methoxyphenoxy)methyl)-4,7-dihydrothieno[2,3-c]pyridin-6(5H)-yl)methanone (25b).
General procedure XIII was followed using compound 24a (25 mg, 0.091 mmol), EDCI (17 mg, 0.11 mmol), DMAP (13 mg, 0.11 mmol), and 5-chlorothiophene-2-carboxylic acid (16 mg, 0.10 mmol) in DCM (0.4 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–70% EtOAc/hexanes gradient) to afford the title compound as rotamers in solution and a clear oil (10 mg, 26%). Rf (1:1 EtOAc/Hex): 0.90; 1H NMR (600 MHz, CDCl3, probe temp: −25 °C) δ 7.41 (dd, J = 15.3, 1.8 Hz, 0.7H), 7.30 (d, J = 5.1 Hz, 0.7H), 7.24 (d, J = 5.1 Hz, 0.3H), 7.08 (dd, J = 15.3, 1.9 Hz, 0.3H), 6.89–6.73 (m, 6H), 6.02 (t, J = 5.2 Hz, 0.3H), 5.47 (dd, J = 8.9, 4.2 Hz, 0.7H), 4.91 (dd, J = 13.3, 4.7 Hz, 0.7H), 4.27 (dd, J = 9.4, 4.9 Hz, 0.3H), 4.22–4.15 (m, 2H), 4.12 (dd, J = 13.6, 4.3 Hz, 0.3H), 3.77 (s, 2H), 3.76 (s, 1H), 3.05 (td, J = 13.1, 4.3 Hz, 0.7H), 2.89–2.73 (m, 2H). 13C NMR (151 MHz, CDCl3, probe temp: −25 °C) δ 163.8, 162.9, 154.0, 153.8, 152.1, 151.6, 136.6, 134.1, 132.0, 130.0, 129.8, 129.7, 129.4, 129.4, 129.3, 129.3, 129.3, 129.2, 129.2, 129.0, 128.0, 127.3, 126.7, 125.0, 124.7, 123.5, 121.8, 115.4, 114.8, 114.6, 114.4, 69.9, 55.7, 55.7, 54.6, 51.0, 42.1, 36.1, 26.6, 25.4. HRMS calcd for C20H19O3NClS2, 420.04894 [M + H]+; found 420.04910 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 2.68 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 0.76 min).
(E)-4,4,4-Trifluoro-1-(7-((4-methoxyphenoxy)methyl)-4,7-dihydrothieno[2,3-c]pyridin-6(5H)-yl)but-2-en-1-one (25c).
General procedure XIII was followed using compound 24a (25 mg, 0.091 mmol), EDCI (17 mg, 0.11 mmol), DMAP (13 mg, 0.11 mmol), and (E)-4,4,4-trifluorobut-2-enoic acid (14 mg, 0.10 mmol) in DCM (0.4 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–70% EtOAc/hexanes gradient) to afford the title compound as rotamers in solution and a yellow oil (12 mg, 33%). Rf (1:1 EtOAc/Hex): 0.71; 1H NMR (600 MHz, CDCl3, probe temp: −25 °C) δ 7.62 (d, J = 3.9 Hz, 0.5H), 7.28–7.24 (m, 1H), 7.15 (d, J = 3.8 Hz, 0.5H), 6.95–6.77 (m, 6H), 5.91 (t, J = 5.0 Hz, 0.5H), 5.81 (dd, J = 8.5, 5.0 Hz, 0.5H), 4.74 (dd, J = 13.4, 5.1 Hz, 0.5H), 4.47 (dd, J = 14.0, 4.8 Hz, 0.5H), 4.39–4.34 (m, 0.5H), 4.31–4.17 (m, 1.5H), 3.78 (s, 1.5H), 3.76 (s, 1.5H), 3.74–3.67 (m, 0.5H), 3.19 (td, J = 12.9, 3.5 Hz, 0.5H), 3.02–2.87 (m, 1H), 2.86–2.79 (m, 0.5H), 2.73 (dd, J = 16.4, 3.0 Hz, 0.5H). HRMS calcd for C19H19O3NF3S, 398.10323 [M + H]+; found 398.10325 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 2.00 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 0.64 min).
(3-Chlorophenyl)(2-methoxy-4-((4-methoxyphenoxy)methyl)-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)methanone (25d).
General procedure IX was followed using compound 24b (25 mg, 0.082 mmol), triethylamine (23 μL, 0.16 mmol), and 3-chlorobenzoyl chloride (13 μL, 0.10 mmol) in DCM (1.3 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a mixture of rotamers in solution and a yellow oil (13 mg, 36%). Rf (1:1 EtOAc/Hex): 0.63; 1H NMR (600 MHz, CDCl3, probe temp: −25 °C) δ 7.65 (bs, 0.5H), 7.44–7.32 (m, 3.5H), 7.29–7.24 (m, 0.5H), 6.90–6.76 (m, 4.5H), 6.01 (s, 0.5H), 5.79 (t, J = 4.6 Hz, 0.5H), 5.79 (s, 0.5H), 5.03 (dd, J = 9.5, 3.0 Hz, 0.5H), 4.94 (dd, J = 13.2, 5.6 Hz, 0.5H), 4.37–4.26 (m, 1H), 4.10 (t, J = 9.8 Hz, 0.5H), 3.94–3.86 (m, 1H), 3.86 (s, 1.5H), 3.81 (s, 1.5H), 3.77 (s, 3H), 3.73–3.67 (m, 0.5H), 3.24 (td, J = 12.8, 4.2 Hz, 0.5H), 3.06–2.97 (m, 0.5H), 2.85–2.76 (m, 0.5H), 2.68 (dd, J = 16.2, 3.6 Hz, 0.5H), 2.63–2.57 (m, 0.5H). 13C NMR (151 MHz, CDCl3, probe temp: −25 °C) δ 172.5, 170.6, 169.7, 164.5, 164.5, 153.8, 153.7, 152.5, 152.0, 137.7, 137.6, 134.7, 134.2, 130.3, 130.0, 129.9, 129.8, 129.1, 127.9, 127.7, 126.8, 125.7, 124.7, 121.9, 119.7, 115.3, 114.8, 114.5, 114.4, 101.0, 100.8, 69.4, 68.2, 60.1, 60.1, 56.1, 55.7, 55.7, 51.0, 43.5, 36.1, 25.2, 24.1. HRMS calcd for C23H23O4NClS, 444.10308 [M + H]+; found 444.10290 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 2.33 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 0.69 min).
(3-Chlorophenyl)(4-((4-methoxyphenoxy)methyl)-2-methyl-6,7-dihydrothiazolo[5,4-c]pyridin-5(4H)-yl)methanone (25e).
General procedure IX was followed using compound 24c (5.0 mg, 0.014 mmol), triethylamine (3.8 μL, 0.028 mmol), and 3-chlorobenzoyl chloride (2.1 μL, 0.017 mmol) in DCM (0.2 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as rotamers in solution and a yellow oil (5.5 mg, 93%). Rf (1:1 EtOAc/Hex): 0.61; 1H NMR (600 MHz, CDCl3, probe temp: −25 °C) δ 7.58 (s, 0.2H), 7.46–7.34 (m, 3H), 7.32–7.27 (m, 0.8H), 6.92–6.77 (m, 4H), 6.02–5.97 (m, 0.8H), 5.26–5.21 (m, 0.2H), 4.95 (dd, J = 13.3, 5.7 Hz, 0.2H), 4.34 (dd, J = 9.0, 4.6 Hz, 0.8H), 4.19–4.13 (m, 1H), 3.99–3.91 (m, 1H), 3.76 (s, 2H), 3.75 (s, 1H), 3.65–3.56 (m, 0.8H), 3.28–3.19 (m, 0.2H), 3.14–3.04 (m, 0.2H), 2.93–2.81 (m, 1.8H), 2.68 (s, 2H), 2.67 (s, 1H). 13C NMR (151 MHz, CDCl3, probe temp: −25 °C) δ 172.5, 170.4, 169.6, 165.8, 154.0, 153.9, 152.0, 151.6, 149.9, 148.0, 137.2, 137.1, 134.8, 134.5, 130.3, 130.3, 130.2, 130.2, 127.6, 127.0, 125.6, 125.2, 124.6, 123.7, 115.3, 114.9, 114.5, 114.5, 69.1, 68.9, 55.7, 54.4, 49.6, 43.4, 36.4, 27.6, 26.7, 19.5, 19.4. HRMS calcd for C22H22O3N2ClS, 429.10342 [M + H]+; found 429.10342 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 1.69 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 0.65 min).
