

Abstract
Ethylene-forming enzyme (EFE) is an ambifunctional iron(II)- and 2-oxoglutarate-dependent (Fe/2OG) oxygenase. In its major (EF) reaction, it converts carbons 1, 2, and 5 of 2OG to CO2 and carbons 3 and 4 to ethylene, a four-electron oxidation drastically different from the simpler decarboxylation of 2OG to succinate mediated by all other Fe/2OG enzymes. EFE also catalyzes a minor reaction, in which the normal decarboxylation is coupled to oxidation of L-arginine (a required activator for the EF pathway), resulting in its conversion to L-glutamate semialdehyde and guanidine. Here we show that, consistent with precedent, the L-Arg-oxidation (RO) pathway proceeds via an iron(IV)-oxo (ferryl) intermediate. Use of 5,5-[2H2]-L-Arg slows decay of the ferryl complex by >16-fold, implying that RO is initiated by hydrogen-atom transfer (HAT) from C5. That this large substrate deuterium kinetic isotope effect has no impact on the EF:RO partition ratio implies that the same ferryl intermediate cannot be on the EF pathway; the pathways must diverge earlier. Consistent with this conclusion, the variant enzyme bearing the Asp191Glu ligand substitution accumulates ~ four times as much of the ferryl complex as the wild-type enzyme and exhibits a ~ 40-fold diminished EF:RO partition ratio. The selective detriment of this nearly conservative substitution to the EF pathway implies that it has unusually stringent stereoelectronic requirements. An active-site, like-charge guanidium pair, which involves the L-Arg substrate/activator and is unique to EFE among four crystallographically characterized L-Arg-modifying Fe/2OG oxygenases, may serve to selectively stabilize the transition state leading to the unique EF branch.
Graphical Abstract
INTRODUCTION
Iron(II)- and 2-oxoglutarate-dependent (Fe/2OG) oxygenases use a common co-substrate (2OG) as the source of the two electrons needed to balance the four-electron reduction of dioxygen with the two-electron oxidation of an Fe(II) cofactor to an Fe(IV)-oxo (ferryl) intermediate.1–4 The co-substrate chelates the Fe(II) cofactor via its C1 carboxylate and C2 carbonyl oxygens, contributing to a square-pyramidal coordination sphere that is rounded out by (most often) three facially disposed amino acid ligands – two histidines and one carboxylate – provided by the conserved β-sandwich protein architecture that defines the enzyme family.5–7 Dioxygen adds to the open axial site of this five-coordinate complex, and the co-substrate undergoes oxidative decarboxylation (C1-C2-bond cleavage) to produce succinate. In the process, one of the two oxygen atoms from O2 is incorporated into the C2-derived carboxylate, as the other oxygen remains bonded to iron in the ferryl complex. The ferryl intermediate most often abstracts hydrogen (H•) from an aliphatic carbon of the “prime” substrate (R–H) to form a fleeting Fe(III)-OH/R• intermediate.8 Different fates of this intermediate lead to the varied outcomes – hydroxylation, halogenation, desaturation, cyclization, ring expansion, stereoinversion, endoperoxidation, and others – of which members of the versatile enzyme family are collectively capable.2,3,9,10 Most of these outcomes extract two-electrons from the prime substrate, thus resetting the enzyme for another turnover.
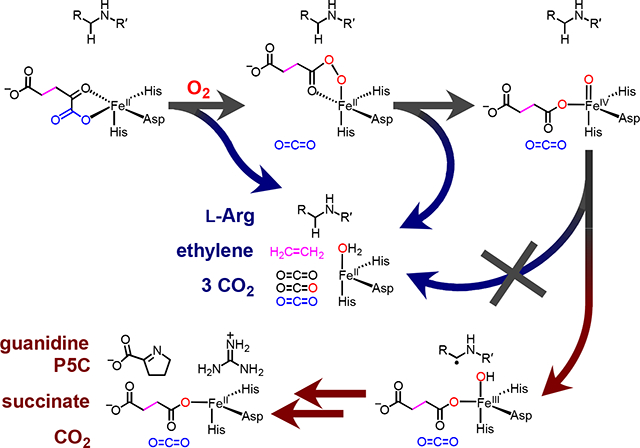
Ethylene-forming enzyme (EFE) from pseudomonads and fungi is a unique, ambifunctional Fe/2OG oxygenase that, in its namesake, major reaction, diverges rather dramatically from the chemical logic that characterizes the enzyme family.11,12 In this reaction (Scheme 1, lower pathway), EFE confers all four oxidizing equivalents of O2 to the co-substrate, globally fragmenting it to three equivalents of CO2 (from carbons 1, 2, and 5) and one equivalent of ethylene (from carbons 3 and 4). The enzyme also catalyzes a minor reaction (upper pathway) that does conform to the family logic, in which oxidation of 2OG to succinate is coupled to the oxidative fragmentation of L-arginine to guanidine and L-glutamate semialdehyde (which cyclizes in solution to pyrroline-5-carboxylate, P5C). Intriguingly, although it is transformed only in the minor reaction, L-Arg is still strictly required for the major reaction.12,13 In essence, the amino acid has distinct roles as prime substrate for the ordinary, L-Arg-oxidizing (RO) reaction and essential effector for the unique, ethylene-forming (EF) reaction.
Scheme 1.

Reactions catalyzed by EFE.
To date, experimental investigations of EFE have been limited to structural characterization of its substrate-bound states, examination of the kinetics and ratios of products (ethylene, pyrroline-5-carboxylate, and succinate) in multiple-turnover assays of wild-type (wt) and variant enzymes, and evaluation of substrate analogs for the ability to support one or both reactions.12–18 By all accounts, wt EFE exhibits an EF:RO partition ratio of ~ 2–3:1 when turning over its native substrates. Substitutions to active-site-proximal residues have, in general, proven deleterious to both reactions, but a number of those examined compromise the EF pathway more profoundly than the RO pathway. By contrast, none has been shown to increase the EF:RO partition ratio relative to that of the wt enzyme. Experiments with L-Arg analogues have suggested a remarkable specificity for the allosteric activator/prime substrate. All analogues that have been tested are, according to these reports, poorer activators of the EF pathway.13,16 From the limited available mechanistic data, it has not yet been possible to answer the central mechanistic question posed by the unusual EF outcome: at which point, and by what novel step, does this pathway diverge from the more canonical RO pathway?
