

Abstract
Glutamatergic plasticity in the nucleus accumbens core (NAcore) is a key neuronal process in appetitive learning and contributes to pathologies such as drug addiction. Understanding how this plasticity factors into cannabis addiction and relapse has been hampered by the lack of a rodent model of cannabis self-administration. We used intravenous self-administration of two constituents of cannabis, Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD) to examine how contingent cannabis use and cue-induced cannabinoid-seeking alters glutamatergic neurotransmission and synaptic plasticity in NAcore. NMDA receptor (NMDAR)–dependent long-term depression (LTD) in the NAcore was lost after cannabinoid, but not sucrose self-administration. Surprisingly, when rats underwent cue-induced cannabinoid seeking, LTD was restored. Loss of LTD was accompanied by desensitization of cannabinoid receptor 1 (CB1R). CB1R are positioned to regulate synaptic plasticity by being expressed on glutamatergic terminals and negatively regulating presynaptic excitability and glutamate release. Supporting this possibility, LTD was restored by promoting CB1R signaling with the CB1 positive allosteric modulator GAT211. These data implicate NAcore CB1R as critical regulators of metaplasticity induced by cannabis self-administration and the cues predicting cannabis availability.
Keywords: addiction, cannabinoids, synaptic plasticity
1 |. INTRODUCTION
The nucleus accumbens core (NAcore) is an essential component of the mesocorticolimbic system and plays a prominent role in motivation and reward.1 GABAergic medium spiny neurons (MSNs) comprise 90% to 95% of all neurons in the striatum and are the sole projection neurons in the accumbens.2 Although MSNs are innervated by many neurotransmitter systems, the acquisition of drug reward associations depends on the convergence of dopamine and glutamate signaling in the NAcore.1 Furthermore, cue-induced reinstatement of drug-seeking is critically driven by glutamatergic inputs to the NAcore from the prelimbic cortex and basolateral amygdala.3–6 Glutamatergic synapses in the NAcore are capable of undergoing many different forms of synaptic plasticity, and drugs of abuse hijack the plasticity machinery that regulates synaptic transmission and synaptic plasticity.7,8
Chronic exposure to drugs of abuse causes neuroadaptations that can alter the strength of glutamatergic transmission (ie, synaptic plasticity)9–12 and induce metaplasticity.13 Metaplasticity is a higher-order plasticity caused by neuronal adaptations that change the threshold or the rules to induce synaptic plasticity. These adaptations occur at an earlier point in time but their persistence affects the capacity to undergo synaptic plasticity in the future.14
Dysfunction in the expression of synaptic plasticity parallels behavioral deficits in many murine models of neuropsychiatric diseases, including addiction.15,16 One form of impaired plasticity associated with chronic use of different drugs is the loss of NMDAR-dependent long-term depression (LTD) induced by a low-frequency pairing protocol in MSNs.17–21 NMDAR-LTD is abolished after cocaine self-administration (SA),17–19 chronic ethanol exposure,20,21 and extinction from heroin SA.22 These studies indicate that, even though drugs of abuse induce distinct neuroadaptations in the NAcore, all classes of drugs of abuse cause enduring impairments in synaptic plasticity.
Cannabis sativa L. derivatives are used for therapeutic and recreational purposes23 and are rapidly becoming decriminalized or legalized across the globe. Δ9-tetrahydrocannabinol (THC) is the major psycho-active component of cannabis and produces its primary pharmacological effects as a partial agonist at the cannabinoid receptor 1 (CB1R).24 Presynaptic CB1R are coupled to Gi/o proteins, which upon activation, dissociate into Gαi/o and Gβϒ subunits. The Gβϒ subunits activate K+ channels, inhibit voltage-gated calcium channels,25 and might also have a direct inhibitory influence on components of the vesicular release machinery.26 The Gαi subunits inhibit adenylyl cyclase (AC). Together, these actions decrease excitability and reduce activity dependent Ca2+ entry into the presynaptic terminal, which decreases the probability of vesicle fusion and transmitter release. Hence, CB1R induce several forms of presynaptic short- and long-term depression27 and modulate synaptic metaplasticity.28,29 Recently, we found that extinction from SA THC combined with cannabidiol (CBN) impairs NMDA-dependent LTD in the NAcore, akin to the SA of other addictive drug classes.30
To examine the mechanisms of cannabinoid-induced metaplasticity, we trained rats to self-administer THC + CBD and performed in vitro recordings of MSNs in the NAcore. We observed the expected loss of NMDAR-dependent LTD after extinction from cannabinoid self-adminstration and found that LTD was reestablished in animals undergoing 30 minutes of cue-induced reinstatement of cannabis-seeking. Dose-response curves for the CB1R agonist WIN 55,212–2 showed that THC-induced loss of LTD was accompanied by desensitization of CB1R. Moreover, we could restore LTD in extinguished rats by promoting CB1R signaling with the CB1-positive allosteric modulator GAT211.
2 |. METHODS
2.1 |. Animals
Male Sprague Dawley rats were obtained from Charles River Laboratories and allowed for 1 week of vivarium acclimation before surgeries and experiments. All animals were maintained on a 12-to-12-hour reverse light–dark cycle, and experiments were performed during the dark cycle. Before surgery, animals were anesthetized with ketamine (100 mg/kg) and xylazine (7 mg/kg, IP) and given the analgesic ketorolac (0.28–0.32 mg/kg, IP) and prophylactic antibiotic (Cefazolin, 200 mg/ml, subcutaneous; West-Ward Pharmaceuticals, NJ). Rats were implanted with indwelling jugular catheters as described previously.30 All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals.
2.2 |. THC + CBD SA and reinstatement
Rats had 1 week of recovery from surgery before starting behavioral training. The SA procedure has been described in detail in our previous publication.30 Briefly, following 5 days of THC + CBD (NIDA, Bethesda, MD, USA) vapor pre-exposure, male and female rats began daily 90-minute SA sessions using a fixed ratio 1 (FR-1) schedule of intravenous THC + CBD delivery in a 10:1 dose ratio (60 μg/infusion infusion on d1–5, 30 μg/infusion on all subsequent days), paired with tone and light cues over a 2-week period. THC + CBD were dissolved in vehicle containing 0.28% to 0.56% ethanol, an equivalent concentration of Tween 80, and saline to volume. Previous studies show that this concentration of intravenous (IV) ethanol alone does not support SA.30,31 However, to control for effects attributed to the vehicle solution, we used a vehicle control group for all experiments where we directly compared control with THC + CBD SA (ie, in Figures 1, 4, and 5). These control animals received comparable numbers of infusions as the animals that underwent THC + CBD SA (see Figure S2). Only animals that reached an average rate of eight infusions or a discrimination index of 0.33 (see Spencer et al30 for further details) for the last 5 days of SA were used in this study. Three of the 30 trained to self-administer THC + CBD were excluded because they did not reach criterion. Following SA, rats were extinguished daily without access to drug or cues for at least 10 days. Tissue was obtained 24 hours after the last extinction session or after 30 minutes of cued reinstatement. This time point was chosen given that morphological and physiological synaptic changes are transient and peak between 15 and 45 minutes after initiating cue-induced reinstatement to other drugs of abuse.32
FIGURE 1.
