

Abstract
Background
Senior–Loken syndrome is a rare genetic disorder that presents with nephronophthisis and retinal degeneration, leading to end‐stage renal disease and progressive blindness. The most frequent cause of juvenile nephronophthisis is a mutation in the nephronophthisis type 1 (NPHP1) gene. NPHP1 encodes the protein nephrocystin‐1, which functions at the transition zone (TZ) of primary cilia.
Methods
We report a 9‐year‐old Senior–Loken syndrome boy with NPHP1 deletion, who presents with bilateral vision decrease and cystic renal disease. Renal function deteriorated to require bilateral nephrectomy and renal transplant. We performed immunohistochemistry, H&E staining, and electron microscopy on the renal sample to determine the subcellular distribution of ciliary proteins in the absence of NPHP1.
Results
Immunohistochemistry and electron microscopy of the resected kidney showed disorganized cystic structures with loss of cilia in renal tubules. Phosphoinositides have been recently recognized as critical components of the ciliary membrane and immunostaining of kidney sections for phosphoinositide 5‐phosphatase, INPP5E, showed loss of staining compared to healthy control. Ophthalmic examination showed decreased electroretinogram consistent with early retinal degeneration.
Conclusion
The decreased expression of INPP5E specifically in the primary cilium, coupled with disorganized cilia morphology, suggests a novel role of NPHP1 that it is involved in regulating ciliary phosphoinositide composition in the ciliary membrane of renal tubular cells.
Keywords: INPP5E, NPHP1, primary cilia, Senior–Loken syndrome, transition zone
Short abstract
In this study, we report a 9‐year‐old Senior‐Loken syndrome patient with NPHP1 deletion. We found the decreased expression of INPP5E specifically in the primary cilium, coupled with disorganized cilia morphology, suggests a novel role of NPHP1 in regulating ciliary phosphoinositide composition in the ciliary membrane of renal tubular cells.
1. INTRODUCTION
The primary cilium is a solitary, immotile microtubule‐based structure extending from the surface of almost all mammalian cell types (Anderson et al., 2008; Berbari et al., 2009; Satir & Christensen, 2007; Veland et al., 2009). Defects in cilia result in a class of multi‐organ diseases that usually include retinal degeneration, renal disease, and cerebral abnormalities collectively called ciliopathies (Halbritter et al., 2013; Hildebrandt et al., 2009). Senior–Loken syndrome is a rare inherited disorder that is characterized by nephronophthisis and retinal degeneration. Nephronophthisis (NPHP) is an autosomal recessive cystic renal ciliopathy characterized by polyuria and anemia. It is also the most frequent genetic cause of end‐stage kidney disease before age 30 (Ahmed & Ali, 2011; Srivastava & Sayer, 2014; Stokman et al., 1993). Deletion of NPHP1, an important ciliary protein residing at the ciliary transition zone (TZ), is associated with the development of Senior–Loken syndrome and NPHP, which mainly affects the kidneys and eyes (Ronquillo et al., 2012).
The transition zone (TZ) is a compartment of the proximal region of the cilium, between the basal body and axoneme. Numerous studies in diverse model systems have shown that the TZ functions as a selective membrane diffusion barrier, regulating ciliary protein entry and exit (Goncalves & Pelletier, 2017; Omran, 2010). Two TZ protein complexes have been identified: the NPHP complex and MKS complex. In mammalian cells, ciliary proteins adenylyl cyclase III (AC3) and polycystic kidney disease 2 (PKD2) are aberrantly located in MKS‐complex‐deficient cilia (Garcia‐Gonzalo et al., 2011). In C. elegans, TRAM protein and membrane‐associated RP2 homologs are abnormal in TZ‐deficient cilia (Williams et al., 2011). Previous studies have demonstrated by LAP‐tagging and in vitro binding that NPHP1, NPHP4, and NPHP8 have a strong mutual interaction (Sang et al., 2011): NPHP4 acts as a bridge that directly binds both NPHP1 and NPHP8 in vitro, whereas NPHP1 and NPHP8 cannot directly bind to each other. Furthermore, NPHP4 and NPHP8 have been shown to localize to the base of the cilia of renal tubular cells in vivo and in vitro (Delous et al., 2009; Sang et al., 2011). Recently phosphoinositides have been recently shown as a critical component of the ciliary membrane; however, the distribution of phosphoinositide enzymes in NPHP deficient cells is not known.
