

Graphical abstract
Abbreviations: AAA, abdominal aortic aneurysm; AD, Alzheimer’s disease; AKI, acute kidney injury; ALI, acute lung injury; ALS, amyotrophic lateral sclerosis; AMD, age-related macular degeneration; CaMKII, Ca2+/calmodulin-dependent protein kinase II; cIAP1/2, cellular inhibitor of apoptosis proteins 1 and 2; COVID-19, coronavirus disease 2019; Cyp-D, cyclophilin-D; DAMPs, damage-associated molecular patterns; ERS, endoplasmic reticulum stress; FADD, fas associated via death domain; FHF, fulminant hepatic failure; GSH, glutathione; HD, Huntington’s disease; HG, high glucose; HI, hypoxia-ischemia; HIV-1, human immunodeficiency virus type 1; HMGB1, high mobility group box 1; I/R, Ischemia-reperfusion; ICH, intracerebral hemorrhage; IDO, indoleamine 2,3-dioxygenase; IL, interleukin; LDL, low density lipoprotein; LMP, lysosomal membrane permeabilization; LPS, lipopolysaccharide; MI, myocardial infarction; MLKL, mixed lineage kinase domain-like protein; MPTP, mitochondrial permeability transition pore; MTH-Trp, methyl-thiohydantoin-tryptophan; mTOR, mechanistic target of rapamycin; Nec-1, Necrostatin-1; Nec-1i, Necrostatin-1 inactive; Nec-1s, Necrostatin-1 stable; NF-κB, nuclear factor kappa B; ox-LDL, oxidized LDL; PARP1, polymerase 1; PCD, programmed cell death; PD, Parkinson’s disease; PRR, pattern recognition receptor; PYGL, glycogen phosphorylase; RDA, RIP1-dependent apoptosis; RGAG, advanced glycation end products; RGCs, retinal ganglion cells; RIA, RIP1-indipendent apoptosis; RIP1, receptor interacting protein 1; RIP3, receptor interacting protein 3; ROS, reactive oxygen species; RPE, retinal pigment epithelial; SAH, subarachnoid Hemorrhage; SCI, spinal cord injury; SIRS, systemic inflammatory response syndrome; TBI, traumatic brain injury; TNF, tumor necrosis factor; TNFR1, TNF receptor 1; TRADD, TNFR-associated death domain; TRAF2, TNFR-associated factor 2
Keywords: necrostatin-1, Necroptosis, Disease models, Anti-Inflammation, COVID-19
Abstract
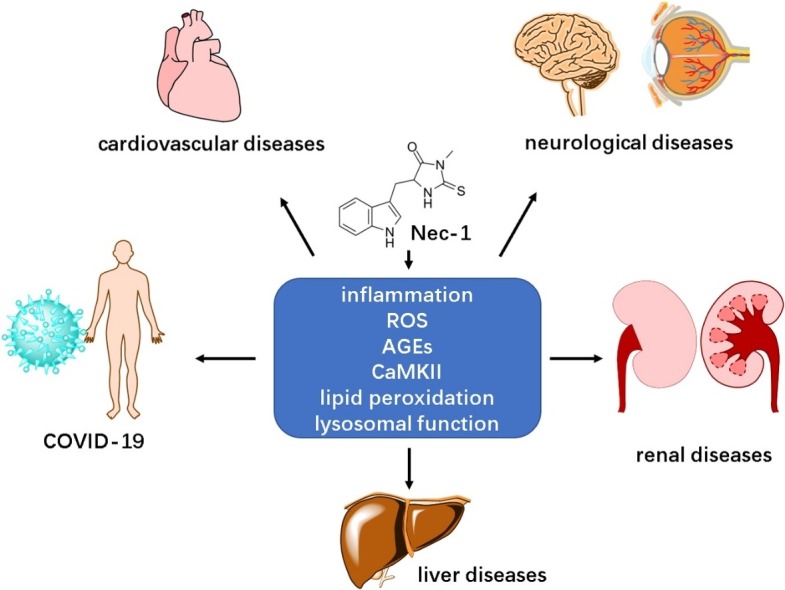
Necrostatin-1 (Nec-1) is a RIP1-targeted inhibitor of necroptosis, a form of programmed cell death discovered and investigated in recent years. There are already many studies demonstrating the essential role of necroptosis in various diseases, including inflammatory diseases, cardiovascular diseases and neurological diseases. However, the potential of Nec-1 in diseases has not received much attention. Nec-1 is able to inhibit necroptosis signaling pathway and thus ameliorate necroptotic cell death in disease development. Recent research findings indicate that Nec-1 could be applied in several types of diseases to alleviate disease development or improve prognosis. Moreover, we predict that Nec-1 has the potential to protect against the complications of coronavirus disease 2019 (COVID-19). This review summarized the effect of Nec-1 in disease models and the underlying molecular mechanism, providing research evidence for its future application.
1. Introduction
Necroptosis is a newly defined form of cell death. According to traditional belief, necrosis is a passive and unregulated cell death process, however, the discovery of necroptosis has overturned this assumption. Similar to necrosis, it is featured by the morphology of necrosis, including cell swelling and rupture; similar to apoptosis, it is controlled by a determined signal pathway which is why it is also called regulated necrosis [1] (other forms of cell death are summarized in Box 1 ). Since the discovery of necroptosis, researchers have been trying to investigate the role and mechanism of necroptosis in various diseases, including cardiovascular diseases [2], neurodegenerative diseases [3] and liver diseases [4]. Although there are plenty of evidences and papers about necroptosis, little attention has been paid to Necrostatin-1 (Nec-1), the inhibitor of necroptosis, and the potential therapeutic capability of Nec-1 has not been completely reviewed. Currently the application of Nec-1 is limited by metabolic instability and off-target effects, but this limitation does not harm the defined protective effects of Nec-1 and its analogues/derivatives in disease models. In fact, Nec-1 provides new prospect for prevention and treatment of multiple diseases. Here, we summarized potential functions that Nec-1 has exhibited in various disease models including typical inflammatory disorders and coronavirus disease 2019 (COVID-19), providing a comprehensive viewpoint on Nec-1 from different aspects.
Box 1. Other forms of Programmed cell death.
Traditionally, programmed cell death (PCD) refers in particular to apoptosis. With more and more researches investigating into cell death, scientists realize that there are other forms of PCD: necroptosis, autophagy-dependent cell death, parthanatos, pyroptosis and ferroptosis. Among these PCD, necroptosis, parthanatos, pyroptosis and ferroptosis are also described as regulated necrosis.
Apoptosis: apoptosis is an important cell death process characterized by cell shrinkage, nuclear and cytoplasmic condensation and chromatin fragmentation. Apoptosis plays an essential role in embryologic development, after birth, and during adulthood. The apoptosis activation is mediated by caspase cascades [5].
Autophagy-dependent cell death: Although used to be termed as ‘autophagic cell death’, autophagy-dependent cell death is not executed by autophagy. In fact, autophagy is a survival mechanism that cellular materials (especially unnecessary or dysfunctional components) to be delivered from cytoplasm to lysosomes for degradation, which contributes to cell homeostasis. However, autophagy is reported to mediate or associate with cell death, and cell demise that has a strict requirement of autophagy is defined as autophagy-dependent cell death [6,7]
Parthanatos: parthanatos is a form of regulated necrosis. It refers to a programmed cell death pathway mediated by the effects of pathogenically high levels of poly (ADP-ribose) polymerase 1 (PARP1) activity. PARP1 is a potent consumer of nuclear NAD+, thus overactivated PARP1 causes cell death via a potentially lethal depletion of NAD+ [8].
Pyroptosis: pyroptosis is a form of regulated necrosis. It was long regarded as caspase-1-mediated monocyte death in response to bacterial insults, regulating innate immune responses (Thus it was often misregarded as apoptosis because of the involvement of caspase-1). Recent studies show that caspase-4, 5, and 11 also induce pyroptosis upon recognition of intracellular lipopolysaccharide (LSP) [9].
Ferroptosis: ferroptosis is a form of regulated necrosis. It is defined as an iron-dependent form of regulated cell death, which occurs when facing oxidative stress like ROS accumulation and lipid peroxidation. Ferroptosis has been reported to paly a role in diseases like cancer and neurodegeneration. It can be inhibited by inhibitors of lipid peroxidation, lipophilic antioxidants and iron chelators [10].
Alt-text: Box 1
2. Necroptosis and Nec-1
2.1. Signaling pathway of necroptosis
The critical mechanism of necroptosis is related to the activation (including ubiquitination and phosphorylation) of receptor interacting protein 1 (RIP1), receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like protein (MLKL) [11,12]. Among researches on the initiation of necroptosis, mechanism of tumor necrosis factor (TNF)-induced necroptosis has been studied the most thoroughly (Fig. 1 .). The combination of TNF-α with TNF receptor 1 (TNFR1) on the cell membrane stimulates different signaling pathways, including nuclear factor kappa B (NF-κB), RIP1-indipendent apoptosis (RIA), RIP1-dependent apoptosis (RDA) and necroptosis. Upon activation, TNF trimers bind to TNFR1 trimers, followed by recruitment of RIP1, TNFR-associated death domain (TRADD), TNFR-associated factor 2 (TRAF2) and cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1/2) [[13], [14], [15]]. These components assemble complex Ⅰ at plasma membrane, activating NF-κB signaling. Subsequently, TRADD and RIP1 dissociate from TNFR1 and lead to the formation of complex II [14], including complex Ⅱa, Ⅱb and Ⅱc. After dissociation from complex I, TRADD recruits fas associated via death domain (FADD) and caspase-8, forming complex Ⅱa [14,16]. Complex Ⅱa formation is followed by induction of RIA, which is independent of RIP1 and RIP1 kinase activity [16]. During the transition from complex Ⅰ to complex Ⅱ, RIP1 undergoes C-terminal death domain-mediated dimerization [17]. RIP1 dimerization is essential for RIP1 activation and leads to formation of complex Ⅱb (composed of RIP1, FADD and caspase-8) and Ⅱc (composed of RIP1 and RIP3) [18,19]. Complex Ⅱb is formed without TRADD and thus requires RIP1 kinase activity to activate caspase-8 and RDA, which is RIP1-dependent [16,20]. The formation of complex Ⅱa and Ⅱb is followed by the activation of caspase-dependent cell death via apoptosis, while complex Ⅱc induces necroptosis. When activated RIP1 combines with RIP3 and thus leads to the formation of complex Ⅱc (or known as necrosome), cell death shifts to a caspase-independent way, followed by the phosphorylation and oligomerization of MLKL, which marks the beginning of necroptosis [12]. Polymerized MLKL translocates to cell membrane and causes membrane disruption, executing necroptotic cell death [21,22].
Fig. 1.

TNFα-induced signaling pathway of cell survival, apoptosis and necroptosis. The combination of TNF-α and TNFR1 on the cell membrane stimulates different signaling pathways, including NF-κB, RIA, RDA and necroptosis. TNFR1 exists in the cell membrane and its subunits can spontaneously trimerize and bind to ligands, allowing their cytosolic tails to recruit multiple proteins and generate a complex (complex I). Ubiquitylation of RIP1 could stabilize complex I and contribute to NF-κB release. Additionally, deubiquitylated RIP1 cooperates with its cognate kinase RIP3 for recruitment of another complex (complex II). The formation of complex Ⅱa and Ⅱb is followed by the activation of caspase-dependent cell death via apoptosis, while complex Ⅱc induces necroptosis. When activated RIP1 combines with RIP3 and thus leads to the formation of complex Ⅱc, cell death shifts to a caspase-independent way, followed by the phosphorylation and oligomerization of MLKL, which marks the beginning of necroptosis.
2.2. Nec-1 and its specificity
The patho and physiological relevance of necroptosis used to be underestimated or ignored because of the absence of a defined and convenient biochemical marker or indicator. The identification of necrostatins has changed the situation and enabled researchers to investigate more about the molecular mechanism of necroptosis. Necrostatins is a group of compounds named for their capability to prevent necroptosis, among which Nec-1 has been used to study the contribution of necroptosis and target RIP1 kinase activity in a wide range of pathological cell death events. Nec-1 (Fig. 2 A), a type of alkaloid with small molecule weight, was first identified as an inhibitor of necrotic cell death in 2005 [23] and was found to be a specific inhibitor of RIP1 in further studies [24]. By interacting with the T-loop, an essential structure for death domain receptor engagement, Nec-1 inhibits RIP1 kinase activity [24]. It binds allosterically to the hydrophobic pocket of kinase domain near ATP-binding active center, with RIP1 adopting a DLG-out inactive conformation [24]. After binding to the activation loop, Nec-1 can potently inhibit RIP1 autophosphorylation [25]. RIP1 autophosphorylation is a crucial process during TNF-induced necroptosis signaling and several RIP1 autophosphorylation sites have been reported by researchers, including Ser14/15, Ser20, Ser161 and Ser166 [24]. RIP1 phosphorylation leads to the recruitment of RIP3 to RIP1 and subsequent formation of RIP1-RIP3 complex [26], which then phosphorylates MLKL. Therefore, Nec-1 efficiently blocks RIP1-RIP3-MLKL signal transduction by inhibiting RIP1 phosphorylation.
Fig. 2.

The effects of Nec-1 in programmed cell death. (A) 2D and 3D structures of Nec-1. Nec-1 is a type of alkaloid with small molecule weight, identified as an inhibitor of RIP1 and IDO. (B) Nec-1 halts the necroptosis signaling pathways by inhibiting RIPI signaling cascades. Furthermore, Nec-1 also affects necroptosis by targeting ROS. Nec-1 inhibits apoptosis by targeting RIP1 when the cell undergoes signaling pathway of RDA, yet it loses this anti-apoptosis effect when the pathway is RIA.