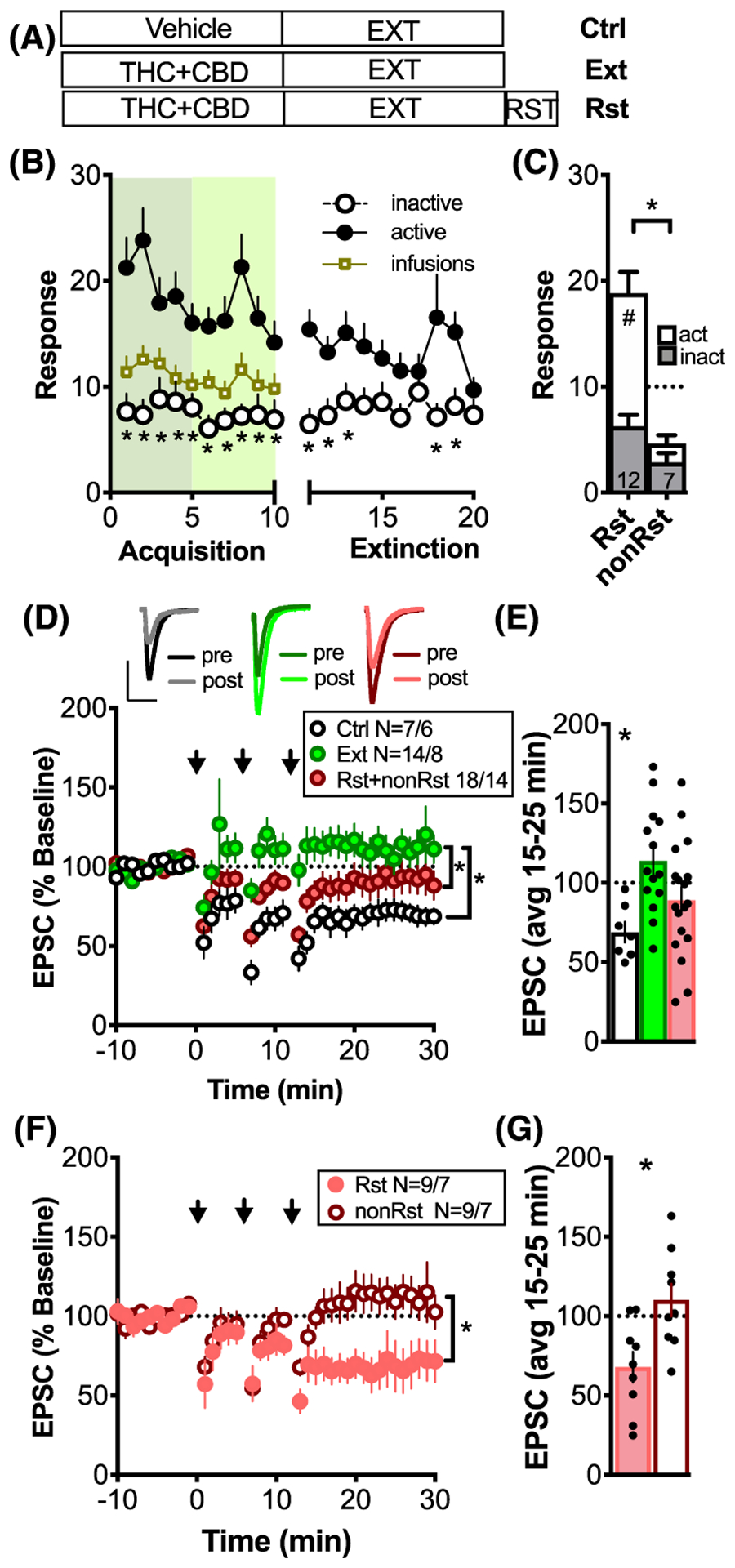
THC + CBD self-administration and cue-induced reinstatement induces a loss and restoration of LTD, respectively. A, The three treatment groups used throughout the study: vehicle-extinguished (Ctrl), THC + CBD-extinguished (Ext), THC + CBD-reinstated (Rst). B, SA of THC + CBD followed by extinction training. Note that the discrimination between active and inactive levers during SA is no longer apparent by day 4 of extinction (except for day 8 + 9) *P < .05, comparing active and inactive lever, N = 27. C, Active and inactive lever pressing during 30 minutes of cued-reinstatement. Dashed line indicates reinstatement criterion of greater than10 active lever presses. *P < .05, comparing Rst and non-Rst; #P < .05, comparing active and inactive lever presses. D, Time-course of synaptic response before and after LTD induction (arrows). Inset above: representative average pre- and post-traces for the three treatment groups; scale 200 pA and 20 ms. NAcore MSNs showed robust LTD in Ctrl (N = 7 cells/6 rats). Extinction of THC + CBD-induced metaplasticity (14 cells/8 rats) that was partly rectified by 30 minutes of cue-induced reinstatement (18 cells/14 rats). *P < .05, 2-way ANOVA, main effect of time and treatment groups. E, Bar graph summarizing the average response 15 to 25 minutes after LTD induction. *P < .05, comparing baseline response to 15 to 25 postinduction. F, Time course of synaptic response before and after LTD induction for animals grouped into nonreinstaters (non-RST; N = 9 cells/7 animals) and reinstaters (RST; N = 9 cells/7 animals) according to criterion in Figure 1C. *P < .05, 2-way ANOVA, main effect of time and treatment groups. G, Bar graph illustrating significant LTD expression in Rst but not non-RST. *P < .05, comparing baseline response to response 15 to 25 postinduction
FIGURE 4.
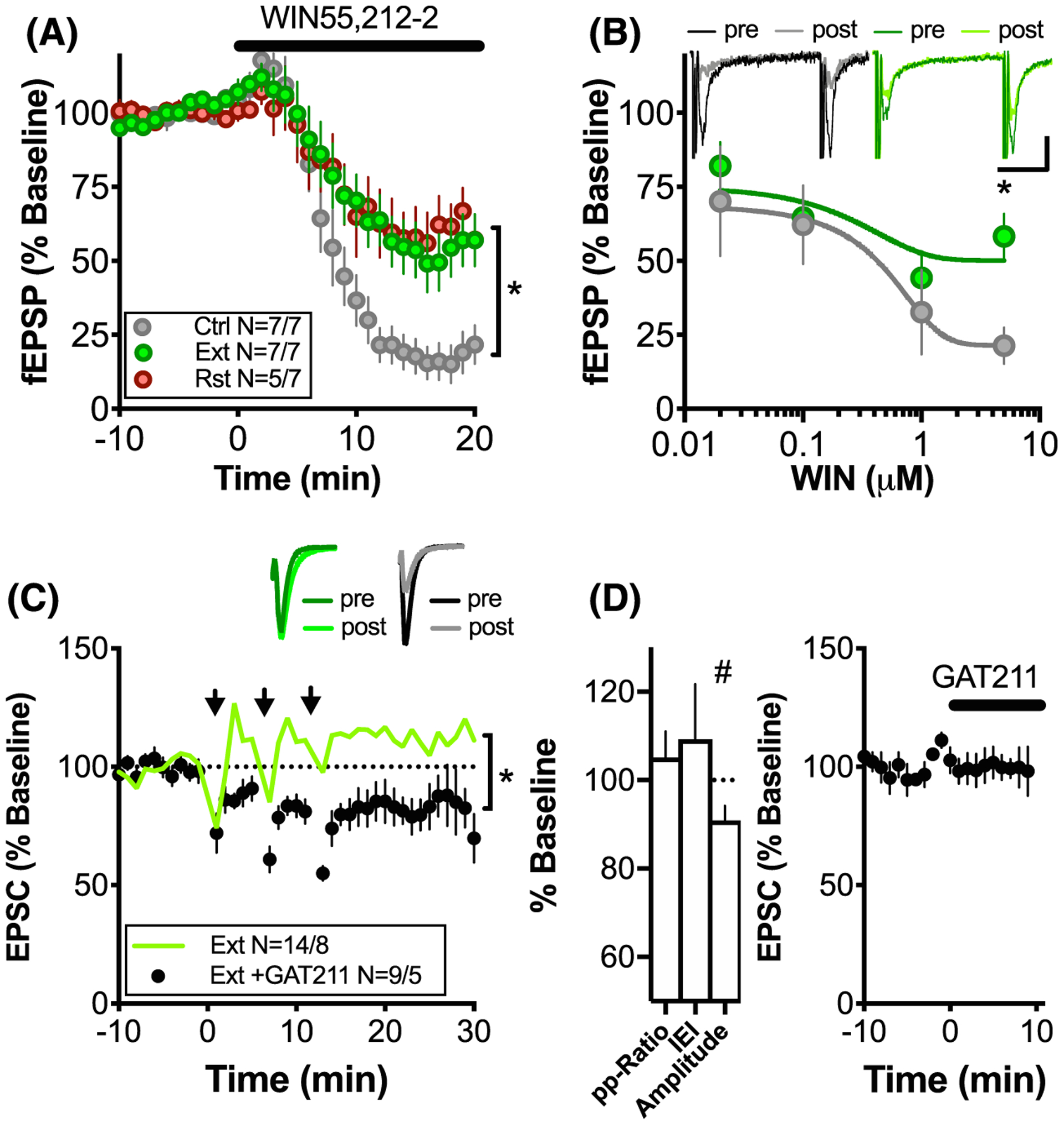
THC + CBD-induced metaplasticity can be reversed by CB1R PAM GAT-211. A, Average time course of CB1 agonist WIN55,212–2-dependent inhibition of fEPSPs in Ctrl, Ext, and Rst groups. *P < .05, 2-way ANOVA, main effect of time and treatment groups. B, Dose-response curves for WIN55,212–2. The inhibition induced by the 5-μM dose was significantly stronger for control as compared with Ext and RST rats. Four to seven slices were used to generate each point. *P < .05, multiple t tests with Holm-Sidak post hoc. C, GAT211, a positive allosteric Modulator of CB1, rescues LTD after THC + CBD self-administration. Green, open symbols represent average response for extinguished animals from Figure 1D as comparison. D, Bar graph illustrating change in PP-Ratio, sEPSC IEI, and amplitude as compared with baseline. Significant reduction in sEPSC amplitude indicates postsynaptic locus of LTD. #P = .029, Student’s t test. E, GAT211 had no effect on baseline synaptic transmission N = 12/4
FIGURE 5.
