

Abstract
Summary
To address the limited software options for performing survival analyses with millions of SNPs, we developed gwasurvivr, an R/Bioconductor package with a simple interface for conducting genome-wide survival analyses using VCF (outputted from Michigan or Sanger imputation servers), IMPUTE2 or PLINK files. To decrease the number of iterations needed for convergence when optimizing the parameter estimates in the Cox model, we modified the R package survival; covariates in the model are first fit without the SNP, and those parameter estimates are used as initial points. We benchmarked gwasurvivr with other software capable of conducting genome-wide survival analysis (genipe, SurvivalGWAS_SV and GWASTools). gwasurvivr is significantly faster and shows better scalability as sample size, number of SNPs and number of covariates increases.
Availability and implementation
gwasurvivr, including source code, documentation and vignette are available at: http://bioconductor.org/packages/gwasurvivr.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Genome-wide association studies (GWAS) are population-level experiments that investigate genetic variation in individuals to observe single nucleotide polymorphism (SNP) associations with a phenotype. Genetic variants tested for association are genotyped on an array and imputed from a reference panel of sequenced genomes, e.g. 1000 Genomes Project or Haplotype Reference Consortium (HRC) (Das et al., 2016; Genomes Project et al., 2015). Imputed SNPs can be tested for association with binary outcomes (case/control) and quantitative outcomes (e.g. height) using a range of available software packages, including SNPTEST (Marchini et al., 2007) or PLINK (Purcell et al., 2007). However, existing software options for performing survival analyses, genipe (Lemieux Perreault et al., 2016), SurvivalGWAS_SV (Syed et al., 2017) and GWASTools (Gogarten et al., 2012) either require user interaction with raw output, were not initially designed for survival and/or have long run times. For these reasons, we developed an R/Bioconductor package, gwasurvivr, for genome-wide survival analyses of imputed data in multiple file formats with flexible analysis and output options.
2 Implementation
2.1 Data structure
Gwasurvivr can analyze data in IMPUTE2 format (Howie et al., 2009), in VCF files derived from Michigan (Das et al., 2016) or Sanger imputation servers (McCarthy et al., 2016), and directly genotyped PLINK format (Purcell et al., 2007). Data from each are prepared in gwasurvivr by leveraging existing Bioconductor packages GWASTools (Gogarten et al., 2012) or VariantAnnotation (Obenchain et al., 2014) depending on the imputation file format. Input file formats for gwasurvivr include IMPUTE2, VCF and PLINK. IMPUTE2 (Howie et al., 2009) format is a standard genotype (.gen) file which store genotype probabilities (GP). We utilized GWASTools in R to compress files into genomic data structure (GDS) format (Gogarten et al., 2012). This allows for efficient, iterative access to subsets of the data, while simultaneously converting GP into dosages (DS) for use in survival analyses. VCF files generated from Michigan or Sanger servers include a DS field and server-specific meta-fields (INFO score [Sanger] or r2 [Michigan]), as well as reference panel allele frequencies that are iteratively read in by VariantAnnotation (Obenchain et al., 2014). Plink bed files contain genotype information encoded in binary format. Fam and bim files include phenotype information and marker location, respectively (Purcell et al., 2007).
2.2 Survival analysis
Gwasurvivr implements a Cox proportional hazards regression model (Cox, 1972) to relate the SNP to survival time, allowing for covariates and/or SNP-covariate interactions. To decrease the number of iterations needed for convergence when optimizing the parameter estimates in the Cox model we modified the R package survival (Therneau and Grambsch, 2000). Covariates in the model are first fit without the SNP, and those parameter estimates are used as initial points for analyses with each SNP. If no additional covariates are added to the model, the parameter estimation optimization begins with null initial value (Supplementary Fig. S1).
