

Abstract
Tumor necrosis factor alpha (TNF-α) was initially recognized as a factor that causes the necrosis of tumors, but it has been recently identified to have additional important functions as a pathological component of autoimmune diseases. TNF-α binds to two different receptors, which initiate signal transduction pathways. These pathways lead to various cellular responses, including cell survival, differentiation, and proliferation. However, the inappropriate or excessive activation of TNF-α signaling is associated with chronic inflammation and can eventually lead to the development of pathological complications such as autoimmune diseases. Understanding of the TNF-α signaling mechanism has been expanded and applied for the treatment of immune diseases, which has resulted in the development of effective therapeutic tools, including TNF-α inhibitors. Currently, clinically approved TNF-α inhibitors have shown noticeable potency in a variety of autoimmune diseases, and novel TNF-α signaling inhibitors are being clinically evaluated. In this review, we briefly introduce the impact of TNF-α signaling on autoimmune diseases and its inhibitors, which are used as therapeutic agents against autoimmune diseases.
Keywords: TNF-α, autoimmune diseases, rheumatoid arthritis, inflammatory bowel disease, psoriatic arthritis, TNF-α inhibitors
1. Introduction
Tumor necrosis factor alpha (TNF-α) is a cytokine that has pleiotropic effects on various cell types. It has been identified as a major regulator of inflammatory responses and is known to be involved in the pathogenesis of some inflammatory and autoimmune diseases [1]. Structurally, TNF-α is a homotrimer protein consisting of 157 amino acids, mainly generated by activated macrophages, T-lymphocytes, and natural killer cells [2]. It is functionally known to trigger a series of various inflammatory molecules, including other cytokines and chemokines. TNF-α exists in a soluble and transmembrane form. The transmembrane TNF-α (tmTNF-α) is the initially synthesized precursor form and is required to be processed by TNF-α-converting enzyme (TACE), a membrane-bound disintegrin metalloproteinase, to be released as the soluble TNF-α (sTNF-α) [3]. The processed sTNF-α then facilitates various biological activities through type 1 receptors (TNFR1, also known as TNFRSF1A, CD120a, and p55) and type 2 receptors (TNFR2, also known as TNFRSF1B, CD120b, and p75) [4,5,6,7]. tmTNF-α also acts on both TNFR1 and TNFR2, but its biological activities are expected to be mediated mainly through TNFR2 [8]. TNFR1 is expressed by all human tissues and is the key signaling receptor for TNF-α. TNFR2 is generally expressed in immune cells and facilitates limited biological responses [9]. In general, TNF-α binds to its receptors, mainly TNFR1 and TNFR2, and then transmits molecular signals for biological functions such as inflammation and cell death. TNFR1 is activated by both sTNF-α and tmTNF-α, and it processes a death domain (DD) that interacts with the TNFR1-associated death domain (TRADD) adaptor protein [10].
The activation of TNFR1 can trigger the formation of different signaling complexes, referred to as complexes I, IIa, IIb, and IIc, which result in distinct cellular responses [11,12]. During the assembly of complex I, the activated TNFR1 binds to TRADD, followed by the association and interaction of various components, including receptor-interacting serine/threonine-protein kinase 1 (RIPK1), TNFR-associated factor 2 or 5 (TRAF2/5), cellular inhibitor of apoptosis protein 1 or 2 (cIAP1/2), and the linear ubiquitin chain assembly complex (LUBAC) [13]. This signaling pathway results in the activation of nuclear factor κB (NF-κB) and mitogen-activated protein kinases (MAPKs) [14,15]. The functional outcome of complex I signaling is known to be the induction of inflammation, tissue, cell survival and proliferation, and the immune defense against pathogens [12,16]. Unlike complex I, which is assembled at the plasma membrane, complexes IIa, IIb, and IIc are assembled in the cytoplasm [17]. Complex IIa consists of TRADD, RIPK1, TRAF2, cIAP1/2, pro-Caspase-8, and Fas-associated protein with death domain (FADD) [18,19]. Complex IIb has the same composition as complex IIa with the addition of RIPK3 [18]. The formation of complexes IIa and IIb (also known as apoptosome) activates caspase-8 and results in apoptosis. Complex IIc (known as necrosome) is known to be formed by RIPK1 and RIPK3, which bind to form the complex when they remain uncleaved. Complex IIc activates the mixed lineage kinase domain-like protein (MLKL) via RIPK3-mediated phosphorylation, which induces necroptosis and inflammation (Figure 1) [13,20].
Figure 1.
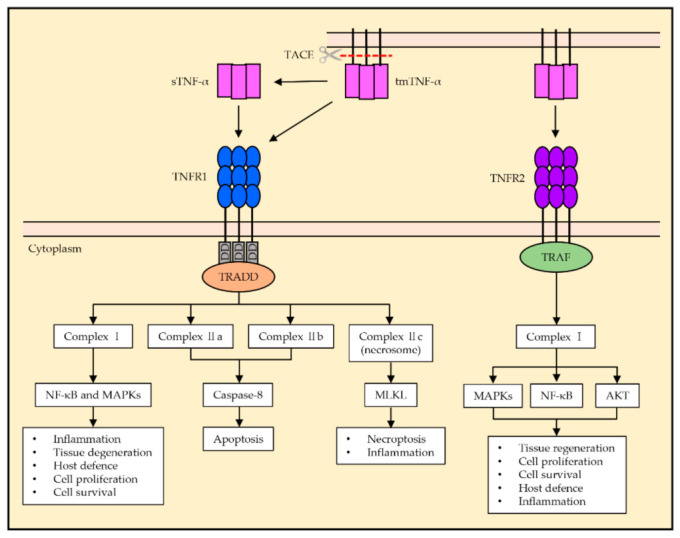
General tumor necrosis factor alpha (TNF-α) signaling pathway of TNFR1 and TNFR2. The TNFR1 signaling pathway is activated by the ligation of sTNF-α and tmTNF-α, and the death domain of TNFR1 recruits TRADD. Complex I activates NF-κB and MAPKs, which results in inflammation, tissue degeneration, host defense, cell proliferation, and cell survival. Complexes IIa and IIb activate caspase-8 and induce apoptosis. Complex IIc is known to induce necroptosis and inflammation via the activation of MLKL. The TNFR2 signaling pathway is mainly activated by tmTNF-α. TNFR2 does not possess a death domain and recruits TRAF via its TRAF domain, which activates the formation of complex I, resulting in NF-κB and MAPKs and AKT activation. TNFR2 activation is associated with homeostatic bioactivities such as tissue regeneration, cell proliferation, and cell survival, as well as host defense and inflammation.
TNFR2 is suggested to be fully activated by tmTNF-α, primarily in the context of cell-to-cell interactions. Unlike TNFR1, TNFR2 lacks a death domain and thus is unable to induce programmed cell death directly (Figure 2) [8]. During assembly, TNFR2 recruits TRAF2 along with TRAF1, cIAP1, and cIAP2, and this complex allows the downstream activation of NF-κB, MAPKs, and protein kinase B (AKT) [17]. Functionally, TNFR2 is mainly associated with homeostatic bioactivities, including tissue regeneration, cell proliferation, and cell survival [21]. However, this pathway is also known to trigger inflammatory responses and host defense against pathogens. Overall, TNFR1 is essential to induce cytotoxic and proinflammatory TNF-α responses, while TNFR2 may largely mediate cell activation, migration, or proliferation (Figure 1).
Figure 2.