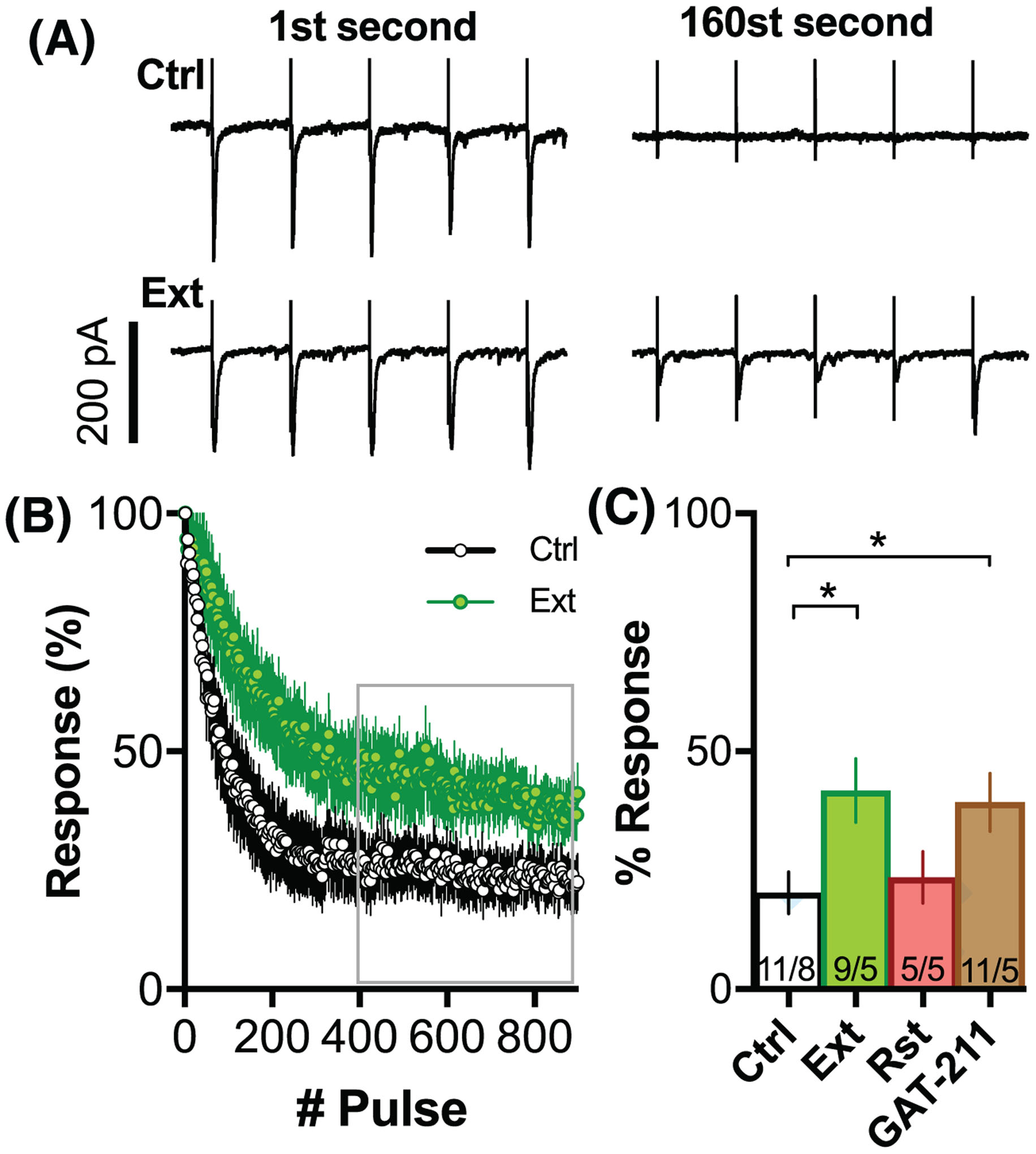
Deficient THC-induced short-term depression during 5 Hz LTD induction is not reversed by GAT-211. A, Representative traces of postsynaptic responses of the first and 160st second of the 5-Hz stimulation (eg, LTD induction protocol). Upper trace Ctrl, lower trace Ext animal. B, Average temporal dynamic of postsynaptic activation during the 5-Hz LTD induction for ctrl and ext. C, Bar graph summarizing the average response to pulses 400 to 900 of LTD induction. N = shown as cells/animals recorded. *P < .05 comparing treatment groups
2.3 |. In vitro whole-cell patch-clamp recordings
Fresh coronal NAcore slices (250 μm; VT1200S Leica vibratome) were collected into a vial containing aCSF (in mM: 126 NaCl, 1.4 NaH2PO4, 25 NaHCO3, 11 glucose, 1.2 MgCl2, 2.4 CaCl2, 2.5 KCl, 2.0 sodium pyruvate, 0.4 ascorbic acid, bubbled with 95% O2 and 5% CO2). For cutting and storage, a mixture of 5 mM kynurenic acid (abcam) and 50 μM D-APV (abcam) was added to the aCSF. Slices were kept at 22°C to 24°C until they were used for recordings and were constantly perfused with oxygenated aCSF heated to 32°C (TC-344B, Warner Instruments). GABAA synaptic transmission was blocked with 100 μM picrotoxin (abcam). Neurons were visualized with a Zeiss Axioscope 2 FS plus microscope with a 40× objective and voltage clamp recordings (Multiclamp 700B, Molecular Devices) performed from visualized MSNs in the medial NAcore near the anterior commissure. Glass microelectrodes (1.5–2.5 ΩM) were prepared using a PC-10 vertical puller (Narishige) and filled with internal solution as follows (in mM: 124 cesium methanesulfonate, 10 HEPES potassium, 1 EGTA, 1 MgCl2, 10 NaCl, 2.0 MgATP, and 0.3 NaGTP, 1 QX-314, pH 7.2–7.3, 290 mOsm). To evoke postsynaptic currents, a bipolar stimulating electrode was placed 300 μm dorso-medial of the recorded cell. Data was acquired at 10 kHz and filtered at 2 kHz using AxographX software (Axograph Scientific).