Survival analyses are run using genetic data in either VCF or IMPUTE2 (Howie et al., 2009) formats and a phenotype file, which contains survival time, survival status and additional covariates; both files are indexed by sample ID. In addition to genomic data, the VCF files contain both sample IDs and imputation quality metrics (INFO score or r2), while IMPUTE2 (Howie et al., 2009) come in separate files (.gen, .sample and .info). Gwasurvivr functions for IMPUTE2 (impute2CoxSurv or gdsCoxSurv) and VCF (michiganCoxSurv or sangerCoxSurv) include arguments for the survival model (event of interest, time to event and covariates) and arguments for quality control that filter on minor allele frequency (MAF) or imputation quality (michiganCoxSurv and sangerCoxSurv only). INFO score filtering using impute2CoxSurv can be performed by accessing the .info file from IMPUTE2 results and subsequently providing the list of SNPs to the ‘exclude.snps’ argument to gwasurvivr. Users can also provide a list of sample IDs for gwasurvivr to internally subset the data. Gwasurvivr outputs two files: (i) .snps_removed file, listing all SNPs that failed QC parameters and (ii) .coxph file with the results from the analyses, including parameter estimates, p-values, MAF, the number of events and total sample N for each SNP. Gwasurvivr also allows the number of cores used during computation on Windows and Linux to be specified. Users can keep compressed GDS files after the initial run by setting keepGDS argument to TRUE when analyzing IMPUTE2 data (Howie et al., 2009). On successive runs, gdsCoxSurv can then be used instead of impute2CoxSurv to avoid compressing the data on each GWAS run.
3 Simulations and benchmarking
Computational runtimes for gwasurvivr were benchmarked against existing software comparing varying sample sizes and SNP numbers, with 4, 8 or 12 covariates and for a single chromosome with 15 000–25 000 individuals. In addition, we evaluated time for gwasurvivr for a GWAS (∼6 million SNPS) for 3000, 6000 and 9000 samples. All benchmarking experiments were performed using IMPUTE2 format (comparison packages do not take VCF from either Sanger or Michigan servers). Descriptions of simulated genotype and phenotype data are in the Supplementary Data.
4 Results
Gwasurvivr was faster than genipe (Lemieux Perreault et al., 2016), SurvivalGWAS_SV (Syed et al., 2017) and GWASTools (Gogarten et al., 2012) for 100 000 SNPs at N = 100, and 5000, with the exception of SurvivalGWAS_SV at N = 1000 (Fig. 1A). Similarly, increasing the number of covariates for gwasurvivr has minimal effects on runtime versus other software (Fig. 1B). Gwasurvivr computes for large sample sizes, however, compression time increases with increasing sample size, and likely will be limited by available RAM on a machine or cluster (Fig. 1C). The keepGDS argument helps address this and results in reduced run times (Fig. 1C and D), i.e. <3 h for a GWAS of N = 9000. A ∼6 million SNP GWAS can be run in <10 h for 9000 samples when using separately scheduled jobs on a supercomputer (Fig. 1D). However, gwasurvivr overcomes memory limitations often attributed to R by processing subsets of the entire data, and thus it is possible to conduct genome-wide survival analyses on a typical laptop computer.
Fig. 1.
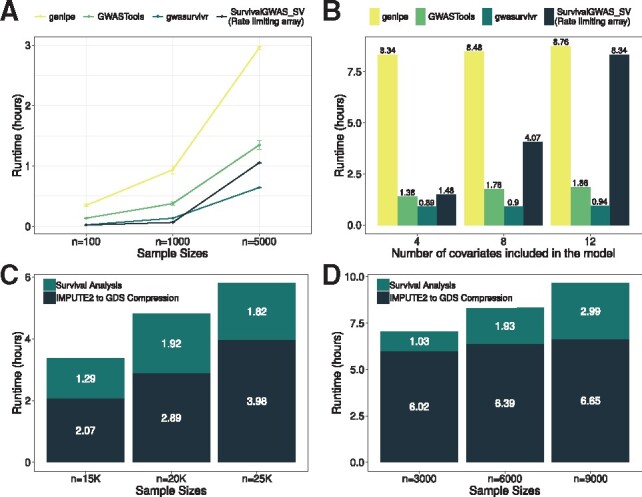
Runtime for survival analyses. Analyses were run with identical CPU constraints of 1 node and 8 cores. All SurvivalGWAS_SV runtimes are for 100 batched jobs with 1000 SNPs in an array index. The run time for the rate-limiting array is the array index that had the longest runtime, which translates to the shortest possible time for SurvivalGWAS_SV to complete if submitted jobs start at the same time. (A) The x-axis shows the three sample sizes with 100 000 SNPs. The y-axis is the total runtime in hours. Mean and 95% confidence intervals (CI) are show for genipe (yellow), GWASTools (light green), SurvivalGWAS_SV (dark blue) and gwasurvivr (dark green). Confidence intervals were calculated for 3 simulations for each n and m combination. (B) Genipe (yellow), GWASTools (light green), SurvivalGWAS_SV (dark blue) and gwasurvivr (dark green) run with 4, 8 and 12 covariates (n = 5000, m = 100 000). (C) Gwasurvivr was run on IMPUTE2 data simulated from chromosome 22 (m ∼ 117 000 SNPs) for n = 15 000, n = 20 000 and n = 25 000. (D) Full GWAS runtimes for varying N for the chromosome that took longest to complete. This corresponds to the full time for a GWAS when using a job scheduler on a cluster. For (C) and (D), the dark blue is elapsed time for compressing to GDS format and dark green is the computational time to run the survival analysis alone
Gwasurvivr is a fast, efficient and flexible program well suited for multicore processors and easily run in a computing cluster environment.