Here we report the clinical–pathological study of a 9‐year‐old patient with Senior–Loken syndrome. He developed renal failure and early signs of retinal degeneration, with decreases on electroretinogram consistent with rod photoreceptor dysfunction. Renal histology showed corticomedullary cysts, tubular basement membrane disruption, and tubulointerstitial nephropathy. Transmission electron microscopic studies of the diseased kidney samples revealed thickened tubular membrane and decreased cilia number and length. Immunofluorescence imaging showed disorganized cilia in renal tubular cells and aberrant expression of phosphoinositide 5‐phosphatase. Our findings are the first report of the NPHP patient‐derived cilia which showed disorganized cilia morphology and decreased expression of phosphoinositide 5‐phosphatase INPP5E from the remaining cilia, suggesting that NPHP1 plays a role in regulating ciliary phosphoinositide composition by controlling the phosphoinositide 5‐phosphatases entry into cilia.
2. MATERIALS AND METHODS
2.1. Ethical compliance
This research was approved by the IRB Committee of Indiana University and Stanford University.
2.2. Reagents
Anti‐acetylated α‐tubulin rabbit antibody (5335) was purchased from Cell Signaling Technology. Anti‐Arl13b mouse antibody (75‐287) was purchased from Antibodies Inc. Anti‐intraflagellar transport protein 88 (IFT88) polyclonal antibodies were purchased from Proteintech (13967‐1‐AP). INPP5E mouse antibody (ab221117) was purchased from Abcam. Anti‐NPHP4 and NPHP8 mouse antibodies were purchased from Santa Cruz. Anti‐NPHP1 rabbit antibody (SAB1401267) was purchased from Sigma‐Aldrich. Secondary antibodies (1:200) were Cy3‐conjugated donkey anti‐mouse IgG and Alexa‐Fluor 488 or 546‐conjugated donkey anti‐mouse IgG (Jackson ImmunoResearch Laboratories, Inc.).
2.3. Family description and genetic analysis
This study included a 2‐generation family with one affected offspring. The disease is displayed in an autosomal recessive manner. The diagnosis of NPHP1 deletion was based on genetic analysis. The patient, a 9‐year‐old boy, was examined at the Indiana University School of Medicine. The research protocol was approved by the Indiana University Institutional Review Board and was in accordance with the tenets of the Declaration of Helsinki. All subjects were informed of the purpose of the examination and all the participating individuals signed written informed consent. Genomic DNA was extracted from the peripheral blood of the patient, NPHP1 deletion mutation in the homozygous state was determined by electrophoretic analysis of PCR amplification products generated from genomic DNA (Athena Diagnostics).
2.4. Ocular examination
Ophthalmic examination included fundus photography, visual acuity testing on Humphrey visual field, and full‐field electroretinography (ERG). Fundus photography was performed after pupil dilation with topical 1% tropicamide. Full‐field ERG was performed using Ganzfeld stimulation as described by the International Society for Clinical Electrophysiology of Vision (ISCEV) (McCulloch et al., 2015).
2.5. Human renal samples and H&E staining
The de‐identified human renal sample was collected at the Department of Pathology, Indiana University School of Medicine. The nephrectomy section was fixed in 4% paraformaldehyde (PFA) solution and then processed for paraffin embedding. Sections were stained with hematoxylin and eosin (H&E) and examined by microscopy. Additional sections were used for immunostaining.
2.6. Transmission electron microscopy (TEM)
Tissue for electron microscopy was fixed in 2.5% glutaraldehyde in PBS and subsequently dehydrated with ethanol, followed by fixation in epoxy resin. The sections were stained with uranyl acetate and examined with a Philips TEM400 transmission electron microscope.
2.7. Immunofluorescence microscopy
For immunofluorescence staining, preparation of the renal samples has been previously described (Luo et al., 2014). Briefly, tissue was permeabilized with 0.2% Triton X‐100 in phosphate‐buffered saline (PBS) for 10 min and incubated in blocking buffer (1% bovine serum albumin, 10% normal goat serum in PBS) for 30 min at room temperature followed by overnight incubation with the primary antibody appropriately diluted in blocking buffer at 4°C. After rinsing three times in PBS, samples were treated for 1 h with secondary antibodies (diluted to 1:200 in blocking buffer) at room temperature. Nuclei were treated with DAPI. Imaging was performed with a Zeiss LSM 880 laser scanning confocal microscope.
2.8. Statistical analysis.
All statistical analyses were performed using Graphpad8 (Prism) software. Results are expressed as mean values ± SEM. Statistical analyses were performed using paired t‐test to compare data between two groups. p value of <.05 was considered statistically significant.