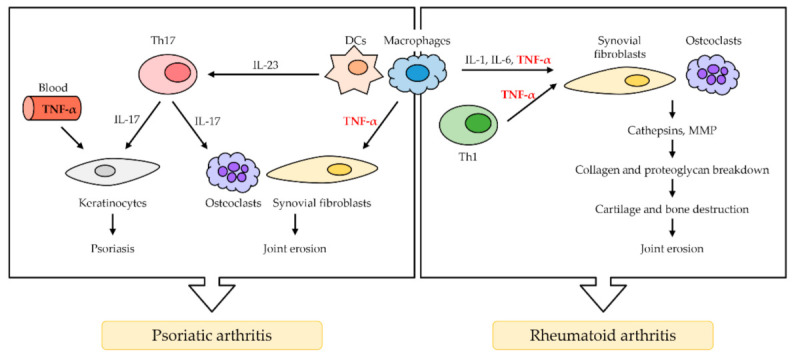
The role of TNF-α in rheumatoid arthritis (RA) and psoriatic arthritis (PsA). TNF-α is mainly secreted by macrophage and Th1 cells. In RA, TNF-α activates synovial fibroblasts, which causes the overproduction of cathepsins and MMP. The breakdown of collagen and proteoglycan follows, resulting in cartilage and bone destruction, as well as joint erosion. Osteoclasts in RA induce synovial hyperplasia and angiogenesis. In PsA, activated dendritic cells (DCs) and macrophages secrete TNF-α and IL-23 excessively. IL-23 induces T cells to differentiate in Th17 cells, which secretes IL-17. IL-17 and TNF-α in the blood activate keratinocytes, resulting in psoriasis (PS). TNF-α also activates osteoclasts, leading to synovial fibroblast, which results in joint erosion.
Physiologically, TNF-α is a crucial component for a normal immune response. TNF-α can activate the immune system to regulate; however, the inappropriate or excessive production of TNF-α can be harmful and may lead to disease. Rheumatoid arthritis (RA) [22], inflammatory bowel disease (IBD) [23,24,25], psoriatic arthritis (PsA), psoriasis (PS) [26], and noninfectious uveitis (NIU) are induced by the abnormal secretion of TNF-α; thus, TNF-α can be classified as a key factor in the pathological development.
Due to the involvement of TNF-α in the pathogenesis of autoimmune diseases, TNF-α inhibitors have been successfully developed and applied in the clinical treatment of autoimmune diseases such as Crohn’s disease (CD) and RA [27,28]. Therapeutic drugs act as antagonists by blocking the interaction of TNF-α with TNFR1/2 or, in some cases, as agonists by stimulating reverse signaling, causing the apoptosis of TNF-α producing immune cells [29,30,31]. Several TNF-α inhibitors have been approved for clinical use: etanercept, infliximab, adalimumab, golimumab, and certolizumab. The impact of TNF-α signaling on each type of autoimmune disease will be introduced in this review, as well as the evaluation of current TNF-α inhibitors utilized as therapeutic drugs against autoimmune diseases.
2. TNF-α Signaling in Autoimmune Diseases
2.1. Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a chronic autoimmune disorder that primarily affects the joints and typically results in redness, swelling, and arthralgia. In serious cases, it limits the range of motion [32]. RA affects nearly 1% of the population, and it is characterized by the inflammation of synovial tissue, leading to progressive damage, including the erosion of adjacent cartilage and bone, which can lead to chronic disability [33]. TNF-α is considered the major inflammatory cytokine involved in the pathogenesis of RA and is found in high frequencies in patients with the disease. Inflammation is associated with the accumulation of inflammatory cells, predominantly type 1 helper T cells (Th1) and macrophages but also B cells, plasma cells, and dendritic cells (DCs) [22]. In RA, TNF-α secreted from Th1 cells and macrophages activates synovial fibroblasts, promotes epidermal hyperplasia, and recruits inflammatory cells [34]. After activation by various cytokines, including IL-1, IL-6, and TNF-α [35], synovial fibroblasts overexpress cathepsins and matrix metalloproteinases (MMPs), followed by collagen and proteoglycan breakdown. As a result, cartilage and bone are destroyed, and finally, joint erosion occurs. Osteoclasts are also important for the progression of RA pathology throughout TNF-α activation, and activated osteoclasts in RA induce synovial hyperplasia and angiogenesis (Figure 2) [36].
2.2. Psoriatic Arthritis
Psoriatic arthritis (PsA) is a long-term autoimmune arthropathy distinct from RA [37]. It typically occurs in people affected by cutaneous psoriasis. Similar to RA, PsA is relatively common, affecting approximately 1% of the population [38]. The representative feature of PsA is the swelling of fingers and toes, leading to a sausage-like appearance [39]. The major cells involved in the PsA pathogenesis are DCs, macrophages, type 17 helper T cells (Th17), and keratinocytes [39,40]. DCs and macrophages activated by TNF-α produce TNF-α and IL-23 excessively. IL-23 causes differentiation of naïve T cells into Th17 cells, which overproduce IL-17. Then, IL-17 and TNF-α activate keratinocytes, which promote epidermal hyperplasia and recruit inflammatory cells such as DCs. TNF-α induces the proliferation and anti-apoptosis of keratinocytes via the NF-κB signal pathway, eventually leading to the formation of microabscess by enhancing the recruitment of inflammatory cells in psoriasis (Figure 2) [41].
2.3. Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) is a group of autoimmune diseases localized in the gastrointestinal tract. CD and ulcerative colitis (UC) are the primary types of IBD, which are distinguished by the location at which the disease occurs. CD mostly occurs in the small and large intestine, but it can also affect the mouth, esophagus, stomach, and anus, while CD commonly affects the colon and the rectum [42,43]. In both CD and CD, TNF-α is secreted from Th1 cells along with other cytokines, including IL-1, IL-6, and IL-17 [44,45]. These cytokines are capable of accumulating intestinal fibroblasts, neutrophils, and macrophages in the gut. Accumulated intestinal fibroblasts cause intestinal fibrosis, which leads to the formation of stricture in the intestine. Accumulated neutrophils in the intestine secrete elastase to induce matrix degradation. Finally, accumulated macrophages in the intestine produce TNF-α, IL-1, and IL-6, which eventually induce intestinal matrix degradation, epithelial damage, endothelial activation, and vascular disruption (Figure 3) [23,24,25].
Figure 3.
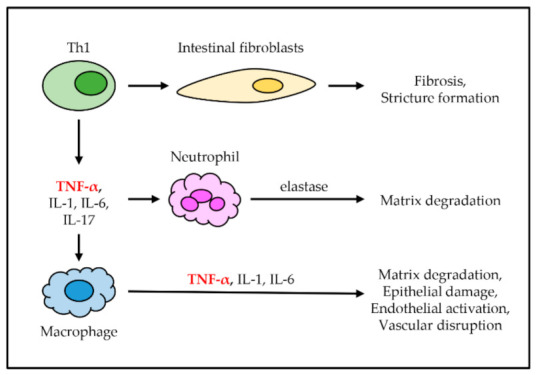
The role of TNF-α in inflammatory bowel disease (IBD). TNF-α is secreted from Th1 cells along with other cytokines. These cytokines cause the accumulation of immune cells, including intestinal fibroblasts, neutrophils, and macrophages in the gut. Intestinal fibroblasts cause fibrosis and stricture formation. Neutrophils secrete elastase, which causes intestinal matrix degradation. Macrophages produce more inflammatory cytokines, which causes intestinal matrix degradation, epithelial damage, endothelial activation, and disruption.