The conformity of the RO pathway to the characteristic Fe/2OG-oxygenase logic suggests a mechanism involving ferryl-mediated abstraction of H• from C5 of L-Arg to initiate either (i) hydroxylation followed by C5-N6 fragmentation12,13 or (ii) C5–N6 desaturation followed by hydrolysis.19 Two recent x-ray crystal structures confirmed the required proximity of C5 to the metal center [N-oxalylglycine-coordinated Fe(II) or 2OG-coordinated Mn(II)], but no direct evidence for the targeting of C5 by a ferryl complex has been reported. It is less obvious that the major EF pathway should proceed through a ferryl intermediate; mechanisms with and without such a complex can be proposed. Indeed, two of the aforementioned experimental studies posited different ferryl complexes – in one case, a succinate-coordinated ferryl initiating ethylene production16 and, in the second case, an oxalate-coordinated ferryl forming concomitantly with ethylene17 – whereas a recent computational study proposed an EF pathway not proceeding through a ferryl intermediate.20
In the reactions of other Fe/2OG oxygenases, ferryl intermediates have been demonstrated on the basis of three hallmarks: (i) their increased UV absorption (typically monitored at ~ 320 nm) relative to the enzyme•Fe(II)•2OG•substrate and enzyme•Fe(II)•succinate•product complexes; (ii) their characteristic Mössbauer isomer shifts (δ ~ 0.3 mm/s) and quadrupole splitting (ΔEQ ~ −1 mm/s) parameters;21,22 and (iii) the large substrate deuterium kinetic isotope effects (D-KIEs) on their decay by H•/D• transfer, which results in increased intensity and longevity of these spectral signatures.3,8,23 The weak absorption feature of the reactant complexes at 500–530 nm (ε ~ 200 M−1cm−1) – arising from a metal-to-ligand [Fe(II)-to-2OG] charge-transfer (MLCT) transition5 – has also proven useful in transient-kinetic monitoring of the reactions. The ferryl complexes are approximately isosbestic in this region, but the enzyme•Fe(II)•succinate•product complexes (referred to in the original work on TauD as a “second intermediate”21) are transparent, with the result that this optical feature provides an additional way to monitor either conversion of the reactant complex to the product complex in cases of a non-accumulating ferryl intermediate or decay of the ferryl intermediate to the product(s) complex when the high-valent species accumulates substantially.24,25
In this study, we used stopped-flow absorption (SF-Abs) and freeze-quench Mössbauer (FQ-Möss) spectroscopies along with substrate deuterium labeling, quantitative product analysis, and site-directed mutagenesis to test for a ferryl complex in either or both pathways of the EFE reaction, and we solved the structure of the authentic EFE•Fe(II)•2OG•L-Arg reactant complex. The kinetic and spectroscopic results confirm the expectation that the RO reaction proceeds via the canonical ferryl intermediate but also reveal that the detected complex forms after the branchpoint of the two pathways, meaning that it is not on the EF pathway. The ability of a nearly conservative Asp → Glu ligand substitution to almost completely derail the EF pathway while barely affecting the RO pathway is consistent with unusually stringent stereoelectronic requirements for the major reaction, and structural features of the EFE reactant complex that distinguish it from those of several other L-Arg-modifying Fe/2OG oxygenases characterized previously – most notably, the flipped orientation of L-Arg16 and resulting like-charge guanidium pair involving its side-chain and an active-site Arg residue – suggest how the transition state leading to the unique outcome might be selectively stabilized.
RESULTS
Activity and Partition Ratio of Our Preparations of EFE.
To verify the integrity of our preparations of Pseudomonas savastanoi pv. phaseolicola EFE, we quantified the products of multiple-turnover reactions by liquid chromatography with mass spectrometric detection (LC-MS) and gas chromatography with flame-ionization detection (GC-FID). We detected pyrroline-5-carboxylate (P5C), a product of the RO pathway, by first reducing it to proline with sodium cyanoborohydride and then appending an Fmoc protecting group prior to LC-MS analysis. Our results corroborate previous reports12,13 that, with unlabeled L-Arg as co-substrate, ~ 70 % of the 2OG consumed by wild-type EFE yields ethylene, while the remaining ~ 30 % is converted to succinate (Figure 1). The rates ([product]•time−1•[EFE]−1) of succinate and ethylene production that we measured (~ 15 min−1 and ~ 40 min−1, respectively) are also consistent with published reports.13,14,26
Figure 1.

Yields of succinate, Fmoc-proline (P5C), and ethylene after incubation of 10 μM wt or D191E EFE with 40 μM (NH4)2Fe(SO4)2, 200 μM 2OG, and 500 μM of unlabeled L-arginine (L-Arg), perdeuterated L-arginine (d7-L-Arg), or 5,5-d2-L-arginine (d2-L-Arg) in air-saturated 40 mM sodium HEPES (pH 7.5) buffer at 21°C for ~10 minutes. 2OG was completely consumed during the reaction. P5C was derivatized to Fmoc-proline prior to quantification. Ethylene analysis was performed by GC-FID; succinate and Fmoc-proline were quantified by LC-MS. Error bars correspond to standard errors of the mean values for two experiments. Detailed procedures are provided in the Supporting Information.
Evidence for an L-Arg-Oxidizing Ferryl Intermediate in the EFE Reaction.
To test for accumulation of a ferryl intermediate in the EFE reaction, we mixed the anoxic EFE•Fe(II)•2OG•L-Arg complex with O2-containing buffer in a stopped-flow apparatus. Weak UV absorption developed within ~ 0.07 s and decayed by more than 90 % within 1 s (Figure 2A, solid black circles). The loss of absorbance at 510 nm in this same time regime (Figure 2B, solid black circles) signified conversion of the reactant state to the transparent Fe(II)-containing product state(s) via a barely accumulating (presumptive) ferryl complex. Use of L-Arg bearing deuterium at C5 (here, 2,3,3,4,4,5,5-[2H7]-L-Arg or d7-L-Arg) resulted in 4–5-fold increases in both the upward deflection in the ΔA318-versus-time trace and the time for its return back to the initial absorbance (Figure 2A, open black circles). Use of deuterated L-Arg also resulted in a modest decrease in the downward deflection of the ΔA510-versus-time trace (Figure 2B, open black circles), but, importantly, the temporal delay and diminution of the amplitude of this trace were not as profound as one would have expected in view of the markedly greater accumulation of the presumptive ferryl complex, which, as noted, absorbs significantly in the visible regime in other Fe/2OG enzymes. This observation is explained by the facts that (i) only ~ one-third of the reaction flux (the RO pathway) is sensitive to the presence of deuterium in L-Arg and (ii) the other two-thirds (the EF pathway) converts the reactant complex to a transparent product complex without substantial accumulation of an intervening, 510-nm-absorbing state (as explained more below). These observations are consistent with a modest but detectable accumulation of the ferryl intermediate with the protium-containing substrate and enhanced accumulation with d7-L-Arg as a result of a large, normal D-KIE on HAT to the high-valent complex.
Figure 2.

Kinetic analysis of the wt and D191E EFE reactions with protium- and deuterium-bearing L-Arg substrates by stopped-flow absorption spectroscopy. The change in absorbance (A) at 318 nm and (B) at 510 nm was monitored after mixing (at 5 °C) of an anoxic solution containing 0.7 mM wt EFE (black) or D191E EFE (red), 0.6 mM Fe(II), 8 mM 2OG, and 8 mM L-Arg (filled circles) or d7-L-Arg (open circles) in 40 mM sodium HEPES (pH 7.5) buffer with an equal volume of the same buffer containing ~ 0.48 mM O2. The solid lines are the results of global fitting of the data to a kinetic model in which two rapidly-equilibrating reactant complexes react with O2, as described in the Supporting Information and shown in Scheme S1.
To confirm the expectation that the H• donor to the presumptive ferryl complex is C5, we also examined the reaction with site-specifically C5-deuterated L-Arg (5,5-[2H2]-L-Arg or d2-L-Arg). As anticipated, the increased accumulation and slower decay seen with the d7-L-Arg substrate was also observed with the specifically labeled substrate (Figure S3). The fact that the kinetics of decay with d2-L-Arg and d7-L-Arg were essentially identical implies that the ferryl complex cannot abstract H• from a different site with an appreciable rate constant, a conclusion supported by our inability to detect any other L-Arg-derived product in the reaction with d2-L-Arg.