3. RESULTS
3.1. Clinical presentation and physical examination
The ocular involvement in Senior–Loken syndrome is variable and includes different degrees of retinal dystrophy and cataracts (Aggarwal et al., 2013). The most frequently reported ophthalmologic manifestations of NPHP have been retinal degeneration and ocular‐motor apraxia (OMA) (Aggarwal et al., 2013; Hemachandar, 2014). In this study, we report the clinical phenotype of a 9‐year‐old male patient. The ophthalmic exam showed that his visual acuity was 20/20 in both eyes. On slit‐lamp examination, the anterior segment of both eyes was also normal. Color fundus photographs of both eyes were unremarkable, without pigmentary changes, lesions or scars in the macula or periphery (Figure 1a). Visual field examination showed no focal defects in both eyes (Figure 1b). Color fundus photographs in NPHP1 patients are typically normal despite bilateral visual field loss. Full‐field ERG, which provides an assessment of overall retinal function, was recorded after 30 minutes of dark adaptation. The scotopic and photopic flash ERG showed a decreased rod and cone response in both eyes (Figure 1c). The amplitude of the b‐wave was reduced for the rod response (0.01, 3.0, and 10.0 scotopic) without an implicit time delay. The b value of the dark‐adapted ERG (0.01) was 36 µV OD compared with 140 µV in the control patient. ERG (3.0) was 120 µV OD compared with 212 µV in the control patient. ERG (10.0) was 152 µV OD compared with 217 µV in the control patient. The amplitude of both a‐ and b‐waves of the cone response was bilaterally reduced compared to the age‐matched control. The b value of the light‐adapted ERG was 50 µV OD in the patient and 143 µV in the control patient. No remarkable changes were found in 30‐Hz flicker in either eye (data not shown). In summary, the patient with NPHP1 deletion demonstrated diminished rod responses and overall impaired cone function.
FIGURE 1.

Early retinal degeneration and kidney cyst in a patient with NPHP1 deletion. (a) Representative color fundus photographs of the patient with NPHP1 deletion showing normal fundus in both eyes without optic disc pallor or retinal dystrophy. (b) Visual field examination demonstrating bilateral visual field loss in this patient. The grayscale plots present as a cluster of paracentral points with decreased sensitivity and scotoma. (c) Full‐field standard ERG of OD showing increased implicit time and reduced amplitude in scotopic 0.01 ERG, and in photopic 3.0 ERG. (d) Representative hematoxylin and eosin (H&E)‐stained kidney sections showing the histological pattern in normal human and the patient with NPHP1 deletion. Kidney specimen is shown at low (left) and high (right) magnification. Kidney histology of the NPHP1 patient illustrates the characteristic triad of corticomedullary cysts, tubular basement membrane disruption, and tubulointerstitial nephropathy. Scale bars = (left) 200 μm; (right) 50 μm. Transmission electron micrographs of primary cilia and TBM in kidney epithelial cells from the NPHP1 deletion patient. TEM reveals a thickened tubular membrane and decreased cilia. Scale bars = 2 μm
3.2. Histopathology of NPHP1 kidney
To determine the renal phenotype of this patient, histopathological analysis of his kidney obtained by nephrectomy was performed alongside an identical analysis of a healthy human kidney (Figure 1d). H&E staining of the patient's kidney sections showed a diffuse sclerosing tubulointerstitial process with a predominance of tortuous and atrophic tubules at the corticomedullary junction. Enlarged cortical cyst formation was present in the corticomedullary region. Higher magnification highlighted a thickened and multilayered tubular basement membrane (TBM). Transmission electron microscopy was used to determine the impact of NPHP1 mutation on human primary cilia and TBM of renal epithelial cells. Electron microscopy revealed an irregular TBM pattern, consisting frequently of two or three membrane layers which were folded excessively, while the lining of the tubular cysts was flattened. As shown in Figure 1d, fewer primary cilia were seen on the apical side of the tubular epithelial cells, indicating renal cilia dysfunction.
3.3. Disorganized cilia in renal tubular cells of NPHP1 deficient kidney
The TZ, a cilia sub‐compartment just distal of the basal body, has a unique membrane content and has also been implicated in regulating what proteins enter the cilia signaling compartment in diverse model systems (Awata et al., 2014; Fliegauf et al., 2006; Sang et al., 2011; Shi et al., 2017; Szymanska & Johnson, 2012; Williams et al., 2011). It has been previously demonstrated that nephrocystin proteins localize to the ciliary TZ of renal tubular epithelia in mice and cultured cells. To our knowledge, cilia frequency, morphology, and content of renal primary cilia in a human patient with NPHP1 deletion have not been reported. Here, we assess primary cilia in a kidney biopsy of a patient with nephronophthisis. We examined the renal biopsy with confocal immunofluorescence microscopy and compared the findings to those in relevant control samples from a healthy kidney. Typical ciliary markers, acetylated α‐tubulin (an axoneme marker), Arl13b (a cilia membrane marker), and Intraflagellar Transport 88 (a cilia specific IFT marker), were employed to assess primary cilia. Compared with control kidney primary cilia, NPHP1 patient renal cilia were disorganized and shortened (Figure 2a). Arl13b positive cilia were quantified to determine cilia frequency and showed that the loss of NPHP1 significantly reduced the proportion of ciliated cells from 78.5 ± 12.3% in the normal patient to 60 ± 7.3% in the NPHP1 deletion patient, indicating that NPHP1 influences ciliogenesis in human renal tubular cells (Figure 2e). In addition, quantification of immunofluorescently labeled primary cilia revealed that ciliary length was reduced from 5.6 ± 1.2 µm in the healthy patient to 3.2 ± 1.0 µm in the NPHP1 patient (Figure 2f). Taken together, our findings suggest that NPHP1 may be required for normal cilia maintenance and function in the human kidney.
