

Abstract
Cardiac injury and dysfunction occur in COVID-19 patients and increase the risk of mortality. Causes are ill defined but could be through direct cardiac infection and/or inflammation-induced dysfunction. To identify mechanisms and cardio-protective drugs, we use a state-of-the-art pipeline combining human cardiac organoids with phosphoproteomics and single nuclei RNA sequencing. We identify an inflammatory “cytokine-storm”, a cocktail of interferon gamma, interleukin 1β, and poly(I:C), induced diastolic dysfunction. Bromodomain-containing protein 4 is activated along with a viral response that is consistent in both human cardiac organoids (hCOs) and hearts of SARS-CoV-2-infected K18-hACE2 mice. Bromodomain and extraterminal family inhibitors (BETi) recover dysfunction in hCOs and completely prevent cardiac dysfunction and death in a mouse cytokine-storm model. Additionally, BETi decreases transcription of genes in the viral response, decreases ACE2 expression, and reduces SARS-CoV-2 infection of cardiomyocytes. Together, BETi, including the Food and Drug Administration (FDA) breakthrough designated drug, apabetalone, are promising candidates to prevent COVID-19 mediated cardiac damage.
Keywords: inflammation, COVID-19, organoids, heart, drug discovery, Bromodomain and extraterminal family inhibitors
Graphical abstract
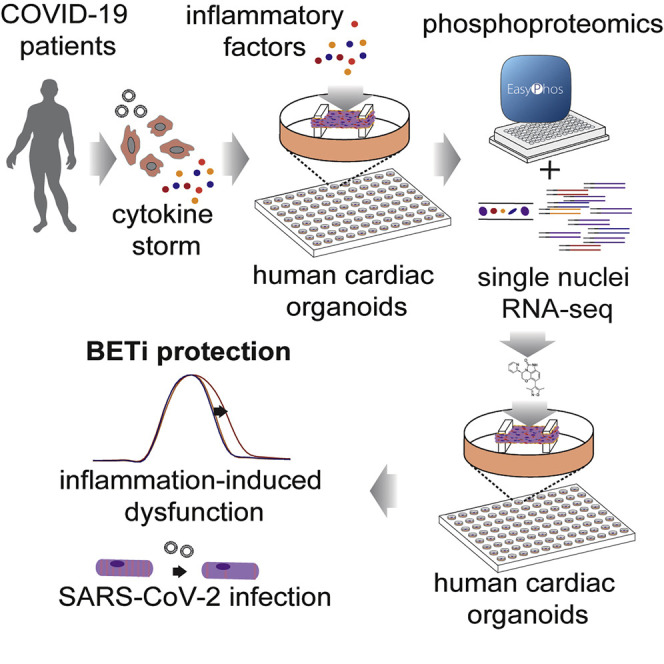
COVID-19 causes cardiac injury, although mechanisms and effective therapeutics are lacking. In this study, Mills et al., show that cytokines elevated in COVID-19 patients drive cardiac dysfunction. These responses are mapped using phosphoproteomics and single nuclei RNA sequencing, enabling a targeted drug screen to identify therapeutics for rapid repurposing. BET inhibitors were identified as leading candidates to block cardiac dysfunction and decrease SARS-CoV-2 cardiac infection.
Introduction
SARS-CoV-2 infection leads to cardiac injury and dysfunction in 20%–30% of hospitalized patients (Guo et al., 2020) and higher rates of mortality in patients with pre-existing cardiovascular disease (Shi et al., 2020; Wu and McGoogan, 2020). The cardiac sequelae reported in patients with COVID-19 include acute coronary syndromes, cardiomyopathy, acute pulmonary heart disease, arrhythmias, and heart failure (Gupta et al., 2020). Furthermore, 68%–78% have sustained cardiac dysfunction, primarily right ventricle dysfunction and left ventricular diastolic dysfunction (Puntmann et al., 2020; Szekely et al., 2020). Yet, clear mechanistic insight is currently lacking.
In the absence of infection, well known inflammatory mediators such as tumor necrosis factor (TNF) are associated with heart failure and have been demonstrated to induce systolic dysfunction (Feldman et al., 2000). Therefore, severe inflammation may play a key role in cardiac injury and dysfunction (Chen et al., 2020). In support of this, there is a severe inflammatory response in 5% of COVID-19 patients associated with septic shock (Wu and McGoogan, 2020). Due to the septic shock and a drop in blood pressure, ~30% of hospitalized patients with COVID-19 require vasopressors to improve blood pressure (Goyal et al., 2020). Mediating these responses in some patients is a cytokine storm of similar magnitude to that induced by CAR-T cell-associated cytokine storms (Del Valle et al., 2020). Additionally, severe COVID-19 is associated with sepsis and bacterial products in the serum (Arunachalam et al., 2020), which are known drivers of cardiac pathology and dysfunction. Cardiac dysfunction may also result in further exacerbation of infectious diseases, via inadequate organ perfusion and/or immune cell infiltration. Thus, preventing cytokine-induced cardiac dysfunction may limit severe outcomes in inflammatory diseases. However, targeted treatment strategies, particularly in severe infections such as COVID-19, are currently lacking.
Several anti-inflammatory agents have shown clinical benefit for the acute management of COVID-19. Dexamethasone improved 28-day mortality in COVID-19 patients receiving invasive mechanical ventilation or oxygen at randomization (Horby et al., 2020). However, systemic immunosuppression may impede viral clearance thus potentially exacerbating disease (Mangalmurti and Hunter, 2020). To circumvent this, we aimed to identify cardiac-specific inflammatory targets that trigger cardiac dysfunction in response to the cytokine storm, reasoning that these might provide a safe and effective therapeutic option.
Here, we utilize multi-cellular human pluripotent stem cell (hPSC)-derived cardiac organoids (hCOs) combined with phosphoproteomics and single nuclei RNA sequencing (RNA-seq) to identify therapeutic targets and treatments for cardiac dysfunction. We recently adapted our hCO system (Mills et al., 2017, 2019) to include co-culture with endothelial cells that form enhanced branched endothelial structures surrounded by pericytes (Figure S1 A; H.K.V. L.T.R, B.L. Parker, G.A.Q.-R., P.R.J.F., E.M., C.E. Friedman, M. Francois, N.J. Palpant, E.J. Needham, M.P. Lopez, G. del Monte-Nieto, L.K. Jones, I.M. Smyth, V. Janbandhu, E. Yao, R.P. Harvey, J.J.H. Chong, D.A.E., E.G. Stanley, S. Wiszniak, Q. Schwarz, D.E.J., R.J.M., E.R.P., and J.E.H., unpublished data). This protocol results in a complex mixture of self-organizing cells including epicardial, fibroblasts/pericytes, endothelial cells, and cardiomyocytes. This was combined together with an optimized culture environment that reflects a maturation state; mimicking the postnatal metabolic environment (Mills et al., 2017, 2019) followed by reversion to a more adult metabolic substrate provision (see STAR Methods). This platform enabled rapid screening of cytokine combinations that recapitulate the COVID-19-induced cytokine storm (Mangalmurti and Hunter, 2020) and cardiac dysfunction, with the subsequent application of -omic assays and drug screening.
Figure S1.

Expression of immunomodulatory receptors and signaling mediators, related to Figure 1
A) Whole-mount immunofluorescent images of human cardiac organoids stained with CD31 (endothelial cells), NG2 (pericytes), cardiac troponin T (cardiomyocytes) and Hoescht33342. Scale = 50 μm.
B) Comparison of immunomodulatory receptors and signaling mediators in human cardiac organoids relative to human adult heart using existing bulk RNA-sequencing data (n = 4 experiments for hCO) (Mills et al., 2017). All were identified in human cardiac organoids except CSF1R which is leukocyte specific.
C) Cell type specificity of immunomodulatory receptors and signaling mediators in adult mouse hearts using existing bulk RNA-sequencing data (n = 4 experiments) (Quaife-Ryan et al., 2017).
D) Normalized expression of genes in human cardiac organoids that mark the human heart sub-populations defined in Tucker et al., 2020.
E) UMAP clustering of single nuclei RNA-sequencing of human cardiac organoids using the enhanced protocol (H.K.V. et al., unpublished data). Location of key markers for different cell populations are also highlighted.
F) Principal component analysis of our single nuclei RNA-sequencing in comparison to purified bulk RNA-sequencing of purified human cardiomyocyte nuclei (Gilsbach et al., 2018).
G) Expression of immunomodulatory receptors and signaling mediators in different cell populations in the human cardiac organoids.
Data presented as mean ± SEM hCO – human cardiac organoids. Human pluripotent stem cell-derived cardiac cells- AA line. Endothelial cells- RM3.5 line.
Results
Cytokine-induced cardiac dysfunction
We began by examining the effects of a range of pro-inflammatory cytokines elevated in COVID-19 patients (Huang et al., 2020) on cardiac function in our hCOs (Mills et al., 2017). Inflammatory molecules tested were likely candidates in COVID-19 including: TNF, interleukin (IL)-1β, interferon (IFN)-γ, IL-6, IL-17A, and G-CSF, as well as pathogen-associated molecular patterns including poly(I:C) to mimic double-stranded RNA (dsRNA), and lipopolysaccharide (LPS) to mimic TLR4 activation and septic responses. Using our RNA-seq data (Mills et al., 2017), we identified that the receptor genes IL1R1, TNFRSF1A, TNFRSF1B, IFIH1, MYD88, IL6ST, IFNAR1, IL6R, TMEM173, IL17RA, IL17RB, IL17RC, IL17RD, IL17RE, IFNGR1, TLR3, and TLR4 were expressed at similar or higher abundance in our hCOs compared to adult human heart (Figure S1B). In adult mouse hearts, many of these are enriched in non-myocyte populations (Quaife-Ryan et al., 2017; Figure S1C). We used single nuclei RNA sequencing (snRNA-seq) to assess cell specificity in our enhanced hCO (H.K.V. et al., unpublished data). Mapping to human heart, snRNA-seq (Tucker et al., 2020) revealed the presence of pro-epicardial/epicardial cells, fibroblasts, activated fibroblasts/pericytes, and cardiomyocytes (Figures S1D and S1E). Some cardiomyocytes were fetal-like, however, there was a distinct sub-cluster that mapped adjacent to adult ventricular cardiomyocytes from human hearts (Gilsbach et al., 2018; Figure S1F). The cytokine/pro-inflammatory receptors were expressed in the different cell types and were more highly expressed in epicardial cells and fibroblasts (Figure S1G). We screened inflammatory factors in all pairwise combinations in hCOs with multiple functional measurements including contractile force, rate, activation kinetics, and relaxation kinetics (Figure 1 A). TNF caused a reduction in force, whereas IL-1β, IFN-γ, poly(I:C), and LPS caused diastolic dysfunction characterized by a preserved contractile force but prolonged time from peak to 50% relaxation (Figures S2 A–S2E). A secondary full-factorial screen of TNF, IFN-γ, IL-1β, and poly(I:C) once again revealed that TNF induced systolic dysfunction (Figures 1B and 1D) with a EC50 of 1 ng/mL at 48 h (Figure S2F). A combination of IL-1β, IFN-γ, and poly(I:C) induced diastolic dysfunction (Figures 1C and 1E), however, it also decreased the beating rate that may influence the kinetics of contraction (Figure S3 A;Videos S1 and S2). Changes in rate were not responsible for increased relaxation time, as hCOs paced at 1 Hz retained the severe diastolic dysfunction phenotype (Figure 1F; Videos S3 and S4). Individually, IFN-γ and IL-1β caused concentration-dependent diastolic dysfunction with an EC50 of 0.8 ng/mL at 48 h and 3 ng/mL at 24 h, respectively, whereas poly(I:C) alone did not induce dysfunction (Figures S2G–S2I). These results were confirmed in an independent hPSC line, where the combination of IFN-γ, IL-1β, and poly(I:C) induced the most consistent, robust diastolic dysfunction (Figures S3A–S3E). Taken together, TNF induces systolic dysfunction consistent with previous in vitro (Vasudevan et al., 2013) and in vivo (Kubota et al., 1997) studies, and the combination of IFN-γ, IL-1β, and poly(I:C) induces severe diastolic dysfunction in hCOs. The dominant factor identified that causes diastolic dysfunction, IFN-γ (Figure S3C), is generally elevated in heart failure patients but with contradictory effects in animal models (Levick and Goldspink, 2014).
Figure 1.

Identification of pro-inflammatory factors driving cardiac dysfunction
(A) Schematic of experiments. Mean of n = 1,100 human cardiac organoids from 9 experiments.
(B) Impact of inflammatory modulators on force (systolic function). n = 3–5 human cardiac organoids from 1 experiment.
(C) Impact of inflammatory modulators on time to 50% relaxation (diastolic function). n = 3–5 human cardiac organoids from 1 experiment.
(D) TNF causes systolic dysfunction. n = 37 and 63 human cardiac organoids for CTRL and TNF conditions, respectively, from 6 experiments.
(E) Cardiac cytokine storm (CS) causes diastolic dysfunction n = 49 and 73 human cardiac organoids for CTRL and CS conditions, respectively, from 6 experiments.
(F) Representative force curve of human cardiac organoids under CTRL and CS conditions (1 Hz) 48 h after treatment. Time to 50% relaxation under paced conditions (1 Hz) 48 h after treatment. n = 15 and 17 human cardiac organoids from 3 experiments.
Data presented as mean ± SEM. Cardiac CS: IL-1β, IFN-γ, and poly(I:C). Ta, time from 50% activation to peak; Tr, time from peak to 50% relaxation. Human pluripotent stem cell (hPSC)-derived cardiac cells AA line (B and C) and HES3 line (D–F). Endothelial cells RM3.5 line (B and C) and RM3.5 or CC lines (D–F). Bold outline indicates p < 0.05 using a one-way ANOVA with Dunnett’s multiple comparisons test comparing each condition to CTRL at the respective time points (B and C). ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, using Student’s t test (D, E, and F). See additional functional data in Figures S1, S2, and S3. Inflammatory screen in (B)–(E) repeated in an additional cell line in Figure S3.
Figure S2.

Screening for the impact of pro-inflammatory factors on human cardiac organoid function, related to Figure 1
A) Contraction force
B) Contraction rate
C) Time from 50% activation to peak
D) Time from peak to 50% relaxation
(A-D) Human cardiac organoid function normalized to baseline contraction parameters at 0 hr. # Due to a microscope camera shutter issue at the 0 h time point, IL-1β & IL-17A is normalized to the 1 h time point. Bold outline indicates p < 0.05 using a one-way ANOVA with Dunnett’s multiple comparisons test comparing each condition to CTRL at comparable time point. n = 2-5 human cardiac organoids for treatments, n = 7-9 human cardiac organoids for CTRL from 1 experiment. Data are presented as mean ± SEM. Bold outline indicates p < 0.05 using a one-way ANOVA with Dunnett's multiple comparisons test comparing each condition to CTRL at the respective time points. Human pluripotent stem cell-derived cardiac cells- AA line. Endothelial cells- RM3.5 line.
E) Coefficients of linear regression performed using binary predictors (cytokine presence/absence) with time from peak to 50% relaxation as the outcome variable at 24 h. Coefficients represent the mean change in the response given one unit change in the predictor. Sign of the coefficient represents the direction of the change between predictor and response. Overall, presence of IFN-γ, IL-1β, poly(I:C) all lead to increased relaxation times while presence of TNF leads to a reduced relaxation time. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001 using regression modeling with a two-tailed t tests.
F) Dose-response curves for TNF, force of contraction
G) Dose-response curves for IFN-γ, time to 50% relaxation
H) Dose-response curves for IL-1β, time to 50% relaxation
I) Dose-response curves for poly(I:C), time to 50% relaxation
(F-I) n = 4-5 human cardiac organoids per condition per concentration from 1 experiment. Data are presented as mean ± SEM. Human pluripotent stem cell-derived cardiac cells- HES3 line. Endothelial cells- RM3.5 line. Dose-response curve was generated using nonlinear regression (variable slope model, sigmoidal- 4 parameter logistic) to determine cytokine EC50.
Figure S3.

