

Abstract
Background
Autosomal dominant hearing loss (ADHL) accounts for about 20% of all hereditary non‐syndromic HL. Truncating mutations of the EYA4 gene can cause either non‐syndromic ADHL or syndromic ADHL with cardiac abnormalities. It has been proposed that truncations of the C‐terminal Eya domain lead to non‐syndromic HL, whereas early truncations of the N‐terminal variable region cause syndromic HL with cardiac phenotype.
Methods
The proband and all the other hearing impaired members of the family underwent a thorough clinical and audiological evaluation. The cardiac phenotype was examined by ECG and echocardiography. Their DNA was subjected to target exome sequencing of 129 known deafness genes. The sequencing data were analyzed and the candidate variants were interpreted following the ACMG guidelines for clinical sequence interpretation. The effect of candidate variant on EYA4 gene expression was assessed by quantitative PCR and western blot of gene production in blood.
Results
We report a Chinese family cosegregating post‐lingual onset, progressive ADHL with a novel nonsense mutation NM_004100.4:c.543C>G (p.Tyr181Ter) of EYA4. Two affected members show no cardiac abnormalities at least until now revealed by electrocardiography and echocardiography. The overall expression level of the EYA4 gene in the proband was lower than that in his unaffected relative.
Conclusion
This report expands the mutational spectrum of the EYA4 gene and highlights the fact that more data are needed to elucidate the complex genotype–phenotype correlation of EYA4 mutations.
Keywords: autosomal dominant hearing loss, EYA4, next‐generation sequencing
It has been proposed that truncations of the C‐terminal Eya domain of EYA4 lead to non‐syndromic HL, whereas early truncations of the N‐terminal variable region cause syndromic HL with cardiac phenotype. We report a Chinese family cosegregating post‐lingual onset, progressive HL without cardiac abnormalities with a novel nonsense mutation NM_004100.4:c.543C>G (p.Tyr181Ter) of EYA4, encoding an early truncated protein with partial N‐terminal variable region. This report expands the mutational spectrum of the EYA4 gene and highlights the fact that more data are needed to elucidate the complex genotype–phenotype correlation of EYA4 mutations.

1. INTRODUCTION
Hearing loss is the most common sensory defect in humans. The prevalence of permanent HL at birth is 1.07/1000 (Morton & Nance, 2006). HL is a heterogeneous disorder caused by genetic and environmental factors, yet half of the cases tend to be genetic (Nance, 2003). If the pathology is genetically related, it can be classified according to the pattern of inheritance (autosomal dominant, autosomal recessive, and X‐linked), and the presence (syndromic) or absence (non‐syndromic) of abnormalities in other parts of the body. It has been estimated that approximately 80% of genetic deafness is non‐syndromic, where 80% of non‐syndromic HL have been inherited in an autosomal recessive manner (Morton & Nance, 2006). In contrast to the autosomal recessive HL, which is congenital and prelingual, patients with autosomal dominant non‐syndromic HL (ADNSHL) often have a family history of post‐lingual HL.
So far, a total of 47 genes have been identified as causal genes for ADNSHL (http://hereditaryhearingloss.org), including DIAPH1 (Lynch et al., 1997), KCNQ4 (Kubisch et al., 1999), and GJB3 (Xia et al., 1998). Autosomal dominant deafness 10 (DFNA10, OMIM #601316) locus was first identified and mapped on chromosome 10 in 1996 in a large American family as a causal gene for ADNSHL (O'Neill et al., 1996). In 2001, Wayne et al. identified nonsense mutations in the EYA4 gene (OMIM: 603550) in two unrelated families segregating at DFNA10, and defined EYA4 (Eyes Absent Homolog 4) as the causative gene for DFNA10 (Wayne et al., 2001). The EYA4 gene is composed of 21 exons encoding a 640 amino acid protein, which contains a highly conserved region (eya homologous region, eyaHR) and a more divergent proline‐serine‐threonine (PST)‐rich transactivation domain (eya variable region, eyaVR) (Borsani et al., 1999). EYA4 is a member of the vertebrate eya gene family, a group of transcriptional activators that interact with other proteins to ensure the healthy development of multiple organs, including the eye, muscle, kidney, inner ear, and heart (Wang et al., 2008; Wayne et al., 2001). Although some mutations in EYA4 cause syndromic HL with dilated cardiomyopathy (DCM) as described in a family (Schonberger et al., 2000, 2005) and a Japanese patient (Abe et al., 2018), the majority of mutations in this gene are associated with ADNSHL. Hearing impairment for EYA4 mutations is characterized by post‐lingual and progressive sensorineural HL. To date, 34 diseases causing EYA4 mutations have been described, including four nonsense mutations, 12 missense mutations, nine small indels, six splicing mutations, and three gross deletions (Stenson et al., 2012).