2.4 |. LTD protocol
MSNs were voltage clamped at −80 mV, and a stable AMPA baseline response was recorded for at least 10 minutes. For LTD induction, cells were clamped to −50 mV for 3 minutes during which afferents were stimulated at 5 Hz. This sequence was repeated 3× with intermittent 5-minute baseline recordings at −80 mV.17,18,33
2.5 |. Dose/response curves
For extracellular field recordings, the recording pipette was filled with ACSF. After stable baseline recordings of electrically evoked field excitatory postsynaptic potentials (fEPSPs), CB1R agonist WIN55,212–2 (Tocris, 0.01–5 μM) was bath applied for 30 minutes. The glutamatergic nature of the extracellular fEPSP was confirmed via application of the non-NMDA ionotropic glutamate receptor antagonist CNQX (10 μM), which completely blocked the synaptic N2 component without altering the nonsynaptic N1 component (not shown).34 The fEPSP amplitude was measured, and dose/response curves were computed based on changes from baseline after 20 minutes of drug application.
2.6 |. Reagents
Reagents for in vitro experiments included SR141716A (Rimonabant hydrochloride, 5 μM, Sigma), WIN55,212–2 (Tocris), GAT211 (5 μM, Sigma) were dissolved in 0.5% DMSO.
2.7 |. Statistics
All data were analyzed using Prism, version 7.0 (GraphPad Software, La Jolla, CA, USA), and outliers were removed using the ROUT module of Prism using a maximum false discovery rate of 2% (Q-value). Using this criterion, we removed one outlier from the dataset in Figure 3A,B. Behavioral data were analyzed using 2-way and 1-way ANOVAs, as specified. Electrophysiological data were analyzed using cell values as the determinant population. Two-way and 1-way ANOVAs were used to test for differences between treatment groups. For multiple comparisons, P values were adjusted using the Holm-Sidak post hoc test. One-sample t test was used to test for long-term plasticity. All values are given as mean ± standard error, n/N values represent individual cells/animals, and statistical significance was set at P < .05.
FIGURE 3.
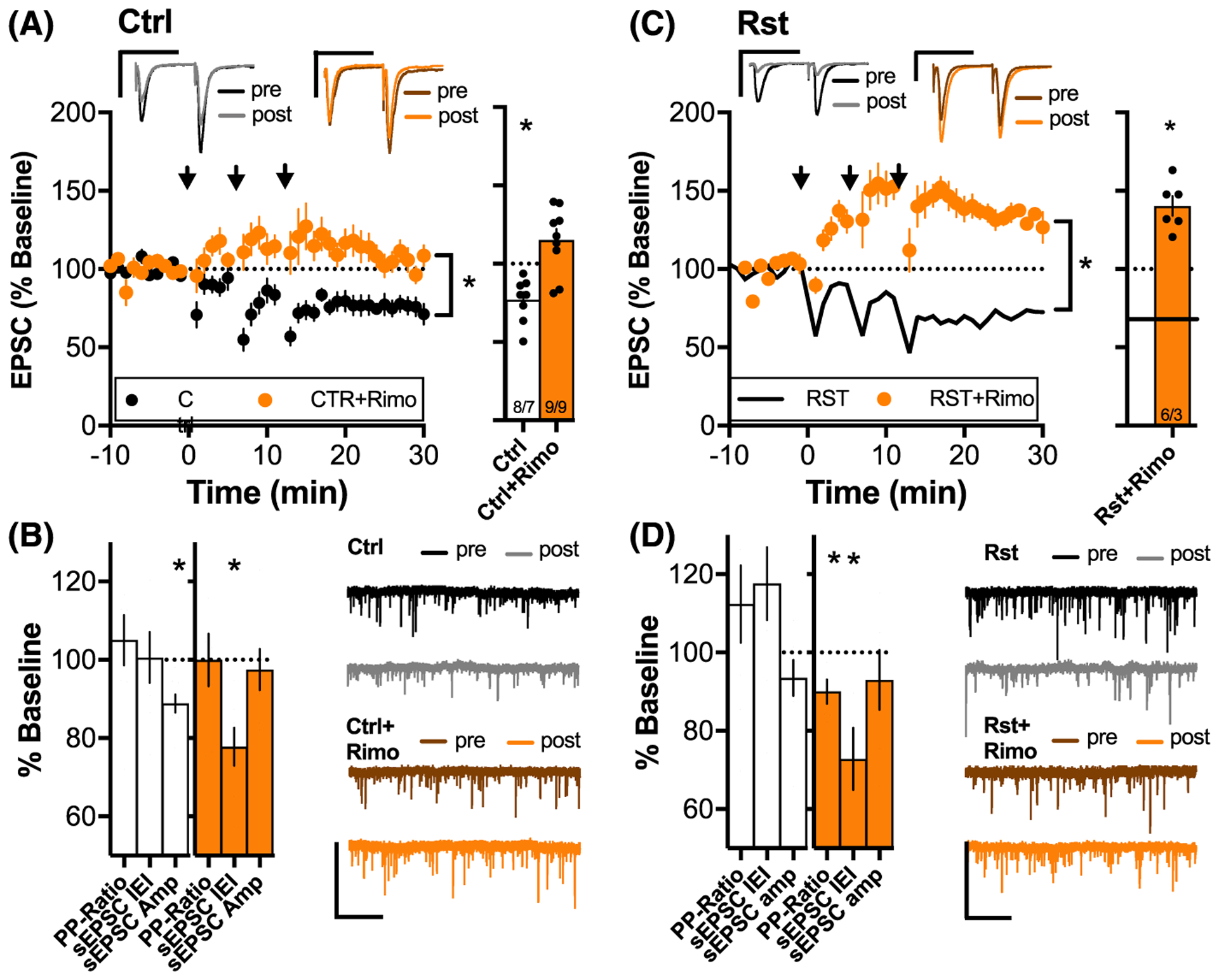
LTD in control and reinstated animals depends on CB1R. A, (left) Average time course of synaptic response before and after LTD induction for slices from drug-naïve animals with and without Rimonabant (5 μM). Inset above: sample average pre- and post-traces for the two treatment groups. *P < .05, 2-way ANOVA, main effect of time and treatment groups. Note that these animals did not receive vehicle treatment but expressed similar LTD amplitudes as compared with the vehicle controls in Figure 1D, indicating that intensive handling of the rats is not necessary for LTD expression. (right) Bar graph summarizes the average response 15 to 25 minutes after LTD induction. MSNs showed robust LTD in untreated slices that was blocked by Rimonabant. *P < .05, comparing baseline response with response 15 to 25 postinduction. B, Bar graph summarizing changes of PP-ratio, sEPSP IEI, and amplitude 15 to 25 minutes after LTD induction. LTD in untreated slices was accompanied by a significant decrease in sEPSC amplitude (white). Incubation in Rimonabant decreased in sEPSC IEI (orange). *P < .05, comparing baseline response with response 15 to 25 postinduction. C, (left) Average time course of synaptic response before and after LTD induction for slices from animals that underwent cue-induced reinstatement with and without incubation in Rimonabant. *P < .05, 2-way ANOVA, main effect of time and treatment groups. Inset above: sample average pre- and post-traces for the two treatment groups. (right) Bar graph summarizing the average response 15 to 25 minutes after LTD induction. MSNs showed robust LTD in untreated slices but LTD was blocked by incubation with the CB1R antagonist Rimonabant. *P < .05, comparing baseline response with response 15 to 25 postinduction. D, Bar graph summarizing changes of PP-Ratio, sEPSP IEI, and amplitude 15 to 25 minutes after LTD induction. LTP in slices incubated with Rimonabant was accompanied by a significant decrease in PP-Ratio and sEPSC IEI (orange). *P < .05, comparing baseline response with response 15 to 25 postinduction N shown as cells/animals recorded
3 |. RESULTS
3.1 |. THC + CBD SA and extinction
Figure 1A illustrates the treatment groups employed throughout the study: vehicle + extinction (Ctrl), THC + CBD + extinction (Ext), and THC + CBD + extinction + cued reinstatement (Rst). Figure 1B summarizesTHC + CBD acquisition. A 2-way ANOVA revealed effects of lever (F (1,52) = 26.28, P < .001), days (F (19,988) = 2.88, P < .001), and interaction (F (19,988) = 2.532, P < 0.001). The average rate of total infusions was equivalent between treatment groups (Figure S1). The bar graph in Figure 1C illustrates lever presses for animals that underwent 30 minutes of cue-induced reinstatement. Cued reinstatement did not occur in all rats. Thus, animals were divided according to whether they reached criterion for reinstatement (Rst) or did not reach criterion (non-Rst). The criterion of RST was greater than 10 active lever presses during the 30-minute reinstatement session. The Rst group showed lever discrimination and greater reinstated active lever pressing compared with the non-Rst group (F (3,34) = 24.75, P < .001, one-way ANOVA; post-hoc: Rst-active vs Rst-inactive P < .001, non-Rst-active vs non-Rst-inactive P = .71, Rst-active vs non-Rst-active P < .001).