Validation of pro-inflammatory factor screen in an additional cell line and regression analysis, related to Figure 1
A) Validation of functional inflammatory modulator screening parameters in an additional cell line. n = 2-6 human cardiac organoids for treatments, n = 7 human cardiac organoids for CTRL from 1 experiment.
B) Overall impact of inflammatory modulators on time to 50% relaxation (diastolic function) at 24 h for both lines tested. n = 6-12 human cardiac organoids from 2 experiments.
C) Coefficients of linear regression performed (order = 2) using binary predictors (cytokine presence/absence) with relaxation time as the outcome variable. Coefficients represent the mean change in the response given one unit change in the predictor. Sign of the coefficient represents the direction of the change between predictor and response. The presence of IFN-γ, poly(I:C) and IL-1β lead to increased time to 50% relaxation.
D) Validation of TNF systolic dysfunction in an additional cell line. n = 23-25 from 3 experiments.
E) Validation of cardiac cytokine storm (CS) induced diastolic dysfunction in an additional cell line. n = 19-20 human cardiac organoids from 3 experiments
Data presented as mean ± SEM. Cardiac cytokine storm (CS): IL-1β, IFN-γ and poly(I:C). Human pluripotent stem cell-derived cardiac cells - HES3 (A), HES3 and AA (B) or AA (D,E) lines. Endothelial cells – CC (A), RM3.5 (B), or RM3.5 and CC (D,E) lines. Bold outline indicates p < 0.05 using one-way ANOVA with Dunnett’s multiple comparisons test comparing each condition to CTRL at its’ time point (A). ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 using using one-way ANOVA with Dunnett’s multiple comparisons test comparing each condition to CTRL (B), regression modeling (C) or Students t test (D,E).
The video was taken over a period of 10 s and is displayed in real time (50 frames/s).
The video was taken over a period of 10 s and is displayed in real time (50 frames/s).
The video was taken over a period of 5 s and is displayed in real time (50 frames/s).
The video was taken over a period of 5 s and is displayed in real time (50 frames/s).
Mechanisms driving cardiac cytokine storm-induced dysfunction
The most common types of cardiac dysfunction observed in hospitalized COVID-19 patients are right ventricular dysfunction or left ventricular diastolic dysfunction (Szekely et al., 2020). Therefore, we focused on diastolic dysfunction induced by IFN-γ, IL-1β, and poly(I:C), which we refer to as “cardiac cytokine storm” (CS). Protein phosphorylation is linked with all biological functions (Needham et al., 2019), and thus we measured the global phosphoproteome in hCOs. Leveraging our latest phosphoproteomics technology (Humphrey et al., 2015, 2018), we identified over 7,000 phosphosites in each sample. We accurately pinpointed 7,927 phosphorylation sites to a single amino acid residue on ~3,000 different phosphoproteins from single-run measurements (Figure 2 A). Preliminary studies on TNF-treated hCOs identified several known effects including decreased phosphorylation of protein kinase A and increased phosphorylation of beta-adrenergic receptor kinase 1 (also known as GRK2), supporting our approach (data not shown). CS treatment induced 91 phosphosites that were consistently elevated (Figure 2B). These sites were enriched for transcriptional responses with 35 sites found on transcription factors or chromatin-binding proteins and 13 associated with the biological process term “cell proliferation” (false discovery rate [FDR] <0.05, Fisher’s exact test). Among these was phosphorylation of signal transducer and activator of transcription 1 (STAT1) S727 (median 13.9-fold), as well as two sites on bromodomain-containing protein 4 (BRD4) S469 and S1083 (median 7.4- and 12.3-fold, respectively) (Figures 2B and 2C). In light of the availability of specific small molecule inhibitors for each of these targets or their upstream regulators, we focused on these proteins in subsequent functional assays.
Figure 2.

Phosphoproteomics reveals signaling driving cardiac dysfunction
(A) Schematic of the experiment.
(B) Enriched phosphopeptides in human cardiac organoids following CS treatment after 1 h. TF/TA circles depict transcription factors and transcriptional activators.
(C) Phosphorylation sites induced by CS on STAT1 and BRD4 proteins.
CS, cardiac cytokine storm; AA line, human pluripotent stem cell-derived cardiac cells; RM3.5 line, endothelial cells.
We assessed activation of individual cell populations in hCOs using snRNA-seq (Figure 3 A) with mapping as described above (Figures S1D and S1E, CTRL, and S4A and S4B, CS). In the CS condition, there was an increase in fibroblast and activated fibroblast number (Figure 3B). KEGG pathway analysis revealed a transcriptional response dominated by a viral response in both cardiomyocytes (CM1, CM2, and CM3 pooled) and fibroblasts (epicardial/fibroblast and pericyte/activated fibroblasts) (Figures 3C and 3D). There were fewer downregulated genes, which were predominantly in the fibroblasts and dominated by extracellular matrix (ECM) genes including COL1A1, COL3A1, and COL4A5 (Figure 3E). The top predicted mediators of the transcriptional response were STAT1 and general epigenetic activation by EP300 (Figure 3F). This is consistent with our phosphoproteomic data (Figure 2), given EP300 has been shown to share up to 78% of DNA binding regions with BRD4 in chromatin immunoprecipitation studies (Williams et al., 2020). These analyses together revealed a robust viral response in the heart in multiple cell populations (Figures 3G and 3H), predicted to be mediated via STAT1 and epigenetic activation including BRD4.
Figure 3.

Single nuclei RNA-sequencing reveals cardiac cytokine storm activates viral responses in human cardiac organoids
(A) Schematic of experiment.
(B) Cell compositions identified in single nuclei RNA-sequencing.
(C) Differential normalized log2 expression in cardiomyocytes and fibroblasts following cardiac cytokine storm (CS) treatment (all populations pooled for each cell type).
(D) Activation of viral responses in cardiomyocytes and fibroblasts revealed using KEGG pathway analysis of upregulated genes. Size represents number of genes regulated and the pathways of the colored circles are highlighted by the text.
(E) Repression of extracellular matrix processes in fibroblasts revealed using KEGG pathway analysis of downregulated genes. Size represents number of genes regulated and the pathways of the colored circles are highlighted by the text.
(F) STAT1 and EP300 are predicted as key transcriptional mediators. Values presented are adjusted p values, number of genes regulated by the transcription factor/number of genes regulated, and % of genes regulated over the total. The size of the colored slices represent the fraction of genes regulated (180° = 100%), and overlaps for each transcription factor are also depicted.
(G) Key upregulated genes (see Figure 5Q) in CS-treated human cardiac organoids.
(H) UMAP of CTRL and CS-treated human cardiac organoid subpopulations and expression of key regulated genes.
hCO, human cardiac organoid; CM, cardiomyocyte; Prlf, proliferating; EpC, epicardial cells; Fib, fibroblasts; Per, pericytes; Afib, activated fibroblasts; HES3 line, human pluripotent stem cell-derived cardiac cells; RM3.5 line, endothelial cells.
See also Figure S4.
Figure S4.

Populations in human cardiac organoids treated with cardiac cytokine storm, related to Figure 3
A) Normalized expression of genes in human cardiac organoids that mark the human heart sub-populations defined in Tucker et al., 2020.
B) UMAP clustering of single nuclei RNA-sequencing data for cardiac cytokine storm treated human cardiac organoids. Location of key markers for different cell populations are highlighted.
Drugs for the prevention and treatment of cardiac dysfunction
We next screened drugs that could potentially treat cardiac dysfunction caused by either TNF-induced systolic dysfunction or CS-driven diastolic dysfunction (Figure 4 A). TNF is known to induce systolic dysfunction via GRK2-mediated repression of β-adrenergic receptor signaling (Vasudevan et al., 2013). The selective serotonin reuptake inhibitor, paroxetine hydrochloride, can inhibit GRK2 (Schumacher et al., 2015), but we found that it was toxic at effective in vitro concentrations (Guo et al., 2017; Figures S5 A and S5B). GRK2 promotes clathrin-mediated endocytosis (Evron et al., 2012), and baricitinib was recently identified as a potential AP2-associated protein kinase 1 (AAK1)-mediated endocytosis inhibitor using machine learning (Richardson et al., 2020). Baricitinib prevented TNF-induced dysfunction in hCOs (Figures 4B, S5A, and S5B). However, baricitinib was only protective against TNF-induced systolic dysfunction when co-administered with TNF and was not effective after 24 h TNF treatment (Figure 4C). Additionally, hCOs did not recover quickly from TNF-induced systolic dysfunction after the removal of TNF (Figure 4C) indicating that secondary remodeling events may have occurred.
Figure 4.

Discovery of drugs that improve cardiac function
(A) Schematic of experiment.
(B) Protection against systolic dysfunction (force of contraction) by baricitinib. n = 9–32 human cardiac organoids from 2–3 experiments.
(C) Assessment of human cardiac organoid recovery from TNF and baricitinib treatment. n = 6–12 human cardiac organoids from 1–2 experiments.
(D) Protection against diastolic dysfunction (time to 50% relaxation time) by INCB054329. n = 8–43 human cardiac organoids from 2–4 experiments.
(E) Assessment of human cardiac organoid recovery from CS and INCB054329 treatment. n = 6–11 human cardiac organoids from 1–2 experiments.
(F) BRD4 is expressed in all cell populations in human cardiac organoids.
CS, cardiac cytokine storm. Data presented as mean ± SEM. HES3, human pluripotent stem cell-derived cardiac cells; RM3.5, endothelial cells. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 using one-way ANOVA with Dunnett’s multiple comparisons test compared to TNF (B) or compared to CS (D). #p < 0.05 compared to CTRL at the same time-point, and ∗p < 0.05 compared to specific condition at 0 h with color indicating comparison, using two-way ANOVA with Dunnett’s multiple comparisons test compared to CTRL (C and E). Drug screening was confirmed across in an additional cell line, and additional data are provided in Figures 6 and S5.
Figure S5.

Drugs and targets protecting against inflammation-driven dysfunction in human cardiac organoids, related to Figure 4
A) Human cardiac organoids were concurrently treated with 100 ng/ml TNF and inhibitors and then functionally assessed at 24 h. n = 4-6 human cardiac organoids from 1 experiment.
B) Validation in an additional cell line. Human cardiac organoids were concurrently treated with 100 ng/ml TNF and inhibitors and then functionally assessed at 24 h. n = 3-23 human cardiac organoids from 1-2 experiments.
(C-F) Human cardiac organoids were concurrently treated with the cardiac cytokine storm and CDK8-STAT1 S727 inhibitors, and then functionally assessed at 24 h. n = 9-21 human cardiac organoids from 2 experiments.
C) Contraction force.
D) Contraction rate.
E) Time from 50% activation to peak.
F) Time to 50% relaxation.
G) Validation of INCB054329 protection in an additional cell line. Time to 50% relaxation. Human cardiac organoids were concurrently treated with the cardiac cytokine storm and INCB054329, and then functionally assessed at 24 h. n = 4-12 human cardiac organoids from 1-2 experiments.
H) Multiple bromodomain extraterminal protein inhibition prevents cardiac cytokine storm induced diastolic dysfunction, presented as change relative to increased relaxation time. n = 8-43 human cardiac organoids from 2-4 experiment.
I) Validation of results in an additional cell line. Multiple bromodomain extraterminal protein inhibition prevents cardiac cytokine storm induced diastolic dysfunction, presented as change relative to increased relaxation time. n = 14-15 for CTRL and cardiac cytokine storm conditions and 4-6 human cardiac organoids from 1-2 experiments.
J) Assessment of INCB054329 efficacy in conditions with cardiac cytokine storm with the addition of TNF. n = 6-16 human cardiac organoids from 1-2 experiments.
K) BRD4 knockdown prevents cardiac cytokine storm induced diastolic dysfunction, presented as normalized relaxation time. n = 5-8 human cardiac organoids from 1 experiment.
L) Representative force trace of a CTRL human cardiac organoid.
M) Representative force trace of different types of arrhythmias in human cardiac organoids treated with cardiac cytokine storm.
N) Arrhythmic events in human cardiac organoids per experiment. n = 4-7 experiments.
CS – cardiac cytokine storm. Data presented as mean ± SEM. Human pluripotent stem cell-derived cardiac cells- HES3 (A,C-F,H,J,K [with no endothelial cells], L-N) or AA (B,G,I) lines. Endothelial cells- RM3.5 (A,C-F,H,J,L-N) or CC(B,G,I) lines. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, using a one-way ANOVA with Dunnett’s multiple comparisons test compared to TNF (A,B) CS (C-I), CS + AAV6-shSCR (scramble control) (K) or with Tukey’s multiple comparison test (J) or with Kruskal-Wallis comparisons test to CTRL (N).
A key signature of diastolic dysfunction under CS conditions was the elevated phosphorylation of transcriptional regulators. STAT1-S727 (Figure 2C) is associated with assembly into chromatin and is required for STAT1 transcriptional and biological activity in response to IFN-γ (Sadzak et al., 2008). The putative STAT1-S727 kinase is CDK8 (Bancerek et al., 2013), so we next tested two CDK8 inhibitors SEL120-34A (Rzymski et al., 2017) and BI-1347 (Hofmann et al., 2020) previously shown to reduce STAT1-S727 phosphorylation. We also tested two inhibitors of the JAK/STAT pathway, baricitinib and ruxolitinib and a broader spectrum CDK inhibitor, flavopiridol. However, none of these compounds prevented the CS-induced diastolic dysfunction, noting that flavopiridol was toxic, reducing force and hence all kinetic parameters (Figures S6 A–S6H). Notably, SEL120-34A and BI-1347 specifically attenuated the rate and activation time defects under CS conditions (Figures S6B, S6C, S6F, and S6G), which we validated in additional experiments (Figures S5C–S5F), and may still have clinical utility in this setting.
Figure S6.

