

Rhombencephalosynapsis is a rare cerebellar malformation of unknown cause. Mak et al. report the identification of a cluster of de novo truncating mutations in the MN1 gene in patients with a novel syndrome that comprises midface hypoplasia, severe language delay and distinctive brain malformations including an atypical form of rhombencephalosynapsis.
Keywords: MN1, MCTT syndrome, craniofacial development, intellectual disability, rhombencephalosynapsis
Abstract
MN1 encodes a transcriptional co-regulator without homology to other proteins, previously implicated in acute myeloid leukaemia and development of the palate. Large deletions encompassing MN1 have been reported in individuals with variable neurodevelopmental anomalies and non-specific facial features. We identified a cluster of de novo truncating mutations in MN1 in a cohort of 23 individuals with strikingly similar dysmorphic facial features, especially midface hypoplasia, and intellectual disability with severe expressive language delay. Imaging revealed an atypical form of rhombencephalosynapsis, a distinctive brain malformation characterized by partial or complete loss of the cerebellar vermis with fusion of the cerebellar hemispheres, in 8/10 individuals. Rhombencephalosynapsis has no previously known definitive genetic or environmental causes. Other frequent features included perisylvian polymicrogyria, abnormal posterior clinoid processes and persistent trigeminal artery. MN1 is encoded by only two exons. All mutations, including the recurrent variant p.Arg1295* observed in 8/21 probands, fall in the terminal exon or the extreme 3′ region of exon 1, and are therefore predicted to result in escape from nonsense-mediated mRNA decay. This was confirmed in fibroblasts from three individuals. We propose that the condition described here, MN1 C-terminal truncation (MCTT) syndrome, is not due to MN1 haploinsufficiency but rather is the result of dominantly acting C-terminally truncated MN1 protein. Our data show that MN1 plays a critical role in human craniofacial and brain development, and opens the door to understanding the biological mechanisms underlying rhombencephalosynapsis.
Introduction
The use of trio whole exome sequencing (WES) has dramatically increased our ability to provide a molecular diagnosis for individuals with sporadic developmental disorders (Deciphering Developmental Disorders Study, 2015). Recently, global data exchange platforms such as GeneMatcher have been created to facilitate the validation of suspected disease-causing variants, identified predominantly by WES, in rare disorders (Sobreira et al., 2015). Despite the success of these approaches, the genetic causes of many presumed Mendelian conditions remain elusive. Rhombencephalosynapsis (RES), a brain malformation characterized by total or partial absence of the cerebellar vermis with apparent fusion of the cerebellar hemispheres is one of these conditions (Ishak et al., 2012). Individuals with RES have prominent neurodevelopmental features including motor delays, abnormal eye movements, stereotypies, hyperactivity, executive dysfunction, and a broad range of cognitive ability that correlates somewhat with the degree of RES on imaging (Ishak et al., 2012; Tully et al., 2013). Syndromic associations with RES have been reported (Tully et al., 2012), including Gomez-Lopez-Hernandez syndrome (MIM 601853) and VACTERL association (MIM 192350); however, no definitive genetic causes of RES have been identified (Aldinger et al., 2018).
Here, we report the use of ‘genotype-first’ approaches for the identification of clustered C-terminal truncating mutations in MN1 proto-oncogene, transcriptional regulator (MN1; MIM 156100) as the cause of a highly recognizable neurodevelopmental disorder with distinctive craniofacial features. Although large chromosomal deletions of a region including this gene has been reported in patients with variable developmental delay, a distinct phenotype is evident in patients with C-terminal truncating mutations. Furthermore, systematic review of the brain imaging for individuals with MN1 C-terminal truncating mutations revealed a highly characteristic pattern of brain malformation, including a focal variant of RES, thus shedding light on a poorly understood brain malformation.
Materials and methods
Comparative genomic hybridization, and whole exome and genome sequencing
WES or whole genome sequencing (WGS) was performed for 25 individuals with undiagnosed, syndromic intellectual disability or developmental delay and dysmorphic facial features in 15 independent research or diagnostic laboratories. WES was performed using methods described previously as follows: Strauss et al. (2018) for Individual 1; Poirier et al. (2017) for Individual 3; Choi et al. (2009) for Individual 9; Lelieveld et al. (2016) for Individual 10; Farwell et al. (2015) for Individual 13; Deciphering Developmental Disorders Study, (2017) for Individuals 16 and 19; Hempel et al. (2015) for Individual 20; Van De Weghe et al. (2017) for Individual 21 and Louie et al. (2017) for Individual 24. For Individual 15, Medical EmExome sequencing was performed by EGL Genetics. Clinical WGS was performed for Individual 18 using 2 × 150 bp reads on an Illumina sequencer, with a mean coverage of 30× in the target region. For Individuals 4–7, 11, 12, 14, 17, 23 and 25, WES was performed by GeneDx, as follows. Using genomic DNA from the proband and parents, exonic regions and flanking splice junctions were captured using the Clinical Research Exome kit (Agilent Technologies), the SureSelect Human All Exon V4 (50 Mb) kit or the IDT xGen Exome Research Panel v1.0 and sequenced on an Illumina system with ≥100 bp paired-end reads. Reads were aligned to human genome build GRCh37/UCSC hg19, and analysed for sequence variants using a custom-developed analysis tool. Additional sequencing technology and variant interpretation protocols used by GeneDx have been previously described (Tanaka et al., 2015). The general assertion criteria for variant classification by GeneDx are publicly available on the GeneDx ClinVar submission page. WES for Individuals 8 and 22 was performed using Illumina sequencing followed by standard protocols for read mapping and variant calling. WES for Individual 26 was performed using the Agilent SureSelectXT Human all Exon v5 capture kit followed by sequencing on a NextSeq 500 (Illumina) and analysed using an in-house sequence analysis pipeline (Modular GATK-Based Variant Calling Pipeline, MAGPIE), with LOVDplus (Leiden Genome Technology Center, LUMC, Leiden) used for interpretation of variants. The deletion in Individual 27 was identified by array comparative genomic hybridization (CGH) using peripheral blood DNA on the Cytochip Oligo 4x180K v1.0 (Fa. Illumina/Bluegnome) and confirmed by qPCR with primers covering the deleted 22q region. The deletion in Individual 28 (Patient 7807 in Friedman et al., 2006, 2009) was identified using an Affymetrix 100K CGH platform (Friedman et al., 2006) and the breakpoints refined on an Affymetrix 500K platform (Friedman et al., 2009). Genetic research was performed according to approved institutional ethical guidelines and consent was obtained from all families.