We next confirmed by freeze-quench Mössbauer spectroscopy that the UV-absorbing intermediate is, in fact, a ferryl complex. The spectrum of the frozen EFE•57Fe(II)•2OG•d7-L-Arg reactant solution acquired at 4.2 K without an applied magnetic field (Figure 3A, i, solid lines) consists of a quadrupole doublet with isomer shift (δ) and quadrupole splitting (ΔEQ) parameters characteristic of N/O-coordinated, high-spin Fe(II) ions (δ ~ 1.1 mm/s and ΔEQ ~ 2.6 mm/s).27 This reactant solution was mixed at 5 °C with an equal volume of buffer containing ~ 1.8 mM O2 and frozen after 0.34 s, the reaction time at which the UV-absorbing complex was seen to accumulate maximally (Figure S4). The 4.2-K/zero-field spectrum of this sample (Figure 3A, ii, solid lines) exhibits a broadened doublet for the Fe(II) species (indicated by black arrows) and a new peak at ~ 0.8 mm/s (indicated by the red arrow). The latter feature is at the position typical for the high-energy line associated with the high-spin ferryl complexes detected in other Fe/2OG enzymes.8 Both sets of features are less sharp in the spectra acquired at the same temperature (4.2 K) with a 53 mT magnetic field oriented parallel to the γ-beam (Figure 3A, i and ii, vertical bars), as has also been seen in studies of other Fe/2OG enzymes. Subtraction of the zero-field spectrum of the frozen reactant solution from the zero-field spectrum of the 0.34-s reaction sample resulted in a difference spectrum that can be simulated as a superposition of three quadrupole doublets (Figure 3A, iii, vertical bars and black solid line). One of the downward-pointing quadrupole doublets (which arise from the species that form in the 0.34-s incubation) has parameters typical of a high-spin ferryl complex (δ = 0.26 mm/s, ΔEQ = 0.96 mm/s, Γ = 0.25 mm/s, red line) and contributes 11 % of the total intensity. The 4.2-K/53-mT Mössbauer spectrum of a sample reacted with O2 for 5 s – by which time the 318-nm-absorbing intermediate was observed to decay in SF-Abs experiments – lacks the line at 0.8 mm/s, demonstrating decay of the ferryl complex and the transient UV absorption on the same timescale (Figure 3A, iv). Together, the SF-Abs and FQ-Möss results establish the accumulation of a ferryl intermediate that can initiate L-Arg oxidation by abstracting H• from C5.
Figure 3.
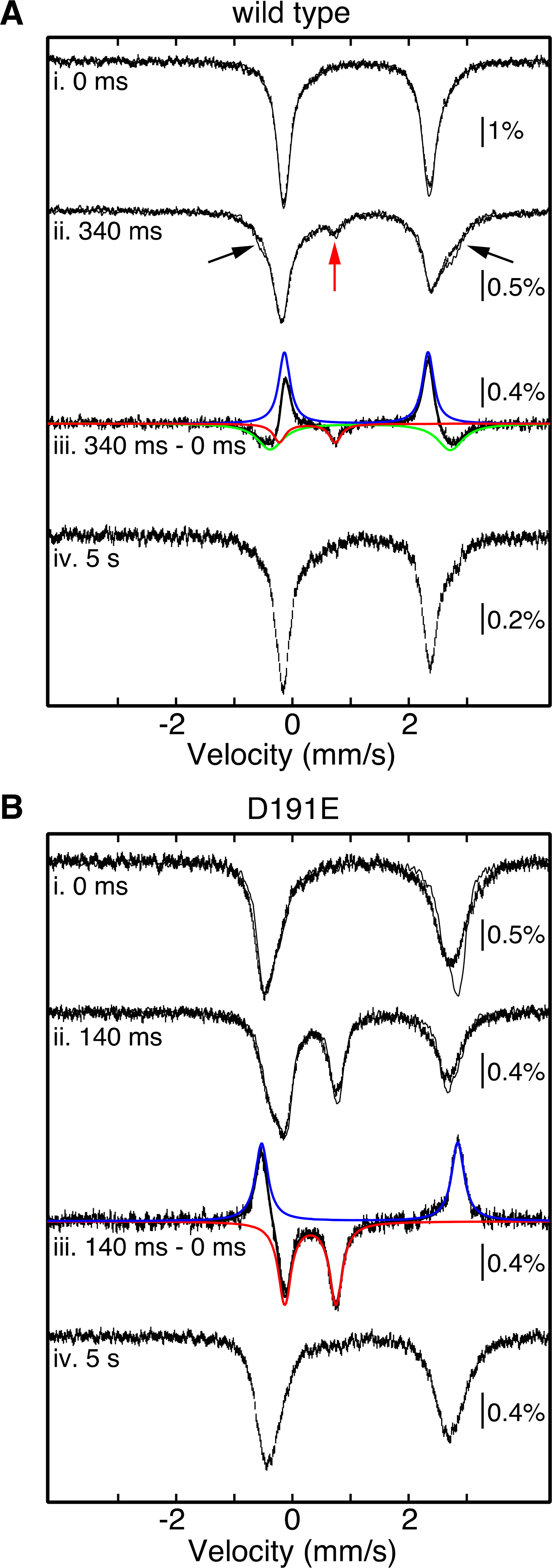
Analysis of the reactions of (A) wt and (B) D191 EFE by freeze-quench Mössbauer spectroscopy. Experimental spectra (i, ii, and iv) were collected at 4.2 K with either no applied magnetic field (solid lines) or a field of 53 mT applied parallel to the γ-beam (vertical bars). In preparation of the freeze-quenched samples (spectra ii and iv), an anoxic solution containing 1.8 mM EFE (wt or D191E), 1.4 mM 57Fe(II), 5 mM 2OG, and 5 mM d7-L-Arg in 40 mM sodium HEPES buffer (pH 7.5) containing 5 % glycerol was mixed at 5 °C with an equal volume of O2-saturated (~ 1.8 mM O2) buffer, and the reaction was allowed to proceed for the indicated time before being terminated by rapid freezing in the cryosolvent (2-methylbutane at ~ −150 °C). For samples of the reactant complexes (spectra i), the same anoxic protein reactant solution was manually frozen (wt, panel A) or diluted with an equal volume of O2-free buffer before being manually frozen (D191E, panel B). Spectra iii (vertical bars) result from subtraction of the experimental zero-field spectra (ii - i) to illustrate the changes that occur at early time. These difference spectra can be accounted for as summations (black lines) of either three (wt, panel A) or two (D191E, panel B) quadrupole-doublet components (colored lines) corresponding to features that form (downward) and decay (upward) over short reaction times. The colored theoretical spectra are associated with the ferryl intermediate (red), the Fe(II)-containing reactant complex (blue), and the Fe(II)-containing product complex of the EF pathway (green in panel A only). The parameters used to generate these theoretical spectra are given in the text and Table S8.
Evidence that the Ferryl Intermediate is Specific to the RO Pathway.
