

Abstract
Summary
Large scale genomic studies produce millions of sequence variants, generating datasets far too massive for manual inspection. To ensure variant and genotype data are consistent and accurate, it is necessary to evaluate variants prior to downstream analysis using quality control (QC) reports. Variant call format (VCF) files are the standard format for representing variant data; however, generating summary statistics from these files is not always straightforward. While tools to summarize variant data exist, they generally produce simple text file tables, which still require additional processing and interpretation. VariantQC fills this gap as a user friendly, interactive visual QC report that generates and concisely summarizes statistics from VCF files. The report aggregates and summarizes variants by dataset, chromosome, sample and filter type. The VariantQC report is useful for high-level dataset summary, quality control and helps flag outliers. Furthermore, VariantQC operates on VCF files, so it can be easily integrated into many existing variant pipelines.
Availability and implementation
DISCVRSeq's VariantQC tool is freely available as a Java program, with the compiled JAR and source code available from https://github.com/BimberLab/DISCVRSeq/. Documentation and example reports are available at https://bimberlab.github.io/DISCVRSeq/.
1 Introduction
High throughput sequencing advancements have accelerated genomic studies of human diseases and these large-scale sequencing studies typically produce millions of sequence variants (Gonzaga-Jauregui et al., 2012). After data processing and variant discovery, it is prudent to evaluate a call set prior to downstream analysis. Quality control (QC) evaluation ensures derived genotype data are consistent and accurate, thus reducing the chance of incorrect genotypes impacting association tests and study results (Carson et al., 2014; Nielsen et al., 2011). Although powerful tools to summarize variants exist, such as GATK’s VariantEval (Van der Auwera et al., 2013), summarizing a complex variant call format (VCF) file (Danecek et al., 2011) still requires multiple executions of those tools, which generates multiple tables stored as individual text files. Interpreting these data remains a challenge, and there is a need for visual QC reports to concisely summarize millions of variants. Some tools provide visual summary reports, such as SnpEff (http://snpeff.sourceforge.net/); however, these are generally focused on the domain of that tool. To provide a more general purpose solution, we developed VariantQC, a tool to generate a user-friendly, interactive QC report to summarize variants, highlight outliers and identify potential data concerns. A fairly basic yet powerful feature of VariantQC is the stratification of variant statistics by multiple levels, including per sample and per chromosome, which aids in the identification of outliers.
2 Materials and methods
VariantQC is a command-line Java tool, included as part of DISCVR-seq Toolkit (https://bimberlab.github.io/DISCVRSeq/). Basic input requires a VCF file and an indexed genome FASTA file. The pedigree PED file is optional and only used to determine gender information of the samples, which is included in the final report. The input VCF is parsed and evaluated to generate summary statistics, aggregating the data using four stratifications. Each stratification produces a set of statistics summarizing variants by: the entire VCF, chromosome, sample, or filter type. These summary statistics are generated by internally utilizing GATK4’s VariantEval (Van der Auwera et al., 2013). VariantQC will generate multiple sets of summary data from a single scan of the input VCF. This results in faster execution, avoids multiple VariantEval executions, and the need to merge resulting tables. These summary statistics are used to produce an HTML-based report with interactive tables and bar graphs. The Javascript and HTML templates used for this report are, with permission, heavily based on MultiQC (Ewels et al., 2016).
As shown in Figure 1A, all VariantQC reports have the following four main stratification levels: Entire VCF, By Contig, By Sample and By Filter. To improve rendering performance for large datasets, each stratification is organized as a separate section and only rendered on-demand when the user loads that section. Within each stratification section, there are multiple summary reports identifying the total variants by type (single nucleotide variants (SNVs), insertions, deletions, etc.), a summary of genotypes, Ti/Tv data, a summary of variants by contig and a summary of variants by filter type. Summary statistics are presented as interactive tables or bar graphs (Fig. 1B). For all tabular reports, users can sort and/or customize the set of columns displayed. To easily identify outliers, most tables display a bar graph behind the raw value, based on the range of that column. Additionally, values that are two standard deviations from the column mean are highlighted in red for easy identification. Data are also presented as bar graphs, which support hover and tooltips to provide the user with more information. Together, these allow the user to quickly visualize summary statistics for trends or filter items for specific analyses. All data can be exported as tab-delimited files.
Fig. 1.