cDNA sequencing and real-time PCR
For Individual 2, RNA was extracted from cultured fibroblasts using an RNeasy® Mini Kit (Qiagen) and cDNA was generated with a Verso™ cDNA Synthesis Kit (Thermo Scientific). For Individual 10, cDNA was generated from TRIzol®-extracted fibroblast RNA using the SuperScript™ III First-Strand Synthesis System (Invitrogen). MN1 cDNA was amplified by PCR using a sense primer in exon 1 and an antisense primer in exon 2 followed by Sanger sequencing. Real-time PCR for Individual 10 was performed in a 7900HT Fast Real-Time PCR System with Fast 96-Well Block Module, using PrimeTime qPCR primer and probe-based assays (Integrated DNA Technologies), with wild-type and mutant c.3870_3879dup MN1 probes specific to each sequence. See Supplementary Table 2 for primer and probe sequences.
RNA sequencing
For RNA-Seq of skin fibroblasts from Individual 10, libraries were constructed using Illumina TruSeq RNA sample Prep Kit v2, and sequenced on an Illumina Hiseq 2500. Trimmed FASTQ files were aligned to the NCBI RefSeq human reference genome (GRCh37/hg19) and human transcriptome by Strand NGS software (Strand Life Sciences Private Limited) based on the COBWEB aligner with a gap percentage of 15 bp. Variants with a minimum base quality and mapping quality of 20 were then called by Strand NGS software to obtain all variants in the MN1 gene. For RNA-Seq of fibroblasts from Individual 21, total RNA was extracted using TRIzol® and purified with an RNA Clean & Concentrator-5 kit (Zymo Research). Libraries were prepared using a TruSeq Stranded mRNA kit and sequenced on an Illumina NovaSeq instrument, yielding 100 bp paired-end reads. Trimmed fastq files were processed according to GATK’s RNA-Seq best practices and aligned to the human reference genome (GRCh37/hg19) using the two-pass method of STAR (v2.5).
Data availability
Raw data are available upon reasonable request.
Results
Identification of C-terminal truncating variants in MN1
Twenty-two probands sequenced by WES or WGS (Individuals 1, 3, 4, and 8–26) harboured de novo predicted protein-truncating (stop or frameshift) variants in MN1 (Fig. 1, Supplementary Fig. 1 and Supplementary Table 1). Because of strong clinical similarities noted between Individual 2 and other probands in the cohort, we performed Sanger sequencing of MN1 in Individual 2, and identified the variant p.(Glu1249*), which was absent in the father (the mother was unavailable for testing). Individual 7 is the mildly affected father of affected siblings (Individuals 5 and 6). The mutation p.(Gln1273*) was present in all three by WES, but with wild-type:mutant read imbalance in the father (137:87), and Sanger sequencing of blood DNA further supported somatic mosaicism of the variant in the father (Supplementary Fig. 2). The MN1 gene comprises two exons and encodes a protein of 1320 amino acids. The large first exon codes for amino acids 1–1260 and the second encodes the remaining 60 C-terminal amino acids. All truncating variants in Individuals 1–23 are located within the second exon or at the extreme 3′ end of exon 1 (Fig. 1). Individuals 24–26 harboured de novo truncating mutations in a much more N-terminal region of MN1 (Fig. 1), and did not display dysmorphic facial features typical of those patients with C-terminal truncations (see below). None of the above MN1 variants have been reported in the Genome Aggregation Database (gnomAD; data accessed May 2019). Given that all the variants in Individuals 1–23 create premature stop codons in the final exon or the final 37 nucleotides of exon 1, all are predicted to result in MN1 transcripts that escape nonsense-mediated mRNA decay, given the rule that premature stop codons more than 50–55 nucleotides upstream of the final exon-exon junction trigger nonsense-mediated mRNA decay (Nagy and Maquat, 1998). cDNA sequencing, real-time PCR and/or RNA-Seq demonstrated expression of the mutant transcript at levels similar to that of wild-type in cultured fibroblasts from Individual 2 (harbouring a premature stop codon within the 3′ terminus of exon 1) and Individuals 10 and 21 (harbouring premature stops in exon 2) (Supplementary Figs 3 and 4). These results suggest that the MN1 variants identified in Individuals 1–23 may lead to the expression of C-terminally truncated protein with pathogenic effect. Unique phenotypes associated with C-terminal truncating mutations that escape nonsense-mediated mRNA decay have similarly been reported for several genes (White et al., 2016; Jansen et al., 2017). Strikingly, 8 of 21 probands with C-terminal truncations harboured an identical MN1 variant, p.(Arg1295*), suggesting a mutational hotspot (Fig. 1). This bias may at least in part be due to underlying nucleotide composition; p.(Arg1295*) is generated from a C to T transition at a CpG dinucleotide. This mutational signature is overrepresented among human germline mutations (Acuna-Hidalgo et al., 2016), and there are no other CpGs with potential to generate a stop codon by C to T transition in exon 2 or in the final 55 nucleotides of exon 1 of MN1.
Figure 1.

MN1 mutations and sequence conservation. (A) Distribution of C-terminal truncating mutations identified in MN1 in 21 probands (red arrows). Beneath the schema of the gene, black arrows indicate the positions of loss-of-function variants in the gnomAD database and p.(Ser179*), p.(Pro365Thrfs*120) and p.(Ser472*) identified in Individuals 24, 25 and 26, respectively. (B) MN1 C-terminal sequence conservation. Multi-species sequence alignment of the entire exon 2 coding region of MN1. Sequences were obtained from the following RefSeq or Ensembl transcripts: human, NM_002430.2; mouse, NM_001081235.1; Xenopus, NM_001100202.1; zebrafish, ENSDART00000129197. Red arrows indicate the positions of the mutations identified in exon 2.