2.4. Psoriasis
Psoriasis (PS) is a chronic inflammatory disease with a strong genetic diathesis and autoimmunity characterized by a specific region of abnormal skin. There are five types of PS-laque, guttate, inverse, pustular, and erythrodermic, and about 90% of PS is plaque-type PS. Plaque-type PS classically presents pruritic plaques with white scales on the top [46,47]. The lesions of PS reflect the epidermal hyperplasia, angiogenesis, and inflammatory infiltration of leukocytes into the dermis as a result of the dysregulation of skin immune responses [48]. The inflammatory pathways involved in the pathogenesis of plaque psoriasis overlap the rest of the clinical variants [46]. Stressed keratinocytes excessively secreted TNF-α along with IL-1 and IL-6. Those cytokines activate DCs. IL-12 from activated DCs differentiates naïve T cells into Th1 cells, and IL-23 differentiates naïve T cells into Th17 cells [40]. These two kinds of cells overproduce specific cytokines, and Th1 cells secrete TNF-α and IFN-γ, whereas Th17 cells secrete IL-17 immoderately [46]. This abnormal immune reaction makes TNF-α activate DCs continuously. TNF-α, IFN-γ, and IL-17 ultimately lead to keratinocyte hyperproliferation and epidermal change, such as acanthosis, parakeratosis, and hypogranulosis (Figure 4) [40].
Figure 4.
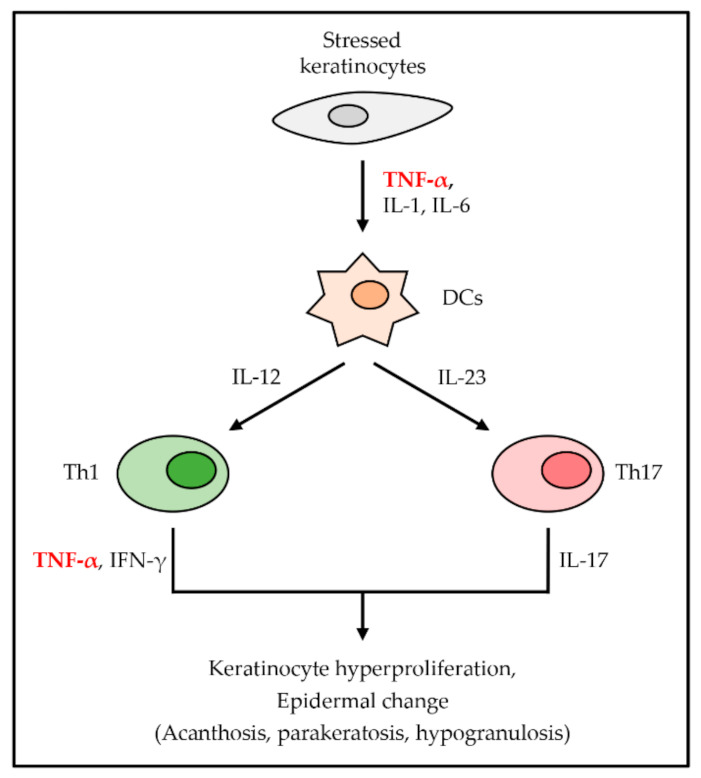
The role of TNF-α in PS. TNF-α is first secreted from stressed keratinocytes with other proinflammatory cytokines. Secreted cytokines activate DCs, which induce the differentiation of T cells into Th1 cells and Th17 cells. They secrete TNF-α, IFN-γ, and IL-17, respectively, which cause keratinocyte hyperproliferation and several epidermal changes.
2.5. Noninfectious Uveitis
Noninfectious uveitis (NIU), also known as autoimmune uveitis, is an autoimmune disease inside the eye that aims at the neuroretina [49]. NIU can be associated with other autoimmune diseases, including RA, IBD, and PS. Patients with NIU present an expanded risk of retinal detachment, cataracts, developing glaucoma, and visual distribution, along with blindness or low vision [50]. The start of NIU is an overproduced TNF-α from macrophages, as well as other various cytokines such as IL-6, IL-10, IL-12, and IL-23. DCs are activated TNF-α and other cytokines [51,52]. Th1 cells are differentiated from naïve T cells by immoderate IL-12, which activated DCs produce. Th17 cells are differentiated by excessively secreted IL-6 and TGF-β from DCs. Activated Th1 cells and Th17 cells migrate and infiltrate inside the uvea, which is the middle layer of the eye and provides blood to the retina [53]. Migrated Th1 cells and Th17 cells generate damage to the blood–retinal barrier, trigger the retinal vasculature, and recruit nonspecific blood-circulating leukocytes such as monocytes, lymphocytes, and polymorphonuclear leukocytes (PMNs). The inflammation that makes swelling and destroys uvea eventually occurs in NIU (Figure 5) [54].
Figure 5.
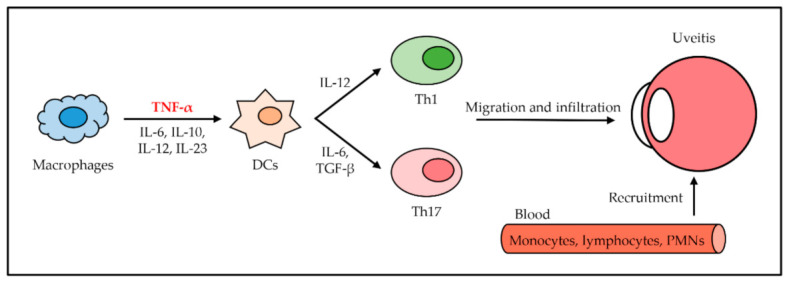
The role of TNF-α in uveitis. TNF-α is secreted from macrophages with other cytokines and activates DCs. Activated DCs secrete IL-12, IL-6, and TGF-β to induce differentiation of Th1 cells and Th17 cells from naïve T cells, respectively. Th1 cells and Th17 cells arrive at the uvea through migration and infiltration. Several leukocytes from blood are recruited by Th1 cells and Th17 cells. NIU occurs due to inflammation.
3. TNF-α Inhibitors as Therapeutic Drugs
3.1. Infliximab (Remicade®)
Infliximab is a recombinant chimeric monoclonal antibody containing a murine variable region and a human IgG1 constant region. It is specific for all forms of TNF-α in humans and effectively blocks the binding of TNF-α to its soluble and transmembrane receptors [55]. After infliximab treatment for IBD patients, the lysis of cell lines expressing TNF-α by the complement-dependent and antibody-dependent cytotoxicity was promoted and reduced inflamed tissue [56]. In addition to generating apoptosis, infliximab blocks IFN-γ production in colonic and stimulated T cells, and anti-inflammation occurs [57]. Furthermore, infliximab downregulates intracellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VACM-1) and modulates the MMPs/tissue inhibitor of metalloproteinases (TIMPs) balance [58,59]. The half-life of infliximab is approximately 8–10 days and can be maintained by administering doses every eight weeks [60]. Infliximab was initially FDA approved for the therapy of CD in 1998 and later also for RA. It was further approved for the treatment of ankylosing spondylitis (AS), PsA, CD, and PS (Figure 6) [61].
Figure 6.

Timeline of the approval dates of TNF-α inhibitors in autoimmune diseases.