FIGURE 2.
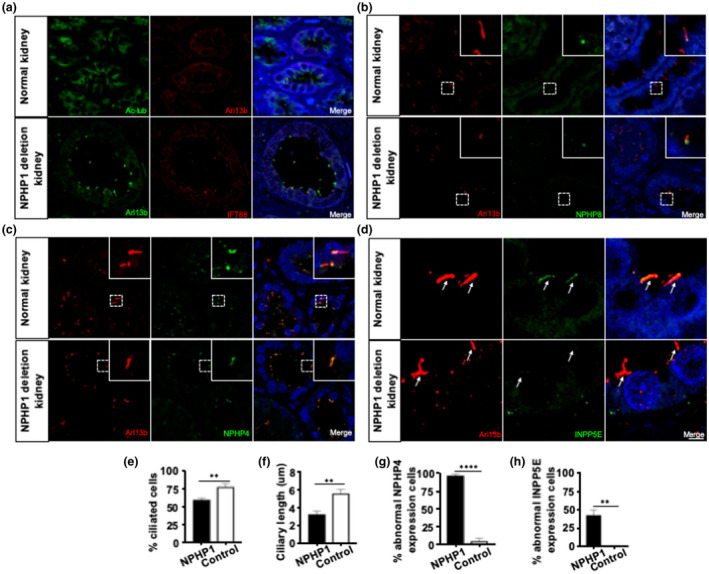
Disorganized cilia with abnormal ciliary protein expression in renal tubular cells of a patient with NPHP1 deletion. (a) Disorganized cilia in renal tubular cells of a patient with NPHP1 deletion. Confocal immunomicroscopy of normal and patient kidney sections stained with Arl13b and Ac‐tubulin antibodies to detect primary cilia and with IFT88 to detect basal body. Primary cilia appear shorter and disorganized in epithelial cells of patient kidney sections. (e and f) Quantification of ciliation and ciliary length (n = 200). (b) Normal expression of NPHP8 and abnormal expression of (c) NPHP4 in cilia of renal tubular cells from the NPHP1 deletion patient. Confocal immunomicroscopy of patient kidney sections stained for NPHP8 and NPHP4. (g) Quantification of cells with abnormal expression of NPHP4 (n = 50). (d) Aberrant expression of phosphoinositide 5‐phosphatases in cilia of renal tubular cells from the NPHP1 deletion patient. Confocal immunomicroscopy of patient kidney sections stained for INPP5E. (h) Quantification of cells with abnormal expression (n = 50). Statistical analysis in E‐H was performed using Student's test, p < 0.05 was considered statistically significant. Scale bars: (a–c) 10 μm; (d) magnified images 40 μm. (Arrow, marks primary cilia)
3.4. Abnormal ciliary protein expression in the cilia of NPHP1 deficient kidney
Based on these studies, we hypothesized that the loss of NPHP1 would directly affect NPHP4 localization in vivo, but would not affect NPHP8. To test this hypothesis, we co‐stained renal sections from the patient with NPHP1 deletion and healthy controls for either NPHP4 or NPHP8 and the ciliary marker Arl13b. Analysis of confocal images revealed no alteration in NPHP8 localization, indicating that NPHP1 exerts its function independently of NPHP8 (Figure 2b). In healthy control cilia, NPHP4 localized only at the base of the cilia. In contrast, in renal sections from the patient with NPHP1 deletion, NPHP4 was significantly increased not only at the base of the cilia but also in the ciliary axoneme (Figure 2c). Quantitative analysis showed that 97.78% ± 3.85% renal epithelial cells in the NPHP1 deletion group demonstrated an abnormal enrichment of NPHP4, compared to only 4.2% ± 7.2% in the control group (Figure 2g). These results suggest that the NPHP1‐NPHP4 interaction is conserved and that it plays a role in controlling NPHP4 localization in the primary cilia of human renal epithelial cells.