Clinical features in individuals with MN1 C-terminal truncating variants
The major clinical features and their frequencies for individuals with C-terminal truncations in MN1 (Individuals 1–6 and 8–23; Individual 7 was not included in the phenotype frequency analysis because of mosaicism of the mutation) are summarized in Table 1 and all available clinical details are provided in Supplementary Table 1. While complete clinical data were not available for all individuals, neurodevelopmental deficits were frequent, with intellectual disability present in 16/17 patients and delayed or absent expressive speech in 18/20. Gross motor development was delayed in 19/20 cases. Hypotonia was frequent (n = 17/18) and feeding difficulties occurred in 12/18. Non-standardized photographs were reviewed for all except Individuals 1, 8, 9 and 15, revealing a distinctive facial gestalt, characterized by midface hypoplasia (n = 21/22), hypertelorism (n = 19/21), downslanting palpebral fissures (n = 15/21) and a short, upturned nose especially in infancy (n = 21/22) (Fig. 2). Skull shape anomalies were frequent (n = 16/21), and consisted of brachycephaly, plagiocephaly, turricephaly, dolicocephaly and/or bi-temporal narrowing, with craniosynostosis documented in three individuals (Fig. 3). Mild exorbitism, suggesting shallow orbits, was apparent in the majority of patients, and there was a trend for frontal bossing in infancy and a tall forehead at later ages. A high-arched palate occurred in 15/21 patients (Fig. 2). Only one patient had a cleft palate (submucous), which was associated with a bifid uvula. Ear anomalies were present in 22/22 patients and included low-set, posteriorly-rotated and/or small ears, with the upper portion of the pinna frequently dysplastic (Supplementary Fig. 5). Hearing loss, conductive or sensorineural, occurred in 16/20 individuals. Anomalies occurring in a minority of the 22 patients included: oculomotor defects in nine (including Duane anomaly, nystagmus and strabismus); spinal anomalies, either clinical (lordosis, scoliosis, kyphosis) or radiographic, in eight; atrial or ventricular septal defects in six; seizures in six; and congenital diaphragmatic hernia in two. Growth parameters tended to remain within the normal range. Individual 7 carrying a mosaic MN1 mutation did not have major neurodevelopmental or craniofacial anomalies other than mildly dysplastic ears and a high palate. The identification of a truncating variant in MN1 in Individual 2, who was tested because of clinical similarities with other individuals in the series, demonstrates the recognizability of this disorder. A computational composite created through the Face2Gene application (Gurovich et al., 2019) using multiple patient photos highlights the facial gestalt (Supplementary Fig. 6). We name this condition MN1 C-terminal truncation (MCTT) syndrome.
Table 1.
Frequency of major clinical findings associated with C-terminal truncations of MN1
| Phenotype | Affected individuals, n (%) |
|---|---|
| Hypertelorism | 19/21 (90) |
| Downslanting palpebral fissures | 15/21 (71) |
| Midface hypoplasia | 21/22 (95) |
| Cranial shape defects (plagiocephaly, brachycephaly, turricephaly, dolicocephaly, bitemporal narrowing) | 16/21 (76) |
| Short, upturned nose | 21/22 (95) |
| High-arched palate | 15/21 (71) |
| Dysplastic ears | 22/22 (100) |
| Hearing loss (conductive or sensorineural) | 16/20 (80) |
| Speech delay | 18/20 (90) |
| Intellectual disability | 16/17 (94) |
| Motor delay | 19/20 (95) |
| Feeding difficulties | 12/18 (67) |
| Hypotonia | 17/18 (94) |
Note that only Individuals 1–6 and 8–23 were considered for the frequencies listed (Individual 7 was excluded because mosaicism of the MN1 mutation).
Figure 2.

Facial features of individuals with C-terminal truncating mutations in MN1. Oral view for Individuals 18 and 22 indicates high and narrow palate. Individual identification numbers are indicated at the top left of each panel.
Figure 3.
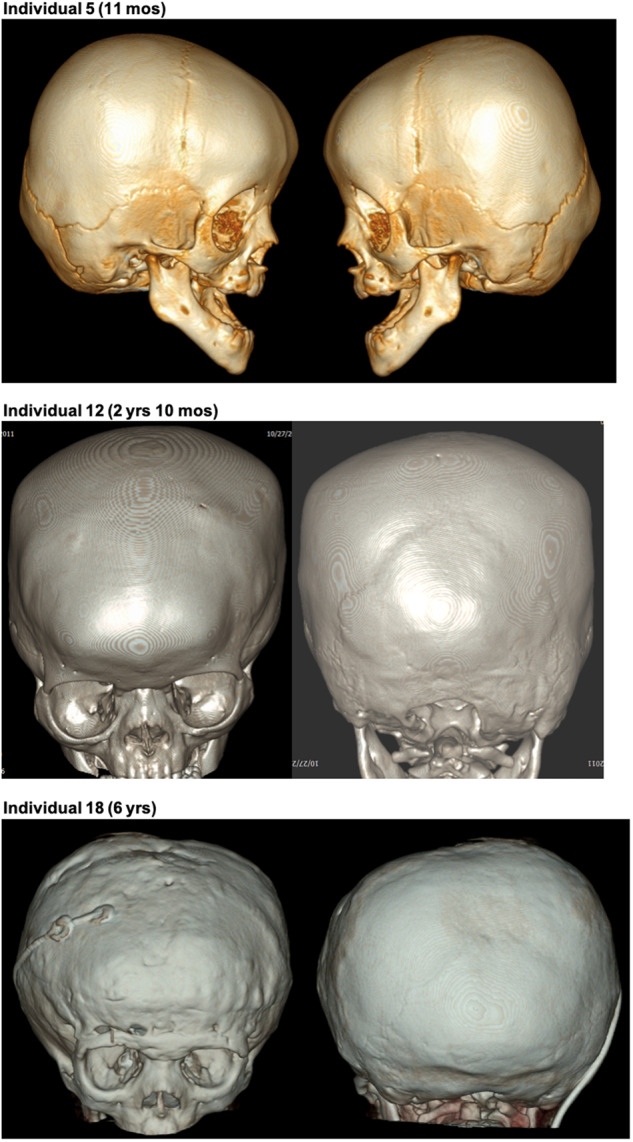
3D CT scans indicating craniosynostosis in individuals with C-terminal truncating mutations in MN1. For Individual 5, images show bilateral partial craniosynostosis of squamosal, frontosphenoid and coronal sutures. For Individuals 12 and 18, images show closure of the metopic, coronal and sagittal sutures and partial closure of lambdoid sutures. The images of Individual 18 are subsequent to fronto-orbital advancement surgery and placement of a ventriculoperitoneal shunt.