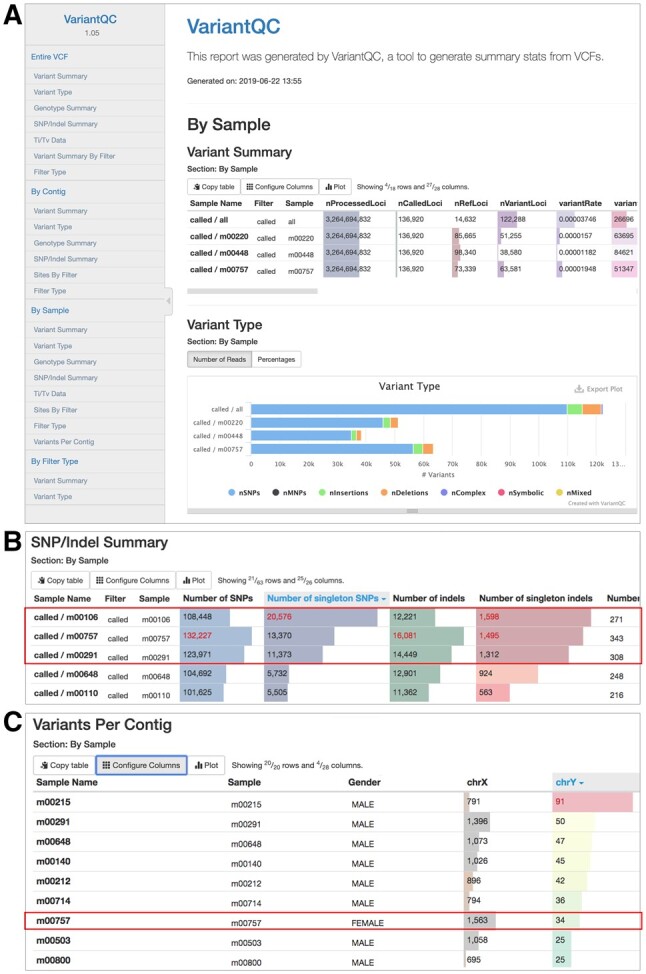
Representative VariantQC Report. (A) HTML report for the ‘By Sample’ stratification, displaying summary statistics as interactive tables and bar graphs. Left gray panel lists four primary stratifications and their corresponding set of summary reports beneath. (B) Using the ‘SNP/Indel Summary’ table from the ‘By Sample’ stratification, the number of singleton SNVs was sorted to quickly identify the outlier m00106, which was flagged in red, along with two other potential outliers. (C) Using the ‘Variants Per Contig’ table from the ‘By Sample’ stratification, samples were sorted by the number of chrY variants, allowing easy detection of a potential QC issue for sample m00757, which was listed as female, but has a high number of chrY variants
3 Results
VariantQC has been successfully used on large VCFs, including a dataset with 341 whole genome samples or 1369 genotyping-by-sequencing samples. For these datasets, the ability of VariantQC to stratify data by sample allowed rapid identification of subjects with high numbers of private SNVs (Fig. 1B) and subjects with mismatched gender assignment based on X and Y genotype calls (Fig. 1C). The interactive visual QC report for variant evaluation is suitable for large datasets. Furthermore, any project that generates variant or genotype data using the common VCF format can easily integrate VariantQC for summary and QC analysis.
Acknowledgements
The authors thank Philip Ewels for his permission to adapt MultiQC’s code for the VariantQC report. The authors also thank the Broad Institute, particularly Chris Norman, for assistance with the migration of GATK’s VariantEval tool from GATK3 to GATK4.
Funding
This work was supported by the National Institutes of Health [R24 OD021324, P51 OD011092].
Conflict of Interest: none declared.
Contributor Information
Melissa Y Yan, Division of Genetics, Beaverton, OR, USA.
Betsy Ferguson, Division of Genetics, Beaverton, OR, USA; Division of Neuroscience, Oregon National Primate Research Center, Oregon Health & Science University, Beaverton, OR, USA; Molecular and Medical Genetics Department, Oregon Health & Science University, Portland, OR, USA.
Benjamin N Bimber, Division of Genetics, Beaverton, OR, USA; Division of Pathobiology, Oregon National Primate Research Center, Oregon Health & Science University, Beaverton, OR, USA.
References
- Carson A.R. et al. (2014) Effective filtering strategies to improve data quality from population-based whole exome sequencing studies. BMC Bioinformatics, 15, 125.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P. et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P. et al. (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzaga-Jauregui C. et al. (2012) Human genome sequencing in health and disease. Annu. Rev. Med., 63, 35–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R. et al. (2011) Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet., 12, 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera G.A. et al. (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform., 43, 11 10 11–33. [DOI] [PMC free article] [PubMed] [Google Scholar]