NPHP proteins are the important regulators of ciliary entrance and exit for a diverse array of components (Omran, 2010). Therefore, the regulation of phosphoinositides, which recently have been recognized as a critical component of the ciliary membrane, could also be influenced by these complexes residing in the ciliary transition zone. Several groups have shown that a mutation of inositol 5‐phosphatase (INPP5E), which is found to localize in the axoneme of the primary cilium, is responsible for ciliary instability in Joubert syndrome (Chavez et al., 2015; Dyson et al., 2017; Jacoby et al., 2009). Based on the previously described distribution of NPHP1 in cilia, we hypothesized that NPHP1 may be critical for controlling phosphoinositide composition within the cilia of human renal epithelial cells. Using a monoclonal antibody against INPP5E, we assessed the distribution of INPP5E within the primary cilia of renal tissue from both a patient with NPHP1 deletion and healthy control. While INPP5E was expressed along the axoneme of primary cilia in normal tissue, it was absent from the cilia of the NPHP1 deletion patient (Figure 2d). Quantitative analysis showed that up to 42.40% ± 13.83% of renal epithelial cells in the NPHP1 deletion patient had abnormal INPP5E expression, compared to healthy tissue (Figure 2h). This data indicates a critical role for NPHP1 in phosphoinositide regulation.
4. DISCUSSION
In this study, we presented a young patient who was diagnosed with Senior–Loken syndrome according to the clinical manifestations (renal failure and retinal degeneration) and genetic analysis. Immunohistochemical analysis of renal biopsy showed disorganized cilia and a significant decrease in ciliation. Furthermore, partially retained cilia in the renal tubular cells suggested NPHP1 functions as a key gating component regulating NPHP4 at the TZ and INPP5E recruitment to axoneme of cilia, providing a potential mechanism underlying Senior–Loken syndrome with defective ciliary gating.
Kidneys of NPHP1 patients display a characteristic triad of corticomedullary cysts, tubular basement membrane disruption, and tubulointerstitial nephropathy (Srivastava & Sayer, 2014; Stokman et al., 1993; Wolf, 2015). This child initially presented with progressive renal failure at age 2. Subsequent ultrasound findings were consistent with cystic kidney disease. He went on to develop renal failure and early signs of retinal degeneration. The patient presented with polyuria and polydipsia, then progressed to end‐stage renal disease, for which he underwent bilateral nephrectomy and a renal transplant. In addition to retinal degeneration and kidney defects, clinical manifestations of Senior–Loken syndrome may also include skeletal, dermatological and cerebellar anomalies, small hands (short metacarpals), and madarosis (Aggarwal et al., 2013; Ronquillo et al., 2012). Currently, no treatment is available to prevent disease pathogenesis. Deletion in the NPHP1 gene represents the leading genetic abnormality reported in nephrophthisis patients to date (Hildebrandt et al., 2009; Wolf, 2015). Similar to other NPHP gene products, the NPHP1 protein has been found to localize at the basal body and TZ of primary cilia in renal tubular cells and retinal photoreceptors (Fliegauf et al., 2006; Garcia‐Gonzalo & Reiter, 2017; Gogendeau et al., 2020; Goncalves & Pelletier, 2017; Omran, 2010). NPHP1 has also been found in the areas of cell‐to‐cell contact, including tight junctions, adherens junctions, and focal adhesions (Hildebrandt et al., 2009; Zhou et al., 2012).
Interestingly, nephronophthisis and Joubert syndrome are two types of ciliopathies with overlapping clinical manifestations, including cystic kidney disease and retinal degeneration. Previous publications showed that NPHP1 and INPP5E mutations are responsible for Joubert syndrome (Constable et al., 2020; Jacoby et al., 2009). Our study shows that NPHP1 deletion results in the abnormal expression of INPP5E, which supports the hypothesis that Joubert syndrome and nephronophthisis represent the same biological disorder but with different versions of clinical manifestations. Revealing the mechanism by which NPHP1 regulates the amount of INPP5E at the vertebrate TZ and elucidating the relevant signaling pathways represent important steps toward developing novel therapeutic targets.
Previous studies have shown that mice with NPHP1 deletion do not develop pathological characteristics of nephronophthisis (Jiang et al., 2008). However, NPHP1 knockout significantly decreases ciliation and ciliary length in MDCK cells, which is consistent with our data for patient‐derived cilia (Delous et al., 2009). This difference in phenotype might be due to species differences in gene function (Williams et al., 2010). Recent data have also begun to demonstrate that TZ function may be cell type and tissue specific, with mutations in some TZ members having specific phenotypes (Lewis et al., 2019).
Because its defects are associated with many severe ciliopathies, the function of the TZ is an active field of biomedical research. Numerous publications have demonstrated that TZ proteins serve as the ciliary gatekeeper, but the exact mechanisms by which they regulate ciliary gating are still not clear. Although previous studies have shown that NPHP1 acts as a ciliary gate in C. elegans (Williams et al., 2011), NPHP1 deficiency did not alter ciliary levels of Arl13b or Sstr3 in vertebrate cells, suggesting that NPHP1 is not involved in governing the transition of these proteins in vertebrate cilia (Lin et al., 2018). More recent studies have proposed that the TZ could act as a specific ciliary gate for lipids (Goncalves & Pelletier, 2017). Supporting this notion, the location of INPP5E within vertebrate cilia has been shown to depend on many MKS complex proteins, such as TCTN1 and MKS1. RPGRIP1L/MKS5, which directly interacts NPHP4/NPHP1 and MKS complex proteins, has also been reported to play a role in regulating the lipids within cilia. In our study, we found the abnormal localization of NPHP4 in NPHP1 mutant cells, which may directly or indirectly affect the MKS complex, resulting in abnormal INPP5E localization in renal tubular epithelial cells.