Screening for compounds that prevent cardiac cytokine storm induced diastolic dysfunction, related to Figure 4
A) Contraction force in human cardiac organoids.
B) Contraction rate in human cardiac organoids.
C) Time to 50% activation in human cardiac organoids.
D) Time to 50% relaxation in human cardiac organoids.
E) Contraction force in human cardiac organoids from an additional line.
F) Contraction rate in human cardiac organoids from an additional line.
G) Time to 50% activation in human cardiac organoids from an additional line.
H) Time to 50% relaxation in human cardiac organoids from an additional line.
Human cardiac organoids were concurrently treated with the cardiac cytokine storm (CS) and compounds, and then functionally assessed at 24 h. (A-D) n = 4-11 and (E-F) n = 3-9 hCOs per condition from 1 experiment. Data presented as mean ± SEM. Human pluripotent stem cell –derived cardiac cells - HES3 (A-D) or AA (E-H) lines. Endothelial cells – RM3.5 (A–D) or CC (E–H) lines. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, using one-way ANOVA with Dunnett’s multiple comparisons test compared to cardiac cytokine storm.
We observed elevated phosphorylation of the epigenetic regulator BRD4 and other epigenetic regulators in our CS-treated hCOs phosphoproteome, consistent with our snRNA-seq analysis. We have previously shown that bromodomain extraterminal inhibitors (BETi) reduce relaxation time in immature hCOs (Mills et al., 2019), so we next evaluated three BETi available in a Food and Drug Administration (FDA) compound library, INCB054329 (Stubbs et al., 2019), JQ-1 (Filippakopoulos et al., 2010), and ABBV-744 (Faivre et al., 2020). Strikingly, INCB054329 prevented CS-induced diastolic dysfunction in a dose-dependent manner (Figures 4D and S5G) without affecting force or rate (Figures S6A–S6H; Video S5). JQ-1 also showed improved diastolic function in one hPSC line at the highest concentration (Figure S6H), so additional higher concentrations for both JQ-1 and ABBV-744 were tested. JQ-1 protected hCOs against CS-induced diastolic dysfunction, although INCB054329 was the most efficacious (Figures S5H and S5I). In contrast, ABBV-744 increased diastolic dysfunction in the hCOs, potentially via its dual actions as an androgen receptor inhibitor, which is associated with prolonged QTc in patients undergoing androgen deprivation therapy (Gagliano-Jucá et al., 2018). To validate BRD4 as a target, we used adeno-associated virus 6 (AAV6)-mediated delivery of short hairpin (shRNA) and demonstrated that ~74% knockdown could also reduce diastolic dysfunction in CS-treated hCOs (Figure S5K).
The video was taken over a period of 10 s and is displayed in real time (50 frames/s).
INCB054329-mediated BETi rescued dysfunctional hCO and restored diastolic function following 24 h of CS conditions (Figure 4E). This is potentially because CS-induced diastolic dysfunction is reversible and is driven by the presence of the inflammatory mediators, demonstrated by partial hCO recovery 24 h after removing CS factors (Figure 4E). In patients, all inflammatory factors may be present simultaneously, and we found that INCB054329 attenuated diastolic dysfunction with all four dysfunction inducing factors, TNF, IFN-γ, IL-1β, and poly(I:C), present (Figure S5J). Taken together, CS mediates diastolic dysfunction via BRD4-dependent mechanisms that can be blocked using BETi. Because BRD4 is broadly expressed in our hCOs (Figure 4F), it may also be responsible for the multi-cellular response observed.
INCB054329 reduces the host response to SARS-CoV-2 infection in K18-hACE2 mouse hearts
We next assessed the response to SARS-CoV-2 infection in vivo. Because mice are not susceptible to SARS-CoV-2 infection, we used a recently described K18-hACE2 model (Oladunni et al., 2020) to study the response and effects of BETi (Figure 5 A). SARS-CoV-2-infected mice had severe lung pathology and substantial viral RNA reads in the lungs at 4–5 days post infection, confirming successful lung infection (Figures 5B and 5C). RNA-seq of the lungs revealed increased expression of 419 genes (Figure 5D) strongly associated with a viral response (Figure 5E; Table S1A). Concordant with our hCO data, Stat1 and Ep300 were the top predicted transcriptional regulators (Figure 5F; Table S1B). Interestingly, there was only negligible infection of the heart (Figure 5C) and no obvious pathology including necrosis, immune cell infiltration, or fibrosis (Figure 5G). However, there was a substantial and robust upregulation of a viral response in the heart with ECM repression observed (Figures 5H and 5I; Table S1A) including Col1a1, Col3a1, and Col4a2. This was again enriched for Stat1 and Ep300 as top predicted transcriptional regulators (Figure 5J; Table S1B), indicating a robust systemic response in the hearts of SARS-CoV-2-infected mice. This response could be partially blocked by INCB054329 treatment with repression of 91 genes that were enriched for the viral response (Figures 5K–5M; Table S1A). This response was more specific to the heart, because INCB054329 did not regulate any genes in the lungs (data not shown). The repression by INCB054329 was predicted to be primarily mediated via Ep300 rather than Stat1 (Figure 5N; Table S1B). These results were further supported by ingenuity pathway analysis of upstream regulators revealing strong activation signatures for IFNG, poly rI:rC-RNA, and Stat1 in both lungs and hearts of K18-hACE2 SARS-CoV-2-infected mice, which INCB054329 strongly inhibited (Table S1C).
Figure 5.

SARS-CoV-2 activates viral responses in the heart repressed by INCB054329
(A) Schematic of the experiment.
(B) Lungs of SARS-CoV-2-infected K18-hACE2 mice 5 days post infection. Infection causes sloughing of bronchial epithelium, and white arrowheads show (A) collapse of alveolar spaces and (B) bronchiolar lumen.
(C) Severe lung infection with no/negligible heart infection at 4 days post infection. n = 5 mice per group.
(D) Lung RNA-sequencing reveals a robust upregulation (logFC >0.5) of 419 genes and downregulation (logFC <−0.5) of 98 genes, both FDR <0.05. n = 5 mice per group.
(E) Activation of viral responses in lungs revealed using KEGG pathway analysis of upregulated genes. Size represents number of genes regulated and the pathways of the colored circles are highlighted by the text.
(F) Stat1 and Ep300 are predicted as key transcriptional mediators of infection in the lungs.
(G) Hearts of SARS-CoV-2-infected K18-hACE2 mice 5 days post infection. Relatively normal, with no significant necrosis, fibrosis (Masson’s Tri-chrome not shown), or immune infiltrates.
(H) Heart RNA-sequencing reveals a robust upregulation (logFC >0.5) of 249 and downregulation (logFC <−0.5) of 159 genes, both FDR <0.05. n = 5 mice per group.
(I) Activation of viral responses in hearts revealed and repression of ECM using KEGG pathway analysis of upregulated genes and downregulated genes. Size represents number of genes regulated and the pathways of the colored circles are highlighted by the text.
(J) Stat1 and Ep300 are predicted as key transcriptional mediators in the heart.
(K) PCA of heart RNA-sequencing samples. n = 4–5.
(L) Heart RNA-sequencing reveals a robust upregulation (logFC >0.5) of 11 genes and downregulation (logFC <−0.5) of 91 genes, both FDR <0.05 by INCB054329. n = 4–5 mice per group.
(M) Repression of viral responses in hearts revealed using KEGG pathway analysis of downregulated genes. Size represents number of genes regulated and the pathways of the colored circles are highlighted by the text.
(N) Ep300 is predicted as the key transcriptional mediator of INCB054329 effects in the heart.
(O) Cross-analysis of the transcriptional responses in human cardiac organoids with hearts of SARS-CoV-2-infected K18-hACE2 mice.
(P) Co-regulated genes in (O) reveal a consistent activation of viral responses in both models using KEGG pathway analysis of upregulated genes. Size represents number of genes regulated, and the pathways of the colored circles are highlighted by the text.
(Q) Genes induced by both CS in human cardiac organoids and SARS-CoV-2-infected K18-hACE2 mouse hearts that are also repressed by INCB054329.
(R) Severe weight loss by 4–5 days post infection in SARS-CoV-2-infected K-18-hACE2 mice is due to severe lung infection and brain infection, and euthanasia is required.
d.p.i., days post infection; CS, cardiac cytokine storm. Data presented as mean ± SEM. ∗∗p < 0.01 using Mann-Whitney, ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001 using two-way ANOVA with Sidak’s post hoc test compared to CTRL. (D, H, and L) Red dots are regulated as per the described cut-offs and gray dots are not. (F, J, and N) Values presented are adjusted p values, number of genes regulated by the transcription factor/number of genes regulated, and % of genes regulated over the total. The size of the colored slices represent the fraction of genes regulated (180° = 100%), and overlaps for each transcription factor are also depicted. Additional data bioinformatic analysis is provided in Tables S1A–S1C.
Potentially important mediators and markers of the response were found by integrating the multiple datasets. The CS induced response in the hCOs (either fibroblasts or cardiomyocytes) and hearts of SARS-CoV-2-infected K18-hACE2 mice shared 32 regulated genes (Figure 5O) that were enriched for the viral response (Figure 5P). The consistent transcriptional program in CS-treated hCOs (both fibroblasts and cardiomyocytes) and SARS-CoV-2-infected K18-hACE2 mice, which were also downregulated genes by INCB054329 treatment in vivo, revealed 5 key targets. These comprise the key inflammatory genes Nmi, Tap1, B2m, Stat1, and Lgals3bp (Figure 5Q). Of particular interest is LGALS3BP (galectin-3 binding protein), because it has been shown to be a top-predictor of COVID-19 severity in humans (Messner et al., 2020) and we therefore interrogated its expression in our subsequent models.
INCB054329 protects against inflammatory mediated dysfunction in vivo and by COVID-19 serum
The study into the efficacy of SARS-CoV-2-related cytokine storm therapeutics on the heart in vivo is technically challenging in biosafety level 3 and because the severe lung/brain infection in K18-hACE2 mice causes a rapid decrease in weight and requires euthanasia (Figure 5R). We therefore used surrogate models.
We used a LPS-induced cytokine storm mouse model (Figure 6 A). LPS induced pro-inflammatory cytokines TNF, IL-1β, and IFN-γ, which were elevated in the plasma (Figure 6B), along with Lgals3bp in the heart (Figure 6C). Treatment with INCB054329 blocked the LPS-induced pro-inflammatory cytokine production (Figure 6B) and Lgals3bp induction in the heart (Figure 6C). We observed a marked improvement in mortality, whereby all INCB054329-treated mice survived after 24 h of the LPS-challenge, compared with only 25% in the control group (Figure 6D). To determine whether BETi could treat an established LPS-induced cytokine storm, we delayed injection of INCB054329 1.5 h after LPS injection and assessed cardiac function at 6 h (Figure 6E). INCB054329 fully prevented the decrease in cardiac function observed after LPS injection (Figure 6F). Together, these findings demonstrate that BETi using INCB054329 has robust effects in preventing inflammatory-induced cardiac dysfunction in vivo.
Figure 6.

INCB054329 prevents cardiac dysfunction in a mouse lipopolysaccharide-induced cytokine storm model and in response to COVID-19 patient serum
(A) Schematic for (B)–(D).
(B) INCB054329 blocks cytokine induction 6 h after lipopolysaccharide injection. n = 5–6 mice.
(C) INCB054329 blocks induction of Lgals3bp 6 h after lipopolysaccharide injection. n = 5–6 mice.
(D) Kaplan-Meier curve of survival after lipopolysaccharide injection. n = 12 control and 11 INCB054329 treatment (67 mg/kg).
(E) Schematic for (F).
(F) INCB054329 prevents the reduction in ejection fraction 6 h after lipopolysaccharide injection. n = 3–4 mice at 0 and 1.5 h, and n = 8 at 6 h.
(G) Schematic for (H)–(K).
(H) IFN-γ was not higher in patients with elevated cardiac troponin I (CTNI >0.5 ng/mL). n = 27.
(I) IFN-γ was higher in patients with elevated brain natriuretic peptide (BNP >0.3 ng/mL). n = 27.
(J) Serum from COVID-19 patients with elevated brain natriuretic peptide induces diastolic dysfunction. Orange highlights human cardiac organoids with elevated force of contraction. Blue highlights dysfunctional human cardiac organoids.
(K) Diastolic dysfunction induced by COVID-19 patient 6 serum is prevented by 1 μM INCB054329. n = 4–9 human cardiac organoids from 1 experiment.
(L) LGALS3BP is induced by CS and repressed by bromodomain and extraterminal protein inhibition in human cardiac organoids. n = 3 each (2 human cardiac organoids pooled per n).
LPS, lipopolysaccharide; CS, cardiac cytokine storm. Data presented as mean ± SEM. HES3 line, human pluripotent stem cell-derived cardiac cells; RM3.5 line, endothelial cells. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001, using one-way ANOVA with Tukey’s multiple comparisons test (B and C), with Dunnett’s multiple comparisons test compared to CTRL serum (J and K) or CS (L), or using two-way ANOVA with Sidak’s multiple comparisons test (F) or Mann-Whitney (I). p value calculated using Gehan-Breslow-Wilcoxon test (D). Additional patient data are provided in Tables S2A and S2B.
The factors present in COVID-19 patient serum are more complex than our CS conditions. We assessed the impact of this serum on hCOs (Figure 6G; Tables S2A and S2B). Our most potent CS factor, IFN-γ, correlates with COVID-19 disease progression and is elevated in patient serum in one of the most comprehensive profiling studies to date (Ren et al., 2021). We found that IFN-γ was higher in patients with elevated BNP as a marker of cardiac stress (>0.3 ng/mL) but not CTNI as a marker of acute injury (>0.5 ng/mL) (Figures 6H and 6I). Factors in human serum can alter hCO function, because patients receiving noradrenaline as inotropic support had elevated contractile force in hCOs (Figure 6J). Diastolic dysfunction was induced in hCOs by serum with elevated BNP (Figure 6J) with no viral infection detected (data not shown), and INCB054329 could prevent this response (Figure 6K). We also found that LGALS3BP induction could be prevented by treatment with multiple BETi (Figure 6L).
Collectively, these data indicate that BET inhibition with INCB054329 prevents cardiac dysfunction in multiple complex inflammatory models, as well as repressing the key COVID-19 severity marker LGALS3BP.
INCB054329 decreases hACE2 expression and reduces SARS-CoV-2 in hPSC-cardiac cells
SARS-CoV-2 potentially infects human hearts and has been shown to infect human pluripotent stem cell-derived cardiac cells (hPSC-CM) (Sharma et al., 2020). Recently, other investigators have also demonstrated that BETi reduced Ace2 in vivo and SARS-CoV-2 infection (Qiao et al., 2020). We sought to determine whether BETi blocked infection (Figure S7 A).
Figure S7.

Pre-treatment with INCB054329 prevents SARS-CoV-2 infection of cardiac cells, related to Figure 7
A) Schematic of the experiments.
B) Optimization of loading with increasing cell death of 2D cardiac cells with increasing SARS-CoV-2 infection.
C) Infection at low multiplicity of infection (0.01) results in viral replication and eventually death following SARS-CoV-2 infection of 2D cardiac cells. n = 6 from 3 experiments.
D) ACE2 expression in 2D cultured cardiac cells pre-treated with 1 μM INCB054329 for 3 days.
E) Immunostaining of cardiomyocytes and SARS-CoV-2 reveals that 1 μM INCB054329 reduces viral loading.
F) Pre-treatment with 1 μM INCB054329 for 3 days reduces SARS-CoV-2 infection. E-gene expression in 2D cultured cardiac cells 3 days after infection. n = 6 from 2 experiments.
G) Pre-treatment with 3 μM JQ-1 for 3 days reduces SARS-CoV-2 infection. E-gene expression in 2D cultured cardiac cells 3 days after infection. n = 8 from 2 experiments.
H) INCB054329 decreases endogenous mAce2 expression in hearts in vivo. n = 4-5 mice.
I) Flow cytometry analysis of ACE2 on cardiomyocytes (CD90 negative) and CD90 positive stromal cells. Analysis was performed in 2 different cells lines in separate experiments.
J) Flow cytometry analysis of spike protein binding on cardiomyocytes (CD90 negative) and CD90 positive stromal cells. Analysis was performed in 2 different cells lines in separate experiments.
K) Immunostaining of cardiomyocytes and SARS-CoV-2 reveals that 30 μM apabetalone preserves cardiomyocyte structures and reduces viral loading.
All scale bars = 20 μm. TCID50 - Fifty-percent tissue culture infective dose. Data presented as mean ± SEM. Human pluripotent stem cell-derived cardiac cells – HES3 (C,D,K), AA (E,F) or HES3 and AA (G,I,J) lines. ∗p < 0.05, ∗∗∗∗p < 0.0001, using one-way ANOVA with Dunnett’s multiple comparisons test (C - compared day 0 and F,G – compared to DMSO) and Mann-Whitney (H).
We confirmed previous findings using 2D cultured hPSC-cardiac cell infection studies, where increasing the MOI increased the degree of cell death (Figure S7B). Infection with a low MOI (0.01) was sufficient for viral replication and cell death over 7 days (Figure S7C). A 3-day pre-incubation of INCB054329 was sufficient to reduce ACE2 expression ~4-fold (Figure S7D). Consequently, pre-treatment with INCB054329 reduced SARS-CoV-2 N protein expression (Figure S7E) and intracellular viral RNA (Figure S7F). In addition to INCB054329, the widely used BETi JQ-1 reduced SARS-CoV-2 RNA (Figure S7G). In our SARS-CoV-2 K-18 mouse infection studies, INCB054329 treatment reduced endogenous Ace2 in hearts in vivo (Figure S7H). Thus, BETi also has potential to block SARS-CoV-2 infection of cardiac cells in addition to preventing dysfunction.
BETi for translation to the clinic
We assessed the ability of all commercially available BETi compounds to prevent CS-induced diastolic dysfunction in hCOs. We found that all compounds prevented dysfunction except for ABBV-744 (Figure 7 A). BETi with dual bromodomain 1 (BD1) and bromodomain 2 (BD2) activities display side effects (Gilan et al., 2020); as such, it is critical that we determine the bromodomain selectivity of the response. BD2-selective drugs, such as ABBV-744 and apabetalone, have limited side effects. Apabetalone has been used for up to 3 years in >1,700 humans at risk of cardiac disease, with efficacy in preventing heart failure and a favorable safety profile (Nicholls et al., 2021; Ray et al., 2020). ABBV-744 elevated diastolic dysfunction (Figure 7A), and we suspect its lack of efficacy was due to its on-target inhibition of the androgen receptor (AR). BD2-specific efficacy was confirmed using a BD2-specific molecule, RXV-2157 (Figure 7B), and we also confirmed efficacy with the BD-2 selective apabetalone (Figures 7A and 7B). Additionally, analysis of plasma from the ASSURE phase IIb clinical trial indicated that BD2-selective apabetalone reduced LGALS3BP in patients with cardiovascular disease (Figure 7C), a marker of COVID-19 severity (Messner et al., 2020).
Figure 7.