(3-Chlorophenyl)(7-((4-methoxyphenoxy)methyl)-4,7-dihydrofuro[2,3-c]pyridin-6(5H)-yl)methanone (25f).
General procedure IX was followed using compound 24d (28 mg, 0.108 mmol), triethylamine (30 μL, 0.21 mmol), and 3-chlorobenzoyl chloride (17 μL, 0.13 mmol) in DCM (1.7 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as rotamers in solution and a clear oil (19 mg, 44%). Rf (1:1 EtOAc/Hex): 0.44; 1H NMR (600 MHz, CDCl3, probe temp: −25 °C) δ 7.62 (s, 0.3H), 7.44–7.32 (m, 4H), 7.28–7.24 (m, 0.7H), 6.86–6.77 (m, 4H), 6.35–6.30 (m, 1H), 5.86 (t, J = 4.1 Hz, 0.7H), 5.17 (dd, J = 8.8, 2.3 Hz, 0.3H), 4.89 (dd, J = 13.3, 5.3 Hz, 0.3H), 4.49 (dd, J = 10.1, 3.2 Hz, 0.7H), 4.40 (dd, J = 10.1, 4.2 Hz, 0.7H), 4.11 (t, J = 9.7 Hz, 0.3H), 4.05 (dd, J = 10.1, 3.2 Hz, 0.3H), 3.83 (dd, J = 13.7, 4.9 Hz, 0.7H), 3.77 (s, 1H), 3.77 (s, 2H), 3.75–3.68 (m, 0.7H), 3.25–3.13 (m, 0.3H), 2.88–2.78 (m, 0.3H), 2.67–2.58 (m, 0.7H), 2.58–2.45 (m, 1H). 13C NMR (151 MHz, CDCl3, probe temp: −25 °C) δ 170.9, 169.9, 153.8, 153.7, 152.5, 151.9, 145.7, 144.8, 142.6, 142.3, 137.7, 137.4, 134.6, 134.3, 130.2, 130.0, 129.9, 129.9, 127.8, 126.8, 125.6, 124.7, 118.5, 116.9, 115.3, 114.8, 114.5, 114.4, 110.5, 110.1, 68.5, 67.4, 55.7, 54.9, 50.1, 44.4, 36.7, 23.2, 22.0. HRMS calcd for C22H21O4NCl, 398.11536 [M + H]+; found 398.11536 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 2.11 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 0.67 min).
(3-Chlorophenyl)(4-((4-methoxyphenoxy)methyl)-1-methyl-1,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridin-5-yl)methanone (25g).
General procedure IX was followed using compound 24e (49 mg, 0.18 mmol), triethylamine (50 μL, 0.36 mmol), and 3-chlorobenzoyl chloride (28 μL, 0.38 mmol) in DCM (2.8 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as rotamers in solution and a white solid (71 mg, 96%). Rf (1:1 EtOAc/Hex): 0.64; 1H NMR (600 MHz, CDCl3, probe temp: −25 °C) δ 7.62 (s, 0.5H), 7.47 (s, 0.5H), 7.44–7.31 (m, 3H), 7.29–7.23 (m, 0.5H), 7.18 (dd, J = 7.5, 3.9 Hz, 0.5H), 6.89–6.76 (m, 4H), 5.96 (d, J = 5.4 Hz, 0.5H), 5.18 (dd, J = 9.5, 4.0 Hz, 0.5H), 4.98 (dd, J = 13.4, 5.8 Hz, 0.5H), 4.26 (dd, J = 9.4, 5.4 Hz, 0.5H), 4.20 (dd, J = 9.4, 5.7 Hz, 0.5H), 4.04 (t, J = 9.8 Hz, 0.5H), 3.93 (dd, J = 13.9, 5.3 Hz, 0.5H), 3.84 (dd, J = 10.0, 4.1 Hz, 0.5H), 3.79 (s, 1.5H), 3.79 (s, 1.5H), 3.76 (s, 3H), 3.63–3.53 (m, 0.5H), 3.17 (td, J = 13.0, 4.3 Hz, 0.5H), 2.93 (ddd, J = 17.5, 12.0, 5.9 Hz, 0.5H), 2.79–2.68 (m, 1H), 2.65 (dd, J = 15.5, 3.5 Hz, 0.5H). 13C NMR (151 MHz, CDCl3, probe temp: −25 °C) δ 171.0, 169.9, 153.8, 153.7, 152.4, 151.9, 137.8, 137.6, 137.5, 135.9, 135.6, 134.9, 134.7, 134.3, 130.3, 130.1, 130.0, 130.0, 129.1, 128.3, 127.8, 126.7, 125.5, 125.4, 124.6, 115.3, 114.8, 114.5, 114.4, 113.8, 112.8, 69.4, 68.7, 55.7, 53.0, 47.8, 42.4, 36.1, 35.1, 22.5, 21.7, 21.4. HRMS calcd for C22H23O3N3Cl, 412.14225 [M + H]+; found 412.14237 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 1.53 min). Method 2: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 0.82 min).
(3-Chlorophenyl)(4-((4-methoxyphenoxy)methyl)-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)methanone (25h).
General procedure IX was followed using compound 24f (50 mg, 0.18 mmol), triethylamine (51 μL, 0.36 mmol), and 3-chlorobenzoyl chloride (28 μL, 0.22 mmol) in DCM (2.8 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–70% EtOAc/hexanes gradient) to afford the title compound as rotamers in solution and a white foam (49 mg, 65%). Rf (1:1 EtOAc/Hex): 0.77; 1H NMR (600 MHz, CDCl3, probe temp: −25 °C) δ 7.67 (bs, 0.5H), 7.45–7.34 (m, 3H), 7.31–7.27 (m, 0.5H), 7.19 (dd, J = 16.2, 5.2 Hz, 1H), 6.99 (d, J = 5.2 Hz, 0.5H), 6.91–6.78 (m, 4H), 6.75 (d, J = 5.2 Hz, 0.5H), 5.99 (t, J = 4.8 Hz, 0.5H), 5.22 (dd, J = 9.6, 3.5 Hz, 0.5H), 4.99 (dd, J = 13.1, 5.4 Hz, 0.5H), 4.35 (qd, J = 9.8, 5.1 Hz, 1H), 4.11 (t, J = 9.8 Hz, 0.5H), 3.97–3.90 (m, 1H), 3.77 (s, 3H), 3.73–3.65 (m, 0.5H), 3.25 (td, J = 12.7, 4.0 Hz, 0.5H), 3.19–3.07 (m, 0.5H), 2.97–2.89 (m, 1H), 2.84 (dd, J = 16.0, 3.1 Hz, 0.5H). 13C NMR (151 MHz, CDCl3, probe temp: −25 °C) δ 170.7, 169.8, 153.8, 153.7, 152.5, 152.0, 137.7, 137.6, 136.3, 134.7, 134.3, 133.9, 132.3, 130.9, 130.3, 130.0, 129.9, 129.9, 128.0, 126.8, 125.7, 125.6, 125.0, 124.7, 123.9, 123.8, 69.5, 68.4, 56.2, 55.7, 51.1, 43.3, 36.1, 25.8, 24.8. HRMS calcd for C44H40O6N2Cl2NaS2, 849.15970 [2M + Na]+; found 849.16095 [2M + Na]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 2.33 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 0.69 min).
