

Abstract
The pathological alterations that manifest during the early embryonic development due to inherited and acquired factors trigger various neurodevelopmental disorders (NDDs). Besides major NDDs, there are several rare NDDs, exhibiting specific characteristics and varying levels of severity triggered due to genetic and epigenetic anomalies. The rarity of subjects, paucity of neural tissues for detailed analysis, and the unavailability of disease-specific animal models have hampered detailed comprehension of rare NDDs, imposing heightened challenge to the medical and scientific community until a decade ago. The generation of functional neurons and glia through directed differentiation protocols for patient-derived iPSCs, CRISPR/Cas9 technology, and 3D brain organoid models have provided an excellent opportunity and vibrant resource for decoding the etiology of brain development for rare NDDs caused due to monogenic as well as polygenic disorders. The present review identifies cellular and molecular phenotypes demonstrated from patient-derived iPSCs and possible therapeutic opportunities identified for these disorders. New insights to reinforce the existing knowledge of the pathophysiology of these disorders and prospective therapeutic applications are discussed.
Keywords: Rare neurodevelopmental disorders, Induced pluripotent stem cells, Directed differentiation, CRISR/Cas9 technology, 3D brain organoids, Fragile X syndrome, Rett syndrome, Dravet syndrome, Friedreich’s ataxia, Phelan-McDermid syndrome, Spinal muscular atrophy, Miller dieker syndrome, Angelman syndrome, Huntington’s disease, Alexander disease
1. Introduction
The dynamic process of neurodevelopment commences with the neural tube formation from neural progenitor cells. As the brain matures, all of its characteristic features emerge through a highly organized cascade of events, including propagation, migration, differentiation, and functional maturation of the cells through molecular regulation (Sur and Rubenstein, 2005; Stiles and Jernigan, 2010; Tau and Peterson, 2010; Silbereis et al., 2016). Any disruption in this highly coordinated molecular event/s could impede healthy brain development, depending on the severity and cause of the insult, leading to the manifestation of neurodevelopmental disorder. While multiple factors are known to trigger developmental disorders, modifications in genes involved intimately in development could contribute significantly to the etiology of the pathogenesis (Deciphering Developmental Disorders Study, 2017). Neurodevelopmental disorders (NDDs) comprise a large group of various complex and diverse disorders, including intellectual disability, autism spectrum disorder, attention deficit hyperactivity disorder, cerebral palsy, and epilepsy, affecting brain development (Surén et al., 2012; Mullin et al., 2013; Owen and O’Donovan, 2017; Niemi et al., 2018). Besides, several rare NDDs exhibit specific characteristics and varying levels of severity triggered due to genetic and epigenetic anomalies. These disorders may be individually rare and of low prevalence, but collectively, a substantial proportion of the world population is affected.
Currently, there is no broadly established definition for a rare disease, but each country or region has its definition. As per the United States definition, any disorder that affects less than 200,000 people is considered a rare disease, while in Japan and Australia, the prevalence should be less than 50,000 and 2,000, respectively (Lavandeira, 2002). In European Union, life-threatening or chronically debilitating conditions with a prevalence of <5 in 10,000 people and requiring special combined efforts to prevent significant morbidity and mortality are considered rare diseases (Baldovino et al., 2016). One of the fundamental challenges in treating rare NDDs is the limited information about natural disease history as well as its incompletely characterized pathophysiology. The rarity of subjects and scarcity of neural tissues for detailed analysis hampers the progress of a detailed understanding of these disorders. Apart from the unavailability of disease-specific animal models, data obtained from post-mortem tissues provided insights mostly on the end-stage of the disease. However, with the advent of patient-derived induced pluripotent stem cells (iPSCs), it is now possible to understand the molecular progression of rare NDDs at the cellular, tissue, and organ level, as well as to develop potential therapeutics.
2. Human iPSCs in modeling rare neurodevelopmental disorders
The discovery of iPSCs have helped researchers evade most of the ethical issues associated with the use of embryonic stem cells and provided the tremendous opportunity and a valuable resource to understand monogenic and polygenic disorders through molecular characterization (Albani and Prakken, 2009; Bell et al., 2017; Kim et al., 2019b; a). With direct differentiation protocols efficiently generating major types of functional neuronal and non-neuronal cells, patient-derived iPSCs have provided an excellent opportunity to understand defective molecular events contributing to altered brain development in rare NDDs (Fig. 1). Various rare NDDs have been modeled in 2-dimensional (2D) cultures, and also a few studies have been performed with 3D cultures, using the iPSC technology under a controlled laboratory environment combined with gene-editing tools like CRISPR/Cas9, transcription activator-like effector nucleases (TALENS), or zinc-finger nucleases (ZFNs) (Hotta and Yamanaka, 2015; Bell et al., 2017; Huong Le et al., 2019). Nonetheless, the human brain is an extraordinarily complex and dynamic system with a vast number of specialized neurons and glia that communicate with each other through intricate pathways. Thus, it is very perplexing to extrapolate in vitro findings to the developing human brain. In this review, we gathered and deliberated the available information on altered brain development in rare NDDs gleaned from patient-derived iPSC modeling. Furthermore, we proposed future studies that could reinforce the existing knowledge in rare NDDs as well as studies on many unexplored rare NDDs affecting brain development.
Fig. 1.
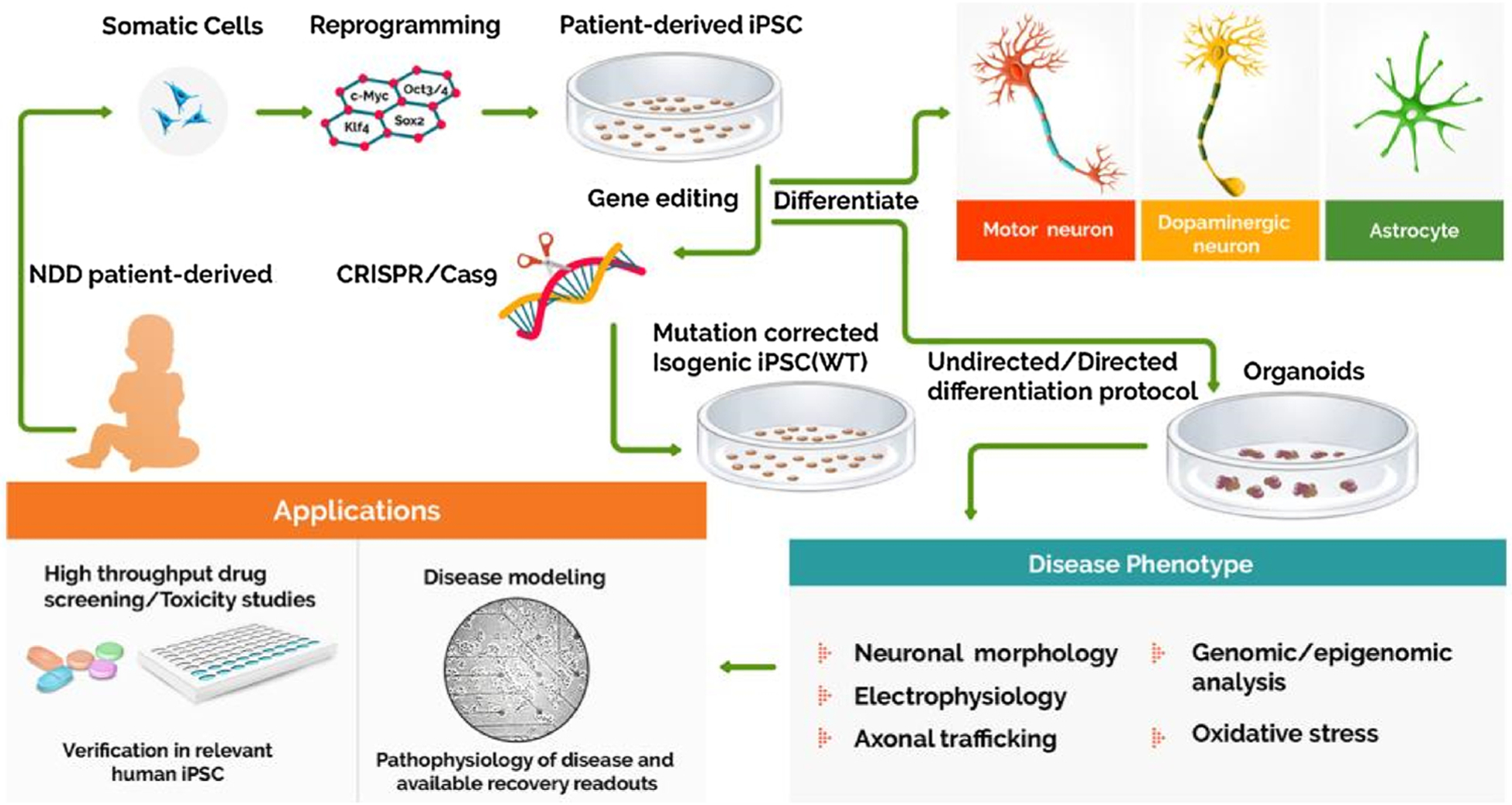
Application of iPSCs in modeling rare neurodevelopmental disorders. Patient-derived somatic cells reprogrammed into iPSCs can be differentiated into 2D neurons and glial cultures, or 3D organoids. Alternatively, the isogenic iPSC lines can be derived via the genome editing techniques like CRISPR/Cas9 that aid in precisely assessing the pathogenic mechanisms caused by the gene mutation. These neural cells can be critically employed in analyzing the disease pathology, drug screening, and toxicity studies to assess the efficacy of newly developed compounds.
2.1. Fragile X syndrome
Fragile X Syndrome (FXS), an X-linked inherited trait, affects approximately one in 4000 males and one in 8000 females, but the prevalence varies in different populations (Turner et al., 1996; Crawford et al., 1999; Patsalis et al., 1999; Tzeng et al., 2005). The symptoms of FXS comprise intellectual disability and behavioral deficits resembling autistic features (Lewis et al., 2006; Lubala et al., 2018). While up to 55 CGG repeats are typical and 55–200 are seen in premutation conditions, over 200 CGG repeats imply a full mutation and lead to epigenetic gene silencing of the 5′-untranslated region of the fragile X mental retardation 1 (FMR1, located at Xq27.3) gene, and decreased translation of FMR1 protein (FMRP) (Pieretti et al., 1991; Verkerk et al., 1991).
2.1.1. iPSCs from Fragile X syndrome patients display hypermethylation of FMR1
In iPSCs generated from FXS patients (FXS-iPSCs), FMR1 remains transcriptionally inactive, unlike embryonic stem cells where the downregulation of FMR1 RNA and protein expression occurs only during the differentiation of pluripotent cells (Eiges et al., 2007; Urbach et al., 2010). Reprogramming of fibroblasts carrying active unmethylated FMR1 gene into iPSCs triggers the generation of clones with inactive hypermethylated FMR1 gene (De Esch et al., 2014). FXS-iPSCs exhibit hypermethylation, typified by intact methylation at H3K9, no histone acetylation and H3K4 methylation, the features consistent with the inactive transcriptional state of FMR1 (Urbach et al., 2010; Sheridan et al., 2011). Genome-wide analysis validated that FMR1 hypermethylation in FXS-iPSCs is locus-specific as the rest of the genome is not differentially methylated (Alisch et al., 2013). Although FXS-iPSCs display transcriptionally inactive FMR1, iPSCs generated from patients with premutation retain transcriptionally active FMR1 with varying expression (Sheridan et al., 2011; Liu et al., 2012; Colak et al., 2014; Zhou et al., 2016).
2.1.2. Demethylation of CGG repeats in FXS-iPSC-derived neurons restores FMRP expression
The neurons generated from FXS-iPSCs with methylated and inactive FMR1 undergo an aberrant differentiation process with defective neurite outgrowth characterized by aberrant initiation and extension, implying the importance of normal FMRP expression in early neurodevelopment before synaptogenesis (Sheridan et al., 2011; Doers et al., 2014). Also, glial cell generation varied among iPSCs from different FXS patients (Sheridan et al., 2011). Removal of CGG repeats in FXS-iPSCs through CRISPR/Cas9 gene-editing resulted in the reinstatement of FMR1 with the demethylation of hypermethylated FMR1 promoter (Xie et al., 2016) and chromatin modification. Also, neurons generated from these iPSCs displayed reactivation of FMRP (Park et al., 2015). Studies from this group proposed that, for silencing FMR1, the presence of >200 CGG repeats is necessary, but not sufficient (Urbach et al., 2010; Bar-Nur et al., 2012; Park et al., 2015). Furthermore, the demethylation of CGG repeats resulted in CpG island hypomethylation with increased H3K27 acetylation and H3K4 trimethylation, and decreased H3K9 trimethylation at the FMR1 promoter. Such changes nullified the epigenetic silencing of the FMR1 gene (Gerhardt, 2017), which restored FMRP expression in FXS-iPSCs and facilitated the normal differentiation of neurons with no spontaneous hyperactivity. The demethylation of CGG repeats in post-mitotic FXS neurons also rescued their function with reactivation of the FMR1 (Liu et al., 2018). Likewise, treatment of FXS iPSCs with a combination of chromatin remodeling agents such as DNA methyltransferase inhibitor 5-azadC, and S-adenosyl homocysteine hydrolase inhibitor DZNep, potentiated the reactivation of FMR1 in neural progenitor cells as well as neurons (Vershkov et al., 2019).
2.1.3. Role of mGluR and REST in abnormal differentiation of FXS-iPSC-derived neurons
The NPCs derived from FXS-iPSCs are influenced by metabotropic glutamate receptor (mGluR) signaling, which results in an increased calcium influx and impaired differentiation (Achuta et al., 2017). These NPCs display Ca2+-permeable AMPA receptors and reduced neurite growth. The permeability of AMPAR’s to calcium depends on the presence of the GluA2 subunit, and its absence or reduced levels leads to increased Ca2+ influx through CP-AMPARs, resulting in neuronal differentiation defects and aberrant neural circuits (Achuta et al., 2018). Also, increased Ca2+ influx through L-type voltage-gated Ca2+ channels contributes to the differentiation and migration defects of NPCs derived from FXS-iPSCs (Danesi et al., 2018). Moreover, neurons derived from FXS-iPSCs lack retinoic acid (RA) signaling and homeostatic synaptic plasticity. These features contribute to a significant synaptic dysfunction because RA signaling plays a critical role in homeostatic synaptic plasticity, stabilizing firing rates required for network activity during developmental changes and learning (Turrigiano, 2012). Interestingly, CRISPR/Cas9 mediated repair of mutant FMR1 rescued defective RA signaling with the restoration of responsiveness of excitatory and inhibitory synapses to RA and normalized the synaptic function in FXS (Zhang et al., 2018). Furthermore, the RE-1 silencing transcription factor (REST), a key signaling molecule that gets repressed during neuronal differentiation and axonogenesis, is upregulated in FXS-iPSC derived NPCs due to FMRP deficiency mediated downregulation of the microRNA-382. Remarkably, overexpression of mirR-382 in FXS-iPSC derived neurons resulted in the downregulation of REST with concomitant upregulation of axon guidance genes (Halevy et al., 2015).
Additionally, cortical neurons generated from FXS-iPSCs showed differentiation defects, with longer primary neurites and immature synaptic networks, indicative of defective early dorsal forebrain development. Global gene expression and methylation profiling revealed defective developmental signaling, the extracellular matrix rearrangement, and neuronal maturation and migration defects in FXS (Boland et al., 2017). It was also found that impaired neuronal differentiation and maturation in neurons derived from FXS-iPSCs was associated with upregulation of early neuronal differentiation genes and downregulation of potassium channel genes and impaired temporal regulation of SHANK1 and NNAT (Lu et al., 2016). Also, in the absence of FMRP, FXS-iPSC derived excitatory neurons showed hyperactivity, but the reinstatement of 5% of the overall FMRP can prevent neuronal hyperactivity. Further studies showed that in a mosaic population of neurons, the presence of 20% FMRP expressing neurons is adequate for preventing the hyperactivity of neurons derived from FXS patients (Graef et al., 2020). Additional studies in NPCs from FMR1 gene knockout hiPSCs demonstrated enhanced expression of astrocyte marker GFAP. These FMRP-deficient NPCs upon differentiation at two weeks exhibited persistent GFAP expression, with abnormal firing properties resulting from reduced spontaneous calcium burst. The protein kinase inhibitor LX7101 reduced GFAP levels and corrected neuronal firing defects, likely through modulation of the AKT-mTOR and BMPR2-LIMK1-cofilin pathways (Sunamura et al., 2018), as increase in BMPR2 and LIMK1 are associated with FMRP loss and aberrant synaptic connectivity (Kashima et al., 2016).
Collectively, multiple studies performed on iPSCs derived from FXS patients have unraveled several molecular mechanisms underlying the abnormal differentiation and function of neurons. Approaches such as restoration of FMRP through the editing of mutant FMR1, repression of REST during neuronal differentiation via overexpression of mirR-382 have shown promise for apt neuronal differentiation from FXS-iPSCs. Furthermore, as FXS-iPSCs remain completely methylated while retaining the epigenetic memory, they provide an efficient model for high throughput screening of compounds for elevating the FMRP expression in FXS patient-derived neurons (Kaufmann et al., 2015; Kumari et al., 2015).
2.2. Rett syndrome
Rett syndrome (RTT), an X-linked dominant neurodegenerative syndrome found almost exclusively in females, exhibits a host of comorbidities with striking heterogeneity. RTT has an incidence rate of one in 10,000 and is typified mainly by severe mental retardation (Hagberg, 1985, 1985; Fehr et al., 2011). Mutation in the methyl-CpG binding protein 2 (MECP2 at Xq28) gene is the primary cause of this disorder (Amir et al., 1999; Webb et al., 1998). MeCP2 regulates the transcription of several genes, many of which are still being identified (Horvath and Monteggia, 2017, 2018). The symptoms of RTT commences between 6–18 months of age with decelerated head growth leading to microcephaly. The other symptoms include loss of previously acquired skills, social withdrawal, gait ataxia, stereotypic movement of the hands, recurrent seizures, autonomic dysfunction leading to cardiovascular or respiratory arrest with death occurring between 13 years of age to mid-twenties (Amir et al., 2000; Chahrour and Zoghbi, 2007; Freilinger et al., 2010).
2.2.1. Functional loss of MeCP2 in RTT-iPSCs leads to synaptic dysfunction in neurons
Studying neurons generated from RTT patient-derived iPSCs exhibiting mutations in MECP2 or FOXG1 has been an area of intense research (Rastegar et al., 2009; Marchetto et al., 2010; Ananiev et al., 2011; Williams et al., 2014; Hunihan et al., 2017). The neural progenitor cells (NPCs) from RTT-iPSCs carrying mutant MeCP2 promote L1 retrotransposon activity, which leads to somatic mutations in the brain contributing to RTT pathology (Muotri et al., 2010). Several studies have also demonstrated defects in neuronal branching and synaptic dysfunction in iPSC-RTT neurons. These neurons show significantly smaller nuclear and soma size, with reduced dendritic complexity, cell capacitance, and impaired differentiation compared to neurons generated from control-iPSCs (Ananiev et al., 2011; Cheung et al., 2011; Fernandes et al., 2015; Djuric et al., 2015; Ohashi et al., 2018). MECP2 promoter regulates L1 expression accounting for its role in RTT (Yoo et al., 2017), and decreased neuritogenesis in RTT-iPSC-derived NPCs could be restored by expressing L1. BLOC-1, a multisubunit protein complex expressed ubiquitously, plays a vital role in the biogenesis of specialized organelles of the endolysosomal system and vesicular trafficking. The BLOC-1 complex subunits include dysbindin, pallidin, muted, snapin, and cappuccino, found in both pre and postsynaptic compartments. In neurons, MeCP2 regulates BLOC-1 components such as the dysbindin and pallidin, and hence its deficiency in RTT-iPSCs contributes to synaptic dysfunction (Larimore et al., 2013).
2.2.2. Glutamatergic neuron differentiated from RTT-iPSCs reveal morphological, electrophysiological and synaptic defects
The dysfunction of glutamatergic synapses is one of the key traits of the RTT phenotype. The NPCs generated from RTT-iPSCs showed normal cellular proliferation. However, their differentiation into glutamatergic neurons resulted in distinct RTT-associated phenotypes, including smaller soma, reduced number of dendritic spines and synapses, electrophysiological defects, and altered calcium signaling. In RTT-iPSC clones derived from 3 different MECP2 mutations, reduced VGLUT1 puncta on glutamatergic neurons strongly denote the ratelimiting role of MeCP2 in regulating the number of glutamatergic synapses in human neurons (Marchetto et al., 2010). Moreover, neurons generated from the congenital variant of RTT-iPSCs from FOXG1+/− patients reveals a reduction in excitatory presynaptic VGLUT1 and postsynaptic GluA1, GluN1, and PSD-95 levels (Patriarchi et al., 2016).
2.2.3. Dysregulation of GABA-ergic neurons derived from RTT-iPSCs
Mutant MECP2 patient-derived iPSCs express reduced levels of GRID1, which encodes for GluD1, a synaptic adhesion molecule, unlike the high GLUD1 level typically expressed in normal NPCs and neurons (Livide et al., 2015). Neurons generated from the congenital variant of RTT-iPSCs from FOXG1+/− patients showed increased GluD1 protein levels known to enhance inhibitory fate during synaptic differentiation. Such changes resulted in a visible increase in the inhibitory synapse markers such as presynaptic GAD67 and postsynaptic GABAA receptor subunit α1, along with reduced excitatory synaptic markers (Patriarchi et al., 2016). However, in another study, MeCP2 deficiency resulted in delayed development of the inhibitory system. MeCP2 deficient neurons differentiated from RTT-iPSCs exhibited a delay in GABA functional switch from excitation to inhibition. MeCP2 regulated neuronal GABA switch is controlled through REST mediated expression of neuron-specific membrane transporter KCC2. A key observation was that late-onset expression of KCC2 during brain development caused the delayed onset of RTT symptoms (Tang et al., 2016). Upregulation of GABA receptors and GABAergic circuits has also been seen in MECP2 mutant neurons derived from other RTT-iPSCs with overexpression of HDAC6, resulting in reduced acetylated α-tubulin in neurons (Landucci et al., 2018).
