

Abstract
Background
Grenada is a small, resource‐limited Caribbean country with a high incidence of sickle cell disease (SCD). Since little is known about the challenges facing individuals living with SCD in the West Indies, we sought to assess barriers to healthcare and the impact of SCD on quality of life in Grenada.
Methods
Both adults aged 18+ (n = 19) and caregivers of children aged 2–17 (n = 26) completed validated survey measures regarding barriers to care and quality of life, along with a genetics knowledge questionnaire. Caregivers also completed a caregiver burden scale. Survey scores were calculated, and responses were analyzed for an association between demographic variables.
Results
The Barriers to Care Questionnaire, in which lower scores indicate more barriers, revealed that both adults (mean = 69.9) and children (mean = 75.5) with SCD experienced reduced access to care. The Adult Sickle Cell Quality of Life Measurement Information System indicated increased depression and loneliness in adults, with the lowest scores in the Emotional subscale. However, the Pediatric Quality of Life Inventory answered by caregivers of children with SCD showed the lowest scores in the Physical Functioning subscale. Further analysis using the Caregiver Burden Scale‐Zarit Burden Interview revealed that 53.8% of caregivers of children with SCD indicated “little to no burden,” which may reflect a difference in cultural expectations of a caregiver between high‐income countries and Grenada. Finally, ~80% of respondents knew that SCD was a genetic condition; however, 61%–84% could not correctly indicate recurrence risks, demonstrating a need for additional education.
Conclusion
These data provide new insights regarding the experience of living with SCD in Grenada and support the need for further investigations into specific barriers to healthcare delivery, which could also improve education and well‐being for those affected by SCD in Grenada and in the broader Caribbean community.
Keywords: barriers to care, Grenada, quality of life, resource‐limited, sickle cell disease
Grenada is a small, resource‐limited Caribbean country with a high incidence of sickle cell disease (SCD). Despite the prevalence, little is known about the challenges facing individuals living with SCD. In this study, caregivers of children with SCD and adults with SCD reported a variety of barriers to care and reduced quality of life that highlight the need for improvements to infrastructure, including education, facilities, and personnel, to facilitate earlier diagnosis and improved lifelong therapeutic management for individuals with SCD.

1. INTRODUCTION
Sickle cell disease (SCD) is an inherited genetic condition that affects the structure of hemoglobin, the protein in red blood cells that carries oxygen. A pathogenic variant in the gene encoding beta globin (HBB) causes an alteration in the structure of hemoglobin which causes hemoglobin to polymerize, distorting red blood cells into a sickle or crescent shape. Individuals with SCD experience anemia, recurrent infections, and debilitating pain crises caused by vaso‐occlusive events. Other life‐threatening complications may also occur, such as acute chest syndrome or stroke. SCD is inherited in an autosomal recessive manner, meaning that both parents of an individual with SCD are heterozygous carriers of the HBB pathogenic variant. The sickle cell variant may be seen in individuals from all parts of the world but is most common in those of African descent.
In the United States (USA), SCD can be well managed. Newborn screening programs in the U.S. provide early identification of both the presence of disease and trait (Wethers et al., 1989). These individuals are notified of their status and receive education about the inheritance of SCD, prevention of symptoms, and management when symptoms do occur. Newborns in the USA may be referred to specialized centers that provide ongoing state‐of‐the‐art care for individuals with hemoglobinopathies. Several medical advancements in the treatment of SCD are currently being developed, including gene editing using CRISPR‐Cas9, which is now in clinical trials (Salinas Cisneros and Thein, 2020). However, many individuals with SCD, especially those in underserved communities, are still managing their symptoms with treatments such as folic acid supplementation, immunizations, red blood cell transfusions, and hydroxyurea. Hydroxyurea is a chemotherapeutic agent that helps prevent acute vaso‐occlusive events in severely affected individuals by inducing the expression of fetal hemoglobin (Rowe, 1995). Even with multiple preventative measures, acute pain crises can occur, which can severely impact someone's quality of life. Previous studies have shown that the pain associated with SCD has a significant impact on mood and enjoyment of life (Shah et al., 2014).
In underserved or resource‐limited communities, newborn screening may not be available and individuals with SCD may not discover their status until complications manifest. Additionally, once symptoms do occur, education may not be provided, and preventative treatment options might be more difficult to obtain. One such community is Grenada, a small Caribbean country which consists of the islands of Grenada, Carriacou, and Petite Martinique. This tri‐island nation has a population of ~111,764 and a 37% poverty rate, with only 7% of persons covered by private insurance (Grenada, 2019) SCD is prevalent in Grenada, affecting ~1 in 160 individuals (Knight‐Madden et al., 2019). Grenada has a special history with SCD, as Grenada was the birthplace of Walter Clement Noel, the first individual recognized as having the condition (Steensma et al., 2010). Even if early screening and identification exist, in resource‐limited nations like Grenada, individuals with SCD and their families might not receive the appropriate education about SCD or proper treatment.
The exact barriers to care that individuals with SCD in Grenada face or how their quality of life is impacted by the disease is unknown. Previous studies have analyzed quality of life in other Caribbean countries, such as Jamaica, where respondents reported frustration with the lack of control that the condition imposed on them and stigmatization from healthcare providers, who accused them of seeking drugs (Anderson and Asnani, 2013). However, no other studies have looked at barriers to care in conjunction with quality of life for individuals with SCD in the Caribbean. The purpose of this study is to investigate the experience of this population, specifically in Grenada, with the goal of improving the delivery of health care to people affected by SCD in all resource‐limited countries. Survey measures that were validated in other populations were used to assess the perceived quality of life of both adults and children (by caregiver proxy) with SCD and the caregiver burden of those caring for individuals with SCD, along with barriers to care.
2. METHODS
2.1. Ethical compliance
This research study was a collaboration between Baylor College of Medicine, Texas, USA, and St. George's University, Grenada. The protocol, an exploratory cross‐sectional design, was approved by Institutional Review Boards (IRB) at both institutions, and permission to carry out the study was granted by the Grenada Ministry of Health. Data were collected from September to December 2019.