Brain imaging features in individuals with MN1 C-terminal truncating variants
Initial clinical interpretation of brain MRIs from MCTT syndrome patients identified varied anomalies in several individuals, including polymicrogyria, dysmorphic corpus callosum and anomalies of the cerebellum. Prior to detection of the de novo variant in MN1, Individual 21 had a long-standing diagnosis of RES. This prompted detailed review of the brain imaging for 11 individuals in the cohort by D.D., G.I. and W.B.D., revealing a highly characteristic pattern of brain malformation (Table 2). Eight of ten patients had focal crossing of the cerebellar folia with loss of vermis morphology consistent with a partial form of RES (Fig. 4). The most extensive involvement resembles more classic partial RES but without fusion of the central white matter or complete loss of vermis landmarks on sagittal view. In addition, 9/10 individuals had polymicrogyria of the insula, which sometimes extended more broadly in the perisylvian region. Similar to other individuals with RES, 3/11 had ventriculomegaly (one shunted), and 4/8 had hypoplastic olfactory bulbs. In contrast to other individuals with RES, 6/11 MCTT patients had above average thickness of the rostral corpus callosum and none had absent septum, although one had a cavum velum interpositum (Fig. 4C, Individual 14). We also noted a persistent medial primitive trigeminal artery in 7/10 (six unilateral, one bilateral), often associated with prominent posterior clinoid processes that sometimes were fused across the midline (Figs 4 and 5). Persistent trigeminal artery is an embryonic connection between the anterior and posterior cerebral circulation that has been detected in 0.1–0.6% of individuals undergoing vascular imaging (O’uchi and O’uchi, 2010), so the 70% prevalence in association with MN1 variants is far greater than expected by chance. The pituitary did not appear abnormal in any of the patients.
Table 2.
Brain imaging features in individuals with C-terminal truncating MN1 variants
| Feature | Prevalence |
|---|---|
| RES | 8/10 |
| Cerebellar dysplasia (other than RES) | 4/10 |
| Perisylvian polymicrogyria | 9/10 |
| Subcortical heterotopia | 2/11 |
| Ventriculomegaly | 3/11 |
| Thick rostral corpus callosum | 6/11 |
| Hypoplastic olfactory bulbs | 4/8 |
| Persistent trigeminal artery | 7/10 |
| Prominent posterior clinoid process | 7/10 |
| Hypertelorism | 10/11 |
The frequencies are based uniquely on MRIs that were reviewed as a series at one centre.
Figure 4.

Brain findings in patients with C-terminal truncating MN1 variants. (A) Inferior cerebellum (axial view): foliar dysplasia with indistinct vermis and abnormal folia crossing the midline (Individuals 2, 14, 17, 20, 21); normal inferior cerebellar anatomy (Individuals 24 and 28). (B) Superior cerebellum (axial view): small (Individuals 17, 20 and 21) or almost absent (Individuals 2 and 14) vermis with abnormal folia crossing the midline especially ventrally; normal superior cerebellar anatomy with intact vermis (Individuals 24 and 28). Arrow in Individual 17 indicates persistent trigeminal artery. (C) Insula (axial view): polymicrogyria interior to yellow bars (Individuals 14, 17, 20 and 21), normal appearance (Individuals 2, 24 and 28). Note that Individual 14 has a cavum velum interpositum (asterisk) and all individuals with C-terminal truncating variants have unusual head shape with bitemporal narrowing. (D) Midline (sagittal view): Tall, flat forehead and thickened rostral corpus callosum (Individuals 2, 14, 17, 20 and 21), abnormal vermis lobulation with indistinct primary and horizontal fissures (Individuals 2, 14, 17, 20 and 21), persistent trigeminal artery (arrow in Individual 14) and prominent posterior clinoid process (arrowheads in Individuals 17 and 20).
Figure 5.
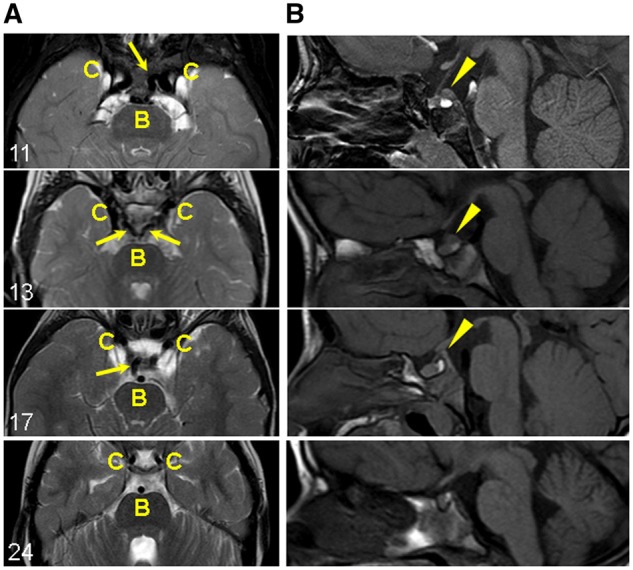
Persistent trigeminal artery and prominent posterior clinoid process in patients with C-terminal truncating MN1 variants. (A) Carotid and basilar arteries (axial view): Persistent trigeminal artery flow-voids (dark signal) connecting the carotid (C) and basilar (B) artery flow-voids (arrows). The persistent trigeminal arteries are unilateral in Individuals 11 and 17, and bilateral in Individual 13. Individual 24 with an early truncating variant does not have persistent trigeminal arteries (shown for comparison). (B) Prominent posterior clinoid process (sagittal view): abnormal tissue just superior to the posterior pituitary bright spot and continuous with the posterior clinoid process (arrowheads). Individual 24 without the abnormal tissue is shown for comparison.