2.2.4. MeCP2 deficiency affects astrocyte differentiation
Astrocytes also play an essential role in RTT pathogenesis. Conflicting reports are available from patient-derived iPSCs regarding the astrocyte involvement in RTT pathophysiology. It was demonstrated that NPCs differentiated from RTT-iPSCs showed neuronal maturation defects, with astrocytic differentiation remaining unaltered (Kim et al., 2011). Also, NSCs derived from MeCP2 deficient RTT-iPSCs exhibited enhanced astrocytic differentiation compared to neuronal differentiation, possibly due to the absence of direct binding of MeCP2 to GFAP, which resulted in enhanced expression and GFAP and drove cells towards astrocytic lineage (Andoh-Noda et al., 2015). Furthermore, differentiated astrocytes from RTT- iPSCs adversely affected the morphology and function of wild-type neurons (Williams et al., 2014). Besides, MeCP2 deficiency in astrocytes resulted in reduced tubulin acetylation, impaired microtubule dynamics, and microtubule-dependent dysfunction in vesicular transport (Delépine et al., 2016). Paradoxically, in another study, reduced MeCP2 was found to be associated with reduced astrocytic differentiation. The NPCs from RTT-iPSCs showed enhanced expression of the gene LIN28 critical for cell fate specification and developmental timing, which suppressed astrocyte differentiation with synaptic defects (Kim et al., 2019b; a).
2.2.5. Molecular profile through transcriptome analysis unravels RTT phenotype
Molecular studies using RTT-iPSCs, NPCs, and neurons revealed the mechanistic insights of the RTT phenotype. Transcriptome analysis of MECP2 mutant RTT-iPSCs revealed that MeCP2 regulated very early embryonic genes in undifferentiated cells. MeCP2 regulated several genes encoding mitochondrial membrane proteins along with the derepression of inactive X chromosome genes (Tanaka et al., 2014). Defective neuronal and mitochondrial function in neurons generated from RTT-iPSCs have been attributed to reduced levels of CREB and phosphorylated CREB in neurons, wherein MeCP2 regulated WNT2 expression through modulating CREB (Bu et al., 2017). Molecular profiling of these neurons revealed defective dendritic branching, with both NPCs and neurons showing increased expression of p53, p21, and H2AX deposition along with senescence phenotype (Ohashi et al., 2018). Quantitative mass spectrometry studies demonstrated lower protein levels related to axon regeneration and filopodium assembly, dendritic morphogenesis, axonal guidance, and synaptic function, during very early developmental stages and elevated levels of proteins associated with insulin receptor pathway and neuronal apoptosis. Moreover, protein dysregulation in interleukin −12 signaling and calcium homeostasis, along with attenuated levels of proteins related to cholesterol biosynthesis, fatty acid oxidation, proteolysis, and mRNA stability were evident. Network analysis of MeCP2 revealed reduced RNA/DNA binding proteins, of which MeCP2 and CAT were the most downregulated (Varderidou-Minasian et al., 2019). A recent study using RTT-iPSC-derived neurons revealed translational dysregulation of >2100 genes affecting the mTOR signaling and ubiquitin pathway, which reduced the translation of NEDD4-Family E3-ubiquitin ligases and K48-ubiquitin linked chains. It resulted in reduced proteasomal degradation and concomitant accumulation of undegraded protein targets in RTT-iPSC derived neurons (Rodrigues et al., 2020).
2.2.6. Exosomes from RTT-iPSC derived neurons are deficient in several essential proteins
In addition to changes in intracellular regulatory proteins, exosomes secreted from RTT-iPSC derived neurons did not contain many essential proteins compared to the exosomes released from a typical neuron. The quantity of ~ 237 proteins in exosomes was altered due to mutation in MECP2. Neurons derived from isogenic RTT-iPSCs with corrected MECP2 mutation, released exosomes loaded with all the essential proteins, however (Sharma et al., 2013, 2019). Transferring exosomes derived from neurons of isogenic lines to neurons derived from RTT-iPSCs enhanced their synaptic connectivity, highlighting new realms for therapeutic opportunities (Sharma et al., 2013, 2019).
2.2.7. Neurogenesis disrupting miRNAs are upregulated in MeCP2 deficient RTT-iPSCs
The role of individual miRNAs could also be ascertained from the neuropathology of RTT. Studies on 2-D and 3-D cultures of RTT-iPSC-derived neurons demonstrated that MeCP2-regulated miRNAs such as miR-199 and miR-214 with their downstream targets PAK4 and PTEN might impact neurogenesis through modulation of AKT and ERK signaling pathways. A significant upregulation in these two miRNAs was seen in RTT patients as well as MeCP2-deficient neuronal progenitors, suggesting their critical role in early neuronal development. Inhibition of miR-199 and miR-214 ameliorated the dysregulation in neuronal differentiation and rescued the neurons (Mellios et al., 2018).
2.2.8. Treatment modalities to augment MeCp2 levels in RTT-iPSCs
Wide-ranging translational research opportunities have been suggested from the studies using RTT-iPSCs. Therapies targeting the initial neurodevelopmental stages may help to regulate the downstream pathways induced by MECP2 mutation. CRISPR/Cas9 mediated repair of the endogenous MECP2 mutant alleles restored the physiological level of MeCP2 (Le et al., 2019). Pharmacological application of IGF-1 and gentamycin in optimal concentration rescued RTT-hiPSC derived glutamatergic neurons by enhancing MeCP2 levels and restoring the synaptic plasticity (Marchetto et al., 2010; Williams et al., 2014). It appeared that the IGF-1 mediated its activity through the thyroid hormone receptor (de Souza et al., 2017). Furthermore, treatment with selective HDAC6 inhibitors enhanced the acetylated α-tubulin that was disrupted by dysregulated GABA-ergic circuits and overexpression of HDAC6 (Landucci et al., 2018). Many treatment approaches have been proposed based on findings in RTT-iPSCs. These include choline supplementation (Chin et al., 2016), drug treatment that facilitates the increase in KCC2 (Tang et al., 2016; Tang et al., 2019) and phosphorylated CREB levels (Bu et al., 2017), microtubule-stabilization (Delépine et al., 2016), LIN28 downregulation (Kim et al., 2019b; a), and inhibition of p53 pathway (Ohashi et al., 2018). Also, selective ubiquitin variants that target specific E3 ligases (Rodrigues et al., 2020) have been suggested.
Another interesting study revealed that the synaptic connectivity of RTT-iPSC-derived neurons could be enhanced through a simple transfer of exosomes derived from isogenic lines (Sharma et al., 2013, 2019), implying the therapeutic value of exosomes in RTT. This also highlights the requirement of a MECP2 mutation-specific therapeutic approach as different mutations in MECP2 affect a set of different genes that result in different RTT phenotypes (Tanaka et al., 2014). Overall, these findings have great potential from the translational perspective, as they provide avenues for reducing RTT phenotypic abnormalities with repurposed drugs, new classes of known drugs, and novel drugs or through supplementation with specific natural products or nutraceuticals. In line with these findings, vigorous research needs to be carried out for reproducible and reliable outcomes in promoting interventional studies in RTT. Furthermore, how these therapeutic molecules could be delivered to the brain with stability, and functional integrity needs to be explored.
2.3. Dravet syndrome
Dravet syndrome (DS), commonly known as SMEI (severe myoclonic epilepsy of infancy), is a catastrophic form of pediatric epilepsy characterized by the occurrence of intractable seizures during early infancy progressing throughout childhood (Wolff et al., 2006; Dravet, 2011; Dravet and Oguni, 2013; Villas et al., 2017; Sun and Dolmetsch, 2018). It is a rare neurodevelopmental disorder with the incidence ranging from one in 15,000 to one in 41,000 children (Hurst, 1990; Brunklaus et al., 2012; Bayat et al., 2015; Rosander and Hallböök, 2015; Wu et al., 2015). The clinical hallmarks include developmental comorbidities such as impaired psychomotor development as well as impaired autonomic and communicative functions. Severe mental retardation develops before the age of six in most children (Wolff et al., 2006; Villas et al., 2017; Sun and Dolmetsch, 2018). De novo haploinsufficiency mutation in the SCN1A, the gene encoding alpha 1 pore-forming subunit of the Nav1.1 voltage-gated sodium channel, is the underlying cause of DS (Escayg et al., 2001; Sugawara et al., 2002; Nabbout et al., 2003; Mulley et al., 2005; Fujiwara, 2006; Shi et al., 2009).
2.3.1. Glutamatergic neurons exhibit hyper excitability in DS-iPSCs derived neurons
Several studies have been carried out in DS-patient-derived iPSCs (DS-iPSCs), with distinct mutations in SCN1A. Three DS-iPSCs generated from patients carrying a distinct mutation in the SCN1A gene, namely c.5502–5509dupGCTTGAAC, c.2965 G > C, and c.651C > G have been characterized (Schuster et al., 2019a). Neural progenitor cells derived from DS-iPSCs displayed forebrain identity and differentiated into GABA-ergic (80–90 %) and glutamatergic (~10 %) neurons. These glutamatergic neurons formed functional excitatory synapses with enhanced expression of several voltage-gated sodium channels. Electrophysiological studies revealed increased amplitude and delayed inactivation of voltage-gated sodium channels, with increased sodium current density, which resulted in hyperexcitability associated with epileptic features (Liu et al., 2013). These impairments were also seen in neurons generated directly from DS patient fibroblasts.
2.3.2. SCN1A mutation leads to functional decline in GABAergic inhibition
It is well known that SCN1A encoding Nav1.1 is expressed predominantly in the GABA-ergic inhibitory neurons (Yu et al., 2006; Ogiwara et al., 2007). Incorporation of a nonsense mutation (c.4933C > T substitution) in SCN1A in DS-iPSCs resulted in their differentiation into >50 % GABA-ergic neurons and <1% glutamatergic neurons. These neurons elicited reduced action potential (AP) with attenuated amplitude in response to larger sustained currents, mimicking pathophysiological mechanisms seen in DS. Thus, a functional decline in GABA-ergic inhibition appeared to be the major driving force contributing to epileptogenesis in DS (Higurashi et al., 2013). Several studies have corroborated these findings (Schuster et al., 2019b). For example, telencephalic interneurons generated from DS-iPSCs with p.S1328 P mutation in Nav1.1 exhibited reduced sodium currents and greatly impaired AP output upon stimulation with larger currents. The excitatory neurons remained unaffected, however. Moreover, electrophysiological studies in DS-iPSCs generated from 2 patients carrying distinct missense (c.4261 G > T) and a nonsense mutation (c.3576_3580 del TCAAA), revealed the differential impact of sodium currents in forebrain GABA-ergic neurons. Intriguingly, GABA-ergic neurons derived from DS-iPSCs carrying missense mutation elicited reduced APs and sodium current density compared to DS-iPSCs with nonsense mutation (Kim et al., 2018). Such changes likely reflect substitution of glycine with the tryptophan, which can hamper the sodium channel pore and impede the sodium permeability, resulting in altered excitability of the neuron (Meisler and Kearney, 2005; Meisler et al., 2010).
2.3.3. Effect of histone proteins and tyrosine hydroxylase on DS patient derived neurons
The factors other than the SCN1A mutation could also contribute to the vast phenotypic differences among individuals with SCN1A mutations. Transcriptomic analysis of NPCs and GABA-ergic neurons revealed deregulated histone and cell cycle protein expression along with transcriptional changes and aberrant chromatin remodeling pathways in Nav1.1 haploinsufficient GABA-ergic neurons (Schuster et al., 2019b). Quantitative PCR and western blot analysis revealed elevated tyrosine hydroxylase in the mutant form compared to isogenic (wild type) neurons, suggesting that perturbation in the dopaminergic system could be responsible for the neurocognitive abnormalities observed in DS patients (Maeda et al., 2016).
2.3.4. Application of DS-iPSC derived neurons in evaluating therapeutic efficacy of drugs
Pharmacological treatment of DS-iPSC-derived neurons with antiepileptic drug phenytoin prevented the hyperexcitability from being observed due to persistent sodium channel activation (Jiao et al., 2013). Cannabidiol (CBD) as an adjunct therapy, is being used to treat convulsive seizures with other standard care (Devinsky et al., 2019; Upadhya et al., 2018). CBD enhanced the excitability of DS-iPSC-derived GABA-ergic neurons and inhibited the excitability of glutamatergic neurons at low concentrations. Such effects supported CBD’s translational value for treating DS through modulation of network excitability (Sun and Dolmetsch, 2018). In another study, the ectopic expression of Nav1.1 reversed the impairment in inhibitory neurons (Sun et al., 2016). The development of new drugs to combat neurological deficits seen in DS is a challenging field. The various drugs that can attenuate epileptogenesis and modulate GABA-ergic inhibition can be successfully tested in DS-iPSC-derived neurons and hence promises a better outlook for developing apt drug therapy for DS.
2.4. Friedreich’s Ataxia
Friedreich Ataxia (FRDA) is an autosomal recessive genetic disorder caused due to homozygous hyper expansion of Guanine-adenine adenine (GAA) motif in several hundreds of copies, within the first intron of the frataxin gene (FXN) located on chromosome 9 (Campuzano et al., 1996; Dürr et al., 1996; Herman et al., 2006; Long et al., 2017). The FXN gene encodes frataxin, a mitochondrial protein, involved in the biosynthesis of iron-sulfur (Fe-S) and iron metabolism (Martelli et al., 2007; Colin et al., 2013). In FRDA patients, the frataxin mRNA and protein levels were significantly reduced compared to those of healthy individuals. Reduced frataxin level mediated reduction in the activity of Fe-S cluster enzymes and iron accumulation in mitochondria resulted in a significant sensory neuronal death in dorsal root ganglia and cerebellar dentate nucleus (Koeppen and Mazurkiewicz, 2013). Nearly 60 % of death in FRDA occurs due to cardiac dysfunction during early adulthood (Tsou et al., 2011).
2.4.1. FRDA patient derived iPSCs exhibit reduced frataxin levels and mitochondrial dysfunction
Neurons generated from FRDA patient-derived iPSCs (FRDA-iPSCs) highlighted mitochondrial dysfunction and cell death as standard features occurring due to reduced levels of FXN (Ku et al., 2010; Liu et al., 2011; Du et al., 2012; Eigentler et al., 2013; Hick et al., 2013; Igoillo-Esteve et al., 2015). The intrinsic cell death pathway in these neurons is activated by the induction of a pro-apoptotic protein Bim (Igoillo-Esteve et al., 2015). The cortical and sensory neurons generated from FRDA-iPSCs carry lower levels of Fe–S cluster proteins, display defective iron metabolism and excessive oxidative stress with higher levels of mitochondrial SOD2 and ROS, associated with reduced GSH levels (Codazzi et al., 2016; Hu et al., 2017). Contrary to this, another study conducted in FRDA-iPSCs with lower FXN levels showed normal mitochondrial function as well as no increase in cell death. The grafting of these NPCs into the cerebellar region of the adult rodent brain also resulted in differentiation and integration into the host brain (Bird et al., 2014).
2.4.2. Therapeutic strategies for elevating FXN levels
Various therapeutic strategies have been evaluated to enhance FXN expression and reduce the FRDA phenotype (Schreiber et al., 2019). Deletion of expanded GAA repeats in FRDA-iPSCs using ZFN rescued neuronal phenotype such as reduced aconitase activity and intracellular ATP levels by resuming FXN expression (Li et al., 2015). Studies with NPCs generated from FRDA-iPSCs demonstrated that electroporation mediated entry of antisense oligonucleotide could enhance FXN expression (Shen et al., 2019). Furthermore, FXN expression in FRDA-iPSC derived cells was activated by using GAA- specific synthetic transcription elongation factors (Syn-TEF1) (Erwin et al., 2017). Remarkably, intrinsic pathway-mediated apoptosis in FRDA-iPSC derived neurons could be prevented by treating neurons with cAMP activator forskolin or glucagon-like peptide (GLP-1) analog exenatide through increased FXN expression, reduced oxidative stress, and enhanced mitochondrial function (Igoillo-Esteve et al., 2015, 2020).
Because of the efficiency in reversing FXN silencing, HDAC inhibitors could be effectively used to express FXN as a treatment for FRDA. Several classes of HDAC inhibitors have been tested for their efficacy in FRDA. For reversing FXN silencing, HDAC1, 2, and 3 should be necessarily targeted, and the key residue for regulating FXN transcription is lysine 9 of H3 histone (Soragni et al., 2014). Among several HDAC inhibitors, only compounds targeting HDACs 1 and 3 mediated a slow-on/slow-off effects for the HDAC enzymes (Soragni and Gottesfeld, 2016). The reprogramming of FRDA patient fibroblasts into iPSCs in the presence of small molecule inhibitors such as HDAC inhibitor sodium butyrate and monoamine oxidase inhibitor tranylcypromine corrected repressive histone modifications at FXN locus and enhanced FXN expression. Nonetheless, long term treatment resulted in progressive and persistent expansions of the GAA repeat and re-silencing of the FXN gene leading to therapeutic inefficacy (Ku et al., 2010; Polak et al., 2016). Interestingly, the treatment of these neurons with 2-aminobenzamide HDAC inhibitor (HDACi 109, RG2833) increased the FXN levels in a dose-dependent manner. Importantly, increased FXN transcription by this inhibitor was not associated with enhanced GAA repeat instability or induction of GAA-repeat RNA foci, implying its high translation value for treating FRDA (Soragni et al., 2014). Defective iron metabolism and oxidative stress-mediated mitochondrial dysfunction and apoptosis of cortical neurons derived from FRDA-iPSCs could also be rescued with HDACi 109 through the upregulation of FXN expression (Codazzi et al., 2016). With this treatment, increased FXN expression was also seen in primary sensory neurons (Hu et al., 2017). Furthermore, HDACi 109 partially rescued deregulated pathways linked to neuronal function, transcriptional regulation, organization of extracellular matrix, and apoptosis in neurons derived from FRDA-iPSCs (Lai et al., 2019). Another 2-aminobenzamide HDAC inhibitor (compound 106) also showed effectiveness for increasing FXN expression in NSCs through modulation of transcriptional and translational regulation of several genes, including transcription elongation factor TCEB2 (Shan et al., 2014).
Overall, besides helping in understanding the pathophysiology, FRDA-iPSCs provided excellent material for new drug discovery such as HDAC inhibitors, antisense oligonucleotides, GAA-specific synthetic transcription elongation factors, and cAMP activators for enhancing FXN expression. Although a few drugs mediated the expression of FXN in FXN deficient FRDA-iPSC-derived neurons with progressive and persistent expansions of the GAA repeat and re-silencing of the FXN gene, the other drugs showed great promise with a high translational value. The neurons derived from FRDA-iPSCs are also useful for predicting novel drugs’ translational efficacy before their clinical trials (Georges et al., 2019).
2.5. Phelan-McDermid syndrome
Phelan-McDermid syndrome (PMDS), also known as 22q13.3 deletion syndrome is a contiguous gene disorder, caused due to terminal deletion of chromosome 22q13 (Phelan and McDermid, 2012). The majority of the deletions in this region include SHANK3 gene that encodes synaptic scaffolding proteins located at glutamatergic synapses connecting membrane-bound receptors to the actin cytoskeleton (Durand et al., 2007; Phelan, 2008; Gong et al., 2012; Phelan and McDermid, 2012; Monteiro and Feng, 2017). The symptoms include universal developmental delay, varying levels of intellectual impairment, speech impairment, and neonatal hypotonia with > 50 % patients showing features of ASD (Phelan, 2008; Phelan and McDermid, 2012; Sarasua et al., 2014).
2.5.1. PMDS-iPSCs derived neurons reveal reduced SHANK3 levels with synaptic deficits
Aberrant synaptic connectivity is a common feature observed in neurons derived from PMDS patient-derived iPSCs (PMDS-iPSCs). These neurons displayed reduced SHANK3 expression and defects in excitatory synaptic transmission, while the inhibitory synapses were unaffected (Fig. 2). These neurons generated smaller currents in response to either AMPA or NMDA application due to a reduced expression of GluA1 and GluN1 protein, respectively, while the amplitude of GABA-evoked currents was unaltered. Restoration of the SHANK3 expression ameliorated these synaptic defects in excitatory neurons (Shcheglovitov et al., 2013). The defects during the early stages of the differentiation could lead to synaptic defects in PMDS. The neurons derived from PMDS-iPSCs with SHANK3 mutations exhibited reduced soma size, and longer neurites during the early stages, while mature neurons displayed a reduced number of synaptic puncta with 30 fold reduction in SHANK3 expression, which could be restored by the overexpression of SHANK3 (Kathuria et al., 2018; Gouder et al., 2019). RNA sequencing of NPCs and neurons generated from PMDS-iPSCs revealed that SHANK3 deficiency resulted in the downregulation of genes related to Wnt signaling, protein translation, and embryonic development. In contrast, genes related to pre and post-synaptic density, synaptic plasticity, and potassium channel activity were upregulated. (Breen et al., 2020).
Fig. 2.