(3-Chlorophenyl)(1-((4-methoxyphenoxy)methyl)-3,4-dihydropyrrolo[1,2-a]pyrazin-2(1H)-yl)methanone (25i).
General procedure IX was followed using compound 24g (50 mg, 0.19 mmol), triethylamine (54 μL, 0.39 mmol), and 3-chlorobenzoyl chloride (30 μL, 0.23 mmol) in DCM (3.0 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–70% EtOAc/hexanes gradient) to afford the title compound as rotamers in solution and a white foam (67 mg, 87%). Rf (1:1 EtOAc/Hex): 0.58; 1H NMR (600 MHz, CDCl3, probe temp: −25 °C) δ 7.69 (bs, 0.7H), 7.48–7.31 (m, 3H), 7.26–7.22 (m, 0.3H), 6.88–6.74 (m, 4H), 6.72–6.68 (m, 0.7H), 6.67–6.62 (m, 0.3H), 6.24 (t, J = 3.1 Hz, 0.3H), 6.21 (t, J = 3.1 Hz, 0.7H), 6.17 (t, J = 4.9 Hz, 0.3H), 6.12 (d, J = 3.4 Hz, 0.3H), 5.94 (d, J = 2.4 Hz, 0.7H), 5.44 (dd, J = 10.2, 3.8 Hz, 0.7H), 4.97 (dd, J = 13.7, 3.9 Hz, 0.7H), 4.40 (dd, J = 9.8, 4.2 Hz, 0.3H), 4.28 (dd, J = 9.8, 5.8 Hz, 0.3H), 4.20–4.07 (m, 2H), 4.04–3.84 (m, 2H), 3.77 (s, 3H), 3.51–3.42 (m, 0.7H). 13C NMR (151 MHz, CDCl3, probe temp: −25 °C) δ 170.7, 169.5, 153.8, 153.7, 152.5, 151.9, 137.2, 136.8, 134.8, 134.3, 130.4, 130.2, 130.1, 129.9, 128.1, 126.9, 125.9, 124.8, 124.6, 123.2, 120.2, 119.5, 115.4, 114.8, 114.5, 114.4, 108.8, 108.6, 105.0, 104.8, 70.2, 68.7, 55.7, 54.3, 49.4, 45.0, 44.3, 43.5, 35.9. HRMS calcd for C22H22O3N2Cl, 397.13135 [M + H]+; found 397.13144 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 1.87 min). Method 2: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 0.96 min).
(1-((4-Methoxyphenoxy)methyl)-3,4-dihydropyrrolo[1,2-a]-pyrazin-2(1H)-yl)(3-(trifluoromethyl)phenyl)methanone (25j).
General procedure IX was followed using compound 24g (100 mg, 0.387 mmol), triethylamine (108 μL, 0.774 mmol), and 3-chlorobenzoyl chloride (70 μL, 0.46 mmol) in DCM (6.5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–70% EtOAc/hexanes gradient) to afford the title compound as rotamers in solution and a white foam (0.10 g, 62%). Rf (1:1 EtOAc/Hex): 0.60; 1H NMR (600 MHz, CDCl3, probe temp: −25 °C) δ 8.01 (bs, 0.7H), 7.80 (d, J = 7.2 Hz, 0.7H), 7.72 (d, J = 7.6 Hz, 1H), 7.65 (s, 0.3H), 7.63–7.54 (m, 1.3H), 6.89–6.75 (m, 4H), 6.74–6.70 (m, 0.7H), 6.66 (m, 0.3H), 6.28–6.17 (m, 1H), 6.14 (d, J = 3.3 Hz, 0.3H), 5.95 (d, J = 2.5 Hz, 0.7H), 5.42 (dd, J = 10.4, 3.8 Hz, 0.7H), 5.00 (dd, J = 13.7, 4.0 Hz, 0.7H), 4.43 (dd, J = 9.9, 4.2 Hz, 0.3H), 4.34–4.29 (m, 0.3H), 4.23–4.09 (m, 2H), 4.06–3.95 (m, 1H), 3.89 (dd, J = 10.1, 4.0 Hz, 1H), 3.77 (s, 3H), 3.50 (td, J = 13.5, 4.1 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 170.8, 169.5, 153.9, 153.7, 152.5, 151.8, 136.3, 136.0, 131.2, 130.8, 130.6, 130.0, 129.6, 129.1, 126.9, 125.1, 124.7, 124.6, 123.8, 123.0, 122.9, 120.3, 119.5, 115.4, 114.8, 114.5, 114.4, 108.9, 108.6, 105.1, 104.9, 70.2, 68.6, 55.7, 54.3, 49.5, 44.9, 44.3, 43.5, 35.9. HRMS calcd for C23H22O3N2F3, 431.15770 [M + H]+; found 431.15762 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 1.90 min). Method 2: 95% MeOH in water over 3 min at 1 mL/min (retention time = 0.90 min).
(3-Chlorophenyl)(4-((4-methoxyphenoxy)methyl)-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)methanethione (26).
General procedure XIV was followed using compound 25a (48 mg, 0.12 mmol) and Lawesson’s reagent (80 mg, 0.20 mmol) in toluene (1.5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–40% EtOAc/hexanes gradient) to afford the title compound as a clear oil (11 mg, 18%). Rf (1:1 EtOAc/Hex): 0.77; 1H NMR (600 MHz, CDCl3) δ 7.24–7.15 (m, 3H), 7.06–7.01 (m, 2H), 6.83–6.77 (m, 3H), 6.76–6.71 (m, 3H), 6.49 (d, J = 15.2 Hz, 1H), 4.91 (dd, J = 6.9, 4.7 Hz, 1H), 4.82 (d, J = 15.2 Hz, 1H), 4.53 (d, J = 19.6 Hz, 1H), 4.28 (d, J = 19.6 Hz, 1H), 4.11–4.03 (m, 2H), 3.81 (s, 3H), 3.76 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 199.4, 159.9, 154.4, 151.8, 137.2, 134.6, 133.4, 130.1, 127.9, 127.7, 126.7, 125.5, 123.1, 115.5, 114.7, 113.3, 111.6, 70.6, 62.7, 56.2, 55.7, 55.4, 47.7. HRMS calcd for C25H25O3NClS, 454.12382 [M + H]+; found 454.12380 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 2.84 min). Method 2: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 1.41 min).
Methyl 2-(1H-Pyrrol-1-yl)acetate (27).
Glycine methyl ester hydrochloride (26) (5.00 g, 39.8 mmol, 1 equiv) and sodium acetate (4.90 g, 59.7 mmol, 1.5 equiv) were dissolved in a 2:1 (v/v) mixture of acetic acid (50 mL) and water (25 mL). 2,5-Dimethoxytetrahydrofuran (5.66 mL, 43.8 mmol, 1.1 equiv) was then added, and the resulting mixture was heated to 100 °C for 4 h before cooling to room temperature. The crude mixture was poured into water, washed with EtOAc, and the aqueous phase was neutralized with solid Na2CO3. This was then extracted with EtOAc, washed with brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–50% EtOAc/hexanes gradient) to afford the title compound as a clear oil (3.76 g, 68%). Rf (1:1 EtOAC/Hex): 0.68; 1H NMR (399 MHz, CDCl3) δ 6.70 (t, J = 2.1 Hz, 2H), 6.24 (t, J = 2.1 Hz, 2H), 4.67 (s, 2H), 3.79 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.3, 121.8, 109.1, 52.5, 50.7. HRMS calcd for C7H10O2N, 140.07061 [M + H]+; found 140.07063 [M + H]+.