3.2 |. THC + CBD-induced metaplasticity
Whole-cell patch-clamp recordings of MSNs revealed that THC + CBD exposure induced metaplasticity that was highly dependent on treatment group (Figure 1D: two-way ANOVA revealed effects of time F (27, 1917) = 11.66, P < .001, and treatment group F (7, 71) = 9.18, P < .001; Holm-Sidak post hoc: Ctrl vs Ext P < .001, Ext vs Rst + non-Rst P = .0143; see Table S3 for detailed statistics of post hoc comparisons between all the eight treatment groups used in Figures 1–4). In control slices, the induction protocol produced robust LTD that was abolished in slices from THC + CBD-extinguished animals (Figure 1e: t(6) = 4.81, P = .003 vs t(13) = 1.58, P = .138; one-sample t test comparing 100% baseline response vs 15–25 minutes after LTD induction). To examine whether cue-induced reinstatement affected THC + CBD-induced metaplasticity and the induction of LTD, slices containing the NAcore were prepared after 30 minutes of cue-induced reinstatement session. LTD was not restored in rats exposed to THC + CBD-associated cues (t(17)=1.28, P = .2176). However, when we divided rats according to the reinstatement criterion (Figure 1C), we found a significant difference between the two groups (Figure 1F; Holm-Sidak post hoc: Rst vs non-Rst, P = .043; see Table S3). Animals showing reinstatement demonstrated a robust restoration of LTD, while those that did not reach criterion showed loss of LTD akin to Ext rats (Figure 1G; t(8) = 3.351, P = .010 vs t(8) = 0.9879, P = .3521; one-sample t test comparing 100% baseline response vs 15–25 minutes after LTD induction).
The failure to induce LTD after extinction training may be due either to impaired LTD induction or the occlusion of LTD expression. Alternatively, if cues potentiated glutamatergic synapses, this could facilitate the depotentiation of synapses in reinstated animals. To distinguish between these two scenarios, we quantified basal levels of paired-pulse ratio (PP-Ratio), spontaneous inter-event intervals (sEPSC), and amplitude (Figure S4). No differences in basal glutamatergic transmission were found between the treatment groups, indicating that no preexisting potentiation or depotentiation influenced the loss of LTD during extinction or the restoration of LTD after cue-induced reinstatement.
3.3 |. Lack of sucrose-induced metaplasticity
To determine if the loss of LTD was related to the operant procedure, rats were trained to self-administer the natural reward sucrose, then extinguished using the same procedure as for THC + CBD SA (Figure 2A). Rats that underwent sucrose SA and extinction expressed LTD that was not different from LTD in control animals (Holm-Sidak post hoc: Ctrl vs Suc P > .809; see Table S3 for detailed statistics), demonstrating that loss of LTD is specific to a history of voluntary THC + CBD SA and extinction and not the result of operant reward learning and extinction (Figure 2B,C).
FIGURE 2.
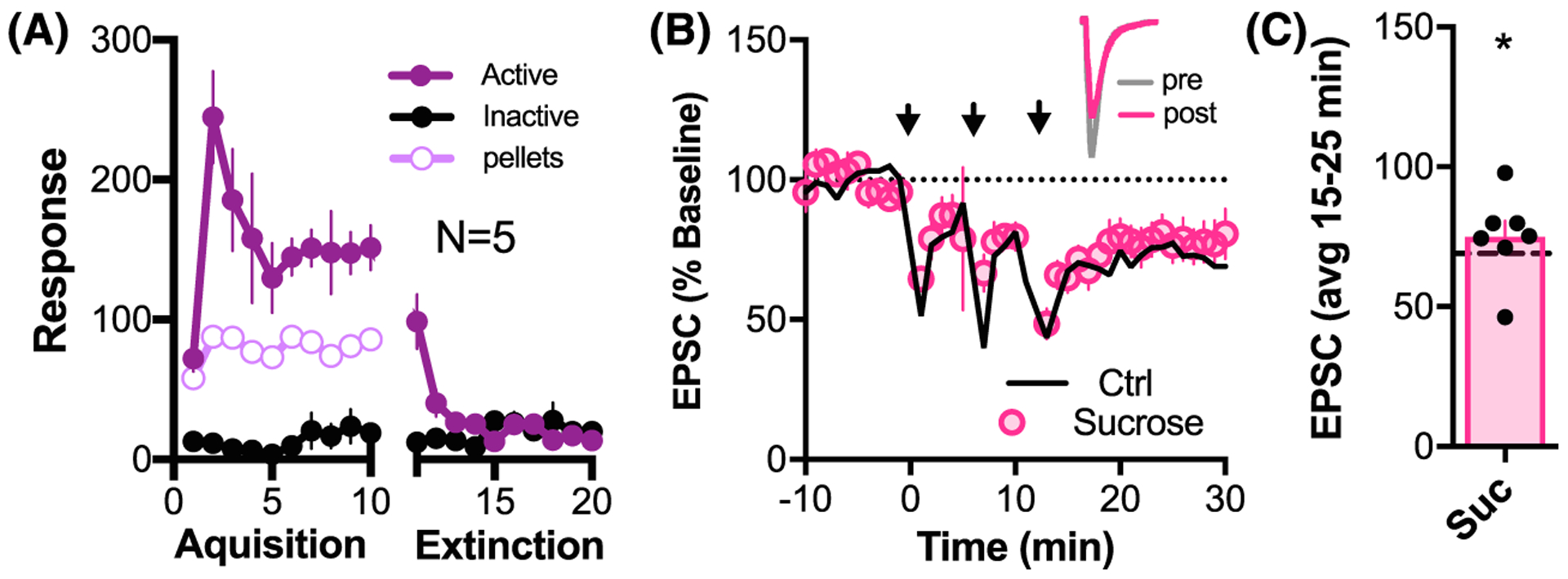
Sucrose SA and extinction training did not alter the capacity to induce LTD. A, Sucrose SA and extinction behavior, N = 5. B, Time course of synaptic response to LTD induction. Black line corresponds to the mean response to the LTD protocol in vehicle controls (Ctrl) from Figure 1D. Inset above: sample average pre- and post-traces for LTD experiment in a cell from a sucrose-treated animal. NAcore MSNs showed robust LTD in sucrose-extinguished animals which was comparable with Ctrl (Holm-Sidak post hoc: Ctrl vs Suc, P = .809; see Table S3). C, Bar graph illustrating significant LTD expression in the Sucrose group (average LTD in control group indicated as dashed line) *P < .05, t(6) = 4.331 comparing baseline response with 15 to 25 minutes after LTD induction protocol
3.4 |. CB1R dependence of metaplasticity
Endocannabinoids can induce a canonical form of presynaptic LTD in NAcore29 and can also modulate other forms of synaptic plasticity28 via presynaptic CB1R activation. The fact that chronic noncontigent THC exposure causes functional tolerance of CB1R in the striatum34–36 could explain THC-induced loss of LTD. Accordingly, we probed the neccessity of CB1R function for the induction of LTD. The CB1R antagonist rimonabant blocked the induction of LTD in drug naïve rats (Figure 3A: two-way ANOVA revealed effects of interaction F (27,405) = 1.924, P = .004, time F (27,405) = 1.994, P = .003, and treatment group F (1,15) = 19.48, P < .001; Figure 3B: one-sample t test comparing 100% baseline response vs 15–25 minutes after LTD induction Ctrl: t(7) = 4.633, P = .002; Rimo: t(8) = 2.127, P = .066). Given the presynaptic action of CB1R37,38 and the postsynaptic expression of NMDAR-LTD,15,33 this result is surprising. We investigated the locus of the LTD by quantifying changes in PP-Ratio, inter-event intervals (IEI), and amplitude of sEPSC 15 to 25 minutes after induction of LTD. The significant decrease of sEPSC amplitude (t(7) = 4.857 P = .001) without any change in sEPSC IEI (t(7) = 0.09443 P = .9275) or PP-Ratio (t(7) = 0.7901 P = .4554) confirmed the postsynaptic locus of LTD in drug naïve animals (left bar chart in Figure 3B). The loss of LTD in slices incubated with rimonabant was accompanied by a decrease in sEPSC IEI (ie, higher spontaneous events; t(6) = 4.562, P = .004), indicating the LTD protocol induced an increase in glutamate release probability when CB1 receptors were blocked. sEPSC amplitude and PP-Ratio remained unchanged (t(6) = 0.4719, P = .454 and t(7) = 0.004, P = .997; right bar chart in Figure 3B). Blocking CB1R in slices from rats that underwent cue-induced reinstatement (16 ± 4 active lever presses; all animals reached the ≥10 active press reinstatement criterion) converted LTD to long-term potentiation (LTP) (Figure 3C: for 2-way ANOVA, see Table S3; Figure 3B: Rst: t(8) = 3.351, P < .001; Rst-Rimo: t(5) = 6.419, P < .001; one-sample t test comparing 100% baseline response vs 15–25 minutes after LTD induction). This LTP was accompanied by a significant decrease in PP-Ratio and sEPSC IEIs (ie, more spontaneous events) (t(5) = 3.238, P = .0230 and t(5) = 3.424, P = .0188) with no change in sEPSC amplitude (t(5) = 3.238, P = .4049), indicating a presynaptic locus of potentiation (Figure 3D).