Bromodomain and extraterminal protein inhibitors targeting bromodomain 2 are effective therapeutic candidates
(A) All bromodomain and extraterminal protein inhibitors (except ABBV-744) used in clinical trials prevent CS-induced diastolic dysfunction. n = 12–19 human cardiac organoids from 3 experiments.
(B) Bromodomain and extraterminal protein inhibitors specific (RVX-2157) or selective (apabetalone) for bromodomain 2 prevent CS-induced diastolic dysfunction. n = 12–18 human cardiac organoids from 3 experiments.
(C) Apabetalone decreases serum LGALS3BP in the phase IIb ASSURE clinical trial. Data are changes from baseline. n = 47 both groups.
(D) Bromodomain and extraterminal protein inhibitors specific (RVX-2157) or selective (apabetalone) for bromodomain 2 decrease ACE2 expression after 3 days.
(E) Pre-treatment for 3 days with bromodomain and extraterminal protein inhibitors specific (RVX-2157) or selective (apabetalone) for bromodomain 2 reduce SARS-CoV-2 infection. E-gene expression in 2D cultures 3 days after infection. n = 6–8 from 2 experiments.
(F) Apabetalone 3-day pre-treatment to reduce SARS-CoV-2 infection. E-gene expression in 2D cultures 3 days after infection. n = 6 from 1 experiment.
(G) Apabetalone or JQ-1 3-day pre-treatment reduces SARS-CoV-2 titer in hPSC-CM 3 days after infection. n = 6 from 1 experiment.
CS, cardiac cytokine storm; TCID50, 50% tissue culture infective dose. Human pluripotent stem cell-derived cardiac cells (HES3 line) (no endothelial cells) (A–C, F, and G) or HES3 and AA lines (E). Data presented as mean ± SEM for all plots except for (C), which is median ± interquartile range. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, using one-way ANOVA with Dunnett’s multiple comparisons test (A and B, compared to CS; E–G, compared to DMSO) or Mann-Whitney (C).
See also Figure S7.
We confirmed the BD2-specificity of blocking SARS-CoV-2 infection. The BD2-specific RXV-2157 and BD2-selective apabetalone molecules downregulated hACE2 (Figure 7D), which led to decreased surface expression (Figure S7I) and SARS-CoV-2 spike protein binding (Figure S7J). These compounds also reduced SARS-CoV-2 loading (Figures 7E, 7F, and S7K) and viral titer, including a 2.6-fold decrease in viral titer with apabetalone (Figure 7G).
Together, this demonstrates that BD2-selective BETi drugs are lead candidates for rapid clinical translation to prevent COVID-19 injury in the heart.
Discussion
We define CS conditions resulting in severe diastolic dysfunction with 20%–50% increases in time to 50% relaxation in hCOs. This is consistent with ~13%–18% increases in cardiomyocytes derived from patients with heart failure with preserved ejection fraction (HFpEF), with similar absolute increases of 100–150 ms (Runte et al., 2017). It is also consistent with diastolic dysfunction reported in COVID-19 patients (Szekely et al., 2020), indicating our hCO model recapitulates key clinical features of diastolic dysfunction.
Our transcriptional profiling revealed a striking similarity between the inflammatory response in CS-treated hCOs and hearts of SARS-CoV-2-infected K18-hACE2 mice. CS also elicited a more pronounced transcriptional response in the fibroblasts within hCOs (Figures 3C and 3D). This was with negligible viral infection in our models, meaning that inter-organ and intra-organ signaling appears to play a key role in inflammation-induced cardiac dysfunction. It will be important to decipher the systemic and intra-organ drivers of dysfunction in other organs, as COVID-19 and many other inflammatory diseases can result in multi-organ dysfunction.
Our data establish BET inhibition as a viable therapeutic strategy to attenuate cytokine storm-induced cardiac dysfunction. Previously, BETi compounds have shown efficacy in small animal cytokine storm models (Nicodeme et al., 2010). There is also compelling evidence implicating bromodomain proteins as key mediators in pathological pro-fibrotic signaling in heart failure (Duan et al., 2017; Stratton et al., 2019), in experimental models of pressure overload and myocardial infarction-induced HF (Anand et al., 2013), and in genetic cardiomyopathies (Antolic et al., 2020; Auguste et al., 2020). However, this study is instrumental in establishing BET inhibition as a therapeutic intervention to prevent cardiac dysfunction caused by inflammation.
Clinical data from COVID-19 patients also point to additional cardiac pathologies. Microthrombi were reported in the hearts of 14 out of 40 patients (35%) that died from COVID-19, which was associated with areas of myocardial necrosis (Pellegrini et al., 2021). Consistent with these observations, we observed that “complement and coagulation cascades” were enriched in the lungs of K18-hACE2 mice with SARS-CoV-2 infection (Figure 5E; Table S1) and were likely related to the viral-induced inflammatory response. Serpine 1 is a key inhibitor of tissue-type plasminogen activator and urokinase-type plasminogen activator and is required for fibrinolysis downregulation and degradation of blood clots. Concordantly, serpine1 (also known as plasminogen activator inhibitor 1), was upregulated in both the lungs (3.9-fold) and in the heart (2.8-fold, both FDR <0.05) of SARS-CoV-2-infected K18-hACE2 mice, which was indeed abrogated in the heart upon treatment with the BETi INCB054329 (FDR <0.05). In addition, arrhythmic events have been widely reported in COVID-19 patients (Nishiga et al., 2020). Indeed, we observed that arrhythmic events increased in hCOs with CS, for which INCB054329 also conferred protection (Figures S5L–S5N). These data suggest that BET inhibition may be effective in attenuating multiple deleterious aspects of systemic inflammation on the heart that warrant further investigation.
We demonstrated that BETi are attractive therapeutic candidates, however, the side effect profiles of some BETi may preclude their use in the clinic. Genetic ablation studies have shown that BRD4 plays an integral homeostatic role in cardiomyocytes, suggesting that the loss of BET proteins may have detrimental effects on mitochondrial energy production (Kim et al., 2020; Padmanabhan et al., 2020). Emerging evidence dissecting the roles of BD1 and BD2 bromodomains in inflammatory disease models has indicated that BD2-selective inhibition preferentially blocks the induction of gene expression while minimally affecting established transcription programs (Gilan et al., 2020). More recently, BD2-selective drugs such as ABBV-744 and apabetalone have been developed to overcome these side-effect profiles. Although ABBV-744 was not effective in our hCO model (potentially due to its targeting of AR), we demonstrate that BD-2 selective compounds RXV-2157 and apabetalone demonstrate efficacy. This underscores the need for careful BETi selection, despite broad ability to modulate critical target genes (Figure 6L) and utility for a variety of clinical conditions (Cochran et al., 2019). Importantly, BD2-selective BETi apabetalone reduced CS-induced diastolic dysfunction and downregulated ACE2 and reduced viral infection (Figure 7). Taken together, the efficacy and known safety profile of apabetalone make it a prime candidate to protect against cardiac injury for inflammatory diseases such as COVID-19.
The overlap in risk factors for HFpEF and COVID-19 mortality suggests that our findings may also have broader implications. HFpEF risk factors including diabetes and obesity are also associated with chronic inflammation. Recent studies have shown that elevated inflammatory markers are associated with worsening heart function in HFpEF (Sanders-van Wijk et al., 2020), thus indicating that inflammation may drive dysfunction across multiple cardiac diseases, and BET inhibitors are putative therapeutic candidates.
Limitations of study
Human COVID-19 patient serum and CS directly impacted hCO function. There is evidence that the heart can be inflamed in patients with COVID-19 (Kotecha et al., 2021; Puntmann et al., 2020), but whether direct cardiac inflammation is required for functional impact or whether the systemic environment is a driver of cardiac dysfunction remains to be determined. Larger studies are required to ascertain the full extent of the inflammatory effects on heart function in the clinic, in particular on diastolic function as found in some echocardiography studies (Szekely et al., 2020). It will be important to determine whether cardiac inflammation and its functional effects (that may be sub-clinical in some cases) are prolonged following acute infection and whether this predisposes patients to future risk of cardiovascular events.
Our hCO model is free from an active immune system and the secondary effects of neurohormonal compensation present in vivo. Our hCO work, the lack of response in the lungs of INCB054329-treated SARS-CoV-2-infected K18-hACE2 mice, and improvement of heart function with delayed INCB054329 treatment in the LPS-treated mice (Figure 6F) all indicates robust cardiac-specific effects. However, BETi within an in vivo setting may also reduce immune responses and cytokine induction (e.g., Figure 6B), thus, we cannot rule this out as a potential mechanism for cardiac protection in mouse studies. In order to elucidate cardiac-specific effects, genetic studies could be useful. However, these may be difficult because (1) BRD4 knockout has different effects to small molecule inhibitors (Kim et al., 2020; Padmanabhan et al., 2020), and (2) BETi drugs bind multiple BRD family members, which may be more potent that targeting one member (Gilan et al., 2020). Further mechanistic insight into the key transcriptional targets of BET proteins is important. These targets could then be manipulated using cardiac-specific genetic approaches to validate cardiac specificity of BETi. However, the feasibility of this approach will be dependent on the number of targets that are critical for BETi efficacy, as multiple targets may require simultaneous genetic manipulation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SUPPLIER | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-human CD31 | Dako | RRID: AB_2114471 |
| Neural/Glial Antigen 2 | Life Technologies | RRID: AB_10870987 |
| Rabbit anti-cardiac Troponin T | Abcam | RRID: AB_956386 |
| Mouse IgG2a anti-Human CD90 | RnD Systems | RRID: AB_2203306 |
| Human ACE-2 Alexa Fluor 647-conjugated antibody | RnD Systems | CAT# FAB933R |
| Goat IgG Alexa Fluor 647-conjugated Antibody | RnD Systems | CAT# IC108R |
| Goat anti-human ACE2 polyclonal antibody | RnD Systems | RRID: AB_355722 |
| SARS-CoV-2 Nucleocapsid Antibody, Mouse mAb | Sino Biological | RRID: AB_2827977 |
| Mouse Anti-GAPDH Monoclonal Antibody | Cell Signaling Technology | RRID:AB_2756824 |
| Goat anti-Mouse IgG1 Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher Scientific | RRID: AB_2535764 |
| Goat anti-Mouse IgG2a Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | ThermoFisher Scientific | RRID: AB_2535776 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher Scientific | RRID: AB_2534069 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Alexa Fluor 555 | ThermoFisher Scientific | RRID: AB_2535844 |
| F(ab’)2-Goat anti-Human IgG Fc Secondary Antibody | ThermoFisher Scientific | RRID: AB_2536548 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | ThermoFisher Scientific | RRID: AB_2535849 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 633 use 1:400 | ThermoFisher Scientific | RRID:AB_2535731 |
| IRDye800CW Goat anti-Mouse IgG Secondary Antibody | LI-COR Biotechnology | RRID AB_621842 |
| IRDye680RD Donkey anti-Goat IgG Secondary Antibody | LI-COR Biotechnology | RRID AB_2650427 |
| Proteins | ||
| rSARS-CoV-2 Spike RB | RnD Systems | CAT# 10499CV100 |
| ELISA | ||
| Human CNTI ELISA | RayBiotech | CAT# ELH-CTNI-1 |
| BNP ELISA | Abcam | CAT# ab193694 |
| IFN-γ ELISA | RnD Systems | CAT# DIF50C |
| Mouse Inflammation Kit Cytokine Bead Array | BD Biosciences | CAT# 552364 |
| Deposited data | ||
| Phospho-Proteomics data of vascularised cardiac organoids | This study | PRIDE: PXD020994 |
| RNA-seq of mouse heart maturation | Gilsbach et al., 2018 | BioProject ID: PRJNA353755 |
| Bulk RNA-seq of SARS-CoV2 infected K-18 mice | This study | ENA: PRJEB43658 |
| snRNA-seq of vascularised cardiac organoids | This study | EGA: EGAS00001005174 |
| Experimental models: cell lines | ||
| Human embryonic stem cell line HES3 | WiCell | RRID: CVCL_7158 |
| Human induced pluripotent stem cell line RM3.5 | Murdoch Children’s Research Institute | N/A |
| Human induced pluripotent stem cell line AA | CIRM hPSC Repository | CW30382A |
| Human induced pluripotent stem cell line CC | CIRM hPSC Repository | CW30318C |
| Experimental models: organisms/strains | ||
| C57BL/6J mice | N/A | RRID: MGI: 3028467 |
| K18-hACE2 C57BL/6J mice (strain B6.Cg-Tg(K18-ACE2)2Prlmn/J) | N/A | RRID:IMSR_JAX:034860 |
| SARS-CoV2 (QIMR Berghofer) | N/A | hCoV-19/Australia/QLD02/2020 |
| SARS-CoV2 (The Doherty Institute) | N/A | CoV/Australia/VIC01/2020 |
| Oligonucleotides | ||
| Primers for qPCR | N/A | see STAR Methods—Quantitative RT-PCR |
| Software and algorithms | ||
| Pole tracking analysis | Mills et al., 2017 | N/A |
| MaxQuant | Cox and Mann, 2008 | RRID:SCR_014485 |
| Perseus | Tyanova and Cox, 2018 | RRID:SCR_015753 |
| STAR aligner | Dobin et al., 2013 | RRID:SCR_015899 |
| CellRanger | N/A | RRID:SCR_017344 |
| Cutadapt | Martin, 2011 | RRID:SCR_011841 |
| RNA-SeQC | DeLuca et al., 2012 | RRID:SCR_005120 |
| RSEM | Li and Dewey, 2011 | RRID:SCR_013027 |
| Scanpy | Wolf et al., 2018 | RRID:SCR_018139 |
| Bioconductor R | Huber et al., 2015 | RRID: SCR_001905 |
| Bioconductor packages edgeR | Robinson et al., 2010 | RRID:SCR_012802 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, James E Hudson james.hudson@qimrberghofer.edu.au
Materials availability
This study did not generate new unique reagents except for RVX-2157 for which requests should be addressed to Resverlogix.
Data and code availability
Mass spectrometry-based proteomics data reported in this paper have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Deutsch et al., 2017) with the dataset identifier PXD020994. snRNA-seq reported in this paper has been deposited to the European Genome-phenome Archive (EGA) with the dataset identifier EGAS00001005174. Bulk RNA-seq data reported in this paper have been deposited to the European Nucleotide Archive (ENA) with the dataset identifier PRJEB43658. All MATLAB m-files will be provided upon request as they require custom training.
Experimental model and subject details
Mice
Mouse work was undertaken in accordance with the Australian Code for Care and Use of Animals for Scientific Purposes, as outlined by the National Health and Medical Research Council of Australia. Animal work was approved by the QIMR Berghofer Medical Research Institute and University of Queensland Animal Ethics Committees.