Methyl 2-(2-(2-Chloroacetyl)-1H-pyrrol-1-yl)acetate (28).
General procedure XV was followed using 27 (6.80 g, 48.9 mmol), DBN (0.91 g, 7.3 mmol), and 2-chloroacetyl chloride (4.70 mL, 58.6 mmol) in PhMe (82 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 40 g column, 0–100% EtOAc/hexanes gradient) to afford the title compound as a white solid (8.8 g, 83%). Rf (1:1 EtOAC/Hex): 0.60; 1H NMR (500 MHz, CDCl3) δ 7.06 (dd, J = 4.2, 1.6 Hz, 1H), 6.92 (dd, J = 2.4, 1.7 Hz, 1H), 6.26 (dd, J = 4.2, 2.6 Hz, 1H), 5.03 (s, 2H), 4.51 (s, 2H), 3.76 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 181.6, 168.8, 132.3, 127.7, 120.4, 109.5, 51.0, 45.5. HRMS calcd for C9H13O3NCl, 216.04220 [M + H]+; found 216.04225 [M + H]+.
Methyl 2-(2-(2-(4-Methoxyphenoxy)acetyl)-1H-pyrrol-1-yl)-acetate (29).
General procedure XVI was followed using 28 (0.84 g, 3.9 mmol), potassium iodide (0.77 g, 4.6 mmol), 4-methoxyphenol (0.94 g, 4.6 mmol), and potassium carbonate (2.14 g, 15.5 mmol) in acetone (19 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 24 g column, 0–70% EtOAc/hexanes gradient) to afford the title compound as a white solid (1.6 g, quant.). Rf (1:1 EtOAC/Hex): 0.76; 1H NMR (399 MHz, CDCl3) δ 7.17 (dd, J = 4.1, 1.6 Hz, 1H), 6.90 (dd, J = 2.5, 1.5 Hz, 1H), 6.89–6.76 (m, 4H), 6.26 (dd, J = 4.2, 2.6 Hz, 1H), 5.05 (s, 2H), 5.00 (s, 2H), 3.75 (s, 3H), 3.74 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 185.5, 168.9, 154.2, 152.3, 131.6, 127.8, 119.6, 115.7, 114.6, 109.4, 70.8, 55.7, 52.4, 51.0. HRMS calcd for C16H18O5N, 304.11795 [M + H]+; found 304.11780 [M + H]+.
1-((4-Methoxyphenoxy)methylene)-2-(3-(trifluoromethyl)-benzyl)-1,2-dihydropyrrolo[1,2-a]pyrazin-3(4H)-one (30a).
This procedure was run at a concentration of 1 M due to incomplete conversion of the starting material to product. General procedure XVII was followed using compound 29 (0.20 g, 0.66 mmol), 3-(trifluoromethyl)benzylamine (113 μL, 0.790 mmol), tetraisopropoxytitanium (0.58 mL, 2.0 mmol), and sodium triacetoxyborohydride (0.42 g, 2.0 mmol) in DCE (0.8 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 12 g column, 0–35% EtOAc/hexanes gradient) to afford the title compound as a mixture of E/Z isomers and a white solid (89 mg, 32%). HRMS calcd for C23H20O3N2F3, 429.14205 [M + H]+; found 429.14178 [M + H]+.
2-(3-Fluorobenzyl)-1-((4-methoxyphenoxy)methylene)-1,2-dihydropyrrolo[1,2-a]pyrazin-3(4H)-one (30b).
This procedure was run at a concentration of 1 M due to incomplete conversion of the starting material to product. General procedure XVII was followed using compound 29 (50 mg, 0.16 mmol), 3-fluorobenzylamine (28 μL, 0.20 mmol), tetraisopropoxytitanium (0.15 mL, 0.49 mmol), and sodium triacetoxyborohydride (0.10 g, 0.49 mmol) in DCE (0.16 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–35% EtOAc/hexanes gradient) to afford the title compound as a mixture of E/Z isomers and a white solid (24 mg, 38%). HRMS calcd for C22H20O3N2F, 379.14525 [M + H]+; found 379.14551 [M + H]+.
1-((4-Methoxyphenoxy)methyl)-2-(3-(trifluoromethyl)benzyl)-1,2-dihydropyrrolo[1,2-a]pyrazin-3(4H)-one (31a).
General procedure XVIII was followed using compound 30a (55 mg, 0.13 mmol) and Pd/C (20 mg, 0.019 mmol) in EtOAc (5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–35% EtOAc/hexanes gradient) to afford the title compound as a white foam (20 mg, 36%). Rf (1:1 EtOAc/Hex): 0.62; 1H NMR (500 MHz, CDCl3) δ 7.54–7.50 (m, 2H), 7.45 (d, J = 7.7 Hz, 1H), 7.42–7.37 (m, 1H), 6.82–6.75 (m, 2H), 6.71–6.66 (m, 2H), 6.65 (dd, J = 2.7, 1.6 Hz, 1H), 6.22 (dd, J = 3.5, 2.8 Hz, 1H), 6.00 (dd, J = 3.5, 1.5 Hz, 1H), 5.44 (d, J = 15.4 Hz, 1H), 4.87 (d, J = 17.0 Hz, 1H), 4.83 (dd, J = 4.5, 3.3 Hz, 1H), 4.77 (d, J = 17.0 Hz, 1H), 4.49 (d, J = 15.4 Hz, 1H), 4.15 (dd, J = 9.7, 3.2 Hz, 1H), 4.04 (dd, J = 9.7, 4.7 Hz, 1H), 3.75 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 166.3, 154.5, 152.0, 137.4, 131.2, 131.1 (q, J = 31.7 Hz, 2J), 129.35, 124.80 (q, J = 3.8 Hz, 3J), 124.6 (q, J = 3.8 Hz, 3J), 124.0, 123.9 (q, J = 272.5 Hz, 1J), 118.9, 115.6, 114.7, 109.6, 103.9, 72.4, 55.7, 55.0, 49.0, 48.6. HRMS calcd for C23H22O3N2F3, 431.15770 [M + H]+; found 431.15735 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 1.82 min). Method 2: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 0.87 min).
2-(3-Fluorobenzyl)-1-((4-methoxyphenoxy)methyl)-1,2-dihydropyrrolo[1,2-a]pyrazin-3(4H)-one (EU-1180–453).
General procedure XVIII was followed using compound 30b (24 mg, 0.063 mmol) and Pd/C (10 mg, 0.010 mmol) in EtOAc (5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–35% EtOAc/hexanes gradient) to afford the title compound as a white solid (14 mg, 58%). Rf (1:1 EtOAc/Hex): 0.68; mp 87–88 °C, 1H NMR (400 MHz, CDCl3) δ 7.30–7.20 (m, 1H), 7.03 (d, J = 7.9 Hz, 1H), 7.00–6.90 (m, 2H), 6.83–6.74 (m, 2H), 6.73–6.65 (m, 2H), 6.64 (dd, J = 2.7, 1.6 Hz, 1H), 6.22 (dd, J = 3.3, 2.8 Hz, 1H), 5.99 (dd, J = 3.5, 1.5 Hz, 1H), 5.43 (d, J = 15.4 Hz, 1H), 4.86 (d, J = 17.0 Hz, 1H), 4.82 (d, J = 3.7 Hz, 2H), 4.75 (d, J = 17.0 Hz, 1H), 4.37 (d, J = 15.4 Hz, 1H), 4.15 (dd, J = 9.7, 3.2 Hz, 1H), 4.02 (dd, J = 9.7, 4.5 Hz, 1H), 3.75 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.2, 163.0(d, J = 246.9 Hz, 1J), 154.3, 152.0, 138.7 (d, J = 7.1 Hz, 3J), 130.33 (d, J = 8.2 Hz, 3J), 124.1, 123.5 (d, J = 2.9 Hz, 4J), 118.8, 115.6, 114.9 (d, J = 21.9 Hz, 2J), 114.7 (d, J = 21.2 Hz, 2J), 114.6, 109.5, 103.8, 72.2, 55.7, 54.6, 49.0, 48.2. HRMS calcd for C22H22O3N2F, 381.16090 [M + H]+; found 381.16132 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 1.41 min). Method 2: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 0.80 min).
1-((4-Ethoxyphenoxy)methyl)-2-(3-(trifluoromethyl)benzyl)-1,2-dihydropyrrolo[1,2-a]pyrazine-3(4H)-thione (32).
General procedure XIV was followed using compound 31a (13 mg, 0.030 mmol) and Lawesson’s reagent (18 mg, 0.045 mmol) in toluene (0.5 mL). The crude product was purified via flash column chromatography (ISCO, Redisep 4 g column, 0–20% EtOAc/hexanes gradient) to afford the title compound as a yellow oil (4.3 mg, 32%). Rf (1:1 EtOAc/Hex): 0.83; 1H NMR (399 MHz, CDCl3) δ 7.57–7.48 (m, 2H), 7.43–7.37 (m, 2H), 6.84–6.69 (m, 4H), 6.67 (dd, J = 2.7, 1.6 Hz, 1H), 6.40 (d, J = 15.3 Hz, 1H), 6.22 (dd, J = 3.5, 2.8 Hz, 1H), 6.00 (dd, J = 3.5, 1.4 Hz, 1H), 5.44 (d, J = 18.0 Hz, 1H), 5.15 (d, J = 18.0 Hz, 1H), 5.07 (dd, J = 6.0, 3.4 Hz, 1H), 4.93 (d, J = 15.3 Hz, 1H), 4.17 (dd, J = 10.0, 3.4 Hz, 1H), 4.10 (dd, J = 10.0, 6.1 Hz, 1H), 3.76 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 195.2, 154.6, 151.7, 135.7, 131.2 (q, J = 32.5 Hz, 2J), 130.8, 124.9 (q, J = 3.7 Hz, 3J), 124.7 (q, J = 3.7 Hz, 3J), 124.4 (q, J = 271.8 Hz, 1J), 122.8, 118.7, 115.6, 114.7, 109.4, 104.1, 77.2, 72.0, 58.2, 56.6, 55.7. HRMS calcd for C23H21O2N2F3S, 445.12031 [M − H]+; found 445.12034 [M − H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (4.6 mm × 50 mm, 3.5 μm). Method 1: 75–95% MeOH in water over 3 min at 1 mL/min (retention time = 2.59 min). Method 2: 85–95% MeOH in water over 5 min at 1 mL/min (retention time = 1.17 min).
Separation of 25i Enantiomers.
Semipreparative separation of enantiomers from racemic 25i (0.020 g) was done using a ChiralPak AD-H (30 mm × 250 mm) with the following conditions: 20 mL/min flow rate, 10 mL injection volume (2 mg/1 mL), 90% hexanes/10% IPA over 90 min to afford R-(+)-25i, tR 59.2 min; S-(−)-25i, tR 70.7 min. The enantiomeric excess (ee) was determined using an Agilent pump on a ChiralPak OD-H column (4.6 mm × 150 mm, 5 μm) with the following conditions: 1 mL/min flow rate, 10 μL injection volume, 90% hexanes/10% IPA. R-(+)-25i: tR 31.5 min; >99% ee; . S-(−)-25i: tR 41.4 min, >99% ee; .
Separation of 25j Enantiomers.
Semipreparative separation of 25j enantiomers from racemic 25j (0.020 g) was done using a ChiralPak AD-H (30 mm × 250 mm) with the following conditions: 20 mL/min flow rate, 10 mL injection volume (2 mg/1 mL), 90% hexanes/10% IPA over 60 min to afford R-(+)-25j, tR 41.4 min; S-(−)-25j, tR 50.5 min. The enantiomeric excess (ee) was determined using an Agilent pump on a ChiralPak OD-H column (4.6 mm × 150 mm, 5 μm) with the following conditions: 1 mL/min flow rate, 10 μL injection volume, 90% hexanes/10% IPA. R-(+)-25j: tR 27.1 min; >99% ee; . S-(–)-25j: tR 32.4 min, >99% ee; .
Separation of 31a Enantiomers.
Semipreparative separation of 31a enantiomers from racemic 31a (0.020 g) was done using a ChiralPak AD-H (30 mm × 250 mm) with the following conditions: 20 mL/min flow rate, 10 mL injection volume (2 mg/1 mL), 90% hexanes/10% IPA over 60 min to afford S-(−)-31a, tR 33.8 min; R-(+)-31a, tR 47.2 min. The enantiomeric excess (ee) was determined using an Agilent pump on a ChiralPak OD-H column (4.6 mm × 150 mm, 5 μm) with the following conditions: 1 mL/min flow rate, 10 μL injection volume, 90% hexanes/10% IPA. S-(−)-31a: tR 21.4 min;>99% ee; . R-(+)-31a: tR 30.1 min, >99% ee; .
Separation of EU-1180–453 Enantiomers.
Semipreparative separation of EU-1180–453 enantiomers from racemic EU-1180–453 (0.020 g) was done using a ChiralPak AD-H (30 mm × 250 mm) with the following conditions: 20 mL/min flow rate, 10 mL injection volume (2 mg/1 mL), 75% hexanes/25% IPA over 60 min to afford S-(−)-EU-1180–453, tR 24.3 min; R-(+)-EU-1180–453, tR 37.5 min. The enantiomeric excess (ee) was determined using an Agilent pump on a ChiralPak AD-H column (4.6 mm × 150 mm, 5 μm) with the following conditions: 1 mL/min flow rate, 10 μL injection volume, 80% hexanes/20% IPA. S-(−)-EU-1180–453: tR 14.5 min; >99% ee; . R-(+)-EU-1180–453: tR 22.1 min, >99% ee; .
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH-NINDS (NS065371, NS111619 SFT). The authors thank Janssen Research & Development, LLC, Bartosz Balana, Robert Neff, and Tim Lovenberg for their support and help with pharmacokinetic analysis. They also thank Jing Zhang for excellent technical assistance. The opinions expressed in this publication are those of the author(s) and do not necessarily reflect the views of the NIH.
ABBREVIATIONS
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ACN
acetonitrile
- ATP
adenosine triphosphate
- AMPAR
AMPA receptor
- AUC
area under the curve
- ABD
agonist binding domain
- ATD
amino terminal domain
- BAPTA-AM
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid tetraacetoxymethyl ester
- CTD
carboxy terminal domain
- CNS
central nervous system
- CIQ
(3-chlorophenyl)-(6,7-dimethoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroi-soquinolin-2(1H)-yl)methanone
- DNA
deoxyribonucleic acid
- DBN
1,5-diazabicyclo[4.3.0]non-5-ene
- DCE
dichloroethy-lene
- DCM
dichloromethane
- DMAP
4-dimethylaminopyridine
- DMSO
dimethyl sulfoxide
- EtOAc
ethyl acetate
- EDCI
1-ethyl-(3-dimethylaminopropyl)-carbonyldiimide hydrochloride
- EDTA
Ethylenediaminetetraacetic acid
- FDA
Food and Drug Administration
- GABA
γ-aminobutyric acid
- HPLC
high-performance liquid chromatography
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IACUC
Institutional Animal Care and Use Committee
- IP
intraperitoneal
- iGluR
ionotropic glutamate receptor
- LCMS
liquid chromatography–mass spectrometry
- LLE
lipophilic ligand efficiency
- NAM
negative allosteric modulator
- TCN
N-(4-(2-benzoyl-hydrazine-1-carbonyl)benzyl)-3-chloro-4-fluorobenzenesulfonamide
- NMDA
N-methyl-d-aspartic acid
- NMDAR
NMDA receptor
- NIMH
National Institute for Mental Health
- NMR
nuclear magnetic resonance
- PFP
pentafluorophenyl
- PAM
positive allosteric modulator
- PEG
poly(ethylene glycol)
- K-BAPTA
potassium 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- PS
pregnenolone sulfate
- PDSP
Psychoactive Drug Screening Program
- RNA
ribonucleic acid
- RPM
rotations per minute
- SEM
standard error of the mean
- SAR
structure–activity relationship
- THF
tetrahydrofuran
- TEVC
two-electrode voltage clamp
- TIQ
tetrahydroisoquinoline
- TMD
transmembrane domain
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01733.