The modest (11 %) accumulation of the ferryl intermediate arises from two factors. First, despite the retarding effect of the large D-KIE, decay of the intermediate in the d7-L-Arg reaction is still not fully kinetically resolved from its formation, and this competition suppresses accumulation. Second, intermediacy of the ferryl complex in only the minor, RO pathway diminishes the quantity that can accumulate to only ~ 0.3 times (the fraction proceeding down the RO branch) the limit imposed by the former effect. This more mechanistically informative origin of ferryl suppression is established by the quantitative, global simulations of the stopped-flow data (Figure 2, Table 1, and Supporting Information), but it can also be understood qualitatively by comparing the deflections in the ΔA510- and ΔA318-versus-time traces from the reactions with the unlabeled and d7-L-Arg substrates (Figure 2). From anoxic titrations of the enzyme with Fe(II) in the presence of the other substrates (Figure S1), we determined a molar absorption coefficient at 510 nm (ε510) of 110 ± 10 M−1cm−1 for the EFE•Fe(II)•2OG•L-Arg complex. This value is somewhat greater than that reported by Hausinger and co-workers (~80 M−1cm−1),26 but problems with determination of enzyme concentration13,26 in that study allay our concern over the discrepancy. With the protium-bearing substrate, the ferryl complex accumulates minimally, and the ΔA510-versus-time trace (Figure 2B, solid black circles) reflects (primarily) a cycle of net conversion of the absorbing reactant complex to the transparent product complex(es) followed by regeneration of the reactant. The downward deflection of 0.011 in this trace implies net loss of 0.10 mM of the reactant complex (0.3 iron equiv) at ~ 0.85 s, the minimum of the trace. This shortfall from the maximum imposed by the [O2] of 0.24 mM is accounted for by kinetic competition between decay and regeneration of the absorbing reactant complex. At the approximately equal concentrations of EFE•Fe(II)•2OG•L-Arg and O2, the comparable rates of the two steps, combined with the second-order nature of the first step, suppresses accumulation of the product complexes (i.e., net loss of the reactant) to ~ 40 % the concentration of the limiting reactant. The ferryl complexes that have been characterized in other Fe/2OG enzymes all have molar absorption coefficients (ε318) of ~ 1,500 M−1cm−1, and our global kinetic model (see Supporting Information) is consistent with an equivalent ε318 for the EFE ferryl complex. The upward deflection of the ΔA318-versus-time trace from the d7-L-Arg reaction of 0.05 at 0.55 s thus implies accumulation of a maximum of only 0.033 mM of the ferryl intermediate (0.11 equiv), consistent with the quantity detected by freeze-quench Mössbauer spectroscopy. The timescales for decay of the ferryl complex in the d7-L-Arg reaction and regeneration of the reactant complex in the reaction with unlabeled substrate differ by no more than a factor of 1.5, and so a kinetic origin of the 3.6-fold shortfall in ferryl accumulation relative to reactant decay can be ruled out. The explanation is that the ferryl intermediate accumulates only in the minor, RO pathway.
Table 1.
Rate constants for formation and decay of the RO ferryl complex obtained by fitting the ΔA318-versus-time data shown in Figure 2 to an A + B → C → D model using KinTek Explorer. Initial reactant and O2 concentrations were reduced by a factor that reflects flux through the RO pathway (i.e., divided by 3.5 for wt and 1.05 for D191E). ε318 and ε510 values for the reactant complexes were ~ 40 and 90 M−1 cm−1, respectively, and ε318 and ε510 for the ferryl complexes were ~1450 and 190 M−1 cm−1, respectively.
| substrate | kformation, net (103 M−1 s−1) | kdecay (s−1) | kH/kD | |
|---|---|---|---|---|
| wt | L-Arg | 54 | 16 | |
| wt | d7-L-Arg | 54 | 1 | 16 |
| D191E | L-Arg | 31 | 42 | |
| D191E | d7-L-Arg | 31 | 1.6 | 26 |
The Mössbauer spectra from the freeze-quench experiment provide additional evidence for this conclusion. In addition to the downward features for the ferryl complex that develops (11 % of total intensity), the difference spectrum showing the changes that occur in the first 0.34 s of the reaction (Figure 3A, iii, vertical bars) also exhibits upward features associated with loss of the Fe(II)-containing reactant complex (δ = 1.10 mm/s, ΔEQ = 2.47 mm/s, line width Γ = 0.24 mm/s, blue line) corresponding to 39 % of the total intensity. The quantitative mismatch in these contributions is accounted for by the signature of a new Fe(II) species (δ = 1.16 mm/s, ΔEQ = 3.10 mm/s, Γ = 0.54 mm/s, 32 % of total intensity, green line) that develops along with the ferryl complex. The combined analysis of SF-Abs and FQ Mössbauer data reveals that only ~ one-third of the reactant complex that decays in this short reaction time yields the ferryl intermediate, while ~ two-thirds yields a new Fe(II)-containing species, which is presumably the product complex in the EF pathway.
Implications of the L-Arg C5 D-KIE on Ferryl Decay and Absence of its Effect on the EF:RO Partition Ratio.
Further analysis lends even greater weight to the conclusion that the detected ferryl complex is specific to the RO pathway and not a branchpoint between the pathways. In a scenario involving partitioning of a common ferryl intermediate between HAT from C5 of L-Arg and succinate fragmentation (e.g., by proton-coupled electron transfer or PCET) to ethylene and two (additional) equivalents of CO2 (Scheme 2, orange vs purple or blue dotted arrows), a large D-KIE on the C5-HAT step should impact the partition ratio, making the EF pathway even more dominant. We verified that, to the contrary, the presence of deuterium does not measurably impact the EF:RO partition ratio (Figure 1). The observed partition ratio (FEF/FRO, where F is “fraction”) would reflect the relative magnitudes of the rate constants for the two competing steps, kHAT and kPCET, and the observed rate constant for decay of the ferryl complex (kobs) would be the sum of the rate constants for the two steps (Eqs. 1–4). In this case, with succinate oxidation being faster
| (1) |
| (2) |
| (3) |
| (4) |
than L-arginine oxidation by a factor of ~ 2.5 (kPCET = 2.5•kHAT), the intrinsic D-KIE on the HAT step (kHAT/kDAT) would be only minimally “expressed” in the observed D-KIE on decay of the ferryl complex (kobs,H/kobs,D) as a consequence of the insensitivity of the other step (PCET) to deuterium substitution in L-Arg. Thus, even an extremely large intrinsic D-KIE on the HAT step giving a very small value of kDAT would yield an observed effect of only ≤ 1.4 (Eqs. 5–6). By con-
| (5) |
| (6) |
trast, the global simulations summarized in Table 1 and the Supporting Information imply an observed D-KIE of at least 16 on decay of the intermediate with either the d7- or the d2-L-Arg substrate. This D-KIE is simply too large for there to be more than a few percent of flux (ferryl decay) through another step that competes with HAT from C5 to the intermediate complex (at least for the case of the protium-bearing L-Arg substrate). The failure of these predictions of Scheme 2 rules out this latest possible EF/RO branchpoint, with the ferryl complex as a common intermediate.
Scheme 2.

Potential mechanisms of ethylene formation by EFE.
Demonstration of a Selectively Compromised EF Pathway and Enhanced Ferryl Accumulation in the D191E Ligand Variant.
The conclusion that the detected ferryl complex is specific to the RO pathway raised the question of whether perturbations impacting the EF:RO partition ratio (e.g., amino acid substitutions or substrate modifications) might also impact the quantity of ferryl complex to accumulate. We were encouraged to address this question by previous studies, in which single substitutions near the cofactor were reported to alter the partition ratio, compromising ethylene production to a greater extent than L-Arg oxidation.17 However, none of these variant proteins has (to the best of our knowledge) been examined in the transient state for detriments to O2 activation leading to intermediate formation, which, in past work, has generally been seen for such active-site-proximal substitutions.28,29 Seeking a minimal structural perturbation to alter the partition ratio without causing such unwanted, global kinetic effects, we substituted the facial-triad ligand, Asp191, with Glu. LC-MS/GC-FID assays showed that the D191E variant degrades L-Arg and 2OG to P5C (again detected after reduction to proline and Fmoc derivatization) and succinate nearly stoichiometrically, with only ~ 5 % of the co-substrate being fragmented to ethylene (Figure 1). As with the wt enzyme, mixing of the anoxic EFE-D191E•Fe(II)•2OG•L-Arg complex with O2 resulted in development of transient absorption at 318 nm in tens of milliseconds and subsequent decay over seconds (Figure 2A, solid red circles), and use of d7-L-Arg in place of the protium-bearing substrate increased the amplitude and decay time by a factor of ~ 4 (open red circles). Importantly, the deflections in the ΔA318-versus-time traces were, for both substrates, ~ 3.5 times as large as for wt EFE under the same reaction conditions, suggesting that the variant protein accumulates more of the ferryl complex.