Given the CB1R dependence of LTD in drug-naïve and reinstated rats (Figure 3), the loss of LTD after extinction from THC + CBD SA could be explained by deficient CB1R signaling. Therefore, CB1R function was examined by measuring dose response curves for the CB1R agonist WIN55,212–22 (Figure 4A,B). Consistent with previous studies examining early withdrawal from daily noncontingent THC administration,34,35 we found that the maximum ability of WIN55,212–22 to inhibit glutamatergic transmission was reduced in NAcore slices from THC + CBD-extinguished animals and reinstated animals compared with drug naïve animals (Figure 4A: two-way ANOVA revealed effects of interaction, F (40, 320) = 3.755, P < .001; time F (20,320) = 60.53, P < .001; and treatment group F (2,16) = 4.009, P = 0.039; Holm-Sidak post hoc: Ctrl vs Ext, P = 0.047; Ctrl vs Rst, P = .047). Figure 4B further illustrates the significant reduction in response to 5 μM but not lower doses of WIN55,212–22 (two-way ANOVA revealed effects of dose, F (3,33) = 6.312, P = .002 and treatment group F (1,33) = 4.584, P = .04; Holm-Sidak post hoc test P = .007 for 5 μM).
We next tested whether the loss of LTD in THC + CBD-extinguished animals resulted from functional tolerance of CB1R. CB1R signaling was up-regulated by administering the positive allosteric modulator GAT211 (5 μM).39 GAT211 augmented the inhibition of glutamate transmission produced by WIN-55–212 (Figure S6). Incubation with GAT211 rescued LTD in slices from THC + CBD-extinguished animals (Figure 4C: for 2-way ANOVA, see Table S3; t(8) = 2.61, P < .031; one-sample t test comparing 100% baseline response vs 15–25 minutes after LTD induction). Similar to control animals, this LTD was accompanied by stable PP-ratio and sEPSC IEI but a significant decrease in sEPSC amplitude after LTD induction, indicating a postsynaptic locus of expression (Figure 4D, t(8) = 0.896, P = .3964, t(7) = 0.7429, P = .4817, t(7) = 2.744, P = .0289). GAT211 had no effect on baseline transmission (Figure 4E, t(11) = 0.2119, P = .8360).
CB1R activation could limit the amount of glutamate release during the 5-Hz induction protocol. A dysfunction of CB1R signaling after THC + CBD could therefore lead to excess release of glutamate during plasticity induction and change the plasticity rules. To test this hypothesis, we examined whether a CB1R-dependent short-term depression modulated the magnitude of postsynaptic responses during the 5-Hz induction protocol. Figure 5A shows representative examples of postsynaptic responses during the first and 160th second of the 5-Hz stimulation of drug-naïve (Ctrl, upper traces) and THC + CBD-extinguished (Ext, lower traces) rats. Figure 5B illustrates the average time course of EPSCs during the course of the 3-minute 5-Hz induction protocol. Short-term depression was significantly reduced in THC + CBD-extinguished animals. Figure 5C shows that the extent of average reduction of postsynaptic responses for pulses 400 to 900 depended on the treatment group (F (3,31) = 3.351, P = .0315, one-way ANOVA). Although GAT211 rescued LTD in THC + CBD-extinguished animals (Figure 4), it did not rescue short-term depression during the 5-Hz induction (GAT211 vs Ext; P = .8304, Holm Sidak post hoc comparison). These results indicate that the short-term depression during stimulus induction is not necessary for the expression of LTD.
4 |. DISCUSSION
Behavioral flexibility is required to control reward-seeking behaviors, and the loss of this control is a core symptom of addiction.40 Deficits in synaptic plasticity and metaplasticity might prevent the reward circuitry from updating to changed environmental contingencies and thereby contribute to perseverative drug-seeking and relapse. One form of impaired plasticity associated with chronic drug use is loss of NMDA-dependent LTD induced by a low-frequency pairing in NAcore MSN. In vivo and in vitro LTD is abolished after withdrawal or extinction from cocaine SA17–19 and in vivo after extinction from heroin SA.22 Here, we confirm that cannabis can be added to the list of drugs causing a loss of NMDAR-dependent LTD in NAcore MSNs30 and demonstrate that cue-induced reinstatement rectified this loss of plasticity. We furthermore identified CB1R as a key regulator of cannabinoid-induced metaplasticity.
4.1 |. CB1Rs regulate drug-induced metaplasticity
Drug-induced metaplasticity can be caused by many different adaptations at glutamatergic synapses that affect the induction and expression of synaptic plasticity. Two prominent examples are drug-induced increases in the expression of GluN2B-containing NMDARs and calcium-permeable AMPARs (GluA2 lacking AMPAR), both of which can modify plasticity threshold and induction.13 Although postsynaptic adaptations have to date been the focus in addiction research, drug-induced presynaptic adaptations can also affect both presynaptic and postsynaptic plasticity. Previous studies indicate that repeated noncontingent THC injections cause CB1R internalization and uncoupling of CB1R from Gi/o proteins.41–43 Here, we demonstrate for the first time that the loss of NMDAR-LTD is accompanied by functional tolerance of CB1R in rats that underwent cannabinoid SA and extinction. Surprisingly, we also observed that CB1R signaling is necessary for the induction of a form of LTD that is expressed at the postsynapse. Furthermore, GAT211, a novel positive allosteric modulator (PAM) for CB1R,39,44 rescued LTD in slices from THC + CBD-extinguished animals. This indicates that deficient CB1R signaling plays a central role in THC + CBD-induced metaplasticity and corroborates a functional role for CB1R as regulators of metaplasticity.28
4.2 |. How can predominantly presynaptic CB1R regulate postsynaptic LTD?