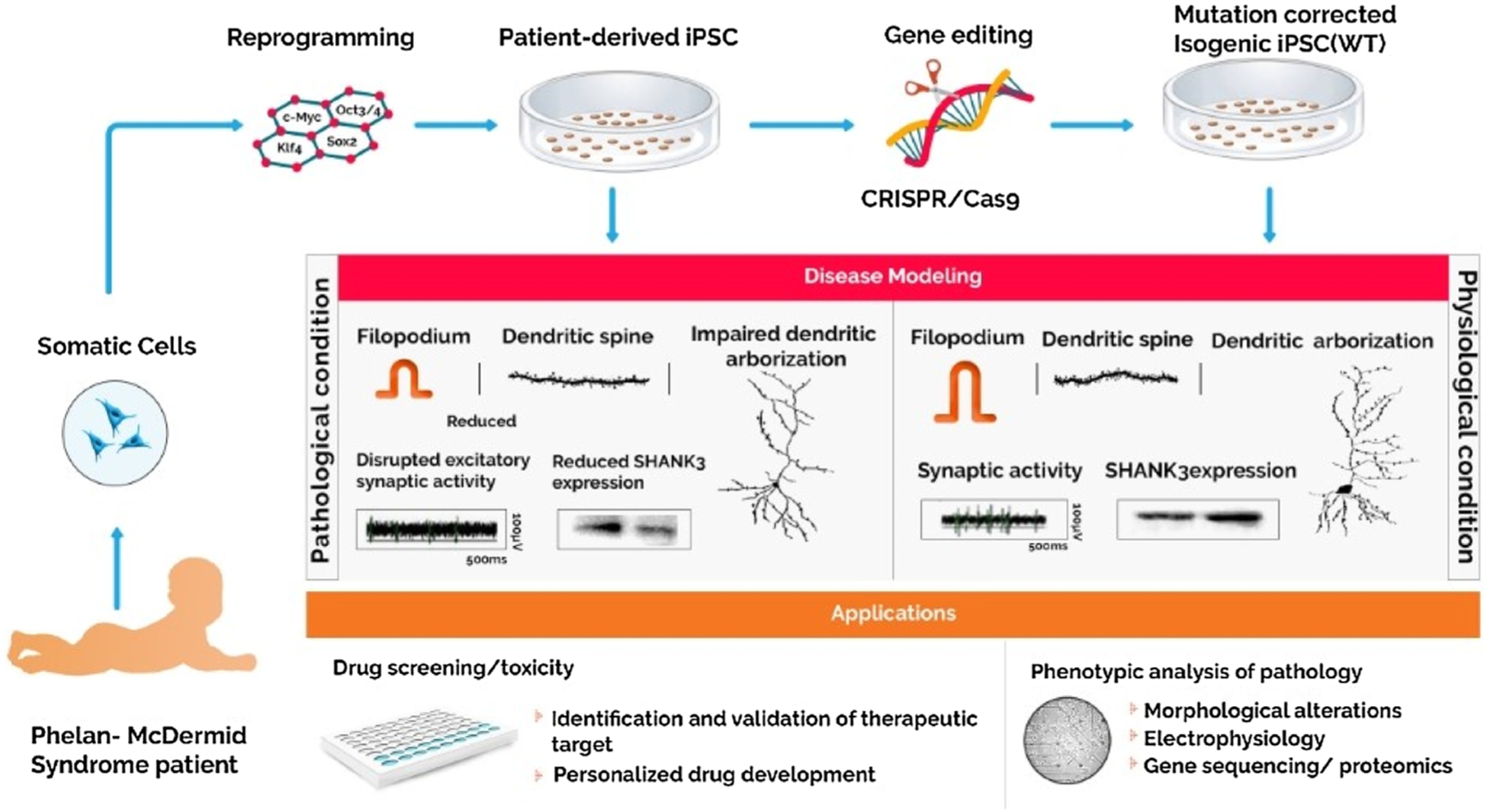
Phelan-McDermid Syndrome patient-derived iPSCs obtained after reprogramming have been utilized for disease modeling along with their isogenic control lines obtained via genomic correction of the mutation. The differentiated neural cells allow for in vitro study of the disease phenotype and can be used for high throughput drug screening and validation of drug targets that can be translated into clinical practice.
2.5.2. Treatment with IGF-1/Akt activators/CLK2 inhibitors can restore synaptic defects
Treatment with IGF-1 increased the number of synaptic NMDA and AMPA receptors and restored excitatory synaptic transmission (Shcheglovitov et al., 2013). Further, Akt activation and inhibition of the Cdc2-like kinase 2 (CLK2) also rescued synaptic deficits in PMDS-iPSC derived neurons (Bidinosti et al., 2016). Overall, it appeared that SHANK3 haploinsufficiency mediated compromised structural integrity during early neuronal development leads to synaptic defects in mature neurons, but treatment with 1GF-1 or Akt activators or CLK2 inhibitors could improve PMDS phenotype.
2.6. Spinal muscular atrophy
Spinal muscular atrophy (SMA) is a fatal motor neuron disorder characterized by lower motor neuron degeneration in the spinal cord’s anterior horn and is known to affect one in 11,000 live births (Mercuri et al., 2018). Clinical features include muscle weakness, paralysis, respiratory failure, and death (Crawford and Pardo, 1996; Jablonka et al., 2000; Jablonka and Sendtner, 2003; Lefebvre et al., 1997). Of the four types of SMA (SMA I-IV) based on severity (Munstat and Davies, 1992), the most severe and frequent type, (D’Amico et al., 2011) is SMA type 1 (Werdnig-Hoffmann disease), caused due to reduced survival motor neuron (SMN) protein levels in motor neurons, encoded by SMN1 gene, located in chromosome 5q11.2-q13.3 (Bürglen et al., 1996; Lefebvre et al., 1995). Since SMN protein is expressed ubiquitously, apart from motor neurons, other types of cells may also get affected. However, sensory neurons are not responsible for motor neuron loss in SMA (Schwab and Ebert, 2014).
2.6.1. SMN deficiency leads to mitochondrial dysfunction and ER stress in SMA-iPSCs
The patient-derived iPSCs have been generated from SMA type 1-IV (Boza-Morán et al., 2015; Ebert et al., 2009; Heesen et al., 2016; Valetdinova et al., 2019). In SMA, the motor neurons are more vulnerable than other neural types. Reduced dendritic and axonal length with increased selective motor neuron death, a defect mimicking disease phenotype has been recapitulated in motor neurons generated SMA-iPSCs (Ebert et al., 2009; Chang et al., 2011; Ohuchi et al., 2016; Son et al., 2019). SMN being a critical component of the core splicing machinery, its deficiency leads to DNA damage in SMA motor neurons (Jangi et al., 2017), probably leading to massive motor neuron death. Endoplasmic reticulum and mitochondrial dynamics are also drastically altered in SMN deficiency. Generation of mitochondrial ROS and oxidative stress-mediated cell death has been reported in these motor neurons (Ando et al., 2017). Endoplasmic reticulum and mitochondrial dynamics are also drastically altered in SMN deficiency. RNA sequencing of motor neurons from SMN deficient SMA-iPSCs demonstrated the upregulation of apoptotic genes and selective hyper-activation of the ER stress pathway indicating the critical role of ER in the SMA phenotype. ER stress is caused due to activated unfolded protein response due to SMN deficiency. Furthermore, ER stress surges in mature motor neurons, not linked to apoptosis. ER stress showed an inverse link with SMN levels (Ng et al., 2015). The immature motor neurons derived from SMA-iPSCs with SMN deficiency were unaffected, but with maturation, neurons demonstrated impaired mitochondrial dynamics with reductions in mitochondrial number, area, and axonal transport (Xu et al., 2016).
2.6.2. Reduced motor neuron survival due to SMN deficiency
The motor neurons generated from iPSCs of SMAI and SMAIIIa/IV individuals showed normal differentiation. However, the survival rate was compromised due to the progressive decline in SMN protein. Further, actin-binding and bundling protein plastin 3 (PLS3) expressed abundantly in growth cones of WT motor neurons was significantly reduced in these neurons, increasing their vulnerability to SMA (Boza-Morán et al., 2015; Heesen et al., 2016). Moreover, motor neurons generated from SMAI-iPSCs exhibited hyperexcitability due to enhanced sodium channel activity (Liu et al., 2015), but such changes were absent in neurons derived from SMAIII-iPSCs (Lin et al., 2017). Mass spectrometry studies revealed reduced ubiquitin-activating enzyme 1 protein and ubiquitin carboxyl-terminal esterase L1 linked to neurodevelopment and differentiation, highlighting a defective ubiquitin pathway in SMA (Fuller et al., 2016). The ventral spinal organoids generated using SMA-iPSCs revealed abnormal cell cycle reactivation induced by p53 activation due to SMN deficiency in motor neurons (Hor et al., 2018). Furthermore, RNA sequencing studies demonstrated a deficiency in neuron-specific presynaptic cell adhesion protein neurexin 2 (NRXN2), a critical protein supporting motor neuron survival and function (Rizzo et al., 2019).
2.6.3. Role of miRNAs and SnRNAs in motor neuron functions in SMA
Altered function of individual miRNAs supporting neuronal survival has been observed in SMA-iPSC derived neurons. The motor neurons expressed reduced NCAM and motor neuron-specific markers (Murdocca et al., 2016). Profiling of miRNA in motor neurons derived from SMA-iPSCs demonstrated selective downregulation of miR-23a, and restoration of miR-23a expression in motor neurons protected them from degenerating, which implied the crucial role of miR-23a on SMA phenotype (Kaifer et al., 2019). Moreover, astrocytes generated from SMA-iPSCs activated phenotypes (Ohuchi et al., 2016), which increased the expression of miR-146a and induced the death of motor neurons. Interestingly, such motoneuron death could be rescued with the inhibition of astrocytic miR-146a (Sison et al., 2017). Also, a delicate balance was found between multifunctional small noncoding RNAs (SnRNA) such as U1 small nuclear (sn) RNA (U1) and variant (v)U1 in human cells. In SMN deficient SMA-iPSC derived motor neurons, this pattern of expression was specifically deregulated, leading to an imbalance in the vU1/U1 ratio and neuromuscular dysfunction phenotype (Vazquez-Arango et al., 2016).
2.6.4. Evaluation of neuromuscular junction defects using SMA-iPSC
The various defects in neuromuscular junction have been modeled using hiPSCs to evaluate the pathophysiology and drug discovery in vitro by expression of myogenic differentiation 1 protein. Following their long term differentiation, iPSCs developed into a functional neuromuscular junction (NMJ) like structures containing neurons, myotubes, and Schwann cells. In an isogenic SMN deficient NMJ model, SMN deficiency caused abnormal NMJ with functional defects consistent with the SMA phenotype (Lin et al., 2019). Additionally, the NMJ-like structures of motor neurons developed from SMA-iPSCs revealed significant impairment in the clustering of acetylcholine receptors (AChR) (Yoshida et al., 2015).
2.6.5. Role of astrocytes and oligodendrocytes in SMA phenotype
The studies using SMA-iPSCs revealed that astrocytes and oligodendrocytes also contribute to the SMA phenotype. The functional and morphological alterations, along with the upregulation of GFAP and nestin expression through activation of astrocytes, possibly leads to motor neuron death. The basal calcium levels were dysregulated in these astrocytes with an attenuated response to the ATP stimulation (Mcgivern et al., 2013). Although SMN deficiency contributed to these astrocytes’ functional defects, astrocyte activation did not appear to be related to the oxidative stress induced by SMN deficiency (Patitucci and Ebert, 2016). SMA was also associated with the impaired differentiation of oligodendrocytes. The impairments were evident from the downregulation of NG2, a marker of the oligodendrocyte precursor cells, and MBP, a marker of myelinating oligodendrocytes (Ohuchi et al., 2019).
2.6.6. Multiple translational approach for improving motor neuron survival in SMA
Several therapeutic opportunities are being explored using SMA-iPSC derived motor neurons. Since mitochondrial function is compromised in SMA, exposure to free radical scavengers such as edaravone, levetiracetam (Ando et al., 2019, 2017) and antioxidant N-acetylcysteine (Xu et al., 2016) reversed the mitochondrial ROS and oxidative stress-induced cell death in these motor neurons. Interestingly, motor neuron survival was enhanced using ER stress inhibitors such as Salubrinal or Guanabenz by reversing the SMA phenotype of motor neurons without altering SMN levels (Ng et al., 2015). Various therapeutic agents that enhance cell survival have also been investigated using SMA-iPSCs. Ras mimetic rigosertib (Son et al., 2019), caspase-3 specific inhibitor Z-DVED-FMK and anti-Fas monoclonal antibody (Sareen et al., 2012), overexpression of motor neuron survival specific presynaptic cell adhesion protein neurexin 2 as well as RNA binding protein SYNCRIP (Rizzo et al., 2019) are some of the agents known to attenuate apoptosis. Furthermore, treatment with neuroprotective recombinant chemokine monocyte chemoattractant protein 1 (MCP1) enhanced neurite growth of motor neurons derived from SMA-iPSCs (Martin et al., 2017). Restoration of miR-23a expression in these motor neurons (Kaifer et al., 2019) and inhibition of astrocytic miR-146a (Sison et al., 2017) also aided in combating the disease phenotype. Using a 3-D spinal organoid model derived from SMA-iPSCs, studies showed that the cell cycle inhibitor CDK4/6 rescued the motor neuron from degeneration (Hor et al., 2018). Treatment with valproic acid and antisense oligonucleotides could reinstate the neuromuscular junction defects through enhancing SMN proteins (Yoshida et al., 2015). Using the in vitro model of NMJ developed from SMA-iPSCs, therapeutic molecules that help in normalizing NMJ function in SMA could be identified (Lin et al., 2019).
Gene therapy mediated therapeutic modules for SMA have also been investigated. Using CRISPR/Cpf1 homology-directed repair or ASO variants such as morpholinos, the aberrant SMN1 paralogous SMN2 gene was converted to an SMN1-like gene in SMA-iPSCs (Ramirez et al., 2018; Zhou et al., 2018; Valetdinova et al., 2019). Motor neurons from SMA-iPSCs revealed long non-coding RNAs derived from the antisense strand of SMN. Hence targeted degradation of these antisense strands with ASOs transcriptionally activating SMN2 enhanced the SMN expression (Nizzardo et al., 2015; d’Ydewalle et al., 2017). Moreover, several cyclic tetrapeptide histone deacetylase inhibitors and thyrotropin-releasing hormone (TRH) analog also enhanced SMN2 expression. (Ohuchi et al., 2016; Lai et al., 2017). Even with a cell therapy approach, motor neurons generated from SMA-iPSCs demonstrated SMA specific features. Correction of genetic defect improved motor neurons defects, and transplantation of these cells improved the disease phenotype and extended life span in a mouse model of SMA (Corti et al., 2012).
Thus, extensive studies on SMA-iPSCs have aided the understanding of molecular pathophysiology, drug screening, and investigation of the promise of cell and gene therapy. The studies showed that reduced SMN protein was closely linked to reduced dendritic growth, ER and mitochondrial impairments, and motor neuron death. The studies have also unraveled the promise of several drugs and new therapeutic avenues for alleviating the SMA phenotype’s adverse effects. Furthermore, SMA-iPSCs-derived neurons are being utilized for assessing drug response in SMA patients (Garbes et al., 2013).
2.7. Miller Dieker syndrome
Miller Dieker syndrome (MDS), caused by a submicroscopic deletion of 17p13.3, is characterized by smooth cerebral surface and severe lissencephaly. The dominant defective gene is LISI, along with YWHAE gene coding for 14.3.3ε with phosphoserine/threonine binding ability, to diverse signaling proteins and/or CRK that codes for an adaptor signaling protein (Reiner et al., 1993; Cardoso et al., 2003). LISI protein, in association with NDEL1 and 14.3.3ε, forms an intracellular multiprotein complex and regulates several cellular processes, including cell division, neuronal survival, proliferation, and migration. Their deficiency leads to aberrant cortical development with attenuated gyrification (Sheen et al., 2006a, 2006b; Wynshaw-Boris, 2007; Nagamani et al., 2009). The significant symptoms include developmental delay, intellectual disability, abnormal spasticity, and craniofacial dysmorphisms and seizures (Blazejewski et al., 2018).
2.7.1. Forebrain organoids from MDS-iPSCs exhibit abnormal cortical organization
For the evaluation of the impact of MDS mutations on specific biological processes and multiple cell types comparable to the first trimester of cortical development, cerebral organoids were cultured from MDS patient-derived iPSCs (MDS-iPSCs). Forebrain organoids obtained from MDS-iPSCs were significantly smaller in size, exhibited asymmetric cell division of the ventricular zone radial glial cells (vRGCs) with the abnormal cortical organization. Furthermore, alterations in the microtubule network of vRGCs disrupted the structural organization leading to aberrant N-cadherin/ β-catenin/ Wnt signaling axis. (Iefremova et al., 2017). In yet another study, cerebral organoids cultured from three MDS patient-derived iPSCs recapitulated several MDS phenotypes such as smaller sized organoids, aberrant neocortical expansion due to cytokinesis defects in outer radial glial cells, and defects in neuronal migration (Bershteyn et al., 2017). The smaller organoid size was due to severe apoptosis-induced cell death in founder neuroepithelial stem cells in ventricular zone-like regions, but the proliferation of neuroepithelial cells was unaffected. Also, an increased incidence of horizontal division instead of vertical cleavage was demonstrated in neuroepithelial stem cells. Mitosis was delayed due to cytokinesis defects in outer radial glial cells that are critical for human neocortical expansion (Bershteyn et al., 2017).
2.7.2. Wnt activation and correction of MDS causative chromosomal deletion rescues MDS patient derived organoids
Pharmacological activation of Wnt signaling with GSK3-β inhibitor resulted in a homogenous generation of cortical niche and subsequently ameliorated aberrant growth in MDS patient-derived organoids (Iefremova et al., 2017). The correction of MDS causative chromosomal deletion and restoration of the LIS1 and 14−3-3ε proteins ameliorated the neuronal migration defects (Bershteyn et al., 2017). In summary, organoids derived from MDS-iPSC patients recapitulated most of the disease phenotype and reinstated LIS1 and 14−3-3ε proteins through correction of the genetic defects, which ameliorated the aberrant cortical development and neuronal migration defects.
2.8. Angelman syndrome
Angelman syndrome, a rare neurodevelopmental disorder, affects 1 in 15,000 individuals. The loss of UBE3A gene function that is exclusively expressed from maternal allele in the neurons is the cause of the disease (Jiang et al., 1999; Fink et al., 2017). Symptoms include intellectual disability, developmental delays, language impairments, ataxia and recurrent seizures (Williams, 2005; Mabb et al., 2011).
2.8.1. AS-iPSC derived neurons undergo paternal UBE3A allele repression during neural differentiation
Functional neurons have been generated from AS patient-derived iPSCs (AS-iPSCs). Following reprogramming from control and AS patient fibroblasts, control iPSCs maintained original methylation imprint, carrying both the methylated maternal allele and an unmethylated paternal allele, whereas the AS- iPSCs demonstrated only an unmethylated paternal allele. Upon neuronal differentiation imprinting of UBE3A is established, with the repression of paternal UBE3A allele, associated with up-regulation of a UBE3A antisense transcript (Chamberlain et al., 2010)
2.8.2. Late-onset of paternal UBE3A silencing is observed in AS- iPSC derived neurons
Characterization of AS-iPSCs derived from a patient carrying three-base pair deletion in UBE3A revealed a strong SNHG14 induction, leading to imprinted expression of UBE3A, followed by silencing of paternal UBE3A expression only late during neuronal differentiation. Hence iPSCs derived from AS patients can be used to delineate the molecular pathways affected by UBE3A deletion (Stanurova et al., 2016). Parallel studies using AS-iPSCs depicted imprinting defects leading to loss of DNA methylation at chromosome 15 imprinting center, a region involved in controlling the maternal-specific expression of UBE3A(Neureiter et al., 2018).
2.8.3. Loss of UBE3A function disrupts synaptic activity in AS derived iPSC neurons
Neurons generated from AS-iPSCs exhibit enhanced depolarized resting membrane potential and an immature firing of the action potential. Further, attenuated spontaneous excitatory synaptic activity and the diminished activity-dependent synaptic plasticity was also demonstrated. The loss of UBE3A is the causative factor that leads to the given phenotypic differences. Knocking out of UBE3A in the isogenic control cell lines recapitulated these defects (Fink et al., 2017). Unsilencing the paternal UBE3A expression through pharmacological intervention such as topoisomerase inhibitor topotecan could help to recoup from these phenotype by restoring the endogenous UBE3A levels (Fink et al., 2017). Studies with 3D human cerebral organoid (hCO) revealed an early transition of UBE3A from the cytoplasm to the nucleus. Moreover, hCO exhibited a cortex-like development that recapitulates the specific time window during which UBE3A is imprinted or undergoes changes in subcellular localization (Sen et al., 2019). Thus, UBE3A imprinting defects during neuronal differentiation and UBE3A functional loss observed in AS-iPSC derived neurons contribute to the understanding of AS phenotype.
2.9. Huntington’s disease
Huntington’s disease (HD) is a monogenic neurodevelopmental disorder caused due to CAG trinucleotide expansion of ≥36 repeats in exon 1 of the HTT gene and is inherited in an autosomal dominant manner (Walker, 2007; Ross et al., 2014; McColgan and Tabrizi, 2018). Epidemiological studies revealed that 3–7 in 100,000 individuals are affected by HD (Ohlmeier et al., 2019; Pringsheim et al., 2012). HD patients exhibit a triad of motor, cognitive, and behavioral deficits that manifests over time (Roos, 2010; Singer, 2012). Classic sign involves chorea, leading to psychomotor retardation. Although typified as a neurodevelopmental disorder, a plethora of evidence suggest that abnormal neurodevelopment might play a critical role in triggering the disorder.
2.9.1. HD-iPSC derived neurons typify CAG repeat expansion related phenotype
Microarray profiling of iPSCs derived from HD patients (HD-iPSCs) and controls depicted CAG expansion-associated changes in gene expression that segregated HD cell lines from controls and the early versus late onset of the disease. The HD derived neural progenitor cells carrying medium and long CAG repeat expansions elicited typical features, including alterations in electrophysiological, cell adhesion, metabolic and cell death phenotype controls (HD iPSC Consortium, 2012). HD-iPSCs also exhibited an increase in lysosomal activity (Camnasio et al., 2012). Neural cultures derived from HD NSCs that were patterned towards a striatal fate elicited a high mortality rate due to expanded CAG repeats. Since BDNF is significantly reduced in HD patients, leading to striatal degeneration, withdrawal of this trophic factor resulted in increased caspase 3/7 activity and increased mortality rate. The HD cells demonstrated a distinct CAG expansion dependent phenotype in calcium signal dysfunction when subjected to glutamate-induced excitotoxicity. Moreover, treatment with toxic cell stressors such as H2O2 (oxidative stress) and 3-MA (autophagy inhibition) led to enhanced cell death in HD lines compared to controls (HD iPSC Consortium, 2012). Although HD-iPSCs and control iPSCs can differentiate into neuron/glial cells, the HD derived cells expressed a higher number of nestin-positive neural progenitor cells than controls. This accumulated neural progenitor cell population from the juvenile onset HD cell line is highly vulnerable to BDNF withdrawal due to the loss of TrkB receptor-mediated signaling. One possible mechanism by which BDNF withdrawal leads to enhanced cytotoxicity is glutamate-mediated excitotoxicity, as shown by enhanced NR2B upregulation. The application of glutamate receptor inhibitors rescued the neurons (Mattis et al., 2015). HD-iPSCs when corrected using CRISPR-Cas9 and differentiated into forebrain neurons, these isogenic iPSCs rescued the neurons from the HD phenotypic abnormalities such as increased vulnerability to BDNF withdrawal, aberrant neural rosette formation, and mitochondrial dysfunction. Hence, isogenic controls derived from corrected HD iPSCs and devoid of phenotypic abnormalities could serve as pertinent control for disease modeling (Xu et al., 2017).