As for pharmacokinetics, Nec-1 can quickly get into blood or circulatory system. Geng et al. found that plasma concentration of Nec-1 reaches its peak at 1 h after oral administration in rats [27], and Nec-1 could completely dissolve in 95 % ethanol with absolute bioavailability of more than 50 %. These characteristics endows Nec-1 with promising potentials of clinical application. One major drawback of Nec-1 is its short half-life, which is about 1−2 h in rats (1.8 ± 0.9 h for intravenous route and 1.2 ± 0.3 h for oral route, tested by Geng et al) [27]. Another limitation is its off-target effect. In fact, the original name of Nec-1 was methyl-thiohydantoin-tryptophan (MTH-Trp), defined as an inhibitor of indoleamine 2,3-dioxygenase (IDO) [28]. Thus Nec-1 application not only inhibits necroptosis but also IDO and this off-target effect has been criticized by many researchers [25,29]. It is true that off-target action makes it more difficult for researchers to define RIP1 kinase function by using Nec-1, and limits its clinical application of targeting RIP1 specifically. However, as a classic and potent RIP1 inhibitor, the broad researches investigating the role of Nec-1 in various disease models still cannot be ignored, nor its potential clinical significance. In fact, IDO plays a significant role in inflammation [30], therefore, the off-target effect enables Nec-1 to protect against inflammatory diseases by inhibiting both necroptosis-induced and IDO-mediated inflammation. Although currently there is no direct evidence proving this (perhaps because of difficulty in distinguishing between necroptosis-induced and IDO-mediated inflammation), RIP1-independent inhibitory effect of Nec-1 has been demonstrated [31,32]. Therefore, off-target effect may endow Nec-1 with stronger anti-inflammation ability.
3. Role of Nec-1 in disease models
3.1. Molecular mechanisms of Nec-1 in disease models
Necroptosis has been verified to participate in various diseases [33], and the protection of Nec-1 has also been authenticated in many disease models, including cardiovascular, neurological and renal diseases (Table 1 ). The basic molecular mechanism of this protective effect is blocking necroptotic cell death by targeting RIP1. Therefore, Nec-1 administration usually results in alleviated cell death and improved cell viability. In fact, Nec-1 not only inhibits RIP1, but also reduces RIP3 expression and phosphorylation, which has been demonstrated in many disease models, including myocardial infarction (MI), mental diseases and intestinal inflammation [[34], [35], [36]]. However, no direct inhibitory effect of Nec-1 on RIP3 has been reported [25] and Nec-1 does not block RIP3 autophosphorylation [25], therefore this influence on RIP3 is indirect. Nec-1 reduces the interaction between RIP1 and RIP3, inhibits the formation and/or reduces the stability RIP1-RIP3 complex, thus affects downstream RIP3 activity [37].
Table 1.
Role of Nec-1 in disease models.
| Diseases | Role of Nec-1 | References |
|---|---|---|
| Cardiovascular Diseases | ||
| High glucose (HG) -induced cardiac cell injury | Pretreatment of Nec-1 ameliorates HG-induced injuries, reduces ROS production and causes mitochondrial membrane potential loss; Nec-1 attenuates HG-induced downregulation of ALDH2 activity, downregulates expression of RIP1, RIP3 and MLKL | [38,39,40,41] |
| Cardiomyocyte hypertrophy | Pretreatment of Nec-1 attenuates cardiomyocyte hypertrophy, reduces phosphorylation level of AKT and mTOR | [42] |
| Atherosclerosis | Nec-1 reduces lesion size and markers of plaque instability in Apoe(-/-) atherosclerosis mice | [2] |
| Cardiac ischemia/ reperfusion (I/R) injury | Nec-1 reduces cardiomyocyte necrosis, improves cardiac output and prolongs cardiac allograft survival time | [43] |
|
Myocardial infarction (MI) |
Nec-1 inhibits myocardial tissue necroptosis and improves cardiac function in acute MI models of rat and pig | [34,44] |
| Myocarditis | Nec-1 protects heart tissues from myocardial injury by downregulating RIP1/RIP3 expression in CVB3-induced myocarditis mouse model | [45] |
| Neurological Diseases | ||
| Traumatic brain injury (TBI) | Pretreatment of Nec-1 decreases RIP1/RIP3/MLKL expression, inhibits cell death, reduces edema and restores learning and behavior performance | [46,47] |
| Spinal cord injury (SCI) | Nec-1 alleviates tissue damage, increases surviving neurons and improves functional recovery | [48,49] |
| Cerebral I/R injury | Nec-1 inhibits neuronal death by preventing RIP3 upregulation and nuclear translocation | [50,51] |
| Neonatal hypoxia-ischemia (HI) | Nec-1 decreases ERS-related biochemical markers after neonatal HI, providing delayed neuroprotection against neonatal HI | [52] |
| Intracerebral hemorrhage (ICH) | Nec-1 inhibits cell death induced by hemin in HT-22 cells, improves cell viability and neurological function, and attenuated brain edema in mice after ICH | [37,53,54] |
| Subarachnoid Hemorrhage (SAH) | Nec-1 decreases RIP1/RIP3/MLKL expression, inhibits cell death in basal cortex, reduces pro-inflammatory cytokines and inflammasome, alleviates brain edema and blood-brain barrier disruption | [54,55] |
| Postoperative cognitive dysfunction | Nen-1 reduces neuroinflammation and attenuates postoperative cognitive dysfunction in d-Galactose-induced aged mice | [56] |
| Ischemic stroke | Pretreatment of Nec-1promotes survival of oligodendrocyte precursor cells, alleviates white matter injury, and improves cognitive function after transient cerebral ischemia | [57] |
| Alzheimer's disease (AD) | Nec-1 directly targets Abeta and tau proteins, alleviates brain cell death and ameliorates cognitive impairment in AD models | [58] |
|
Parkinson’s disease (PD) |
Necrostatin-1 improves cell viability, stabilizes mitochondrial membrane potential, inhibits excessive autophagy, upregulates apoptosis signaling pathway | [59] |
| Huntington’s disease (HD) | Nec-1 rescues striatal cell death in striatal cell model of HD and maintains motor function in R6/2 mouse model of HD | [60] |
| Amyotrophic lateral sclerosis (ALS) | Nec-1 prevents cell loss of motor neurons | [61] |
| Age-related macular degeneration (AMD) | Subretinal/ retro-orbital injection of Nec-1 protects against retinal pigment epithelial (RPE) cell death and suppressed macrophage infiltration | [62,63] |
| Glaucoma | Nec-1 inhibits cell death of retinal ganglion cells (RGCs) | [64] |
| Retinal detachment | Nec-1 reduces retinal histopathological damage, attenuates acute plasmalemma permeability, prevents photoreceptor necroptotic cell death and contributes to objective functional improvement | [65] |
| Retinal I/R injury | Nec-1 increases RGC survival, reduces RGC necrosis, decreases pro-inflammatory markers, preserves in inner retina thickness/histoarchitecture and improves function | [66] |
| Renal Diseases | ||
| Acute kidney injury (AKI) | Nec-1 reduces necroptotic cell death, ameliorates kidney dysfunction and protects renal tubular epithelial cells | [67,68,69] |
| Chronic kidney disease | Nec-1 improves renal pathologic changes and renal function in a remnant-kidney rat model | [70] |
| Unilateral ureteral obstruction | Nec-1 ameliorates kidney inflammation and interstitial fibrosis in unilateral ureteral obstruction mouse model | [71] |
| Renal graft injury | Nec-1 attenuates remote lung injury after receiving ischemic renal allografts | [72] |
| Renal I/R injury | Nec-1 reduces serum creatinine/urea concentrations in renal I/R mouse models | [73,74] |
| Liver Diseases | ||
| Liver I/R Injury. | Nec-1 improves liver function and activates apoptosis signaling pathway | [75,76] |
| Acute liver failure/ fulminant hepatic failure (FHF) | Nec-1 protects against hepatic injury and alleviates hepatotoxicity in acute liver failure mouse model induced by d-Galactosamine /LPS or Acetaminophen | [77,78,79] |
| Trauma induced hemorrhagic shock | Nec-1 improves liver function and remarkably reduces morality in a rat model of hemorrhagic shock | [80] |
| Chronic hepatitis C virus infection | Nec-1 improves cell viability of cells infected by hepatitis C virus | [81] |
| Skeletal System Diseases | ||
| Osteoarthritis | Nec-1 ameliorates destruction of osteoarthritis cartilage | [82] |
| Cartilage thinning | Co-treatment of Nec-1 and z-VAD-fmk alleviates force-mediated cartilage thinning and cell death of chondrocyte | [83] |
| Osteonecrosis | Nec-1 markedly decreases the osteonecrosis rate in a rat model | [84] |
| Osteoporosis | Nec-1 improves bone formation and alleviates trabecular bone in a glucocorticoid-induced osteoporosis rat model; Nec-1 inhibits osteocyte necroptotic cell death and attenuates trabecular bone deterioration in a rat model of postmenopausal osteoporosis | [85,86] |
| Other Diseases | ||
| Colitis-associated cancer | Nec-1 evidently ameliorates colitis-related symptoms and inhibits tumorigenesis by downregulating JNK/c-Jun signaling | [36] |
| Intestinal I/R injury | Nec-1 inhibits necroptotic cell death in intestinal epithelial cells; Co-treatment of Neec-1 and z-VAD-fmk alleviates intestinal I/R injury in a rat model |
[87] |
| Inflammatory bowel disease | Nec-1 inhibitsd intestinal inflammation in inflammatory bowel disease | [88] |
| Systemic inflammatory response syndrome (SIRS) | Pretreatment with Nec-1 protects against SIRS | [25,89] |
| Sepsis | Nec-1 improves liver function and ameliorates pathological damage in septic rats | [90] |
| Abdominal aortic aneurysm (AAA) | Nec-1 lessens aortic expansion and improves aortic structure pathologically in AAA mouse model | [91] |
| Acute respiratory distress syndrome | Nec-1 ameliorates inflammatory response and improves pulmonary function in rat model induced by oleic acid | [92] |
| Human immunodeficiency virus type 1 (HIV-1) infection | Nec-1 alleviates HIV-1-induced cytopathic effect and interestingly and inhibits the formation of HIV-induced syncytia | [93] |
| Systemic autoimmunity | Nec-1 inhibits pro-inflammatory cytokine secretion | [94] |
Necroptosis and apoptosis share part of the same upstream pathway, and RIP1 kinase activity also drives apoptosis, which is defined as RDA. Therefore, Nec-1 also targets RIP1-mediated apoptosis. Increasing studies show that Nec-1 not only suppresses necroptosis, but also inhibits apoptosis [82,95]. Besides, Han et al. found that Nec-1 can enhance leukemia cell apoptosis induced by Shikonin [96], showing an anti-cancer effect; Jie et al. found that Nec-1 could specifically induce neutrophils apoptosis rather than blockage [97], which implies an anti-inflammatory effect. Despite researches mentioned above, some researchers claim that Nec-1 does not affect apoptosis [38,48], which is probably RIA. When apoptosis is mainly RIP1-dependent, Nec-1 inhibits RIP1 kinase activity and formation of complex Ⅱa, and thus apoptosis is blocked; however, when apoptosis is RIP1-independent, Nec-1 has no inhibitory effect. As for the opposite results of Han and Jie, it can be explained by the shift between necroptosis and apoptosis: apoptosis can be induced when necroptosis is inhibited by Nec-1, and co-treatment with the pan-caspase inhibitor z-VAD-fmk leads to a shift from apoptosis to necroptosis [98]. These different research results indicate that the underlying mechanism among RIP1, necroptosis and apoptosis is a complicated network, which is why Nec-1 exhibits different effects in different situations/diseases models.
Reactive oxygen species (ROS) is a group of highly reactive chemical species containing oxygen, which is usually the natural byproduct of metabolism [99]. However, ROS levels could increase dramatically under harmful conditions, causing damage to cell structures or even cell death [100]. Studies have demonstrated that the application of Nec-1 attenuates the elevated intracellular ROS production in disease models of acute liver failure, spinal cord injury, and I/R injury etc [43,79,101]. This effect of downregulation on ROS generation could even result in a similar ROS level as normal cells [79]. Although the specific mechanism underlying the inhibitory effect of Nec-1 on ROS has not been demonstrated very clearly, it is verified that the ROS production is probably RIP1-dependent [102]. In other researches demonstrating the relation between ROS and necroptosis, ROS increases the expression of RIP1/RIP3 and improves the stabilization of RIP1-RIP3 complex [103,104]. Such an interactive relation between necroptosis and ROS is still not very clear of definite, remaining to be supported and further investigated by more researches. The underlying mechanism of Nec-1 in diseases is summarized in Fig. 2B.
3.2. Inflammation and inflammatory diseases
Inflammation is a defensive response in reaction to tissue or cell damage caused by endogenous or exogenous injuries, which involves multiple cells including endothelial cells, mononuclear macrophages, fibroblasts and platelets. The relation between inflammation and necroptosis has gained attention since the observation of necroptosis. The necrotic characteristics of necroptosis is marked by cell rupture and release of intracellular immunogenic contents, which initiate inflammatory responses and indicate the pro-inflammatory effect of necroptosis. And, conversely, inflammation induces necroptosis by pro-inflammatory mediators or mechanism of direct contact with immune cells [105], promoting cell death. This vicious circle emphasizes the crucial role of necroptosis in various inflammatory diseases, including infectious diseases [106], osteoarthritis [82] and atherosclerosis [2].
The key components of necroptosis, RIP1, RIP3 and MLKL are verified to participate in inflammation. Compared with MLKL, RIP1 and RIP3 have a more significant role in inflammation [107], and here the focus will be on RIP1. Before the identification of RIP1 as a key target of necroptosis [24], RIP1 was firstly identified as a mediator during NF-κB signaling induced by TNF-α [108]. Therefore, RIP1 promotes inflammatory responses through two ways: necroptotic cell death and inflammation independent of cell death. Upon pro-necroptotic stimuli (e.g., TNF, Fas), RIP1 activates RIP3 and then phosphorylates MLKL, which leads to cell membrane pores [109] and release of death-associated molecular patterns (DAMPs) [110]. DAMPs are a group of endogenous molecules that can initiate non-infectious inflammatory responses, for example, cytokine family interleukin (IL)-1. DAMPs released from necroptotic cells sensitize neighboring cells to necroptosis and can be modified by ROS or endoplasmic reticulum stress (ERS). The role of necroptosis-associated DAMPs in inflammation (during bacterial and viral infection) is reviewed by Kaczmarek [110]. Apart from inducing pro-inflammatory necroptotic cell death, recent evidence suggests that RIP1 and RIP3 directly induce inflammation by production of pro-inflammatory cytokines, which is independent of cell death. Zhu et al. found that cytokine expression level of necroptosis directly induced by MLKL oligomerization was much lower than that of necroptosis induced by TNF-α, which involved RIP1 and RIP3 activation [111]. Similar result was also proved by Saleh et al. that production of INF-β induced by lipopolysaccharide (LPS) requires RIP1 and RIP3 but not MLKL [112]. Besides, RIP1 activation was proved to be crucial for the secretion of IL-1α which is independent of RIP3-mediated necroptosis [113], and for the production of TNF /TNF mRNA which is independent of NF-κB [114].