In this study, we report on a case of a three‐generation family with late‐onset progressive sensorineural HL. The genetic cause was investigated using targeted sequencing of 129 known deafness genes. A novel nonsense variant, NM_004100.4:c.543C>G (p.Tyr181Ter) in exon 8 of the EYA4 gene, was identified in the family. The onset age and audiograms were compared with other studies, and the impact of reported mutations on cardiac phenotype has been discussed. This study expands the mutational spectrum of the EYA4 gene and suggests that next‐generation sequencing is effective in elucidating the etiology of familiar progressive HL.
2. MATERIALS AND METHODS
2.1. Ethical compliance
This study was conducted according to the Helsinki Declaration and approved by the local institutional ethics committee in human research of Zhengzhou University (reference no. 2018008). Written informed consent for genetic testing and the submission of the study was obtained from all the studied individuals.
2.2. Subjects and clinical evaluation
The proband was a 28‐year‐old male Chinese, who was referred to the Department of Otolaryngology at the Second Affiliated Hospital of Zhengzhou University in Zhengzhou. Detailed family history questioning revealed the other four affected members across three generations with AD inheritance patterns (Figure 1). All affected family members underwent a medical history interview that included questions on onset age and degree of HL, other clinical manifestations, and environmental factors (viral infection, noise, and ototoxic drugs). A complete clinical evaluation, audiological tests, and cardiac examination were performed in the affected family members who agreed to participate in this study. The level of HL was classified into four tiers in terms of pure tone average (averaged over 0.5, 1.0, 2.0, and 4.0 kHz): mild (21–40 dB), moderate (41–70 dB), severe (71–90 dB), and profound (>91 dB). .
FIGURE 1.
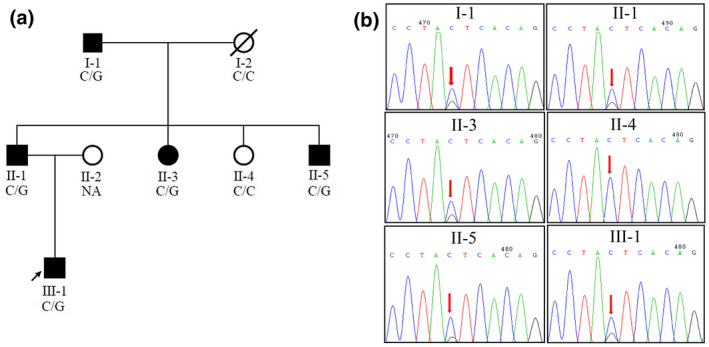
Pedigree of the family with non‐syndromic autosomal dominant sensorineural hearing loss segregating EYA4 c.543C>G (canonical transcript NM_172105.3.). (a) Family pedigree. Open symbols unaffected; filled black symbols affected; diagonal line deceased. (b) Sanger sequencing. The red arrow indicates the EYA4 c.543C>G mutation
2.3. Target exome sequencing of known deafness genes
Genomic DNA was isolated from peripheral blood leukocytes using the GenMagBio Genomic DNA Purification kit (GenMagBio, Changzhou, China) following the manufacturer's standard procedures. Genomic DNA from affected and unaffected members was fragmented to an average size of 250 bp, and end repair, adapter ligation as well as PCR enrichment were performed following the protocol for VAHTS TM Universal DNA Library Prep Kit for Illumina V3 (Vazyme Biotech Co., Ltd). The enriched DNA libraries were captured with the Human Deafness Panel oto‐DA3 (Otogenetics Corporation) with 129 known HL genes, following the manufacturer's protocol. The resulting libraries were sequenced on an Illumina HiSeq 4000 sequencer (Illumina Inc.) with the paired‐end of 150 bp at the Precision Medicine Center of Zhengzhou University, Zhengzhou, China.