3.2. Etanercept (Enbrel®)
Etanercept is a fusion protein that includes two identical TNFR2 extracellular regions connected to the Fc fragment of human IgG1 [62]. There are diverse variants of each etanercept group with very few differences [63]. Etanercept binds to sTNF-α or tmTNF-α and inactivates them by blocking the interaction with receptors [56]. It only enables it to bind to the active trimeric TNF-α since its binding site is placed in the cleft between subunits [64]. Etanercept has a half-life of 3–3.5 days after subcutaneous injection [60]. It relieves symptoms of arthritis and prevents the progression of RA in patients [65]. Etanercept also appears to modulate the proinflammatory genes such as NF-κB in plaque PS, resulting in a significant decrease in the production of TNF-α. Furthermore, it causes the apoptosis of large amounts of DCs in plaque, which stops positive feedback related to TNF-α by early apoptotic cell death before the activation and maturation of DCs [66]. It was permitted by FDA in 1998 for the treatment of juvenile idiopathic arthritis (JIA) and PsA, RA, PS, and AS (Figure 6) [67]. For articular involvement, favorable results have been reported, but there is a limited outcome in ocular inflammation such as uveitis [68].
3.3. Adalimumab (Humira®)
Adalimumab is a fully human IgG1 monoclonal antibody capable of specifically blocking the binding of human TNF-α to the receptors because its function and structure are identical to that of natural human IgG1 [69]. Adalimumab needs a less frequent subcutaneous administration because of its comparatively longer half-life, which is about 10 to 13 days [70]. Furthermore, adalimumab shows lower immunogenicity than infliximab [71]. Because adalimumab has better tolerance and lower immunogenicity, it is also effectively used for patients with CD and can be administered to patients who have had an allergic reaction to infliximab. In PA patients treated with adalimumab, declines in the levels of TNF-α and IL-6, as well as acute-phase reactants of inflammation, are observed [72]. Additionally, it was found that the treatment of RA with adalimumab can inhibit IL-17, which is oversecreted by Th17 cells by increasing the number of regulatory T cells (Treg) compared to untreated patients [73]. Adalimumab was approved by the FDA in 2002 for the treatment of RA, followed by approval for the treatment of PsA, PS, AS, JIA, and CD, as well as uveitis (Figure 6) [71,74].
3.4. Certolizumab Pegol (Cimzia®)
Certolizumab pegol (CDP87) is a humanized monoclonal antibody that has a polyethylene glycosylated (PEG) Fab fragment and lacks the Fc region [75]. Due to the lack of the Fc region, it does not include complement or antibody-dependent cytotoxicity. It is a novel TNF-α inhibitor with a remarkable mechanism of action in comparison to other TNF-α inhibitors. Because of the PEGylation, certolizumab pegol can be more significantly distributed into inflamed tissues compared to infliximab and adalimumab [76]. The distinct structure of the inhibitor might be the reason behind the higher efficacy of certolizumab pegol in comparison with other TNF-α inhibitors [77]. The PEGylation of certolizumab pegol enhances its half-life to two weeks, which may be attributed to the high concentration of the inhibitor examined in inflamed tissues [78]. Certolizumab pegol was approved for the treatment of CD, RA, PsA, As, and plaque PS by the FDA by 2018 (Figure 6) [79].
3.5. Golimumab (Simponi®we)
Golimumab is a completely humanized IgG1 monoclonal antibody that has functional specificity for human TNF-α [80]. Compared to infliximab and adalimumab, golimumab has a higher affinity and is more potent in the neutralization of sTNF-α and tmTNF-α, thus it can effectively inhibit the biological activity of TNF-α [81]. It also blocks the leukocyte infiltration by prevention of cell adhesion proteins such as E-selectin, ICAM-1, and VCAM-1, as well as proinflammatory cytokine secretion [82]. Golimumab’s half-life is about 7–20 days [83]. Golimumab was approved by the FDA in 2009 for the treatment of RA when injected in a mixture with methotrexate. It was also approved for UC, AS, and PsA treatment (Figure 6) [84].
3.6. TNF-α Inhibitor Biosimilars
Since the initial approval of each TNF-α inhibitor, several kinds of biosimilars that are not generic products but are highly similar to the originators have received FDA approval. These have only minor differences in clinically inactive components in the molecular structure, which are about the same in terms of purity, safety, and efficacy [85]. While biosimilars for infliximab, etanercept, and adalimumab have received FDA approval within the last five years, no biosimilars approved for certolizumab pegol and golimumab to date [86]. However, out of 12 approved biosimilars, only infliximab-axxq (Avsola®), infliximab-abda (Renflexis®), and infliximab-dyyb (Inflectra®) with infliximab as a reference product are launched and available in the US market (Table 1) [87].
Table 1.
TNF-α inhibitor originators and FDA-approved biosimilars by the end of 2020.
| Originators (Reference Products) |
Biosimilars (FDA-Approved Products) |
Approval Date | Launch Date (US) |
|---|---|---|---|
| Infliximab (Remicade®) | Infliximab-axxq (Avsola®) | December 2019 | July 2020 |
| Infliximab-qbtx (IxifiTM) | December 2017 | Not launching in US | |
| Infliximab-abda (Renflexis®) | May 2017 | July 2018 | |
| Infliximab-dyyb (Inflectra®) | April 2016 | November 2016 | |
| Etanercept (Enbrel®) | Etanercept-ykro (EticovoTM) | April 2019 | - |
| Etanercept-szzs (Erelzi®) | August 2016 | - | |
| Adalimumab (Humira®) | Adalimumab-fkjp (HulioTM) | July 2020 | - |
| Adalimumab-afzb (AbriladaTM) | November 2019 | - | |
| Adalimumab-bwwd (HadlimaTM) | July 2019 | - | |
| Adalimumab-adaz (Hyrimoz®) | October 2018 | - | |
| Adalimumab-adbm (Cyltezo®) | August 2017 | - | |
| Adalimumab-atto (AmjevitaTM) | September 2016 | - | |
| Certolizumab pegol (Cimzia®) | No biosimilars approved | ||
| Golimumab (Simponi®) | No biosimilars approved | ||
3.7. New Anti-TNF-α Agents
New anti-TNF-α agents are currently being developed to overcome shortcomings such as the nonresponse of existing TNF-α inhibitors. Ozoralizumab is a humanized monoclonal antibody designed for the treatment of inflammatory diseases. In a Phase 2 study, clinical activity and safety were evaluated in patients worldwide and in Japan. When 80 mg of ozoralizumab was injected, effective and well-tolerated treatment was possible as a novel TNF-α inhibitor (NCT01007175). Due to the specific molecular characteristics of the nanobody (small size, low immunogenicity potential, manufacturability), most patients showed remarkable improvement in disease activity, and once induced, remission can be maintained at a dose of less than 80 mg per month, which shows the desired treatment result. In addition, since Phase 3 is currently in progress, ozoralizumab is highly likely to be used as a new TNF-α inhibitor.
Saddala and Huang established a new drug model, ZINC09609430, that could specifically inhibit TNF-α. It was screened based on the Zinc library using structure-based pharmacophore modeling, virtual screening, and molecular docking with in silico absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis. As a result, the identified new small compound could serve as structurally various novel inhibitors for TNF-α and could potentially be used as anti-inflammatory agents to treat related diseases. Although ZINC09609430 is safe and active in silico ADMET results, it is worthy of further evaluation for its safety and efficacy in vitro and in vivo [88].
TNF blocking treatment for CD is safe and effective, but due to the high nonresponse rate in patients, Kwak et al. conducted an in silico study to suggest a novel drug treatment for anti-TNF refractory CD. Among them, CP-69033401 showed high drug response in the cluster of patients with anti-TNF refractory CD. These findings could provide new options for anti-TNF treatment (Table 2) [89].
Table 2.