2.9.2. HD-iPSCs exhibit the disease phenotype during early stages of development
HD-iPSCs, as well as iPSCs from juvenile-HD patients, exhibited several changes in cellular processes similar to HD phenotype (Jeon et al., 2012). Aberrant changes in the oxidative stress response gene SOD1, a two-fold reduction in phosphorylated ERK1/2 and disrupted Wnt signalling pathways were detected. Furthermore, dysregulation in p53 expression was also evident in HD iPSCs. Nevertheless, multiple signaling pathways known to be dysregulated in HD are obvious in undifferentiated pluripotent cells, suggesting that HD pathogenesis might be triggered during the early neurodevelopmental stage (Chae et al., 2012; Szlachcic et al., 2015).
2.9.3. Karyotypic studies indicate genomic instability in HD iPSCs
Karyotypic analysis revealed genomic abnormalities in HD iPSCs. During reprogramming, profound genomic instability was observed in HD cells, and the severity of the instability depends on the repeat length. Chromosomal aberrations during the reprogramming of fibroblast to a pluripotent state by p53 knockdown are associated with the disease-causing mutation in HD (Tidball et al., 2016).
2.9.4. FOXO regulates proteasome activity and has therapeutic potential
The FOXO factor plays a vital role in regulating chronic stress response and cell survival in normal and diseased cells. HD-iPSCs exhibit enhanced proteasome activity and elevated FOXO1 and FOXO4 protein levels. Knockdown of FOXO4 in HD-iPSC derived NPCs reduced the proteasome activity, whereas its overexpression drastically elevated the proteasome activity, compared to FOXO1. HD-iPSC derived NPCs differentiated into DARPP32 positive neurons when subjected to oxidative stress formed Htt aggregates, making them more vulnerable to cell death. The HD iPSC derived neurons exhibited reduced proteasome activity and FOXO4 expression with higher AKT activity compared to WT-iPSC derived neurons. AKT plays a vital role in modulating the FOXO levels, and inhibition of AKT activity enhances proteasome activity and FOXO levels (Liu et al., 2017). Apart from FOXO, other enzymes regulate the proteasome activity in HD. For example, UBR5 is a ubiquitin ligase involved in proteasomal degradation of Htt aggregates. Loss of UBR5 function leads to Htt accumulation and increases polyQ-expanded aggregation in HD-iPSCs. Conversely, overexpression of UBR5 triggers polyubiquitination and breakdown of mutant HTT, thereby, reducing the polyQ-expanded aggregate, suggesting that UBR5 modulates proteasome activity in HD-iPSCs (Koyuncu et al., 2018).
2.10. Alexander disease
Alexander disease (AxD) is a fatal neurodevelopmental disorder caused by a missense mutation in the glial fibrillary acidic protein (GFAP) gene. It has an incidence of 1: 2,700,000 individuals with infantile, juvenile, and adult-onset subtypes (Yoshida et al., 2011). The primary hallmark is Rosenthal fibers, which are astrocytic cytoplasmic inclusions capable of triggering neurological manifestations (Iwaki et al., 1989; Johnson and Bettica, 1989; Brenner et al., 2001). The disease is typified by clinical phenotypes such as macrocephaly, motor dysfunction and encephalopathy, gait ataxia, frontal leukodystrophy, abnormal myelination, and epileptic seizures (Brenner et al., 2001; Prust et al., 2011).
2.10.1. AxD-iPSC generated astrocytes exhibit GFAP aggregates, enhance inflammation and reduce myelination of oligodendrocytes
AxD patient-derived iPSCs (AxD-iPSCs) showed that GFAP mutations unaffected astrocyte differentiation, but mature astrocytes displayed typical AxD phenotype such as cytoplasmic aggregates of GFAP as well as molecular chaperons like alpha- β-crystallin (Kondo et al., 2016; Jones et al., 2018; Li et al., 2018). Furthermore, these astrocytes showed altered ER, vesicle regulation, as well as metabolism. Alteration in the localization of lysosomes and ER and disruption of their function hampered calcium wave propagation and attenuated ATP release in AXD astrocytes. In CRISPR/Cas9 corrected isogenic lines, pathological features demonstrated in AxD patient-derived astrocytes were significantly diminished (Jones et al., 2018). Upon co-culturing of AxD patient-derived astrocytes with iPSC-derived oligodendrocyte progenitor cells (OPCs), AxD astrocytes inhibited OPC proliferation and myelination possibly through CHI3L, while OPCs survival was unaffected. Furthermore, expression of genes related to cytokine activity, immune response, and cellular membranes was elevated in AxD astrocytes, whereas expression of genes related to synaptic transmission and ion transport were reduced (Li et al., 2018). This study partially identified the cellular and molecular mechanisms responsible for myelination defect in AxD. In another study, AxD-iPSC-derived astrocytes showed mTOR activation and phosphorylation of 4E-BPs. Besides, these astrocytes secreted higher levels of inflammatory cytokines such as TNFα, IL6, GM-CSF, and IL5, compared to healthy controls (Kondo et al., 2016). Overall, the studies demonstrated GFAP accumulation, ER and lysosomal abnormalities, and mechanisms underlying the myelination defects in AxD, such as astrocyte-mediated inhibition of OPC proliferation, and inflammation (Fig. 3).
Fig. 3.
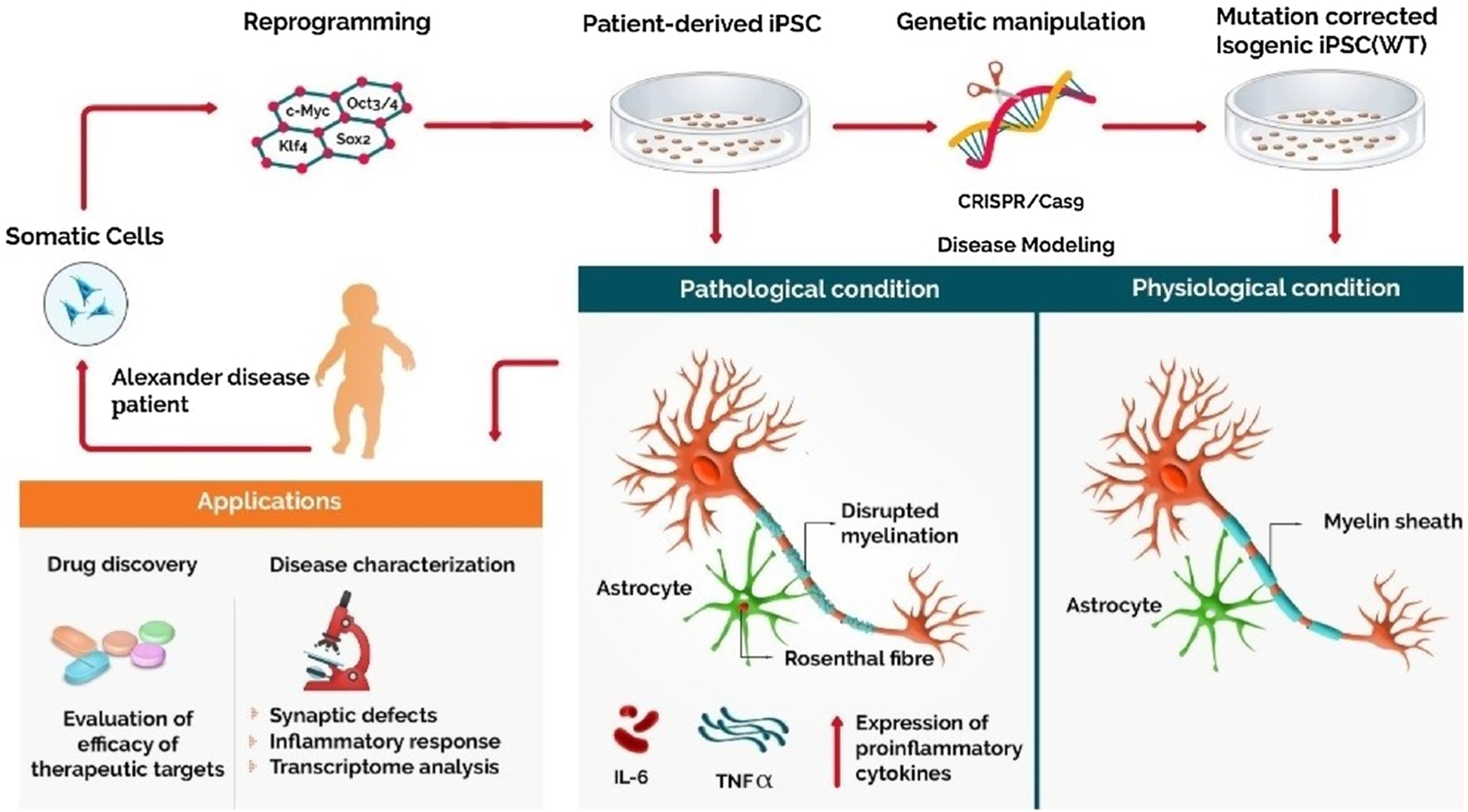
Use of patient-derived iPSC in modeling Alexander disease. Somatic cells obtained from AxD patients are reprogrammed to their pluripotent state using specific transcription factors. Neurons and astrocytes differentiated from these iPSCs, aid in recapitulating the relevant disease phenotype showing unique cytoplasmic inclusions called Rosenthal fibers in astrocytes, reduced myelination, and increased inflammatory response. These iPS cells serve as a promising platform to evaluate the efficacy of various therapeutic drugs and personalized drug treatment.
2.11. Other rare NDDs
Apart from the above studies, Timothy syndrome, Williams–Beuren syndrome, Prader Willi syndrome, Microcephaly, X-linked adrenoleu-kodystrophy, Pelizaeus-Merzbaucher disease, and Lowe Syndrome have been studied with patient-derived iPSCs to certain extent (Table 1).
Table 1.
Patient-derived iPSC used to model rare neurodevelopmental disorders and their disease-relevant phenotypes.
| Condition | Key Gene/ Prevalence/ Mutation type | Phenotypic characterization | Reference |
|---|---|---|---|
| Timothy syndrome | CACNA1C 1:1000000 Dominant |
Perturbation in calcium signaling, abnormalities in differentiation, and expression of tyrosine hydroxylase. |
Paşca et al., 2011. Krey et al., 2013. Tian et al., 2014. Birey et al., 2017. Panagiotakos et al., 2019. |
| TS mutation leads to Cav1.2 inactivation, preventing Cav1.2 mediated recruitment of Gem, eventually leading to RhoA activation and dendritic retraction. | |||
| Transcriptome analysis using WGCNA on neural progenitor cells and neurons from TS patient-derived iPSCs reveal altered Ca2+ signaling leads to TS-associated transcriptional changes that are co-regulated by the calcium-dependent transcriptional regulators NFAT, MEF2, FOXO, and CREB. | |||
| Spheroids derived from TS hPSCs display abnormalities in the interneuron saltatory migration. Calcium imaging studies reveal increased calcium residue following depolarization in TS derived hCS and hSS neurons. | |||
| Cortical differentiation in TS is altered by disruption of calcium splicing patterns that are developmentally regulated. | |||
| Williams–Beuren syndrome | 7q111.23 1:10,000 Deletion |
Increase in dendritic spines and total dendritic length in cortical layer V/VI pyramidal neurons, higher Ca2+ transient frequency, altered electrophysiological properties, and downregulation of Wnt genes. |
Chailangkarn and Muotri, 2017. Chailangkarn et al., 2016. Khattak et al., 2015. Adamo et al., 2015. |
| WS-iPSCs showed marked electrophysiological alterations due to deficits in voltage-activated K+ currents. | |||
| Dosage imbalance in GTF2I involved in encoding key transcription factor at 7q11.23 is associated with transcriptionally repressive chromatin modifier LSD1 (histone demethylase) and HDAC2 (histone deacetylase) | |||
| Prader Willi syndrome | Chromosome 15q11–13 1:15,000 Deletion |
iPSCs derived from PWS patients preserve the molecular signature of the disease phenotype and retain the DNA methylation at the PWS locus even after reprogramming into iPSC as well as neuronal differentiation. |
Burnett et al., 2016. Pόlvora-Brandão et al., 2018 Soeda et al., 2019. |
| Loss of methylation at the maternal PWS imprinting center PWS-IC results in putative imprinting defects. | |||
| iPSC from PWS patients exhibit defects in neuronal differentiation. | |||
| Primary Microcephaly | CDK5RAP2MCPH1, WDR62 1.3–150:100,000 Autosomal recessive |
Premature differentiation of NPCs due to retarded cilium disassembly and delayed cell cycle re-entry in iPSC derived brain organoids from patients carrying a mutation in the centrosomal protein CPAP. |
Gabriel and Gopalakrishnan, 2017. Zhang et al., 2019. |
| WDR62-CEP179-KIF2A protein pathway promotes cilium disassembly, and disruption of this pathway leads to primary microcephaly with reduced organoid size. | |||
| X-linked adrenoleukodystrophy | ABCD1 1:20,000 X-linked recessive |
X-ALD iPSC showed abnormal VLCFA accumulation in oligodendrocytes and this was ameliorated by the upregulation of ABCD2 gene expression. |
Jang et al., 2011. Wang et al., 2012. Baarine et al., 2015. Son et al., 2017. Lee et al., 2018. |
| In iPSC from CCALD patients, genes related to peroxisome proliferation (PEX11) and activation of macrophages (CD200) was reduced. | |||
| CCALD oligodendrocytes express the excessive accumulation of saturated VLCFA along with high levels of proinflammatory cytokines in astrocytes. | |||
| Generation of two X-ALD iPSC lines from adrenomyeloneuropathy patients. | |||
| iPSCs, differentiated into induced brain microvascular endothelial cells (iBMECs), reveal significantly reduced trans-endothelial electrical resistance (TEER), along with lipid droplet accumulation. | |||
| Treatment with PEO-PPO block copolymers could ameliorate these defective properties. | |||
| Pelizaeus -Merzbaucher disease | PLP1 1:90,000–1:750,000 X-linked recessive |
PMD iPSC derived oligodendrocytes exhibit PLP1 protein mislocalization to ER, enhanced ER stress, associated with dysmyelination, aberrant ER morphology, and an increase in apoptosis. |
Numasawa-Kuroiwa et al., 2014 Nevin et al., 2017. Elitt et al., 2018. |
| hiPSC derived oligodendrocytes generated from 12 PMD patients demonstrated abnormal expression of PLP1 mRNA, defects in OPC development, and reduced myelin formation. | |||
| High throughput phenotypic screening using human PMD oligocortical spheroids reveals Ro25–6981 compound modulates ER stress and enhances oligodendrocyte survival. | |||
| Lowe Syndrome | OCRL gene 1:1,000,000–10:1,000,000 X-linked recessive |
LS patient-derived iPSCs and their isogenic lines have been generated. LS-iPSC derived neurons are deficient in F-actin fiber production due to abnormal expression of WAVE-1 involved in actin polymerization. |
Barnes et al., 2018. Liu et al., 2020. |
| Transcriptome analysis of NPCs derived from Lowe Syndrome iPSCs reveals 16 differentially expressed genes. |
3. Limitations of patient-derived iPSCs for studying rare NDDs and future challenges
Human iPSCs have undoubtedly provided us with an unprecedented prospect of decoding the etiology of many rare NDDs at the cellular and molecular levels. Their ability to exhibit clinically relevant phenotypes, easy accessibility, and origin from human patients have made them an excellent prototype in deciphering the functional impairment of specific genes involved in various rare NDDs. Despite these potential benefits, several hurdles need to be overcome to efficiently exploit iPSCs to their full potential in clinical settings and the effective translation of findings. 1. The development of specific patient-derived iPSCs in large cohorts is quite challenging due to the dearth of subjects in rare NDDs with specific genotypes. 2. It involves high cost and time as well as expertise for reprogramming and differentiation of iPSCs into specific cell types. 3. Although iPSCs display identical genotypes, genetic instability and alteration in the epigenetic signature due to reprogramming, maintenance, and environmental factors could lead to altered gene expression during the generation of specific patient-derived iPSC clones and differentiated cells. 4. Given that rare NDDs comprises intellectual disability, cognitive deficits, and other psychiatric comorbidities, it is currently not possible to recapitulate behavioral phenotypes using in vitro systems. Hence, parallel studies using appropriate animal models are required for corroborating the behavioral phenotype associated with rare NDDs. 5. Human iPSC based disease models have been used to understand the etiology of early onset of rare NDDs, but such studies have failed to recapitulate late-onset disease, due to a lack of complete maturation by iPSC-derived cells. The 3-dimensional brain organoids could serve as an excellent model for longitudinal studies to unravel the early brain maldevelopment during neurogenesis and neuronal migration involved in various rare NDDs, which is quite challenging to investigate in 2-D cultures. However, their slow maturation and absence of vascular and immune systems could limit their application in understanding later stages of brain development and neuropathology. Besides, the lack of microvasculature also results in a limited supply of nutrients and oxygen to the inner core of the cerebral organoids, triggering apoptotic cell death (Lancaster et al., 2013; Qian et al., 2016; Yin et al., 2016). Advancements in growing a functional vascular (Pham et al., 2018) and immune systems (Ormel et al., 2018) within organoids might resolve some of the limitations in recapitulating the human brain cytoarchitecture to make them a more suitable archetype to comprehend various rare NDDs that have not been contemplated so far.
Several translational research approaches have been made using human patient-derived iPSCs for rare NDDs. Patient-derived iPSC models and gene-corrected cell lines have facilitated the testing of repurposed drugs, new classes of drugs, small molecule inhibitors, natural products, or nutraceuticals to reduce neuronal phenotype abnormalities. Research with natural bioactive compounds and small molecule modulators possessing antioxidant and/or antiinflammatory activity needs to be further evaluated to reduce the severity of the rare NDD phenotype. Along with antioxidants and antiinflammatory compounds, epigenetic modulators, DNA/RNA based therapeutics, or modulating agents need to be ramped up for finalizing preclinical studies for efficacy and safety in animal models, which could potentially bring them to phase 1 clinical trials. The primary question arises with the stable delivery of these therapeutics directly to the brain for their physiological action or integration. Careful administration of therapeutics through the intranasal route offers an exceptional direct delivery to the brain, which circumvents degradation in the digestive system and hepatic presystemic metabolism, ensuring the requirement of lower doses as well as with reduced systemic effects. Further, patient cell-derived extracellular vesicles that cross the blood-brain barrier could be evaluated for their efficacy in carrying these therapeutic molecules to the brain for safe delivery.
The demonstration of neuronal phenotype rescue with gene-corrected iPSCs provided a useful foundation for future research, bringing outcomes to the clinic. Grafting of cells generated from patient-iPSCs with gene corrections provides huge promise for replenishing cells, proteins, enzymes, neurotrophins, or neurotransmitters in the brain to improve the phenotype of rare NDDs. Grafting of the iPSC-derived progenitor cells has shown efficacy in preclinical models of NDDs (Doerr et al., 2015; Meneghini et al., 2017; Upadhya et al., 2019). Further, certain rare NDDs show phenotypes of inflammation (Cortelazzo et al., 2014; Leoncini et al., 2015; Auvin et al., 2018; Nachun et al., 2018). In such cases, anti-inflammatory MSCs or MSC derived exosomes could relieve the inflammatory burden safely and effectively (Long et al., 2017; Riordan et al., 2019; Upadhya and Shetty, 2019).
The various factors, such as the patient’s genetic background, epigenetic factors, and inter-individual differences, could contribute to the heterogeneity of iPSCs generated. Isogenic lines generated using gene editing systems like ZFNs, TALENs, and CRISPR could help overcome the intrinsic genetic background and variability between the iPSC clones. However, developing isogenic lines using these techniques come with their drawbacks, such as unwanted mutations in the genome due to off-target effects. Moreover, it is not feasible to generate isogenic lines in some genetic mutations, forcing researchers to use related or nonrelated controls, which might sometimes lead to incorrect conclusions.
Patient-derived iPSCs with specific genotype and differentiated cells could be used in a high throughput screening of drugs for rare NDDs (Kaufmann et al., 2015; Kumari et al., 2015; Li et al., 2017). Moreover, the 2-D and 3-D culture models should be adequately explored for patient-specific drug screening as patients with rare NDDs frequently need to try multiple drugs before finding a drug regimen that works best. Further, patient-iPSC models should be rigorously explored for genotype-specific transcriptomics based biomarker discovery for prognostic purposes.
The exploitation of iPSCs from a larger cohort of patients would likely offer a much broader perception of the molecular pathophysiology, genotype-phenotype correlations, and the efficacy of multiple therapeutic strategies because of higher heterogeneity between patients. The creation of cell bank repositories for each rare disease with major genotypes would provide a powerful resource for conducting well-designed studies using multiple cell lines with specific mutations. Cell banks for a few rare NDDs have already been instituted, and the cells are available to investigators for research (Brick et al., 2014; Li et al., 2017). Undoubtedly, the scientific benefit will overshadow the economic impact of maintaining such repositories.
Acknowledgements
This work is supported by grants from Science and Engineering Research Board (SERB-EMR/2017/005213 to D.U) and Department of Biotechnology, Govt of India (BT/PR26814/NNT/28/1476/2017 to D. U), Intramural grant from Manipal Academy of Higher Education, Manipal (MAHE/PDF/2019 to S.K.R), R01 grant from the National Institutes of Health - National Institute of Neurological Disorders and Stroke (NIH-NINDS R01NS106907-01 to A.K.S.
References
- Achuta VS, Grym H, Putkonen N, Louhivuori V, Kärkkäinen V, Koistinaho J, Roybon L, Castrén ML, 2017. Metabotropic glutamate receptor 5 responses dictate differentiation of neural progenitors to NMDA-responsive cells in fragile X syndrome. Dev. Neurobiol 77 (4), 438–453. 10.1002/dneu.22419. [DOI] [PubMed] [Google Scholar]
- Achuta VS, Möykkynen T, Peteri UK, Turconi G, Rivera C, Keinänen K, Castrén ML, 2018. Functional changes of AMPA responses in human induced pluripotent stem cell-derived neural progenitors in fragile X syndrome. Sci. Signal 11 (513), eaan8784. 10.1002/dneu.22419. [DOI] [PubMed] [Google Scholar]
- Adamo A, Atashpaz S, Germain PL, Zanella M, D’Agostino G, Albertin V, Chenoweth J, Micale L, Fusco C, Unger C, Augello B, Palumbo O, Hamilton B, Carella M, Donti E, Pruneri G, Selicorni A, Biamino E, Prontera P, McKay R, et al. , 2015. 7q11.23 dosage-dependent dysregulation in human pluripotent stem cells affects transcriptional programs in disease-relevant lineages. Nat. Genet 47 (2), 132–141. 10.1038/ng.3169. [DOI] [PubMed] [Google Scholar]
- Albani S, Prakken B, 2009. The advancement of translational medicine-from regional challenges to global solutions. Nat. Med 15, 1006–1009. 10.1038/nm0909-1006. [DOI] [PubMed] [Google Scholar]
- Alisch RS, Wang T, Chopra P, Visootsak J, Conneely KN, Warren ST, 2013. Genome-wide analysis validates aberrant methylation in fragile X syndrome is specific to the FMR1 locus. BMC Med. Genet 14, 18. 10.1186/1471-2350-14-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY, 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet 23 (2), 185–188. 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Schultz R, Malicki DM, Tran CQ, Dahle EJ, Philippi A, Timar L, Percy AK, Motil KJ, Lichtarge O, Smith EO, Glaze DG, Zoghbi HY, 2000. Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann. Neurol 47 (5), 670–679. . [DOI] [PubMed] [Google Scholar]
- Ananiev G, Williams EC, Li H, Chang Q, 2011. Isogenic pairs of wild type and mutant induced pluripotent stem cell (iPSC) lines from Rett syndrome patients as in vitro disease model. PLoS One 6 (9), e25255. 10.1371/journal.pone.0025255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando S, Funato M, Ohuchi K, Kameyama T, Inagaki S, Seki J, Kawase C, Tsuruma K, Shimazawa M, Kaneko H, Hara H, 2017. Edaravone is a candidate agent for spinal muscular atrophy: In vitro analysis using a human induced pluripotent stem cells-derived disease model. Eur. J. Pharmacol 814, 161–168. 10.1016/j.ejphar.2017.08.005. [DOI] [PubMed] [Google Scholar]
- Ando S, Funato M, Ohuchi K, Inagaki S, Sato A, Seki J, Kawase C, Saito T, Nishio H, Nakamura S, Shimazawa M, Kaneko H, Hara H, 2019. The protective effects of Levetiracetam on a human iPSCs-Derived spinal muscular atrophy model. Neurochem. Res 44 (7), 1773–1779. 10.1007/s11064-019-02814-4. [DOI] [PubMed] [Google Scholar]
- Andoh-Noda T, Akamatsu W, Miyake K, Matsumoto T, Yamaguchi R, Sanosaka T, Okada Y, Kobayashi T, Ohyama M, Nakashima K, Kurosawa H, Kubota T, Okano H, 2015. Differentiation of multipotent neural stem cells derived from Rett syndrome patients is biased toward the astrocytic lineage. Mol. Brain 8, 31. 10.1186/s13041-015-0121-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auvin S, Jeljeli M, Desnous B, Soussi-Yanicostas N, Dournaud P, Sterkers G, 2018. Altered vaccine-induced immunity in children with Dravet syndrome. Epilepsia. 59 (4), e45–e50. 10.1111/epi.14038. [DOI] [PubMed] [Google Scholar]
- Baarine M, Khan M, Singh A, Singh I, 2015. Functional characterization of IPSC-Derived brain cells as a model for X-Linked adrenoleukodystrophy. PLoS One 10 (11), e0143238. 10.1371/journal.pone.0143238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldovino S, Moliner AM, Taruscio D, Daina E, Roccatello D, 2016. Rare diseases in Europe: from a wide to a local perspective. Isr. Med. Assoc. J 18 (6), 359–363. [PubMed] [Google Scholar]
- Barnes J, Salas F, Mokhtari R, Dolstra H, Pedrosa E, Lachman HM, 2018. Modeling the neuropsychiatric manifestations of Lowe syndrome using induced pluripotent stem cells: defective F-actin polymerization and WAVE-1 expression in neuronal cells. Mol. Autism 9, 44. 10.1186/s13229-018-0227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Nur O, Caspi I, Benvenisty N, 2012. Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell Biol 4 (3), 180–183. 10.1093/jmcb/mjs007. [DOI] [PubMed] [Google Scholar]
- Bayat A, Hjalgrim H, Møller RS, 2015. The incidence of SCN1A-related Dravet syndrome in Denmark is 1:22,000: a population-based study from 2004 to 2009. Epilepsia 56 (4), e36–e39. 10.1111/epi.12927. [DOI] [PubMed] [Google Scholar]
- Bell S, Peng H, Crapper L, Kolobova I, Maussion G, Vasuta C, Yerko V, Wong TP, Ernst C, 2017. A rapid pipeline to model rare neurodevelopmental disorders with simultaneous CRISPR/Cas9 gene editing. Stem Cells Transl. Med 6 (3), 886–896. 10.1002/sctm.16-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershteyn M, Nowakowski TJ, Pollen AA, Di Lullo E, Nene A, Wynshaw-Boris A, Kriegstein AR, 2017. Human iPSC-Derived cerebral organoids model cellular features of lissencephaly and reveal prolonged mitosis of outer radial glia. Cell Stem Cell 20 (4), 435–449. 10.1016/j.stem.2016.12.007 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidinosti M, Botta P, Krüttner S, Proenca CC, Stoehr N, Bernhard M, Fruh I, Mueller M, Bonenfant D, Voshol H, Carbone W, Neal SJ, McTighe SM, Roma G, Dolmetsch RE, Porter JA, Caroni P, Bouwmeester T, Lüthi A, Galimberti I, 2016. CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science 351 (6278), 1199–1203. 10.1126/science.aad5487. [DOI] [PubMed] [Google Scholar]
- Bird MJ, Needham K, Frazier AE, van Rooijen J, Leung J, Hough S, Denham M, Thornton ME, Parish CL, Nayagam BA, Pera M, Thorburn DR, Thompson LH, Dottori M, 2014. Functional characterization of Friedreich ataxia iPS-derived neuronal progenitors and their integration in the adult brain. PLoS One 9 (7), e101718. 10.1371/journal.pone.0101718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, Fan HC, Metzler K, Panagiotakos G, Thom N, O’Rourke NA, Steinmetz LM, Bernstein JA, Hallmayer J, Huguenard JR, Paşca SP, 2017. Assembly of functionally integrated human forebrain spheroids. Nature 545 (7652), 54–59. 10.1038/nature22330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazejewski SM, Bennison SA, Smith TH, Toyo-Oka K, 2018. Neurodevelopmental genetic diseases associated with microdeletions and microduplications of chromosome 17p13.3. Front. Genet 9, 80. 10.3389/fgene.2018.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland MJ, Nazor KL, Tran HT, Szücs A, Lynch CL, Paredes R, Tassone F, Sanna PP, Hagerman RJ, Loring JF, 2017. Molecular analyses of neurogenic defects in a human pluripotent stem cell model of fragile X syndrome. Brain 140 (3), 582–598. 10.1093/brain/aww357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boza-Morán MG, Martlnáz-Hernandez R, Bernal S, Wanisch K, Also-Rallo E, Le Heron A, Alías L, Denis C, Girard M, Yee JK, Tizzano EF, Yáñez-Muñoz RJ, 2015. Decay in survival motor neuron and plastin 3 levels during differentiation of iPSC-derived human motor neurons. Sci. Rep 5, 11696. 10.1038/srep11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen MS, Browne A, Hoffman GE, Stathopoulos S, Brennand K, Buxbaum JD, Drapeau E, 2020. Transcriptional signatures of participant-derived neural progenitor cells and neurons implicate altered Wnt signaling in Phelan-McDermid syndrome and autism. Mol. Autism 11 (1), 53. 10.1186/s13229-020-00355-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A, 2001. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat. Genet 27 (1), 117–120. 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- Brick DJ, Nethercott HE, Montesano S, Banuelos MG, Stover AE, Schutte SS, O’Dowd DK, Hagerman RJ, Ono M, Hessl DR, Tassone F, Schwartz PH, 2014. The autism Spectrum disorders stem cell resource at Children’s Hospital of Orange County: implications for disease modeling and drug discovery. Stem Cells Transl. Med 3 (11), 1275–1286. 10.5966/sctm.2014-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunklaus A, Ellis R, Reavey E, Forbes GH, Zuberi SM, 2012. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain 135 (Pt 8), 2329–2336. 10.1093/brain/aws151. [DOI] [PubMed] [Google Scholar]
- Bu Q, Wang A, Hamzah H, Waldman A, Jiang K, Dong Q, Li R, Kim J, Turner D, Chang Q, 2017. CREB signaling is involved in rett syndrome pathogenesis. J. Neurosci 37 (13), 3671–3685. 10.1523/JNEUROSCI.3735-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürglen L, Lefebvre S, Clermont O, Burlet P, Viollet L, Cruaud C, Munnich A, Melki J, 1996. Structure and organization of the human survival motor neurone (SMN) gene. Genomics. 32 (3), 479–482. 10.1006/geno.1996.0147. [DOI] [PubMed] [Google Scholar]
- Burnett LC, LeDuc CA, Sulsona CR, Paull D, Eddiry S, Levy B, Salles JP, Tauber M, Driscoll DJ, Egli D, Leibel RL, 2016. Induced pluripotent stem cells (iPSC) created from skin fibroblasts of patients with Prader-Willi syndrome (PWS) retain the molecular signature of PWS. Stem Cell Res. 17 (3), 526–530. 10.1016/j.scr.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camnasio S, Delli Carri A, Lombardo A, Grad I, Mariotti C, Castucci A, Rozell B, Lo Riso P, Castiglioni V, Zuccato C, Rochon C, Takashima Y, Diaferia G, Biunno I, Gellera C, Jaconi M, Smith A, Hovatta O, Naldini L, Di Donato S, et al. , 2012. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol. Dis 46 (1), 41–51. 10.1016/j.nbd.2011.12.042. [DOI] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, et al. , 1996. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271 (5254), 1423–1427. 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Cardoso C, Leventer RJ, Ward HL, Toyo-Oka K, Chung J, Gross A, Martin CL, Allanson J, Pilz DT, Olney AH, Mutchinick OM, Hirotsune S, Wynshaw-Boris A, Dobyns WB, Ledbetter DH, 2003. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am. J. Hum. Genet 72 (4), 918–930. 10.1086/374320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae JI, Kim DW, Lee N, Jeon YJ, Jeon I, Kwon J, Kim J, Soh Y, Lee DS, Seo KS, Choi NJ, Park BC, Kang SH, Ryu J, Oh SH, Shin DA, Lee DR, Do JT, Park IH, Daley GQ, et al. , 2012. Quantitative proteomic analysis of induced pluripotent stem cells derived from a human Huntington’s disease patient. Biochem. J 446 (3), 359–371. 10.1042/BJ20111495. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY, 2007. The story of Rett syndrome: from clinic to neurobiology. Neuron. 56 (3), 422–437. 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chailangkarn T, Muotri AR, 2017. Modeling Williams syndrome with induced pluripotent stem cells. Neurogenesis. 4 (1), e1283187. 10.1080/23262133.2017.1283187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chailangkarn T, Trujillo CA, Freitas BC, Hrvoj-Mihic B, Herai RH, Yu DX, Brown TT, Marchetto MC, Bardy C, McHenry L, Stefanacci L, Järvinen A, Searcy YM, DeWitt M, Wong W, Lai P, Ard MC, Hanson KL, Romero S, Jacobs B, et al. , 2016. A human neurodevelopmental model for Williams syndrome. Nature 536 (7616), 338–343. 10.1038/nature19067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, Lalande M, 2010. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl. Acad. Sci. U. S. A 107 (41), 17668–17673. 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T, Zheng W, Tsark W, Bates S, Huang H, Lin RJ, Yee JK, 2011. Brief report: phenotypic rescue of induced pluripotent stem cell-derived motoneurons of a spinal muscular atrophy patient. Stem Cells 29 (12), 2090–2093. 10.1002/stem.749. [DOI] [PubMed] [Google Scholar]
- Cheung AY, Horvath LM, Grafodatskaya D, Pasceri P, Weksberg R, Hotta A, Carrel L, Ellis J, 2011. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum. Mol. Genet 20 (11), 2103–2115. 10.1093/hmg/ddr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin EW, Marcy G, Yoon SI, Ma D, Rosales FJ, Augustine GJ, Goh EL, 2016. Choline ameliorates disease phenotypes in human iPSC models of rett syndrome. Neuromolecular Med. 18 (3), 364–377. 10.1007/s12017-016-8421-y. [DOI] [PubMed] [Google Scholar]
- Codazzi F, Hu A, Rai M, Donatello S, Salerno Scarzella F, Mangiameli E, Pelizzoni I, Grohovaz F, Pandolfo M, 2016. Friedreich ataxia-induced pluripotent stem cell-derived neurons show a cellular phenotype that is corrected by a benzamide HDAC inhibitor. Hum. Mol. Genet 25 (22), 4847–4855. 10.1093/hmg/ddw308. [DOI] [PubMed] [Google Scholar]
- Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, Disney MD, Jaffrey SR, 2014. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343 (6174), 1002–1005. 10.1126/science.1245831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin F, Martelli A, Clémancey M, Latour JM, Gambarelli S, Zeppieri L, Birck C, Page A, Puccio H, Ollagnier de Choudens S, 2013. Mammalian frataxin controls sulfur production and iron entry during de novo Fe4S4 cluster assembly. J. Am. Chem. Soc 135 (2), 733–740. 10.1021/ja308736e. [DOI] [PubMed] [Google Scholar]
- Cortelazzo A, De Felice C, Guerranti R, Signorini C, Leoncini S, Pecorelli A, Zollo G, Landi C, Valacchi G, Ciccoli L, Bini L, Hayek J, 2014. Subclinical inflammatory status in Rett syndrome. Mediators Inflamm. 2014, 480980 10.1155/2014/480980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti S, Nizzardo M, Simone C, Falcone M, Nardini M, Ronchi D, Donadoni C, Salani S, Riboldi G, Magri F, Menozzi G, Bonaglia C, Rizzo F, Bresolin N, Comi GP, 2012. Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci. Transl. Med 4 (165), 165ra162. 10.1126/scitranslmed.3004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford TO, Pardo CA, 1996. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis 3 (2), 97–110. 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- Crawford DC, Meadows KL, Newman JL, Taft LF, Pettay DL, Gold LB, Hersey SJ, Hinkle EF, Stanfield ML, Holmgreen P, Yeargin-Allsopp M, Boyle C, Sherman SL, 1999. Prevalence and phenotype consequence of FRAXA and FRAXE alleles in a large, ethnically diverse, special education-needs population. Am. J. Hum. Genet 64 (2), 495–507. 10.1086/302260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amico A, Mercuri E, Tiziano FD, Bertini E, 2011. Spinal muscular atrophy. Orphanet J. Rare Dis 6, 71. 10.1186/1750-1172-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Ydewalle C, Ramos DM, Pyles NJ, Ng SY, Gorz M, Pilato CM, Ling K, Kong L, Ward AJ, Rubin LL, Rigo F, Bennett CF, Sumner CJ, 2017. The antisense transcript SMN-AS1 regulates SMN expression and is a novel therapeutic target for spinal muscular atrophy. Neuron 93 (1), 66–79. 10.1016/j.neuron.2016.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danesi C, Achuta VS, Corcoran P, Peteri UK, Turconi G, Matsui N, Albayrak I, Rezov V, Isaksson A, Castrén ML, 2018. Increased calcium influx through L-type calcium channels in human and mouse neural progenitors lacking fragile X Mental retardation protein. Stem Cell Reports 11 (6), 1449–1461. 10.1016/j.stemcr.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Esch CE, Ghazvini M, Loos F, Schelling-Kazaryan N, Widagdo W, Munshi ST, van der Wal E, Douben H, Gunhanlar N, Kushner SA, Pijnappel WW, de Vrij FM, Geijsen N, Gribnau J, Willemsen R, 2014. Epigenetic characterization of the FMR1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Reports 3 (4), 548–555. 10.1016/j.stemcr.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza JS, Carromeu C, Torres LB, Araujo BH, Cugola FR, Maciel RM, Muotri AR, Giannocco G, 2017. IGF1 neuronal response in the absence of MECP2 is dependent on TRalpha 3. Hum. Mol. Genet 26 (2), 270–281. 10.1093/hmg/ddw384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study, 2017. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542 (7642), 433–438. 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delépine C, Meziane H, Nectoux J, Opitz M, Smith AB, Ballatore C, Saillour Y, Bennaceur-Griscelli A, Chang Q, Williams EC, Dahan M, Duboin A, Billuart P, Herault Y, Bienvenu T, 2016. Altered microtubule dynamics and vesicular transport in mouse and human MeCP2-deficient astrocytes. Hum. Mol. Genet 25 (1), 146–157. 10.1093/hmg/ddv464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Nabbout R, Miller I, Laux L, Zolnowska M, Wright S, Roberts C, 2019. Long-term cannabidiol treatment in patients with Dravet syndrome: an open-label extension trial. Epilepsia. 60 (2), 294–302. 10.1111/dmcn.13623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djuric U, Cheung A, Zhang W, Mok RS, Lai W, Piekna A, Hendry JA, Ross PJ, Pasceri P, Kim DS, Salter MW, Ellis J, 2015. MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol. Dis 76, 37–45. 10.1016/j.nbd.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerr J, Böckenhoff A, Ewald B, Ladewig J, Eckhardt M, Gieselmann V, Matzner U, Brüstle O, Koch P, 2015. Arylsulfatase a overexpressing human iPSC-derived neural cells reduce CNS sulfatide storage in a mouse model of metachromatic leukodystrophy. Mol. Ther 23 (9), 1519–1531. 10.1038/mt.2015.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, Zhang SC, Abbeduto L, Bhattacharyya A, 2014. iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 23 (15), 1777–1787. 10.1089/scd.2014.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravet C, 2011. The core Dravet syndrome phenotype. Epilepsia. 52 (Suppl 2), 3–9. 10.1111/j.1528-1167.2011.02994.x. [DOI] [PubMed] [Google Scholar]
- Dravet C, Oguni H, 2013. Dravet syndrome (severe myoclonic epilepsy in infancy). Handb. Clin. Neurol 111, 627–633. 10.1016/B978-0-444-52891-9.00065-8. [DOI] [PubMed] [Google Scholar]
- Du J, Campau E, Soragni E, Ku S, Puckett JW, Dervan PB, Gottesfeld JM, 2012. Role of mismatch repair enzymes in GAA·TTC triplet-repeat expansion in Friedreich ataxia induced pluripotent stem cells. J. Biol. Chem 287 (35), 29861–29872. 10.1016/B978-0-444-52891-9.00065-00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Rogé B, Héron D, Burglen L, et al. , 2007. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet 39 (1), 25–27. 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M, 1996. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N. Engl. J. Med 335 (16), 1169–1175. 10.1056/NEJM199610173351601. [DOI] [PubMed] [Google Scholar]
- Ebert AD, Yu J, Rose FF Jr., Mattis VB, Lorson CL, Thomson JA, Svendsen CN, 2009. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 457 (7227), 277–280. 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigentler A, Boesch S, Schneider R, Dechant G, Nat R, 2013. Induced pluripotent stem cells from friedreich ataxia patients fail to upregulate frataxin during in vitro differentiation to peripheral sensory neurons. Stem Cells Dev. 22 (24), 3271–3282. 10.1089/scd.2013.0126. [DOI] [PubMed] [Google Scholar]
- Eiges R, Urbach A, Malcov M, Frumkin T, Schwartz T, Amit A, Yaron Y, Eden A, Yanuka O, Benvenisty N, Ben-Yosef D, 2007. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 1 (5), 568–577. 10.1016/j.stem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Elitt MS, Shick HE, Madhavan M, Allan KC, Clayton B, Weng C, Miller TE, Factor DC, Barbar L, Nawash BS, Nevin ZS, Lager AM, Li Y, Jin F, Adams DJ, Tesar PJ, 2018. Chemical screening identifies enhancers of mutant oligodendrocyte survival and unmasks a distinct pathological phase in pelizaeus-merzbacher disease. Stem Cell Reports 11 (3), 711–726. 10.1016/j.stemcr.2018.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin GS, Grieshop MP, Ali A, Qi J, Lawlor M, Kumar D, Ahmad I, McNally A, Teider N, Worringer K, Sivasankaran R, Syed DN, Eguchi A, Ashraf M, Jeffery J, Xu M, Park P, Mukhtar H, Srivastava AK, Faruq M, et al. , 2017. Synthetic transcription elongation factors license transcription across repressive chromatin. Science 358 (6370), 1617–1622. 10.1126/science.aan6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg A, Heils A, MacDonald BT, Haug K, Sander T, Meisler MH, 2001. A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus–and prevalence of variants in patients with epilepsy. Am. J. Hum. Genet 68 (4), 866–873. 10.1086/319524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr S, Bebbington A, Nassar N, Downs J, Ronen GM, DE Klerk N, Leonard H, 2011. Trends in the diagnosis of Rett syndrome in Australia. Pediatr. Res 70 (3), 313–319. 10.1203/PDR.0b013e3182242461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes TG, Duarte ST, Ghazvini M, Gaspar C, Santos DC, Porteira AR, Rodrigues GM, Haupt S, Rombo DM, Armstrong J, Sebastião AM, Gribnau J, Garcia-Cazorla À, Brüstle O, Henrique D, Cabral JM, Diogo MM, 2015. Neural commitment of human pluripotent stem cells under defined conditions recapitulates neural development and generates patient-specific neural cells. Biotechnol. J 10 (10), 1578–1588. 10.1002/biot.201400751. [DOI] [PubMed] [Google Scholar]
- Fink JJ, Robinson TM, Germain ND, Sirois CL, Bolduc KA, Ward AJ, Rigo F, Chamberlain SJ, Levine ES, 2017. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat. Commun 8, 15038. 10.1038/ncomms15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freilinger M, Bebbington A, Lanator I, De Klerk N, Dunkler D, Seidl R, Leonard H, Ronen GM, 2010. Survival with Rett syndrome: comparing Rett’s original sample with data from the Australian Rett Syndrome Database. Dev. Med. Child Neurol 52 (10), 962–965. 10.1111/j.1469-8749.2010.03716.x. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, 2006. Clinical spectrum of mutations in SCN1A gene: severe myoclonic epilepsy in infancy and related epilepsies. Epilepsy Res. 70 (Suppl 1), S223–S230. 10.1016/j.eplepsyres.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Fuller HR, Mandefro B, Shirran SL, Gross AR, Kaus AS, Botting CH, Morris GE, Sareen D, 2016. Spinal muscular atrophy patient iPSC -derived motor neurons have reduced expression of proteins important in neuronal development. Front. Cell. Neurosci 9 (506) 10.3389/fncel.2015.00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel E, Gopalakrishnan J, 2017. Generation of iPSC-derived human brain organoids to model early neurodevelopmental disorders. Journal of visualized experiments. JoVE (122), 55372. 10.3791/55372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbes L, Heesen L, Hölker I, Bauer T, Schreml J, Zimmermann K, Thoenes M, Walter M, Dimos J, Peitz M, Brüstle O, Heller R, Wirth B, 2013. VPA response in SMA is suppressed by the fatty acid translocase CD36. Hum. Mol. Genet 22 (2), 398–407. 10.1093/hmg/dds437. [DOI] [PubMed] [Google Scholar]
- Georges P, Boza-Moran MG, Gide J, Pêche GA, Forêt B, Bayot A, Rustin P, Peschanski M, Martinat C, Aubry L, 2019. Induced pluripotent stem cells-derived neurons from patients with Friedreich ataxia exhibit differential sensitivity to resveratrol and nicotinamide. Sci. Rep 9 (1), 14568. 10.1038/s41598-019-49870-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt J, 2017. Epigenetic modifications in human fragile X pluripotent stem cells; Implications in fragile X syndrome modeling. Brain Res. 1656, 55–62. 10.1016/j.brainres.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Gong X, Jiang YW, Zhang X, An Y, Zhang J, Wu Y, Wang J, Sun Y, Liu Y, Gao X, Shen Y, Wu X, Qiu Z, Jin L, Wu BL, Wang H, 2012. High proportion of 22q13 deletions and SHANK3 mutations in Chinese patients with intellectual disability. PLoS One 7 (4), e34739. 10.1371/journal.pone.0034739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouder L, Vitrac A, Goubran-Botros H, Danckaert A, Tinevez JY, Andŕe-Leroux G, Atanasova E, Lemìere N, Biton A, Leblond CS, Poulet A, Boland A, Deleuze JF, Benchoua A, Delorme R, Bourgeron T, Cloëz-Tayarani I, 2019. Altered spinogenesis in iPSC-derived cortical neurons from patients with autism carrying de novo SHANK3 mutations. Sci. Rep 9 (1), 94. 10.1038/s41598-018-36993-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef JD, Wu H, Ng C, Sun C, Villegas V, Qadir D, Jesseman K, Warren ST, Jaenisch R, Cacace A, Wallace O, 2020. Partial FMRP expression is sufficient to normalize neuronal hyperactivity in Fragile X neurons. Eur. J. Neurosci 51 (10), 2143–2157. 10.1111/ejn.14660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg B, 1985. Rett’s syndrome: prevalence and impact on progressive severe mental retardation in girls. Acta Paediatr. Scand 74 (3), 405–408. 10.1111/j.1651-2227.1985.tb10993.x. [DOI] [PubMed] [Google Scholar]
- Halevy T, Czech C, Benvenisty N, 2015. Molecular mechanisms regulating the defects in fragile X syndrome neurons derived from human pluripotent stem cells. Stem Cell Reports 4 (1), 37–46. 10.1016/j.stemcr.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HD iPSC Consortium, 2012. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 11 (2), 264–278. 10.1016/j.stem.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesen L, Peitz M, Torres-Benito L, Hölker I, Hupperich K, Dobrindt K, Jungverdorben J, Ritzenhofen S, Weykopf B, Eckert D, Hosseini-Barkooie SM, Storbeck M, Fusaki N, Lonigro R, Heller R, Kye MJ, Brüstle O, Wirth B, 2016. Plastin 3 is upregulated in iPSC-derived motoneurons from asymptomatic SMN1-deleted individuals. Cell. Mol. Life Sci 73 (10), 2089–2104. 10.1007/s00018-015-2084-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM, 2006. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol 2 (10), 551–558. 10.1038/nchembio815. [DOI] [PubMed] [Google Scholar]
- Hick A, Wattenhofer-Donzé M, Chintawar S, Tropel P, Simard JP, Vaucamps N, Gall D, Lambot L, André C, Reutenauer L, Rai M, Teletin M, Messaddeq N, Schiffmann SN, Viville S, Pearson CE, Pandolfo M, Puccio H, 2013. Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Dis. Model. Mech 6 (3), 608–621. 10.1242/dmm.010900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higurashi N, Uchida T, Lossin C, Misumi Y, Okada Y, Akamatsu W, Imaizumi Y, Zhang B, Nabeshima K, Mori MX, Katsurabayashi S, Shirasaka Y, Okano H, Hirose S, 2013. A human Dravet syndrome model from patient induced pluripotent stem cells. Mol. Brain 6, 19. 10.1186/1756-6606-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hor JH, Soh ES, Tan LY, Lim V, Santosa MM, Winanto Ho BX, Fan Y, Soh BS, Ng SY, 2018. Cell cycle inhibitors protect motor neurons in an organoid model of Spinal Muscular Atrophy. Cell Death Dis. 9 (11), 1100. 10.1038/s41419-018-1081-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath PM, Monteggia LM, 2017. Engineering MeCP2 to spy on its targets. Nat. Med 23 (10), 1120–1122. 10.1038/nm.4425. [DOI] [PubMed] [Google Scholar]
- Horvath PM, Monteggia LM, 2018. MeCP2 as an activator of gene expression. Trends Neurosci. 41 (2), 72–74. 10.1016/j.tins.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta A, Yamanaka S, 2015. From genomics to gene therapy: induced pluripotent stem cells meet genome editing. Annu. Rev. Genet 49, 47–70. 10.1146/annurev-genet-112414-054926. [DOI] [PubMed] [Google Scholar]
- Hu A, Rai M, Donatello S, Pandolfo M, 2017. Oxidative stress and loss of Fe-S proteins in Friedreich ataxia induced pluripotent stem cell-derived PSNs can be reversed by restoring FXN expression with a benzamide HDAC inhibitor. BioRxiv. 10.1101/221242. [DOI] [Google Scholar]
- Hunihan L, Brown J, Cacace A, Fernandes A, Weston A, 2017. Generation of a clonal induced pluripotent stem cell (iPSC) line expressing the mutant MECP2 allele from a Rett Syndrome patient fibroblast line. Stem Cell Res. 20, 67–69. 10.1016/j.scr.2017.02.017. [DOI] [PubMed] [Google Scholar]
- Hurst DL, 1990. Epidemiology of severe myoclonic epilepsy of infancy. Epilepsia. 31 (4), 397–400. 10.1111/j.1528-1157.1990.tb05494.x. [DOI] [PubMed] [Google Scholar]
- Iefremova V, Manikakis G, Krefft O, Jabali A, Weynans K, Wilkens R, Marsoner F, Brändl B, Müller FJ, Koch P, Ladewig J, 2017. An organoid-based model of cortical development identifies non-cell-Autonomous defects in wnt signaling contributing to miller-dieker syndrome. Cell Rep. 19 (1), 50–59. 10.1016/j.celrep.2017.03.047. [DOI] [PubMed] [Google Scholar]
- Igoillo-Esteve M, Gurgul-Convey E, Hu A, Romagueira Bichara Dos Santos L, Abdulkarim B, Chintawar S, Marselli L, Marchetti P, Jonas JC, Eizirik DL, Pandolfo M, Cnop M, 2015. Unveiling a common mechanism of apoptosis in β-cells and neurons in Friedreich’s ataxia. Hum. Mol. Genet 24 (8), 2274–2286. 10.1093/hmg/ddu745. [DOI] [PubMed] [Google Scholar]
- Igoillo-Esteve M, Oliveira AF, Cosentino C, Fantuzzi F, Demarez C, Toivonen S, Hu A, Chintawar S, Lopes M, Pachera N, Cai Y, Abdulkarim B, Rai M, Marselli L, Marchetti P, Tariq M, Jonas JC, Boscolo M, Pandolfo M, Eizirik DL, Cnop M, 2020. Exenatide induces frataxin expression and improves mitochondrial function in Friedreich ataxia. JCI Insight 5 (2), e134221. 10.1172/jci.insight.134221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaki T, Kume-Iwaki A, Liem RK, Goldman JE, 1989. Alpha B-crystallin is expressed in non-lenticular tissues and accumulates in Alexander’s disease brain. Cell. 57 (1), 71–78. 10.1016/0092-8674(89)90173-6. [DOI] [PubMed] [Google Scholar]
- Kim J, Nonis D, Gabriela Otero M, Mark Pierson T, 2019a. Modeling rare pediatric neurogenetic disorders with IPSCs. Aims Cell Tissue Eng. 3 (1), 1–25. 10.3934/celltissue.2019.1.1. [DOI] [Google Scholar]
- Kim JJ, Savas JN, Miller MT, Hu X, Carromeu C, Lavallée-Adam M, Freitas B, Muotri AR, Yates JR 3rd, Ghosh A, 2019b. Proteomic analyses reveal misregulation of LIN28 expression and delayed timing of glial differentiation in human iPS cells with MECP2 loss-of-function. PLoS One 14 (2), e0212553. 10.1371/journal.pone.0212553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonka S, Sendtner M, 2003. Molecular and cellular basis of spinal muscular atrophy. Amyotroph. Lateral Scler. Other Motor Neuron Disord 4 (3), 144–149. 10.1080/14660820310011296. [DOI] [PubMed] [Google Scholar]
- Jablonka S, Rossoll W, Schrank B, Sendtner M, 2000. The role of SMN in spinal muscular atrophy. J. Neurol 247 (Suppl 1), I37–I42. 10.1007/s004150050555. [DOI] [PubMed] [Google Scholar]
- Jang J, Kang HC, Kim HS, Kim JY, Huh YJ, Kim DS, Yoo JE, Lee JA, Lim B, Lee J, Yoon TM, Park IH, Hwang DY, Daley GQ, Kim DW, 2011. Induced pluripotent stem cell models from X-linked adrenoleukodystrophy patients. Ann. Neurol 70 (3), 402–409. 10.1002/ana.22486. [DOI] [PubMed] [Google Scholar]
- Jangi M, Fleet C, Cullen P, Gupta SV, Mekhoubad S, Chiao E, Allaire N, Bennett CF, Rigo F, Krainer AR, Hurt JA, Carulli JP, Staropoli JF, 2017. SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc. Natl. Acad. Sci. U. S. A 114 (12), E2347–2356. 10.1073/pnas.1613181114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon I, Lee N, Li JY, Park IH, Park KS, Moon J, Shim SH, Choi C, Chang DJ, Kwon J, Oh SH, Shin DA, Kim HS, Do JT, Lee DR, Kim M, Kang KS, Daley GQ, Brundin P, Song J, 2012. Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells 30 (9), 2054–2062. 10.1002/stem.1135. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Lev-Lehman E, Bressler J, Tsai TF, Beaudet AL, 1999. Genetics of angelman syndrome. Am. J. Hum. Genet 65 (1), 1–6. 10.1086/302473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J, Yang Y, Shi Y, Chen J, Gao R, Fan Y, Yao H, Liao W, Sun XF, Gao S, 2013. Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Hum. Mol. Genet 22 (21), 4241–4252. 10.1093/hmg/ddt275. [DOI] [PubMed] [Google Scholar]
- Johnson AB, Bettica A, 1989. On-grid immunogold labeling of glial intermediate filaments in epoxy-embedded tissue. Am. J. Anat 185 (2–3), 335–341. 10.1002/aja.1001850228. [DOI] [PubMed] [Google Scholar]
- Jones JR, Kong L, Hanna MG 4th, Hoffman B, Krencik R, Bradley R, Hagemann T, Choi J, Doers M, Dubovis M, Sherafat MA, Bhattacharyya A, Kendziorski C, Audhya A, Messing A, Zhang SC, 2018. Mutations in GFAP disrupt the distribution and function of organelles in human astrocytes. Cell Rep. 25 (4), 947–958. 10.1016/j.celrep.2018.09.083 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaifer KA, Villalón E, O’Brien BS, Sison SL, Smith CE, Simon ME, Marquez J, O’Day S, Hopkins AE, Neff R, Rindt H, Ebert AD, Lorson CL, 2019. AAV9-mediated delivery of miR-23a reduces disease severity in Smn2B/-SMA model mice. Hum. Mol. Genet 28 (19), 3199–3210. 10.1093/hmg/ddz142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima R, Roy S, Ascano M, Martinez-Cerdeno V, Ariza-Torres J, Kim S, Louie J, Lu Y, Leyton P, Bloch KD, Kornberg TB, Hagerman PJ, Hagerman R, Lagna G, Hata A, 2016. Augmented noncanonical BMP type II receptor signaling mediates the synaptic abnormality of fragile X syndrome. Sci. Signal 9 (431), ra58. 10.1126/scisignal.aaf6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria A, Nowosiad P, Jagasia R, Aigner S, Taylor RD, Andreae LC, Gatford N, Lucchesi W, Srivastava DP, Price J, 2018. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol. Psychiatry 23 (3), 735–746. 10.1038/mp.2017.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann M, Schuffenhauer A, Fruh I, Klein J, Thiemeyer A, Rigo P, Gomez-Mancilla B, Heidinger-Millot V, Bouwmeester T, Schopfer U, Mueller M, Fodor BD, Cobos-Correa A, 2015. High-throughput screening using iPSC-Derived neuronal progenitors to identify compounds counteracting epigenetic gene silencing in fragile X syndrome. J. Biomol. Screen 20 (9), 1101–1111. 10.1177/1087057115588287. [DOI] [PubMed] [Google Scholar]
- Khattak S, Brimble E, Zhang W, Zaslavsky K, Strong E, Ross PJ, Hendry J, Mital S, Salter MW, Osborne LR, Ellis J, 2015. Human induced pluripotent stem cell derived neurons as a model for Williams-Beuren syndrome. Mol. Brain 8 (1), 77. 10.1177/1087057115588287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KY, Hysolli E, Park IH, 2011. Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc. Natl. Acad. Sci. U. S. A 108 (34), 14169–14174. 10.1073/pnas.1018979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HW, Quan Z, Kim YB, Cheong E, Kim HD, Cho M, Jang J, Yoo YR, Lee JS, Kim JH, Kim YI, Kim DS, Kang HC, 2018. Differential effects on sodium current impairments by distinct SCN1A mutations in GABAergic neurons derived from Dravet syndrome patients. Brain Dev. 40 (4), 287–298. 10.1016/jbraindev.2017.12.002. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Mazurkiewicz JE, 2013. Friedreich ataxia: neuropathology revised. J. Neuropathol. Exp. Neurol 72 (2), 78–90. 10.1097/NEN.0b013e31827e5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Funayama M, Miyake M, Tsukita K, Era T, Osaka H, Ayaki T, Takahashi R, Inoue H, 2016. Modeling Alexander disease with patient iPSCs reveals cellular and molecular pathology of astrocytes. Acta Neuropathol. Commun 4 (1), 69. 10.1186/s40478-016-0337-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyuncu S, Saez I, Lee HJ, Gutierrez-Garcia R, Pokrzywa W, Fatima A, Hoppe T, Vilchez D, 2018. The ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington’s disease patients. Nat. Commun 9 (1), 2886. 10.1038/s41467-018-05320-05323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey JF, Paşca SP, Shcheglovitov A, Yazawa M, Schwemberger R, Rasmusson R, Dolmetsch RE, 2013. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci 16 (2), 201–209. 10.1038/nn.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku S, Soragni E, Campau E, Thomas EA, Altun G, Laurent LC, Loring JF, Napierala M, Gottesfeld JM, 2010. Friedreich’s ataxia induced pluripotent stem cells model intergenerational GAA·TTC triplet repeat instability. Cell Stem Cell 7 (5), 631–637. 10.1016/j.stem.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari D, Swaroop M, Southall N, Huang W, Zheng W, Usdin K, 2015. High-throughput screening to identify compounds that increase fragile X Mental retardation protein expression in neural stem cells differentiated from fragile X syndrome patient-derived induced pluripotent stem cells. Stem Cells Transl. Med 4 (7), 800–808. 10.5966/sctm.2014-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JI, Leman LJ, Ku S, Vickers CJ, Olsen CA, Montero A, Ghadiri MR, Gottesfeld JM, 2017. Cyclic tetrapeptide HDAC inhibitors as potential therapeutics for spinal muscular atrophy: screening with iPSC-derived neuronal cells. Bioorg. Med. Chem. Lett 27 (15), 3289–3293. 10.1016/j.bmcl.2017.06.027. [DOI] [PubMed] [Google Scholar]
- Lai JI, Nachun D, Petrosyan L, Throesch B, Campau E, Gao F, Baldwin KK, Coppola G, Gottesfeld JM, Soragni E, 2019. Transcriptional profiling of isogenic Friedreich ataxia neurons and effect of an HDAC inhibitor on disease signatures. J. Biol. Chem 294 (6), 1846–1859. 10.1074/jbc.RA118.006515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA, 2013. Cerebral organoids model human brain development and microcephaly. Nature 501 (7467), 373–379. 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landucci E, Brindisi M, Bianciardi L, Catania LM, Daga S, Croci S, Frullanti E, Fallerini C, Butini S, Brogi S, Furini S, Melani R, Molinaro A, Lorenzetti FC, Imperatore V, Amabile S, Mariani J, Mari F, Ariani F, Pizzorusso T, Meloni I, 2018. iPSC-derived neurons profiling reveals GABAergic circuit disruption and acetylated α-tubulin defect which improves after iHDAC6 treatment in Rett syndrome. Exp. Cell Res 368 (2), 225–235. 10.1016/j.yexcr.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimore J, Ryder PV, Kim KY, Ambrose LA, Chapleau C, Calfa G, Gross C, Bassell GJ, Pozzo-Miller L, Smith Y, Talbot K, Park IH, Faundez V, 2013. MeCP2 regulates the synaptic expression of a Dysbindin-BLOC-1 network component in mouse brain and human induced pluripotent stem cell-derived neurons. PLoS One 8 (6), e65069. 10.1371/journal.pone.0065069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavandeira A, 2002. Orphan drugs: legal aspects, current situation. Haemophilia 8 (3), 194–198. 10.1046/j.1365-2516.2002.00643.x. [DOI] [PubMed] [Google Scholar]
- Le T, Tran NT, Dao T, Nguyen DD, Do HD, Ha TL, Kühn R, Nguyen TL, Rajewsky K, Chu VT, 2019. Efficient and precise CRISPR/Cas9-Mediated MECP2 modifications in human-induced pluripotent stem cells. Front. Genet 10, 625. 10.3389/fgene.2019.00625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Seo HS, Armien AG, Bates FS, Tolar J, Azarin SM, 2018. Modeling and rescue of defective blood-brain barrier function of induced brain microvascular endothelial cells from childhood cerebral adrenoleukodystrophy patients. Fluids Barriers CNS 15 (1), 9. 10.1186/s12987-018-0094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, 1995. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80 (1), 155–165. 10.1016/0092-8674(95)90460-90463. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J, 1997. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet 16 (3), 265–269. 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- Leoncini S, De Felice C, Signorini C, Zollo G, Cortelazzo A, Durand T, Galano JM, Guerranti R, Rossi M, Ciccoli L, Hayek J, 2015. Cytokine dysregulation in MECP2- and CDKL5-Related rett syndrome: relationships with aberrant redox homeostasis, inflammation, and ω−3 PUFAs. Oxid. Med. Cell. Longev 2015, 421624 10.1155/2015/421624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis P, Abbeduto L, Murphy M, Richmond E, Giles N, Bruno L, Schroeder S, 2006. Cognitive, language and social-cognitive skills of individuals with fragile X syndrome with and without autism. J. Intellect. Disabil. Res 50 (Pt 7), 532–545. 10.1111/j.1365-2788.2006.00803.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Polak U, Bhalla AD, Rozwadowska N, Butler JS, Lynch DR, Dent S, Napierala M, 2015. Excision of expanded GAA repeats alleviates the molecular phenotype of Friedreich’s Ataxia. Mol. Ther 23 (6), 1055–1065. 10.1038/mt.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Zhao H, Ananiev GE, Musser MT, Ness KH, Maglaque DL, Saha K, Bhattacharyya A, Zhao X, 2017. Establishment of reporter lines for detecting fragile X Mental retardation (FMR1) gene reactivation in human neural cells. Stem Cells 35 (1), 158–169. 10.1002/stem.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Tian E, Chen X, Chao J, Klein J, Qu Q, Sun G, Sun G, Huang Y, Warden CD, Ye P, Feng L, Li X, Cui Q, Sultan A, Douvaras P, Fossati V, Sanjana NE, Riggs AD, Shi Y, 2018. GFAP mutations in astrocytes impair oligodendrocyte progenitor proliferation and myelination in an hiPSC model of alexander disease. Cell Stem Cell 23 (2), 239–251. 10.1016/j.stem.2018.07.009 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Li JJ, Qian WJ, Zhang QJ, Wang ZF, Lu YQ, Dong EL, He J, Wang N, Ma LX, Chen WJ, 2017. Modeling the differential phenotypes of spinal muscular atrophy with high-yield generation of motor neurons from human induced pluripotent stem cells. Oncotarget 8 (26), 42030–42042. 10.18632/oncotarget.14925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Yoshida M, Li LT, Ikenaka A, Oshima S, Nakagawa K, Sakurai H, Matsui E, Nakahata T, Saito MK, 2019. iPSC-derived functional human neuromuscular junctions model the pathophysiology of neuromuscular diseases. JCI Insight 4 (18), e124299. 10.1172/jci.insight.124299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Verma PJ, Evans-Galea MV, Delatycki MB, Michalska A, Leung J, Crombie D, Sarsero JP, Williamson R, Dottori M, Pébay A, 2011. Generation of induced pluripotent stem cell lines from Friedreich ataxia patients. Stem Cell Rev Rep. 7 (3), 703–713. 10.1007/s12015-010-9210-x. [DOI] [PubMed] [Google Scholar]