2.4. Bioinformatics analysis and variant interpretation
Sequencing adapters and low‐quality reads were trimmed from raw reads with Trimmomatic (Bolger et al., 2014). Clean reads were aligned to the human reference genome (version GRCh37) using Burrow‐Wheeler Aligner (version 0.7.17‐r1188) (Li, 2013). Duplicate reads were flagged by sambamba (version 0.6.6) (Tarasov et al., 2015). Single nucleotide variants (SNVs) and small indels were characterized using the Genome Analysis Toolkit version 4 (GATK4) HaplotypeCaller (DePristo et al., 2011). Variant annotation was performed using Vcfanno (Pedersen et al., 2016) with several variant frequency databases for healthy population, such as 1000 Genomes Project database (Genomes Project et al., 2015), dbSNP (Sherry et al., 2001), Exome Aggregation Consortium (ExAC) (Lek et al., 2016), Genome Aggregation Database (gnomAD) (Karczewski et al., 2019), ClinVar (Landrum et al., 2018), InterVar (Li & Wang, 2017), and dbNSFP (Liu et al., 2016), which compiled mutation prediction scores from many algorithms (Ioannidis et al., 2016; Kumar et al., 2009; Schwarz et al., 2014). All analysis steps described above were performed in the framework of bcbio‐nextgen (https://github.com/bcbio/bcbio‐nextgen), which provides best practice pipelines for variant calling, annotation, and validation. We filtered out variants with minor allele frequency >0.05 in any general continental population in which at least 2000 alleles were observed in the gnomAD database, except the ones on ACMG benign stand‐alone exception list or those linked to diseases in ClinVar database (Ghosh et al., 2018). Variant interpretation was done by an expert panel of an otorhinolaryngologist, a bioinformatician, and a molecular geneticist following the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG) guidelines for clinical sequence interpretation (Richards et al., 2015). Variant nomenclature was based on EYA4 canonical transcript NM_172105.3. The phenotype and pathogenic variant were submitted to the LOVD database (https://databases.lovd.nl/shared/individuals/00320009).
2.5. Sanger sequencing
To confirm candidate variants detected by next‐generation sequencing, we performed PCR amplification and Sanger sequencing. The following primers were designed using Primer‐BLAST software: forward, 5′‐CATGGTGAGTGTGAATTTGCTCA‐3′; reverse, 5′‐GTGTAGCTACTTACCTGGCATT‐3′. PCR products were purified by a PCR purification kit (Life Sciences) and sequenced using a SeqStudio Genetic Analyzer (Applied Biosystems/Life Technologies).