Funding
This work was supported by the NIH/NHLBI R01HL102278 (to L.S.C. and T.H.), NIH/NCI R03CA188733 (to L.S.C. and T.H.), The Ohio State University and the Translational Data Analytics Initiative (L.S.C.). This work was also supported in part by the Pelotonia Fellowship Program (to E.K.). Any opinions, findings and conclusions expressed in this material are those of the author(s) and do not necessarily reflect those of the Pelotonia Fellowship Program or The Ohio State University. This work was performed in part at the University at Buffalo’s Center for Computational Research.
Conflict of Interest: none declared.
Supplementary Material
Contributor Information
Abbas A Rizvi, Division of Pharmaceutics and Pharmaceutical Chemistry, College of Pharmacy, The Ohio State University, Columbus, OH, USA.
Ezgi Karaesmen, Division of Pharmaceutics and Pharmaceutical Chemistry, College of Pharmacy, The Ohio State University, Columbus, OH, USA.
Martin Morgan, Department of Biostatistics and Bioinformatics, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA.
Leah Preus, Division of Pharmacy Practice and Science, College of Pharmacy, The Ohio State University, Columbus, OH, USA.
Junke Wang, Division of Pharmacy Practice and Science, College of Pharmacy, The Ohio State University, Columbus, OH, USA.
Michael Sovic, Division of Pharmacy Practice and Science, College of Pharmacy, The Ohio State University, Columbus, OH, USA.
Theresa Hahn, Department of Medicine, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA.
Lara E Sucheston-Campbell, Division of Pharmacy Practice and Science, College of Pharmacy, The Ohio State University, Columbus, OH, USA; Department of Veterinary Biosciences, College of Veterinary Medicine, The Ohio State University, Columbus, OH 43210, USA.
References
- Cox D.R. (1972) Regression Models and Life Tables. Journal of Royal Statistical Society. Series B (Methodological), 34, 187–220. [Google Scholar]
- Das S., et al. (2016) Next-generation genotype imputation service and methods. Nat. Genet., 48, 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C., et al. (2015) A global reference for human genetic variation. Nature, 526, 757168–757174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogarten S.M., et al. (2012) GWASTools: an R/Bioconductor package for quality control and analysis of genome-wide association studies. Bioinformatics, 28, 3329–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B.N., et al. (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet., 5, e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux Perreault L.P., et al. (2016) genipe: an automated genome-wide imputation pipeline with automatic reporting and statistical tools. Bionformatics, 32, 3661–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S., et al. (2016) A reference panel of 6,976 haplotypes for genotype imputation. Nat. Genet., 48, 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini J., et al. (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet., 39, 906–913. [DOI] [PubMed] [Google Scholar]
- Obenchain V., et al. (2014) VariantAnnotation: a Bioconductor package for exploration and annotation of genetic variants. Bionformatics, 30, 2076–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S., et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed H., et al. (2017) SurvivalGWAS_SV: software for the analysis of genome-wide association studies of imputed genotypes with ‘time-to-event’ outcomes. BMC Bionformatics, 18, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therneau T.M., Grambsch P.M. (2000) Modeling Survival Data: Extending the Cox Model. Springer, New York. ISBN 0-387-98784-3. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.