As a RIP1-target inhibitor of necroptosis, Nec-1 is proved to exhibit significant protective effect in various inflammatory diseases, including acute and chronic inflammatory responses. In the LPS-induced disease models of fulminant hepatic failure (FHF) and acute lung injury (ALI) [77,115], injection of LPS remarkably upregulated the expression and phosphorylation of RIP1 and RIP3, which was attenuated by Nec-1. Apart from prolonged survival time of mice, Nec-1 also showed great anti-inflammatory ability, inhibiting the activation of NF-κB in both models. In LPS-induced FHF, application of Nec-1 attenuated cell death, IL-33 release and RAGE (the receptor for advanced glycation endproducts) interaction, suggesting the protective mechanism was related to necroptosis and DAMPs-mediated pattern recognition receptor (PRR) pathways. However, in LPS-induced ALI, pretreatment with Nec-1 resulted in lower level of inflammatory cytokines including TNF-α, IL-6 and IL-8, but had no effect on cell viability. This suggests that Nec-1 also exhibits protection against inflammation independent of necroptosis. In systemic inflammatory response syndrome (SIRS) mouse model induced by TNF, Nec-1 prevented mice from hypothermia and death, but the authors inferred that NF-κB was not affected [89], which conflicts with NF-κB inhibition observed in FHF and ALI mouse models. In fact, Degterev et al. also reported no effect of Nec-1 on NF-κB in their early study [24]. This divergence could be explained by a recent research that RIP1 does not necessarily participate in activation of NF-κB [116]. Despite crucial role of RIP1 in NF-κB signaling reported by many researches [117,118], Wong et al. found that canonical NF-κB was activated in Rip1−/− MEFs upon TNF application, confirming that RIP1 is not obligate in NF-κB pathway [116]. Bertrand et al. proposed an explanation that the role of RIP1 in NF-κB activation is probably cell-type-specific [119]. Therefore, the RIP1 inhibitor Nec-1 has different effects on NF-κB in different disease models. Besides, Nec-1 can also attenuate chronic inflammatory responses like autoimmune disease. Autoimmune disease is caused by an abnormal immune response that mistakenly attacks human body. Researches have proved that the pathogenesis of autoimmune disease (e.g., autoimmune arthritis and vasculitis) involves necroptosis, with upregulated key factors of necroptosis [120,121]. Nec-1/Nec-1 analogues suppress autoimmune disease not only by inhibiting necroptosis, but also by suppressing apoptosis [122,123]. The underlying molecular mechanism is not clear, perhaps it is due to Nec-1 inhibition on necroptosis-induced DAMPs and pro-inflammatory cytokines. Besides, Nec-1 also targets RDA in a chronic inflammatory model of osteoarthritis, protecting against inflammation by suppressing apoptosis via RIP1/HMGB1/TLR4 pathway [82].
3.3. Ischemia-reperfusion injury and related diseases
Ischemia-reperfusion (I/R) injury is a type of tissue damage caused by reperfusion of blood after a period of anoxia or hypoxia [124], which is involved in various clinical diseases, including MI, cerebral infarction and gastrointestinal dysfunction. The pathophysiology of I/R injury consists of two stages. Firstly, the process of ischemia causes damages to cells and a shortage of oxygen, inducing oxidative stress and ensuing inflammatory responses. Then, when the stage of reperfusion begins, activated endothelial cells overproduce ROS, ROS cause oxidative stress and subsequent inflammatory injuries, finally resulting in cell death including necroptosis [125]. Nec-1 is reported to protect against several types of I/R injuries, including cardiac, cerebral and renal I/R injuries [44,57,73,74,126]. Here, we will discuss it from different mechanisms that Nec-1 protection involves.
Bundles of researches have been conducted to investigate the specific role of necroptosis in I/R injuries and the underlying mechanism, and it has been obvious that RIP1-mediated necroptosis is involved in I/R injuries, marked by increased levels of TNF-α, TNFR1 and RIP1/RIP3 phosphorylation [87,127]. Necroptosis occurs during both ischemia and reperfusion, causing cell death, tissue damage and finally organ dysfunction. Nec-1 can protect against I/R injury by inhibiting necroptotic pathway and reducing necroptotic cell death. The expression and phosphorylation of RIP1, RIP3 and MLKL is suppressed after application of Nec-1, usually accompanied by attenuated cell death/tissue injury and improved prognosis. For example, Nec-1 reduces infarct size and prevents adverse remodeling in cardiac I/R models [44,127], improves memory and cognitive function in cerebral I/R models [57,126], and reduces serum creatinine/urea concentrations in renal I/R models [73,74]. This protective effect of Nec-1 is also closely related to its anti-inflammation ability. In the liver I/R models of normal liver or fatty liver, Nec-1 notably suppressed liver inflammatory response, reducing expression of inflammation-related genes (e.g., NF-κB, JNK and ERK) [128]. The necroptosis downstream protein high mobility group Box 1 (HMGB1), a mediator of necroptosis-induced inflammation, is reported to participate in I/R injury [87,129]. As mentioned in Section 3.1, HMGB1 can be downregulated by Nec-1 [82], therefore it is also a key target (although indirect) for Nec-1 to alleviate inflammation in I/R injury [43]. The other models of liver and cardiac I/R injury also observed decreased inflammation or pro-inflammatory cytokines [127,130].
Another critical pathophysiological process during I/R injury is mitochondrial dysfunction or mitochondrial-related response. Ischemia causes hypoxia and great oxidative stress, and reperfusion intrigues the second wave of mitochondrial-mediated response. After the treatment or pre-treatment of Nec-1, I/R-induced ROS production decreases notably [127,128]. RIP1-mediated necroptosis is closely related to ROS production: necroptotic cell death causes oxidative stress/ROS overproduction [102,131], and ROS promotes necroptosis by increasing RIP1/RIP3 expression and RIP1-RIP3 complex stabilization [103,104]. Therefore, it is possible that Nec-1 prevents I/R injury by inhibiting ROS through two ways: decreased ROS level alleviates mitochondrial dysfunction, or unstable necrosome blocks necroptotic cell death. Besides, Nec-1 also targets Cyp-D/MPTP during I/R injury. Mitochondrial permeability transition pore (MPTP) has already been demonstrated as a crucial factor in the pathophysiology of I/R injury [132,133] and it also interacts with necroptosis pathway [134]. Lim et al. demonstrated the significant regulatory function of cyclophilin-D (Cyp-D) in the activation of MPTP by using a Cyp-D deficient mouse model, and found that Nec-1 showed no significant protective effect in Cyp-d-/- mice, indicating the mechanism of Nec-1 protection is MPTP-dependent [135].
Apart from I/R injury, the protective effect of Nec-1 has been observed in other disease models like brain hemorrhage and traumatic injury, which is related to I/R injury or I/R disease mechanism. In animal models of intracerebral/subarachnoid hemorrhage (ICH/SAH), application of Nec-1 attenuates cell death, reduces hematoma volume and improves neurological outcomes [37,55,136,137]. Besides, both pre-treatment and post-treatment of Nec-1 exerts protection against brain hemorrhage [54,55], indicating a broad therapeutic window. The underlying molecular mechanism is still not definite, but it involves oxidative stress and inflammation: for example, Nec-1 alleviates glutathione (GSH) depletion in hemin-induced cell death and inhibits NLRP3 inflammasome activation; the attenuated oxidation and inflammation ameliorates brain swelling and blood-brain barrier disruption, finally inhibits the formation of brain edema [54,55,138]. In researches of traumatic brain injury (TBI) and spinal cord injury (SCI), necroptosis plays a significant role in neural cell death and participates in both primary and secondary injury [47,48]. The Nec-1 protection is associated with Akt/mTOR activation, mitochondrial dysfunction and ERS [46,49,139]. Several researches reported that Nec-1 also inhibited apoptosis/autophagy [95,101], yet contradicted results were reported by Liu et al. that Nec-1 did not alter apoptosis [48], suggesting that there is crosstalk among necroptosis, apoptosis and autophagy in the pathogenesis of traumatic injury.
3.4. Metabolism-related cardiovascular diseases
In cardiovascular diseases, the death of cardiomyocytes is proved to be irreversible, which means poor prognosis. Previously, necrosis and apoptosis are regarded as main forms of cardiac cell death. With more and more studies investigating into programmed cell death, necroptosis has been proved to be another important pathway of cell death in cardiovascular diseases [140]. For example, expression levels of RIP1, RIP3 and MLKL are upregulated in clinical samples of atherosclerosis, [141], RIP3 mediates adverse remodeling after MI [142], and RIP1-RIP3 pathway is activated in acute myocarditis [45]. Since acute cardiovascular events like acute MI are inextricably linked with I/R, which has been discussed above (Section 3.3), here we would like to focus on metabolism-related cardiovascular diseases, including diabetic cardiomyopathy and atherosclerosis.
Hyperglycemia is a risk factor of cardiovascular diseases, and people with diabetes face higher risk of developing cardiovascular disease, including atrial fibrillation and coronary heart disease [143]. High glucose (HG) is closely associated with vascular endothelial dysfunction, causing damage to endothelial cells. Since necroptosis has been so active in pathological processes of cardiovascular diseases like cardiac I/R, as demonstrated above in Section 3.3, it is natural to question whether it participates in HG-induced endothelial dysfunction. Liang et al. firstly reported the contribution of necroptosis in HG-induced injury in cardiac cells and role of Nec-1 in reducing cytotoxicity, oxidative stress and dissipation of mitochondrial membrane potential (MMP) [39]. Similar results were obtained by later researches in both cardiac cells and endothelial cells [38,40,41]. However, current studies only provide an elusive description on this Nec-1 protective effect, and the underlying molecular mechanism remains to be investigated. Here, we would like to discuss the potential mechanisms related to advanced glycation end products (AGEs) and Ca2+/calmodulin-dependent protein kinase II (CaMKII). One of the major modifications under hyperglycemic conditions is the glycation of proteins or lipids, leading to the formation of AGEs. In the pathogenesis of hyperglycemia-induced cardiovascular dysfunction, AGE pathway has been proved to be a crucial mediator of HG-induced detrimental effects [143]. When AGEs combine to the receptor for advanced glycation end products (RGAG), NADPH oxidase is activated, leading to excessive generation of ROS [144]. Cellular oxidative stress induces, or at least promotes necroptosis signaling pathway under HG condition (as observed by Liang et al. [40]). According to the study of Zhang et al., RIP3 interacts with and allosterically activates glycogen phosphorylase (PYGL), a key metabolic enzyme of glycogenolysis [145]. PYGL catalyzes the breakdown of glycogen into glucose-1-phosphate, and glucose-1-phosphate is subsequently converted into glucose-6-phosphate [1,146]. Then glycolysis begins and each molecule of glucose is split into two molecules of pyruvate [1,146]. Pyruvate produces lactate, which induces ROS [1]. Besides, necroptosis-induced glycogenolysis also produces methylglyoxal, which covalently binds to proteins and forms AGEs [147]. From above, we can see that there is an active interaction among AGE pathway, necroptosis pathway and ROS production. Therefore, we speculate the application of Nec-1 inhibits necroptotic signaling, ameliorates ROS, and finally reduces AGEs in HG-induced cell injury and diabetic cardiomyopathy. Another target of Nec-1 might be CaMKII. CaMKII is a key substrate of RIP3 [148] and is abundant in cardiac tissues, involved in myocardial injuries by regulating l-type calcium channel and function of sarcoplasmic reticulum [149,150]. Although currently there is no proof that Nec-1 directly targets CaMKII, Szobi et al. found that inhibition of CaMKII normalizes the upregulated RIP1 levels and Reventun et al. reported reduced p-CaMKII expression after Nec-1 application [151], indicating a potential interaction (direct or indirect) between RIP1 and CaMKII [152].
Atherosclerosis is closely related to lipid metabolism, and people with hyperlipemia are more likely to develop atherosclerosis [153]. Among the factors of dyslipidemia, the most noticeable one is excess low density lipoprotein (LDL), especially oxidized LDL (ox-LDL) [154]. The accumulation of ox-LDL is a crucial stimuli of cell death, inducing not only apoptosis but also necrosis, which finally leads to the formation of intravascular necrotic cores [155]. Moreover, ox-LDL promotes the formation of foam cells from macrophages [156]. In 1997, Crisby et al. highlighted in their study that necrosis (they referred to it as oncosis) was a more common form of cell death than apoptosis in atherosclerotic plaques [157]. Further study of Grootaert et al. found that deletion of Caspase-3 promoted atherosclerotic plaque progression in Apoe-/- mice, rather than inhibiting it [158]. This suggests that anti-apoptosis is not a favorable strategy but anti-necrosis might be. However, necrosis is probably not an ideal target because it is largely uncontrollable and a single necrosis inhibitor cannot efficiently block all necrosis stimuli. Therefore, necroptosis could be a better target. In 2013, Lin et al. reported the proatherogenic role of RIP3-mediated necrosis [159]. They found that knocking out Rip3 in Ldlr-/- and Apoe-/- background mice significantly reduced the size of atherosclerotic lesions. Later in 2016, Karunakaran et al. innovatively demonstrated the macrophage necroptosis pathway could be applied as a both diagnostic and therapeutic tool to treat atherosclerosis [2]. Using a novel radiotracer developed with Nec-1, they found that radiolabeled Nec-1 localized specifically to atherosclerotic plaques in Apoe -/- mice, and Nec-1 uptake was correlated to lesion areas [2]. These exciting findings provide a new insight into the promising Nec-1 therapy of atherosclerosis, because this therapy appears to be nontoxic and shows efficiency in established atherosclerotic lesions. Another in vitro study further found that Nec-1 treatment could ameliorate eNOS/NO reduction, reduce vascular adhesion molecules (VCAM-1 and E-selectin) and inhibit NF-κB pathway in ox-LDL induced endothelial injury [160]. The role of necroptosis/Nec-1 in atherosclerosis is probably also associated with lipid peroxidation (necroptosis-mediated lipid peroxidation has been well discussed in the review of Vandenabeele et al. [1]): lipid peroxidation increases ox-LDL accumulation and ox-LDL induces more necroptotic cell death. Moreover, the strong pro-inflammatory effect of necroptosis (compared with other forms of cell death, necroptosis is more pro-inflammatory) further promotes atherosclerotic development. Therefore, targeting necroptosis in atherosclerosis with Nec-1 is a promising strategy, targeting both oxidative stress and inflammatory responses.