Impact of MN1 N-terminal truncating variants and whole gene deletions
The clustering of de novo truncating mutations at the C-terminus of MN1, associated with a specific phenotype, is suggestive of a pathogenic effect of the mutations that is not equivalent to haploinsufficiency. We investigated individuals with other types of lesions at the MN1 locus in order to provide support for this argument, and thereby to establish specific genotype-phenotype relationships. In gnomAD, which is composed of variants identified in individuals unlikely to have severe paediatric disease, five loss-of-function MN1 variants have been reported (each with an allele count of one; data accessed May 2019), all of which are positioned >55 bp upstream of the 3′ end of exon 1 (Fig. 1), suggesting all would provoke nonsense-mediated mRNA decay, in contrast to the more C-terminal variants. In addition to the C-terminal truncating variants in MCTT patients, we also identified, through trio WES, three de novo truncating variants in the N-terminal third of MN1, in patients with a phenotype partly overlapping, but distinct from that of MCTT syndrome: p.(Ser179*) in Individual 24 (with dyspraxia, mild conductive hearing loss, facial asymmetry and submucous cleft palate); p.(Pro365Thrfs*120) in Individual 25 (with social interaction issues, speech delay, mild conductive hearing loss and a prominent nose); and p.(Ser472*) in Individual 26 (with speech delay, mild conductive hearing loss and non-specific facial features) (Figs 1 and 6 and Supplementary Table 1). These three variants are also predicted to induce nonsense-mediated mRNA decay. Although all three patients had conductive hearing loss and a range of speech defects, none had significant intellectual disability or a facial gestalt reminiscent of MCTT syndrome.
We also report here the first known microdeletion harbouring MN1 but no other genes (130 kb at chr22:287 1057 465–287 2357 360), which occurred de novo in Individual 27 (with microcephaly, intellectual disability, speech and motor delays and mildly dysmorphic facial features) (Fig. 6 and Supplementary Table 1). In the DECIPHER (Firth et al., 2009) database and the literature, large deletions containing MN1 and other genes have been described in individuals with variable neurodevelopmental and facial anomalies. The most frequent findings reported in these individuals are cleft or high-arched palate, micro- and/or retrognathia, hypertelorism, either depressed or prominent nasal bridge, low set and/or dysplastic ears, hypoplastic corpus callosum, mild to moderate developmental delay and intellectual disability including delayed speech (we include here the cases described in: Bruder et al., 2001; Barbi et al., 2002; Said et al., 2011; Davidson et al., 2012; Beck et al., 2015; Breckpot et al., 2016) and DECIPHER Individuals 999, 4110, 290785, 294749 and 331398, for a total of 19 individuals). Although some of these features are observed in MCTT syndrome, the majority (16/19) of these individuals have deletions >2 Mb and that contain at least 10 MIM genes, making it difficult to judge the contribution of MN1 haploinsufficiency to the phenotype. For one of the smallest previously reported deletions harbouring MN1, DECIPHER 999 (Individual 28 here; see Supplementary Table 1 for clinical details), facial features are shown in Fig. 6 (photographs have not been previously published). Based on photographs of Individuals 27 and 28 and those available in the literature, individuals with deletions of MN1 do not exhibit a consistent facial gestalt, and they do not resemble individuals with MCTT syndrome. Furthermore, review of brain imaging for Individuals 24, 27 and 28 did not reveal RES or the other findings frequently observed in patients with C-terminal truncating variants (Figs 4 and 5). Collectively, the above data suggest that haploinsufficiency for MN1 may lead to neurodevelopmental anomalies, palatal defects and facial dysmorphisms with variable expressivity and penetrance but without significant similarity to the craniofacial gestalt or brain malformations of MCTT syndrome. We speculate that these differences are due to the stable expression of C-terminally truncated MN1 protein in the latter, which may act in a dominant negative or gain-of-function manner.
Figure 6.

Facial features of individuals with N-terminal truncating mutations in MN1 or whole deletions of MN1. Individual identification numbers are indicated at the top left of each panel.
Discussion
MN1: a poorly characterized gene implicated in craniofacial skeletal development
MN1 was initially named because of its disruption by a balanced translocation in a meningioma (Lekanne Deprez et al., 1995), and at the same time was identified in translocations in myeloproliferative disorders (Buijs et al., 1995), resulting in fusions with TEL, encoding an ETS family transcription factor (now known as ETV6; MIM 600618). While the relevance of the initial association with meningioma has remained unclear, MN1 has been strongly associated with leukaemia. Overexpression of wild-type MN1 in mice results in acute myeloid leukaemia (AML) or myeloproliferative disease (Carella et al., 2007; Heuser et al., 2007) and elevated MN1 expression is a marker of poor prognosis of AML in humans (Heuser et al., 2006). Note that none of the patients reported here have cancer. MN1 contains no recognized protein domains or homology to other proteins, making predictions of its molecular functions difficult. It localizes to the nucleus (Buijs et al., 2000) and contains proline-rich regions and poly-glutamine tracts. In vitro reporter assays have shown that MN1 can act as a transcriptional activator, synergizing in some contexts with retinoic acid receptors, nuclear receptor coactivators or p300 (Buijs et al., 2000; van Wely et al., 2003; Sutton et al., 2005; Liu et al., 2008; Zhang et al., 2009), but MN1 is not thought to directly bind DNA (van Wely et al., 2003; Meester-Smoor et al., 2007). Further evidence for a role in transcriptional regulation comes from ChIP-Seq (chromatin immunoprecipitation-sequencing) studies that identified co-occupancy of MN1 and the HOX co-factor MEIS1 (MIM 601739) at chromatin regions containing putative direct target genes of both factors in leukaemic cells (Heuser et al., 2011).
Structure-function studies have highlighted several broad regions of MN1, in either the N- or C-terminal halves of the protein, involved in its ability to regulate haematopoietic cell fate and leukaemogenesis (Kandilci et al., 2013; Lai et al., 2014); however, to the best of our knowledge, no specific activity has been attributed to the 60 C-terminal amino acids encoded by exon 2. Interestingly, within exon 2, the final 20 amino acids (which are fully or partially deleted in all individuals with MCTT syndrome) are particularly highly conserved across vertebrate species (Fig. 1B). A key area for future studies will involve investigation of the molecular function of this highly conserved C-terminus of MN1.