We confirmed this implication by freeze-quench Mössbauer spectroscopy. Figure 3B shows the difference spectrum (iii, vertical bars) obtained by subtracting the 4.2-K/zero-field spectrum of the frozen D191E-EFE•57Fe(II)•2OG•d7-L-Arg reactant complex (i, solid line) from the corresponding spectrum of a sample frozen after this complex was exposed to O2 for 0.14 s (ii, solid line). A developing quadrupole doublet from the high-spin ferryl complex appears as downward features contributing 43 % of the total intensity (red line). The upwards features arising from the decaying reactant complex have essentially the same relative intensity (40 %, blue line). Interestingly, the Mössbauer parameters of the ferryl complexes in the variant (δ = 0.32 mm/s, ΔEQ = 0.88 mm/s, Γ = 0.27 mm/s) and wt (see above) enzymes are not identical, and those of their reactant complexes differ to an even greater extent (Table S8). As in the reaction of the wt enzyme, the ferryl complex in the D191E variant was seen to decay within 5 s, as indicated by the absence of a feature at ~ 0.8 mm/s in the 4.2K/53-mT spectrum of the sample frozen at this reaction time (Figure 3B, iv).
The Mössbauer spectra of Figure 3 confirm the implication of the stopped-flow absorption data that 3–4 times as much of the ferryl complex accumulates in the reaction of the D191E variant as in the reaction of wt EFE. This difference does not have a kinetic origin in formation and decay rates, which are similar for the two reactions. Rather, it is explained by the almost complete elimination of flux through the EF pathway and the consequent ~ 3.5-fold increase in flux through the RO pathway. This explanation is further supported by the observations that, in the Mössbauer difference spectrum associated with the reaction of the D191E variant protein, the contribution from the developing ferryl complex quantitatively matches that from the decaying reactant complex (~ 40 %), and no new features for an Fe(II)-containing product complex are seen. This behavior contrasts with that of the wt EFE reaction, in which loss of the reactant complex is associated with accumulation of both the ferryl intermediate (11 %) and a new Fe(II)-containing “product” (32 %). With the reaction of the D191E variant proceeding almost homogeneously through the RO pathway, the ferryl-stabilizing effect of deuterium at C5 of L-Arg results in a simple net conversion of the reactant complex to the intermediate – without significant “loss” to the non-absorbing Fe(II) product states – over short reaction times. This simpler conversion has a noteworthy manifestation also in the ΔA520-versus-time traces – an initial rise phase as a result of the fact that the ferryl complex, which in other Fe/2OG enzymes is nearly isosbestic with its precursor, here has a greater molar absorptivity than the (unusually hypochromic) EFE•Fe(II)•2OG•L-Arg reactant complex. This fact would not have been obvious solely from the data on wild-type EFE, because the net conversion of the reactant complex to the ferryl complex is, in that case, accompanied by ~ 2.5 times as much flux directly to a transparent Fe(II)-containing product complex. These observations on the variant protein corroborate that the detected ferryl complex is specific to the RO pathway.
Structural Basis for Effect of the D191E Substitution on the EF:RO Partition Ratio.
To address why the EF:RO partition ratio is so markedly perturbed by the nearly conservative ligand substitution, we solved crystal structures of the wt and D191E variant proteins in complex with Fe(II), 2OG and L-Arg. Crystals were grown under anoxic conditions to prevent turnover, and structures were solved to resolutions of 1.83 Å (wt) and 1.97 Å (D191E). Our structure (the first) of the authentic reactant complex of wt EFE closely matches both the A conformation in the structure of the EFE•Fe(II)•N-oxalylglycine•L-Arg complex reported by Zhang et al. (PDB accession code 5LUN, rmsd ~0.31 Å over 329 Cα atoms) and the structure of the EFE•Mn(II)•2OG•L-Arg complex presented by Martinez et al. (PDB accession code 5V2Y, rmsd ~0.40 Å over 329 Cα atoms, Figure 4A).16,17 Comparison of our structural models of the wt and D191E EFE•Fe(II)•2OG•L-Arg complexes reveals no global differences between the two proteins (root-mean-square deviation of ≤ 0.12 Å over 328 Cα atoms). The longer tether of the ligand in the D191E variant causes the only significant deviation in the active site. For all monomers in the asymmetric unit, the new glutamate is modeled best in two different, approximately equally prevalent sidechain conformations (Figure 4D), of which one is analogous to that of the Asp ligand in the wild-type protein (Figure 4B). In the other conformation, the carboxylate remains coordinated to the cofactor and hydrogen bonded with the L-Arg substrate, but the projection of both carboxylate oxygen atoms toward the substrate enables the engagement in an additional hydrogen bond with the guanidino group (Figure 4F). The ligand substitution does not, however, detectably perturb the position of either substrate. Moreover, neither of our data sets suggests the presence of the alternative L-Arg binding mode that Zhang et al. posited to enable the EF pathway.16
Figure 4.

X-ray crystallographic models of the EFE active site with 2OG and L-Arg bound. (A) Comparison of the 1.83-Å-resolution wild-type EFE•Fe•2OG•L-Arg structure (PDB accession code 6VP4) to the published 1.43-Å-resolution EFE•Mn•2OG•L-Arg structure (PDB accession code 5V2Y) reveals no significant differences. (B) One of two conformations of the carboxylate side chain observed in the D191E EFE•Fe•2OG•L-Arg structure (conformation A) closely resembles that observed in the wild-type enzyme. In (C) and (D), 2Fo-Fc maps contoured to 1.0σ are shown in black for wild-type and D191E EFE, respectively. Conformation A of the E191 side chain forms a single hydrogen bond with the L-Arg substrate (E), whereas Conformation B forms two hydrogen bonds (F).
DISCUSSION
Regioselectivity of HAT by EFE in comparison to other L-Arg-oxidizing Fe/2OG oxygenases.
The inability of deuterium-bearing isotopologues to generate alternative products of L-Arg oxidation and the identical rates of ferryl decay observed with perdeuterated L-Arg and 5,5-d2-L-Arg demonstrate site-specificity in the HAT step by EFE that contrasts with the behavior of several other ferryl-utilizing nonheme-iron enzymes. In many of these systems the ferryl complexes can be redirected to neighboring sites (leading to different outcomes) when challenged by substrates bearing deuterium only at the preferred H•-donor sites.19,30–34 The different behavior of EFE can be explained, at least in part, by the fact that the binding orientation of the amino acid is essentially opposite (rotated by ~ 180°) to that in these other enzymes. Zhang et al. previously noted this flipped L-Arg orientation in the EFE active site relative to its orientation in the L-Arg 3-hydroxylase, VioC, from the viomycin biosynthetic pathway35,36 which process L-Arg itself and clavaminate synthase, CAS, which acts on a β-lactam derivative of L-Arg.16,37,38 Figure 5 illustrates the intriguing fact that the VioC/CAS-like orientation of the amino acid is, in fact, conserved in two additional, structurally characterized L-Arg modifying enzymes – the 3,4-dihydroxylase, OrfP, from the streptolidine biosynthetic pathway and the 4,5-desaturase, NapI, from the naphthyridinomycin pathway19,39,40 Indeed, the flipped binding mode appears, at present, to be unique to EFE. The comparison with NapI is particularly germane, because the 4,5-desaturase also targets C5 to initiate its reaction but can, unlike EFE, be redirected to C4 and perhaps even C3 by the presence of deuterium at the preferred HAT site(s).19 In the NapI binding mode, the guanidinium functional group of the side chain is poised over His146 of the His2Asp “facial triad,” and the propyl tether (C5-C3) to the α-carbon (C2) projects away. Consequently, the propyl moiety projects over the cofactor in space that is not occupied by protein ligands and is thus potentially available for the H•-abstracting ferryl oxo ligand to take up residence. Although it has been posited that, in general, the oxo ligand can be located trans to either the more C-terminal His ligand (the so-called inline position) or the more N-terminal His ligand (offline),2 the vector of the C5-C3 chain potentially exposes each of these carbons for HAT to a ferryl complex in either configuration. By contrast, the EFE binding mode centers C5 above the cofactor but projects C4-C2 toward the His2Asp facial triad, effectively immunizing these carbons from hydrogen abstraction. Its unique L-Arg binding mode nicely rationalizes the unusually strict regiochemistry of HAT to the ferryl complex in EFE. Might it also hold clues to the mechanism of the novel EF outcome?