For LPS experiments wild-type (WT) C57BL/6 were purchased from Walter and Eliza Hall Institute for Medical Research, the Australian Research Centre in Western Australia or bred in house at QIMR Berghofer Medical Research Institute. Mice used in this study were older than 6 weeks and were sex-matched. The number of mice in each group of treatment for each experiment is indicated in the figure legends. No mice were excluded based on pre-established criteria and randomization was applied immediately prior to treatment in therapy experiments.
For SARS-CoV-2 infection studies, heterozygous K18-hACE2 C57BL/6J mice (strain B6.Cg-Tg(K18-ACE2)2Prlmn/J) were purchased from The Jackson Laboratory, USA, and bred in house at QIMR Berghofer Medical Research Institute and genotyped using standard PCR as per Jackson Labs genotyping protocol. Female mice were used for experiments.
Cell lines
Ethical approval for the use of human embryonic stem cells (hESCs) was obtained from QIMR Berghofer’s Ethics Committee and was carried out in accordance with the National Health and Medical Research Council of Australia (NHMRC) regulations. hESCs utilized were female HES3 (WiCell). A male RM3.5 iPSC line was used (generated by Edouard Stanley (Murdoch Children’s Research Institute, Melbourne, Australia)). The following cell lines were obtained from the CIRM hPSC Repository funded by the California Institute of Regenerative Medicine: CW30382A (male, designated AA) and CW30318C (female, designated CC) which were both obtained from FujiFilm. hPSC lines were maintained in mTeSR-1 (Stem Cell Technologies)/Matrigel (Millipore) and passaged using TrypLE (ThermoFisher Scientific) or ReLeSR (Stem Cell Technologies). Quality control was performed with Karyotyping and mycoplasma testing.
Human COVID-19 plasma and serum
Plasma samples were obtained from individuals with PCR confirmed COVID-19 infection in the community or hospital as a part of the COVID-19 Biobank (Alfred Human Research and Ethics Committee - Project 182/20). Individuals consented to provide additional blood that was processed within 24 h of collection for plasma and peripheral blood mononuclear cells. Whole blood was centrifuged at 1000 x g for 10 min at 22°C-24°C then plasma removed within 5 mm of the buffy coat. Plasma aliquots were transferred to 2 mL cryovials for long term storage at –80°C. Plasma was thawed and immediately use for ELISA for CTNI, IFN-γ and BNP. Calcium was added to 10 mM to normalize calcium levels of citrated plasma and clot. The supernatant was removed (serum) and used for the hCO experiments (50% serum/50% WM). No viral RNA was detected in hCO treated with human COVID-19 serum.
Human ASSURE trial plasma
The design and rationale of the ASSURE trial is described in ClinicalTrials.gov identifier NCT01067820. Patients with established cardiovascular disease received 200 mg apabetalone daily for 26 weeks on top of standard of care, which included statins. Baseline and end of study EDTA plasma samples were analyzed using SOMAScan™.
SARS-CoV-2 stock production and titration at QIMR Berghofer
SARS-CoV-2 infection studies at QIMR Berghofer were conducted in a dedicated PC3 (BSL3) suite, with safety approval from the QIMR Safety Committee (P3600). The SARS-CoV-2 virus was isolated from a patient and was a kind gift from Queensland Health Forensic & Scientific Services, Queensland Department of Health; the isolate, hCoV-19/Australia/QLD02/2020; has been sequenced as is available at GISAID (https://www.gisaid.org/). Virus stock was generated by infecting Vero E6 cells (C1008, ECACC, Wiltshire, England; Sigma Aldridge, St. Louis, MO, USA) and after 3 days culture supernatant was clarified by centrifugation at 3000 x g for 15 min at 4°C, and was aliquoted and stored at −80°C. Virus titer was determined using standard TCID50 assay by infecting Vero E6 cells with 10-fold serial dilutions of virus stock and measuring cytopathic effect with titer calculation by the method of Spearman and Karber. Virus was determined to be mycoplasma free (La Linn et al., 1995) and fetal calf serum used for culture determined to be endotoxin free (Johnson et al., 2005).
SARS-CoV-2 stock production at The Doherty Institute
SARS-CoV-2 isolate CoV/Australia/VIC01/2020, provided by the Victorian Infectious Diseases Reference Laboratory (VIDRL) was amplified in Vero cells and stock vials were stored at −80°C. The amplified virus was sequenced to confirm that there were no mutations resulting from passage in Vero cells. All work with infectious virus was performed inside a biosafety cabinet, in a biosafety containment level 3 facility, and personnel wore powered air-purifying respirators (3M TR-315A VERSAFLO Cat# RPPKTR315A, from Safetyquip) or P2 masks. Vero cells were obtained from VIDRL and maintained in Minimum Essential Medium (MEM, Media Preparation Unit, Peter Doherty Institute) with 5% FBS, Penicillin-Streptomycin, GlutaMax (and 7.5ml HEPES (all ThermoFisher Scientific).
Method details
Cardiac differentiation
Cardiac differentiation was performed as previously described (Hudson et al., 2012; Mills et al., 2017; Voges et al., 2017). hPSCs were seeded on Matrigel-coated flasks at 2 × 104 cells/cm2 and cultured in mTeSR-1 for 4 days. To induce cardiac mesoderm, hPSCs were cultured in RPMI B27-medium (RPMI 1640 GlutaMAX+ 2% B27 supplement without insulin, 200 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Sigma) and 1% Penicillin/Streptomycin (ThermoFisher Scientific), supplemented with 5 ng/ml BMP-4 (RnD Systems), 9 ng/ml Activin A (RnD Systems), 5 ng/ml FGF-2 (RnD Systems) and 1 μM CHIR99021 (Stem Cell Technologies). Mesoderm induction required daily medium exchanges for 3 days. This was followed by cardiac specification using RPMI B27- containing 5 μM IWP-4 (Stem Cell Technologies) for another 3 days, and then further 7 days using 5 μM IWP-4 RPMI B27+ (RPMI1640 Glutamax + 2% B27 supplement with insulin, 200 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate and 1% Penicillin/Streptomycin) with media change every 2-3 days. For the final 2 days of differentiation, hPSCs were cultured in RPMI B27+. Harvest of differentiated cardiac cells involved enzymatic digestion, first in 0.2% collagenase type I (Sigma) containing 20% fetal bovine serum (FBS) in PBS (with Ca2+ and Mg2+) at 37°C for 1 h, and second in 0.25% trypsin-EDTA at 37°C for 10 minutes. Cells were filtered through a 100 μm mesh cell strainer (BD Biosciences), centrifuged at 300 x g for 3 min, and resuspended in α-MEM Glutamax, 10% FBS, 200 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate and 1% Penicillin/Streptomycin. Previous flow cytometry analysis indicated that differentiated cardiac cells were ~70% α-actinin+/CTNT+ cardiomyocytes, ~30% CD90 stromal cells (Voges et al., 2017).
Endothelial differentiation
Endothelial cell differentiation was performed following a protocol modified from Orlova et al. (2014). hPSCs were seeded onto Matrigel-coated T-25 or T-75 tissue culture flasks at the density 5 × 103 cells/cm2 and cultured in mTeSR-1 for 3 days. Mesoderm was induced with RPMI B27- (RPMI 1640 GlutaMAX+ 2% B27 supplement without insulin, 200 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Sigma) and 1% Penicillin/Streptomycin (ThermoFisher Scientific), and the small molecules 25 ng/ml Activin A (R&D systems), 30 ng/ml Bone morphogenic protein-4 (BMP4) (R&D systems), 1.5 μM CHIR99021 (Stemgent), and 50 ng/ml Vascular Endothelial Growth Factor type A (VEGF-A) (Peprotech) for 3 days (no media changes). Endothelial cell fate was further specified with RPMI B27- medium supplemented with 50 ng/ml VEGF-A and 10 μM SB431542 with media changes every 2 to 3 days until day 8.
FACS sorting endothelial cells
Endothelial cells were harvested after 8 d of differentiation using TrypLE (ThermoFisher Scientific). Single cells were separated using a 100 μm strainer and labeled with CD31 antibody (1:200, M082329-2, DAKO) at 4°C for 45 min followed by 30 min staining with a goat anti-mouse secondary antibody conjugated to AlexaFluor 488 or 555 (1:400, A-11001 and A-21422, ThermoFisher Scientific). Cells were analyzed using Becton Dickinson FACSAria II, gated on forward and side scatter. Single cells were identified and sorted based on CD31+ expression. CD31+ endothelial cells were expanded in EGM-2 (Lonza) in Matrigel flasks and cryopreserved.
hCO fabrication
hCO culture inserts were fabricated using SU-8 photolithography and PDMS molding (Mills et al., 2017). Differentiated cells were mixed at ratio of 20% endothelial cells and 80% cardiomyocytes/fibroblasts to form hCO. Acid-solubilized bovine collagen 1 (Devro) was salt balanced using 10x DMEM (ThermoFisher Scientific) and pH neutralized using 0.1M NaOH before combining with Matrigel and then the cell suspension on ice. Each hCO contained 5 × 104 cells, a final concentration of 2.6 mg/ml collagen I and 9% Matrigel. 3.5 μL of suspension was pipetted into the hCO culture insert and incubated at 37°C with 5% CO2 for 45 min in order to gel. After gelling, α-MEM GlutaMAX (ThermoFisher Scientific), 10% fetal bovine serum (FBS), 200 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Sigma) and 1% Penicillin/Streptomycin (ThermoFisher Scientific) was added. hCO were subsequently cultured in maturation media (MM) (Mills et al., 2017) with medium changes every 2 to 3 days for 5 days (7 days old hCO). To better approximate adult metabolic provisions a ‘weaning medium’ (WM) was utilized. hCO were cultured in WM containing 4% B27 – insulin, 5.5 mM glucose, 1 nM insulin, 200 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate, 1% P/S, 1% GlutaMAX (100x), 33 μg/mL aprotinin and 10 μM palmitate (conjugated to bovine serum albumin in B27) in DMEM without glucose, glutamine and phenol red (ThermoFisher Scientific) with media changes every 2-3 days.
Force analysis of hCO
The elasticity of the Heart Dyno poles enables the contractile properties to be determined via tracking pole deflection, which directly correlates with force (Mills et al., 2017). Videos of 10 s were made of each hCO with the Nikon ANDOR WD Revolution Spinning Disk microscope (magnification 4x). While imaging, hCO were incubated at 37°C, 5% CO2 to prevent changes in contractile behavior. For pacing, hCOs were electrically stimulated at 1 Hz with 5 ms square pulses with 20 mA current using a Panlab/Harvard Apparatus Digital Stimulator. Videos were then analyzed with a custom written MATLAB program (Mills et al., 2017). This facilitated the analysis of the contractile properties of the organoids and the production of time-force graphs (Mills et al., 2017). Moreover, data was obtained regarding additional important functional parameters including the contraction rate and the activation and relaxation time of the organoids.
Immunostaining of hCO
hCO were fixed with 1% paraformaldehyde (Sigma) for 1 h. Cells were stained with primary antibodies CD31 (1:200, M082329-2, DAKO), NG2 (1:200, 14-6504-82, ThermoFisher Scientific) and cardiac troponin T (1:400, ab45932, Abcam) in 5% FBS and 0.25% Triton X-100 Blocking Buffer at 4°C overnight on a rocker. Cells were washed twice for 1 h with Blocking Buffer and labeled with secondary antibodies goat anti-mouse IgG1 AlexaFluor 488 (1:400, A-21121), goat anti-mouse IgG2a AlexaFluor 555 (1:400, A-21137) and goat anti-rabbit IgG AlexaFluor 633 (1:400, A-21070) and Hoechst33324 (all ThermoFisher Scientific) at 4°C overnight on a rocker. Cells were again washed with Blocking Buffer twice for 1 h and mounted on microscope slides using ProLong Glass (ThermoFisher Scientific).
Phosphoproteomics
Phosphoproteomics experiments were performed with biological triplicates. Phosphopeptides were enriched from 20 pooled hCO, yielding approximately 100 μg of total protein per sample. The high-sensitivity EasyPhos workflow was employed as previously described (Humphrey et al., 2018). Briefly, pooled organoids were lysed in SDC buffer (4% Sodium deoxycholate, 100 mM Tris pH 8.5) and immediately heated for 5 min at 95°C. Lysates were cooled on ice, and sonicated with a tip-probe sonicator (50% output power, 30 s). An aliquot of lysate was taken and diluted 1:5 in 8 M Urea from which protein concentration was determined by BCA assay (Thermo Fisher Scientific). Aliquots corresponding to 100 μg of protein were subsequently diluted in SDC buffer into a 96-well deep-well plate, reduced and alkylated at 45°C for 5 min by the addition of 10 mM Tris (2-carboxyethyl)phosphine (TCEP)/40 mM 2-Chloroacetamide (CAA) pH 8, and digested by the addition of 1:100 Lys-C and Trypsin overnight at 37°C with agitation (1,500 rpm). After digestion phosphopeptides were enriched in parallel according to the EasyPhos workflow as described (Humphrey et al., 2018). Eluted phosphopeptides were dried in a SpeedVac concentrator (Eppendorf) and resuspended in MS loading buffer (0.3% TFA/2% acetonitrile) prior to LC-MS/MS measurement.
LC-MS/MS Measurement
Phosphopeptides were loaded onto a 40 cm column fabricated in-house with 75 μM inner diameter fused silica packed with 1.9 μM C18 ReproSil particles (Dr. Maisch GmBH). A column oven (Sonation) was used to maintain column temperature at 60°C, and a U3000 HPLC system (Dionex, Thermo Fisher Scientific) was connected to a Q Exactive HF X benchtop Orbitrap mass spectrometer (Thermo Fisher Scientific) with a NanoSpray Flex ion source (Thermo Fisher Scientific). For all samples, peptides were separated using a binary buffer system of 0.1% (v/v) formic acid (buffer A) and 80% (v/v) acetonitrile/0.1% (v/v) formic acid (buffer B). Peptides were eluted at a flow rate of 400 nl/min and separated with a gradient of 3 – 19% buffer B over 40 minutes, followed by 19 – 41% buffer B over 20 minutes, and peptides were analyzed with a full scan (350 – 1,400 m/z; R = 60,000 at 200 m/z) at a target of 3e6 ions, followed by up to ten data-dependent MS2 scans using HCD (target 1e5; max. IT 50 ms; isolation window 1.6 m/z; NCE 27%; min. AGC target 2e4), detected in the Orbitrap mass analyzer (R = 15,000 at 200 m/z). Dynamic exclusion (30 s) and Apex trigger (2 to 4 s) were enabled.
MS data processing
RAW MS data was processed in the MaxQuant software environment (Cox and Mann, 2008) (version 1.6.0.9), searching against the Human UniProt Reference database (December 2019 release), using default settings with the addition of ‘Phospho(STY)’ as a variable modification and ‘Match between runs’ switched on for all analyses. Data analysis was performed using the Perseus software package (Tyanova and Cox, 2018).
Single nuclei RNA-sequencing of hCO
Pooled hCO (~40) were homogenized in 4 mL lysis buffer (300 mM sucrose, 10 mM Tris-HCl (pH = 8), 5 mM CaCl2, 5 mM magnesium acetate, 2 mM EDTA, 0.5 mM EGTA, 1 mM DTT)(all Sigma-Aldrich) with 30 strokes of a dounce tissue grinder (Wheaton). Large pieces of hCO were allowed to settle and homogenate was passed through pre-wetted 40 μm cell strainers (Becton Dickinson). Remaining hCO material in the douncer was resuspended in 4 mL and the douncing and filtering steps were repeated twice. All steps of the homogenization were performed on ice. The filtered homogenate was centrifuged at 1500 x g for 5 min at 4°C. Nuclei pellets were then re-suspended in PBS. A fraction of resuspended nuclei were then stained with Hoechst33324 nuclear stain (1:500 dilution) and counted on a haemocytometer under fluorescent microscope.