2.2. Study population and recruitment
The study population consisted of both adults and parents/caregivers of children with SCD in Grenada. Potential participants included adults aged 18 or older with SCD living in Grenada or caregivers of children aged 2–17 with SCD. Individuals with sickle cell trait were excluded from this study. Eligible participants were identified through the Sickle Cell Association of Grenada (SCAG) membership and called by the investigators or approached in person at the local monthly health clinic for children with SCD and asked to participate in the survey. Participants were informed of the study objectives, consent procedures, and offered the option to complete the survey on paper, over the telephone, or be sent a weblink to fill out the survey on an online platform (www.SurveyMonkey.com). Both participants that began the survey in person and over the phone were given the opportunity to separate the survey into two sessions and finish at a later point over the phone.
2.3. Survey design
The survey for parents/caregivers of children with SCD included four components: demographic information with questions about genetics knowledge, Barriers to Care Questionnaire (BCQ; Seid et al., 2004), Caregiver Burden Scale‐Zarit Burden Interview (CBS‐ZBI; Zarit et al., 1980), and the Pediatric Quality of Life Inventory (PedsQL; Varni et al., 2001) The survey for adults with SCD included three components: the same demographic questions and Barriers to Care Questionnaire, along with the Adult Sickle Cell Quality of Life Measurement Information System (ASCQ‐ME; Treadwell et al., 2014) Following the BCQ, respondents were asked to provide any additional comments about barriers to healthcare. These responses were not formally analyzed for themes.
2.3.1. Demographic questionnaire
The demographic questionnaire consists of 12 questions related to demographics, including age, gender, race/ethnicity, highest level of education, and employment status.
2.3.2. Barriers to care questionnaire
The barriers to care questionnaire consists of 39 items with five domains: Pragmatics, Skills, Expectations, Marginalization, and Knowledge and Beliefs. Response choices were on a five‐point Likert scale with scores ranging from 100 (no problem) to 0 (almost always a problem), with higher scores indicating fewer barriers. Response choice language was adapted from a previous study of barriers to care in a population of individuals with SCD (Jacob et al., 2015). The BCQ total scale has been shown to have internal consistency reliability at 0.95 (Seid et al., 2004).
2.3.3. Adult sickle cell quality of life measurement information system
The ASCQ‐ME consists of 30 items with six domains: Emotional Impact, Social Functioning Impact, Sleep Impact, Stiffness Impact, Pain Impact, and Pain Episodes. The five “impact” domains are each scored from 5 (never) to 1 (always), with higher scores representing better quality of life. Only the impact domains were utilized for the purpose of this study. The ASCQ‐Me has been shown to have good internal consistency for each item (≥0.92; Keller et al., 2014).
2.3.4. Pediatric quality of life inventory
The PedsQL consists of four Generic Core Scales: Physical, Emotional, Social, and School. It is separated into four age groups with age‐appropriate language: toddlers (age 2–4), young children (age 5–7), children (age 8–12), and teens (age 13–17). The PedsQL for toddlers is 21 questions; the PedsQL for children and teens is 23 questions. The items are scored 0 to 100, with higher scores indicated higher perceived quality of life. Each age‐specific PedsQL survey has a caregiver‐proxy survey that can be filled out on the behalf of the child, which was used for the purposes of this protocol. The caregiver‐proxy PedsQL total scale across the age subgroups has been shown to approach or exceed 0.90 for internal consistency reliability (Varni et al., 2007). The PedsQL has been used to assess quality of life with respect to several chronic conditions, including type I diabetes (Özyazıcıoğlu et al., 2017) and cystic fibrosis (Zink et al., 2019).
2.3.5. Caregiver Burden Scale‐Zarit Burden Interview
The Caregiver Burden Scale consists of 22 items rated on a 5‐point Likert scale that ranges from 0 (never) to 4 (nearly always), with higher scores indicating greater burden. While originally developed for caregivers of individuals with dementia, this scale has been shown to have good internal consistency in other populations (0.92; Al‐Rawashdeh et al., 2016). The CBS‐ZBI has been previously utilized in a population of individuals with sickle cell disease (Silva et al., 2012).
2.3.6. Genetics knowledge
This unvalidated measure assessed genetics knowledge with the use of 2 true‐false questions and 3 multiple‐choice questions. These questions assessed understanding that SCD is a genetic condition, cannot be “caught” later in life, and asked participants to identify recurrence risk related to SCD for 3 scenarios.
2.4. Data analysis
Each BCQ (Seid et al., 2004), CBS‐ZB I (Keller et al., 2014), PedsQL (Varni et al., 2001), and ASCQ‐ME (Treadwell et al., 2014) was analyzed individually based on the validated survey's scoring measure. One‐way ANOVAS and t‐tests were conducted to determine demographic variables associated with negative quality of life and specific barriers to care; chi square tests were conducted to determine the demographic variables associated with caregiver burden. One‐way ANOVAs, t‐tests, and chi square tests were also utilized to determine an association between incorrect responses to genetic questions and negative quality of life, barriers to care, and caregiver burden. Analysis was performed using Stata 16. A p < 0.05 was considered statistically significant.
3. RESULTS
3.1. Participant demographics
During the data collection period of September–December 2019, 46 individuals were identified and consented to participate in the survey (Tables 1 and 2). The participants consisted of 19 adults with SCD and 26 caregivers of children with SCD. One caregiver was excluded from analysis because the child was above the age of 17.
TABLE 1.
Participant demographics of adults with SCD.
| Demographic variable | Participants |
|---|---|
| n = 19, n (%) | |
| Age (years) | |
| 18–34 | 12 (63.2) |
| 35–55+ | 7 (36.8) |
| Sex | |
| Male | 5 (26.3) |
| Female | 14 (73.7) |
| Ancestry a | |
| Afro‐Caribbean | 10 (52.6) |
| Black/African | 11 (57.9) |
| Prefer not to say | 1 (5.3) |
| Education | |
| Primary | 4 (21.1) |
| Secondary | 7 (36.8) |
| College | 8 (42.1) |
| Employment | |
| Less than full time | 9 (47.4) |
| Full time | 10 (52.6) |
| Age at diagnosis (years) | |
| 0–4 | 6 (31.6) |
| 5–12 | 9 (47.4) |
| 13–17 | 4 (21.1) |
| Other medical conditions | |
| Yes | 3 (15.8) |
| No | 16 (84.2) |
| Children | |
| No | 11 (57.9) |
| Yes | 8 (42.1) |
| Children with SCD | |
| 0 | 17 (89.5) |
| 1 | 2 (10.5) |
Participants selected all that apply; each response was counted separately.