New anti-TNF-α agents currently under development.
| Drugs | Research institute | Form | Indications | Progress | Ref. |
|---|---|---|---|---|---|
| Ozoralizumab (TS-152) | Taisho | Monoclonal antibody | RA | Phase 3 (NCT04077567) Est. End Date: Dec. 2022 | - |
| ZINC09609430 | University of Missouri | Small-molecule drug compound |
Unknown | In silico study | [88] |
| CP-690334-01 | Kyung Hee University College of Medicine | Small-molecule drug compound |
CD | In silico study | [89] |
4. Conclusions and Discussion
Here, we introduced the association of TNF-α signaling in some autoimmune diseases and the current TNF-α inhibitors used as therapeutic drugs for these diseases. TNF-α, which is commonly known as a proinflammatory cytokine, has also been shown to apply pleiotropic effects on various cell models and has been identified as a significant factor in the pathogenesis of autoimmune diseases (Figure 7). The role of TNF-α in these diseases has not been entirely understood; however, it is generally known to contribute to the progression of disease when excessively produced by activating and accumulating fibroblasts, causing joint erosion, fibrosis, and stricture formation. Presently, five TNF-α inhibitors are clinically utilized to treat several inflammatory diseases. These include infliximab, adalimumab, etanercept, certolizumab pegol, and golimumab, which are monoclonal antibodies, and, lastly, etanercept, which is a fusion protein. These inhibitors are specific for all forms of TNF-α in humans, and effectively block the binding of TNF-α to its receptors. Compared with the earlier TNF-α inhibitors, golimumab is a monoclonal antibody that has a higher affinity to TNF-α and can neutralize the biological activity of TNF-α effectively.
Figure 7.
The association of TNF-α in various autoimmune diseases.
Regarding safety issues, there are several side effects of TNF-α inhibitors. General side effects of all TNF-α inhibitors involve headaches, rashes, anemia, transaminitis, infectious, and, most commonly, an injection site reaction through a subcutaneous route [90,91]. The worsening of heart failure and increased risk of developing tuberculosis as well as lymphoma are significantly observed in patients who have autoimmune diseases compared to healthy people [92,93,94]. Thus, it is recommended that all individuals be screened for these before beginning the treatment, and preventive therapy must be performed by priority if they are detected.
Currently, more TNF-α signaling inhibitors for autoimmune diseases are being clinically assessed, and even biosimilars of approved therapeutic drugs are being developed by many research institutes and companies, as the market for TNF-α inhibitors shows great promise. Furthermore, TNF-α inhibitors are used in off-label indications as well as the approved therapies for autoimmune diseases. Case reports have demonstrated that infliximab, etanercept, and adalimumab are effective against diseases such as Pyoderma gangrenous, Granuloma annulare, and Pityriasis rubra pilaris [95]. Although studies on these are still lacking, it suggests that the applicability of the TNF-α inhibitors could be expanded.
Therefore, it can be predicted that the understanding of TNF-α signaling will be much more emphasized in the near future to develop effective tools for the treatment of other autoimmune diseases as well as a wide range of diseases involving TNF.
Abbreviations
| ADMET | Absorption, distribution, metabolism, excretion and toxicity |
| AKT | Protein kinase B |
| AS | Ankylosing spondylitis |
| CD | Crohn’s disease |
| cIAP1/2 | Cellular inhibitor of apoptosis protein 1 or 2 |
| DCs | Dendritic cells |
| DD | Death domain |
| FADD | Fas-associated protein with death domain |
| IBD | Inflammatory bowel disease |
| ICAM-1 | Intracellular adhesion molecule 1 |
| JIA | Juvenile idiopathic arthritis |
| LUBAC | Linear ubiquitin chain assembly complex |
| MAPKs | Mitogen-activated protein kinases |
| MLKL | Mixed-lineage kinase domain-like protein |
| MMP | Matrix metalloproteinases |
| NF-κB | Nuclear factor κB |
| NIU | Noninfectious uveitis |
| PEG | Polyethylene glycosylated |
| PMNs | Polymorphonuclear leukocytes |
| PS | Psoriasis |
| PsA | Psoriatic arthritis |
| RA | Rheumatoid arthritis |
| RIPK1/3 | Receptor-interacting serine/threonine-protein kinase 1 or 3 |
| sTNF-α | Soluble TNF-α |
| TACE | TNF-α-converting enzyme |
| Th1 | Type 1 helper T cells |
| Th17 | Type 17 helper T cells |
| TIMPs | Tissue inhibitor of metalloproteinases |
| tmTNF-α | Transmembrane TNF-α |
| TNF-α | Tumor necrosis factor alpha |
| TNFR1 | TNF receptor type 1 |
| TNFR2 | TNF receptor type 2 |
| TRADD | TNFR1-associated death domain |
| TRAF2/5 | TNFR-associated factor 2 or 5 |
| Treg | Regulatory T cells |
| UC | Ulcerative colitis |
| VACM-1 | Vascular cell adhesion molecule 1 |
Author Contributions
Conceptualization, D.-i.J. and S.-H.Y.; Visualization, D.-i.J., A.-H.L., and H.-Y.S.; Writing—original draft, D.-i.J., A.-H.L., H.-Y.S., T.-B.K., and S.-H.Y.; Writing—review and editing, D.-i.J., A.-H.L., H.-R.S., J.-H.P., and S.-H.Y.; Funding acquisition, S.-R.L. and S.-H.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (2015R1A6A3A04058568); by a National Research Foundation of Korea (NRF) grant, funded by the government of Korea (MSIT, 2020R1F1A1076240); and by Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ014297) of the Rural Development Administration in the Republic of Korea.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bradley J.R. TNF-Mediated inflammatory disease. J. Pathol. 2008;214:149–160. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- 2.Horiuchi T., Mitoma H., Harashima S., Tsukamoto H., Shimoda T. Transmembrane TNF-alpha: Structure, function and interaction with anti-TNF agents. Rheumatology. 2010;49:1215–1228. doi: 10.1093/rheumatology/keq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang Y., Yu M., Hu X., Han L., Yang K., Ba H., Zhang Z., Yin B., Yang X.P., Li Z., et al. STAT1 mediates transmembrane TNF-alpha-induced formation of death-inducing signaling complex and apoptotic signaling via TNFR1. Cell Death Differ. 2017;24:660–671. doi: 10.1038/cdd.2016.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vandenabeele P., Declercq W., Beyaert R., Fiers W. Two tumour necrosis factor receptors: Structure and function. Trends Cell Biol. 1995;5:392–399. doi: 10.1016/S0962-8924(00)89088-1. [DOI] [PubMed] [Google Scholar]
- 5.Bazzoni F., Beutler B. The tumor necrosis factor ligand and receptor families. N. Engl. J. Med. 1996;334:1717–1725. doi: 10.1056/NEJM199606273342607. [DOI] [PubMed] [Google Scholar]