In summary, we report a diagnosed case of Senior–Loken syndrome, which derives from a deletion of a key TZ protein. The patient had renal failure and retinal degeneration. We show that primary cilia in the patient's renal biopsy exhibit morphological abnormalities that likely impaired normal function. Importantly, we also show that the ciliary localization of INPP5E is altered in the sample from the patient with NPHP1 deletion. Based on human NPHP1 deficient tissue, we detected an abnormal expression of NPHP4, which suggests that NPHP1 acts as a structural scaffold for the NPHP4 present in the TZ. We bring into focus the input of NPHP1 in forming a TZ in which NPHP4 functions normally, which in turn regulates INPP5E transition and its ability to influence cilia stability.
5. ETHICS STATEMENT
The study was conducted in accordance with the principles of the Declaration of Helsinki. All study procedures were defined, and patient consent was obtained as specified by the IRB protocol 45037 approved by the Institutional Review Board at Stanford University.
CONFLICT OF INTEREST
The authors declare no financial/non‐financial competing interest.
AUTHOR CONTRIBUTIONS
KN wrote the manuscript, ES and YS designed and carried out all experiments. BS and PP contributed to editing the manuscript. KC, AG, JA, and BW contributed to the experimental design and data analysis. NB and HY provided valuable advice and experimental suggestions for this study. YS supervised the whole project.
ACKNOWLEDGMENTS
We thank Dr. Tia Kowal for the critical review of the manuscript.
Funding informationThis work was supported by NIH/NEI K08‐EY022058 (YS), R01‐EY025295 (YS), VA Merit CX001298 (YS), Maternal Children's Health Research Institute Award (YS). Research for Prevention of Blindness Unrestricted grant (Stanford Ophthalmology). P30 Vision Center grant (Stanford Ophthalmology) YS is a Laurie Kraus Lacob Faculty Scholar in Pediatric Translational Medicine. R01‐EY‐023295 (Y.H.) R01‐EY024932 (Y.H.). International Retinal Research Foundation‐PR810542 (K.N.).
DATA AVAILABILITY STATEMENT
All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors.
REFERENCES
- Aggarwal, H. K. , Jain, D. , Yadav, S. , Kaverappa, V. , & Gupta, A. (2013). Senior‐loken syndrome with rare manifestations: A case report. The Eurasian Journal of Medicine, 45(2), 128–131. 10.5152/eajm.2013.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, C. T. , Castillo, A. B. , Brugmann, S. A. , Helms, J. A. , Jacobs, C. R. , & Stearns, T. (2008). Primary cilia: Cellular sensors for the skeleton. The Anatomical Record: Advances in Integrative Anatomy and Evolutionary Biology, 291(9), 1074–1078. 10.1002/ar.20754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awata, J. , Takada, S. , Standley, C. , Lechtreck, K. F. , Bellve, K. D. , Pazour, G. J. , Fogarty, K. E. , & Witman, G. B. (2014). NPHP4 controls ciliary trafficking of membrane proteins and large soluble proteins at the transition zone. Journal of Cell Science, 127(Pt 21), 4714–4727. 10.1242/jcs.155275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari, N. F. , O'Connor, A. K. , Haycraft, C. J. , & Yoder, B. K. (2009). The primary cilium as a complex signaling center. Current Biology, 19(13), R526–R535. 10.1016/j.cub.2009.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez, M. , Ena, S. , Van Sande, J. , de Kerchove d'Exaerde, A. , Schurmans, S. , & Schiffmann, S. N. (2015). Modulation of ciliary phosphoinositide content regulates trafficking and sonic hedgehog signaling output. Developmental Cell, 34(3), 338–350. 10.1016/j.devcel.2015.06.016 [DOI] [PubMed] [Google Scholar]
- Constable, S. , Long, A. B. , Floyd, K. A. , Schurmans, S. , & Caspary, T. (2020). The ciliary phosphatidylinositol phosphatase Inpp5e plays positive and negative regulatory roles in Shh signaling. Development, 147(3), 10.1242/dev.183301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delous, M. , Hellman, N. E. , Gaudé, H.‐M. , Silbermann, F. , Le Bivic, A. , Salomon, R. , Antignac, C. , & Saunier, S. (2009). Nephrocystin‐1 and nephrocystin‐4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Human Molecular Genetics, 18(24), 4711–4723. 