3.5. Neurodegenerative diseases
Neurodegeneration refers to a process of losing neurological function as a result of aging or pathological changes, which is due to neural cell degeneration and death. Some neurodegenerative diseases are common among the elderly population, like Alzheimer's disease (AD) and Parkinson's disease (PD), and some neurodegenerative diseases are heritable, like Huntington’s disease (HD). Whether age-related or genetics-related, neural cell death plays an important role in the pathogenesis of these neurodegenerative diseases. For example, neuronal cell death in hippocampus and cortex is defined as a part of the etiology of AD. The role of apoptosis in neurodegenerative diseases like AD has been investigated by many researches during last two decades [161,162], and recent researches demonstrate that necroptosis also participate. Several neurodegenerative disorders have been proved to corelate with activation of necroptosis, including AD [163,164], PD [165], amyotrophic lateral sclerosis (ALS) [61], Huntington’s disease (HD) [60], glaucoma [166] and retinitis pigmentosa [167]. Since the role of necroptosis in AD, PD, ALS and HD has been summarized in several reviews [3,33], we would like to discuss retinal degeneration in the following paragraph, focusing on the Nec-1 protection in retinal degeneration.
Retinal degeneration is a pathological process of progressive retinal cell death, which may finally result in vision loss. There are several causes of retinal degeneration, including aging, heredity, diabetic retinopathy and retinal detachment [168]. Murakami el al reported that necroptosis was the major mechanism of cell loss in a dsRNA-induced mouse model of retinal degeneration, which is contrary to previous studies attributing apoptosis as a main mediator of retinal pigment epithelial (RPE) cell death in response to prooxidants [62]. Their finding was confirmed by Hanus et al. in both in vitro [169] and in vivo [63] studies, highlighting the mechanism of necroptosis in RPE degeneration. In the experiments of both Murakami el al and Hanus et al., subretinal/ retro-orbital injection of Nec-1 protects RPE from degeneration [62,63], providing therapeutic prospects in RPE degeneration-related diseases like age-related macular degeneration (AMD). The mechanism of this interplay among retinal degeneration, necroptosis and Nec-1 has not yet been well investigated, current researches could only provide evidence for the involvement of oxidative stress and neuroinflammation [170,171]. Here we speculate that lysosomal dysfunction may also be an important factor. In the development of retinal degenerative diseases like AMD, lysosomal dysfunction is closely related to RPE degeneration, especially deregulation of cathepsins, a group of lysosomal hydrolases/proteases [172]. Under normal conditions, cathepsins regulate intracellular homeostasis by degrading proteins, however, imbalanced cathepsins is correlated with AMD. Cathepsin d-deficient mice were observed with hallmarks of AMD, which might be a result of impaired degradation of rod outer segments [173,174]. However, contrary result was reported by Ogawa et al. that mRNA and protein levels of cathepsin S was upregulated in the RPE/choroid of aged mice [175]. These findings underline the significance of a balanced lysosomal function. Necroptosis execution is associated with lysosomal dysfunction (as reviewed by Vandenabeele et al. [1]). Necroptosis-induced calpains and lipid peroxidation cause lysosomal membrane destabilization, or to be more specific, lysosomal membrane permeabilization (LMP) [1]. LMP leads to the spillage of cathepsins, and this might be the mechanism of necroptosis-induced injury in RPE. Therefore, Nec-1 treatment could protect against retinal degeneration not only by anti-oxidation/anti-inflammation, but also by restoring lysosomal function. The protection of Nec-1 in other retinal degenerative disease models like glaucoma, retinal detachment and retinal I/R injury are summarized in Table 1.
Apart from retinal degeneration, the protective effect of Nec-1 has been observed in other neurological disease models as well (like PD [176] and axonal degeneration [177]). It is also reported that Nec-1 could ameliorate neural cell injury induced by aluminum, a common metal material which can be transported to the brain and contribute to AD etiology [178]. Although the application of Nec-1 may face challenges like relatively short half-time for effective work in neurological system, the significance and potential therapeutic effects of Nec-1 should not be ignored.
4. Role of Nec-1 analogues in disease models
Since the discovery of necroptosis in driving inflammation and disease pathology, gradually it has been verified as a promising therapeutic target. The ability to inhibit or block necroptosis is the main characteristic of necrostatin family members, and here is a short list of Nec-1 and Nec-1 analogues (Table 2 ). Nec-1 is regarded as a potent necroptosis inhibitor, but it needs further optimization due to off-target effect and poor metabolic stability. Although not every necrostatin family member has been thoroughly studied, the key target has been proved to be RIP1, whose kinase activity is inhibited when necrostatins are applied. With more and more researches investigating the pharmacologic features of Nec-1, there are also several papers focused on Nec-1 analogues, providing a basic overview of these chemicals, including chemical structures and pharmacologic\toxicological features of Nec-1 analogues [25,29]. However, the potential role of Nec-1 analogues in disease models has not been systematically reviewed or summarized yet. Nec-1 analogues share some properties in common with Nec-1, which means they probably have similar functions (like anti-inflammation or anti-oxidation effects) when applied in diseases; on the other hand, Nec-1 analogues have their own unique attributes in aspects to molecule activity and stability compared with Nec-1, which could result in different effects and perhaps make them a better alternative of Nec-1 in some diseases or circumstances. Thus, we will give a quick glimpse of Nec-1 analogues and focus on two analogues: Nec-1i and Nec-1 s.
Table 2.
Compounds of necrostatin family.
| Name | Target | CAS Number | Molecular Formula | Structural Formula |
|---|---|---|---|---|
| Necrostatin-1 | RIP1 | 4311−88-0 | C13H13N3OS | 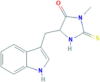 |
|
Necrostatin-1, Inactive Control (Nec-1i) |
RIP1 | 64419−92-7 | C12H11N3OS | 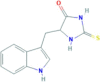 |
|
Necrostatin 2 racemate (Nec-1 s) |
RIP1 | 852391−15-2 | C13H12ClN3O2 | 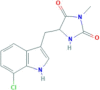 |
| Necrostatin 2 | RIP1 | 852391−19-6 | C13H12ClN3O2 | 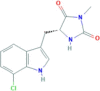 |
| Necrostatin 2 S enantiomer | RIP1 | 852391−20-9 | C13H12ClN3O2 | 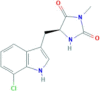 |
| Necrostatin-5 | RIP1 | 337349−54-9 | C19H17N3O2S2 | 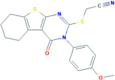 |
| Necrostatin-7 | Not reported | 351062−08-3 | C16H10FN5OS2 | 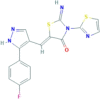 |
4.1. Nec-1i
Necrostatin-1 inactive (Nec-1i) is the demethylated variant of Nec-1, with minor inhibitory effect on the phosphorylation of RIP1, performing more than 100-fold less inhibitory activity compared with Nec-1 [25]. Possessing the characteristics mentioned above, Nec-1i has been used as a vehicle of Nec-1 in many disease models [152,179,180]. However, recent researches found that Nec-1i is actually not as “inactive” as its name suggests. Takahashi et al. proved that both Nec-1 and Nec-1i have inhibition effects on IDO enzyme activity, which involves immune regulation. In a mouse necroptosis assay, the inhibitory effect of Nec-1i on RIP1 was only 10 times less than Nec-1, although Nec-1i has no effect on necroptosis in human cells. Moreover, in vivo studies suggest that Nec-1i has the capability to protect against SIRS induced by TNF and the effect is almost equipotent to Nec-1-induced inhibitory effect in SIRS [25]. These findings are just some preliminary researches on Nec-1i, whether it has any clinical implications still needs more investigation.
4.2. Nec-1s
Necrostatin-1 stable (Nec-1 s), or 7Cl-O-Nec-1, is a stable form of Nec-1 analogues, exhibiting inhibitive effect of RIP1 in a dose-dependent manner which is more potent than Nec-1 [24,181]. Although Nec-1 has already been used in various studies and is proved to have protective effect on plenty necroptosis-related diseases, it has following shortcomings: (1) Nec-1 has off-target effect on IDO; (2) Nec-1 has a short half-life of about 1 h. Compared to Nec-1 and Nec-1i, Nec-1 s does not inhibit IDO, proved to be a more specific RIP1 inhibitor. Besides, Nec-1 s is able to protect against SIRS induced by TNF without paradoxical sensitizing effect [25]. In view of the above-mentioned facts, Nec-1 s with improved stability can be used as a superb alternative of Nec-1. In the folic acid-induced AKI mouse model, both pretreatment and posttreatment of Nec-1 s protected against the second wave of cell death in AKI [68]. Wang et al. [91] conducted a research on abdominal aortic aneurysm (AAA) and found that Nec-1 was not able to lessen the aortic expansion significantly in elastase-induced AAA mouse model, whereas the mean aortic diameter of Nec-1 s group was evidently smaller compared with DMSO group. In the study of progression of existing aneurysms, Nec-1 s was proved to resolve the inflammatory response, marked by mitigated macrophage infiltration and reduced MMP9 [91]. More surprisingly, the histological result of Nec-1 s treated mice showed almost normal looking arterial structure, without progressive elastin degradation [91]. The reports about Nec-1 s in disease models are summarized in Table 3 . These researches demonstrated the potential of Nec-1 s to be a stronger therapy with improved performance.
Table 3.
Role of Nec-1 s in diseases.
| Diseases | Role of Nec-1s | References |
|---|---|---|
| Acute kidney injury (AKI) | Nec-1 s ameliorates cell death and kidney dysfunction in AKI | [68] |
| Abdominal aortic aneurysm (AAA) | Nec-1 s lessens aortic expansion and improves aortic structure pathologically in AAA mouse model | [91] |
| Colon cancer | Nec-1 s prevents 2-methoxy-6-acetyl-7-methyljuglone-induced cell death in HCT116 and HT29 colon cancer cells | [182] |
| I/R injury | Nec-1 s protects Kupffer cells and mitigates inflammation in IR | [75] |
| SIRS | Nec-1 s prevents mortality in TNF-induced SIRS mouse model | [25] |
| Multiple sclerosis | Nec-1 reduces myelin loss and inflammation | [183] |
| Rheumatoid arthritis | Nec-1 alleviates rheumatoid arthritis in the collagen-induced arthritis mouse model with lower arthritis score and arthritis incidence | [122] |
5. Nec-1 and COVID-19
As of now, coronavirus disease 2019 (COVID-19) has swept the globe, impacting everyone and every nation. Upon coronavirus infection, the immune system is quickly activated, and continuous infection causes continuous inflammatory response. The inflammatory response begins with an initial recognition of coronavirus, and then mediates immune cells recruitment and tissue repair. However, coronavirus may induce excessive and prolonged cytokine responses, namely cytokine storm in some severe individuals [184]. Cytokine storm causes acute respiratory distress syndrome or multiple-organ dysfunction, which leads to physiological deterioration and death [185]. Therefore, cytokine storm is probably the reason why some patients with mild symptoms suddenly become critically ill, especially for young men with strong immune system. The subsequent cytokine storm leads to SIRS, causing damages to kidney [186], liver [187] and myocardium [188] and finally resulting in multiple organ dysfunction syndrome. According to latest researches and clinical data, cytokine storm is closely related to prognosis of patients. Serum cytokine and chemokine levels are significantly higher in ICU-patients than patients with mild to moderate clinical symptoms [189]. Thus, cytokine storm is charged with being the mechanism explaining the morbidity and mortality of current COVID-19 cases. Therefore, seeking for new strategies to control cytokine storm in severe COVID-19 patients becomes a matter of great urgency.
Although many researchers believe the timely control of cytokine storm in its early stage could be the key to improve prognosis, there is no effective drug to lower mortality of COVID-19 patients. As demonstrated above, necroptosis is a key factor in inflammatory diseases, and Nec-1 is an anti-inflammation compound by targeting RIP1. Necroptotic cell death causes pro-inflammatory responses, releasing cytokines including IL-6, TNF-α, and IL-1β [190], which have been proved as key cytokines in disease development of COVID-19 [191,192]. Therefore, targeting necroptosis by using Nec-1 could be a potential strategy to fight against COVID-19. In the research of Zelic et al. [193], RIP1 kinase-inactive knock-in mice were observed to be resistant to SIRS model and cytokine storm, indicating the essential role of necroptosis and RIP1 in SIRS, thus researchers further predicted that RIP1 inhibitors may be beneficial to sepsis or systemic inflammation patients [193,194]. In fact, Nec-1 is indeed associated with systemic inflammation. In the rat model of acute pancreatitis, rats treated with Nec-1 displayed significant milder symptoms and pathophysiological characteristics, and Nec-1 alleviated the extent of systemic inflammatory response caused by acute pancreatitis, the underlying mechanism of this protective effect is probably related to RIP1/NF-κB p65/AQP8 axis [195]. Although there is no research directly showing the relation between necroptosis and COVID-19, or the effect of Nec-1 in COVID-19, the strong anti-inflammation ability of Nec-1 makes us predict that Nec-1/Nec-1 analogues might have the capability to alleviate cytokine storm, systemic inflammation and thus COVID-19. Since Nec-1 can inhibit both RIP1 and IDO, its promising anti-inflammation effect in COVID-19 may undergo multiple pathways. By targeting RIP1, it can alleviate both necroptotic and apoptotic cell death, reduce DAMPs release and pro-inflammatory cytokines, inhibit inflammatory pathway NF-κB and ameliorate oxidative stress; by targeting IDO, it may also reduce inflammatory responses. Besides, coronaviruses is a group of RNA viruses, and it has been proved that RNA virus can promote inflammasome activation via RIP1/RIP3/DRP1 pathway, which can be inhibited by Nec-1 and Nec-1 s [196]. Thus, it is possible that Nec-1 or its analogues can alleviate inflammation and cytokine release induced by COVID-19. Furthermore, Nec-1 has protective effects on kidney, liver and cardiovascular system (Table 1), which means that Nec-1 or its derivatives might also help to prevent and protect against multi-organ complications of COVID-19 patients.
Some studies suggest that necroptosis is an alternate cell death form of apoptosis during virus infection [197]. Apoptosis is important in host defense against viral infection, damaged cells undergo apoptotic cell death upon infection, which inhibits virus replication [198]. It is observed that some viruses could prevent apoptosis by inhibiting caspase-8, thus necroptosis is initiated as an alternative form of cell death [199] (some viruses could even inhibit necroptosis [200,201]). So here comes the question: will Nec-1 inhibition of necroptosis promote virus infection? It is indeed that necroptosis plays a role in cell demise after viral infection, but whether it favors host antiviral responses or aggravates tissue inflammation and damage is not validated, or at least controversial. It is possible that necroptosis inhibits viral replication by inducing cell death during the early stage of virus infection, but it may also promote virus spread by cell rupture in the following stages. What’s more, researchers found that the necroptotic cell death activated by influenza A virus infection does not depend on RIP1 [202] (yet it is usually RIP1-dependent in inflammatory responses). Therefore, using Nec-1 to control cytokine storm, which usually happens during an advanced stage, does not necessarily influence host antiviral immunity. Besides, it is observed in patients that T lymphocytes become functionally exhausted with disease progression [203]. Thus Nec-1 may also ameliorate T cell exhaustion of COVID-19 patients by regulating host defense.