TABLE 2.
Participant demographics of caregivers of children with SCD.
| Demographic variable | Participants |
|---|---|
| n = 26, n (%) | |
| Age of caregiver (years) | |
| 18–34 | 11 (42.3) |
| 35+ | 15 (57.7) |
| Sex | |
| Male | 3 (11.5%) |
| Female | 23 (88.5%) |
| Ancestry a | |
| Afro‐Caribbean | 21 (80.8) |
| Black/African | 7 (26.9) |
| White/Caucasian | 2 (7.7) |
| Indian | 2 (7.7) |
| Middle Eastern | 1 (3.8) |
| Education | |
| Primary | 6 (23.1) |
| Secondary | 11 (42.3) |
| College+ | 9 (34.6) |
| Employment | |
| Less than full time | 10 (38.5) |
| Full time | 16 (61.5) |
| Number of children | |
| 1–2 | 13 (50.0) |
| 3+ | 13 (50.0) |
| Children with SCD | |
| 1 | 22 (84.6) |
| 2 | 4 (15.4) |
| Age of children with SCD(years) b | |
| 2–4 | 6 (20.7) |
| 8–12 | 14 (48.3) |
| 13–17 | 9 (31.0) |
| Age at diagnosis (years) b | |
| 0–1 | 15 (51.7) |
| 2–4 | 9 (31.0) |
| 5–13 | 5 (17.2) |
| Other medical conditions | |
| Yes | 7 (26.9) |
| No | 19 (73.1) |
Participants selected all that apply; each response was counted separately.
n = 29 total children with SCD.
Of the adult participants (Table 1), most were between the ages of 18–34 (63.2%), female (73.7%), and Afro‐Caribbean and/or Black/African (94.7%). Most individuals (78.9%) completed secondary school or higher education and were employed full time (52.6%). The age at diagnosis for adults with SCD varied from infancy to adolescence. Most participants had children (57.9%), and 2 (10.5%) participants had a child with SCD (Table 1). Other medical conditions (15.8%) included diabetes and unspecified eye problems.
Of the caregiver participants (Table 2), most were over the age of 35 (42.3%), female (88.5%), and Afro‐Caribbean and/or Black/African (83.8%). Most individuals (76.9%) completed secondary school or higher education and were employed full time (61.5%). Half of the participants (50.0%) had 3 or more children, with 15.4% having 2 children with SCD. The age of the children with SCD varied between 2 and 17 years old. For the purposes of statistical analysis, children were grouped into 3 age categories: toddlers (ages 2–4), children (ages 8–12), and teenagers (13–17). No one with children aged 5–7 participated in this study. The majority (51.7%) were diagnosed under the age of 2 (Table 2); however, diagnosis ranged between at birth and 13 years old. Other medical conditions (26.9%) included allergies and eczema.
3.2. Barriers to care
The BCQ was administered to both adults with SCD and caregivers of children with SCD to measure the perceived level and type of barriers to healthcare that each participant encounters. An individual's BCQ was calculated by averaging each subscale, then, taking the average of the subscales to determine the mean total score. Higher scores on the BCQ indicate fewer barriers to care.
For the adults with SCD, the average total BCQ was 69.9 ± 10.9. There was little variation between subscales, with the greatest barriers in Expectations (60.0) and the fewest barriers in Knowledge (72.7), suggesting that each subscale has a similar level of effect as a healthcare barrier (Table 3a). Analysis of BCQ scores with respect to demographic variables (Table S1) suggested that women experience greater barriers to care (mean = 66.9) compared to men (78.6; t‐test, p = 0.04). The contributing subscale was Skills (p = 0.04), with women reporting greater barriers in response to “having enough information about how the health care system works” (52 vs. 80). Individuals with the highest education indicated as primary school reported fewer barriers (83.6) compared to those with secondary education (68.2) or college education (64.2). This variation in reported barriers was also statistically significant (ANOVA, p = 0.01), with both Marginalization and Expectations subscales as the contributing factors (p = 0.02 and p = 0.01, respectively). Additionally, participants that worked full time reported greater barriers (65.6) than those that worked less than full time (76.1; t‐test, p = 0.04). The contributing subscale was Expectations (p = 0.04), with those that worked full time reporting greater barriers in response to “doctors treating the symptom without finding the cause of the illness” (47.5 vs. 77.8). There were no statistically significant differences observed for the demographic variables of age, age at diagnosis, or whether or not they have children.
TABLE 3a.
Barriers to Care survey scores of adults with SCD a .
| Score (n = 19) | Minimum | Maximum | Mean | SD |
|---|---|---|---|---|
| Total | 55.1 | 96.8 | 69.9 | 10.9 |
| Skills | 56.3 | 96.9 | 72.2 | 11.4 |
| Marginalization | 25 | 95.5 | 69.5 | 17.7 |
| Expectations | 46.4 | 100 | 68 | 15.9 |
| Knowledge | 43.8 | 100 | 72.7 | 18.9 |
| Pragmatics | 55.6 | 94.4 | 68.7 | 10.2 |
Responses for each survey question were scored from 0 to 100, with 0 as “always a problem” and 100 as “never a problem.” The scores for each participant were averaged for each subscale (Skills, Marginalization, Expectations, Knowledge, and Pragmatics). Higher scores represent fewer barriers to care.
For the caregivers of children with SCD, the average total BCQ was 75.5 ± 13.1. The greatest barriers were observed in the subscale Pragmatics (mean = 67.2), while the fewest barriers were reported in Skills (80.4; Table 3b). Analysis across this cohort revealed that toddlers had greater barriers (mean = 51.8) than children (79.8) or teenagers (72.4; ANOVA, p = 0.02), with Pragmatics as the primary contributing subscale (p = 0.02; Table S2). There were no particular questions within the subscale of Pragmatics that contributed to this difference. No statistical differences between age of diagnosis for the child or age, sex, education, employment, number of children, or number of children with SCD for the caregivers.