Figure 5.

Comparison of the active sites of EFE and three other L-Arg-modifying Fe/2OG oxygenases, NapI (PDB: 6DAW), OrfP (PDB: 4M27), and VioC (PDB: 6ALM), showing a distinct L-Arg binding mode in EFE that features (i) a stacked guanidium pair involving the substrate and Arg171 and (ii) projection of C3 and C4 over His189 to shield them from HAT.
Implications for the Mechanism of Ethylene Formation.
The failure of the large D-KIE on HAT from L-Arg to alter the EF:RO partition ratio implies that the EFE•Fe(IV)-oxo•succinate•L-Arg complex that initiates RO cannot be the branchpoint of the two pathways, but it does not rule out the possibility that a different, fleeting (non-accumulating) succinate-ligated ferryl complex – possibly in an “offline” configuration and originating from earlier pathway bifurcation – initiates ethylene production. Indeed, Zhang et al. proposed a ferryl-mediated Grob-like fragmentation of succinate involving migration of electron density over seven atomic centers (this type of concerted fragmentation more typically involves only five centers), in which the Fe(IV) center would function, uncharacteristically, as nucleofuge by accepting two electrons from the coordinated succinate carboxylate (Scheme 2).16 An alternative means by which a distinct, undetected ferryl complex could initiate ethylene production would involve single-electron transfer (most likely coupled to proton transfer) to the Fe(IV) center from the succinate carboxylate (purple dotted arrow in Scheme 2) followed by radical β-scission of the C1-C2 bond, a mechanism akin to those of the electrolytic Kolbe reaction and the oxidation of 4-hydroxyphenylacetate initiated by a thiyl radical in hydroxyphenylacetate decarboxylase (HPAD).41
We consider it more likely that an unusual fragmentation of an intermediate that would normally be a precursor to the succinate-coordinated ferryl complex, such as the unstable Fe(IV)-peroxyhemiketal complex or Fe(II)-persuccinate intermediate, preempts its formation in the EF pathway (Scheme 2). In a mechanism envisaged by Martinez, et al.,13,17 the unstable Fe(IV)-peroxyhemiketal complex would fragment between C2 and C3 rather than between C1 and C2, thus generating an oxalate- rather than succinate-coordinated ferryl complex. Oxidative fragmentation of the oxalate to the C1- and C2-derived CO2 molecules would proceed by inner-sphere ET to the ferryl complex, decarboxylation of the oxalyl radical, and reduction of the Fe(III) center by the formate radical, similar to the mechanism of the manganese-dependent oxalate oxidase.42,43 In principle, the unique C2-C3 fragmentation could occur by a polar or radical mechanism (Scheme 2), but the heterolytic pathway would require the strained bicyclic complex – which is already thought to be unstable44 – to adopt a conformation with appropriate orbital alignment across the seven atomic centers involved, and the homolytic pathway would generate an Fe(V) complex, unprecedented for the Fe/2OG-oxygenase class. Interestingly, in proposing the Fe(IV)-peroxyhemiketal complex as the likely branchpoint and thereby implying that pathway bifurcation precedes ferryl formation, Martinez et al. also posited that encroachment of the Phe283 side chain toward the open coordination site of the cofactor upon L-Arg binding could promote ethylene production by impeding rearrangement of an “offline” ferryl complex to an inline configuration required for L-Arg hydroxylation. According to this hypothesis, displacement of the active-site Phe caused by the increased length/bulk of the Glu ligand in the D191E EFE variant might, in principle, cause the change in partition ratio. However, the position of the Phe283 side chain is essentially unchanged by the ligand substitution (Figure S6). Given that we do not discern an obvious structural rationale for the marked shift in the EF:RO partition ratio, it is possible that the basis is more dynamic than structural or, alternatively, arises from structural changes too subtle to detect.
In our view, the crystallographically validated Fe(II)-persuccinate complex,45 which canonically undergoes O-O-bond heterolysis to generate the succinate-coordinated ferryl intermediate, is a more likely branch point between the RO and EF pathways than the Fe(IV)-peroxyhemiketal complex (Scheme 2). One possible mechanism of ethylene formation from the Fe(II)-persuccinate intermediate – a seven-center Grob-like fragmentation, wherein the nucleofugal oxygen is bound to the iron center – would seem likely to require concomitant protonation of this oxygen to avoid formation of an extremely basic Fe(II)-oxo complex. An extensive hydrogen bonding network, which includes the co-substrate along with Arg171, Glu84 and the L-Arg substrate, could deliver the requisite protons. Alternatively, as has been explored extensively in studies of heme enzymes and model complexes,46 homolysis of the iron-coordinated peroxide could compete with the canonical heterolysis, generating the Fe(III)-(hydr)oxo/succin-1-yl state that would break down as previously delineated. Given that neither of these two alternative fragmentations of the Fe(II)-peroxysuccinate complex is known to occur in any other Fe/2OG enzyme, unique electrostatic features within the EFE active site would seem to be required to counteract the favored polarity of the O–O cleavage step. In this respect, it seems possible that the flipped L-Arg orientation could have greater import than merely ensuring strict regiochemistry for the ferryl-mediated HAT step in the minor RO pathway. In this orientation, the substrate guanidinium stacks with the side chain guanidinium of Arg171 (Figure 5). This counterintuitive pairing of like-charged Arg side chains in peptides and proteins has received considerable attention over the last decade and has been explained computationally.47-50 Its uniqueness to EFE among the L-Arg processing Fe/2OG oxygenases, the reported sensitivity of the EF pathway to modifications to L-Arg and substitutions of residues that interact with it, and the close proximity of the guanidium pair to the cofactor (especially to the expected location of the coordinated peroxide of the peroxysuccinate intermediate) all suggest that this feature of the EFE•substrates complex might have a role in lowering the barrier for a non-canonical fragmentation of the intermediate that commits to ethylene production.
CONCLUSIONS
A ferryl complex accumulates in the reaction of EFE, but quantitative analysis of time-dependent absorption and Mössbauer spectra implies that it forms in only ~ one-third of events. The 16-fold extension of its lifetime by the presence of deuterium at C5 of L-Arg – without impact to the EF:RO partition ratio – proves that the ferryl complex forms only along the RO pathway, beyond the reaction branchpoint. Thus, the D191E ligand substitution, which virtually abolishes the EF reaction, also promotes accumulation of ~ four times as much of the ferryl complex. The identity of the branch-point intermediate and mechanism of the step leading to ethylene production remain to be established, but it is clear from results presented here and elsewhere that the marginal, selective stabilization of the transition state for this step by the wt enzyme is quite fragile. The basis for the differential robustness of the two pathways, and whether an even greater degree of relative stabilization of the ethylene-committing transition state is possible, are intriguing questions for the future.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the Office of Basic Energy Science within the Department of Energy Office of Science (BES Award DE-SC0016255). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). Data was also collected at the Advanced Light Source, a Department of Energy Office of Science User Facility under Contract No. DE-AC02-05CH11231. The Berkeley Center for Structural Biology is supported by the Howard Hughes Medical Institute and the National Institutes of Health, National Institute of General Medical Sciences, grant P30 GM124169. K.M.D. is grateful for support from the National Institutes of Health, Pathway to Independence Award (4R00GM129460). We thank Dr. Benjamin Allen for technical assistance and Prof. Todd Sowers at the Pennsylvania State Earth and Environmental Systems Institute for GC-FID analysis.