6. Conclusion
Although necroptosis was defined as a new form of cell death just fifteen years ago, scientists have achieved much progress. From the fundamental mechanism to its role in various diseases, the picture of necroptosis is becoming clear and detailed. The clinical significance of targeting necroptosis is supported by more and more researches, thus necroptotic inhibitors have been attached with great significance for both scientific and clinical researches. Since the discovery of necroptosis, the specific necroptotic inhibitor Nec-1 has been applied in many disease models and it is actually the most used one among all necroptotic inhibitors. Furthermore, lots of studies have proved the therapeutic effects of Nec-1 in various disease models, therefore we propose that Nec-1 and its derivatives could have great clinical potential. Currently, some other RIP1 inhibitors are now undergoing clinical trials [204,205]. Although Nec-1 has drawbacks like metabolic instability and off-target effect, its protection in disease models and potential for optimization cannot be ignored. In fact, some classic drugs like aspirin is also characterized by short half-life [206], and off-target effect on IDO may endow Nec-1 with more potent anti-inflammation ability. In this review, we summarized the effects Nec-1 exhibits in disease models and research progress of recent studies. Apart from RIP1-dependent mechanism, we discussed other possible RIP1-independent factors (such as RIA and IDO) of Nec-1 protection. After probing into inflammation and I/R injury, we innovatively reviewed the role of Nec-1 in disease models of diabetic cardiomyopathy, atherosclerosis and retinal degeneration. Finally, we predicted that Nec-1 might help to ameliorate cytokine storm in COVID-19. Although many researchers have thrown spotlight on necroptosis inhibition, some questions remain unclear. For example, the effect of Nec-1 on apoptosis is perplexing. Some studies observed inhibitory effect on apoptosis, while the other claimed no influence. Here we attribute this divergence to different underlying pathways of RDA and RIA. Nec-1 inhibits apoptosis by targeting RIP1 when the cell undergoes signaling pathway of RDA, yet it loses this anti-apoptosis effect when the pathway is RIA. Considering the effects on necroptosis, apoptosis and inflammatory responses, here is another open question: does Nec-1 affect one or multiple RIP1-dependent pathways? The findings and remaining questions suggest that the molecular network of Nec-1 administration in diseases is quite complicated, remaining to be explored deeper by scientists. There is still a long road of scientific researches before the clinical practice of Nec-1 or its derivatives, which is hopeful to be an attractive strategy.
Funding
This work was funded by the National Natural Science Foundation of China (81803269) and the Science and Technology Commission of Shanghai Municipality (18YF1412100, 2019Y0150).
CRediT authorship contribution statement
Liyuan Cao: Conceptualization, Writing - original draft, Writing - review & editing. Wei Mu: Conceptualization, Funding acquisition.
Declaration of Competing Interest
The authors declare no conflict of interest.
Footnotes
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.phrs.2020.105297.
Appendix A. Supplementary data
The following is Supplementary data to this article:
References
- 1.Vandenabeele P., Galluzzi L., Vanden Berghe T., Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature reviews. Mol. cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 2.Karunakaran D., Geoffrion M., Wei L., Gan W. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2016;2 doi: 10.1126/sciadv.1600224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang S., Tang M.B., Luo H.Y., Shi C.H., Xu Y.M. Necroptosis in neurodegenerative diseases: a potential therapeutic target. Cell Death Disease. 2017;8:e2905. doi: 10.1038/cddis.2017.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saeed W.K., Jun D.W., Jang K., Koh D.H. Necroptosis signaling in liver diseases: an update. Pharmacol. Res. 2019;148 doi: 10.1016/j.phrs.2019.104439. [DOI] [PubMed] [Google Scholar]
- 5.Kaczanowski S. Apoptosis: its origin, history, maintenance and the medical implications for cancer and aging. Phys. Biol. 2016;13 doi: 10.1088/1478-3975/13/3/031001. [DOI] [PubMed] [Google Scholar]
- 6.Kroemer G., Levine B. Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denton D., Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019;26:605–616. doi: 10.1038/s41418-018-0252-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenwald S.H., Pierce E.A. Parthanatos as a cell death pathway underlying retinal disease. Adv. Exp. Med. Biol. 2019;1185:323–327. doi: 10.1007/978-3-030-27378-1_53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi J., Gao W., Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 2017;42:245–254. doi: 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Hirschhorn T., Stockwell B.R. The development of the concept of ferroptosis. Free Rad. Biol. Med. 2019;133:130–143. doi: 10.1016/j.freeradbiomed.2018.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho Y.S., Challa S., Moquin D., Genga R., Ray T.D., Guildford M., Chan F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L., Wang H., Wang Z., He S., Chen S., Liao D., Wang L., Yan J., Liu W., Lei X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 13.Harper N., Hughes M., Macfarlane M., Cohen G.M. Fas-associated death domain protein and Caspase-8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during tumor necrosis factor-induced apoptosis. J. Biol. Chem. 2003;278:25534–25541. doi: 10.1074/jbc.M303399200. [DOI] [PubMed] [Google Scholar]
- 14.Micheau O., Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 15.Haas T.L., Emmerich C.H., Gerlach B., Schmukle A.C., Walczak H. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-Mediated gene induction. Mol. Cell. 2009;36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 16.Wang L., Du F., Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;113:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 17.Meng H., Liu Z., Li X., Wang H., Jin T., Wu G., Shan B., Christofferson D.E., Qi C., Yu Q. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc. Natl. Acad. Sci. U. S. A. 2018;115:E2001–E2009. doi: 10.1073/pnas.1722013115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holbrook J., Lara-Reyna S., Jarosz-Griffiths H., Mcdermott M. Tumour necrosis factor signalling in health and disease. F1000 Res. 2019;8:111. doi: 10.12688/f1000research.17023.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grootjans S., Vanden Berghe T., Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Different. 2017;24:1184–1195. doi: 10.1038/cdd.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dondelinger Y., Aguileta M.A., Goossens V., Dubuisson C., Grootjans S., Dejardin E., Vandenabeele P., Bertrand M.J. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013;20:1381–1392. doi: 10.1038/cdd.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H., Sun L., Su L., Rizo J., Liu L., Wang L.F., Wang F.S., Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 22.Chen X., Li W., Ren J., Huang D., He W.T., Song Y., Yang C., Li W., Zheng X., Chen P. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–121. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G.D., Mitchison T.J., Moskowitz M.A., Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 24.Degterev A., Hitomi J., Germscheid M., Ch’en I.L., Korkina O., Teng X., Abbott D., Cuny G.D., Yuan C., Wagner G. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi N., Duprez L., Grootjans S., Cauwels A., Nerinckx W., DuHadaway J.B., Goossens V., Roelandt R., Van Hauwermeiren F., Libert C. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Disease. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laurien L., Nagata M., Schunke H., Delanghe T., Wiederstein J.L., Kumari S., Schwarzer R., Corona T., Kruger M., Bertrand M.J.M. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat. Commun. 2020;11:1747. doi: 10.1038/s41467-020-15466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geng F., Yin H., Li Z., Li Q., He C., Wang Z., Yu J. Quantitative analysis of necrostatin-1, a necroptosis inhibitor by LC-MS/MS and the study of its pharmacokinetics and bioavailability. Biomed. Pharmacotherapy. 2017;95:1479. doi: 10.1016/j.biopha.2017.09.063. [DOI] [PubMed] [Google Scholar]
- 28.Muller A.J., DuHadaway J.B., Donover P.S., Sutanto-Ward E., Prendergast G.C.I. Nhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005;11:312. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 29.Degterev A., Maki J.L., Yuan J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2013;20:366. doi: 10.1038/cdd.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prendergast G.C., Smith C., Thomas S., Mandik-Nayak L., Laury-Kleintop L., Metz R., Muller A.J. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunotherapy : CII. 2014;63:721–735. doi: 10.1007/s00262-014-1549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho Y., McQuade T., Zhang H., Zhang J., Chan F.K. RIP1-dependent and independent effects of necrostatin-1 in necrosis and T cell activation. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamura Y., Chiba Y., Tanioka T., Shimizu N., Shinozaki S., Yamada M., Kaneki K., Mori S., Araki A., Ito H. NO donor induces Nec-1-inhibitable, but RIP1-independent, necrotic cell death in pancreatic beta-cells. FEBS Lett. 2011;585:3058–3064. doi: 10.1016/j.febslet.2011.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou W., Yuan J. Necroptosis in health and diseases. Seminars Cell Developmental Biol. 2014;35:14–23. doi: 10.1016/j.semcdb.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y.R., Xu H.M. Protective effect of necrostatin-1 on myocardial tissue in rats with acute myocardial infarction. Genetics Mol. Res. : GMR. 2016;15 doi: 10.4238/gmr.15027298. [DOI] [PubMed] [Google Scholar]
- 35.Nikseresht S., Khodagholi F., Nategh M., Dargahi L. RIP1 inhibition rescues from LPS-Induced RIP3-Mediated programmed cell death, distributed energy metabolism and spatial memory impairment. J. Mol. Neurosci. : MN. 2015;57:219–230. doi: 10.1007/s12031-015-0609-3. [DOI] [PubMed] [Google Scholar]
- 36.Liu Z.Y., Wu B., Guo Y.S., Zhou Y.H., Fu Z.G., Xu B.Q., Li J.H., Jing L., Jiang J.L., Tang J. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. Am. J. Cancer Res. 2015;5:3174–3185. doi: 10.1016/j.biopha.2017.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su X., Wang H., Kang D., Zhu J., Sun Q., Li T., Ding K. Necrostatin-1 ameliorates intracerebral hemorrhage-induced brain injury in mice through inhibiting RIP1/RIP3 pathway. Neurochem. Res. 2015;40:643–650. doi: 10.1007/s11064-014-1510-0. [DOI] [PubMed] [Google Scholar]
- 38.Fang T., Cao R., Wang W., Ye H., Shen L., Li Z., Hu J., Gao Q. Alterations in necroptosis during ALDH2-mediated protection against high glucoseinduced H9c2 cardiac cell injury. Mol. Med. Rep. 2018;18:2807–2815. doi: 10.3892/mmr.2018.9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang W., Chen M., Zheng D., He J., Song M., Mo L., Feng J., Lan J. A novel damage mechanism: contribution of the interaction between necroptosis and ROS to high glucose-induced injury and inflammation in H9c2 cardiac cells. Int. J. Mol. Med. 2017;40:201–208. doi: 10.3892/ijmm.2017.3006. [DOI] [PubMed] [Google Scholar]
- 40.Liang W., Chen M., Zheng D., Li J., Song M., Zhang W., Feng J., Lan J. The opening of ATP-Sensitive K+ channels protects H9c2 cardiac cells against the high glucose-induced injury and inflammation by inhibiting the ROS-TLR4-Necroptosis pathway. Cell. Physiol. Biochem. 2017;41:1020–1034. doi: 10.1159/000461391. [DOI] [PubMed] [Google Scholar]
- 41.Lin J., Chen M., Liu D., Guo R., Lin K., Deng H., Zhi X., Zhang W., Feng J., Wu W. Exogenous hydrogen sulfide protects human umbilical vein endothelial cells against high glucoseinduced injury by inhibiting the necroptosis pathway. Int. J. Mol. Med. 2018;41:1477–1486. doi: 10.3892/ijmm.2017.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao M., Lu L., Lei S., Chai H., Wu S., Tang X., Bao Q., Chen L., Wu W., Liu X. Inhibition of receptor interacting protein kinases attenuates cardiomyocyte hypertrophy induced by palmitic acid. Oxid. Med. Cell. Longevity. 2016;2016 doi: 10.1155/2016/1451676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang A., Mao X., Li L., Tong Y., Huang Y., Lan Y., Jiang H. Necrostatin-1 inhibits Hmgb1-IL-23/IL-17 pathway and attenuates cardiac ischemia reperfusion injury. Transplant Int. 2014;27:1077–1085. doi: 10.1111/tri.12349. [DOI] [PubMed] [Google Scholar]
- 44.Koudstaal S., Oerlemans M.I., Van der Spoel T.I., Janssen A.W., Hoefer I.E., Doevendans P.A., Sluijter J.P., Chamuleau S.A. Necrostatin-1 alleviates reperfusion injury following acute myocardial infarction in pigs. Eur. J. Clin. Invest. 2015;45:150–159. doi: 10.1111/eci.12391. [DOI] [PubMed] [Google Scholar]
- 45.Zhou F., Jiang X., Teng L., Yang J., Ding J., He C. Necroptosis may be a novel mechanism for cardiomyocyte death in acute myocarditis. Mol. Cell. Biochem. 2018;442:11–18. doi: 10.1007/s11010-017-3188-5. [DOI] [PubMed] [Google Scholar]
- 46.Zhu X., Park J., Golinski J., Qiu J., Khuman J., Lee C.C., Lo E.H., Degterev A., Whalen M.J. Role of Akt and mammalian target of rapamycin in functional outcome after concussive brain injury in mice. J. Cerebral blood flow Metabol. 2014;34:1531–1539. doi: 10.1038/jcbfm.2014.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bao Z., Fan L., Zhao L., Xu X., Liu Y., Chao H., Liu N., You Y., Liu Y., Wang X. Silencing of A20 aggravates neuronal death and inflammation after traumatic brain injury: a potential trigger of necroptosis. Front. Mol. Neurosci. 2019;12:222. doi: 10.3389/fnmol.2019.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu M., Wu W., Li H., Li S., Huang L.T., Yang Y.Q., Sun Q., Wang C.X., Yu Z., Hang C.H. Necroptosis, a novel type of programmed cell death, contributes to early neural cells damage after spinal cord injury in adult mice. J. Spinal Cord Med. 2015;38:745–753. doi: 10.1179/2045772314y.0000000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang S., Wu J., Zeng Y.Z., Wu S.S., Deng G.R., Chen Z.D., Lin B. Necrostatin-1 mitigates endoplasmic reticulum stress after spinal cord injury. Neurochem. Res. 2017;42:3548–3558. doi: 10.1007/s11064-017-2402-x. [DOI] [PubMed] [Google Scholar]
- 50.Xu Y., Wang J., Song X., Qu L., Wei R., He F., Wang K., Luo B. RIP3 induces ischemic neuronal DNA degradation and programmed necrosis in rat via AIF. Sci. Rep. 2016;6:29362. doi: 10.1038/srep29362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan L., Wang Z., Liu L., Jian X. Inhibiting histone deacetylase 6 partly protects cultured rat cortical neurons from oxygenglucose deprivationinduced necroptosis. Mol. Med. Rep. 2015;12:2661–2667. doi: 10.3892/mmr.2015.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Northington F.J., Chavez-Valdez R., Graham E.M., Razdan S., Gauda E.B., Martin L.J. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J. Cerebral Blood Flow Metabol. 2011;31:178–189. doi: 10.1038/jcbfm.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su X., Wang H., Lin Y., Chen F. RIP1 and RIP3 mediate hemin-induced cell death in HT22 hippocampal neuronal cells. Neuropsychiatr. Dis. Treat. 2018;14:3111–3119. doi: 10.2147/ndt.s181074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou K., Shi L., Wang Z., Zhou J., Manaenko A., Reis C., Chen S., Zhang J. RIP1-RIP3-DRP1 pathway regulates NLRP3 inflammasome activation following subarachnoid hemorrhage. Exp. Neurol. 2017;295:116–124. doi: 10.1016/j.expneurol.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 55.Chen J., Jin H., Xu H., Peng Y., Jie L., Xu D., Chen L., Li T., Fan L., He P. The Neuroprotective Effects of Necrostatin-1 on Subarachnoid Hemorrhage in Rats Are Possibly Mediated by Preventing Blood-Brain Barrier Disruption and RIP3-Mediated Necroptosis. Cell Transplant. 2019;28:1358–1372. doi: 10.1177/0963689719867285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duan S., Wang X., Chen G., Quan C., Qu S., Tong J. Inhibiting RIPK1 limits neuroinflammation and alleviates postoperative cognitive impairments in D-Galactose-Induced aged mice. Front. Behav. Neurosci. 2018;12:138. doi: 10.3389/fnbeh.2018.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Y., Zhang L., Yu H., Song K., Shi J., Chen L., Cheng J. Necrostatin-1 improves long-term functional recovery through protecting oligodendrocyte precursor cells after transient focal cerebral ischemia in mice. Neuroscience. 2018;371:229–241. doi: 10.1016/j.neuroscience.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 58.Yang S.H., Lee D.K., Shin J., Lee S., Baek S., Kim J., Jung H., Hah J.M., Kim Y. Nec-1 alleviates cognitive impairment with reduction of Abeta and tau abnormalities in APP/PS1 mice. EMBO Mol. Med. 2017;9:61–77. doi: 10.15252/emmm.201606566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu J.R., Wang J., Zhou S.K., Yang L., Yin J.L., Cao J.P., Cheng Y.B. Necrostatin-1 protection of dopaminergic neurons. Neural Regener. Res. 2015;10:1120–1124. doi: 10.4103/1673-5374.160108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu S., Zhang Y., Bai G., Li H. Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington’s disease. Cell Death Disease. 2011;2:e115. doi: 10.1038/cddis.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Re D.B., Le Verche V., Yu C., Amoroso M.W., Politi K.A., Phani S., Ikiz B., Hoffmann L., Koolen M., Nagata T. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81:1001–1008. doi: 10.1016/j.neuron.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murakami Y., Matsumoto H., Roh M., Giani A., Kataoka K., Morizane Y., Kayama M., Thanos A., Nakatake S., Notomi S. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 2014;21:270–277. doi: 10.1038/cdd.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hanus J., Anderson C., Sarraf D., Ma J., Wang S. Retinal pigment epithelial cell necroptosis in response to sodium iodate. Cell Death Discovery. 2016;2:16054. doi: 10.1038/cddiscovery.2016.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Do Y.J., Sul J.W., Jang K.H., Kang N.S., Kim Y.H., Kim Y.G., Kim E. A novel RIPK1 inhibitor that prevents retinal degeneration in a rat glaucoma model. Exp. Cell. Res. 2017;359:30–38. doi: 10.1016/j.yexcr.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 65.Dong K., Zhu H., Song Z., Gong Y., Wang F., Wang W., Zheng Z., Yu Z., Gu Q., Xu X. Necrostatin-1 protects photoreceptors from cell death and improves functional outcome after experimental retinal detachment. Am. J. Pathol. 2012;181:1634–1641. doi: 10.1016/j.ajpath.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 66.Dvoriantchikova G., Degterev A., Ivanov D. Retinal ganglion cell (RGC) programmed necrosis contributes to ischemia-reperfusion-induced retinal damage. Exp. Eye Res. 2014;123:1–7. doi: 10.1016/j.exer.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luo J., Tao Y., Liang X., Chen Y., Zhang L., Jiang F., Liu S., Ye Z., Li Z., Shi W. Flow control effect of necrostatin-1 on cell death of the NRK-52E renal tubular epithelial cell line. Mol. Med. Rep. 2017;16:57–62. doi: 10.3892/mmr.2017.6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Martin-Sanchez D., Fontecha-Barriuso M., Carrasco S., Sanchez-Nino M.D., Massenhausen A.V., Linkermann A. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc. Natl. Acad. Sci. U. S. A. 2018;115:4182–4187. doi: 10.1073/pnas.1716578115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dong W., Li Z., Chen Y., Zhang L., Ye Z., Liang H., Li R., Xu L., Zhang B., Liu S. Necrostatin-1 attenuates sepsis-associated acute kidney injury by promoting autophagosome elimination in renal tubular epithelial cells. Mol. Med. Rep. 2018;17:3194–3199. doi: 10.3892/mmr.2017.8214. [DOI] [PubMed] [Google Scholar]
- 70.Zhu Y., Cui H., Gan H., Xia Y., Wang L., Wang Y., Sun Y. Necroptosis mediated by receptor interaction protein kinase 1 and 3 aggravates chronic kidney injury of subtotal nephrectomised rats. Biochem. Biophys. Res. Commun. 2015;461:575–581. doi: 10.1016/j.bbrc.2015.03.164. [DOI] [PubMed] [Google Scholar]
- 71.Xiao X., Du C., Yan Z., Shi Y., Duan H., Ren Y. Inhibition of necroptosis attenuates kidney inflammation and interstitial fibrosis induced by unilateral ureteral obstruction. Am. J. Nephrol. 2017;46:131–138. doi: 10.1159/000478746. [DOI] [PubMed] [Google Scholar]
- 72.Zhao H., Ning J., Lemaire A., Koumpa F.S., Sun J.J., Fung A., Gu J., Yi B., Lu K., Ma D. Necroptosis and parthanatos are involved in remote lung injury after receiving ischemic renal allografts in rats. Kidney Int. 2015;87:738–748. doi: 10.1038/ki.2014.388. [DOI] [PubMed] [Google Scholar]
- 73.Linkermann A., Brasen J.H., Darding M., Jin M.K., Sanz A.B., Heller J.O., De Zen F., Weinlich R., Ortiz A., Walczak H. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U. S. A. 2013;110:12024–12029. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Linkermann A., Brasen J.H., Himmerkus N., Liu S., Huber T.B., Kunzendorf U., Krautwald S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012;81:751–761. doi: 10.1038/ki.2011.450. [DOI] [PubMed] [Google Scholar]
- 75.Yue S., Zhou H., Wang X., Busuttil R.W. Prolonged ischemia triggers necrotic depletion of tissue-resident macrophages to facilitate inflammatory immune activation in liver ischemia reperfusion injury. J. Immunol. 2017;198:3588–3595. doi: 10.4049/jimmunol.1601428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hong J.M., Kim S.J., Lee S.M. Role of necroptosis in autophagy signaling during hepatic ischemia and reperfusion. Toxicol. Appl. Pharmacol. 2016;308:1–10. doi: 10.1016/j.taap.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 77.Kim S.J., Lee S.M. Necrostatin-1 protects against D-Galactosamine and lipopolysaccharide-induced hepatic injury by preventing TLR4 and RAGE signaling. Neurochem. Res. 2017;40:1912–1923. doi: 10.1007/s10753-017-0632-3. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Y.F., He W., Zhang C., Liu X.J., Lu Y., Wang H., Zhang Z.H., Chen X., Xu D.X. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol. Lett. 2014;225:445–453. doi: 10.1016/j.toxlet.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 79.Takemoto K., Hatano E., Iwaisako K., Takeiri M., Noma N., Ohmae S., Toriguchi K., Tanabe K., Tanaka H., Seo S. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS open bio. 2014;4:777–787. doi: 10.1016/j.fob.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang L., Cui Y., Wang B., Yu J., Wang Y., Wang Y. Protective effect of necrostatin-1 on the liver of rats with trauma induced hemorrhagic shock. Zhonghua wei zhong bing ji jiu yi xue. 2014;26:17–22. [PubMed] [Google Scholar]
- 81.Lim E.J., El Khobar K., Chin R., Earnest-Silveira L., Angus P.W., Bock C.T., Nachbur U., Silke J., Torresi J. Hepatitis C virus-induced hepatocyte cell death and protection by inhibition of apoptosis. J. General Virol. 2014;95:2204–2215. doi: 10.1099/vir.0.065862-0. [DOI] [PubMed] [Google Scholar]
- 82.Liang S., Lv Z.T., Zhang J.M., Wang Y.T., Dong Y.H., Wang Z.G., Chen K., Cheng P., Yang Q., Guo F.J. Necrostatin-1 attenuates trauma-induced mouse osteoarthritis and IL-1beta induced apoptosis via HMGB1/TLR4/SDF-1 in primary mouse chondrocytes. Front. Pharmacol. 2018;9:1378. doi: 10.3389/fphar.2018.01378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang C., Lin S., Li T., Jiang Y., Huang Z., Wen J., Cheng W., Li H. Mechanical force-mediated pathological cartilage thinning is regulated by necroptosis and apoptosis. Osteoarthritis Cartilage. 2017;25:1324–1334. doi: 10.1016/j.joca.2017.03.018. [DOI] [PubMed] [Google Scholar]
- 84.Ichiseki T., Ueda S., Ueda Y., Tuchiya M., Kaneuji A., Kawahara N. Involvement of necroptosis, a newly recognized cell death type, in steroid-induced osteonecrosis in a rabbit model. Int. J. Med. Sci. 2017;14:110–114. doi: 10.7150/ijms.17134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Feng M., Zhang R., Gong F., Yang P., Fan L., Ni J., Bi W., Zhang Y., Wang C., Wang K. Protective effects of necrostatin-1 on glucocorticoid-induced osteoporosis in rats. J. Steroid Biochem. Molecul. Biol. 2014;144(Pt B):455–462. doi: 10.1016/j.jsbmb.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 86.Cui H., Zhu Y., Yang Q., Zhao W., Zhang S., Zhou A., Jiang D. Necrostatin-1 treatment inhibits osteocyte necroptosis and trabecular deterioration in ovariectomized rats. Sci. Rep. 2016;6:33803. doi: 10.1038/srep33803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wen S., Ling Y., Yang W., Shen J., Li C., Deng W., Liu W., Liu K. Necroptosis is a key mediator of enterocytes loss in intestinal ischaemia/reperfusion injury. J. Cell. Mol. Med. 2017;21:432–443. doi: 10.1111/jcmm.12987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Negroni A., Colantoni E., Pierdomenico M., Palone F., Costanzo M., Oliva S., Tiberti A., Cucchiara S., Stronati L. RIP3 AND pMLKL promote necroptosis-induced inflammation and alter membrane permeability in intestinal epithelial cells. Digest. Liver Disease. 2017;49:1201–1210. doi: 10.1016/j.dld.2017.08.017. [DOI] [PubMed] [Google Scholar]
- 89.Duprez L., Takahashi N., Van Hauwermeiren F., Vandendriessche B., Goossens V., Vanden Berghe T., Declercq W., Libert C., Cauwels A., Vandenabeele P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 90.Fan L., Li Z., Fan Z., Wang Y. The effect of necrostatin-1 on expression of liver monocyte chemotactic protein-1 in septic rats. Zhonghua wei zhong bing ji jiu yi xue. 2016;28:262–266. [PubMed] [Google Scholar]
- 91.Wang Q., Zhou T., Liu Z., Ren J., Phan N., Gupta K., Stewart D.M., Morgan S., Assa C., Kent K.C. Inhibition of Receptor-Interacting Protein Kinase 1 with Necrostatin-1s ameliorates disease progression in elastase-induced mouse abdominal aortic aneurysm model. Sci. Rep. 2017;7:42159. doi: 10.1038/srep42159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pan L., Yao D.C., Yu Y.Z., Li S.J., Chen B.J., Hu G.H., Xi C., Wang Z.H., Wang H.Y., Li J.H. Necrostatin-1 protects against oleic acid-induced acute respiratory distress syndrome in rats. Biochem. Biophys. Res. Commun. 2016;478:1602–1608. doi: 10.1016/j.bbrc.2016.08.163. [DOI] [PubMed] [Google Scholar]
- 93.Pan T., Wu S., He X., Luo H., Zhang Y., Fan M., Geng G., Ruiz V.C., Zhang J., Mills L. Necroptosis takes place in human immunodeficiency virus type-1 (HIV-1)-infected CD4+ T lymphocytes. PLoS One. 2014;9 doi: 10.1371/journal.pone.0093944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cuda C.M., Misharin A.V., Gierut A.K., Saber R., Haines G.K., 3rd, Hutcheson J., Hedrick S.M., Mohan C., Budinger G.S., Stehlik C. Caspase-8 acts as a molecular rheostat to limit RIPK1- and MyD88-mediated dendritic cell activation. J. Immunol. (Baltimore, Md. : 1950) 2014;192:5548–5560. doi: 10.4049/jimmunol.1400122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y.Q., Wang L., Zhang M.Y., Wang T., Bao H.J., Liu W.L., Dai D.K., Zhang L., Chang P., Dong W.W. Necrostatin-1 suppresses autophagy and apoptosis in mice traumatic brain injury model. Neurochem. Res. 2012;37:1849–1858. doi: 10.1007/s11064-012-0791-4. [DOI] [PubMed] [Google Scholar]