During embryonic development of animal models, MN1 is expressed in restricted domains relevant to the tissues affected in MCTT syndrome. The zebrafish orthologue mn1b is expressed in the neural plate (presumptive central nervous system) during gastrulation and subsequently is strongly expressed in the midbrain and hindbrain during segmentation stages followed by additional expression in the telencephalon and pharyngeal arches (ZFIN database). In mouse embryos between embryonic day (E)9.5 and 12.5, the major sites of Mn1 expression are the frontonasal prominences, pharyngeal arches and brain, especially the ventricular zone (Liu et al., 2008). At E14.5, strongest expression occurs in the mantle zone of the forebrain, with expression also observed in other regions of the brain (lateral midbrain, cerebellum, pons), head mesenchyme, the palatal shelf and the developing digits and axial skeleton (Eurexpress database) (Diez-Roux et al., 2011). Homozygous Mn1-null mice die shortly after birth due to clefting of the secondary palate (Meester-Smoor et al., 2005; Liu et al., 2008). These mice also display agenesis or hypoplasia of several bones of the ventral and lateral walls of the skull and thinning of bones of the skull roof, but no axial or appendicular skeletal defects nor anomalies of internal organs (Meester-Smoor et al., 2005). In a genome-wide study of single nucleotide polymorphisms associated with variation in skull and mandible shape in mice, Mn1 was contained in the locus with the largest effect size (Pallares et al., 2015). Also, during evolution the MN1 gene is suspected to have arisen at the base of the bony vertebrates (Pallares et al., 2015). Mn1 expression increases during differentiation of primary osteoblasts, and Mn1 is necessary for the proliferation, differentiation and several functions of this cell type (Zhang et al., 2009). Collectively these data suggest that MN1 plays an essential and conserved role in development of the bones of the skull. Although the molecular function of the MN1 alleles identified in MCTT syndrome remains unclear (see below), the above studies are consistent with the finding that alterations in skull and facial shape are a constant feature of MCTT syndrome.
Mutations in MN1 cause partial rhombencephalosynapsis
In addition to distinctive craniofacial features, several MCTT syndrome patients have a focal variant of RES, usually involving less than half of the vermis. Interestingly, previously reported individuals with RES can also present craniofacial features such as high forehead, flat midface and low-set ears (Tully et al., 2012), which are frequent features in patients with MCTT syndrome. No consistent causes of RES have been reported and no animal models exist. Possible RES has been reported in a single individual with a de novo deleterious CHAMP1 variant (Hempel et al., 2015), in one of two siblings with a frameshift mutation in ZIC1 (Vandervore et al., 2018), in two siblings with a 7 bp ZIC2 deletion (Ramocki et al., 2011), and in a foetus with a de novo missense variant in ADGRL2 (Vezain et al., 2018); however, the published imaging is inadequate to unambiguously establish the RES diagnosis in the above cases. In addition, we did not identify likely pathogenic variants in these genes in exome data from 59 individuals with RES (Aldinger et al., 2018) and no additional families have been published, so CHAMP1, ZIC1, ZIC2 and ADGRL2 are unlikely to be strongly associated with RES. Therefore, identifying a role for MN1 in the pathogenesis of RES is a major advance in our understanding of this malformation. Several theories for the developmental basis of RES have been proposed, particularly perturbed dorsal midline patterning due to the association of RES with holoprosencephaly and other midline brain defects in some patients (Ishak et al., 2012). Unfortunately, MN1 does not have a known role in these processes, so further understanding of RES will require additional functional work in human and animal model systems.
Clinical implications
The diagnosis of MCTT syndrome has important clinical implications. It is caused by de novo variants, making the recurrence risk low. Based on the 22 individuals with germline C-terminal mutations in our cohort, the range of neurodevelopmental outcome is broad, but all individuals require educational and therapy support, particularly for communication, and are not expected to be fully independent in adulthood. MCTT syndrome individuals are at risk for craniosynostosis, which may result in increased intracranial pressure. In addition, they have a high frequency of persistent trigeminal artery, which can affect surgical approaches to the skull base and pituitary. Finally, our comparison of the MCTT syndrome phenotype with that of patients harbouring more N-terminal truncating variants indicates that the latter group are likely to have less severe neurodevelopmental outcomes.
Conclusion
In summary, we have identified de novo C-terminal truncating mutations in MN1 as the cause of a severe and recognizable syndrome involving neurodevelopmental deficits and dysmorphic craniofacial features that is distinct from the variable and incompletely penetrant phenotypes associated with haploinsufficiency of this gene. Our findings will improve genetic counselling options for families of patients with MN1 variants and will motivate further basic research into the molecular roles of MN1, in particular regarding the function of its highly conserved C-terminus, and into the poorly defined developmental roles of MN1, especially in the context of CNS and craniofacial skeletal development. Importantly, our work opens the door to understanding the mechanisms underlying RES, a poorly understood cerebellar malformation.
Supplementary Material
Acknowledgements
We are extremely grateful to the families for their participation. We thank David Sweetser, Claire Redin and Michael E. Talkowski for initial discussions, and Shelin Adam who provided genetic counselling and coordinated J.M.F.'s research into pathogenic genomic variants.
Funding
This work was supported by grants from the Université Sorbonne Paris-Cité Pôle de recherche et d'enseignement supérieur (project number SPC/JFG/2013-031), the Agence Nationale de la Recherche [CranioRespiro project and ‘Investissements d’avenir’ program (ANR-10-IAHU-01)], MSDAvenir (DevoDecode project), E-Rare (CRANIRARE project), The Society for the Relief of Disabled Children, Medix Medical Services Asia, the Nachwuchskommission of the Charité Berlin (Rahel-Hirsch scholarship) to N.E., the German Research Foundation (DFG; SFB1315 to A.M.K. and LE 4223/1 to D.L.), the NIH Eunice Kennedy Shriver National Institute of Child Health and Human Development (U54HD083091, Genetics Core) to D.D., the NIH National Institute of Neurological Diseases and Stroke (R01NS050375) to W.B.D., the NIH National Human Genome Research Institute (HG009599) to J.T.S., the NIH National Human Genome Research Institute and the NIH National Heart, Lung and Blood Institute (grants UM1 HG006493 and U24 HG008956) to M.J.B. and D.A.N. (for sequencing provided by the University of Washington Center for Mendelian Genomics) and by private donations from families to D.D. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. F.R.Z. was supported by a Canadian Institute of Health Research fellowship and W.T.G. was supported by the BC Children’s Hospital Research Institute through its intramural IGAP Clinician Scientist Award program. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003). This study makes use of DECIPHER (http://decipher.sanger.ac.uk), which is funded by the Wellcome. See Deciphering Developmental Disorders Study (2015) or www.ddduk.org/access.html for full acknowledgement.