Low-frequency pairing protocols induce an NMDAR-dependent LTD in the nucleus accumbens.45 The expression of this NMDAR-dependent LTD is calcium-dependent45 and expressed via clathrin-dependent AMPAR endocytosis.15 We confirmed the NMDAR-dependence (Figure S5) and postsynaptic locus (Figure 3B) of LTD induced in this study. Given its presynaptic action,37,38 the involvement CB1R in the induction of NMDAR-LTD is surprising. Since the direction of plasticity often depends on the amplitude of postsynaptic calcium signaling,46 CB1R could regulate postsynaptic LTD via modulating glutamate release during LTD induction. Indeed, we demonstrated that a history of THC + CBD use affects postsynaptic activation in response to a 5-Hz LTD induction protocol, resulting in a more sustained response in THC + CBD-extinguished rats as compared with drug-naïve animals. However, although the CB1R PAM GAT211 restored postsynaptic LTD, it did not reverse the deficits in short-term depression during the 5-Hz induction. This result indicates that a more sustained postsynaptic response during LTD induction does not account for the loss of LTD. As demonstrated in Figure 3, animals with a history of THC + CBD SA were more prone to presynaptic LTP, which may be overriding postsynaptic LTD. Given the role of CB1R as regulators of the cannabinoid-induced metaplasticity found here, it will be important in future studies to investigate whether adaptations in the eCB system after chronic use of other drugs result in similar changes.
4.3 |. CB1R-dependent metaplasticity and behavior
Another important result in the present study is the restoration of LTD after cue-induced reinstatement. We showed that endocannabinoid mechanisms were involved since bath-applied CB1R antagonist prevented the induction of LTD in both the CTRL or RST group. However, the mechanism of CB1R involvement remains unstudied. Since extinguished and reinstated animals showed similar functional tolerance of CB1R, the transient restoration of CB1R cannot explain the restoration of LTD. However, it is possible that cue-induced changes in eCB synthesis or catabolism could contribute to restoring LTD in reinstated animals. Importantly, LTD was restored only in rats that reinstated lever pressing while rats not achieving the behavioral reinstatement criterion continued to show loss of LTD. The lack of effect in rats not responding to the THC + CBD-associated cue points towards the involvement of this form of metaplasticity in cue-induced drug-seeking. Future studies may identify multiple underlying mechanisms in addition to the down-regulated CB1R function identified in this study.
4.4 |. Cell-type specificity of metaplasticity?
Ninety percent to ninety-five percent of all neurons in the NAcore are GABAergic MSNs, which can be divided into two subpopulations that express Dopamine1- (D1-MSNs) or Dopamine2- (D2-MSNs) receptors. Furthermore, distinct MSN-type specific adaptations after chronic cocaine and morphine produce the same net shift of the balance between excitatory inputs to D1- and D2-type MSNs.47 Therefore, distinct cell-type-specific adaptations at glutamatergic synapses and metaplasticity after chronic THC could explain the variability in our datasets (eg, extinction group in Figure 1E). Many studies using cell-type-specific expression of activity regulators (optogenetics and chemogenetics) reveal that D1-MSNs promote, whereas D2-MSNs negatively modulate reward seeking behavior.48–50 Cell-type-specific neuroadaptations could therefore promote drug-seeking by selectively shifting the balance of activity in favor of D1- over D2-MSN projections. In the future, it will be important to employ transgenic animals to examine the TH + CBD-induced neuroadaptations described in this study.
5 |. CONCLUSION
This study showed that metaplasticity induced by cannabinoid SA can be restored by promoting CB1R signaling and by cue-induced reinstatement. These results are important for three reasons. First, they show that THC + CBD SA impairs synaptic plasticity akin to other addictive drugs, suggesting these impairments may be a generalizable mechanism in drug addiction. Second, they demonstrate that the loss of LTD is restored by presenting cues previously associated with drug use, a novel finding revealing involvement of transient metaplasticity in drug-seeking behavior. Third, NMDAR-LTD induction is CB1R dependent, indicating that metaplasticity could be caused by the desensitization of CB1R, pointing towards a previously undetected role of CB1R signaling in drug-induced metaplasticity.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Madhura Athreya, Eric Dereschewitz, Constanza Garcia-Keller, and Jeffrey Parrilla-Carrero for advice and technical assistance. This research was funded in part by USPHS grants DA003906, DA012513, DA015369 (P.W.K.), and DA016511, Burroughs Wellcome Fund (S.S.), and NIH grant GM072643 (V.C.).
Funding information
NIH, Grant/Award Numbers: DA003906, DA012513, DA016511, GM072643 and DA015369; Burroughs Wellcome Fund; USPHS, Grant/Award Numbers: DA016511, DA015369, DA012513 and DA003906
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
The authors have nothing to disclose.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Floresco SB. The nucleus accumbens: an interface between cognition, emotion, and action. Annu Rev Psychol. 2015;66(1):25–52. 10.1146/annurev-psych-010213-115159 [DOI] [PubMed] [Google Scholar]
- 2.Smith RJ, Lobo MK, Spencer S, Kalivas PW. Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways). Curr Opin Neurobiol. 2013;23(4):546–552. 10.1016/j.conb.2013.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scofield MD, Heinsbroek JA, Gipson CD, et al. The nucleus accumbens: mechanisms of addiction across drug classes reflect the importance of glutamate homeostasis. Pharmacol Rev. 2016;68(3):816–871. 10.1124/pr.116.012484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalivas PW, Volkow ND. The neural basis of addiciton: a pathology of motivation and choice. Am J Psychiatry. 2005;162(8):1403–1413. 10.1176/appi.ajp.162.8.1403 [DOI] [PubMed] [Google Scholar]
- 5.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35(1):217–238. 10.1038/npp.2009.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stefanik MT, Kalivas PW. Optogenetic dissection of basolateral amygdala projections during cue-induced reinstatement of cocaine seeking. Front Behav Neurosci. 2013;7(December):1–6. 10.3389/fnbeh.2013.00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolf ME. The Bermuda Triangle of cocaine-induced neuroadaptations. Trends Neurosci. 2010;33(9):391–398. 10.1016/j.tins.2010.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69(4): 650–663. 10.1016/j.neuron.2011.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gipson CD, Kupchik YM, Kalivas PW. Rapid, transient synaptic plasticity in addiction. Neuropharmacology. 2014;76(Pt B):276–286. 10.1016/j.neuropharm.2013.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacAskill AF, Little JP, Cassel JM, Carter AG. Subcellular connectivity underlies pathway-specific signaling in the nucleus accumbens. Nat Neurosci. 2012;15(12):1624–1626. 10.1038/nn.3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pascoli V, Terrier J, Espallergues J, Valjent E, O’Connor EC, Lüscher C. Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature. 2014;509(7501):459–464. 10.1038/nature13257 [DOI] [PubMed] [Google Scholar]
- 12.Mameli M, Luescher C. Synaptic plasticity and addiction: learning mechanisms gone awry. Neuropharmacology. 2011;61(7):1052–1059. 10.1016/j.neuropharm.2011.01.036 [DOI] [PubMed] [Google Scholar]
- 13.Neuhofer D, Kalivas P. Metaplasticity at the addicted tetrapartite synapse: a common denominator of drug induced adaptations and potential treatment target for addiction. Neurobiol Learn Mem. 2018;154:97–111. 10.1016/j.nlm.2018.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9(5):387. 10.1038/nrn2356 [DOI] [PubMed] [Google Scholar]