- 96.Han W., Xie J., Fang Y., Wang Z., Pan H. Nec-1 enhances shikonin-induced apoptosis in leukemia cells by inhibition of RIP-1 and ERK1/2. Int. J. Mol. Sci. 2012;13:7212–7225. doi: 10.3390/ijms13067212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jie H., He Y., Huang X., Zhou Q., Han Y., Li X., Bai Y., Sun E. Necrostatin-1 enhances the resolution of inflammation by specifically inducing neutrophil apoptosis. Oncotarget. 2016;7:19367–19381. doi: 10.18632/oncotarget.8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sawai H. Induction of apoptosis in TNF-treated L929 cells in the presence of necrostatin-1. Int. J. Mol. Sci. 2016;17:1678–1689. doi: 10.15252/emmm.201606566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Galadari S., Rahman A., Pallichankandy S., Thayyullathil F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radical Biol. Med. 2017;104:144–164. doi: 10.1016/j.freeradbiomed.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 100.Holzerová E.K., Prokisch H. Mitochondria: much ado about nothing? How dangerous is reactive oxygen species production? Int. J. Biochem. Cell Biol. 2015;63:16–20. doi: 10.1016/j.biocel.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang Y., Wang H., Tao Y., Zhang S., Wang J., Feng X. Necroptosis inhibitor necrostatin-1 promotes cell protection and physiological function in traumatic spinal cord injury. Neuroscience. 2014;266:91–101. doi: 10.1016/j.neuroscience.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 102.Chauhan A.K., Min K.J., Kwon T.K. RIP1-dependent reactive oxygen species production executes artesunate-induced cell death in renal carcinoma Caki cells. Mol. Cell. Biochem. 2017;435:15–24. doi: 10.1007/s11010-017-3052-7. [DOI] [PubMed] [Google Scholar]
- 103.Lu B., Gong X., Wang Z.Q., Ding Y., Wang C., Luo T.F., Piao M.H., Meng F.K., Chi G.F., Luo Y.N. Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation. Acta. Pharmacol. Sin. 2017;38:1543–1553. doi: 10.1038/aps.2017.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schenk B., Fulda S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene. 2015;34:5796–5806. doi: 10.1038/onc.2015.35. [DOI] [PubMed] [Google Scholar]
- 105.Linkermann A., Stockwell B.R., Krautwald S., Anders H.J. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat. Rev. Immunol. 2014;14:759–767. doi: 10.1038/nri3743. [DOI] [PubMed] [Google Scholar]
- 106.Huang Z., Wu S.Q., Liang Y., Zhou X., Chen W., Li L., Wu J., Zhuang Q., Chen C., Li J. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe. 2015;17:229–242. doi: 10.1016/j.chom.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 107.Newton K., Dugger D.L., Maltzman A., Greve J.M., Hedehus M., Martin-Mcnulty B., Carano R.A.D., Cao T.C., Van Bruggen N., Bernstein L. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Different. 2016 doi: 10.1038/cdd.2016.46. 10.1038/cdd.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hsu H. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4 doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 109.Dondelinger Y., Declercq W., Montessuit S., Roelandt R., Goncalves A., Bruggeman I., Hulpiau P., Weber K., Sehon C.A., Marquis R.W. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–981. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 110.Kaczmarek A., Vandenabeele P., Krysko D.V. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 111.Zhu K., Liang W., Ma Z., Xu D., Cao S., Lu X., Liu N., Shan B., Qian L., Yuan J. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Disease. 2018;9:500. doi: 10.1038/s41419-018-0524-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saleh D., Najjar M., Zelic M., Shah S., Nogusa S., Polykratis A., Paczosa M.K., Gough P.J., Bertin J., Whalen M. Kinase Activities of RIPK1 and RIPK3 Can Direct IFN-beta Synthesis Induced by Lipopolysaccharide. J. Immunol. 2017;198:4435–4447. doi: 10.4049/jimmunol.1601717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lukens J.R., Vogel P., Johnson G.R., Kelliher M.A., Iwakura Y., Lamkanfi M., Kanneganti T. RIP1-driven autoinflammation targets IL-1α independently of inflammasomes and RIP3. Rare Diseases. 2014;498:224–227. doi: 10.1038/nature12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Christofferson D.E., Li Y., Hitomi J., Zhou W., Upperman C., Zhu H., Gerber S.A., Gygi S., Yuan J. A novel role for RIP1 kinase in mediating TNFα production. Cell Death Disease. 2012;3:e320. doi: 10.1038/cddis.2012.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Guan E., Wang Y., Wang C., Zhang R., Zhao Y., Hong J. Necrostatin-1 attenuates lipopolysaccharide-induced acute lung injury in mice. Exp. Lung Res. 2017;43:378–387. doi: 10.1080/01902148.2017.1384083. [DOI] [PubMed] [Google Scholar]
- 116.Wong W.W., Gentle I.E., Nachbur U., Anderton H., Vaux D.L., Silke J. RIPK1 is not essential for TNFR1-induced activation of NF-kappaB. Cell Death Different. 2010;17:482–487. doi: 10.1038/cdd.2009.178. [DOI] [PubMed] [Google Scholar]
- 117.Kelliher M.A., Grimm S., Ishida Y., Kuo F., Stanger B.Z., Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- 118.Ea C.K., Deng L., Xia Z.P., Pineda G., Chen Z.J.A. Ctivation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 119.Bertrand M.J.M., Vandenabeele P. RIP1’s function in NF-κB activation: from master actor to onlooker. Cell Death Different. 2010 doi: 10.1038/cdd.2009.213. 10.1038/cdd.2009.213. [DOI] [PubMed] [Google Scholar]
- 120.Schreiber A., Rousselle A., Becker J.U., Mssenhausen A.V., Kettritz R. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc. Natl. Acad. U. S. A. 2017;114:E9618. doi: 10.1073/pnas.1708247114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee S.H., Kwon J.Y., Kim S.Y., Jung K.A., Cho M.L. Interferon-gamma regulates inflammatory cell death by targeting necroptosis in experimental autoimmune arthritis. Sci. Rep. 2017;7:10133. doi: 10.1038/s41598-017-09767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jhun J., Lee S.H., Kim S.Y., Ryu J., Kwon J.Y., Na H.S., Jung K., Moon S.J., Cho M.L., Min J.K. RIPK1 inhibition attenuates experimental autoimmune arthritis via suppression of osteoclastogenesis. J. Translat. Med. 2019;17:84. doi: 10.1186/s12967-019-1809-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang Y., Guo L., Wang J., Shi W., Xia Z., Li B. Necrostatin-1 ameliorates the pathogenesis of experimental autoimmune encephalomyelitis by suppressing apoptosis and necroptosis of oligodendrocyte precursor cells. Exp Ther Med. 2019;18:4113–4119. doi: 10.3892/etm.2019.8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Korthuis R.J., Kalogeris T.J., Krenz M., Baines C.P. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2011;298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wu M.-Y., Yiang G.-T., Liao W.-T., Tsai A.-Y., Cheng Y.-L., Cheng P.-W., Li C.-Y., Li C.-J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018:1650–1667. doi: 10.1159/000489241. 10.1159/000489241. [DOI] [PubMed] [Google Scholar]
- 126.Yang R., Hu K., Chen J., Zhu S., Li L., Lu H., Li P., Dong R. Necrostatin-1 protects hippocampal neurons against ischemia/reperfusion injury via the RIP3/DAXX signaling pathway in rats. Neurosci. Lett. 2017;651:207–215. doi: 10.1016/j.neulet.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 127.Oerlemans M.I., Liu J., Arslan F., den Ouden K., van Middelaar B.J., Doevendans P.A., Sluijter J.P. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res. Cardiol. 2012;107:270. doi: 10.1007/s00395-012-0270-8. [DOI] [PubMed] [Google Scholar]
- 128.Yang F., Shang L., Wang S., Liu Y., Ren H., Zhu W., Shi X. TNFalpha-mediated necroptosis aggravates ischemia-reperfusion injury in the fatty liver by regulating the inflammatory response. Oxid. Med. Cell. Longevity. 2019;2019 doi: 10.1155/2019/2301903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang J., He G.Z., Wang Y.K., Zhu Q.K., Chen W., Guo T. TLR4-HMGB1-, MyD88- and TRIF-dependent signaling in mouse intestinal ischemia/reperfusion injury. World J. Gastroenterol. 2015:108–119. doi: 10.3748/wjg.v21.i27.8314. 10.3748/wjg.v21.i27.8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kanou T., Ohsumi A., Kim H., Chen M., Bai X., Guan Z., Hwang D., Cypel M., Keshavjee S., Liu M. Inhibition of regulated necrosis attenuates receptor-interacting protein kinase 1-mediated ischemia-reperfusion injury after lung transplantation. J. Heart Lung Transplant. 2018;37:1261–1270. doi: 10.1016/j.healun.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 131.Zhou Z., Lu B., Wang C., Wang Z., Luo T., Piao M., Meng F., Chi G., Luo Y., Ge P. RIP1 and RIP3 contribute to shikonin-induced DNA double-strand breaks in glioma cells via increase of intracellular reactive oxygen species. Cancer Lett. 2017;390:77–90. doi: 10.1016/j.canlet.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 132.Halestrap A.P., Richardson A.P. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2015;78:129–141. doi: 10.1016/j.yjmcc.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 133.Morciano G., Bonora M., Campo G., Aquila G., Rizzo P., Giorgi C., Wieckowski M.R., Pinton P. Mechanistic role of mPTP in ischemia-reperfusion injury. Adv. Exp. Med. Biol. 2017;982:169–189. doi: 10.1007/978-3-319-55330-6_9. [DOI] [PubMed] [Google Scholar]
- 134.Zhu P., Hu S., Jin Q., Li D., Tian F., Toan S., Li Y., Zhou H., Chen Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018;16:157–168. doi: 10.1016/j.redox.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lim S.Y., Davidson S.M., Mocanu M.M., Yellon D.M., Smith C.C. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc. Drugs Ther. 2007;21:467–469. doi: 10.1007/s10557-007-6067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.King M.D., Whitaker-Lea W.A., Campbell J.M., Alleyne C.H., Jr., Dhandapani K.M. Necrostatin-1 reduces neurovascular injury after intracerebral hemorrhage. Int. j. Cell Biol. 2014;2014 doi: 10.1155/2014/495817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chang P., Dong W., Zhang M., Wang Z., Wang Y., Wang T., Gao Y., Meng H., Luo B., Luo C. Anti-necroptosis chemical necrostatin-1 can also suppress apoptotic and autophagic pathway to exert neuroprotective effect in mice intracerebral hemorrhage model. J. Mol. Neurosci. : MN. 2014;52:242–249. doi: 10.1007/s12031-013-0132-3. [DOI] [PubMed] [Google Scholar]
- 138.Laird M.D., Wakade C., Alleyne C.H., Jr., Dhandapani K.M. Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free Rad. Biol. Med. 2008;45:1103–1114. doi: 10.1016/j.freeradbiomed.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 139.Wang Y., Wang J., Yang H., Zhou J., Feng X., Wang H., Tao Y. Necrostatin-1 mitigates mitochondrial dysfunction post-spinal cord injury. Neuroscience. 2015;289:224–232. doi: 10.1016/j.neuroscience.2014.12.061. [DOI] [PubMed] [Google Scholar]
- 140.Zhe-Wei S., Li-Sha G., Yue-Chun L. The role of necroptosis in cardiovascular disease. Front. Pharmacol. 2018;9:721. doi: 10.3389/fphar.2018.00721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tian F., Yao J., Yan M., Sun X., Wang W., Gao W., Tian Z., Guo S., Dong Z., Li B. 5-Aminolevulinic Acid-Mediated Sonodynamic Therapy Inhibits RIPK1/RIPK3-Dependent Necroptosis in THP-1-Derived Foam Cells. Sci. Rep. 2016;6:21992. doi: 10.1038/srep21992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Luedde M., Lutz M., Carter N., Sosna J., Jacoby C., Vucur M., Gautheron J., Roderburg C., Borg N., Reisinger F. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc. Res. 2014;103:206–216. doi: 10.1093/cvr/cvu146. [DOI] [PubMed] [Google Scholar]
- 143.Mapanga R.F., Essop M.F. Damaging effects of hyperglycemia on cardiovascular function: spotlight on glucose metabolic pathways. Am. J. Physiol. Heart Circ. Physiol. 2016;310:H153–173. doi: 10.1152/ajpheart.00206.2015. [DOI] [PubMed] [Google Scholar]
- 144.Wautier M.P., Chappey O., Corda S., Stern D.M., Schmidt A.M., Wautier J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001;280:E685–694. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]
- 145.Zhang D.W., Shao J., Lin J., Zhang N., Lu B.J., Lin S.C., Dong M.Q., Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 146.Adeva-Andany M.M., González-Lucán M., Donapetry-García C., Fernández-Fernández C., Ameneiros-Rodríguez E. Glycogen metabolism in humans. BBA Clin. 2016;5:85–100. doi: 10.1016/j.bbacli.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Van Herreweghe F., Mao J., Chaplen F.W., Grooten J., Gevaert K., Vandekerckhove J., Vancompernolle K. Tumor necrosis factor-induced modulation of glyoxalase I activities through phosphorylation by PKA results in cell death and is accompanied by the formation of a specific methylglyoxal-derived AGE. Proc. Natl. Acad. Sci. U. S. A. 2002;99:949–954. doi: 10.1073/pnas.012432399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhang T., Zhang Y., Cui M. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016;22:175–182. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- 149.Di Carlo M.N., Said M., Ling H., Valverde C.A., De Giusti V.C., Sommese L., Palomeque J., Aiello E.A., Skapura D.G., Rinaldi G. CaMKII-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2014;74:274–283. doi: 10.1016/j.yjmcc.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Popescu I., Galice S., Mohler P.J., Despa S. Elevated local [Ca2+] and CaMKII promote spontaneous Ca2+ release in ankyrin-B-deficient hearts. Cardiovasc. Res. 2016;111:287–294. doi: 10.1093/cvr/cvw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Reventun P., Sanchez-Esteban S., Cook A., Cuadrado I., Saura M. Bisphenol a induces coronary endothelial cell necroptosis by activating RIP3 /CamKII dependent pathway. Sci. Rep. 2020;10:4190. doi: 10.1038/s41598-020-61014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Szobi A., Rajtik T., Adameova A. Effects of necrostatin-1, an inhibitor of necroptosis, and its inactive analogue nec-1i on basal cardiovascular function. Physiol. Res. 2016;65:861–865. doi: 10.33549/physiolres.933393. [DOI] [PubMed] [Google Scholar]