TABLE 3b.
Barriers to Care survey scores of caregiver participants.
| Score (n = 26) | Minimum | Maximum | Mean | SD |
|---|---|---|---|---|
| Total | 47.1 | 96.4 | 75.5 | 13.1 |
| Skills | 50 | 100 | 80.4 | 13.8 |
| Marginalization | 34.1 | 100 | 76.9 | 19.3 |
| Expectations | 17.9 | 100 | 75.8 | 18.4 |
| Knowledge | 31.3 | 100 | 78.4 | 18.6 |
| Pragmatics | 22.2 | 100 | 67.2 | 18.7 |
Following the BCQ, respondents were asked to provide any additional comments about their barriers to healthcare in Grenada. Responses included concerns about the ability to travel to the hospital and not enough medical staff or medications in clinic (Table 4). Respondents also raised concern about medical staff not providing enough information about SCD, what it means for an individual's health, and how it is inherited. However, some participants commented that the healthcare system in Grenada has improved. These statements exemplify a dichotomy between those that are satisfied with their healthcare and those that believe they are not receiving adequate treatment.
TABLE 4.
Participant comments about barriers to healthcare in Grenada.
| Participant | Quote a |
|---|---|
| 36 (Caregiver) | "No medication available in hospital." |
| 37 (Adult) | "Hardly any doctors and not enough nurses on shift." |
| 30 (Caregiver) | "The health care system is better than before. Improvements are seen." |
| 28 (Caregiver) | "I wish I could have some help to understand how to deal with a child with SCD." |
| 23 (Adult) | "I feel like the doctors need to have a conversation with the patient about sickle cell and what it means." |
| 40 (Caregiver) | "More information is needed to be shared on how SCD is passed on." |
| 33 (Adult) | "It's hard to get to the hospital. When it takes too long to get there, I get sicker." |
| 31 (Caregiver) | "Translating children's care clinics to adult care clinics would be beneficial." |
Comments are quoted verbatim from either phone interviews or online surveys.
3.3. Quality of life
Two measures of quality of life were utilized: the ASCQ‐ME to measure the quality of life of adults with SCD and the PedsQL to measure the quality of life of children with SCD. An individual's ASCQ‐ME was calculated by taking the sum of each category, then, converting to a t‐score using the ASCQ‐ME User Manual (Varni et al., 2001). The t‐scores for the ASCQ‐ME ranged between 47.6 ± 3.9 for Emotional Impact and 52.3 ± 4.9 for Sleep Impact (Table 5). Analysis of ASCQ‐ME scores with respect to demographic variables (Table S3) revealed that adult participants above the age of 35 had lower quality of life (mean of 44.2) than participants between the ages of 18–34 (52.6) for Social Functioning (t‐test, p = 0.02). For Emotional Functioning, those with the highest level of education as secondary school had the lowest quality of life (44.9) compared to those that completed primary school (47.5) and those that completed college (50.7; ANOVA, p = 0.01). There were no statistically significant differences observed for the demographic variables of sex, age at diagnosis, or whether or not the participant had children.
TABLE 5.
ASCQ‐ME survey scores of adults with SCD a .
| Subscale (n = 19) | Minimum | Maximum | T‐score b | SD |
|---|---|---|---|---|
| Emotional impact | 39.9 | 55.2 | 47.6 | 3.9 |
| Social functioning impact | 29.8 | 59.8 | 49.5 | 7.8 |
| Sleep impact | 43.2 | 59.9 | 52.3 | 4.9 |
| Stiffness impact | 35.3 | 65.4 | 51.5 | 7.7 |
| Pain impact | 44.4 | 63.8 | 51.3 | 5 |
Responses for each survey question were scored from “never” (score of 5) to “always” (score of 1). The sum was calculated for each category: Emotional, Social Functioning, Sleep, Stiffness, and Pain Impact.
The raw scores were then converted to a t‐score using the ASCQ‐ME User Manual (Varni et al., 2001) A t‐score of 50 represents an average population.
Caregivers answered the PedsQL survey for each of their children with SCD (n = 29). Each PedsQL survey was calculated by averaging each subscale, then, taking the average of the subscales to determine the mean total score. The mean total score for the PedsQL was 78.9 ± 6.9 for toddlers, 72.5 ± 13.8 for children, and 70.6 ± 19.1 for teenagers (Figure 1). In this cohort, the average score decreased with increasing age, suggesting that as individuals with SCD age, their quality of life decreases. The standard deviation also increases, suggesting that teenagers with SCD have a more varied experience than infants with SCD. The lowest subscale scores for all age groups were in Physical Functioning with a mean score of 72.4 for age group 2–4, 64.3 for age group 5–12, and 63.0 for age group 13–17; the highest subscale scores for all age groups were in Social Functioning with a mean score of 85.8 for age group 2–4, 85.0 for age group 5–12, and 79.4 for age group 13–17. These results suggest that even though individuals with SCD reported lower quality of life as they age, all age groups experience the greatest or least issues in the same domains.
FIGURE 1.
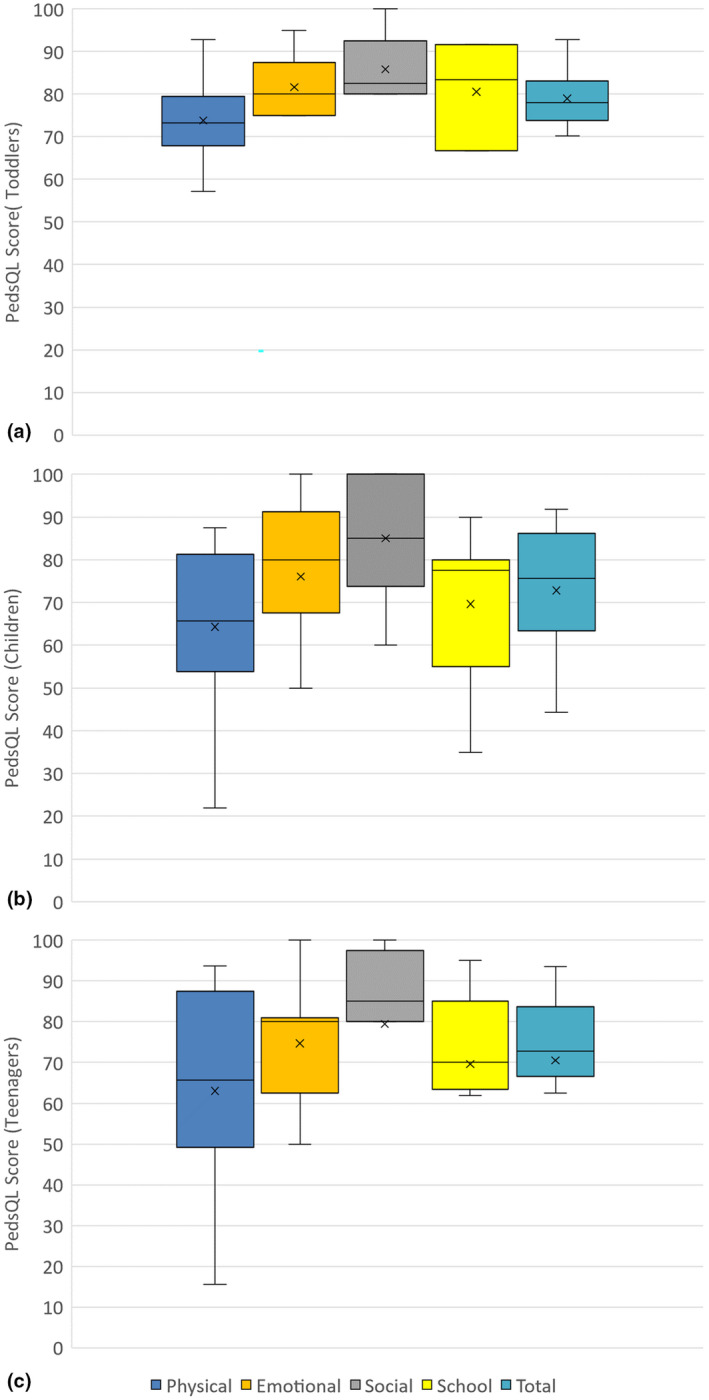
Caregiver reported quality of life for children with SCD. Caregiver‐reported PedsQL scores for (a) Toddlers aged 2–4 (n = 6), (b) Children aged 8–12 (n = 14), and (c) Teenagers 13–17 (n = 9). PedsQL is scored from 0 to 100; higher scores indicate higher perceived quality of life. No caregivers with children aged 5–7 participated in this study.
3.4. Caregiver burden
Each caregiver participant's CBS‐ZBI was calculated by taking the sum of the score of each question. Higher scores indicate greater caregiver burden. The majority of CBS‐ZBI surveys scored on the “little or no burden” scale (54.8%), while no surveys scored on the “severe burden” scale (Table 6). These results were analyzed with respect to demographic variables of both the caregiver and the child with SCD. For the purposes of statistical analysis, the two levels of burden (mild to moderate burden and moderate to severe burden) were collapsed into one level of burden. None of the demographic variables were significantly associated with having caregiver burden (Table S4).
TABLE 6.
Caregiver Burden for Sickle Cell Disease in Grenada a .
| Scale | n (%) |
|---|---|
| Little or no burden (0 – 20) | 14 (53.8) |
| Mild to moderate burden (21 – 40) | 9 (34.6) |
| Moderate to severe burden (41–60) | 3 (11.5) |
| Severe burden (61–88) | 0 (0.0) |
Responses for each survey question were scored from “never” (score of 0) to “nearly always” (score of 4).aResponses for each survey question were scored from “never” (score of 0) to “nearly always” (score of 4).
3.5. Genetics knowledge
All participants completed 6 genetics knowledge questions to determine their understanding of the genetics of SCD. Of the adult participants, most were able to correct identify that SCD is a genetic condition (84.3%) and that an individual cannot catch SCD later in life (68.4%), but the majority were not able to correct identify the recurrence risk (Figure 2a). The inability to correctly answer the genetics knowledge questions were not associated with any demographic variable or negative quality of life. Individuals who did not understand that SCD is a genetic condition perceived fewer barriers in the Barriers to Care subscale of Knowledge (mean = 93.8) compared to those with a correct understanding (68.8; t‐test, p = 0.04; Table S5).
FIGURE 2.
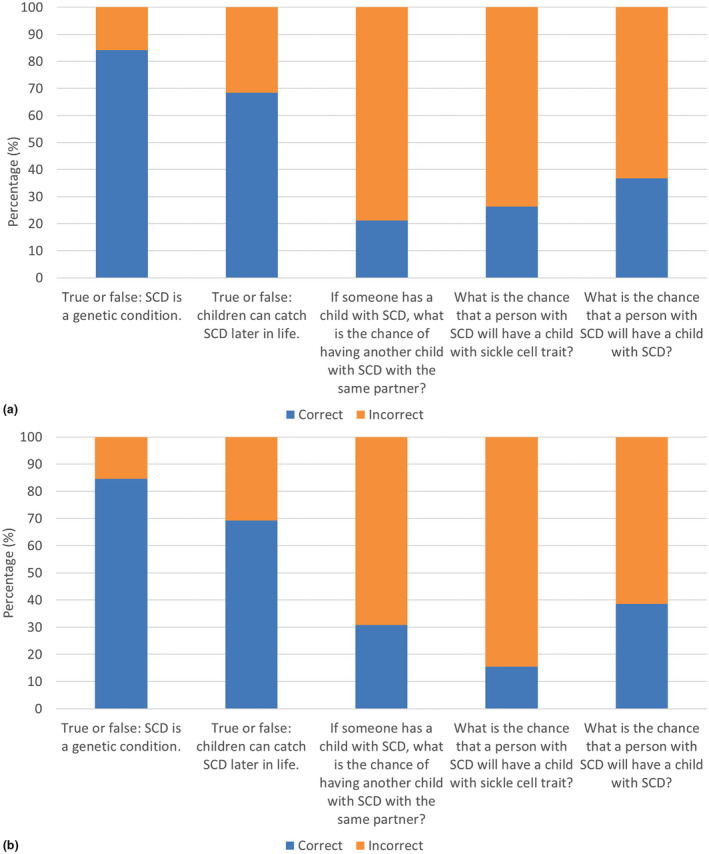
Basic knowledge of SCD genetics. Graded responses to questions about knowledge of sickle cell disease genetics for (a) Adult participants (n = 19) and (b) Caregiver participants (n = 26). The percentage of correct and incorrect answers for each question is shown.