Footnotes
ASSOCIATED CONTENT
Supporting information (Materials & Methods, Figures S1 – S7, Tables S1 – S8, and Schemes S1 – S2) is available free of charge from https://pubs.acs.org.
REFERENCES
- (1).Costas M; Mehn MP; Jensen MP; Que L Jr. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates, Chem. Rev 2004, 104, 939. [DOI] [PubMed] [Google Scholar]
- (2).Hausinger RP Fe(II)/α-ketoglutarate-dependent hydroxylases and related enzymes, Crit. Rev. Biochem. Mol.. Biol 2004, 39, 21. [DOI] [PubMed] [Google Scholar]
- (3).Bollinger JM Jr.; Chang W.-c.; Matthews ML; Martinie RJ; Boal AK; Krebs C In 2-Oxoglutarate-Dependent Oxygenases; Hausinger RP, Schofield CJ, Eds.; Royal Society of Chemistry: London, 2015, p 95. [Google Scholar]
- (4).Kal S; Que L Jr. Dioxygen activation by nonheme iron enzymes with the 2-His-1-carboxylate facial triad that generate high-valent oxoiron oxidants, J. Biol. Inorg. Chem 2017, 22, 339. [DOI] [PubMed] [Google Scholar]
- (5).Pavel EG; Zhou J; Busby RW; Gunsior M; Townsend CA; Solomon EI Circular dichroism and magnetic circular dichroism spectroscopic studies of the non-heme ferrous active site in clavaminate synthase and its interaction with α-ketoglutarate cosubstrate, J. Am. Chem. Soc 1998, 120, 743. [Google Scholar]
- (6).Solomon EI; Brunold TC; Davis MI; Kemsley JN; Lee SK; Lehnert N; Neese F; Skulan AJ; Yang YS; Zhou J Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes, Chem. Rev 2000, 100, 235. [DOI] [PubMed] [Google Scholar]
- (7).Koehntop KD; Emerson JP; Que L Jr. The 2-His-1-carboxylate facial triad: A versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes, J. Biol. Inorg. Chem 2005, 10, 87. [DOI] [PubMed] [Google Scholar]
- (8).Krebs C; Galonić Fujimori D; Walsh CT; Bollinger JM Jr. Non-Heme Fe(IV)–Oxo Intermediates, Acc. Chem. Res 2007, 40, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Martinez S; Hausinger RP Catalytic Mechanisms of Fe(II)- and 2-Oxoglutarate-dependent Oxygenases, J. Biol. Chem 2015, 290, 20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Clifton IJ; McDonough MA; Ehrismann D; Kershaw NJ; Granatino N; Schofield CJ Structural studies on 2-oxoglutarate oxygenases and related double-stranded β-helix fold proteins, J. Inorg. Biochem 2006, 100, 644. [DOI] [PubMed] [Google Scholar]
- (11).Fukuda H; Kitajima H; Fujii T; Tazaki M; Ogawa T Purification and some properties of a novel ethylene-forming enzyme produced by Penicillium digitatum, FEMS Microbiol. Lett 1989, 59, 1. [Google Scholar]
- (12).Fukuda H; Ogawa T; Tazaki M; Nagahama K; Fujiil T; Tanase S; Morino Y Two reactions are simultaneously catalyzed by a single enzyme: The arginine-dependent simultaneous formation of two products, ethylene and succinate, from 2-oxoglutarate by an enzyme from Pseudomonas syringae, Biochem. Biophys. Res. Comm 1992, 188, 483. [DOI] [PubMed] [Google Scholar]
- (13).Martinez S; Hausinger RP Biochemical and Spectroscopic Characterization of the Non-Heme Fe(II)- and 2-Oxoglutarate-Dependent Ethylene-Forming Enzyme from Pseudomonas syringae pv. phaseolicola PK2, Biochemistry 2016, 55, 5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Nagahama K; Yoshino K; Matsuoka M; Tanase S; Ogawa T; Fukuda H Site-directed mutagenesis of histidine residues in the ethylene-forming Enzyme from Pseudomonas syringae, Journal of Fermentation and Bioengineering 1998, 85, 255. [Google Scholar]
- (15).Johansson N; Persson KO; Larsson C; Norbeck J Comparative sequence analysis and mutagenesis of Ethylene Forming Enzyme (EFE) 2-oxoglutarate/Fe(II)-dependent dioxygenase homologs, BMC Biochemistry 2014, 15, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhang Z; Smart TJ; Choi H; Hardy F; Lohans CT; Abboud MI; Richardson MSW; Paton RS; McDonough MA; Schofield CJ Structural and stereoelectronic insights into oxygenase-catalyzed formation of ethylene from 2-oxoglutarate, Proc. Natl. Acad. Sci. USA 2017, 114, 4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Martinez S; Fellner M; Herr CQ; Ritchie A; Hu J; Hausinger RP Structures and Mechanisms of the Non-Heme Fe(II)- and 2-Oxoglutarate-Dependent Ethylene-Forming Enzyme: Substrate Binding Creates a Twist, J. Am. Chem. Soc 2017, 139, 11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Li M; Martinez S; Hausinger RP; Emerson JP Thermodynamics of Iron(II) and Substrate Binding to the Ethylene-Forming Enzyme, Biochemistry 2018, 57, 5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Dunham NP; Chang W.-c.; Mitchell AJ; Martinie RJ; Zhang B; Bergman JA; Rajakovich LJ; Wang B; Silakov A; Krebs C; Boal AK; Bollinger JM Jr. Two Distinct Mechanisms for C-C Desaturation by Iron(II)- and 2-(Oxo)glutarate-Dependent Oxygenases: Importance of α-Heteroatom Assistance, J. Am. Chem. Soc 2018, 140, 7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Xue JQ; Lu JR; Lai WZ Mechanistic insights into a non-heme 2-oxoglutarate-dependent ethylene-forming enzyme: selectivity of ethylene-formation versusl-Arg hydroxylation, Phys. Chem. Chem. Phys 2019, 21, 9957. [DOI] [PubMed] [Google Scholar]
- (21).Price JC; Barr EW; Tirupati B; Bollinger JM Jr.; Krebs C The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: A high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli, Biochemistry 2003, 42, 7497. [DOI] [PubMed] [Google Scholar]
- (22).Sinnecker S; Svensen N; Barr EW; Ye S; Bollinger JM Jr.; Neese F; Krebs C Spectroscopic and computational evaluation of the structure of the high-spin Fe(IV)-oxo intermediates in taurine: α-ketoglutarate dioxygenase from Escherichia coli and its His99Ala ligand variant, J. Am. Chem. Soc 2007, 129, 6168. [DOI] [PubMed] [Google Scholar]
- (23).Bollinger JM Jr.; Krebs C Stalking intermediates in oxygen activation by iron enzymes: Motivation and method, J. Inorg. Biochem 2006, 100, 586. [DOI] [PubMed] [Google Scholar]
- (24).Price JC; Barr EW; Hoffart LM; Krebs C; Bollinger JM Jr. Kinetic dissection of the catalytic mechanism of taurine:α-ketoglutarate dioxygenase (TauD) from Escherichia coli, Biochemistry 2005, 44, 8138. [DOI] [PubMed] [Google Scholar]