- 6.Black R.A., Rauch C.T., Kozlosky C.J., Peschon J.J., Slack J.L., Wolfson M.F., Castner B.J., Stocking K.L., Reddy P., Srinivasan S., et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 7.Moss M.L., Jin S.L., Milla M.E., Bickett D.M., Burkhart W., Carter H.L., Chen W.J., Clay W.C., Didsbury J.R., Hassler D., et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 8.Grell M., Douni E., Wajant H., Lohden M., Clauss M., Maxeiner B., Georgopoulos S., Lesslauer W., Kollias G., Pfizenmaier K., et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 9.Morita C., Horiuchi T., Tsukamoto H., Hatta N., Kikuchi Y., Arinobu Y., Otsuka T., Sawabe T., Harashima S., Nagasawa K., et al. Association of tumor necrosis factor receptor type II polymorphism 196R with Systemic lupus erythematosus in the Japanese: Molecular and functional analysis. Arthritis Rheum. 2001;44:2819–2827. doi: 10.1002/1529-0131(200112)44:12<2819::AID-ART469>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 10.Pobezinskaya Y.L., Liu Z. The role of TRADD in death receptor signaling. Cell Cycle. 2012;11:871–876. doi: 10.4161/cc.11.5.19300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pasparakis M., Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 12.Brenner D., Blaser H., Mak T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015;15:362–374. doi: 10.1038/nri3834. [DOI] [PubMed] [Google Scholar]
- 13.Holbrook J., Lara-Reyna S., Jarosz-Griffiths H., McDermott M. Tumour necrosis factor signalling in health and disease. F1000Research. 2019;8 doi: 10.12688/f1000research.17023.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haas T.L., Emmerich C.H., Gerlach B., Schmukle A.C., Cordier S.M., Rieser E., Feltham R., Vince J., Warnken U., Wenger T., et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell. 2009;36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 15.Gerlach B., Cordier S.M., Schmukle A.C., Emmerich C.H., Rieser E., Haas T.L., Webb A.I., Rickard J.A., Anderton H., Wong W.W., et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 16.Aggarwal B.B., Gupta S.C., Kim J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012;119:651–665. doi: 10.1182/blood-2011-04-325225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalliolias G.D., Ivashkiv L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016;12:49–62. doi: 10.1038/nrrheum.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L., Du F., Wang X. TNF-α induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 19.Wilson N.S., Dixit V., Ashkenazi A. Death receptor signal transducers: Nodes of coordination in immune signaling networks. Nat. Immunol. 2009;10:348–355. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- 20.Cho Y.S., Challa S., Moquin D., Genga R., Ray T.D., Guildford M., Chan F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Probert L. Tnf and Its Receptors in the Cns: The Essential, the Desirable and the Deleterious Effects. Neuroscience. 2015;302:2–22. doi: 10.1016/j.neuroscience.2015.06.038. [DOI] [PubMed] [Google Scholar]
- 22.Choy E.H.S., Panayi G.S. Mechanisms of disease: Cytokine pathways and joint inflammation in rheumatoid arthritis. N. Engl. J. Med. 2001;344:907–916. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- 23.Adegbola S.O., Sahnan K., Warusavitarne J., Hart A., Tozer P. Anti-TNF Therapy in Crohn’s Disease. Int. J. Mol. Sci. 2018;19:2244. doi: 10.3390/ijms19082244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sands B.E., Kaplan G.G. The role of TNF alpha in ulcerative colitis. J. Clin. Pharmacol. 2007;47:930–941. doi: 10.1177/0091270007301623. [DOI] [PubMed] [Google Scholar]
- 25.Levin A.D., Wildenberg M.E., van den Brink G.R. Mechanism of Action of Anti-TNF Therapy in Inflammatory Bowel Disease. J. Crohns Colitis. 2016;10:989–997. doi: 10.1093/ecco-jcc/jjw053. [DOI] [PubMed] [Google Scholar]
- 26.Celis R., Cuervo A., Ramirez J., Canete J.D. Psoriatic Synovitis: Singularity and Potential Clinical Implications. Front. Med.-Lausanne. 2019;6 doi: 10.3389/fmed.2019.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lis K., Kuzawinska O., Balkowiec-Iskra E. Tumor necrosis factor inhibitors—State of knowledge. Arch. Med. Sci. 2014;10:1175–1185. doi: 10.5114/aoms.2014.47827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raychaudhuri S.P., Raychaudhuri S.K. Biologics: Target-specific treatment of systemic and cutaneous autoimmune diseases. Indian J. Dermatol. 2009;54:100–109. doi: 10.4103/0019-5154.53175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang S., Wang J., Brand D.D., Zheng S.G. Role of TNF-TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol. 2018;9:784. doi: 10.3389/fimmu.2018.00784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sedger L.M., McDermott M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014;25:453–472. doi: 10.1016/j.cytogfr.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 31.Steeland S., Libert C., Vandenbroucke R.E. A New Venue of TNF Targeting. Int. J. Mol. Sci. 2018;19:1442. doi: 10.3390/ijms19051442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo Q., Wang Y., Xu D., Nossent J., Pavlos N.J., Xu J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6:15. doi: 10.1038/s41413-018-0016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scutellari P.N., Orzincolo C. Rheumatoid arthritis: Sequences. Eur. J. Radiol. 1998;27(Suppl. 1):S31–S38. doi: 10.1016/S0720-048X(98)00040-0. [DOI] [PubMed] [Google Scholar]
- 34.Koch A.E., Harlow L.A., Haines G.K., Amento E.P., Unemori E.N., Wong W.L., Pope R.M., Ferrara N. Vascular Endothelial Growth-Factor—A Cytokine Modulating Endothelial Function in Rheumatoid-Arthritis. J. Immunol. 1994;152:4149–4156. [PubMed] [Google Scholar]
- 35.Feldmann M., Brennan F.M., Maini R.N. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 36.Goldring S.R. Pathogenesis of bone erosions in rheumatoid arthritis. Curr. Opin. Rheumatol. 2002;14:406–410. doi: 10.1097/00002281-200207000-00013. [DOI] [PubMed] [Google Scholar]
- 37.Merola J.F., Espinoza L.R., Fleischmann R. Distinguishing rheumatoid arthritis from psoriatic arthritis. RMD Open. 2018;4 doi: 10.1136/rmdopen-2018-000656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Al-Awadhi A.M., Hasan E.A., Sharma P.N., Haider M.Z., Al-Saeid K. Angiotensin-Converting enzyme gene polymorphism in patients with psoriatic arthritis. Rheumatol. Int. 2007;27:1119–1123. doi: 10.1007/s00296-007-0349-y. [DOI] [PubMed] [Google Scholar]
- 39.Ritchlin C.T., Colbert R.A., Gladman D.D. Psoriatic Arthritis. N. Engl. J. Med. 2017;376:957–970. doi: 10.1056/NEJMra1505557. [DOI] [PubMed] [Google Scholar]
- 40.Ogawa E., Sato Y., Minagawa A., Okuyama R. Pathogenesis of psoriasis and development of treatment. J. Dermatol. 2018;45:264–272. doi: 10.1111/1346-8138.14139. [DOI] [PubMed] [Google Scholar]
- 41.Gottlieb A.B., Chamian F., Masud S., Cardinale I., Abello M.V., Lowes M.A., Chen F., Magliocco M., Krueger J.G. TNF inhibition rapidly down-regulates multiple proinflammatory pathways in psoriasis plaques. J. Immunol. 2005;175:2721–2729. doi: 10.4049/jimmunol.175.4.2721. [DOI] [PubMed] [Google Scholar]