Genetics knowledge in the caregiver cohort was similar to the adult cohort, with 84.6% of the participants correctly indicating that SCD is a genetic condition, 69.2% indicating that an individual cannot catch SCD later in life, and the majority unable to correctly select the recurrence risk (Figure 2b). While participants with 3 or more children were more likely to answer correctly that SCD is a genetic condition than those with 1–2 children (t‐test, p = 0.03), this response was not dependent upon the number of children with SCD (t‐test, p = 0.35; Table S6). Individuals with an incorrect understanding that SCD is a genetic condition reported greater barriers in the Barriers to Care subscale of Skills (mean = 60.9) compared to those with a correct understanding (83.2; t‐test, p = 0.002; Table S7).
4. DISCUSSION
Despite the prevalence of SCD in Grenada, little is known about the experience of the individuals living with the condition in this population, or the broader Caribbean community. This study explored the perceived quality of life and barriers to care for both adults and children with SCD in Grenada. Barriers to care were evaluated through the BCQ, which was previously used to assess children with SCD in California (Jacob et al., 2015). In California, participants averaged 79.2 on the BCQ, compared to 69.9 for adults and 75.5 for children in our study, demonstrating greater barriers overall in Grenada compared to the USA. Additionally, the California study found the greatest barriers in the subscale of Pragmatics, consistent with the results for the children with SCD in our study. This suggests that both populations have problems accessing clinic services due to transportation and wait times, which was further corroborated by the comments provided by the caregivers (Table 4). For the adults with SCD, all of the subscales had similar scores, suggesting that there is no one factor contributing more than the others. The BCQ has not been reported for adults with SCD, and barriers to care in Caribbean countries have not been assessed. Therefore, these results provide novel information regarding the experience in Grenada. Many of the comments in Table 4 also provide insight into the significant barriers, such as one individual who mentioned how medication was not always available to patients. Even though curative therapies are becoming available in the USA Salinas Cisneros and Thein, 2020), individuals in Grenada lack more traditional interventions, such as hydroxyurea, that have been available in developed countries for several years.
Our study also assessed quality of life for individuals with SCD in Grenada through the ASCQ‐ME for adults and the PedsQL for children with SCD. The ASCQ‐ME is standardized to a “general population” with a t‐score of 50; the general population was derived from responses from individuals with SCD in the USA. The domains of Emotional and Social Functioning Impact were below this cutoff, while Sleep, Stiffness, and Pain Impact were above (Table 5). This indicates that SCD in this population has a significant impact on emotional well‐being, causing depression, loneliness, and isolation from family and friends. Participants perceived less impact on their sleep patterns, level of joint stiffness, or amount of pain compared to the general population. Previous reports have stratified results of the ASCQ‐ME based on reported severity of symptoms for patients with SCD in the USA (Keller et al., 2017), and based on this stratification, our cohort of adults with SCD mimicked results for patients with high severity in Emotional Functioning, patients with medium severity in Social Functioning, and patients with low severity in Sleep, Stiffness, and Pain Impact. In addition, analysis using the ASCQ‐ME in a population of SCD patients in Jamaica resulted in scores in the 50 s for all the domains, with the lowest score as 53.0 in Emotional Impact (Bulgin et al., 2018). The results in our cohort were lower, with the lowest score as 47.5 in Emotional impact. Given these results, adults with SCD in Grenada perceive a poorer quality of life than those in Jamaica. Both cohorts reported the most issues in the Emotional Impact domain, suggesting that those with SCD in the Caribbean struggle the most with depression and loneliness due to their diagnosis.
The quality of life for children with SCD was assessed through the PedsQL Generic Scales by caregiver‐proxy. The PedsQL Sickle Cell Disease Module was not utilized because we believed the terminology was more specific to a high‐income population (Panepinto et al., 2013). Toddlers scored 78.9, children scored 72.5, and teenagers scored 70.6 for the total score of the PedsQL (Figure 1). A previous study attempted to determine a cutoff for the PedsQL, with the total score for ages 2–7 as 83 and for ages 8–17 as 78 (Huang et al., 2009). Our cohort scored below these cutoffs, suggesting poor quality of life. Additionally, our cohort scored 20 points below the cutoff for the Physical subscale, meaning that the caregivers perceived that their affected children had significant limitations with regard to walking, running, and participating in sports. Another study utilizing the PedsQL in children with SCD stratified the results by child (ages 8–17) self‐report and caregiver‐report (Dale et al., 2011). In this study completed in the USA, the total score for the PedsQL by caregiver‐proxy was 62.9, lower than the scores for any of our age groups; the lowest subscale score was in School Functioning, while our cohort's lowest score was in Physical Functioning. These results demonstrate that children with SCD in Grenada have different struggles with regard to quality of life than children with SCD in the USA, but caregivers do not perceive as great of an impact on quality of life in Grenada as in the USA.
We also assessed caregiver burden for those with caring for children with SCD in Grenada. Overall, this cohort scored low on the CBS‐ZBI, with over half (53.8%) of respondents scoring in “little to no burden” (Table 6). This measure was designed for caregivers of individuals with dementia, but has been utilized for caregivers of individuals with SCD in Brazil (Silva et al., 2012). In this study, respondents scored an average of 1.5–2 points per question (responding “sometimes” to most questions), compared to our study in which respondents scored an average of 1 point (responding “rarely” to most questions). These results suggest that caregivers of children with SCD in Grenada do not perceive significant caregiver burden. However, given that the CBS‐ZBI was not validated for this population, it is possible that these results do not accurately reflect the sentiments of the caregivers in our cohort. For example, the CBS‐ZBI results are inconsistent with some of the comments provided by participants, such as the comment by Participant 28: “I wish I could have some help to understand how to deal with a child with SCD.” (Table 4) This statement suggests that even though the participant scored 16 or “little or no burden” on the CBS‐ZBI, she perceives some level of burden with caring for a child with SCD. Additionally, cultural influences in Grenada may decrease the level of perceived caregiver burden. Previous studies, including some in the Caribbean, determined that cultural values, such as sense of obligation and spirituality, had direct effects on positive aspects of caregiving, leading to lower perceived caregiver burden when compared to populations in the USA (Friedemann et al., 2013; Tang, 2011; Scharlach et al., 2006).