- (25).Hoffart LM; Barr EW; Guyer RB; Bollinger JM Jr.; Krebs C A unified mechanism for the α-ketoglutarate-dependent dioxygenases: Evidence for a C-H-cleaving Fe(IV) complex in prolyl-4-hydroxylase, Proc. Natl. Acad. Sci., U.S.A 2006, 103, 14738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Martinez S; Hausinger RP Correction to Biochemical and Spectroscopic Characterization of the Non-Heme Fe(II)- and 2-Oxoglutarate-Dependent Ethylene-Forming Enzyme from Pseudomonas syringae pv. phaseolicola PK2, Biochemistry 2017, 56, 3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Münck E In Physical Methods in Bioinorganic Chemistry; Que L Jr., Ed.; University Science Books: Sausalito, CA, 2000, p 287. [Google Scholar]
- (28).Grzyska PK; Müller TA; Campbell MG; Hausinger RP Metal ligand substitution and evidence for quinone formation in taurine/α-ketoglutarate dioxygenase, J. Inorg. Biochem 2007, 101, 797. [DOI] [PubMed] [Google Scholar]
- (29).Hangasky JA; Taabazuing CY; Martin CB; Eron SJ; Knapp MJ The facial triad in the α-ketoglutarate dependent oxygenase FIH: A role for sterics in linking substrate binding to O2 activation, J. Inorg. Biochem 2017, 166, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Baldwin JE; Abraham E The biosynthesis of penicillins and cephalosporins, Nat. Prod. Rep 1988, 5, 129. [DOI] [PubMed] [Google Scholar]
- (31).Matthews ML; Neumann CS; Miles LA; Grove TL; Booker SJ; Krebs C; Walsh CT; Bollinger JM Jr. Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2, Proc. Natl. Acad. Sci. USA 2009, 106, 17723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tarhonskaya H; Szöllössi A; Leung IKH; Bush JT; Henry L; Chowdhury R; Iqbal A; Claridge TDW; Schofield CJ; Flashman E Studies on deacetoxycephalosporin C synthase support a consensus mechanism for 2-oxoglutarate dependent oxygenases, Biochemistry 2014, 53, 2483. [DOI] [PubMed] [Google Scholar]
- (33).Ushimaru R; Ruszczycky MW; Liu HW Changes in Regioselectivity of H Atom Abstraction during the Hydroxylation and Cyclization Reactions Catalyzed by Hyoscyamine 6β-Hydroxylase, J. Am. Chem. Soc 2019, 141, 1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Pan J; Wenger ES; Matthews ML; Pollock CJ; Bhardwaj M; Kim AJ; Allen BD; Grossman RB; Krebs C; Martin Bollinger J Jr. Evidence for Modulation of Oxygen Rebound Rate in Control of Outcome by Iron(II)- And 2-Oxoglutarate-Dependent Oxygenases, J. Am. Chem. Soc 2019, 141, 15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Yin X; Zabriskie TM VioC is a non-heme iron, α-ketoglutarate-dependent oxygenase that catalyzes the formation of 3S-hydroxy-L-arginine during viomycin biosynthesis, ChemBioChem 2004, 5, 1274. [DOI] [PubMed] [Google Scholar]
- (36).Ju J; Ozanick SG; Shen B; Thomas MG Conversion of (2S)-arginine to (2S,3R)-capreomycidine by VioC and VioD from the viomycin biosynthetic pathway of Streptomyces sp. strain ATCC11861, ChemBioChem 2004, 5, 1281. [DOI] [PubMed] [Google Scholar]
- (37).Zhang Z; Ren J; Stammers DK; Baldwin JE; Marios K; Schofield CJ Structural origins of the selectivity of the trifunctional oxygenase clavaminic acid synthase, Nature Structural Biology 2000, 7, 127. [DOI] [PubMed] [Google Scholar]
- (38).Helmetag V; Samel SA; Thomas MG; Marahiel MA; Essen LO Structural basis for the erythro-stereospecificity of the l-arginine oxygenase VioC in viomycin biosynthesis, FEBS Journal 2009, 276, 3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hiratsuka T; Koketsu K; Minami A; Kaneko S; Yamazaki C; Watanabe K; Oguri H; Oikawa H Core assembly mechanism of quinocarcin/SF-1739: Bimodular complex nonribosomal peptide synthetases for sequential mannich-type reactions, Chemistry and Biology 2013, 20, 1523. [DOI] [PubMed] [Google Scholar]
- (40).Chang CY; Lyu SY; Liu YC; Hsu NS; Wu CC; Tang CF; Lin KH; Ho JY; Wu CJ; Tsai MD; Li TL Biosynthesis of streptolidine involved two unexpected intermediates produced by a dihydroxylase and a cyclase through unusual mechanisms, Angew. Chem. Int. Ed 2014, 53, 1943. [DOI] [PubMed] [Google Scholar]
- (41).Martins BM; Blaser M; Feliks M; Ullmann GM; Buckel W; Selmer T Structural basis for a Kolbe-type decarboxylation catalyzed by a glycyl radical enzyme, J. Am. Chem. Soc 2011, 133, 14666. [DOI] [PubMed] [Google Scholar]
- (42).Tanner A; Bowater L; Fairhurst SA; Bornemann S Oxalate decarboxylase requires manganese and dioxygen for activity: Overexpression and characterization of Bacillus subtilis YvrK and YoaN, J. Biol. Chem 2001, 276, 43627. [DOI] [PubMed] [Google Scholar]
- (43).Zhu W; Easthon LM; Reinhardt LA; Tu C; Cohen SE; Silverman DN; Allen KN; Richards NGJ Substrate Binding Mode and Molecular Basis of a Specificity Switch in Oxalate Decarboxylase, Biochemistry 2016, 55, 2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ye S; Riplinger C; Hansen A; Krebs C; Bollinger JM Jr.; Neese F Electronic Structure Analysis of the Oxygen-Activation Mechanism by Fe- and α-Ketoglutarate (αKG)-Dependent Dioxygenases, Chemistry - A European Journal 2012, 18, 6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Mitchell AJ; Dunham NP; Martinie RJ; Bergman JA; Pollock CJ; Hu K; Allen BD; Chang W.-c.; Silakov A; Bollinger JM Jr.; Krebs C; Boal AK Visualizing the reaction cycle in an Iron(II)- and 2-(Oxo)-glutarate-dependent hydroxylase, J. Am. Chem. Soc 2017, 139, 13830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Huang X; Groves JT Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins, 2018, 118, 2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Vondrášek J; Mason PE; Heyda J; Collins KD; Jungwirth P The Molecular Origin of Like-Charge Arginine - Arginine Pairing in Water, Journal of Physical Chemistry B 2009, 113, 9041. [DOI] [PubMed] [Google Scholar]
- (48).Pednekar D; Tendulkar A; Durani S Electrostatics-defying interaction between arginine termini as a thermodynamic driving force in protein-protein interaction, Proteins: Structure, Function and Bioinformatics 2009, 74, 155. [DOI] [PubMed] [Google Scholar]
- (49).Lee D; Lee J; Seok C What stabilizes close arginine pairing in proteins?, Phys. Chem. Chem. Phys 2013, 15, 5844. [DOI] [PubMed] [Google Scholar]
- (50).Zhang Z; Xu Z; Yang Z; Liu Y; Wang JA; Shao Q; Li S; Lu Y; Zhu W The stabilization effect of dielectric constant and acidic amino acids on arginine-arginine (Arg-Arg) pairings: Database survey and computational studies, Journal of Physical Chemistry B 2013, 117, 4827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.