Chemistry experimental procedures along with crystal structure information (PDF)
Molecular formula strings (CSV)
LCMS and HPLC traces for key compounds (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.9b01733
The authors declare the following competing financial interest(s): SFT is a PI on research grants from Allergan, Biogen, and Janssen to Emory, is a member of the SAB for Sage Therapeutics, the GRIN2B Foundation, CureGRIN Foundation, is a co-founder of NeurOp Inc and AgriThera, and receives licensing fees and royalties for software. DCL is a member of the Board of Directors for NeurOp Inc and co-founder of AgriThera. DCL, DSM, KLS, MPE, SFT are co-inventors on Emory-owned Intellectual Property that includes positive allosteric modulators of NMDA receptor function.
Contributor Information
Matthew P. Epplin, Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States.
Ayush Mohan, Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States.
Lynnea D. Harris, Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States
Zongjian Zhu, Department of Pharmacology and Chemical Biology, Emory University, Atlanta, Georgia 30322, United States.
Katie L. Strong, Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States
John Bacsa, Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States.
Phuong Le, Department of Pharmacology and Chemical Biology, Emory University, Atlanta, Georgia 30322, United States.
David S. Menaldino, Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States
Stephen F. Traynelis, Department of Pharmacology and Chemical Biology, Emory University, Atlanta, Georgia 30322, United States.
Dennis C. Liotta, Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States.
REFERENCES
- (1).Traynelis SF; Wollmuth LP; McBain CJ; Menniti FS; Vance KM; Ogden KK; Hansen KB; Yuan H; Myers SJ; Dingledine R Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev 2010, 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Tang Y-P; Shimizu E; Dube GR; Rampon C; Kerchner GA; Zhuo M; Liu G; Tsien JZ Genetic enhancement of learning and memory in mice. Nature 1999, 401, 63–69. [DOI] [PubMed] [Google Scholar]
- (3).Collingridge GL; Volianskis A; Bannister N; France G; Hanna L; Mercier M; Tidball P; Fang G; Irvine MW; Costa BM; Monaghan DT; Bortolotto ZA; Molnar E; Lodge D; Jane DE The NMDA receptor as a target for cognitive enhacement. Neuropharmacology 2013, 64, 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Rezvani AM Involvement of the NMDA System in Learning and Memory. In Animal Models of Cognitive Impairment; Levin ED; Buccafusco JJ, Eds.; CRC Press/Taylor & Francis: Boca Raton (FL), 2006. [PubMed] [Google Scholar]
- (5).Ewald RC; Cline HT NMDA Receptors and Brain Development. In Biology of the NMDA Receptor; Van Dongen AM, Ed.; CRC Press/Taylor & Francis: Boca Raton (FL), 2009. [PubMed] [Google Scholar]
- (6).Okabe S; Collin C; Auerbach JM; Meiri N; Bengzon J; Kennedy MB; Segal M; McKay RDG Hippocampal synaptic Plasticity in mice overexpresssing an embryonic subunit of the NMDA receptor. J. Neurosci 1998, 18, 4177–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bliss TVP; Collingridge GL A synapti model of memory: long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [DOI] [PubMed] [Google Scholar]
- (8).Coyle JT NMDA receptor and schizophrenia: a brief history. Schizophr. Bull 2012, 38, 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Olney JW; Newcomer JW; Farber NB NMDA receptor hypofunction model of schizophrenia. J. Psychiatr. Res 1999, 33, 523–533. [DOI] [PubMed] [Google Scholar]
- (10).Reisberg B; Doody R; Stöffler A; Schmitt F; Ferris S; Möbius HJ Memantine in moderate-to-severe Alzheimer’s disease. N. Engl. J. Med 2003, 348, 1333–1341. [DOI] [PubMed] [Google Scholar]
- (11).Hallett PJ; Standaert DG Rationale for and use of NMDA receptor antagonists in Parkinon’s disease. Pharmacol. Ther 2004, 102, 155–174. [DOI] [PubMed] [Google Scholar]
- (12).Milnerwood AJ; Raymond LA Early synaptic pathophysiology in neurodegeneration: insights from Huntington’s disease. Trends Neurosci. 2010, 33, 513–523. [DOI] [PubMed] [Google Scholar]
- (13).XiangWei W; Jiang Y; Yuan H De novo mutations and rare variants occurring in NMDA receptors. Curr. Opin. Physiol 2018, 2, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wu LJ; Zhuo M Targeting the NMDA receptor subunit NR2B for the treatment of neuropathic pain. Neurotherapeutics 2009, 6, 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Park CK; Nehls DG; Graham DI; Teasdale GM; McCulloch J The glutamate antagonist MK-801 reduces focal ischemic brain damage in the rat. Ann. Neurol 1988, 24, 543–551. [DOI] [PubMed] [Google Scholar]
- (16).Simon RP; Swan JH; Griffiths T; Meldrum BS Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science 1984, 226, 850–852. [DOI] [PubMed] [Google Scholar]
- (17).Morikawa E; Mori H; Kiyama Y; Mishina M; Asano T; Kirino T Attenuation of focal ischemic brain injury in mice deficient in the epsilon1 (NR2A) subunit of NMDA receptor. J. Neurosci 1998, 18, 9727–9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Berman RM; Cappiello A; Anand A; Oren DA; Heninger GR; Charney DS; Krystal JH Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [DOI] [PubMed] [Google Scholar]
- (19).aan het Rot M; Collins KA; Murrough JW; Perez AM; Reich DL; Charney DS; Mathew SJ Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol. Psychiatry 2010, 67, 139–145. [DOI] [PubMed] [Google Scholar]
- (20).Santangelo RM; Acker TM; Zimmerman SS; Katzman BM; Strong KL; Traynelis SF; Liotta DC Novel NMDA receptor modulators: an update. Expert Opin. Ther. Pat 2012, 22, 1337–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Strong KL; Jing Y; Prosser AR; Traynelis SF; Liotta DC NMDA receptor modulators: an updated patent review (2013–2014). Expert Opin. Ther. Pat 2014, 24, 1349–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Nowak L; Bregestovski P; Ascher P; Herbet A; Prochiantz A Magnesium gates glutamate-activated channels in mouse central neurones. Nature 1984, 307, 462–465. [DOI] [PubMed] [Google Scholar]
- (23).Mayer ML; Westbrook GL; Guthrie PB Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 1984, 309, 261–263. [DOI] [PubMed] [Google Scholar]
- (24).MacDermott AB; Mayer ML; Westbrook GL; Smith SJ; Barker JL NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature 1986, 321, 519–522. [DOI] [PubMed] [Google Scholar]
- (25).Monyer H; Burnashev N; Laurie DJ; Sakmann B; Seeburg PH Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [DOI] [PubMed] [Google Scholar]
- (26).Akazawa C; Shigemoto R; Bessho Y; Nakanishi S; Mizuno N Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Comp. Neurol 1994, 347, 150–160. [DOI] [PubMed] [Google Scholar]
- (27).Watanabe M; Inoue Y; Sakimura K; Mishina M Developmental changes in distribution of NMDA receptor channel subunit mRNAs. NeuroReport 1992, 3, 1138–1140. [DOI] [PubMed] [Google Scholar]