Finally, genetics knowledge was assessed by true‐false and multiple‐choice questions. These questions assessed understanding that SCD was a genetic condition and its recurrence risk. The majority of respondents knew that SCD was a genetic condition; however, 30% still believed that an individual could catch SCD later in life, and 70–85% could not correctly indicate recurrence risk, demonstrating much lower understanding of the inheritance of SCD than found in previous studies (Orelaru et al., 2019; Boyd et al., 2005; Smith & Brownell, 2018). One such study found that all respondents who had a child with SCD knew it was genetic and only 20–30% were not able to indicate the correct recurrence risk (Acharya et al., 2009). In our study, multiple respondents left comments explaining that they wanted to know more about how SCD is inherited (Table 4), suggesting a need for more time to be spent with medical staff explaining the genetics of SCD to patients with the condition and their caregivers rather than simply treating the symptoms and that this should be done as part of routine care in an ongoing basis as the patient ages.
The results of this study demonstrate that SCD has a negative impact on health‐related quality of life for individuals with the condition. This study also identified significant barriers to healthcare, including concerns regarding transportation to hospital, wait times in clinics, and lack of adequate medical treatments. However, this study also has several limitations. The diagnosis of SCD was self‐reported and not confirmed by genetic testing. It is possible that symptomatic carriers were included in the study, but we believe this is unlikely given that most individuals were identified through the SCAG. All of the measures used were originally validated in the USA. Individuals in Caribbean countries likely experience different challenges from those in the USA, and the subscales included in the validated measures may not be most appropriate for this population. This is especially true for the CBS‐ZBI, given the unexpected results. Expectations from the healthcare system may also not be the same for Caribbean populations as they are in North America or the United Kingdom. An important expansion of this study would be a qualitative interview to determine if additional barriers to care or aspects of quality of life significant to this population were not included in the validated measures. Another limitation is that the PedsQL was completed by caregiver‐proxy, which may not be reflective of the child's perceived experience. This study was also limited by the small number of respondents, primarily due to the size of the population of Grenada and the recruitment methods, including knowledge of the monthly sickle cell clinic or the SCAG. The small number of respondents likely limited the significant findings in the data. However, based on the incidence of SCD in Grenada (Shah et al., 2014), ~7% of the total population with SCD in Grenada was surveyed, which can also be seen as a strength of this study.
Another strength was that the experience of adults with SCD was investigated, which has not been previously reported; however, additional studies are needed to further explore the challenges and expectations of this population. Determining where and how often these adult patients receive healthcare, given that clinics for SCD in Grenada are only for children and that the healthcare after age 18 is limited to acute concerns, is a critical need for this population. As mentioned in one of the comments by a survey participant (Table 4), transitional and adult clinics would allow continued surveillance and treatment of older individuals with SCD; thus, qualitative research to investigate specific concerns may provide more comprehensive information. Comparative studies of other populations in the Caribbean could assess if the experience of individuals with SCD in Grenada is similar or distinct. It is also important to evaluate the understanding of SCD by healthcare providers in Grenada to determine if barriers to care exist in that domain. The utilization of the results from this study and further research could improve the delivery of healthcare to people affected by SCD in Grenada. Currently, given the varied age at diagnosis, implementation of a comphrehensive newborn screening program with integrated healthcare for both children and adults could greatly reduce morbidity by allowing earlier intervention, education, and access to care across the lifespan.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
AG, BN, FI, AKS, SHE: conceptualization; AG, SHE: methodology; AG, RS, DB, FI, SHE: data collection; AG: data analysis; AG: manuscript draft preparation; AG, PRF, AKS, BN, SHE: manuscript review and editing.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank St. George’s University for supporting travel to Grenada. We also thank the Sickle Cell Association of Grenada (SCAG) for their support and encouragement throughout this study.
Contributor Information
Andrew K. Sobering, Email: asobering@sgu.edu.
Sarah H. Elsea, Email: sarah.elsea@bcm.edu.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Acharya, K. , Lang, C. W. , & Ross, L. F. (2009). A Pilot Study to Explore Knowledge, Attitudes, and Beliefs about Sickle Cell Trait and Disease. Journal of the National Medical Association, 101(11), 1163–1172. [DOI] [PubMed] [Google Scholar]
- Al‐Rawashdeh, S. Y. , Lennie, T. A. , & Chung, M. L. (2016). Psychometrics of the zarit burden interview in caregivers of patients with heart failure. Journal of Cardiovascular Nursing, 31(6):E21‐E28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, M. , & Asnani, M. (2013). “You Just Have to Live With It": Coping with sickle cell disease in Jamaica. Qualitative Health Research, 23(5), 655–664. [DOI] [PubMed] [Google Scholar]
- Boyd, J. H. , Watkins, A. R. , Price, C. L. , Fleming, F. , & DeBaun, M. R. (2005). Inadequate community knowledge about sickle cell disease among African‐American women. Journal of the National Medical Association, 97(1), 62–67. [PMC free article] [PubMed] [Google Scholar]
- Bulgin, D. , Tanabe, P. , Monika, A. , & Douglas, C. (2018). Health related stigma and quality of life in adults with sickle cell disease in Jamaica. Blood, 132(Supplement 1), 2285. [Google Scholar]
- Dale, J. C. , Cochran, C. J. , Roy, L. , Jernigan, E. , & Buchanan, G. R. (2011). Health‐related quality of life in children and adolescents with sickle cell disease. Journal of Pediatric Health Care, 25(4), 208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedemann, M.‐L. , Buckwalter, K. C. , Newman, F. L. , & Mauro, A. C. (2013). Patterns of caregiving of Cuban, other Hispanic, Caribbean black, and white elders in South Florida. Journal of Cross‐Cultural Gerontology, 28(2), 137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenada, M. C. PAHO/WHO ‐ Home ‐ Pan American Health Organization. 2019. https://www.paho.org/salud-en-las-americas-2012/index.php?option=com_content%26view=article%26id=42:grenada%26Itemid=150%26lang=en
- Huang, I.‐C. , Thompson, L. A. , Chi, Y.‐Y. , Knapp, C. A. , Revicki, D. A. , Seid, M. , & Shenkman, E. A. (2009). The linkage between pediatric quality of life and health conditions: establishing clinically meaningful cutoff scores for the PedsQL. Value Health, 12(5), 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob, E. , Childress, C. , & Nathanson, J. D. (2015). Barriers to care and quality of primary care services in children with sickle cell disease. Journal of Advanced Nursing, 72(6), 1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink, K. K. , Chini, B. , Cowens, J. , Kremer, L. , & Lin, L. (2019). Improving clinical outcomes and quality of life with massage therapy in youth and young adults with cystic fibrosis: A pilot study. International Journal of Therapeutic Massage & Bodywork, 12(1), 4–15. [PMC free article] [PubMed] [Google Scholar]
- Keller, S. , Yang, M. , Treadwell, M. J. , & Hassell, K. L. (2017). Sensitivity of alternative measures of functioning and wellbeing for adults with sickle cell disease: comparison of PROMIS® to ASCQ‐Me℠. Health and Quality of Life Outcomes, 15(1):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, S. D. , Yang, M. , Treadwell, M. J. , Werner, E. M. , & Hassell, K. L. (2014). Patient reports of health outcome for adults living with sickle cell disease: development and testing of the ASCQ‐Me item banks. Health and Quality of Life Outcomes, 12(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight‐Madden, J. , Lee, K. , Elana, G. et al (2019). Newborn screening for sickle cell disease in the Caribbean: an update of the present situation and of the disease prevalence. International Journal of Neonatal Screening, 5(1), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelaru, F. , Bolanle, G. , Tolulope, I. , & Ishmael, J. (2019). Assessing knowledge of sickle cell trait/disease inheritance in metropolitan Detroit. Journal of the National Medical Association, 111(6), 656–664. [DOI] [PubMed] [Google Scholar]
- Özyazıcıoğlu, N. , Avdal, E. Ü. , & Sağlam, H. (2017). A determination of the quality of life of children and adolescents with type 1 diabetes and their parents. International Journal of Nursing Sciences, 4(2), 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panepinto, J. A. , Torres, S. , Bendo, C. B. et al (2013). PedsQL™ sickle cell disease module: Feasibility, reliability, and validity. Pediatric Blood & Cancer, 60(8), 1338–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe, P. (1995). Hydroxyurea for sickle cell disease. Lancet, 345(8945), 311. [Google Scholar]
- Salinas Cisneros, G. , & Thein, S. L. (2020). Recent advances in the treatment of sickle cell disease. Frontiers in Physiology, 11, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharlach, A. E. , Kellam, R. , Ong, N. , Baskin, A. , Goldstein, C. , & Fox, P. J. (2006). Cultural attitudes and caregiver service use. Journal of Gerontological Social Work, 47(1–2), 133–156. [DOI] [PubMed] [Google Scholar]
- Seid, M. , Sobo, E. J. , Gelhard, L. R. , & Varni, J. W. (2004). Parents reports of barriers to care for children with special health care needs: development and validation of the barriers to care questionnaire. Ambulatory Pediatrics, 4(4), 323–331. [DOI] [PubMed] [Google Scholar]
- Shah, P. , Macpherson, C. , & Akpinar‐Elci, M. (2014). Impact of undertreated sickle cell pain in the Caribbean. West Indian Medical Journal Open, 1(2), 63–66. [Google Scholar]
- Silva, L. B. L. D. , Ivo, M. L. , Souza, A. S. D. , Pontes, E. R. J. C. , Pinto, A. M. A. C. , & Araujo, O. M. R. D. (2012). The burden and quality of life of caregivers of sickle cell anemia patients taking hydroxyurea versus those not taking hydroxyurea. Revista Brasileira de Hematologia e Hemoterapia, 34(4), 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, M. , & Brownell, G. (2018). Knowledge, beliefs, attitudes, and behaviors regarding sickle cell disease: Implications for prevention. Social Work in Public Health, 33(5), 299–316. [DOI] [PubMed] [Google Scholar]
- Steensma, D. P. , Kyle, R. A. , & Shampo, M. A. (2010). Walter clement noel—first patient described with sickle cell disease. Mayo Clinic Proceedings, 85(10), e74–e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, M. (2011). Can cultural values help explain the positive aspects of caregiving among Chinese American caregivers? Journal of Gerontological Social Work, 54(6), 551–569. [DOI] [PubMed] [Google Scholar]
- Treadwell, M. J. , Hassell, K. , Levine, R. , & Keller, S. (2014). Adult sickle cell quality‐of‐life measurement information system (ASCQ‐Me). Clinical Journal of Pain, 30(10), 902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varni, J. W. , Limbers, C. A. , & Burwinkle, T. M. (2007). Parent proxy‐report of their children’s health‐related quality of life: an analysis of 13,878 parents reliability and validity across age subgroups using the PedsQL™ 4.0 Generic Core Scales. Health and Quality of Life Outcomes, 5(1), 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varni, J. W. , Seid, M. , & Kurtin, P. S. (2001). PedsQL™ 4.0: Reliability and Validity of the Pediatric Quality of Life Inventory™ Version 4.0 Generic Core Scales in Healthy and Patient Populations. Medical Care, 39(8), 800–812. [DOI] [PubMed] [Google Scholar]
- Wethers, D. , Pearson, H. , & Gaston, M. (1989). Newborn screening for sickle cell disease and other hemoglobinopathies. Pediatrics, 83(5), 813–814. [PubMed] [Google Scholar]
- Zarit, S. H. , Reever, K. E. , & Bach‐Peterson, J. (1980). Relatives of the impaired elderly: correlates of feelings of burden. Gerontologist, 20(6), 649–655. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.