10.1093/hmg/ddp434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson, J. M. , Conduit, S. E. , Feeney, S. J. , Hakim, S. , DiTommaso, T. , Fulcher, A. J. , Sriratana, A. , Ramm, G. , Horan, K. A. , Gurung, R. , Wicking, C. , Smyth, I. , & Mitchell, C. A. (2017). INPP5E regulates phosphoinositide‐dependent cilia transition zone function. Journal of Cell Biology, 216(1), 247–263. 10.1083/jcb.201511055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegauf, M. , Horvath, J. , von Schnakenburg, C. , Olbrich, H. , Müller, D. , Thumfart, J. , Schermer, B. , Pazour, G. J. , Neumann, H. P. H. , Zentgraf, H. , Benzing, T. , & Omran, H. (2006). Nephrocystin specifically localizes to the transition zone of renal and respiratory cilia and photoreceptor connecting cilia. Journal of the American Society of Nephrology, 17(9), 2424–2433. 10.1681/ASN.2005121351 [DOI] [PubMed] [Google Scholar]
- Garcia‐Gonzalo, F. R. , Corbit, K. C. , Sirerol‐Piquer, M. S. , Ramaswami, G. , Otto, E. A. , Noriega, T. R. , Seol, A. D. , Robinson, J. F. , Bennett, C. L. , Josifova, D. J. , García‐Verdugo, J. M. , Katsanis, N. , Hildebrandt, F. , & Reiter, J. F. (2011). A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nature Genetics, 43(8), 776–784. 10.1038/ng.891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Gonzalo, F. R. , & Reiter, J. F. (2017). Open sesame: How transition fibers and the transition zone control ciliary composition. Cold Spring Harb Perspect Biol, 9(2), 10.1101/cshperspect.a028134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogendeau, D. , Lemullois, M. , Le Borgne, P. , Castelli, M. , Aubusson‐Fleury, A. , Arnaiz, O. , Cohen, J. , Vesque, C. , Schneider‐Maunoury, S. , Bouhouche, K. , Koll, F. , & Tassin, A.‐M. (2020). MKS‐NPHP module proteins control ciliary shedding at the transition zone. PLoS Biology, 18(3), e3000640. 10.1371/journal.pbio.3000640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves, J. , & Pelletier, L. (2017). The ciliary transition zone: Finding the pieces and assembling the gate. Molecules and Cells, 40(4), 243–253. 10.14348/molcells.2017.0054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbritter, J. , Porath, J. D. , Diaz, K. A. , Braun, D. A. , Kohl, S. , Chaki, M. , Allen, S. J. , Soliman, N. A. , Hildebrandt, F. , & Otto, E. A. ; Group, G. P. N. S. (2013). Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis‐related ciliopathy. Human Genetics, 132(8), 865–884. 10.1007/s00439-013-1297-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemachandar, R. (2014). Senior‐loken syndrome ‐ A ciliopathy. Journal of Clinical and Diagnostic Research, 8(11), MD04–MD05. 10.7860/JCDR/2014/9688.5120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt, F. , Attanasio, M. , & Otto, E. (2009). Nephronophthisis: Disease mechanisms of a ciliopathy. Journal of the American Society of Nephrology, 20(1), 23–35. 10.1681/ASN.2008050456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby, M. , Cox, J. J. , Gayral, S. , Hampshire, D. J. , Ayub, M. , Blockmans, M. , Pernot, E. , Kisseleva, M. V. , Compère, P. , Schiffmann, S. N. , Gergely, F. , Riley, J. H. , Pérez‐Morga, D. , Woods, C. G. , & Schurmans, S. (2009). INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nature Genetics, 41(9), 1027–1031. 10.1038/ng.427 [DOI] [PubMed] [Google Scholar]
- Jiang, S.‐T. , Chiou, Y.‐Y. , Wang, E. , Lin, H.‐K. , Lee, S.‐P. , Lu, H.‐Y. , Wang, C.‐K. , Tang, M.‐J. , & Li, H. (2008). Targeted disruption of Nphp1 causes male infertility due to defects in the later steps of sperm morphogenesis in mice. Human Molecular Genetics, 17(21), 3368–3379. 10.1093/hmg/ddn231 [DOI] [PubMed] [Google Scholar]
- Lewis, W. R. , Bales, K. L. , Revell, D. Z. , Croyle, M. J. , Engle, S. E. , Song, C. J. , Malarkey, E. B. , Uytingco, C. R. , Shan, D. , Antonellis, P. J. , Nagy, T. R. , Kesterson, R. A. , Mrug, M. M. , Martens, J. R. , Berbari, N. F. , Gross, A. K. , & Yoder, B. K. (2019). Mks6 mutations reveal tissue‐ and cell type‐specific roles for the cilia transition zone. The FASEB Journal, 33(1), 1440–1455. 