The nuclei were then re-centrifuged (1500 x g for 5 min at 4°C) and resuspended at a density to load ~5,000 nuclei per sample. Cells were loaded into the Chromium Controller (10X Genomics) for gel bead emulsion (GEM) formation. Library preparation was conducted according to the manufacturer’s recommended protocol using the Chromium Next GEM Single Cell 3′ GEM, Library & Gel Bead Kit v3.1. Libraries were sequenced on the NextSeq 500/550 v2 (Illumina) with 150 bp reads and were sequenced to ~250,000 reads per cell.
Raw fastq reads for each sample were processed using CellRanger v3.1.0. Default options were used with CellRanger and a custom made pre mRNA reference using GRCh38 v3.0.0 was used to map the reads and for count quantification with the CellRanger counts tool. Following this, the counts were then aggregated together to create a single matrix that contained all the samples. Reads from the single nuclei sequencing data were aligned to a human pre-mRNA GRCh38 reference genome. All pre-processing and filtering steps of the datasets were subsequently carried out via the Python package Scanpy (https://scanpy.readthedocs.io/en/stable/) (Wolf et al., 2018). Briefly, there was an initial filtering step for genes that are expressed in 3 or more cells and cells with at least 200 detected genes, subsequently removing cells displaying high expression of mitochondrial genes using a cut-off of 4%. We then filtered out cells that had a count depth with a threshold of under 2,000 and higher than 40,000 to remove debris and potential doublets. Gene expression was subsequently normalized for each cell by total expression, scaled by 10,000 and log scaled. Highly variable genes were then identified for clustering. Leiden clustering with an initial resolution of 0.2 was performed to identify clusters within the data. A published human cardiac snRNA-seq dataset was then used to identify overlap of marker gene sets for main human heart cell types (e.g., cardiomyocytes, fibroblasts, epicardium, pericytes, etc) with the clusters of our dataset, and were labeled accordingly (Tucker et al., 2020). Further refined clustering was carried out on specific clusters that showed overlap of more than one of the various heart cell types. Visualization of the datasets was primarily carried out using nonlinear dimensionality reduction UMAP plots (Becht et al., 2018). In the snRNA-seq we note the lower than expected percentage of non-myocytes and the loss of endothelial cells (Mills et al., 2017; Voges et al., 2017), indicating the protocol requires further optimization for hCO samples.
hCO comparison to bulk nuclei RNA sequencing data for PCA
Nuclear RNA-seq dataset generated from sorted cardiomyocyte nuclei at two stages (fetal and adult) was obtained from BioProject ID: PRJNA353755 (Gilsbach et al., 2018). RNA-seq dataset was mapped to the human genome (hg38) using STAR aligner (version 2.7.3a). Annotations and genome files (hg38) were obtained from Ensembl (release 102). Uniquely mapped reads were counted across genes with a program in Bioconductor R (Huber et al., 2015) package, featureCounts (version 2.0.1) (Liao et al., 2014). Subsequent analyses of the count data were performed in the R statistical programming language with the Bioconductor packages edgeR (Robinson et al., 2010) and the annotation package org. Hs.eg.db. In this dataset, only genes with > 0.5 counts per million (CPM) in at least 4 samples were retained for statistical analysis. Additionally, ribosomal and mitochondrial genes as well as pseudogenes, and genes with no annotation (Entrez Gene identification) were removed before normalization and statistical analysis.
Principal component analysis (PCA) performed using the intersection of the 25% most highly variable genes of the snRNA-seq dataset and genes expressed in the bulk samples. Each of the two datasets were log transformed and scaled separately before running the dimensionality reduction method.
Pro-inflammatory stimulation of hCO
Cytokines (human) and factors were added individually and in combinations in WM: 100 ng/ml TNF, 10 ng/ml IL-1β, 100 ng/ml IFN-γ, 100 ng/ml IL-6, 100 ng/ml IL-17A, 100 ng/ml G-CSF (Amgen), 10 μg/ml poly(I:C) (HMW, Invivogen) or 1 μg/ml LPS (from Escherichia coli strain 0127:B8, Sigma) (all Peprotech unless noted). Additional concentrations were performed for the dose-response curves for TNF, IL-1β, IFN-γ and poly(I:C) as indicated. The function of hCO was determined before addition of these factors as a baseline (time 0 h) and any changes normalized to the original baseline and to the control. The medium was not exchanged unless noted.
Drug screening
Compounds were sources from MedChem Express (unless noted) and dissolved at 10 mM in DMSO and vehicle controls used. A larger batch of INCB054329 was sourced from Selleckchem. The following compounds were used at 2 or 3 doses previously shown to have in vitro efficacy as per the referenced papers for: JQ-1 (Selleckchem), INCB054329, ABBV-744, ruxolitinib, baricitinib, flavopiridol, SEL120-34A, BI-1347, and paroxetine hydrochloride. Additional compounds tested were molibresib, alobresib, and apabetalone. For some experiments apabetalone and RVX-2157 were sent blinded by Resverogix. Compounds were given at the time of pro-inflammatory factor addition, except for experiments with baricitnib and INCB054329 where recovery of function was also assessed by addition 24 h following addition of inflammatory factors.
Linear regression of cytokine storm responses
To determine factor effects, second order OLS linear regression was performed across all relevant samples using binary predictors (cytokine presence/absence) with force, relaxation/activation times as the outcome variables. p values were determined using two-tailed t tests. Normality (Shipiro-Wilks), heteroskedasticity (Breusch-Pagan), linearity (Harvey-Collier), multicollinearity (condition no.) and skewness/kurtosis (Jarque-Bera) were all checked with the respective tests. The coefficient of determination, Rˆ2 (adjusted), was used to determine goodness of fit.
SARS-CoV-2 K18-hACE2 mouse infection model
Female K18-hACE2 mice were lightly anesthetized using isoflurane and 50 μl of SARS-CoV-2 at 5 × 104 TCID50 per mouse was administered via intranasal inoculation (i.n.). On 1, 2 and 3 dpi, INCB054329 or placebo control was administered via oral gavage. Mice were randomized and received either vehicle 30% (m/v) Kolliphor 15 HS (Sigma) in PBS or 2 mg per 30 g mouse body weight of INCB054329 at 20 mg/ml in the Kolliphor solution (66.7 mg/kg). At 4 or 5 d.p.i., mice were euthanized by cervical dislocation and heart and lung tissue was fixed in 10% formalin for histology. At 4 d.p.i. lungs or hearts were homogenized in TRIzol for RNA extraction and stored at −80°C.
Bulk RNA-seq from SARS-CoV-2 K18-hACE2 mouse infection model
Illumina Stranded Total RNA Prep with Ribo-Zero Plus kits were used to prepare total RNA for sequencing. Libraries were sequenced on the NextSeq 500/550 v2 (Illumina) with 150 bp reads and were sequenced to ~60,000,000 reads per sample. Sequence reads were trimmed for adaptor sequences using Cutadapt version1.9 (Martin, 2011) and aligned using STAR version 2.5.2a (Dobin et al., 2013) to the Mus musculus GRCm38 assembly with the gene, transcript, and exon features of Ensembl (release 102) gene model, and the SARS-CoV-2 Ensembl genome assembly ASM985889v3. Quality control metrics were computed using RNA-SeQC version 1.1.8 (DeLuca et al., 2012) and expression was estimated using RSEM version 1.2.30 (Li and Dewey, 2011). Protein-coding genes with > 5 CPM in ≥ 5 samples were kept for further analysis. Trimmed mean of M-values (TMM) normalization and differential expression analysis were performed using the R package edgeR (Robinson et al., 2010). The glmQLFit() function was used to fit a quasi-likelihood negative binomial generalized log-linear model to the read counts for each gene. Using the glmQLFTest() function, we conducted gene-wise empirical Bayes quasi-likelihood F-tests for a given contrast. Differentially expressed genes (DEGs) were determined using absolute log2 fold change (logFC) > 0.5 and a false discovery rate (FDR) < 0.05. The function prcomp() was used for principal component analysis (PCA). To estimate SARS-CoV-2 replication levels, sequence reads were aligned to SARS-CoV-2 only, and samtools (Li et al., 2009) version 1.9 was used to estimate the mapping rate of the reads to the viral genes.
Comparison of different RNA-seq data
KEGG and Encode TF analyses was performed using Enrichr (Kuleshov et al., 2016). BioMart Ensembl (release 102) was used to obtain the mouse to human orthologs used in comparisons. Networks were generated through the use of Ingenuity Pathway Analysis (IPA) on DEGs (QIAGEN Inc., https://digitalinsights.qiagen.com/products/ingenuity-pathway-analysis). Up-Stream Regulators enriched in differentially expressed genes (no logFC cutoff, FDR < 0.05) in direct and indirect interactions were investigated by performing Core Analysis using (QIAGEN).
Quantitative RT-PCR
RNA was extracted using QIAGEN RNAeasy Micro Kits (QIAGEN) or Trizol. cDNA synthesis using Superscript III (ThermoFisher Scientific) was carried out as per manufacturer’s instructions. Final primer concentration of 200-250 nM was used and gene expression was assessed over 40 cycles on an Applied Biosciences Quant Studio 5. 18S (for viral infection studies) or HPRT1 or hprt (for gene expression) were used as internal controls.
Human
RNA18S5 Fwd GCTGAGAAGACGGTCGAACT Rev CGCAGGTTCACCTACGGAAA
HPRT1 Fwd AACCTCTCGGCTTTCCCG Rev TCACTAATCACGACGCCAGG
LGALS3BP Fwd TGTGGTCTGCACCAATGAAAC Rev CTGCACATTCACGCTGATGG
BRD4 Fwd CTGACAGCGAAGACTCCGA Rev GTGGTGATGATGGTGCTTCTTC
Mouse
Hprt Fwd AGGCCAGACTTTGTTGGATTTGAA Rev CAACTTGCGCTCATCTTAGGCTTT
Lgals3bp Fwd TCTCTTGCTCCCAGGGTTG Rev CCTGGAACCAGCAAGAACAC
SARS-CoV-2
E-Gene Fwd ACAGGTACGTTAATAGTTAATAGCGT Rev ATATTGCAGCAGTACGCACACA
Mouse LPS cytokine storm model
LPS (from Escherichia coli strain 0127:B8, Sigma) suspended in PBS was injected intraperitoneally into mice at 0.6 mg per 30 g mouse body weight for inflammatory cytokine, gene expression and survival studies and 1 mg was used for cardiac function studies. After the injection of LPS mice were randomized and received either vehicle 30% (m/v) Kolliphor 15 HS (Sigma) in PBS or 2 mg per 30 g mouse body weight of INCB054329 at 20 mg/ml in the Kolliphor solution (66.7 mg/kg). Treatment groups were blinded. For the experiments, mice were closely monitored and checked hourly for signs of sepsis.
Mouse LPS plasma cytokine assays
Serum cytokine levels (Ifn-γ, Il-1β and Tnf) were determined with a CBA Flex Set Multiplex Cytokine Bead Array (BD Biosciences).
Cardiac function in vivo
Cardiac function was assessed using a Vevo 2100 ultrasound system fitted with a MS550D transducer, which has a 40 MHz center frequency (Fujifilm Visualsonics). Depilated mice were anaesthetized by isoflurane inhalation (1.5% at 1 L oxygen / min) delivered via a nose cone, kept warm on a heated stage, with respiration and heart rate monitored on ECG pads. B Mode images of the left ventricle were obtained from three short-axis views (proximal, mid and distal positions) and one parasternal long-axis view. Cardiac function parameters were calculated using the Simpson’s tool in Vevolab analysis software v3.2.6 (Fujifilm Visualsonics). Briefly, the endocardial areas were traced from all three short-axis views, and the length of the ventricle was determined, in systole and diastole. These measurements were used to calculate ejection fraction.
ELISA
Human CNTI ELISA (RayBiotech), IFN-γ (RnD Systems) and BNP ELISA (Abcam) was used as per manufacturer’s instructions.
Immunoblotting
Protein from 2D hPSC-cardiac cell cultures was extracted using RIPA lysis buffer supplemented with protease inhibitor cocktail (Roche). Protein lysate concentration was estimated using a BCA assay (ThermoFisher Scientific). 15 μg protein was resolved on 4%–12% Bis-Tris polyacrylamide gel (Invitrogen) at 200V for 20 min and then transferred at 20 V for 1 h onto polyvinylidene difluoride (PVDF) membrane as per manufacturer’s recommendations. After 1 h blocking using a 1:1 mix of LI-COR Odyssey Blocking Buffer (LI-COR Biotechnology) and PBS, membranes were incubated overnight on a platform shaker with primary antibodies for ACE2 (1:200, R&D Systems, AF933) and GAPDH (1:1000, Cell Signaling Technologies, 97166S). Membranes were washed 5 times 3 minutes in PBS with 0.5% Tween, prior to incubation with IRDye® secondary antibodies (1:10000 for IRDye® 800CW Goat anti-Mouse IgG Secondary Antibody, 926-32210, and 680RD Donkey anti-Goat IgG Secondary Antibody, LI-COR Biotechnology, 925-68074) for 1 h at room temperature. Membranes were washed thoroughly (5 × 3 min in PBS + 0.5% Tween) and were then imaged on a LI-COR Odyssey® CLx. Densitometry was performed using ImageStudio Lite (version 4).
Flow Cytometry for ACE2 and SARS-CoV-2 spike binding assays
hPSC-cardiac cells were differentiated as above, then plated at 100,000 per cm2 on gelatin coated plates and cultured for 5 days prior to infection experiments. For 2D cultured hPSC-CM, cells were pre-treated with MM with or without the indicated compounds or DMSO as a vehicle control for 3 days prior to assays. Cells were was 2 x with PBS and detached using 0.25% Trypsin/EDTA (ThermoFisher Scientific) for ~10-15 min at 37°C. This was then neutralized with equivolume 3% bovine serum albumin (Sigma) in PBS (Binding Buffer). Cells were then centriguged at 300 x g for 3 min and the supernatant removed. Cells were then incubated under different conditions.
For ACE2 assays the following was used for control, 1:200 Goat IgG Alexa Fluor 647-conjugated antibody, and assay 1:200 anti-human ACE2 AlexFluor 647 conjugated antibody and 1:200 anti-human CD90 (all RnD Systems) and were incubated for 60 min at 4°C in Binding Buffer. The cells were then washed in Binding Buffer, centrifuged at 300 x g for 3 min and supernatant removed. Both conditions were then incubated with 1:400 goat anti-mouse IgG secondary antibody conjugated to Alexa Fluor 555 (ThermoFisher Scientific) in Binding Buffer for 45 min at 4°C. The cells were then washed in Binding Buffer, centrifuged at 300 x g for 3 min and supernatant removed.
For SARS-CoV-2 binding assays the following was used for control, Binding Buffer only, and assay 1 μg rSARS-CoV-2 Spike RB (per ~100,000 cells) and 1:200 anti-human CD90 (all RnD Systems) and were incubated for 60 min at 4°C in binding buffer. The cells were then washed in Binding Buffer, centrifuged at 300 x g for 3 min and supernatant removed. Both conditions were then incubated with 1:400 F(ab’)2-goat anti-human IgG Fc secondary antibody conjugated to Alexa Fluor 488 and 1:400 goat anti-mouse IgG secondary antibody conjugated to Alexa Fluor 555 (both ThermoFisher Scientific) in Binding Buffer for 45 min at 4°C. The cells were then washed in binding buffer, centrifuged at 300 x g for 3 min and supernatant removed.