Competing interests
M.T.C, S.Y., F.M., I.M.W. and L.B.H. are employees of GeneDx, Inc. C.G-J. is a full-time employee of the Regeneron Genetics Center and receives stock options as part of compensation.
Glossary
Abbreviations
- MCTT =
MN1 C-terminal truncation
- RES =
rhombencephalosynapsis
- WES =
whole exome sequencing
References
- Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol 2016; 17: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldinger KA, Dempsey JC, Tully HM,, Grout ME, Mehaffey MG, Dobyns WB, et al. Rhombencephalosynapsis: Fused cerebellum, confused geneticists. Am J Med Genet C Semin Med Genet 2018; 178: 432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbi G, Rossier E, Vossbeck S, Hummler H, Lang D, Flock F, et al. Constitutional de novo interstitial deletion of 8 Mb on chromosome 22q12.1-12.3 encompassing the neurofibromatosis type 2 (NF2) locus in a dysmorphic girl with severe malformations. J Med Genet 2002; 39: E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Peterson JF, McConnell J, McGuire M, Asato M, Losee JE, et al. Craniofacial abnormalities and developmental delay in two families with overlapping 22q12.1 microdeletions involving the MN1 gene. Am J Med Genet A 2015; 167A: 1047–53. [DOI] [PubMed] [Google Scholar]
- Breckpot J, Anderlid B-M, Alanay Y, Blyth M, Brahimi A, Duban-Bedu B, et al. Chromosome 22q12.1 microdeletions: confirmation of the MN1 gene as a candidate gene for cleft palate. Eur J Hum Genet 2016; 24: 51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruder CE, Hirvelä C, Tapia-Paez I, Fransson I, Segraves R, Hamilton G, et al. High resolution deletion analysis of constitutional DNA from neurofibromatosis type 2 (NF2) patients using microarray-CGH. Hum Mol Genet 2001; 10: 271–82. [DOI] [PubMed] [Google Scholar]
- Buijs A, Sherr S, van Baal S, van Bezouw S, van der Plas D, Geurts van Kessel A, et al. Translocation (12;22) (p13;q11) in myeloproliferative disorders results in fusion of the ETS-like TEL gene on 12p13 to the MN1 gene on 22q11. Oncogene 1995; 10: 1511–9. [PubMed] [Google Scholar]
- Buijs A, van Rompaey L, Molijn AC, Davis JN, Vertegaal AC, Potter MD, et al. The MN1-TEL fusion protein, encoded by the translocation (12;22)(p13;q11) in myeloid leukemia, is a transcription factor with transforming activity. Mol Cell Biol 2000; 20: 9281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carella C, Bonten J, Sirma S, Kranenburg TA, Terranova S, Klein-Geltink R, et al. MN1 overexpression is an important step in the development of inv(16) AML. Leukemia 2007; 21: 1679–90. [DOI] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA 2009; 106: 19096–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson TB, Sanchez-Lara PA, Randolph LM, Krieger MD, Wu S-Q, Panigrahy A, et al. Microdeletion del(22)(q12.2) encompassing the facial development-associated gene, MN1 (meningioma 1) in a child with Pierre-Robin sequence (including cleft palate) and neurofibromatosis 2 (NF2): a case report and review of the literature. BMC Med Genet 2012; 13: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015; 519: 223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017; 542: 433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, et al. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol 2011; 9: e1000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 2015; 17: 578–86. [DOI] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet 2009; 84: 524–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Adam S, Arbour L, Armstrong L, Baross A, Birch P, et al. Detection of pathogenic copy number variants in children with idiopathic intellectual disability using 500 K SNP array genomic hybridization. BMC Genomics 2009; 10: 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JM, Baross A, Delaney AD, Ally A, Arbour L, Armstrong L, et al. Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am J Hum Genet 2006; 79: 500–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurovich Y, Hanani Y, Bar O, Nadav G, Fleischer N, Gelbman D, et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med 2019; 25: 60–4. [DOI] [PubMed] [Google Scholar]
- Hempel M, Cremer K, Ockeloen CW, Lichtenbelt KD, Herkert JC, Denecke J, et al. De Novo mutations in CHAMP1 cause intellectual disability with severe speech impairment. Am J Hum Genet 2015; 97: 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser M, Argiropoulos B, Kuchenbauer F, Yung E, Piper J, Fung S, et al. MN1 overexpression induces acute myeloid leukemia in mice and predicts ATRA resistance in patients with AML. Blood 2007; 110: 1639–47. [DOI] [PubMed] [Google Scholar]
- Heuser M, Beutel G, Krauter J, Döhner K, von Neuhoff N, Schlegelberger B, et al. High meningioma 1 (MN1) expression as a predictor for poor outcome in acute myeloid leukemia with normal cytogenetics. Blood 2006; 108: 3898–905. [DOI] [PubMed] [Google Scholar]
- Heuser M, Yun H, Berg T, Yung E, Argiropoulos B, Kuchenbauer F, et al. Cell of origin in AML: susceptibility to MN1-induced transformation is regulated by the MEIS1/AbdB-like HOX protein complex. Cancer Cell 2011; 20: 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishak GE, Dempsey JC, Shaw DWW, Tully H, Adam MP, Sanchez-Lara PA, et al. Rhombencephalosynapsis: a hindbrain malformation associated with incomplete separation of midbrain and forebrain, hydrocephalus and a broad spectrum of severity. Brain 2012; 135: 1370–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen S, Geuer S, Pfundt R, Brough R, Ghongane P, Herkert JC, et al. De Novo truncating mutations in the last and penultimate exons of PPM1D cause an intellectual disability syndrome. Am J Hum Genet 2017; 100: 650–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandilci A, Surtel J, Janke L, Neale G, Terranova S, Grosveld GC. Mapping of MN1 sequences necessary for myeloid transformation. PLoS ONE 2013; 8: e61706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CK, Moon Y, Kuchenbauer F, Starzcynowski DT, Argiropoulos B, Yung E, et al. Cell fate decisions in malignant hematopoiesis: leukemia phenotype is determined by distinct functional domains of the MN1 oncogene. PLoS ONE 2014; 9: e112671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekanne Deprez RH, Riegman PH, Groen NA, Warringa UL, van Biezen NA, Molijn AC, et al. Cloning and characterization of MN1, a gene from chromosome 22q11, which is disrupted by a balanced translocation in a meningioma. Oncogene 1995; 10: 1521–8. [PubMed] [Google Scholar]