- 42.Baumgart D.C., Carding S.R. Gastroenterology 1—Inflammatory bowel disease: Cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 43.Baumgart D.C., Sandborn W.J. Gastroenterology 2—Inflammatory bowel disease: Clinical aspects and established and evolving therapies. Lancet. 2007;369:1641–1657. doi: 10.1016/S0140-6736(07)60751-X. [DOI] [PubMed] [Google Scholar]
- 44.Ringheanu M., Daum F., Markowitz J., Levine J., Katz S., Lin X.Y., Silver J. Effects of infliximab on apoptosis and reverse signaling of monocytes from healthy individuals and patients with Crohn’s disease. Inflamm. Bowel Dis. 2004;10:801–810. doi: 10.1097/00054725-200411000-00015. [DOI] [PubMed] [Google Scholar]
- 45.Lee S.H., Kwon J.E., Cho M.L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018;16:26–42. doi: 10.5217/ir.2018.16.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rendon A., Schakel K. Psoriasis Pathogenesis and Treatment. Int. J. Mol. Sci. 2019;20:1475. doi: 10.3390/ijms20061475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boehncke W.H., Schon M.P. Psoriasis. Lancet. 2015;386:983–994. doi: 10.1016/S0140-6736(14)61909-7. [DOI] [PubMed] [Google Scholar]
- 48.Griffiths C.E.M., Barker J.N.W.N. Psoriasis 1—Pathogenesis and clinical features of psoriasis. Lancet. 2007;370:263–271. doi: 10.1016/S0140-6736(07)61128-3. [DOI] [PubMed] [Google Scholar]
- 49.Hsu Y.R., Huang J.C.C., Tao Y., Kaburaki T., Lee C.S., Lin T.C., Hsu C.C., Chiou S.H., Hwang D.K. Noninfectious uveitis in the Asia-Pacific region. Eye. 2019;33:66–77. doi: 10.1038/s41433-018-0223-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosenbaum J.T., Bodaghi B., Couto C., Zierhut M., Acharya N., Pavesio C., Tay-Kearney M.L., Neri P., Douglas K., Pathai S., et al. New observations and emerging ideas in diagnosis and management of non-infectious uveitis: A review. Semin. Arthritis Rheum. 2019;49:438–445. doi: 10.1016/j.semarthrit.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 51.Leal I., Rodrigues F.B., Sousa D.C., Duarte G.S., Romao V.C., Marques-Neves C., Costa J., Fonseca J.E. Anti-TNF Drugs for Chronic Uveitis in Adults-A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Med. Lausanne. 2019;6 doi: 10.3389/fmed.2019.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O’Rourke M., Fearon U., Sweeney C.M., Basdeo S.A., Fletcher J.M., Murphy C.C., Canavan M. The pathogenic role of dendritic cells in non-infectious anterior uveitis. Exp. Eye Res. 2018;173:121–128. doi: 10.1016/j.exer.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 53.Gilbert R.M., Zhang X.Z., Sampson R.D., Ehrenstein M.R., Nguyen D.X., Chaudhry M., Mein C., Mahmud N., Galatowicz G., Tomkins-Netzer O., et al. Clinical Remission of Sight-Threatening Non-Infectious Uveitis Is Characterized by an Upregulation of Peripheral T-Regulatory Cell Polarized Towards T-bet and TIGIT. Front. Immunol. 2018;9 doi: 10.3389/fimmu.2018.00907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Forrester J.V., Kuffova L., Dick A.D. Autoimmunity, Autoinflammation, and Infection in Uveitis. Am. J. Ophthalmol. 2018;189:77–85. doi: 10.1016/j.ajo.2018.02.019. [DOI] [PubMed] [Google Scholar]
- 55.Monaco C., Nanchahal J., Taylor P., Feldmann M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015;27:55–62. doi: 10.1093/intimm/dxu102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tracey D., Klareskog L., Sasso E.H., Salfeld J.G., Tak P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008;117:244–279. doi: 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 57.Danese S. Mechanisms of action of infliximab in inflammatory bowel disease: An anti-inflammatory multitasker. Dig. Liver Dis. 2008;40:S225–S228. doi: 10.1016/S1590-8658(08)60530-7. [DOI] [PubMed] [Google Scholar]
- 58.Arijs I., De Hertogh G., Machiels K., Van Steen K., Lemaire K., Schraenen A., Van Lommel L., Quintens R., Van Assche G., Vermeire S., et al. Mucosal Gene Expression of Cell Adhesion Molecules, Chemokines, and Chemokine Receptors in Patients with Inflammatory Bowel Disease Before and After Infliximab Treatment. Am. J. Gastroenterol. 2011;106:748–761. doi: 10.1038/ajg.2011.27. [DOI] [PubMed] [Google Scholar]
- 59.Guo Y., Lu N.H., Bai A.P. Clinical Use and Mechanisms of Infliximab Treatment on Inflammatory Bowel Disease: A Recent Update. Biomed. Res. Int. 2013;2013 doi: 10.1155/2013/581631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chatzantoni K., Mouzaki A. Anti-TNF-α antibody therapies in autoimmune diseases. Curr. Top. Med. Chem. 2006;6:1707–1714. doi: 10.2174/156802606778194217. [DOI] [PubMed] [Google Scholar]
- 61.Melsheimer R., Geldhof A., Apaolaza I., Schaible T. Remicade (R) (infliximab): 20 years of contributions to science and medicine. Biol. Targets Ther. 2019;13:139–178. doi: 10.2147/BTT.S207246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lim H., Lee S.H., Lee H.T., Lee J.U., Son J.Y., Shin W., Heo Y.S. Structural Biology of the TNF alpha Antagonists Used in the Treatment of Rheumatoid Arthritis. Int. J. Mol. Sci. 2018;19:768. doi: 10.3390/ijms19030768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reddy S.P., Shah V.V., Lin E.J., Wu J.J. Therapy for Severe Psoriasis. Elsevier; Amsterdam, The Netherlands: 2016. [Google Scholar]
- 64.Banner D.W., Darcy A., Janes W., Gentz R., Schoenfeld H.J., Broger C., Loetscher H., Lesslauer W. Crystal-Structure of the Soluble Human 55 Kd Tnf Receptor-Human Tnf-Beta Complex—Implications for Tnf Receptor Activation. Cell. 1993;73:431–445. doi: 10.1016/0092-8674(93)90132-A. [DOI] [PubMed] [Google Scholar]
- 65.Moreland L.W., Schiff M.H., Baumgartner S.W., Tindall E.A., Fleischmann R.M., Bulpitt K.J., Weaver A.L., Keystone E.C., Furst D.E., Mease P.J., et al. Etanercept therapy in rheumatoid arthritis—A randomized, controlled trial. Ann. Intern. Med. 1999;130:478. doi: 10.7326/0003-4819-130-6-199903160-00004. [DOI] [PubMed] [Google Scholar]
- 66.Tan J.K., Aphale A., Malaviya R., Sun Y., Gottlieb A.B. Mechanisms of action of etanercept in psoriasis. J. Investig. Dermatol. Symp. Proc. 2007;12:38–45. doi: 10.1038/sj.jidsymp.5650037. [DOI] [PubMed] [Google Scholar]
- 67.Azevedo V.F., Galli N., Kleinfelder A., D’Ippolito J., Urbano P.C.M. Etanercept biosimilars. Rheumatol. Int. 2015;35:197–209. doi: 10.1007/s00296-014-3080-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brambilla A., Simonini G. Handbook of Systemic Autoimmune Diseases. Volume 11 Elsevier; Amsterdam, The Netherlands: 2016. [Google Scholar]