- Liu J, Koscielska KA, Cao Z, Hulsizer S, Grace N, Mitchell G, Nacey C, Githinji J, McGee J, Garcia-Arocena D, Hagerman RJ, Nolta J, Pessah IN, Hagerman PJ, 2012. Signaling defects in iPSC-derived fragile X premutation neurons. Hum. Mol. Genet 21 (17), 3795–3805. 10.1093/hmg/dds207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lopez-Santiago LF, Yuan Y, Jones JM, Zhang H, O’Malley HA, Patino GA, O’Brien JE, Rusconi R, Gupta A, Thompson RC, Natowicz MR, Meisler MH, Isom LL, Parent JM, 2013. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann. Neurol 74 (1), 128–139. 10.1002/ana.23897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Lu J, Chen H, Du Z, Li XJ, Zhang SC, 2015. Spinal muscular atrophy patient-derived motor neurons exhibit hyperexcitability. Sci. Rep 5, 12189. 10.1038/srep12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Qiao F, Leiferman PC, Ross A, Schlenker EH, Wang H, 2017. FOXOs modulate proteasome activity in human-induced pluripotent stem cells of Huntington’s disease and their derived neural cells. Hum. Mol. Genet 26 (22), 4416–4428. 10.1093/hmg/ddx327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, Wu H, Krzisch M, Wu X, Graef J, Muffat J, Hnisz D, Li CH, Yuan B, Xu C, Li Y, Vershkov D, Cacace A, Young RA, Jaenisch R, 2018. Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell. 172 (5), 979–992. 10.1016/j.cell.2018.01.012 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Barnes J, Pedrosa E, Herman NS, Salas F, Wang P, Zheng D, Lachman HM, 2020. Transcriptome analysis of neural progenitor cells derived from Lowe syndrome induced pluripotent stem cells: identification of candidate genes for the neurodevelopmental and eye manifestations. J. Neurodev. Disord 12 (1), 14. 10.1186/s11689-020-09317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livide G, Patriarchi T, Amenduni M, Amabile S, Yasui D, Calcagno E, Lo Rizzo C, De Falco G, Ulivieri C, Ariani F, Mari F, Mencarelli MA, Hell JW, Renieri A, Meloni I, 2015. GluD1 is a common altered player in neuronal differentiation from both MECP2-mutated and CDKL5-mutated iPS cells. Eur. J. Hum. Genet 23 (2), 195–201. 10.1038/ejhg.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long A, Napierala JS, Polak U, Hauser L, Koeppen AH, Lynch DR, Napierala M, 2017. Somatic instability of the expanded GAA repeats in Friedreich’s ataxia. PLoS One 12 (12), e0189990. 10.1371/journal.pone.0189990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Chen X, Feng Y, Zeng Q, Jiang C, Zhu X, Fan G, Xue Z, 2016. Integrated transcriptome analysis of human iPS cells derived from a fragile X syndrome patient during neuronal differentiation. Sci. China Life Sci 59 (11), 1093–1105. 10.1007/s11427-016-0194-0196. [DOI] [PubMed] [Google Scholar]
- Lubala TK, Lumaka A, Kanteng G, Mutesa L, Mukuku O, Wembonyama S, Hagerman R, Luboya ON, Lukusa Tshilobo P, 2018. Fragile X checklists: a meta-analysis and development of a simplified universal clinical checklist. Mol. Genet. Genomic Med 6 (4), 526–532. 10.1002/mgg3.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Judson MC, Zylka MJ, Philpot BD, 2011. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34 (6), 293–303. 10.1016/j.tins.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda H, Chiyonobu T, Yoshida M, Yamashita S, Zuiki M, Kidowaki S, Isoda K, Yamakawa K, Morimoto M, Nakahata T, Saito MK, Hosoi H, 2016. Establishment of isogenic iPSCs from an individual with SCN1A mutation mosaicism as a model for investigating neurocognitive impairment in Dravet syndrome. J. Hum. Genet 61 (6), 565–569. 10.1038/jhg.2016.5. [DOI] [PubMed] [Google Scholar]
- Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR, 2010. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 143 (4), 527–539. 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli A, Wattenhofer-Donzé M, Schmucker S, Bouvet S, Reutenauer L, Puccio H, 2007. Frataxin is essential for extramitochondrial Fe-S cluster proteins in mammalian tissues. Hum. Mol. Genet 16 (22), 2651–2658. 10.1093/hmg/ddm163. [DOI] [PubMed] [Google Scholar]
- Martin JE, Nguyen TT, Grunseich C, Nofziger JH, Lee PR, Fields D, Fischbeck KH, Foran E, 2017. Decreased Motor Neuron Support by SMA Astrocytes due to Diminished MCP1 Secretion. J. Neurosci 37 (21), 5309–5318. 10.1523/JNEUROSCI.3472-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattis VB, Tom C, Akimov S, Saeedian J, Østergaard ME, Southwell AL, Doty CN, Ornelas L, Sahabian A, Lenaeus L, Mandefro B, Sareen D, Arjomand J, Hayden MR, Ross CA, Svendsen CN, 2015. HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum. Mol. Genet 24 (11), 3257–3271. 10.1093/hmg/ddv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColgan P, Tabrizi SJ, 2018. Huntington’s disease: a clinical review. Eur. J. Neurol 25 (1), 24–34. 10.1111/ene.13413. [DOI] [PubMed] [Google Scholar]
- McGivern JV, Patitucci TN, Nord JA, Barabas MA, Stucky CL, Ebert AD, 2013. Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia. 61 (9), 1418–1428. 10.1002/glia.22522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, Kearney JA, 2005. Sodium channel mutations in epilepsy and other neurological disorders. J. Clin. Invest 115 (8), 2010–2017. 10.1172/JCI25466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, O’Brien JE, Sharkey LM, 2010. Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects. J Physiol. 588 (Pt 11), 1841–1848. 10.1113/jphysiol.2010.188482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellios N, Feldman DA, Sheridan SD, Ip J, Kwok S, Amoah SK, Rosen B, Rodriguez BA, Crawford B, Swaminathan R, Chou S, Li Y, Ziats M, Ernst C, Jaenisch R, Haggarty SJ, Sur M, 2018. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatry 23 (4), 1051–1065. 10.1038/mp.2017.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneghini V, Frati G, Sala D, De Cicco S, Luciani M, Cavazzin C, Paulis M, Mentzen W, Morena F, Giannelli S, Sanvito F, Villa A, Bulfone A, Broccoli V, Martino S, Gritti A, 2017. Generation of human induced pluripotent stem cell-derived bona fide neural stem cells for ex vivo gene therapy of metachromatic leukodystrophy. Stem Cells Transl. Med 6 (2), 352–368. 10.5966/sctm.2015-0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri E, Finkel RS, Muntoni F, Wirth B, Montes J, Main M, Mazzone ES, Vitale M, Snyder B, Quijano-Roy S, Bertini E, Davis RH, Meyer OH, Simonds AK, Schroth MK, Graham RJ, Kirschner J, Iannaccone ST, Crawford TO, Woods S, et al. , 2018. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord 28 (2), 103–115. 10.1016/j.nmd.2017.11.005. [DOI] [PubMed] [Google Scholar]
- Monteiro P, Feng G, 2017. SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci 18 (3), 147–157. 10.1038/nrn.2016.183. [DOI] [PubMed] [Google Scholar]
- Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA, 2005. SCN1A mutations and epilepsy. Hum. Mutat 25 (6), 535–542. 10.1038/mp.2017.86. [DOI] [PubMed] [Google Scholar]
- Mullin AP, Gokhale A, Moreno-De-Luca A, Sanyal S, Waddington JL, Faundez V, 2013. Neurodevelopmental disorders: mechanisms and boundary definitions from genomes, interactomes and proteomes. Transl. Psychiatry 3 (12), e329. 10.1038/tp.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muotri AR, Marchetto MC, Coufal NG, Oefner R, Yeo G, Nakashima K, Gage FH, 2010. L1 retrotransposition in neurons is modulated by MeCP2. Nature. 468 (7322), 443–446. 10.1038/nature09544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdocca M, Ciafrè SA, Spitalieri P, Talarico RV, Sanchez M, Novelli G, Sangiuolo F, 2016. SMA human iPSC-derived motor neurons show perturbed differentiation and reduced miR-335–5p expression. Int. J. Mol. Sci 17 (8), 1231. 10.3390/ijms17081231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabbout R, Gennaro E, Dalla Bernardina B, Dulac O, Madia F, Bertini E, Capovilla G, Chiron C, Cristofori G, Elia M, Fontana E, Gaggero R, Granata T, Guerrini R, Loi M, La Selva L, Lispi ML, Matricardi A, Romeo A, Tzolas V, Zara F, 2003. Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology. 60 (12), 1961–1967. 10.1212/01.WNL.0000069463.41870.2F. [DOI] [PubMed] [Google Scholar]
- Nachun D, Gao F, Isaacs C, Strawser C, Yang Z, Dokuru D, Van Berlo V, Sears R, Farmer J, Perlman S, Lynch DR, Coppola G, 2018. Peripheral blood gene expression reveals an inflammatory transcriptomic signature in Friedreich’s ataxia patients. Hum. Mol. Genet 27 (17), 2965–2977. 10.1093/hmg/ddy198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamani SC, Zhang F, Shchelochkov OA, Bi W, Ou Z, Scaglia F, Probst FJ, Shinawi M, Eng C, Hunter JV, Sparagana S, Lagoe E, Fong CT, Pearson M, Doco-Fenzy M, Landais E, Mozelle M, Chinault AC, Patel A, Bacino CA, et al. , 2009. Microdeletions including YWHAE in the Miller-Dieker syndrome region on chromosome 17p13.3 result in facial dysmorphisms, growth restriction, and cognitive impairment. J. Med. Genet 46 (12), 825–833. 10.1136/jmg.2009.067637. [DOI] [PubMed] [Google Scholar]
- Neureiter A, Brändl B, Hiber M, Tandon R, Müller FJ, Steenpass L, 2018. Generation of an iPSC line of a patient with Angelman syndrome due to an imprinting defect. Stem Cell Res. 33, 20–24. 10.1016/j.scr.2018.09.015. [DOI] [PubMed] [Google Scholar]
- Nevin ZS, Factor DC, Karl RT, Douvaras P, Laukka J, Windrem MS, Goldman SA, Fossati V, Hobson GM, Tesar PJ, 2017. Modeling the mutational and phenotypic landscapes of pelizaeus-merzbacher disease with human iPSC-derived oligodendrocytes. Am. J. Hum. Genet 100 (4), 617–634. 10.1016/j.ajhg.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SY, Soh BS, Rodriguez-Muela N, Hendrickson DG, Price F, Rinn JL, Rubin LL, 2015. Genome-wide RNA-Seq of human motor neurons implicates selective ER stress activation in spinal muscular atrophy. Cell Stem Cell 17 (5), 569–584. 10.1016/j.stem.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi M, Martin HC, Rice DL, Gallone G, Gordon S, Kelemen M, McAloney K, McRae J, Radford EJ, Yu S, Gecz J, Martin NG, Wright CF, Fitzpatrick DR, Firth HV, Hurles ME, Barrett JC, 2018. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature. 562 (7726), 268–271. 10.1038/s41586-018-0566-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizzardo M, Simone C, Dametti S, Salani S, Ulzi G, Pagliarani S, Rizzo F, Frattini E, Pagani F, Bresolin N, Comi G, Corti S, 2015. Spinal muscular atrophy phenotype is ameliorated in human motor neurons by SMN increase via different novel RNA therapeutic approaches. Sci. Rep 5, 11746. 10.1038/srep11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numasawa-Kuroiwa Y, Okada Y, Shibata S, Kishi N, Akamatsu W, Shoji M, Nakanishi A, Oyama M, Osaka H, Inoue K, Takahashi K, Yamanaka S, Kosaki K, Takahashi T, Okano H, 2014. Involvement of ER stress in dysmyelination of Pelizaeus-Merzbacher Disease with PLP1 missense mutations shown by iPSC-derived oligodendrocytes. Stem Cell Reports 2 (5), 648–661. 10.1016/j.stemcr.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K, 2007. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying a Scn1a gene mutation. J. Neurosci 27 (22), 5903–5914. 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi M, Korsakova E, Allen D, Lee P, Fu K, Vargas BS, Cinkornpumin J, Salas C, Park JC, Germanguz I, Langerman J, Chronis C, Kuoy E, Tran S, Xiao X, Pellegrini M, Plath K, Lowry WE, 2018. Loss of MECP2 leads to activation of P53 and neuronal senescence. Stem Cell Reports 10 (5), 1453–1463. 10.1016/j.stemcr.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlmeier C, Saum KU, Galetzka W, Beier D, Gothe H, 2019. Epidemiology and health care utilization of patients suffering from Huntington’s disease in Germany: real world evidence based on German claims data. BMC Neurol. 19 (1), 318. 10.1186/s12883-019-1556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohuchi K, Funato M, Kato Z, Seki J, Kawase C, Tamai Y, Ono Y, Nagahara Y, Noda Y, Kameyama T, Ando S, Tsuruma K, Shimazawa M, Hara H, Kaneko H, 2016. Established stem cell model of spinal muscular atrophy is applicable in the evaluation of the efficacy of thyrotropin-releasing hormone analog. Stem Cells Transl. Med 5 (2), 152–163. 10.5966/sctm.2015-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohuchi K, Funato M, Ando S, Inagaki S, Sato A, Kawase C, Seki J, Nakamura S, Shimazawa M, Kaneko H, Hara H, 2019. Impairment of oligodendrocyte lineages in spinal muscular atrophy model systems. Neuroreport 30 (5), 350–357. 10.1097/WNR.0000000000001206. [DOI] [PubMed] [Google Scholar]
- Ormel PR, Vieira de Sá R, van Bodegraven EJ, Karst H, Harschnitz O, Sneeboer M, Johansen LE, van Dijk RE, Scheefhals N, Berdenis van Berlekom A, Ribes Martínez E, Kling S, MacGillavry HD, van den Berg LH, Kahn RS, Hol EM, de Witte LD, Pasterkamp RJ, 2018. Microglia innately develop within cerebral organoids. Nat. Commun 9 (1), 4167. 10.1038/s41467-018-06684-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MJ, O’Donovan MC, 2017. Schizophrenia and the neurodevelopmental continuum: evidence from genomics. World Psychiatry 16 (3), 227–235. 10.1002/wps.20440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagiotakos G, Haveles C, Arjun A, Petrova R, Rana A, Portmann T, Paşca SP, Palmer TD, Dolmetsch RE, 2019. Aberrant calcium channel splicing drives defects in cortical differentiation in Timothy syndrome. ELife 8, e51037. 10.7554/eLife.51037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Halevy T, Lee DR, Sung JJ, Lee JS, Yanuka O, Benvenisty N, Kim DW, 2015. Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X ipsc-derived neurons. Cell Rep. 13 (2), 234–241. 10.1016/j.celrep.2015.08.084. [DOI] [PubMed] [Google Scholar]
- Paşca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Paşca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, Bernstein JA, Hallmayer J, Geschwind DH, Dolmetsch RE, 2011. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med 17 (12), 1657–1662. 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patitucci TN, Ebert AD, 2016. SMN deficiency does not induce oxidative stress in SMA iPSC-derived astrocytes or motor neurons. Hum. Mol. Genet 25 (3), 514–523. 10.1093/hmg/ddv489. [DOI] [PubMed] [Google Scholar]
- Patriarchi T, Amabile S, Frullanti E, Landucci E, Lo Rizzo C, Ariani F, Costa M, Olimpico F, W Hell J, M Vaccarino F, Renieri A, Meloni I, 2016. Imbalance of excitatory/inhibitory synaptic protein expression in iPSC-derived neurons from FOXG1 (+/−) patients and in foxg1 (+/−) mice. Eur. J. Hum. Genet 24 (6), 871–880. 10.1038/ejhg.2015.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsalis PC, Sismani C, Hettinger JA, Boumba I, Georgiou I, Stylianidou G, Anastasiadou V, Koukoulli R, Pagoulatos G, Syrrou M, 1999. Molecular screening of fragile X (FRAXA) and FRAXE mental retardation syndromes in the Hellenic population of Greece and Cyprus: incidence, genetic variation, and stability. Am. J. Med. Genet 84 (3), 184–190. [PubMed] [Google Scholar]
- Pham MT, Pollock KM, Rose MD, Cary WA, Stewart HR, Zhou P, Nolta JA, Waldau B, 2018. Generation of human vascularized brain organoids. Neuroreport. 29 (7), 588–593. 10.1097/WNR.0000000000001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan MC, 2008. Deletion 22q13.3 syndrome. Orphanet J. Rare Dis 3, 14. 10.1186/1750-1172-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan K, McDermid HE, 2012. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol. Syndromol 2 (3–5), 186–201. 10.1159/000334260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL, 1991. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 66 (4), 817–822. 10.1016/0092-8674(91)90125-I. [DOI] [PubMed] [Google Scholar]
- Polak U, Li Y, Butler JS, Napierala M, 2016. Alleviating GAA repeat induced transcriptional silencing of the friedreich’s Ataxia gene during somatic cell reprogramming. Stem Cells Dev. 25 (23), 1788–1800. 10.1089/scd.2016.0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pólvora-Brandão D, Joaquim M, Godinho I, Aprile D, Álvaro AR, Onofre I, Raposo AC, Pereira de Almeida L, Duarte ST, da Rocha ST, 2018. Loss of hierarchical imprinting regulation at the Prader-Willi/Angelman syndrome locus in human iPSCs. Hum. Mol. Genet 27 (23), 3999–4011. 10.1093/hmg/ddy274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N, 2012. The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov. Disord 27 (9), 1083–1091. 10.1002/mds.25075. [DOI] [PubMed] [Google Scholar]
- Prust M, Wang J, Morizono H, Messing A, Brenner M, Gordon E, Hartka T, Sokohl A, Schiffmann R, Gordish-Dressman H, Albin R, Amartino H, Brockman K, Dinopoulos A, Dotti MT, Fain D, Fernandez R, Ferreira J, Fleming J, Gill D, et al. , 2011. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology 77 (13), 1287–1294. 10.1212/WNL.0b013e3182309f72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, Yoon KJ, Jeang W, Lin L, Li Y, Thakor J, Berg DA, Zhang C, Kang E, Chickering M, Nauen D, et al. , 2016. Brain-region-Specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165 (5), 1238–1254. 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A, Crisafulli SG, Rizzuti M, Bresolin N, Comi GP, Corti S, Nizzardo M, 2018. Investigation of new morpholino oligomers to increase survival motor neuron protein levels in spinal muscular atrophy. Int. J. Mol. Sci 19 (1), 167. 10.3390/ijms19010167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastegar M, Hotta A, Pasceri P, Makarem M, Cheung AY, Elliott S, Park KJ, Adachi M, Jones FS, Clarke ID, Dirks P, Ellis J, 2009. MECP2 isoform-specific vectors with regulated expression for Rett syndrome gene therapy. PLoS One 4 (8), e6810. 10.1371/journal.pone.0006810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH, 1993. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature 364 (6439), 717–721. 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- Riordan NH, Hincapié ML, Morales I, Fernández G, Allen N, Leu C, Madrigal M, Paz Rodríguez J, Novarro N, 2019. Allogeneic human umbilical cord mesenchymal stem cells for the treatment of autism spectrum disorder in children: safety profile and effect on cytokine levels. Stem Cells Transl. Med 8 (10), 1008–1016. 10.1002/sctm.19-0010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rizzo F, Nizzardo M, Vashisht S, Molteni E, Melzi V, Taiana M, Salani S, Santonicola P, Di Schiavi E, Bucchia M, Bordoni A, Faravelli I, Bresolin N, Comi GP, Pozzoli U, Corti S, 2019. Key role of SMN/SYNCRIP and RNA-Motif 7 in spinal muscular atrophy: RNA-Seq and motif analysis of human motor neurons. Brain. 142 (2), 276–294. 10.1093/brain/awy330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues DC, Mufteev M, Weatheritt RJ, Djuric U, Ha K, Ross PJ, Wei W, Piekna A, Sartori MA, Byres L, Mok R, Zaslavsky K, Pasceri P, Diamandis P, Morris Q, Blencowe BJ, Ellis J, 2020. Shifts in ribosome engagement impact key gene sets in neurodevelopment and ubiquitination in rett syndrome. Cell Rep. 30 (12), 4179–4196. 10.1016/j.celrep.2020.02.107 e11. [DOI] [PubMed] [Google Scholar]