For flow cytometry cells were resuspended in 300 μl Binding Buffer, put through a 100 μm cell strainer to remove any clumps. Cells were assessed on a BD LSRFortessa Flow Cytometer, gated on FSC-A/SSC-A and then remove doublets using FSC-W/H, and CD90 assessed using YG586/15, ACE2 using R670/14 and SARS-CoV-2 spike protein B530/30. Negative controls were used to draw gates.
hPSC-CM SARS-CoV-2 infection at QIMR Berghofer
hPSC-cardiac cell infection
hPSC-cardiac cells were differentiated as above, then plated at 100,000 per cm2 on gelatin coated plates and cultured for 5 days prior to infection experiments. For 2D cultured hPSC-CM, cells were pre-treated with 1 mL of MM with or without the indicated compounds or DMSO as a vehicle control for 3 days prior to infection. Media was removed before infecting cells with SARS-CoV-2 at MOI 0.01 for 1 h at 37°C. Cells were washed 3 x with MM and replaced with 1 mL of MM with compounds or DMSO. For intracellular RNA cells were washed 3x with PBS before harvesting RNA in Trizol. For supernatant experiments 500 μl of supernatants were harvested each day and replaced with 500 μl of media. Supernatants were frozen at −80°C until they were titerd. Titration was performed in Vero cell monolayers in 96 well plates: inoculated serial 10-fold dilutions in quadruplicate and incubated for 4 days. Cytopathic effect recorded and virus titer in log10 TCID50/ml recorded.
Imaging
For immunostaining cells or hCO were fixed in 4% paraformaldehyde and stained. Cells were stained with primary antibodies Nucleocaspid protein SARS-CoV-2 (1:200, 40143-MM05, Sino Biological) and cardiac troponin T (1:400, ab45932, Abcam) in Blocking Buffer at 4°C for 2 h for 2D or overnight for hCO on a rocker. Cells were washed twice with Blocking Buffer and labeled with secondary antibodies goat anti-mouse IgG AlexaFluor 488 (1:400, A-11001) and goat anti-rabbit IgG AlexaFluor 555 (1:400, A-21428) and Hoechst3332 (all ThermoFisher Scientific) at 4°C for 2 h for 2D or overnight for hCO on a rocker. Cells were again washed with Blocking Buffer twice and then put into PBS and were imaged using a Leica Thunder microscope.
Quantitative RT-PCR
Performed as described above.
hPSC-CM SARS-CoV-2 infection experiments at The Peter Doherty Institute
hPSC-CM infection
The female human embryonic stem cell line HES3 NKX2-5eGFP/w was used for viral infection studies in 2D monolayer cultures (Elliott et al., 2011). Cardiac cells were differentiated as previously described (Anderson et al., 2018), frozen at day 10 following differentiation and stored at −80°C. Cardiac cells were subsequently thawed in RPMI+B27 media (RPMI 1640 supplemented with 2% B-27 Supplement minus vitamin A, 1% GlutaMAX and 1% Penicillin/Streptomycin- All from ThermoFisher Scientific) with Rock inhibitor (Selleck Chemicals) for 24 h. Cardiac cells were then maintained in RPMI + B27 media for an additional 2 days, enriched for cardiomyocytes with lactate purification media - DMEM, no glucose, no glutamine, no phenol red supplemented with 1% GlutaMAX and 1% Penicillin/Streptomycin (Thermo Fisher Scientific) and 5 mM Sodium L-Lactate (Sigma Aldrich) for 2 days, and subsequently cultured in MM from day 15 to day 23 post differentiation prior to viral infection.
The media was removed from cultures of cardiac myocytes and Vero cells in 24 well plates. Vero cell plates were washed with 1ml of serum free media per well. Media was removed, wells were inoculated with 104 TCID50 of virus (MOI = 0.01) in 100 μL serum free media and incubated for 1 h at room temperature. The inoculum for Vero cells incorporated TPCK-treated Trypsin (Worthington Biochemical Corporation) at 1 μg/ml. Following virus adsorption, the inoculum was removed and replaced with 500 μL media. After ~10 minutes, this was removed and stored at −80°C as the day 0 sample. 500 μL per well of media was replenished and plates were incubated at 37°C in 5% CO2. Each day from day 1 through 6 post-infection, 500 μL of culture supernatant was harvested and replenished with 500 μL of media. Supernatants were stored at −80°C till they were titrated.
Virus titration
The amount of infectious virus present in the samples was assayed in Vero cell monolayers in 96 well plates. Samples inoculated into 4 wells were diluted serially in 10-fold dilutions in serum free media containing 1 μg/ml TCPK-treated Trypsin and incubated for at 37°C in 5% CO2. Cytopathic effect was scored on day 4 post-infection and virus titer expressed in log10 TCID50/ml. The lower limit of detection was 1.7 log10 TCID50/ml.
SOMAscan Proteomic Analysis
SOMAScan™ proteomic technology uses Somamers as an affinity reagent (Somalogic Inc.). Plasma samples from 47 patients from each group that received apabetalone or placebo in the ASSURE trial were analyzed and LGALS3BP quantified (Nicholls et al., 2016).
Quantification and statistical analysis
Statistics were performed using GraphPad Prism v8 unless noted with the appropriate tests outlined in the Figure legends.
hCO force experiments were performed on quality controlled hCO (proper formation around the poles, non-arrhythmic, no broken arms, no necking (Mills et al., 2017) across multiple experiments with multiple cell line combinations to ensure reproducibility. Automated force analysis removes the requirement for blinding of hCO experiments. Personnel performing the animal experiments and analyses were blinded to the conditions or treatments.
Acknowledgments
We thank Clive Berghofer and Lyn Brazil (and others) for their generous philanthropic donations; Australian National Fabrication Facility Queensland Node for the fabrication of the Heart-Dyno molds; Dr. I. Anraku (QIMR Berghofer) for assistance in managing the PC3 (BSL3) facility; Dr. Alyssa Pye and Mr. Fredrick Moore (Queensland Health, Brisbane) for providing the SARS-CoV-2 virus; Grace Chojnowski and Michael Rist (QIMR Berghofer) for FACS; Tam Nguyen and Nigel Waterhouse (QIMR Berghofer) for microscopy; Nadine Shultz and Paul Collins (QIMR Berghofer) for the sequencing; Scott Wood, Pamela Mukhopadhyay, John Pearson, Nic Waddell, and Ross Koufariotis for (QIMR Berghofer) bioinformatics assistance; Ben Crossett, Angela Connolly, and Jens Altvater (University of Sydney) for mass spectrometry assistance; Compounds Australia (http://www.griffith.edu.au/science-aviation/compounds-australia) for providing access to compounds; Edouard Stanley (Murdoch Children’s Research Institute) for the RM3.5 iPSC line; Translational Research Institute for Preclinical Imaging and Biological Resources Facility; and Sydney Mass Spectrometry core research facility at the University of Sydney. This work was supported by National Health and Medical Research Council of Australia (to J.E.H., M.J.S., C.R.E., T.B., and A.S.), Heart Foundation of Australia (to J.E.H.), QIMR Berghofer (to J.E.H.), The Stafford Fox Foundation (to E.R.P.), the Royal Children's Hospital Foundation (to E.R.P.), Australian Research Council Strategic Initiative in Stem Cell Science (Stem Cells Australia) (to E.R.P. and J.E.H.), and the Medical Research Future Fund (MRFF9200008 to J.E.H., T.B., M.J.S., K.P.A.M., C.R.E., and E.R.P., APP1132519 and APP1173958 to M.J.S., and APP1173880 to A.S.). Queensland Health supported this research project. The Murdoch Children’s Research Institute is supported by the Victorian Government’s Operational Infrastructure Support Program. T.B. is a member of ImmunoSensation2 supported by the DFG (German Research Foundation) under Germany’s Excellence Strategy EXC2151 390873048. This project received support from Dynomics Snow Medical Fellowship (to J.E.H.).
Author contributions
Investigation, R.J.M., S.J.H., G.A.Q.-R., S.K., M.L., L.T.R., R.R., K.A.S., B.W.C.T., D.J.R., T.L., S.R.F., W.Z., L.L., C.M.-K., D.A.-B., K.K., T.B., and J.E.H.; Methodology, L.D., H.K.V., L.T.R., and E.M.; Formal analysis, R.J.M., S.J.H., P.R.J.F., G.A.Q.-R., M.L., N.R.M., D.G., L.F., E.K., R.L.J., T.D., C.B., B.G., D.T., R.R., K.S., E.R.P., T.B., and J.E.H.; Resources, J.M., C.H., D.G., L.F., S.J.N., J.J., M.S., N.C.W.W., and E.K.; Conceptualization, R.J.M., S.J.H., M.J.S., C.R.E., K.P.A.M., D.G., E.K., T.B., D.A.E., K.S., A.S., D.E.J., and J.E.H.; Supervision, R.J.M., S.J.H., M.J.S., C.R.E., K.P.A.M., D.G., L.F., E.K., R.R., K.S., E.R.P., A.S., T.B., D.E.J., and J.E.H.; Writing – original draft, R.J.M., S.J.H., D.E.J., and J.E.H.; Writing – review & editing, all authors.
Declaration of interests
R.J.M., J.E.H., G.A.Q.-R., D.M.T., and E.R.P. are co-inventors on patents relating to cardiac organoid maturation and cardiac therapeutics. J.E.H. is co-inventor on licensed patents for engineered heart muscle. R.J.M., E.R.P., D.M.T., B.G., and J.E.H. are co-founders, scientific advisors, and stockholders in Dynomics. D.M.T. and B.G. are employees of Dynomics. C.H., D.G., L.F., J.J., M.S., N.C.W.W., and E.K. are employees of Resverlogix. S.J.N. received honoraria and research support from Resverlogix. QIMR Berghofer Medical Research Institute filed a patent on the use of BETi.
Published: March 16, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.03.026.
Supplemental information
References
- Anand P., Brown J.D., Lin C.Y., Qi J., Zhang R., Artero P.C., Alaiti M.A., Bullard J., Alazem K., Margulies K.B., et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013;154:569–582. doi: 10.1016/j.cell.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D.J., Kaplan D.I., Bell K.M., Koutsis K., Haynes J.M., Mills R.J., Phelan D.G., Qian E.L., Leitoguinho A.R., Arasaratnam D., et al. NKX2-5 regulates human cardiomyogenesis via a HEY2 dependent transcriptional network. Nat. Commun. 2018;9:1373. doi: 10.1038/s41467-018-03714-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antolic A., Wakimoto H., Jiao Z., Gorham J.M., DePalma S.R., Lemieux M.E., Conner D.A., Lee D.Y., Qi J., Seidman J.G., et al. BET bromodomain proteins regulate transcriptional reprogramming in genetic dilated cardiomyopathy. JCI Insight. 2020;5:138687. doi: 10.1172/jci.insight.138687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunachalam P.S., Wimmers F., Mok C.K.P., Perera R.A.P.M., Scott M., Hagan T., Sigal N., Feng Y., Bristow L., Tak-Yin Tsang O., et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. 2020;369:1210–1220. doi: 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auguste G., Rouhi L., Matkovich S.J., Coarfa C., Robertson M.J., Czernuszewicz G., Gurha P., Marian A.J. BET bromodomain inhibition attenuates cardiac phenotype in myocyte-specific lamin A/C-deficient mice. J. Clin. Invest. 2020;130:4740–4758. doi: 10.1172/JCI135922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancerek J., Poss Z.C., Steinparzer I., Sedlyarov V., Pfaffenwimmer T., Mikulic I., Dölken L., Strobl B., Müller M., Taatjes D.J., Kovarik P. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity. 2013;38:250–262. doi: 10.1016/j.immuni.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E., McInnes L., Healy J., Dutertre C.-A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- Chen C., Li H., Hang W., Wang D.W. Cardiac injuries in coronavirus disease 2019 (COVID-19) J. Mol. Cell. Cardiol. 2020;145:25–29. doi: 10.1016/j.yjmcc.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran A.G., Conery A.R., Sims R.J., 3rd Bromodomains: a new target class for drug development. Nat. Rev. Drug Discov. 2019;18:609–628. doi: 10.1038/s41573-019-0030-7. [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Del Valle D.M., Kim-Schulze S., Huang H.-H., Beckmann N.D., Nirenberg S., Wang B., Lavin Y., Swartz T.H., Madduri D., Stock A., et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020;26:1636–1643. doi: 10.1038/s41591-020-1051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca D.S., Levin J.Z., Sivachenko A., Fennell T., Nazaire M.D., Williams C., Reich M., Winckler W., Getz G. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–1532. doi: 10.1093/bioinformatics/bts196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E.W., Csordas A., Sun Z., Jarnuczak A., Perez-Riverol Y., Ternent T., Campbell D.S., Bernal-Llinares M., Okuda S., Kawano S., et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017;45(D1):D1100–D1106. doi: 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Q., McMahon S., Anand P., Shah H., Thomas S., Salunga H.T., Huang Y., Zhang R., Sahadevan A., Lemieux M.E., et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med. 2017;9:eaah5084. doi: 10.1126/scitranslmed.aah5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott D.A., Braam S.R., Koutsis K., Ng E.S., Jenny R., Lagerqvist E.L., Biben C., Hatzistavrou T., Hirst C.E., Yu Q.C., et al. NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat. Methods. 2011;8:1037–1040. doi: 10.1038/nmeth.1740. [DOI] [PubMed] [Google Scholar]
- Evron T., Daigle T.L., Caron M.G. GRK2: multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol. Sci. 2012;33:154–164. doi: 10.1016/j.tips.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre E.J., McDaniel K.F., Albert D.H., Mantena S.R., Plotnik J.P., Wilcox D., Zhang L., Bui M.H., Sheppard G.S., Wang L., et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature. 2020;578:306–310. doi: 10.1038/s41586-020-1930-8. [DOI] [PubMed] [Google Scholar]
- Feldman A.M., Combes A., Wagner D., Kadakomi T., Kubota T., Li Y.Y., McTiernan C. The role of tumor necrosis factor in the pathophysiology of heart failure. J. Am. Coll. Cardiol. 2000;35:537–544. doi: 10.1016/s0735-1097(99)00600-2. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., Morse E.M., Keates T., Hickman T.T., Felletar I., et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliano-Jucá T., Travison T.G., Kantoff P.W., Nguyen P.L., Taplin M.-E., Kibel A.S., Huang G., Bearup R., Schram H., Manley R., et al. Androgen Deprivation Therapy Is Associated With Prolongation of QTc Interval in Men With Prostate Cancer. J. Endocr. Soc. 2018;2:485–496. doi: 10.1210/js.2018-00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilan O., Rioja I., Knezevic K., Bell M.J., Yeung M.M., Harker N.R., Lam E.Y.N., Chung C.W., Bamborough P., Petretich M., et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science. 2020;368:387–394. doi: 10.1126/science.aaz8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilsbach R., Schwaderer M., Preissl S., Grüning B.A., Kranzhöfer D., Schneider P., Nührenberg T.G., Mulero-Navarro S., Weichenhan D., Braun C., et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 2018;9:391. doi: 10.1038/s41467-017-02762-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal P., Choi J.J., Pinheiro L.C., Schenck E.J., Chen R., Jabri A., Satlin M.J., Campion T.R., Jr., Nahid M., Ringel J.B., et al. Clinical Characteristics of Covid-19 in New York City. N. Engl. J. Med. 2020;382:2372–2374. doi: 10.1056/NEJMc2010419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S., Carter R.L., Grisanti L.A., Koch W.J., Tilley D.G. Impact of paroxetine on proximal β-adrenergic receptor signaling. Cell. Signal. 2017;38:127–133. doi: 10.1016/j.cellsig.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T., Fan Y., Chen M., Wu X., Zhang L., He T., Wang H., Wan J., Wang X., Lu Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19) JAMA Cardiol. 2020;5:811–818. doi: 10.1001/jamacardio.2020.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A., Madhavan M.V., Sehgal K., Nair N., Mahajan S., Sehrawat T.S., Bikdeli B., Ahluwalia N., Ausiello J.C., Wan E.Y., et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020;26:1017–1032. doi: 10.1038/s41591-020-0968-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M.H., Mani R., Engelhardt H., Impagnatiello M.A., Carotta S., Kerenyi M., Lorenzo-Herrero S., Böttcher J., Scharn D., Arnhof H., et al. Selective and Potent CDK8/19 Inhibitors Enhance NK-Cell Activity and Promote Tumor Surveillance. Mol. Cancer Ther. 2020;19:1018–1030. doi: 10.1158/1535-7163.MCT-19-0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horby P., Lim W.S., Emberson J., Mafham M., Bell J., Linsell L., Staplin N., Brightling C., Ustianowski A., Elmahi E., et al. Effect of Dexamethasone in Hospitalized Patients with COVID-19: Preliminary Report. medRxiv. 2020 doi: 10.1101/2020.06.22.20137273. [DOI] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T., et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015;12:115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson J., Titmarsh D., Hidalgo A., Wolvetang E., Cooper-White J. Primitive cardiac cells from human embryonic stem cells. Stem Cells Dev. 2012;21:1513–1523. doi: 10.1089/scd.2011.0254. [DOI] [PubMed] [Google Scholar]