- (28).Erreger K; Dravid SM; Banke TG; Wyllie DJA; Traynelis SF Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signaling profiles. J. Physiol 2005, 563, 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dravid SM; Prakash A; Traynelis SF Activation of recombinant NR1/NR2C NMDA receptors. J. Physiol 2008, 586, 4425–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wyllie DJA; Behe′ P; Colquhoun D Single-channel activations and concentration jumps: comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. J. Physiol 1998, 510, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Vance KM; Hansen KB; Traynelis SF GluN1 splice variant control of GluN1/GluN2D NMDA receptors. J. Physiol 2012, 590, 3857–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Erreger K; Geballe MT; Kristensen A; Chen PE; Hansen KB; Lee CJ; Yuan H; Le P; Lyuboslavsky PN; Micale N; Jørgensen L; Clausen RP; Wyllie DJA; Snyder JP; Traynelis SF Subunit-specific agonist activity at NR2A-, NR2B-, NR2C-, and NR2D-containing N-methyl-D-aspartate glutamate receptors. Mol. Pharmacol 2007, 72, 907–920. [DOI] [PubMed] [Google Scholar]
- (33).Vicini S; Wang JF; Li JH; Zhu WJ; Wang YH; Luo JH; Wolfe BB; Grayson DR Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J. Neurophysiol 1998, 79, 555–566. [DOI] [PubMed] [Google Scholar]
- (34).Jones S; Gibb AJ Functional NR2B- and NR2D-containing NMDA receptor channels in rat substantia nigra dopaminergic neurones. J. Physiol 2005, 569, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chen PE; Geballe MT; Katz E; Erreger K; Livesey MR; O’Toole KK; Le P; Lee CJ; Snyder JP; Traynelis SF; Wyllie DJ Modulation of glycine potency in rat recombinant NMDA receptors containing chimeric NR2A/2D subunits expressed in Xenopus laevis oocytes. J. Physiol 2008, 586, 227–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Banke TG; Traynelis SF Activation of NR1/NR2B NMDA receptors. Nat. Neurosci 2003, 6, 144–152. [DOI] [PubMed] [Google Scholar]
- (37).Siegler Retchless B; Gao W; Johnson JW A single GluN2 subunit residue controls NMDA receptor channel properties via intersubunit interaction. Nat. Neurosci 2012, 15, 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Paoletti P; Bellone C; Qiang Z NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci 2013, 14, 383–400. [DOI] [PubMed] [Google Scholar]
- (39).Lipton SA Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx 2004, 1, 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Moghaddam B; Javitt D From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012, 37, 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Coyle JT; Tsai G; Goff D Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann. N. Y. Acad. Sci 2003, 1003, 318–327. [DOI] [PubMed] [Google Scholar]
- (42).Gonzalez-Burgos G; Lewis DA NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr. Bull 2012, 38, 950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Won H; Lee H-R; Gee HY; Mah W; Kim J-I; Lee J; Ha S; Chung C; Jung ES; Cho YS; Park S-G; Lee J-S; Lee K; Kim D; Bae YC; Kaang B-K; Lee MG; Kim E Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 2012, 486, 261–265. [DOI] [PubMed] [Google Scholar]
- (44).Schmeisser MJ; Ey E; Wegener S; Bockmann J; Stempel AV; Kuebler A; Janssen A-J; Udvardi PT; Shiban E; Spilker C; Balschun D; Skryabin BV; Dieck S. t.; Smalla K-H; Montag D; Leblond CS; Faure P; Torquet N; Le Sourd A-M; Toro R; Grabrcuker AM; Shoichet SA; Schmitz D; Kreutz MR; Bourgeron T; Gundelfinger ED; Boeckers TM Autistic-liek behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 2012, 486, 256–260. [DOI] [PubMed] [Google Scholar]
- (45).Dalmau J; Gleichman AJ; Hughes EG; Rossi JE; Peng X; Lai M; Dessain SK; Rosenfeld MR; Balice-Gordon R; Lynch DR Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008, 7, 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hughes EG; Peng X; Gleichman AJ; Lai M; Zhou L; Tsou R; Parsons TD; Lynch DR; Dalmau J; Balice-Gordon RJ Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci 2010, 30, 5866–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mikasova LD; De Rossi P; Bouchet D; Georges F; Rogemond V; Didelot A; Meissirel C; Honnorat J; Groc L Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain 2012, 135, 1606–1621. [DOI] [PubMed] [Google Scholar]
- (48).Bannerman DM; Niewoehner B; Lyon L; Romberg C; Schmitt WB; Taylor A; Sanderson DJ; Cottam J; Sprengel R; Seeburg PH; Köhr G; Rawlins JN NMDA receptor subunit NR2A is required for rapidly acquired spatial working memory but not incremental spatial reference memory. J. Neurosci 2008, 28, 3623–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wang D; Cui Z; Zeng Q; Kuang H; Wang LP; Tsien JZ; Cao X Genetic enhancement of memory and long-term potentiation but not CA1 long-term depression in NR2B transgenic rats. PLoS One 2009, 4, No. e7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Khlestova E; Johnson JW; Krystal JH; Lisman J The role of GluN2C-containing NMDA receptors in ketamine’s psychotogenic action and in schizophrenia models. J. Neurosci 2016, 36, 11151–11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Schmitt A; Koschel J; Zink M; Bauer M; Sommer C; Frank J; Treutlein T; Schulze T; Schneider-Axmann T; Parlapani E; Rietschel M; Falkai P; Henn FA Gene expression of NMDA receptor subunits in the cerebellum of elderly patients with schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci 2010, 260, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Meador-Woodruff JH; Hogg AJ Jr; Smith RE Striatal ionotropic glutamate receptor expression in schizophrenia, bipolar disorder, and major depressive disorder. Brain Res. Bull 2001, 55, 631–640. [DOI] [PubMed] [Google Scholar]
- (53).Sapkota K; Mao Z; Synowicki P; Lieber D; Liu M; Ikezu T; Gautam V; Monaghan DT GluN2D N-methyl-D-aspartate receptor subunit contribution to the stimulation of brain activity and gamma oscillations by ketamine: Implications for schizophrenia. J. Pharmacol. Exp. Ther 2016, 356, 702–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Makino C; Shibata H; Ninomiya H; Tashiro N; Fukumaki Y Identification of single-nucleotide polymorphisms in the human N-methyl-D-aspartate receptor subunit NR2D gene, GRIN2D, and association study with schizophrenia. Psychiatr. Genet 2005, 15, 215–221. [DOI] [PubMed] [Google Scholar]
- (55).Lisman JE; Coyle JT; Green RW; Javitt DC; Benes FM; Heckers S; Grace AA Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008, 31, 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Perszyk RE; DiRaddo JO; Strong KL; Low CM; Ogden KK; Khatri A; Vargish GA; Pelkey KA; Tricoire L; Liotta DC; Smith Y; McBain CJ; Traynelis SF GluN2D-containing N-methyl-D-aspartate receptors mediate synaptic transmission in hippocampal interneurons and regulate interneuron activity. Mol. Pharmacol 2016, 90, 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).von Engelhardt J; Bocklisch C; Tönges L; Herb A; Mishina M; Monyer H GluN2D-containing NMDA receptors-mediate synaptic currents in hippocampal interneurons and pyramidal cells in juvenile mice Front. Cell Neurosci [Online]. DOI: 10.3389/fncel.2015.00095. Published Online: Mar 25, 2015, https://www.frontiersin.org/articles/10.3389/fncel.2015.00095/full (accessed Jun 11, 2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Mullasseril P; Hansen KB; Vance KM; Ogden KK; Yuan H; Kurtkaya NL; Santangelo R; Orr AG; Le P; Vellano KM; Liotta DC; Traynelis SF A subunit-selective potentiator of NR2C- and NR2D-containing NMDA receptors. Nat. Commun 2010, 1, No. 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Santangelo Freel RM; Ogden KK; Strong KL; Khatri A; Chepiga KM; Jensen HS; Traynelis SF; Liotta DC Synthesis and structure activity relationship of tetrahydroisoquinoline-based potentiators of GluN2C and GluN2D containing N-methyl-D-aspartate receptors. J. Med. Chem 2013, 56, 5351–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Nouhi M; Zhang X; Yao N; Chergui K CIQ, a positive allosteric modulator of GluN2C/D-containing N-methyl-d-aspartate receptors, rescues striatal synaptic plasticity deficit in a mouse model of Parkinson’s disease. CNS Neurosci. Ther 2018, 24, 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Ogden KK; Khatri A; Traynelis SF; Heldt SA Potentiation of GluN2C/D NMDA receptor subtypes in the amygdala facilitates the retention of fear and extinction learning in mice. Neuropsychopharmacology 2014, 39, 625–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Shelkar GP; Pavuluri P; Gandhi PJ; Ravikrishnan A; Gawande DY; Liu J; Stairs DJ; Ugale RR; Dravid SM Differential effect of NMDA receptor GluN2C and GluN2D subunit ablation on behavior and channel blocker-induced schizophrenia phenotypes. Sci. Rep 2019, 9, No. 7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Strong KL; Epplin MP; Bacsa J; Butch CJ; Burger PB; Menaldino DS; Traynelis SF; Liotta DC The structure-activity relationship of a tetrahydroisoquinoline class of N-methyl-D-aspartate receptor modulators that potentiate GluN2B-containing N-methyl-D-aspartate receptors. J. Med. Chem 2017, 60, 5556–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Wang T-M; Brown BM; Deng L; Sellers BD; Lupardus PL; Wallweber HJA; Gustafson A; Wong E; Volgraf M; Schwarz JB; Hackos DH; Hanson JE A novel NMDA receptor positive allosteric modulator that acts via the transmembrane domain. Neuropharmacology 2017, 121, 204–218. [DOI] [PubMed] [Google Scholar]
- (65).Ceccon M; Rumbaugh G; Vicini S Distinct effect of pregnenolone sulfate on NMDA receptor subtypes. Neuropharmacology 2001, 40, 491–500. [DOI] [PubMed] [Google Scholar]
- (66).Paul SM; Doherty JJ; Robichaud AJ; Belfort GM; Chow BY; Hammond RS; Crawford DC; Linsenbardt AJ; Shu H-J; Izumi Y; Mennerick SJ; Zorumski CF The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J. Neurosci 2013, 33, 17290–17300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Volgraf M; Sellers BD; Jiang Y; Wu G; Ly CQ; Villemure E; Pastor RM; Yuen P; Lu A; Luo X; Liu M; Zhang S; Sun LFY; Lupardus PJ; Wallweber HJA; Liederer BM; Deshmukh G; Plise E; Tay S; Reynen P; Herrington J; Gustafson A; Liu Y; Dirksen A; Dietz MGA; Liu Y; Wang T-M; Hanson JE; Hackos D; Scearce-Levie K; Schwarz JB; et al. Discovery of GluN2A-selective NMDA receptor positive allosteric modulators (PAMs): tuning deactivation kinetics via structure-based design. J. Med. Chem 2016, 59, 2760–2779. [DOI] [PubMed] [Google Scholar]
- (68).Hackos DH; Lupardus PJ; Grand T; Chen Y; Wang T-M; Reynen P; Gustafson A; Wallweber HJA; Volgraf M; Sellers BD; Schwarz JB; Paoletti P; Sheng M; Zhou Q; Hanson JE Positive allosteric modulators of GluN2A-containing NMDARs with distinct modes of action and impacts on circuit function. Neuron 2016, 89, 983–999. [DOI] [PubMed] [Google Scholar]
- (69).Villemure E; Volgraf M; Jiang Y; Wu G; Ly CQ; Yuen PW; Lu A; Luo X; Liu M; Zhang S; Lupardus PJ; Wallweber HJ; Liederer BM; Deshmukh G; Plise E; Tay S; Wang TM; Hanson JE; Hackos DH; Scearce-Levie K; Schwarz JB; Sellers BD GluN2A-selective pyridopyrimidinone series of NMDAR positive allosteric modulators with an improved in vivo profile. ACS. Med. Chem. Lett 2017, 8, 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Zimmerman SS; Khatri A; Garnier-Amblard EC; Mullasseril P; Kurtkaya NL; Gyoneva S; Hansen KB; Traynelis SF; Liotta DC Design, synthesis, and structure-activity relationship of a novel series of GluN2C-seective potentiators. J. Med. Chem 2014, 57, 2334–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Khatri A; Burger PB; Swanger SA; Hansen KB; Zimmerman S; Karakas E; Liotta DC; Furukawa H; Snyder JP; Traynelis SF Structural determinants and mechanism of action of a GluN2C-selective NMDA receptor positive allosteric modulator. Mol. Pharmacol 2014, 86, 548–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Buttar D; Foote KM; Nowak T; Rudge DA; Theoclitou M-E; Thomas AP Pyrimidine derivatives useful in the treatment of cancer. International Patent WO2008001070A12008.
- (73).Waring MJ Lipophilicity in drug discovery. Expert Opin. Drug Discovery 2010, 5, 235–248. [DOI] [PubMed] [Google Scholar]
- (74).Johnson TW; Dress KR; Edwards M Using the golden triangle to optimize clearance and oral absorption. Bioorg. Med. Chem. Lett 2009, 19, 5560–5564. [DOI] [PubMed] [Google Scholar]
- (75).Liu X; Testa B; Fahr A Lipophilicity and its relationship with passive drug permeation. Pharm. Res 2011, 28, 962–977. [DOI] [PubMed] [Google Scholar]
- (76).Yoshida F; Topliss JG QSAR Model for Drug Human Oral Bioavailability. J. Med. Chem 2000, 43, 2575–2585. [DOI] [PubMed] [Google Scholar]
- (77).Moldavski A; Behr J; Bading H; Bengtson CP A novel method using ambient glutamate for the electrophysiological quantification of extrasynaptic NMDA receptor function in acute brain slices. J. Physiol 2020, 598, 633–650. [DOI] [PubMed] [Google Scholar]
- (78).The active enantiomers of this series have been previously shown to be brain penetrant: Gawai P; Upadhyay R; Gakare SG; Sarode L; Dravid S; Ugale RR Antipsychotic-like profile of CIQ isomers in animal models of schizophrenia Behav. Pharmacol 10.1097/FBP.0000000000000532. Published Online: Dec 18, 2019. https://journals.lww.com/behaviouralpharm/Abstract/9000/Antipsychotic_like_profile_of_CIQ_isomers_in.99204.aspx (accessed May 7, 2020). [DOI] [PubMed] [Google Scholar]
- (79).Dravid SM; Erreger K; Yuan H; Nicholson K; Le P; Lyuboslavsky P; Almonte A; Murray E; Mosley C; Barber J; French A; Balster R; Murray TF; Traynelis SF Subunit-Specific Mechanisms and Proton Sensitivity of NMDA Receptor Channel Block. J. Physiol 2007, 581, 107–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Johnson TW; Gallego RA; Edwards MP Lipophilic efficiency as an important metric in drug design. J. Med. Chem 2018, 61, 6401–6420. [DOI] [PubMed] [Google Scholar]
- (81).Letavic MA; Lord B; Bischoff F; Hawryluk NA; Pieters S; Rech JC; Sales Z; Velter AI; Ao H; Bonaventure P; Contreras V; Jiang X; Morton KL; Scott B; Wang Q; Wickenden AD; Carruthers NI; Bhattacharya A Synthesis and Pharmacological Characterization of Two Novel, Brain Penetrating P2X7 Antagonists. ACS Med. Chem. Lett 2013, 4, 419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.