- Roos RA, 2010. Huntington’s disease: a clinical review. Orphanet J. Rare Dis 5, 40. 10.1186/1750-1172-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosander C, Hallböök T, 2015. Dravet syndrome in Sweden: a population-based study. Dev. Med. Child Neurol 57 (7), 628–633. 10.1111/dmcn.12709. [DOI] [PubMed] [Google Scholar]
- Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, Reilmann R, Unschuld PG, Wexler A, Margolis RL, Tabrizi SJ, 2014. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol 10 (4), 204–216. 10.1038/nrneurol.2014.24. [DOI] [PubMed] [Google Scholar]
- Sarasua SM, Dwivedi A, Boccuto L, Chen CF, Sharp JL, Rollins JD, Collins JS, Rogers RC, Phelan K, DuPont BR, 2014. 22q13.2q13.32 genomic regions associated with severity of speech delay, developmental delay, and physical features in Phelan-McDermid syndrome. Genet. Med 16 (4), 318–328. 10.1038/gim.2013.144. [DOI] [PubMed] [Google Scholar]
- Sareen D, Ebert AD, Heins BM, McGivern JV, Ornelas L, Svendsen CN, 2012. Inhibition of apoptosis blocks human motor neuron cell death in a stem cell model of spinal muscular atrophy. PLoS One 7 (6), e39113. 10.1371/journal.pone.0039113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber AM, Misiorek JO, Napierala JS, Napierala M, 2019. Progress in understanding Friedreich’s ataxia using human induced pluripotent stem cells. Expert Opin. Orphan Drugs 7 (2), 81–90. 10.1080/21678707.2019.1562334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster J, Fatima A, Sobol M, Norradin FH, Laan L, Dahl N, 2019a. Generation of three human induced pluripotent stem cell (iPSC) lines from three patients with Dravet syndrome carrying distinct SCN1A gene mutations. Stem Cell Res. 39, 101523 10.1016/j.scr.2019.101523. [DOI] [PubMed] [Google Scholar]
- Schuster J, Laan L, Klar J, Jin Z, Huss M, Korol S, Noraddin FH, Sobol M, Birnir B, Dahl N, 2019b. Transcriptomes of Dravet syndrome iPSC derived GABAergic cells reveal dysregulated pathways for chromatin remodeling and neurodevelopment. Neurobiol. Dis 132, 104583 10.1016/j.nbd.2019.104583. [DOI] [PubMed] [Google Scholar]
- Schwab AJ, Ebert AD, 2014. Sensory neurons do not induce motor neuron loss in a human stem cell model of spinal muscular atrophy. PLoS One 9 (7), e103112. 10.1371/journal.pone.0103112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen D, Voulgaropoulos A, Drobna Z, Keung AJ, 2019. Human cerebral organoids capture the spatiotemporal complexity and disease dynamics of UBE3A. BioRxiv. 10.1101/742213. [DOI] [Google Scholar]
- Shan B, Xu C, Zhang Y, Xu T, Gottesfeld JM, Yates JR 3rd, 2014. Quantitative proteomic analysis identifies targets and pathways of a 2-aminobenzamide HDAC inhibitor in Friedreich’s ataxia patient iPSC-derived neural stem cells. J. Proteome Res 13 (11), 4558–4566. 10.1021/pr500514r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Schiapparelli L, Cline HT, 2013. Exosomes function in cell-cell communication during brain circuit development. Curr. Opin. Neurobiol 23 (6), 997–1004. 10.1016/j.conb.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Mesci P, Carromeu C, McClatchy DR, Schiapparelli L, Yates JR 3rd, Muotri AR, Cline HT, 2019. Exosomes regulate neurogenesis and circuit assembly. Proc. Natl. Acad. Sci. U. S. A 116 (32), 16086–16094. 10.1073/pnas.1902513116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcheglovitov A, Shcheglovitov AO, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF, Dolmetsch RE, 2013. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503 (7475), 267–271. 10.1038/nature12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen VL, Ferland RJ, Harney M, Hill RS, Neal J, Banham AH, Brown P, Chenn A, Corbo J, Hecht J, Folkerth R, Walsh CA, 2006a. Impaired proliferation and migration in human Miller-Dieker neural precursors. Ann. Neurol 60 (1), 137–144. 10.1007/s00401-005-0010-0013. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Ferland RJ, Neal J, Harney M, Hill RS, Banham A, Brown P, Chenn A, Corbo J, Hecht J, Folkerth R, Walsh CA, 2006b. Neocortical neuronal arrangement in Miller Dieker syndrome. Acta Neuropathol. 111 (5), 489–496. 10.1007/s00401-005-0010-0013. [DOI] [PubMed] [Google Scholar]
- Shen X, Beasley S, Putman JN, Li Y, Prakash TP, Rigo F, Napierala M, Corey DR, 2019. Efficient electroporation of neuronal cells using synthetic oligonucleotides: identifying duplex RNA and antisense oligonucleotide activators of human frataxin expression. RNA. 25 (9), 1118–1129. 10.1261/rna.071290.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L, Loring JF, Haggarty SJ, 2011. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One 6 (10), e26203. 10.1371/journal.pone.0026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Yasumoto S, Nakagawa E, Fukasawa T, Uchiya S, Hirose S, 2009. Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome. Brain Dev. 31 (10), 758–762. 10.1016/j.braindev.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N, 2016. The cellular and molecular landscapes of the developing human central nervous system. Neuron. 89 (2), 248–268. 10.1016/j.neuron.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer C, 2012. Comprehensive treatment of Huntington disease and other choreic disorders. Cleve Clin J. Med 79 (Suppl 2), S30–S34. 10.3949/ccjm.79.s2a.06. [DOI] [PubMed] [Google Scholar]
- Sison SL, Patitucci TN, Seminary ER, Villalon E, Lorson CL, Ebert AD, 2017. Astrocyte-produced miR-146a as a mediator of motor neuron loss in spinal muscular atrophy. Hum. Mol. Genet 26 (17), 3409–3420. 10.1093/hmg/ddx230. [DOI] [PubMed] [Google Scholar]
- Soeda S, Saito R, Fujita N, Fukuta K, Taniura H, 2019. Neuronal differentiation defects in induced pluripotent stem cells derived from a Prader-Willi syndrome patient. Neurosci. Lett 703, 162–167. 10.1016/j.neulet.2019.03.029. [DOI] [PubMed] [Google Scholar]
- Son D, Quan Z, Kang PJ, Park G, Kang HC, You S, 2017. Generation of two induced pluripotent stem cell (iPSC) lines from X-linked adrenoleukodystrophy (X-ALD) patients with adrenomyeloneuropathy (AMN). Stem Cell Res. 25, 46–49. 10.1016/j.scr.2017.10.003. [DOI] [PubMed] [Google Scholar]
- Son YS, Choi K, Lee H, Kwon O, Jung KB, Cho S, Baek J, Son B, Kang SM, Kang M, Yoon J, Shen H, Lee S, Oh JH, Lee HA, Lee MO, Cho HS, Jung CR, Kim J, Cho S, et al. , 2019. A SMN2 splicing modifier rescues the disease phenotypes in an in vitro human spinal muscular atrophy model. Stem Cells Dev. 28 (7), 438–453. 10.1089/scd.2018.0181. [DOI] [PubMed] [Google Scholar]
- Soragni E, Gottesfeld JM, 2016. Translating HDAC inhibitors in Friedreich’s ataxia. Expert Opin. Orphan Drugs 4 (9), 961–970. 10.1080/21678707.2016.1215910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soragni E, Miao W, Iudicello M, Jacoby D, De Mercanti S, Clerico M, Longo F, Piga A, Ku S, Campau E, Du J, Penalver P, Rai M, Madara JC, Nazor K, O’Connor M, Maximov A, Loring JF, Pandolfo M, Durelli L, Rusche JR, 2014. Epigenetic therapy for Friedreich ataxia. Ann. Neurol 76 (4), 489–508. 10.1002/ana.24260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanurova J, Neureiter A, Hiber M, de Oliveira Kessler H, Stolp K, Goetzke R, Klein D, Bankfalvi A, Klump H, Steenpass L, 2016. Angelman syndrome-derived neurons display late onset of paternal UBE3A silencing. Sci. Rep 6, 30792. 10.1038/srep30792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles J, Jernigan TL, 2010. The basics of brain development. Neuropsychol. Rev 20 (4), 327–348. 10.1007/s11065-010-9148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Mazaki-Miyazaki E, Fukushima K, Shimomura J, Fujiwara T, Hamano S, Inoue Y, Yamakawa K, 2002. Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology 58 (7), 1122–1124. 10.1212/WNL.58.7.1122. [DOI] [PubMed] [Google Scholar]
- Sun Y, Dolmetsch RE, 2018. Investigating the therapeutic mechanism of cannabidiol in a human induced pluripotent stem cell (iPSC)-Based model of dravet syndrome. Cold Spring Harb. Symp. Quant. Biol 83, 185–191. 10.1101/sqb.2018.83.038174. [DOI] [PubMed] [Google Scholar]
- Sun Y, Paşca SP, Portmann T, Goold C, Worringer KA, Guan W, Chan KC, Gai H, Vogt D, Chen YJ, Mao R, Chan K, Rubenstein JL, Madison DV, Hallmayer J, Froehlich-Santino WM, Bernstein JA, Dolmetsch RE, 2016. A deleterious Nav1.1 mutation selectively impairs telencephalic inhibitory neurons derived from Dravet Syndrome patients. eLife 5, e13073. 10.7554/eLife.13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunamura N, Iwashita S, Enomoto K, Kadoshima T, Isono F, 2018. Loss of the fragile X mental retardation protein causes aberrant differentiation in human neural progenitor cells. Sci. Rep 8 (1), 11585. 10.1038/s41598-018-30025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur M, Rubenstein JL, 2005. Patterning and plasticity of the cerebral cortex. Science. 310 (5749), 805–810. 10.1126/science.1112070. [DOI] [PubMed] [Google Scholar]
- Surén P, Bakken IJ, Aase H, Chin R, Gunnes N, Lie KK, Magnus P, Reichborn-Kjennerud T, Schjølberg S, Øyen AS, Stoltenberg C, 2012. Autism spectrum disorder, ADHD, epilepsy, and cerebral palsy in Norwegian children. Pediatrics. 130 (1), e152–158. 10.1542/peds.2011-3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szlachcic WJ, Switonski PM, Krzyzosiak WJ, Figlerowicz M, Figiel M, 2015. Huntington disease iPSCs show early molecular changes in intracellular signaling, the expression of oxidative stress proteins and the p53 pathway. Dis. Model. Mech 8 (9), 1047–1057. 10.1242/dmm.019406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Kim KY, Zhong M, Pan X, Weissman SM, Park IH, 2014. Transcriptional regulation in pluripotent stem cells by methyl CpG-binding protein 2 (MeCP2). Hum. Mol. Genet 23 (4), 1045–1055. 10.1093/hmg/ddt500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Kim J, Zhou L, Wengert E, Zhang L, Wu Z, Carromeu C, Muotri AR, Marchetto MC, Gage FH, Chen G, 2016. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc Natl Acad Sci U S A. 113 (3), 751–756. 10.1073/pnas.1524013113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Drotar J, Li K, Clairmont CD, Brumm AS, Sullins AJ, Wu H, Liu XS, Wang J, Gray NS, Sur M, Jaenisch R, 2019. Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med 11 (503), eaau0164. 10.1126/scitranslmed.aau0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tau GZ, Peterson BS, 2010. Normal development of brain circuits. Neuropsychopharmacology 35 (1), 147–168. 10.1038/npp.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Voineagu I, Paşca SP, Won H, Chandran V, Horvath S, Dolmetsch RE, Geschwind DH, 2014. Alteration in basal and depolarization induced transcriptional network in iPSC derived neurons from Timothy syndrome. Genome Med. 6 (10), 75. 10.1186/s13073-014-0075-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball AM, Neely MD, Chamberlin R, Aboud AA, Kumar KK, Han B, Bryan MR, Aschner M, Ess KC, Bowman AB, 2016. Genomic instability associated with p53 knockdown in the generation of Huntington’s disease human induced pluripotent stem cells. PLoS One 11 (3), e0150372. 10.1371/journal.pone.0150372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou AY, Paulsen EK, Lagedrost SJ, Perlman SL, Mathews KD, Wilmot GR, Ravina B, Koeppen AH, Lynch DR, 2011. Mortality in Friedreich ataxia. J. Neurol. Sci 307 (1–2), 46–49. 10.1016/j.jns.2011.05.023. [DOI] [PubMed] [Google Scholar]
- Turner G, Webb T, Wake S, Robinson H, 1996. Prevalence of fragile X syndrome. Am. J. Med. Genet 64 (1), 196–197. . [DOI] [PubMed] [Google Scholar]
- Turrigiano G, 2012. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol 4 (1), a005736. 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng CC, Tsai LP, Hwu WL, Lin SJ, Chao MC, Jong YJ, Chu SY, Chao WC, Lu CL, 2005. Prevalence of the FMR1 mutation in Taiwan assessed by large-scale screening of newborn boys and analysis of DXS548-FRAXAC1 haplotype. Am. J. Med. Genet. A 133A (1), 37–43. 10.1002/ajmg.a.30528. [DOI] [PubMed] [Google Scholar]
- Upadhya D, Shetty AK, 2019. Extracellular vesicles as therapeutics for brain injury and disease. Curr. Pharm. Des 25 (33), 3500–3505. 10.2174/1381612825666191014164950. [DOI] [PubMed] [Google Scholar]
- Upadhya D, Castro OW, Upadhya R, Shetty AK, 2018. Prospects of cannabidiol for easing status epilepticus-induced epileptogenesis and related comorbidities. Mol. Neurobiol 55 (8), 6956–6964. 10.1007/s12035-018-0898-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya D, Hattiangady B, Castro OW, Shuai B, Kodali M, Attaluri S, Bates A, Dong Y, Zhang SC, Prockop DJ, Shetty AK, 2019. Human induced pluripotent stem cell-derived MGE cell grafting after status epilepticus attenuates chronic epilepsy and comorbidities via synaptic integration. Proc. Natl. Acad. Sci. U. S. A 116 (1), 287–296. 10.1073/pnas.1814185115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach A, Bar-Nur O, Daley GQ, Benvenisty N, 2010. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 6 (5), 407–411. 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valetdinova KR, Maretina MA, Kuranova ML, Grigor’eva EV, Minina YM, Kizilova EA, Kiselev AV, Medvedev SP, Baranov VS, Zakian SM, 2019. Generation of two spinal muscular atrophy (SMA) type I patient-derived induced pluripotent stem cell (iPSC) lines and two SMA type II patient-derived iPSC lines. Stem Cell Res. 34, 101376 10.1016/j.scr.2018.101376. [DOI] [PubMed] [Google Scholar]
- Varderidou-Minasian S, Hinz L, Hagemans D, Posthuma D, Altelaar M, Heine VM, 2019. Quantitative proteomic alterations of human iPSC-based neuronal development indicate early onset of Rett syndrome. BioRxiv. 10.1101/603647. [DOI] [Google Scholar]
- Vazquez-Arango P, Vowles J, Browne C, Hartfield E, Fernandes HJ, Mandefro B, Sareen D, James W, Wade-Martins R, Cowley SA, Murphy S, O’Reilly D, 2016. Variant U1 snRNAs are implicated in human pluripotent stem cell maintenance and neuromuscular disease. Nucleic Acids Res. 44 (22), 10960–10973. 10.1093/nar/gkw711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, 1991. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 65 (5), 905–914. 10.1016/0092-8674(91)90397-H. [DOI] [PubMed] [Google Scholar]
- Vershkov D, Fainstein N, Suissa S, Golan-Lev T, Ben-Hur T, Benvenisty N, 2019. FMR1 reactivating treatments in fragile X iPSC-Derived neural progenitors in vitro and in vivo. Cell Rep. 26 (10), 2531–2539. 10.1016/j.celrep.2019.02.026 e4. [DOI] [PubMed] [Google Scholar]
- Villas N, Meskis MA, Goodliffe S, 2017. Dravet syndrome: characteristics, comorbidities, and caregiver concerns. Epilepsy Behav. 74, 81–86. 10.1016/j.yebeh.2017.06.031. [DOI] [PubMed] [Google Scholar]
- Walker FO, 2007. Huntington’s disease. Lancet 369 (9557), 218–228. 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- Wang XM, Yik WY, Zhang P, Lu W, Dranchak PK, Shibata D, Steinberg SJ, Hacia JG, 2012. The gene expression profiles of induced pluripotent stem cells from individuals with childhood cerebral adrenoleukodystrophy are consistent with proposed mechanisms of pathogenesis. Stem Cell Res. Ther 3 (5), 39. 10.1186/scrt130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb T, Clarke A, Hanefeld F, Pereira JL, Rosenbloom L, Woods CG, 1998. Linkage analysis in Rett syndrome families suggests that there may be a critical region at Xq28. J. Med. Genet 35 (12), 997–1003. 10.1136/jmg.35.12.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CA, 2005. Neurological aspects of the Angelman syndrome. Brain Dev. 27 (2), 88–94. 10.1016/j.braindev.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Williams EC, Zhong X, Mohamed A, Li R, Liu Y, Dong Q, Ananiev GE, Mok JC, Lin BR, Lu J, Chiao C, Cherney R, Li H, Zhang SC, Chang Q, 2014. Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum. Mol. Genet 23 (11), 2968–2980. 10.1093/hmg/ddu008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff M, Cassé-Perrot C, Dravet C, 2006. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia 47 (Suppl 2), 45–48. 10.1111/j.1528-1167.2006.00688.x. [DOI] [PubMed] [Google Scholar]
- Wu YW, Sullivan J, McDaniel SS, Meisler MH, Walsh EM, Li SX, Kuzniewicz MW, 2015. Incidence of dravet syndrome in a US population. Pediatrics. 136 (5), e1310–e1315. 10.1542/peds.2015-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynshaw-Boris A, 2007. Lissencephaly and LIS1: insights into the molecular mechanisms of neuronal migration and development. Clin. Genet 72 (4), 296–304. [DOI] [PubMed] [Google Scholar]
- Xie N, Gong H, Suhl JA, Chopra P, Wang T, Warren ST, 2016. Reactivation of FMR1 by CRISPR/Cas9-mediated deletion of the expanded CGG-repeat of the Fragile X chromosome. PLoS One 11 (10), e0165499. 10.1111/j.1399-0004.2007.00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu CC, Denton KR, Wang ZB, Zhang X, Li XJ, 2016. Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. Dis. Model. Mech 9 (1), 39–49. 10.1242/dmm.021766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Tay Y, Sim B, Yoon SI, Huang Y, Ooi J, Utami KH, Ziaei A, Ng B, Radulescu C, Low D, Ng A, Loh M, Venkatesh B, Ginhoux F, Augustine GJ, Pouladi MA, 2017. Reversal of phenotypic abnormalities by CRISPR/Cas9-Mediated gene correction in huntington disease patient-derived induced pluripotent stem cells. Stem Cell Reports 8 (3), 619–633. 10.1016/j.stemcr.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Mead BE, Safaee H, Langer R, Karp JM, Levy O, 2016. Engineering stem cell organoids. Cell Stem Cell 18 (1), 25–38. 10.1016/j.stem.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo M, Carromeu C, Kwon O, Muotri A, Schachner M, 2017. The L1 adhesion molecule normalizes neuritogenesis in Rett syndrome-derived neural precursor cells. Biochem. Biophys. Res. Commun 494 (3–4), 504–510. 10.1016/j.bbrc.2017.10.073. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Sasaki M, Yoshida M, Namekawa M, Okamoto Y, Tsujino S, Sasayama H, Mizuta I, Nakagawa M, Alexander Disease Study Group in Japan, 2011. Nationwide survey of Alexander disease in Japan and proposed new guidelines for diagnosis. J. Neurol 258 (11), 1998–2008. 10.1007/s00415-011-6056-3. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Kitaoka S, Egawa N, Yamane M, Ikeda R, Tsukita K, Amano N, Watanabe A, Morimoto M, Takahashi J, Hosoi H, Nakahata T, Inoue H, Saito MK, 2015. Modeling the early phenotype at the neuromuscular junction of spinal muscular atrophy using patient-derived iPSCs. Stem Cell Reports 4 (4), 561–568. 10.1016/j.stemcr.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA, 2006. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci 9 (9), 1142–1149. 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Marro SG, Zhang Y, Arendt KL, Patzke C, Zhou B, Fair T, Yang N, Südhof TC, Wernig M, Chen L, 2018. The fragile X mutation impairs homeostatic plasticity in human neurons by blocking synaptic retinoic acid signaling. Sci. Transl. Med 10 (452), eaar4338. 10.1126/scitranslmed.aar4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yang SL, Yang M, Herrlinger S, Shao Q, Collar JL, Fierro E, Shi Y, Liu A, Lu H, Herring BE, Guo ML, Buch S, Zhao Z, Xu J, Lu Z, Chen JF, 2019. Modeling microcephaly with cerebral organoids reveals a WDR62-CEP170-KIF2A pathway promoting cilium disassembly in neural progenitors. Nat. Commun 10 (1), 2612. 10.1038/s41467-019-10497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Kumari D, Sciascia N, Usdin K, 2016. CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. Mol. Autism 7, 42. 10.1186/s13229-016-0105-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Hu Z, Qiu L, Zhou T, Feng M, Hu Q, Zeng B, Li Z, Sun Q, Wu Y, Liu X, Wu L, Liang D, 2018. Seamless genetic conversion of SMN2 to SMN1 via CRISPR/Cpf1 and single-stranded oligodeoxynucleotides in spinal muscular atrophy patient-specific induced pluripotent stem cells. Hum. Gene Ther 29 (11), 1252–1263. 10.1089/hum.2017.255. [DOI] [PubMed] [Google Scholar]