- 15.Brebner K, Wong TP, Liu L, et al. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science (80-). 2005;310(5752):1340–1343. 10.1126/science.1116894 [DOI] [PubMed] [Google Scholar]
- 16.Pascoli V, Turiault M, Lüscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2012;481(7379):71–75. 10.1038/nature10709 [DOI] [PubMed] [Google Scholar]
- 17.Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A. Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci. 2006;9(7):868–869. 10.1038/nn1713 [DOI] [PubMed] [Google Scholar]
- 18.Kasanetz F, Deroche-Gamonet V, Berson N, et al. Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science. 2010;328(5986):1709–1712. 10.1126/science.1187801 [DOI] [PubMed] [Google Scholar]
- 19.Moussawi K, Pacchioni A, Moran M, et al. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12(2):182–189. 10.1038/nn.2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Renteria R, Jeanes ZM, Morrisett RA. Ethanol attenuation of long-term depression in the nucleus accumbens can be overcome by activation of TRPV1 receptors. Alcohol Clin Exp Res. 2014;38(11):2763–2769. 10.1111/acer.12542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Renteria R, Maier EY, Buske TR, Morrisett RA. Selective alterations of NMDAR function and plasticity in D1 and D2 medium spiny neurons in the nucleus accumbens shell following chronic intermittent ethanol exposure. Neuropharmacology. 2017;112(Pt A):164–171. 10.1016/j.neuropharm.2016.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen H-W, Moussawi K, Zhou W, Toda S, Kalivas PW. Heroin relapse requires long-term potentiation-like plasticity mediated by NMDA2b-containing receptors. Proc Natl Acad Sci U S A. 2011;108(48): 19407–19412. 10.1073/pnas.1112052108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russo EB. History of cannabis and its preparations in saga, science, and sobriquet. Chem Biodivers. 2007;4(8):1614–1648. 10.1002/cbdv.200790144 [DOI] [PubMed] [Google Scholar]
- 24.Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: D 9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. British J Pharm. 2008;153:199–215. 10.1038/sj.bjp.0707442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Atwood BK, Lovinger DM, Mathur BN. Presynaptic long-term depression mediated by Gi/o-coupled receptors. Trends Neurosci. 2014;37(11):663–673. 10.1016/j.tins.2014.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blackmer T G Protein beta gamma subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science (80-). 2001;292(5515):293–297. 10.1126/science.1058803 [DOI] [PubMed] [Google Scholar]
- 27.Chevaleyre V, Takahashi Ka, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. 10.1146/annurev.neuro.29.051605.112834 [DOI] [PubMed] [Google Scholar]
- 28.Melis M, Greco B, Tonini R. Interplay between synaptic endocannabinoid signaling and metaplasticity in neuronal circuit function and dysfunction. Eur J Neurosci. 2014;39(7):1189–1201. 10.1111/ejn.12501 [DOI] [PubMed] [Google Scholar]
- 29.Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A. 2002;99(12):8384–8388. 10.1073/pnas.122149199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spencer S, Neuhofer D, Chioma VC, et al. A model of Δ9-tetrahydrocannabinol self-administration and reinstatement that alters synaptic plasticity in nucleus accumbens. Biol Psychiatry. 2018;84(8): 601–610. 10.1016/j.biopsych.2018.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuzmin A, Semenova S, Zvartau E, De Vry J. Effects of calcium channel blockade on intravenous self-administration of ethanol in rats. Eur Neuropsychopharmacol. 1999;9(3):197–203. 10.1016/S0924-977X(98)00025-X [DOI] [PubMed] [Google Scholar]
- 32.Gipson CD, Kupchik YM, Shen H-W, Reissner KJ, Thomas C a, Kalivas PW. Relapse induced by cues predicting cocaine depends on rapid, transient synaptic potentiation. Neuron. 2013;77(5):867–872. 10.1016/j.neuron.2013.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4(12):1217–1223. 10.1038/nn757 [DOI] [PubMed] [Google Scholar]
- 34.Hoffman AF, Oz M, Caulder T, Lupica CR. Functional tolerance and blockade of long-term depression at synapses in the nucleus accumbens after chronic cannabinoid exposure. J Neurosci. 2003;23(12): 4815–4820. doi:23/12/4815 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mato S, Robbe D, Puente N, Grandes P, Manzoni OJ. Presynaptic homeostatic plasticity rescues long-term depression after chronic Delta 9-tetrahydrocannabinol exposure. J Neurosci. 2005;25(50): 11619–11627. 10.1523/JNEUROSCI.2294-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sim-Selley LJ, Schechter NS, Rorrer WK, et al. Prolonged recovery rate of CB1 receptor adaptation after cessation of long-term cannabinoid administration. Mol Pharmacol. 2006;70(3):986–996. 10.1124/mol.105.019612 [DOI] [PubMed] [Google Scholar]
- 37.Uchigashima M, Narushima M, Fukaya M, Katona I, Kano M, Watanabe M. Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J Neurosci. 2007;27(14):3663–3676. 10.1523/JNEUROSCI.0448-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katona I, Urbán GM, Wallace M, et al. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26(21):5628–5637. 10.1523/JNEUROSCI.0309-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laprairie RB, Kulkarni PM, Deschamps JR, et al. Enantiospecific allosteric modulation of cannabinoid 1 receptor. ACS Chem Nerosci. 2017;8(6):1188–1203. 10.1021/acschemneuro.6b00310 [DOI] [PubMed] [Google Scholar]
- 40.Keramati M, Durand A, Girardeau P, Gutkin B, Ahmed SH. Cocaine addiction as a homeostatic reinforcement learning disorder. Psychol Rev. 2017;124(2):130–153. 10.1037/rev0000046 [DOI] [PubMed] [Google Scholar]
- 41.Romero J, Garcia-Palomero E, Castro JG, Garcia-Gil L, Ramos J a, Fernandez-Ruiz JJ. Effects of chronic exposure to Δ9-tetrahydrocannabinol on cannabinoid receptor binding and mRNA levels in several rat brain regions. Brain Res Mol Brain Res. 1997;46(1–2):100–108. 10.1016/S0169-328X(96)00277-X [DOI] [PubMed] [Google Scholar]
- 42.Mato S, Chevaleyre V, Robbe D, Pazos A, Castillo PE, Manzoni OJ. A single in-vivo exposure to delta 9THC blocks endocannabinoid-mediated synaptic plasticity. Nat Neurosci. 2004;7(6):585–586. 10.1038/nn1251 [DOI] [PubMed] [Google Scholar]
- 43.Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Vogt LJ, Sim-Selley LJ. Chronic Δ9-tetrahydrocannabinol treatment produces a time-dependent loss of cannabinoid receptors and cannabinoid receptor-activated G proteins in rat brain. J Neurochem. 1999;73(6): 2447–2459. http://www.ncbi.nlm.nih.gov/pubmed/10582605 [DOI] [PubMed] [Google Scholar]
- 44.Slivicki RA, Xu Z, Kulkarni PM, et al. Positive allosteric modulation of cannabinoid receptor type 1 suppresses pathological pain without producing tolerance or dependence. Biol Psychiatry. 2017;011(26):1–12. 10.1016/j.biopsych.2017.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas MJ, Malenka RC, Bonci A. Modulation of long-term depression by dopamine in the mesolimbic system. J Neurosci. 2000;20(15): 5581–5586. http://www.ncbi.nlm.nih.gov/pubmed/10908594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lisman JE. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci. 1989;86(23): 9574–9578. 10.1073/pnas.86.23.9574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Graziane NM, Sun S, Wright WJ, et al. Opposing mechanisms mediate morphine-and cocaine-induced generation of silent synapses. Nat Neurosci. 2016;19(7):915–925. 10.1038/nn.4313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lobo MK, Covington HE, Chaudhury D, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science (80-). 2010;330(6002):385–390. 10.1126/science.1188472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calipari ES, Bagot RC, Purushothaman I, et al. In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc Natl Acad Sci U S A. 2016;113(10):2726–2731. 10.1073/pnas.1521238113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heinsbroek JA, Neuhofer D, Griffin WC, et al. Loss of plasticity in the D2-accumbens pallidal pathway promotes cocaine seeking. J Neurosci. 2017;37(4):757–767. 10.1523/JNEUROSCI.2659-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.