- Humphrey S.J., Azimifar S.B., Mann M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat. Biotechnol. 2015;33:990–995. doi: 10.1038/nbt.3327. [DOI] [PubMed] [Google Scholar]
- Humphrey S.J., Karayel O., James D.E., Mann M. High-throughput and high-sensitivity phosphoproteomics with the EasyPhos platform. Nat. Protoc. 2018;13:1897–1916. doi: 10.1038/s41596-018-0014-9. [DOI] [PubMed] [Google Scholar]
- Johnson B.J., Le T.T., Dobbin C.A., Banovic T., Howard C.B., Flores Fde.M., Vanags D., Naylor D.J., Hill G.R., Suhrbier A. Heat shock protein 10 inhibits lipopolysaccharide-induced inflammatory mediator production. J. Biol. Chem. 2005;280:4037–4047. doi: 10.1074/jbc.M411569200. [DOI] [PubMed] [Google Scholar]
- Kim S.Y., Zhang X., Schiattarella G.G., Altamirano F., Ramos T.A.R., French K.M., Jiang N., Szweda P.A., Evers B.M., May H.I., et al. Epigenetic Reader BRD4 (Bromodomain-Containing Protein 4) Governs Nucleus-Encoded Mitochondrial Transcriptome to Regulate Cardiac Function. Circulation. 2020;142:2356–2370. doi: 10.1161/CIRCULATIONAHA.120.047239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha T., Knight D.S., Razvi Y., Kumar K., Vimalesvaran K., Thornton G., Patel R., Chacko L., Brown J.T., Coyle C., et al. Patterns of myocardial injury in recovered troponin-positive COVID-19 patients assessed by cardiovascular magnetic resonance. Eur. Heart J. 2021:ehab075. doi: 10.1093/eurheartj/ehab075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T., McTiernan C.F., Frye C.S., Slawson S.E., Lemster B.H., Koretsky A.P., Demetris A.J., Feldman A.M. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ. Res. 1997;81:627–635. doi: 10.1161/01.res.81.4.627. [DOI] [PubMed] [Google Scholar]
- Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90-7. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Linn M., Bellett A.J., Parsons P.G., Suhrbier A. Complete removal of mycoplasma from viral preparations using solvent extraction. J. Virol. Methods. 1995;52:51–54. doi: 10.1016/0166-0934(94)00136-5. [DOI] [PubMed] [Google Scholar]
- Levick S.P., Goldspink P.H. Could interferon-gamma be a therapeutic target for treating heart failure? Heart Fail. Rev. 2014;19:227–236. doi: 10.1007/s10741-013-9393-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Mangalmurti N., Hunter C.A. Cytokine Storms: Understanding COVID-19. Immunity. 2020;53:19–25. doi: 10.1016/j.immuni.2020.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. [Google Scholar]
- Messner C.B., Demichev V., Wendisch D., Michalick L., White M., Freiwald A., Textoris-Taube K., Vernardis S.I., Egger A.S., Kreidl M., et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020;11:11–24.e4. doi: 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills R.J., Titmarsh D.M., Koenig X., Parker B.L., Ryall J.G., Quaife-Ryan G.A., Voges H.K., Hodson M.P., Ferguson C., Drowley L., et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc. Natl. Acad. Sci. USA. 2017;114:E8372–E8381. doi: 10.1073/pnas.1707316114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills R.J., Parker B.L., Quaife-Ryan G.A., Voges H.K., Needham E.J., Bornot A., Ding M., Andersson H., Polla M., Elliott D.A., et al. Drug Screening in Human PSC-Cardiac Organoids Identifies Pro-proliferative Compounds Acting via the Mevalonate Pathway. Cell Stem Cell. 2019;24:895–907.e6. doi: 10.1016/j.stem.2019.03.009. [DOI] [PubMed] [Google Scholar]
- Needham E.J., Parker B.L., Burykin T., James D.E., Humphrey S.J. Illuminating the dark phosphoproteome. Sci. Signal. 2019;12:eaau8645. doi: 10.1126/scisignal.aau8645. [DOI] [PubMed] [Google Scholar]
- Nicholls S.J., Puri R., Wolski K., Ballantyne C.M., Barter P.J., Brewer H.B., Kastelein J.J., Hu B., Uno K., Kataoka Y., et al. Effect of the BET Protein Inhibitor, RVX-208, on Progression of Coronary Atherosclerosis: Results of the Phase 2b, Randomized, Double-Blind, Multicenter, ASSURE Trial. Am. J. Cardiovasc. Drugs. 2016;16:55–65. doi: 10.1007/s40256-015-0146-z. [DOI] [PubMed] [Google Scholar]
- Nicholls S.J., Schwartz G.G., Buhr K.A., Ginsberg H.N., Johansson J.O., Kalantar-Zadeh K., Kulikowski E., Toth P.P., Wong N., Sweeney M., Ray K.K., BETonMACE Investigators Apabetalone and hospitalization for heart failure in patients following an acute coronary syndrome: a prespecified analysis of the BETonMACE study. Cardiovasc. Diabetol. 2021;20:13. doi: 10.1186/s12933-020-01199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E., Jeffrey K.L., Schaefer U., Beinke S., Dewell S., Chung C.W., Chandwani R., Marazzi I., Wilson P., Coste H., et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiga M., Wang D.W., Han Y., Lewis D.B., Wu J.C. COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020;17:543–558. doi: 10.1038/s41569-020-0413-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oladunni F.S., Park J.G., Pino P.A., Gonzalez O., Akhter A., Allué-Guardia A., Olmo-Fontánez A., Gautam S., Garcia-Vilanova A., Ye C., et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nat. Commun. 2020;11:6122. doi: 10.1038/s41467-020-19891-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova V.V., van den Hil F.E., Petrus-Reurer S., Drabsch Y., Ten Dijke P., Mummery C.L. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat. Protoc. 2014;9:1514–1531. doi: 10.1038/nprot.2014.102. [DOI] [PubMed] [Google Scholar]
- Padmanabhan A., Alexanian M., Linares-Saldana R., González-Terán B., Andreoletti G., Huang Y., Connolly A.J., Kim W., Hsu A., Duan Q., et al. BRD4 (Bromodomain-Containing Protein 4) Interacts with GATA4 (GATA Binding Protein 4) to Govern Mitochondrial Homeostasis in Adult Cardiomyocytes. Circulation. 2020;142:2338–2355. doi: 10.1161/CIRCULATIONAHA.120.047753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini D., Kawakami R., Guagliumi G., Sakamoto A., Kawai K., Gianatti A., Nasr A., Kutys R., Guo L., Cornelissen A., et al. Microthrombi as a Major Cause of Cardiac Injury in COVID-19: A Pathologic Study. Circulation. 2021;143:1031–1042. doi: 10.1161/CIRCULATIONAHA.120.051828. [DOI] [PubMed] [Google Scholar]
- Puntmann V.O., Carerj M.L., Wieters I., Fahim M., Arendt C., Hoffmann J., Shchendrygina A., Escher F., Vasa-Nicotera M., Zeiher A.M., et al. Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered From Coronavirus Disease 2019 (COVID-19) JAMA Cardiol. 2020;5:1265–1273. doi: 10.1001/jamacardio.2020.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y., Wang X.-M., Mannan R., Pitchiaya S., Zhang Y., Wotring J.W., Xiao L., Robinson D.R., Wu Y.-M., Tien J.C.-Y., et al. Targeting transcriptional regulation of SARS-CoV-2 entry factors ACE2 and TMPRSS2. Proc. Natl. Acad. Sci. USA. 2020;118 doi: 10.1073/pnas.2021450118. e2021450118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaife-Ryan G.A., Sim C.B., Ziemann M., Kaspi A., Rafehi H., Ramialison M., El-Osta A., Hudson J.E., Porrello E.R. Multicellular Transcriptional Analysis of Mammalian Heart Regeneration. Circulation. 2017;136:1123–1139. doi: 10.1161/CIRCULATIONAHA.117.028252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray K.K., Nicholls S.J., Buhr K.A., Ginsberg H.N., Johansson J.O., Kalantar-Zadeh K., Kulikowski E., Toth P.P., Wong N., Sweeney M., Schwartz G.G., BETonMACE Investigators and Committees Effect of Apabetalone Added to Standard Therapy on Major Adverse Cardiovascular Events in Patients With Recent Acute Coronary Syndrome and Type 2 Diabetes: A Randomized Clinical Trial. JAMA. 2020;323:1565–1573. doi: 10.1001/jama.2020.3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X., Wen W., Fan X., Hou W., Su B., Cai P., Li J., Liu Y., Tang F., Zhang F., et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell. 2021 doi: 10.1016/j.cell.2021.01.053. Published online February 3, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson P., Griffin I., Tucker C., Smith D., Oechsle O., Phelan A., Rawling M., Savory E., Stebbing J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet. 2020;395:e30–e31. doi: 10.1016/S0140-6736(20)30304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runte K.E., Bell S.P., Selby D.E., Häußler T.N., Ashikaga T., LeWinter M.M., Palmer B.M., Meyer M. Relaxation and the Role of Calcium in Isolated Contracting Myocardium From Patients With Hypertensive Heart Disease and Heart Failure With Preserved Ejection Fraction. Circ. Heart Fail. 2017;10:e004311. doi: 10.1161/CIRCHEARTFAILURE.117.004311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzymski T., Mikula M., Żyłkiewicz E., Dreas A., Wiklik K., Gołas A., Wójcik K., Masiejczyk M., Wróbel A., Dolata I., et al. SEL120-34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget. 2017;8:33779–33795. doi: 10.18632/oncotarget.16810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadzak I., Schiff M., Gattermeier I., Glinitzer R., Sauer I., Saalmüller A., Yang E., Schaljo B., Kovarik P. Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc. Natl. Acad. Sci. USA. 2008;105:8944–8949. doi: 10.1073/pnas.0801794105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders-van Wijk S., Tromp J., Beussink-Nelson L., Hage C., Svedlund S., Saraste A., Swat S.A., Sanchez C., Njoroge J., Tan R.S., et al. Proteomic Evaluation of the Comorbidity-Inflammation Paradigm in Heart Failure With Preserved Ejection Fraction: Results From the PROMIS-HFpEF Study. Circulation. 2020;142:2029–2044. doi: 10.1161/CIRCULATIONAHA.120.045810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher S.M., Gao E., Zhu W., Chen X., Chuprun J.K., Feldman A.M., Tesmer J.J.G., Koch W.J. Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodeling after myocardial infarction. Sci. Transl. Med. 2015;7:277ra231. doi: 10.1126/scitranslmed.aaa0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A., Garcia G., Jr., Wang Y., Plummer J.T., Morizono K., Arumugaswami V., Svendsen C.N. Human iPSC-Derived Cardiomyocytes Are Susceptible to SARS-CoV-2 Infection. Cell Rep. Med. 2020;1:100052. doi: 10.1016/j.xcrm.2020.100052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S., Qin M., Shen B., Cai Y., Liu T., Yang F., Gong W., Liu X., Liang J., Zhao Q., et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020;5:802–810. doi: 10.1001/jamacardio.2020.0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton M.S., Bagchi R.A., Felisbino M.B., Hirsch R.A., Smith H.E., Riching A.S., Enyart B.Y., Koch K.A., Cavasin M.A., Alexanian M., et al. Dynamic Chromatin Targeting of BRD4 Stimulates Cardiac Fibroblast Activation. Circ. Res. 2019;125:662–677. doi: 10.1161/CIRCRESAHA.119.315125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs M.C., Burn T.C., Sparks R., Maduskuie T., Diamond S., Rupar M., Wen X., Volgina A., Zolotarjova N., Waeltz P., et al. The Novel Bromodomain and Extraterminal Domain Inhibitor INCB054329 Induces Vulnerabilities in Myeloma Cells That Inform Rational Combination Strategies. Clin. Cancer Res. 2019;25:300–311. doi: 10.1158/1078-0432.CCR-18-0098. [DOI] [PubMed] [Google Scholar]
- Szekely Y., Lichter Y., Taieb P., Banai A., Hochstadt A., Merdler I., Gal Oz A., Rothschild E., Baruch G., Peri Y., et al. The Spectrum of Cardiac Manifestations in Coronavirus Disease 2019 (COVID-19) - a Systematic Echocardiographic Study. Circulation. 2020;142:342–353. doi: 10.1161/CIRCULATIONAHA.120.047971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker N.R., Chaffin M., Fleming S.J., Hall A.W., Parsons V.A., Jr., Bedi K.C., Jr., Akkad A.D., Herndon C.N., Arduini A., Papangeli I., et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation. 2020;142:466–482. doi: 10.1161/CIRCULATIONAHA.119.045401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S., Cox J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018;1711:133–148. doi: 10.1007/978-1-4939-7493-1_7. [DOI] [PubMed] [Google Scholar]
- Vasudevan N.T., Mohan M.L., Gupta M.K., Martelli E.E., Hussain A.K., Qin Y., Chandrasekharan U.M., Young D., Feldman A.M., Sen S., et al. Gβγ-independent recruitment of G-protein coupled receptor kinase 2 drives tumor necrosis factor α-induced cardiac β-adrenergic receptor dysfunction. Circulation. 2013;128:377–387. doi: 10.1161/CIRCULATIONAHA.113.003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voges H.K., Mills R.J., Elliott D.A., Parton R.G., Porrello E.R., Hudson J.E. Development of a human cardiac organoid injury model reveals innate regenerative potential. Development. 2017;144:1118–1127. doi: 10.1242/dev.143966. [DOI] [PubMed] [Google Scholar]
- Williams L.M., McCann F.E., Cabrita M.A., Layton T., Cribbs A., Knezevic B., Fang H., Knight J., Zhang M., Fischer R., et al. Identifying collagen VI as a target of fibrotic diseases regulated by CREBBP/EP300. Proc. Natl. Acad. Sci. USA. 2020;117:20753–20763. doi: 10.1073/pnas.2004281117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., McGoogan J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020;323:1239–1242. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The video was taken over a period of 10 s and is displayed in real time (50 frames/s).
The video was taken over a period of 10 s and is displayed in real time (50 frames/s).
The video was taken over a period of 5 s and is displayed in real time (50 frames/s).
The video was taken over a period of 5 s and is displayed in real time (50 frames/s).
The video was taken over a period of 10 s and is displayed in real time (50 frames/s).
Data Availability Statement
Mass spectrometry-based proteomics data reported in this paper have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Deutsch et al., 2017) with the dataset identifier PXD020994. snRNA-seq reported in this paper has been deposited to the European Genome-phenome Archive (EGA) with the dataset identifier EGAS00001005174. Bulk RNA-seq data reported in this paper have been deposited to the European Nucleotide Archive (ENA) with the dataset identifier PRJEB43658. All MATLAB m-files will be provided upon request as they require custom training.