- 153.Nimkuntod P., Tongdee P. Association between subclinical atherosclerosis among hyperlipidemia and healthy subjects. J. Med. Assoc. Thai. 2015;98(Suppl 4):S51–57. [PubMed] [Google Scholar]
- 154.Mitra S., Goyal T., Mehta J.L. Oxidized LDL, LOX-1 and atherosclerosis. Cardiovasc. Drugs Ther. 2011;25:419–429. doi: 10.1007/s10557-011-6341-5. [DOI] [PubMed] [Google Scholar]
- 155.Martinet W., Schrijvers D.M., De Meyer G.R. Necrotic cell death in atherosclerosis. Basic Res. Cardiol. 2011;106:749–760. doi: 10.1007/s00395-011-0192-x. [DOI] [PubMed] [Google Scholar]
- 156.Shen C.M., Mao S.J., Huang G.S., Yang P.C., Chu R.M. Stimulation of smooth muscle cell proliferation by ox-LDL- and acetyl LDL-induced macrophage-derived foam cells. Life Sci. 2001;70:443–452. doi: 10.1016/s0024-3205(01)01428-x. [DOI] [PubMed] [Google Scholar]
- 157.Crisby M., Kallin B., Thyberg J., Zhivotovsky B., Orrenius S., Kostulas V., Nilsson J. Cell death in human atherosclerotic plaques involves both oncosis and apoptosis. Atherosclerosis. 1997;130:17–27. doi: 10.1016/s0021-9150(96)06037-6. [DOI] [PubMed] [Google Scholar]
- 158.Grootaert M.O., Schrijvers D.M., Hermans M., Van Hoof V.O., De Meyer G.R., Martinet W. Caspase-3 deletion promotes necrosis in atherosclerotic plaques of ApoE knockout mice. Oxid. Med. Cell. Longevity. 2016;2016 doi: 10.1155/2016/3087469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lin J., Li H., Yang M., Ren J., Huang Z., Han F., Huang J., Ma J., Zhang D., Zhang Z. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013;3:200–210. doi: 10.1016/j.celrep.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 160.An S., Qi Y., Zhang Z., Mo R., Hou L., Yao X., Ge J. Antagonism of receptor interacting protein 1 using necrostatin-1 in oxidized LDL- induced endothelial injury. Biomed. Pharmacotherapy. 2018;108:1809–1815. doi: 10.1016/j.biopha.2018.09.052. [DOI] [PubMed] [Google Scholar]
- 161.Obulesu M., Lakshmi M.J. Apoptosis in alzheimer’s disease: an understanding of the physiology, pathology and therapeutic avenues. Neurochem. Res. 2014;39:2301–2312. doi: 10.1007/s11064-014-1454-4. [DOI] [PubMed] [Google Scholar]
- 162.Behl C. Apoptosis and alzheimer’s disease. J. Neural Transm. 2000;107:1325–1344. doi: 10.1007/s007020070021. [DOI] [PubMed] [Google Scholar]
- 163.Caccamo A., Branca C., Piras I.S., Ferreira E., Huentelman M.J., Liang W.S., Readhead B., Dudley J.T., Spangenberg E.E., Green K.N. Necroptosis activation in alzheimer’s disease. Nat. Neurosci. 2017;20:1236–1246. doi: 10.1038/nn.4608. [DOI] [PubMed] [Google Scholar]
- 164.Koper M.J., Schoor E.V., Ospitalieri S., Vandenberghe R., Thal D.R. Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in alzheimer’s disease. Acta. Neuropathol. 2020;139 doi: 10.1007/s00401-019-02103-y. [DOI] [PubMed] [Google Scholar]
- 165.Iannielli A., Bido S., Folladori L., Segnali A., Cancellieri C., Maresca A., Massimino L., Rubio A., Morabito G., Caporali L. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in parkinson’s disease models. Cell Rep. 2018;22:2066–2079. doi: 10.1016/j.celrep.2018.01.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Thomas C.N., Thompson A.M., Ahmed Z., Blanch R.J. Retinal ganglion cells die by necroptotic mechanisms in a site-specific manner in a rat blunt ocular injury model. Cells. 2019;8 doi: 10.3390/cells8121517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yang H., Fu Y., Liu X., Shahi P.K., Mavlyutov T.A., Li J., Yao A., Guo S.Z., Pattnaik B.R., Guo L.W. Role of the sigma-1 receptor chaperone in rod and cone photoreceptor degenerations in a mouse model of retinitis pigmentosa. Mol. Neurodegener. 2017;12:68. doi: 10.1186/s13024-017-0202-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chinskey N.D., Besirli C.G., Zacks D.N. Retinal cell death and current strategies in retinal neuroprotection. Curr. Opin. Ophthalmol. 2014;25:228–233. doi: 10.1097/icu.0000000000000043. [DOI] [PubMed] [Google Scholar]
- 169.Hanus J., Zhang H., Wang Z., Liu Q., Zhou Q., Wang S. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Disease. 2013;4:e965. doi: 10.1038/cddis.2013.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hanus J., Anderson C., Wang S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 2015;24:286–298. doi: 10.1016/j.arr.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Huang Z., Zhou T., Sun X., Zheng Y., Cheng B., Li M., Liu X., He C. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 2018;25:180–189. doi: 10.1038/cdd.2017.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Im E., Kazlauskas A. The role of cathepsins in ocular physiology and pathology. Exp. Eye Res. 2007;84:383–388. doi: 10.1016/j.exer.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 173.Rakoczy P.E., Zhang D., Robertson T., Barnett N.L., Papadimitriou J., Constable I.J., Lai C.M. Progressive age-related changes similar to age-related macular degeneration in a transgenic mouse model. Am. J. Pathol. 2002;161:1515–1524. doi: 10.1016/s0002-9440(10)64427-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhang D., Brankov M., Makhija M.T., Robertson T., Helmerhorst E., Papadimitriou J.M., Rakoczy P.E. Correlation between inactive cathepsin D expression and retinal changes in mcd2/mcd2 transgenic mice. Investigat. Ophthalmol. Visual Sci. 2005;46:3031–3038. doi: 10.1167/iovs.04-1510. [DOI] [PubMed] [Google Scholar]
- 175.Ogawa T., Boylan S.A., Oltjen S.L., Hjelmeland L.M. Changes in the spatial expression of genes with aging in the mouse RPE/choroid. Mol. Vision. 2005;11:380–386. [PubMed] [Google Scholar]
- 176.Alegre-Cortés E., Muriel-González A., Canales-Cortés S., Uribe-Carretero E., Martínez-Chacón G., Aiastui A., López de Munain A., Niso-Santano M., Gonzalez-Polo R.A., Fuentes J.M. Toxicity of necrostatin-1 in parkinson’s disease models. Antioxidants (Basel) 2020;9 doi: 10.3390/antiox9060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Arrázola M.S., Saquel C., Catalán R.J., Barrientos S.A., Hernandez D.E., Martínez N.W., Catenaccio A., Court F.A. Axonal degeneration Is mediated by necroptosis activation. J. Neurosci. 2019;39:3832–3844. doi: 10.1523/jneurosci.0881-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Qinli Z., Meiqing L., Xia J., Li X., Weili G., Xiuliang J., Junwei J., Hailan Y., Ce Z., Qiao N. Necrostatin-1 inhibits the degeneration of neural cells induced by aluminum exposure. Restor. Neurol. Neurosci. 2013;31:543–555. doi: 10.3233/rnn-120304. [DOI] [PubMed] [Google Scholar]
- 179.Rosentreter D., Funken D., Reifart J., Mende K., Rentsch M., Khandoga A. RIP1-dependent programmed necrosis is negatively regulated by caspases during hepatic ischemia-reperfusion. Shock (Augusta, Ga.) 2015;44:72–76. doi: 10.1097/shk.0000000000000371. [DOI] [PubMed] [Google Scholar]
- 180.Linkermann A., Heller J.O., Prokai A., Weinberg J.M., De Zen F., Himmerkus N., Szabo A.J., Brasen J.H., Kunzendorf U., Krautwald S. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J. Am. Soc. Nephrol. : JASN. 2013;24:1545–1557. doi: 10.1681/asn.2012121169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Teng X., Degterev A., Jagtap P., Xing X., Choi S., Denu R., Yuan J., Cuny G.D. Structure-activity relationship study of novel necroptosis inhibitors. Bioorg. Med. Chem. Lett. 2005;15:5039–5044. doi: 10.1016/j.bmcl.2005.07.077. [DOI] [PubMed] [Google Scholar]
- 182.Sun W., Wu X., Gao H., Yu J., Zhao W., Lu J.J., Wang J., Du G., Chen X. Cytosolic calcium mediates RIP1/RIP3 complex-dependent necroptosis through JNK activation and mitochondrial ROS production in human colon cancer cells. Free Rad. Biol. Med. 2017;108:433–444. doi: 10.1016/j.freeradbiomed.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 183.Ofengeim D., Ito Y., Najafov A., Zhang Y., Shan B., DeWitt J.P., Ye J., Zhang X., Chang A., Vakifahmetoglu-Norberg H. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015;10:1836–1849. doi: 10.1016/j.celrep.2015.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ye Q., Wang B., Mao J. The pathogenesis and treatment of the `Cytokine storm’ in COVID-19. J. Infect. 2020;80:607–613. doi: 10.1016/j.jinf.2020.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ragab D., Eldin H.S., Taeimah M., Khattab R., Salem R. The COVID-19 cytokine storm; What We know so Far. Front. Immunol. 2020;11:1446. doi: 10.3389/fimmu.2020.01446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yang X.H., Sun R.H., Chen D.C. Diagnosis and treatment of COVID-19: acute kidney injury cannot be ignored. Zhonghua Yi Xue Za Zhi. 2020;100:1205–1208. doi: 10.3760/cma.j.cn112137-20200229-00520. [DOI] [PubMed] [Google Scholar]
- 187.Tian D., Ye Q. Hepatic complications of COVID-19 and its treatment. J. Med. Virol. 2020 doi: 10.1002/jmv.26036. 10.1002/jmv.26036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zheng Y.Y., Ma Y.T., Zhang J.Y., Xie X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020;17:259–260. doi: 10.1038/s41569-020-0360-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Huang C., Wang Y., Li X., Ren L., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Deepa S.S., Unnikrishnan A., Matyi S., Hadad N., Richardson A. Necroptosis increases with age and is reduced by dietary restriction. Aging Cell. 2018;17 doi: 10.1111/acel.12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Conti P., Ronconi G., Caraffa A., Gallenga C.E., Ross R., Frydas I., Kritas S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents. 2020;34 doi: 10.23812/conti-e. [DOI] [PubMed] [Google Scholar]
- 192.Zhang C., Wu Z., Li J.W., Zhao H., Wang G.Q. Cytokine release syndrome in severe COVID-19: interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int. J. Antimicrob. Agents. 2020;55 doi: 10.1016/j.ijantimicag.2020.105954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zelic M., Roderick J.E., O’Donnell J.A., Lehman J., Lim S.E., Janardhan H.P., Trivedi C.M., Pasparakis M., Kelliher M.A. RIP kinase 1-dependent endothelial necroptosis underlies systemic inflammatory response syndrome. J. Clinic. Investigat. 2018;128:2064–2075. doi: 10.1172/JCI96147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rickard J.A., O’Donnell J.A., Evans J.M., Lalaoui N., Poh A.R., Rogers T., Vince J.E., Lawlor K.E., Ninnis R.L., Anderton H. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157:1175–1188. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 195.Duan P.Y., Ma Y., Li X.N., Qu F.Z., Ji L., Guo X.Y., Zhang W.J., Xiao F., Li L., Hu J.S. Inhibition of RIPK1-dependent regulated acinar cell necrosis provides protection against acute pancreatitis via the RIPK1/NF-kappaB/AQP8 pathway. Experiment. Molecul. Med. 2019;51:1–17. doi: 10.1038/s12276-019-0278-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wang X., Jiang W., Yan Y., Gong T., Han J., Tian Z., Zhou R. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat. Immunol. 2014;15:1126–1133. doi: 10.1038/ni.3015. [DOI] [PubMed] [Google Scholar]
- 197.Mocarski E.S., Guo H., Kaiser W.J. Necroptosis: the trojan horse in cell autonomous antiviral host defense. Virology. 2015;479-480:160–166. doi: 10.1016/j.virol.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Chattopadhyay S., Yamashita M., Zhang Y., Sen G.C. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J. Virol. 2011;85:3708–3716. doi: 10.1128/jvi.02133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mocarski E.S., Upton J.W., Kaiser W.J. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat. Rev. Immunol. 2011;12:79–88. doi: 10.1038/nri3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Koehler H., Cotsmire S., Langland J., Kibler K.V., Kalman D., Upton J.W., Mocarski E.S., Jacobs B.L. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc. Natl. Acad. Sci. U. S. A. 2017;114:11506–11511. doi: 10.1073/pnas.1700999114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shi F., Zhou M., Shang L., Du Q., Li Y., Xie L., Liu X., Tang M., Luo X., Fan J. EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics. 2019;9:2424–2438. doi: 10.7150/thno.30941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhang T., Yin C., Boyd D.F., Quarato G., Ingram J.P., Shubina M., Ragan K.B., Ishizuka T., Crawford J.C., Tummers B. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell. 2020;180:1115–1129. doi: 10.1016/j.cell.2020.02.050. e1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Diao B., Wang C., Tan Y., Chen X., Liu Y., Ning L., Chen L., Li M., Liu Y., Wang G. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19) Front. Immunol. 2020;(11):827. doi: 10.3389/fimmu.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Harris P.A., Berger S.B., Jeong J.U., Nagilla R., Bandyopadhyay D., Campobasso N., Capriotti C.A., Cox J.A., Dare L., Dong X. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J. Med. Chem. 2017;60:1247–1261. doi: 10.1021/acs.jmedchem.6b01751. [DOI] [PubMed] [Google Scholar]
- 205.Harris P.A., Marinis J.M., Lich J.D., Berger S.B., Chirala A., Cox J.A., Eidam P.M., Finger J.N., Gough P.J., Jeong J.U. Identification of a RIP1 kinase inhibitor clinical candidate (GSK3145095) for the treatment of pancreatic cancer. ACS Med. Chem. Lett. 2019;10:857–862. doi: 10.1021/acsmedchemlett.9b00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Rowland M., Riegelman S., Harris P.A., Sholkoff S.D. Absorption kinetics of aspirin in man following oral administration of an aqueous solution. J. Pharm. Sci. 1972;61:379–385. doi: 10.1002/jps.2600610312. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.