- Lelieveld SH, Reijnders MRF, Pfundt R, Yntema HG, Kamsteeg E-J, de Vries P, et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci 2016; 19: 1194–6. [DOI] [PubMed] [Google Scholar]
- Liu W, Lan Y, Pauws E, Meester-Smoor MA, Stanier P, Zwarthoff EC, et al. The Mn1 transcription factor acts upstream of Tbx22 and preferentially regulates posterior palate growth in mice. Development 2008; 135: 3959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie RJ, Tan QK-G, Gilner JB, Rogers RC, Younge N, Wechsler SB, et al. Novel pathogenic variants in FOXP3 in fetuses with echogenic bowel and skin desquamation identified by ultrasound. Am J Med Genet A 2017; 173: 1219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meester-Smoor MA, Molijn AC, Zhao Y, Groen NA, Groffen CAH, Boogaard M, et al. The MN1 oncoprotein activates transcription of the IGFBP5 promoter through a CACCC-rich consensus sequence. J Mol Endocrinol 2007; 38: 113–25. [DOI] [PubMed] [Google Scholar]
- Meester-Smoor MA, Vermeij M, van Helmond MJL, Molijn AC, van Wely KHM, Hekman ACP, et al. Targeted disruption of the Mn1 oncogene results in severe defects in development of membranous bones of the cranial skeleton. Mol Cell Biol 2005; 25: 4229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 1998; 23: 198–9. [DOI] [PubMed] [Google Scholar]
- O’uchi E, O’uchi T. Persistent primitive trigeminal arteries (PTA) and its variant (PTAV): analysis of 103 cases detected in 16,415 cases of MRA over 3 years. Neuroradiology 2010; 52: 1111–9. [DOI] [PubMed] [Google Scholar]
- Pallares LF, Carbonetto P, Gopalakrishnan S, Parker CC, Ackert-Bicknell CL, Palmer AA, et al. Mapping of craniofacial traits in outbred mice identifies major developmental genes involved in shape determination. PLoS Genet 2015; 11: e1005607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier K, Hubert L, Viot G, Rio M, Billuart P, Besmond C, et al. CSNK2B splice site mutations in patients cause intellectual disability with or without myoclonic epilepsy. Hum Mutat 2017; 38: 932–941. [DOI] [PubMed] [Google Scholar]
- Ramocki MB, Scaglia F, Stankiewicz P, Belmont JW, Jones JY, Clark GD. Recurrent partial rhombencephalosynapsis and holoprosencephaly in siblings with a mutation of ZIC2. Am J Med Genet A 2011; 155A: 1574–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, El-Khechen D, Black MH, Farwell Hagman KD, Tang S, Powis Z. Outcomes of diagnostic exome sequencing in patients with diagnosed or suspected autism spectrum disorders. Pediatr Neurol 2017; 70: 34–43.e2. [DOI] [PubMed] [Google Scholar]
- Said E, Cuschieri A, Vermeesch J, Fryns JP. Toriello-Carey syndrome with a 6Mb interstitial deletion at 22q12 detected by array CGH. Am J Med Genet A 2011; 155A: 1390–2. [DOI] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 2015; 36: 928–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KA, Gonzaga-Jauregui C, Brigatti KW, Williams KB, King AK, Van Hout C, et al. Genomic diagnostics within a medically underserved population: efficacy and implications. Genet Med 2018; 20: 31–41. [DOI] [PubMed] [Google Scholar]
- Sutton ALM, Zhang X, Ellison TI, Macdonald PN. The 1,25(OH)2D3-regulated transcription factor MN1 stimulates vitamin D receptor-mediated transcription and inhibits osteoblastic cell proliferation. Mol Endocrinol 2005; 19: 2234–44. [DOI] [PubMed] [Google Scholar]
- Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C, et al. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am J Hum Genet 2015; 97: 457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully HM, Dempsey JC, Ishak GE, Adam MP, Curry CJR, Sanchez-Lara P, et al. Beyond Gómez-López-Hernández syndrome: recurring phenotypic themes in rhombencephalosynapsis. Am J Med Genet A 2012; 158A: 2393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully HM, Dempsey JC, Ishak GE, Adam MP, Mink JW, Dobyns WB, et al. Persistent figure-eight and side-to-side head shaking is a marker for rhombencephalosynapsis. Mov Disord 2013; 28: 2019–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van De Weghe JC, Rusterholz TDS, Latour B, Grout ME, Aldinger KA, Shaheen R, et al. Mutations in ARMC9, which encodes a basal body protein, cause Joubert syndrome in humans and ciliopathy phenotypes in zebrafish. Am J Hum Genet 2017; 101: 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandervore LV, Schot R, Hoogeboom AJM, Lincke C, de Coo IF, Lequin MH, et al. Mutated zinc finger protein of the cerebellum 1 leads to microcephaly, cortical malformation, callosal agenesis, cerebellar dysplasia, tethered cord and scoliosis. Eur J Med Genet 2018; 61: 783–9. [DOI] [PubMed] [Google Scholar]
- van Wely KHM, Molijn AC, Buijs A, Meester-Smoor MA, Aarnoudse AJ, Hellemons A, et al. The MN1 oncoprotein synergizes with coactivators RAC3 and p300 in RAR-RXR-mediated transcription. Oncogene 2003; 22: 699–709. [DOI] [PubMed] [Google Scholar]
- Vezain M, Lecuyer M, Rubio M, Dupé V, Ratié L, David V, et al. A de novo variant in ADGRL2 suggests a novel mechanism underlying the previously undescribed association of extreme microcephaly with severely reduced sulcation and rhombencephalosynapsis. Acta Neuropathol Commun 2018; 6: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ, Mazzeu JF, Hoischen A, Bayram Y, Withers M, Gezdirici A, et al. DVL3 alleles resulting in a -1 frameshift of the last exon mediate Autosomal-Dominant Robinow syndrome. Am J Hum Genet 2016; 98: 553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Dowd DR, Moore MC, Kranenburg TA, Meester-Smoor MA, Zwarthoff EC, et al. Meningioma 1 is required for appropriate osteoblast proliferation, motility, differentiation, and function. J Biol Chem 2009; 284: 18174–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are available upon reasonable request.