10.1096/fj.201801149R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, H. , Guo, S. , & Dutcher, S. K. (2018). RPGRIP1L helps to establish the ciliary gate for entry of proteins. Journal of Cell Science, 131(20), 10.1242/jcs.220905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, N. , Conwell, M. D. , Chen, X. , Kettenhofen, C. I. , Westlake, C. J. , Cantor, L. B. , Wells, C. D. , Weinreb, R. N. , Corson, T. W. , Spandau, D. F. , Joos, K. M. , Iomini, C. , Obukhov, A. G. , & Sun, Y. (2014). Primary cilia signaling mediates intraocular pressure sensation. Proceedings of the National Academy of Sciences of the United States of America, 111(35), 12871–12876. 10.1073/pnas.1323292111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch, D. L. , Marmor, M. F. , Brigell, M. G. , Hamilton, R. , Holder, G. E. , Tzekov, R. , & Bach, M. (2015). ISCEV Standard for full‐field clinical electroretinography (2015 update). Documenta Ophthalmologica, 130(1), 1–12. 10.1007/s10633-014-9473-7 [DOI] [PubMed] [Google Scholar]
- Omran, H. (2010). NPHP proteins: Gatekeepers of the ciliary compartment. Journal of Cell Biology, 190(5), 715–717. 10.1083/jcb.201008080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquillo, C. C. , Bernstein, P. S. , & Baehr, W. (2012). Senior‐Loken syndrome: A syndromic form of retinal dystrophy associated with nephronophthisis. Vision Research, 75, 88–97. 10.1016/j.visres.2012.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang, L. , Miller, J. J. , Corbit, K. C. , Giles, R. H. , Brauer, M. J. , Otto, E. A. , Baye, L. M. , Wen, X. , Scales, S. J. , Kwong, M. , Huntzicker, E. G. , Sfakianos, M. K. , Sandoval, W. , Bazan, J. F. , Kulkarni, P. , Garcia‐Gonzalo, F. R. , Seol, A. D. , O'Toole, J. F. , Held, S. , … Jackson, P. K. (2011). Mapping the NPHP‐JBTS‐MKS protein network reveals ciliopathy disease genes and pathways. Cell, 145(4), 513–528. 10.1016/j.cell.2011.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satir, P. , & Christensen, S. T. (2007). Overview of structure and function of mammalian cilia. Annual Review of Physiology, 69, 377–400. 10.1146/annurev.physiol.69.040705.141236 [DOI] [PubMed] [Google Scholar]
- Shi, X. , Garcia, G. 3rd , Van De Weghe, J. C. , McGorty, R. , Pazour, G. J. , Doherty, D. , Huang, B. & Reiter, J. F. (2017). Super‐resolution microscopy reveals that disruption of ciliary transition‐zone architecture causes Joubert syndrome. Nature Cell Biology, 19(10), 1178–1188. 10.1038/ncb3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, S. , & Sayer, J. A. (2014). Nephronophthisis. Journal of Pediatric Genetics, 3(2), 103–114. 10.3233/PGE-14086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokman, M. , Lilien, M. , & Knoers, N. (1993). Nephronophthisis. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., & Amemiya A. (Eds.), GeneReviews((R)). NCBI bookshelf. [Google Scholar]
- Szymanska, K. , & Johnson, C. A. (2012). The transition zone: An essential functional compartment of cilia. Cilia, 1(1), 10. 10.1186/2046-2530-1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veland, I. R. , Awan, A. , Pedersen, L. B. , Yoder, B. K. , & Christensen, S. T. (2009). Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiology, 111(3), 39–53. 10.1159/000208212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, C. L. , Li, C. , Kida, K. , Inglis, P. N. , Mohan, S. , Semenec, L. , Bialas, N. J. , Stupay, R. M. , Chen, N. , Blacque, O. E. , Yoder, B. K. , & Leroux, M. R. (2011). MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. Journal of Cell Biology, 192(6), 1023–1041. 10.1083/jcb.201012116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, C. L. , Masyukova, S. V. , & Yoder, B. K. (2010). Normal ciliogenesis requires synergy between the cystic kidney disease genes MKS‐3 and NPHP‐4. Journal of the American Society of Nephrology, 21(5), 782–793. 10.1681/ASN.2009060597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, M. T. (2015). Nephronophthisis and related syndromes. Current Opinion in Pediatrics, 27(2), 201–211. 10.1097/MOP.0000000000000194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, W. , Otto, E. A. , Cluckey, A. , Airik, R. , Hurd, T. W. , Chaki, M. , Diaz, K. , Lach, F. P. , Bennett, G. R. , Gee, H. Y. , Ghosh, A. K. , Natarajan, S. , Thongthip, S. , Veturi, U. , Allen, S. J. , Janssen, S. , Ramaswami, G. , Dixon, J. , Burkhalter, F. , … Hildebrandt, F. (2012). FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nature Genetics, 44(8), 910–915. 10.1038/ng.2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors.