

Abstract
Lentiviruses have been widely used as a means of transferring exogenous DNAs into human cells to treat various genetic diseases. Lentiviral vectors are fundamentally integrated into the host genome, but their integration sites are generally unpredictable, which may increase the uncertainty for their use in therapeutics. To determine the viral integration sites in the host genome, several PCR-based methods have been developed. However, the sensitivities of the PCR-based methods are highly dependent on the primer sequences, and optimized primer design is required for individual target sites. In order to address this issue, we developed an alternative method for genome-wide mapping of viral insertion sites, named CReVIS-seq (CRISPR-enhanced Viral Integration Site Sequencing). The method is based on the sequential steps: fragmentation of genomic DNAs, in vitro circularization, cleavage of target sequence in a CRISPR guide RNA-specific manner, high-throughput sequencing of the linearized DNA fragments in an unbiased manner, and identification of viral insertion sites via sequence analysis. By design, CReVIS-seq is not affected by biases that could be introduced during the target enrichment step via PCR amplification using site specific primers. Furthermore, we found that multiplexed CReVIS-seq, using collections of different single-guide RNAs (sgRNAs), enables simultaneous identification of multiple target sites and structural variations (i.e., circularized viral genome), in both single cell clones and heterogeneous cell populations.
Graphical abstract
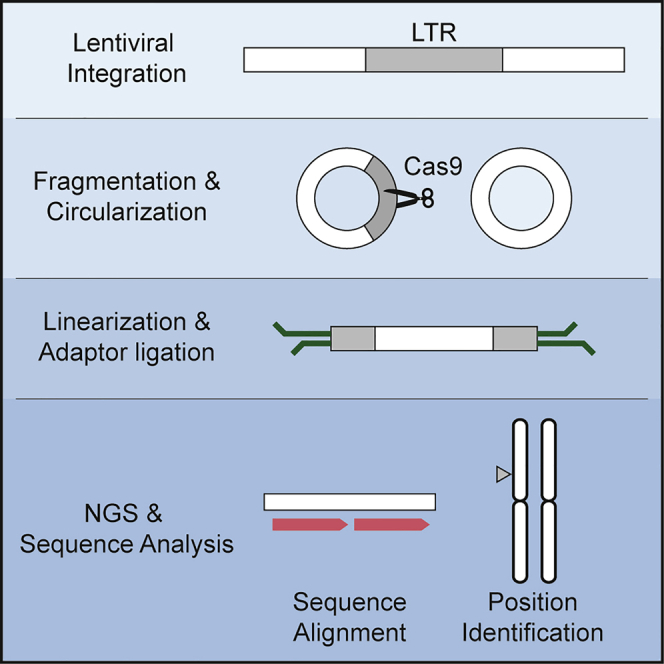
The study demonstrates a new technology “CReVIS-seq” that enables CRISPR-based multiplexed enrichment of genomic regions. The method uses a PCR-free enrichment step based on DNA circularization and precise CRISPR-Cas9 cleavage. The strength of this method was demonstrated by simultaneous enrichment of lentiviral integration sites and genomic sites.
Introduction
Upon lentivirus transduction into host cells, the lentiviral RNA genome undergoes reverse transcription to a complementary DNA (cDNA), which is then integrated into the host cell genome.1,2 As foreign DNAs can be transduced with high efficiency into host cells or organisms using lentiviral gene transfer systems, such systems have been widely used to supply the normal gene to rescue the disease phenotype of various genetic diseases such as β-thalassemia, metachromatic leukodystrophy, and X-linked adrenoleukodystrophy.3, 4, 5, 6 Recently, lentiviruses have also been used to transfer synthetic genes to generate chimeric antigen receptor (CAR) and artificial T cell receptor (TCR) T cells for cancer immunotherapy.7 In addition, because foreign DNAs carried by lentiviruses are integrated into the host cell genome, lentiviral systems have been also applied in genome-wide screening experiments as a tool for stable expression of proteins or RNAs, such as small interfering RNAs or CRISPR single-guide RNAs (sgRNAs) for gene knockdown or knockout, respectively.8,9
While lentivirus-mediated foreign gene integration is quite useful for stable expression of genes of interest, it could affect the cellular phenotype by causing the loss- or gain-of-function mutations in endogenous genes. Genomic studies have revealed that notable risks may arise from the uncertainties related to where the exogenous genes integrate into the host genome. First, viral genome integration could directly cause functional knockout of genes that are essential for cell survival or induce unanticipated pathogenicity via disrupting critical regions of genomic DNA. In lentivirus-mediated gene therapy, for example, insertional activation of proto-oncogenes by viral DNA integration could induce cancer.10 Particularly, distinct from other types of viruses, lentiviruses are known to preferably insert viral cDNAs into gene bodies,11 whereas γ-retroviruses, which are also widely used as viral vectors, have a strong propensity to integrate in the 5′ ends of genes including promoters.12 Second, different genomic loci manifest diverse distribution of protein expression capacities, the expression levels of the inserted transgenes can differ from those of endogenous genes, and vary depending on the integration site.13 The issues pose major potential obstacles in developing medical applications where controlled expression is critical, such as therapeutic enzyme supplementation. Although several studies have reported common integration sites for lentiviral vectors,11,14, 15, 16 the exact locations of foreign DNA insertions are generally unpredictable. Therefore, detailed investigations of lentiviral integration sites in the host genomes are crucial for developing effective and safe lentiviral gene therapy.
For genome-wide identification of lentiviral integration sites, several PCR-based methods, such as ligation-mediated (LM)-PCR17 and linear amplification mediated (LAM)-PCR,18 have generally been utilized. However, since the methods depend on target-specific PCR primer sets, the fidelity of the methods are prone to compromises by ill-designed primer sets that can lead to overlooking rare insertion events among other high copy number insertion events. Furthermore, simultaneous detection to multiple targets are limited by the intrinsic difficulties of multiplex PCR. In this study, we sought to resolve the limitation set by target-specific PCR amplification, and developed a CRISPR-enhanced Viral Integration Site detection method, named CReVIS-seq. Instead of relying on the exponential and primer-specific PCR amplification, CReVIS-seq enriches target DNAs using CRISPR-mediated target DNA cleavage. Our approach is based on in vitro fragmentation and circularization of genomic DNA followed by CRISPR-Cas9 induced cleavage of those DNAs containing lentiviral long terminal repeats (LTRs), which are generally incorporated into the host genome together with other lentiviral sequences. By using CReVIS-seq, we first identified lentiviral insertion sites in clonal populations derived from single cells. Next, we observed that CReVIS-seq is also applicable to identify multiple insertion sites in bulk cell populations. We further found that the CReVIS-seq allows the use of different sgRNAs for multiple CRISPR mediated cleavages to simultaneously acquire the DNA sequence information of multiple target loci in the host genome. Our results show that this method is appropriate for enriching multiple targets, suggesting that the alternative method could be generalized for genome-wide mapping of viral insertion sites including lentivirus.
Results
Strategy for enrichment and unbiased capture of lentiviral integration sites in the host genome
We conceived an in vitro circularization and cleavage method to identify lentiviral integration sites throughout the host genome. The workflow of the CReVIS-seq method is depicted in Figure 1. First, the host genome was sonicated to generate randomly sheared, end-repaired, and circularized by intramolecular ligation as described in a previous study.19 Then, within the circularized DNAs, the desired DNA fragments were selectively linearized by using the CRISPR-Cas9 endonucleases that induce double-strand DNA breaks (DSB) at the target sequences in a guide RNA-specific manner in vitro.20,21 Thus, by targeting viral LTR sequences using Cas9 endonucleases, the circular DNAs containing viral sequences were selectively linearized, whereas most of the host genomic DNA fragments remained circularized. Next, sequencing adaptors were ligated to the linearized DNAs for subsequent library preparation for high-throughput sequencing. Analysis of sequencing data of the linearized DNA allowed genome-wide identification of the junctions between the viral DNAs and genomic sequences. As CReVIS-seq replaces target-specific PCR enrichment step with CRISPR cleavage, the PCR-related biases related to target specific primers are not introduced.
Figure 1.

Schematic representation of CReVIS-Seq
After genomic DNA fragmentation, each DNA sequence is circularized by intra-molecular ligation by T4 ligase. Only linearized DNAs are selected by exonuclease treatment. Circularized DNA, which has our target sequences, are cleaved, linearized via specific Cas9/sgRNA nucleases, and ligated with sequencing adaptor. Prepared libraries are sequenced through Illumina sequencer and viral-host junctions are identified.
Experimental validation of CReVIS-Seq for identifying lentiviral integration sites in human cells
We sought to assess the experimental application of CReVIS-seq to identify lentiviral insertion site in human cells. In order to distinguish different viral insertion events, we prepared lentiviruses containing different DNA barcodes (4,542 sgRNA expression cassettes) instead of a single identical sequence, which was previously used for a CRISPR-based screen (Figure 2).22 We also employed a HeLa-Cas9 cell line that constitutively expresses the Cas9 protein.
Figure 2.

Identification of each lentiviral integration site in each clone
(A) Experimental procedure for obtaining 5 clones derived from single cells. The plasmid library contains 4,542 sgRNA expression cassettes. (B) Chromosome ideogram showing each lentiviral integration site in each clone. Clone #5 has two integration sites, one in in chr2 and one in chr13. (C) Table showing the integrated genome locus and integrated sgRNA cassette in each clone.
We transduced the pooled lentiviruses into HeLa-Cas9 cells at a low multiplicity of infection (MOI; < 0.05), then established single cell clones via serial dilution of the transduced cells into 96-well plates (Figure 2A). Because we treated cells with lentivirus at a low MOI, each cell was anticipated to be infected, on average, by one or fewer lentiviruses. Next, we conducted CReVIS-seq on five selected single cell clones and could exactly pinpoint the integration sites (Figure 2B). Analysis of the sgRNA barcode sequences, of all the single cell clones, confirmed that the numbers of barcodes were identical with the numbers of integration sites (Figure 2C). As expected, we observed a single integration site in four out of five clones; the exception was clone #5, which had two integration sites, one in chromosome 2 and another in chromosome 13, with distinct sgRNA sequences. The results indicated that two different lentiviruses, containing distinct barcodes, were simultaneously transduced into the clone. We further confirmed the integration sites of all clones by Sanger sequencing with PCR primer sets located between the viral sequence and the genomic sequences at the insertion loci (Figure S1). Additionally, we adapted the LM-PCR to confirm integration sites for 4 clones which are identified through CReVIS-seq. Integration sites revealed by both methods were identical (Figure S1C).
CReVIS-Seq observed circular lentiviral genome in human cells
During the CReVIS-seq analysis of the lentiviral integration sites in clones derived from single cells, we observed a distinct lentiviral genome structure in a circular form. As outlined in Figure 1, lentiviral DNAs inserted in the host genome are flanked by long terminal repeats (LTRs) with identical sequences at both ends. Hence, the paired-end deep sequencing results from CReVIS-seq were expected to show LTR sequences that are linked to either viral DNA sequence or host genome sequences; i.e., if one single end read is aligned to either the 5′ or 3′ end of a viral DNA sequence, the other single end read should be aligned to a human genome sequence (Figure S2A). However, for four clones, we found that significant fractions (∼49%) of paired-end sequencing reads were composed of single-end reads that aligned to either the 5′ or 3′ ends of the lentiviral DNA sequences (Figure S2A). Such paired-end sequence compositions were incompatible with the linear lentiviral DNA sequences generated by regular integration in the host genome. On the other hand, the read combinations could be generated from circular forms of lentiviral DNA, known as 1-LTR, that were formed by homologous recombination between the two LTRs.23 In order to assess whether CReVIS-seq detected the presence of 1-LTR, we performed PCR using a specific primer set that could amplify DNA sequences created by 1-LTR, but not from normal integration patterns where lentiviral DNAs are flanked with host genomic sequences (Figure S2B). The PCR assay showed positive amplicon products and correct sequences for all 4 clones that we tested (Figures S2C and S2D), confirming the presence of 1-LTR.
Detection of genome-wide lentiviral integration events
We next assessed whether CReVIS-seq could be applied to detect multiple lentiviral insertion sites heterogenous populations of transduced cells, without clonal selection (Figure 3A). To this end, we performed CReVIS-seq to a pool of lentivirus infected HeLa cells and simultaneously identified 863 independent integration sites (Table S1) by deep sequencing analysis. The summarized results are shown as a circos plot (Figure 3B), a chromosome ideogram (Figure 3C), and a bar plot (Figure 3D). We observed that lentiviral DNA integration sites were relatively uniformly distributed among all chromosomes except the Y chromosome, consistent with HeLa cells being originated from a female donor.
Figure 3.

Identification of lentiviral integration sites in bulk cell populations using CReVIS-Seq
(A) Experimental scheme for CReVIS-seq using genomic DNAs of the population of cells. The pooled sgRNA plasmid library is same to plasmid library in Figure 2A. (B–D) Circos plot (B), chromosome ideogram (C), and bar plot (D) showing the counts of lentivirus integration sites in each region.
Multiplexed CReVIS-Seq for co-identification of multiple genomic sites in human cells
The above results showed that CReVIS-seq could identify distinct lentiviral integration sites in bulk cell populations, as well as in clones derived from single cells by enriching target sites after in vitro circularization and subsequent cleavage of genomic DNAs in an sgRNA-specific manner. Considering the multiplexing activity of CRISPR-Cas9,24 we envisioned that CReVIS-seq could be further harnessed to identify additional orthogonal target sites, via corresponding sgRNAs, simultaneously in addition to the lentiviral DNA integration site.
Accordingly, we asked whether several sgRNAs could be used at once for multiplexed CReVIS-seq to generate DNA cleavages at distinct nonadjacent genomic loci, and to provide more sequence information than just the sites of lentiviral insertion. To this end, we revisited clones #1 and #2 of lentiviral infection, described in Figure 2. We anticipated that, in the transduced Cas9-expressing HeLa cells, DNA mutations would be induced by sgRNAs expressed from the integrated lentiviral sequence that form CRISPR ribonucleotide complexes with the Cas9 protein made in the cells. In addition, from the sequence information of virally-encoded sgRNA in the host genome of each clone (Figure 2C), we had prior knowledge on the corresponding CRISPR genome-editing target sites in the host genome via Cas-OFFinder software.25 Accordingly, we sought to apply multiplexed CReVIS-seq to clones #1 and #2 to simultaneously obtain deep-sequencing data that contained three different pieces of orthogonal sequence information: (1) positions of the lentiviral integration sites, (2) sequences of the integrated sgRNAs, and (3) the mutation patterns at the genomic loci targeted by the integrated sgRNA (Figure 4A). To this end, we performed CReVIS-seq using three different sgRNAs that induce CRISPR-mediated in vitro DNA cleavages in the following corresponding target loci: (1) the LTR sequence, (2) the U6 RNA polymerase III promoter (PU6) sequence upstream of the sgRNA sequence, and (3) genomic sequences proximal to the encoded sgRNA target sites (approximately 50 bp away from each target).
Figure 4.

In-depth analysis to reveal three different pieces of information from clones #1 and #2 via multiplexed CReVIS-Seq
(A) Scheme for multiplexed CReVIS-seq using three sgRNAs that target different sites: (1) the LTR sequence, to identify the lentiviral integration site in the host genome, (2) the U6 promoter sequence, to identify the integrated sgRNA sequence, and (3) the flanking sequence near the Cas9/sgRNA target in the host genome, to identify changes in the genotype after gene editing. (B) Table showing the results of multiplexed CReVIS-seq: integration site, sgRNA sequence, and genotype. Underline, Protospacer; red, PAM; and blue, insertion sequences.
As anticipated, we found that multiplexed CReVIS-seq provided deep-sequencing data that contained sequence information of all three discrete genomic loci: the viral DNA integration sites, the sgRNA sequences encoded in the integrated viral DNA, and the mutation patterns in the host genome caused by Cas9/sgRNA genome editing (Figure 4B). The results of the multiplexed CReVIS-seq were consistent with the results of the monoplex CReVIS-seq (Figure 2C) and Sanger sequencing, supporting the multiplexing ability of CReVIS-seq.
Construction of a web-based CReVIS-Seq analysis tool
To facilitate analysis of the deep-sequencing data, we developed CReVIS-seq analysis software that consists of several statistical and computational analysis programs including a sequence alignment tool. The analysis software that runs offline in the Linux environment is deposited at github (https://github.com/Gue-ho/CReVIS-Seq). Furthermore, to allow easy access to the CReVIS-seq analysis software, we constructed a web-based version (http://www.rgenome.net/crevis-seq). The CReVIS-seq web tool requires input of high-throughput sequencing files along with the basic information including the LTR sequences and in vitro cleavage target sequences (Figure 5A). The CReVIS-seq web tool was constructed with a JavaScript-based algorithm so that large amounts of sequencing data are not required to be uploaded to the server, and the running time is therefore reduced. Furthermore, the operation is almost completely executed at the client-side web browser on-the-fly, including sequence alignment with the human genome (Figure S3). The CReVIS-seq web tool automatically provides a chromoMap and displays a visual table of the major integrated locations (Figure 5B). As our benchmark, we observed that the web tool could fully analyze 379 MB of data in a fastq file within 377 s, using a Ryzen3 3800X central processing unit, indicating that the analysis could be conducted in a reasonable time frame.
Figure 5.

Overview of the CReVIS-Seq web tool
(A) Input panel. The CReVIS-seq web tool requires paired-end NGS files and basic information including the LTR sequence and the sgRNA target sequence for in vitro DNA cleavage. (B) A sample CReVIS-seq web tool results page. The results are displayed as a chromomap and in a table.
Discussion
CReVIS-seq enables the enrichment and detection of target sites of interest, including viral integration sites, via in vitro circularization and subsequent CRISPR-mediated DNA cleavage in a guide RNA-specific manner. We found that CReVIS-seq could effectively identify viral integration sites in clonal cells, without the burden of conducting whole-genome sequencing, which is expensive and time consuming. As shown in Figure S2, CReVIS-seq could reveal circular viral genome, which can’t be done by PCR-based methods. This suggested that CReVIS-seq could be potentially utilized for unbiased detection of structural variations including duplications and inversions at and not limited to the integration sites. Furthermore, we demonstrated that that CReVIS-seq is a generalizable method for mapping genome-wide lentiviral integration sites from bulk cell populations. This method could also be applied to heterogeneous cell population to determine the proportion of clones, which is potentially useful for therapeutic viral delivery as a means of gene therapy. In addition to identification of lentiviral integration sites in both single cell-derived clones and heterogeneous cell populations, CReVIS-seq could be multiplexed to enrich various DNA sequences by simply using multiple sgRNAs for in vitro cleavage of genomic DNAs. Notably, simultaneously sequence analysis of distinct targets have been difficult with previously developed tools. The CRISPR-mediated cleavage by individual sgRNAs intrinsically independent processes and are therefore free of cross-talks. In contrast, the multiplexing method in PCR-based enrichment techniques require precisely designed PCR primer sets that are prone to experimental failure and biases due to sensitivity to GC content and melting temperatures.
While we mainly demonstrated application of CReVIS-seq in viral integration site verification in this study, we anticipate that CReVIS-seq can be adopted in many more applications due to its ease-of-use and multiplexing ability. Currently, multiplexed PCR methods are widely utilized to many areas of research, such as the examinations of potential oncogenes and tumor suppressor genes in cancer panels, and validations of mutations at multiple genomic loci. However, such PCR-based methods are difficult to customize and therefore are largely restricted to pre-defined targets. On the other hand, multiplexed CReVIS-seq is easy to conduct and the design of multiple sgRNAs for in vitro cleavage of host genomes is simple. Sequence optimization for sgRNA is not required, even for multiplexing, and the sgRNA oligonucleotides can be obtained through RNA oligo pool synthesis or in vitro transcription. In addition, CReVIS-seq read counts are proportional to the number of target sites. For each clone in clones #1–4 used in Figure 2, we separately analyzed the read counts of two LTRs, which are located at the 3′ end of genome integration or the 1-LTR by their sequence information. Our quantitative analysis showed that the read counts of two LTR sites were almost identical (1:0.96), implying that CReVIS-seq may be adequate for representing the copy numbers of target DNAs of interest. Furthermore, CReVIS-seq can be utilized to obtain DNA sequence information of wide range in both upstream or downstream of target sites of interest, suggesting that the method could enable detection of various structural rearrangements including DNA duplications in the future.
While CReVIS-seq is versatile tool, there are some technical limitations that we anticipate could be overcome in the future studies. First, we used spCas9 which has -NGG protospacer adjacent motif (PAM), and therefore the target sequences were required to contain the PAM sequences. However, the PAM sequence (NGG) are quite frequent in the genome, once in every 8 bp in average. Moreover, CReVIS-seq also allows the use of other CRISPR systems including engineered Cas9 enzymes (e.g., spCas9-NG and xCas9), which have -NG PAM when we need to expand our targets. Next, the exact sequence information of the end of viral genome (∼100 bp) is required to identify the viral-host junction with the current Illumina deep-sequencing platform, used in this study, that provides short sequence reads. We anticipate that the requirement for prior sequence information would be alleviated by adopting the long-read sequencing platforms (e.g., Oxford Nanopore or Pacific Bioscience) after the target enrichment cleavage step.
Materials and methods
Cell culture and lentivirus infection
HeLa-Cas9 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The cell-line and sgRNA library are from the previous study.22 Briefly, sgRNA library was constructed based on lentiCRISPR v2 (Addgene, #52961). HeLa-Cas9 cells were transduced with each lentivirus at a MOI under 0.05. After 24 h, cells were selected with puromycin. Single-cell-derived lentivirus infected clones were obtained by limiting dilution.
Genomic DNA fragmentation and in vitro circularization
Genomic DNA was purified using a DNeasy Blood & Tissue kit (QIAGEN, Germany). 1 μg of purified high molecular weight DNAs were fragmented to 300 base pairs using a Covaris M220 Focused-ultrasonicator with the following settings: power, 50; duty factor, 20; cycles per burst, 200; time, 65 s; temp, 20°C. Fragmented DNAs were purified using Agencourt AMPure XP beads (Beckman Coulter, USA) and end-repaired using NEBNext End Repair Module (New England Biolabs, UK). 500 ng of end-repaired and phosphorylated DNAs were treated with T4 ligase (Enzynomics, Korea) to circularize them; the remaining linear DNAs were degraded using T5 Exonuclease (New England Biolabs, UK).
CReVIS-Seq
An sgRNA that targets the lentiviral LTR sequence (5′-ACACTGACTAAAAGGGTCTGAGG-3′ or 5′-ACCAGAGTCACACAACAGACGGG-3′) was in vitro transcribed as described before.26 150 ng of circularized DNAs were treated with 4.5 pmol of Cas9 protein (New England Biolabs, UK) and 13.5 pmol of sgRNA for 2 h at 37°C to linearize targeted DNA. The resulting linear fragments were A-tailed using the NEBNext dA-Tailing Module (New England Biolabs, UK) and sequencing adaptors were ligated to these fragments using the NEBNext Ultra II Ligation Module (New England Biolabs, UK). To finalize library preparation, 5 cycles of amplification using NEBNext Ultra II Q5 Master mix (New England Biolabs, UK) was done. DNAs were sequenced using MiSeq or MiniSeq Mid-output kit (Illumina, USA). For multiplexed CReVIS-seq, equal concentrations of 3 different sgRNAs were mixed to give a final concentration of 13.5 pmol.
LM-PCR
Genomic DNA was purified using a DNeasy Blood & Tissue kit (QIAGEN, Germany). 1 μg of purified high molecular weight DNAs were fragmented to 500 base pairs using a Covaris M220 Focused-ultrasonicator with the following settings: power, 50; duty factor, 10; cycles per burst, 200; time, 50 s; temp, 20°C. Fragmented DNAs were purified using Expin PCR SV mini kit (GeneAll Biotechnology, South Korea). The linear fragments were end-repaired, da-tailed, and ligated to sequencing adaptors using NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs, UK). Nested PCR was proceeded to 3 step (1st, 25 cycles; 2nd, 25 cycles; and 3rd, 30 cycles) using KOD multi & Epi (TOYOBO, Japan). Primers were designed to target 3′ LTR and adaptor sequence for nested PCR 1st and 2nd cycle and primers for 3rd PCR was designed as index primers for Illuimna (Table S2). DNAs were sequenced using MiniSeq Mid-output kit (Illumina, USA).
Sequence analysis process
From the received LTR sequence and target sequence, a 15-nt sequence starting from the cleavage site was set as an indicator sequence. The reads in a fastq file were filtered by the indicator sequence allowing up to 1 mismatch, cut off to 30-nt, and consolidated. The consolidated sequences were aligned to the human reference genome (GRCh38) by bwa-fastmap27 and their locations were confirmed from the GRCh38 gff3 file (https://ensembl.org/Homo_sapiens/).
CReVIS-Seq analysis program
The program, which was developed with Python 3, is activated by the following command:
Python3 LTR_sequence target_sequence file1.fastq output.txt -input2 file2.fastq
This program has been uploaded to github (https://github.com/Gue-ho/CReVIS-Seq).
CReVIS-Seq web tool program
The CReVIS-seq web tool was developed using Django (https://www.djangoproject.com/) as a backend program and is displayed using Bootstrap library (https://getbootstrap.com/). The gzipped file is decompressed by using a JavaScript library pako (http://nodeca.github.io/pako). The CReVIS-seq web tool consolidates the next-generation sequencing (NGS) data at the client-side browser using JavaScript and uploads the consolidated data to the server. The uploaded data are aligned by BWA-fastmap and the information about location is retrieved from the database that was constructed from the GRCh38 gff3 file. The analysis results are displayed in a table and visualized by a chromomap using JavaScript library Ideogram (https://github.com/eweitz/ideogram).
Accession number
The high-throughput sequencing data from this study have been submitted to the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) under NCBI: PRJNA602179.
Acknowledgments
We thank Dr. Heather McDonald for editing of this manuscript. This research was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korean Ministry of Education, Science, and Technology (grant numbers 2017R1E1A1A01074529, 2018M3A9H3021707, 2019M3A9H1103783, and 2020R1I1A2075393 to J.K.H. and 2018M3A9H3022412 and 2020M3A9I4036072 to S.B.) and the New Breeding Technologies Development Program (PJ01487401202001) to S.B.
Author contributions
H.S.K. and J.K.H. conceived this project. H.S.K., H.K.L., T.B., and S.-H.P. performed the experiments. G.-H.H. constructed analysis tools for this study. Y.J.K., S.L., and J.-H.P. gave critical comments. H.S.K., G.-H.H., S.B., and J.K.H wrote the manuscript with the approval of all other authors. S.B. and J.K.H. supervised the research.
Declaration of interests
H.S.K. and J.K.H. are co-inventors on a patent application covering the CReVIS-seq method described in this manuscript. The remaining authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.10.012.
Contributor Information
Sangsu Bae, Email: sangsubae@hanyang.ac.kr.
Junho K. Hur, Email: juhur@hanyang.ac.kr.
Supplemental information
References
- 1.Ciuffi A. Mechanisms governing lentivirus integration site selection. Curr. Gene Ther. 2008;8:419–429. doi: 10.2174/156652308786848021. [DOI] [PubMed] [Google Scholar]
- 2.Fanales-Belasio E., Raimondo M., Suligoi B., Buttò S. HIV virology and pathogenetic mechanisms of infection: a brief overview. Ann. Ist. Super. Sanita. 2010;46:5–14. doi: 10.4415/ANN_10_01_02. [DOI] [PubMed] [Google Scholar]
- 3.Milone M.C., O’Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32:1529–1541. doi: 10.1038/s41375-018-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 5.Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., Veres G., Schmidt M., Kutschera I., Vidaud M., Abel U., Dal-Cortivo L., Caccavelli L. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 6.Negre O., Eggimann A.V., Beuzard Y., Ribeil J.A., Bourget P., Borwornpinyo S., Hongeng S., Hacein-Bey S., Cavazzana M., Leboulch P., Payen E. Gene Therapy of the β-Hemoglobinopathies by Lentiviral Transfer of the β(A(T87Q))-Globin Gene. Hum. Gene Ther. 2016;27:148–165. doi: 10.1089/hum.2016.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maude S.L. Future directions in chimeric antigen receptor T cell therapy. Curr. Opin. Pediatr. 2017;29:27–33. doi: 10.1097/MOP.0000000000000436. [DOI] [PubMed] [Google Scholar]
- 8.Ngo V.N., Davis R.E., Lamy L., Yu X., Zhao H., Lenz G., Lam L.T., Dave S., Yang L., Powell J., Staudt L.M. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–110. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- 9.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bokhoven M., Stephen S.L., Knight S., Gevers E.F., Robinson I.C., Takeuchi Y., Collins M.K. Insertional gene activation by lentiviral and gammaretroviral vectors. J. Virol. 2009;83:283–294. doi: 10.1128/JVI.01865-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ronen K., Negre O., Roth S., Colomb C., Malani N., Denaro M., Brady T., Fusil F., Gillet-Legrand B., Hehir K. Distribution of lentiviral vector integration sites in mice following therapeutic gene transfer to treat β-thalassemia. Mol. Ther. 2011;19:1273–1286. doi: 10.1038/mt.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu X., Li Y., Crise B., Burgess S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- 13.Cheng J.K., Lewis A.M., Kim S., Dyess T., Alper H.S. Identifying and retargeting transcriptional hot spots in the human genome. Biotechnol. J. 2016;11:1100–1109. doi: 10.1002/biot.201600015. [DOI] [PubMed] [Google Scholar]
- 14.Biffi A., Bartolomae C.C., Cesana D., Cartier N., Aubourg P., Ranzani M., Cesani M., Benedicenti F., Plati T., Rubagotti E. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood. 2011;117:5332–5339. doi: 10.1182/blood-2010-09-306761. [DOI] [PubMed] [Google Scholar]
- 15.Montini E., Cesana D., Schmidt M., Sanvito F., Bartholomae C.C., Ranzani M., Benedicenti F., Sergi L.S., Ambrosi A., Ponzoni M. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Invest. 2009;119:964–975. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang G.P., Levine B.L., Binder G.K., Berry C.C., Malani N., McGarrity G., Tebas P., June C.H., Bushman F.D. Analysis of lentiviral vector integration in HIV+ study subjects receiving autologous infusions of gene modified CD4+ T cells. Mol. Ther. 2009;17:844–850. doi: 10.1038/mt.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mueller P.R., Wold B. In vivo footprinting of a muscle specific enhancer by ligation mediated PCR. Science. 1989;246:780–786. doi: 10.1126/science.2814500. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt M., Schwarzwaelder K., Bartholomae C., Zaoui K., Ball C., Pilz I., Braun S., Glimm H., von Kalle C. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR) Nat. Methods. 2007;4:1051–1057. doi: 10.1038/nmeth1103. [DOI] [PubMed] [Google Scholar]
- 19.Tsai S.Q., Nguyen N.T., Malagon-Lopez J., Topkar V.V., Aryee M.J., Joung J.K. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeong Y.K., Yu J., Bae S. Construction of non-canonical PAM-targeting adenosine base editors by restriction enzyme-free DNA cloning using CRISPR-Cas9. Sci. Rep. 2019;9:4939. doi: 10.1038/s41598-019-41356-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee S.H., Park Y.H., Jin Y.B., Kim S.U., Hur J.K. CRISPR Diagnosis and Therapeutics with Single Base Pair Precision. Trends Mol. Med. 2020;26:337–350. doi: 10.1016/j.molmed.2019.09.008. [DOI] [PubMed] [Google Scholar]
- 22.Kim H.S., Lee K., Kim S.J., Cho S., Shin H.J., Kim C., Kim J.S. Arrayed CRISPR screen with image-based assay reliably uncovers host genes required for coxsackievirus infection. Genome Res. 2018;28:859–868. doi: 10.1101/gr.230250.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farnet C.M., Haseltine W.A. Circularization of human immunodeficiency virus type 1 DNA in vitro. J. Virol. 1991;65:6942–6952. doi: 10.1128/jvi.65.12.6942-6952.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim D., Kim S., Kim S., Park J., Kim J.S. Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res. 2016;26:406–415. doi: 10.1101/gr.199588.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S., Kim D., Cho S.W., Kim J., Kim J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 arXiv:1303.3997v2. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.