- 69.Mease P.J. Adalimumab in the treatment of arthritis. Ther. Clin. Risk Manag. 2007;3:133–148. doi: 10.2147/tcrm.2007.3.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Simon L.S. The treatment of rheumatoid arthritis (Retracted Article. See vol 22, pg AR3, 2008) Best Pract. Res. Clin. Rheumatol. 2004;18:507–538. doi: 10.1016/j.berh.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 71.Rau R. Adalimumab (a fully human anti-tumour necrosis factor alpha monoclonal antibody) in the treatment of active rheumatoid arthritis: The initial results of five trials. Ann. Rheum. Dis. 2002;61:70–73. doi: 10.1136/ard.61.suppl_2.ii70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kobayashi T., Yokoyama T., Ito S., Kobayashi D., Yamagata A., Okada M., Oofusa K., Narita I., Murasawa A., Nakazono K., et al. Periodontal and Serum Protein Profiles in Patients with Rheumatoid Arthritis Treated with Tumor Necrosis Factor Inhibitor Adalimumab. J. Periodontol. 2014;85:1480–1488. doi: 10.1902/jop.2014.140194. [DOI] [PubMed] [Google Scholar]
- 73.McGovern J.L., Nguyen D.X., Notley C.A., Mauri C., Isenberg D.A., Ehrenstein M.R. Th17 cells are restrained by Treg cells via the inhibition of interleukin-6 in patients with rheumatoid arthritis responding to anti-tumor necrosis factor antibody therapy. Arthritis Rheum. 2012;64:3129–3138. doi: 10.1002/art.34565. [DOI] [PubMed] [Google Scholar]
- 74.LaMattina K.C., Goldstein D.A. Adalimumab for the treatment of uveitis. Expert Rev. Clin. Immunol. 2017;13:181–188. doi: 10.1080/1744666X.2017.1288097. [DOI] [PubMed] [Google Scholar]
- 75.Rivkin A. Certolizumab Pegol for the Management of Crohn’s Disease in Adults. Clin. Ther. 2009;31:1158–1176. doi: 10.1016/j.clinthera.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 76.Acosta-Felquer M.L., Rosa J., Soriano E.R. An evidence-based review of certolizumab pegol in the treatment of active psoriatic arthritis: Place in therapy. Open Access Rheumatol. 2016;8:37–44. doi: 10.2147/OARRR.S56837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Desai R.J., Hansen R.A., Rao J.K., Wilkins T.M., Harden E.A., Yuen A., Jonas D.E., Roubey R., Jonas B., Gartlehner G., et al. Mixed Treatment Comparison of the Treatment Discontinuations of Biologic Disease-Modifying Antirheumatic Drugs in Adults with Rheumatoid Arthritis. Ann. Pharmacother. 2012;46:1491–1505. doi: 10.1345/aph.1R203. [DOI] [PubMed] [Google Scholar]
- 78.Keystone E., van der Heijde D., Mason D., Landewe R., van Vollenhoven R., Combe B., Emery P., Strand V., Mease P., Desai C., et al. Certolizumab Pegol Plus Methotrexate Is Significantly More Effective Than Placebo Plus Methotrexate in Active Rheumatoid Arthritis Findings of a Fifty-Two-Week, Phase III, Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study. Arthritis Rheum. 2008;58:3319–3329. doi: 10.1002/art.23964. [DOI] [PubMed] [Google Scholar]
- 79.Curtis J.R., Mariette X., Gaujoux-Viala C., Blauvelt A., Kvien T.K., Sandborn W.J., Winthrop K., de Longueville M., Huybrechs I., Bykerk V.P. Long-Term safety of certolizumab pegol in rheumatoid arthritis, axial spondyloarthritis, psoriatic arthritis, psoriasis and Crohn’s disease: A pooled analysis of 11 317 patients across clinical trials. RMD Open. 2019;5 doi: 10.1136/rmdopen-2019-000942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shealy D., Cai A., Lacy E., Nesspor T., Staquet K., Johns L., Baker A., Brigham-Burke M., Emmell E., Bugelski P., et al. Characterization of golimumab (CNTO 148), a novel fully human monoclonal antibody specific for human TNFα. Ann. Rheum. Dis. 2007;66:151. [Google Scholar]
- 81.Shealy D., Cai A., Staquet K., Baker A., Lacy E.R., Johns L., Vafa O., Gunn G., Tam S., Sague S., et al. Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor α. mAbs. 2010;2:428–439. doi: 10.4161/mabs.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oldfield V., Plosker G.L. Golimumab: In the treatment of rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. BioDrugs. 2009;23:125–135. doi: 10.2165/00063030-200923020-00005. [DOI] [PubMed] [Google Scholar]
- 83.Zhou H.H., Jang H.S., Fleischmann R.M., Bouman-Thio E., Xu Z.H., Marini J.C., Pendley C., Jiao Q., Shankar G., Marciniak S.J., et al. Pharmacokinetics and safety of golimumab, a fully human anti-TNF-alpha monoclonal antibody, in subjects with rheumatoid arthritis. J. Clin. Pharmacol. 2007;47:383–396. doi: 10.1177/0091270006298188. [DOI] [PubMed] [Google Scholar]
- 84.Mazumdar S., Greenwald D. Golimumab. mAbs. 2009;1:422–431. doi: 10.4161/mabs.1.5.9286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kirchhoff C.F., Wang X.Z.M., Conlon H.D., Anderson S., Ryan A.M., Bose A. Biosimilars: Key regulatory considerations and similarity assessment tools. Biotechnol. Bioeng. 2017;114:2696–2705. doi: 10.1002/bit.26438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.FDA-Biosimilar. [(accessed on 18 February 2021)]; Available online: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information.
- 87.McGowan S., Jesse M. Biosimilars Pipeline Report. AmerisourceBergen; Chesterbrook, PA, USA: 2021. [Google Scholar]
- 88.Saddala M.S., Huang H. Identification of novel inhibitors for TNFα, TNFR1 and TNFα-TNFR1 complex using pharmacophore-based approaches. J. Transl. Med. 2019;17:215. doi: 10.1186/s12967-019-1965-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kwak M.S., Lee H.H., Cha J.M., Shin H.P., Jeon J.W., Yoon J.Y. Novel candidate drugs in anti-tumor necrosis factor refractory Crohn’s diseases: In silico study for drug repositioning. Sci. Rep. 2020;10:10708. doi: 10.1038/s41598-020-67801-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Quartuccio L., Zabotti A., Del Zotto S., Zanier L., De Vita S., Valent F. Risk of serious infection among patients receiving biologics for chronic inflammatory diseases: Usefulness of administrative data. J. Adv. Res. 2019;15:87–93. doi: 10.1016/j.jare.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mocci G., Marzo M., Papa A., Armuzzi A., Guidi L. Dermatological adverse reactions during anti-TNF treatments: Focus on inflammatory bowel disease. J. Crohns Colitis. 2013;7:769–779. doi: 10.1016/j.crohns.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 92.Kotyla P.J. Bimodal Function of Anti-TNF Treatment: Shall We Be Concerned about Anti-TNF Treatment in Patients with Rheumatoid Arthritis and Heart Failure? Int. J. Mol. Sci. 2018;19:1739. doi: 10.3390/ijms19061739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sartori N.S., Picon P., Papke A., Neyeloff J.L., Chakr R.M.D. A population-based study of tuberculosis incidence among rheumatic disease patients under anti-TNF treatment. PLoS ONE. 2019;14:e0224963. doi: 10.1371/journal.pone.0224963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dahmus J., Rosario M., Clarke K. Risk of Lymphoma Associated with Anti-TNF Therapy in Patients with Inflammatory Bowel Disease: Implications for Therapy. Clin. Exp. Gastroenter. 2020;13:339–350. doi: 10.2147/CEG.S237646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patel R., Cafardi J.M., Patel N., Sami N., Cafardi J.A. Tumor necrosis factor biologics beyond psoriasis in dermatology. Expert Opin. Biol. Ther. 2011;11:1341–1359. doi: 10.1517/14712598.2011.590798. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.