2.6. Functional study of EYA4 nonsense mutation
Whole peripheral blood from the proband (III‐1) and his unaffected aunt (II‐4) was collected in EDTA tubes, and total RNA was extracted from blood mononuclear cells ~12 hours after collection using the TRIzol method (Cat. No. 15596026). RNA was reverse transcribed into cDNA using the HiScript III RT SuperMix for qPCR (Vazyme). The forward and reverse primers for EYA4 were 5′‐TCCCCACAGCTGTATCCTTC‐3′ and 5′‐AACTGAGGCAGCCACTCTGT‐3′ as validated in a previous study (Zou et al., 2005). The primers for human GAPDH (internal control, PrimerBank accession number: 378404907c3) were 5′‐CTGGGCTACACTGAGCACC‐3′ and 5'‐AAGTGGTCGTTGAGGGCAATG‐3'. The amplification length for EYA4 and GAPDH was 250 bp and 101 bp, respectively. Relative expression levels of wild‐type and mutant EYA4 transcripts were measured by quantitative real‐time PCR (qPCR) using AceQ qPCR SYBR Green Master Mix (Vazyme) and the ABI StepOnePlus Real‐Time PCR System (Thermo Fisher Scientific). Each condition was replicated for three times and the ΔΔCt method was used to quantify qPCR results.
The serum was removed from anticoagulated whole blood by centrifugation and the red blood cells were lysed using the red blood cell lysis buffer (Solarbio, cat no. R1010). Then, RIPA cell lysate (Solarbio, cat no. R0010) and protease inhibitor (MedChemExpress, cat no. HY‐K0010) were used to rupture mononuclear cells to obtain total protein. The extracted protein was quantified by BCA protein assay kit (Beyotime, cat no. P0010S). The total protein was separated in a 10% gel with SDS‐PAGE, and then transferred to PVDF membrane (Beyotime, cat no. FFP39). The PVDF membrane was blocked with 5% non‐fat milk in TBST solution at room temperature and then incubated with the primary EYA4 antibody (univ‐bio, cat no. abs111928). Beta‐actin served as a loading control. The membranes were incubated with a horseradish peroxidase‐conjugated secondary antibody (Transgene, cat no. HS101‐01).
3. RESULTS
3.1. Clinical features of the family
The three‐generation family had five affected individuals, with age ranging from 28 (III‐1) to 80 (I‐1). The family exhibited a typical autosomal dominant inheritance pattern (Figure 1a). The self‐reported onset age of HL ranged from the second to the fourth decade. The age of onset was 25 years old for the proband with mild sensorineural HL involving all frequencies; the average auditory threshold of air conduction measurements for the right and left ear was 26 dBHL and 33 dBHL, respectively (Figure 2). In the fifth decade, the severity of the HL generally became severe‐to‐profound across all frequencies. The affected individuals reported the use of hearing aids in the third or fourth decade, as II‐1, II‐3, and II‐5. No family members received cochlear implants, and in most cases, the HL was not severe enough to warrant such intervention. The grandpa of the proband (I‐1, 80 years old) manifested severe sensorineural HL, involving entire frequencies. No tinnitus or vestibular dysfunction (such as balance disorders, vertigo, or Meniere disease) was observed in any of the patients. All members denied any family history of cardiomyopathy, and the electrocardiogram and echocardiography conducted on two affected individuals (II‐1 and III‐1) showed no evidence of DCM or other abnormalities (Figure 3). The clinical examinations ruled out the probability of syndromic HL with heart problems.
FIGURE 2.
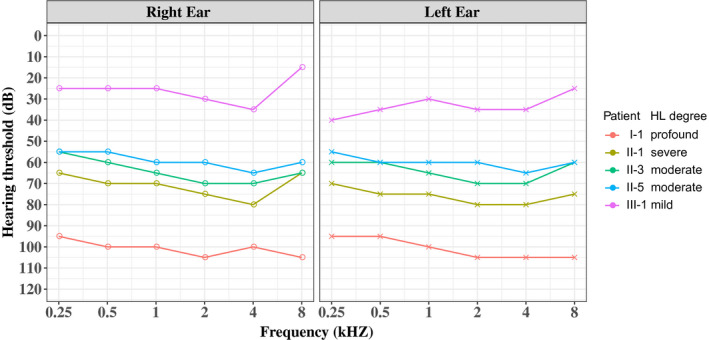
Audiometric phenotypes of five affected members in the family. Pure‐tone air conduction thresholds are shown for right and left ears, and the HL severity is summarized for the ear with better hearing
FIGURE 3.
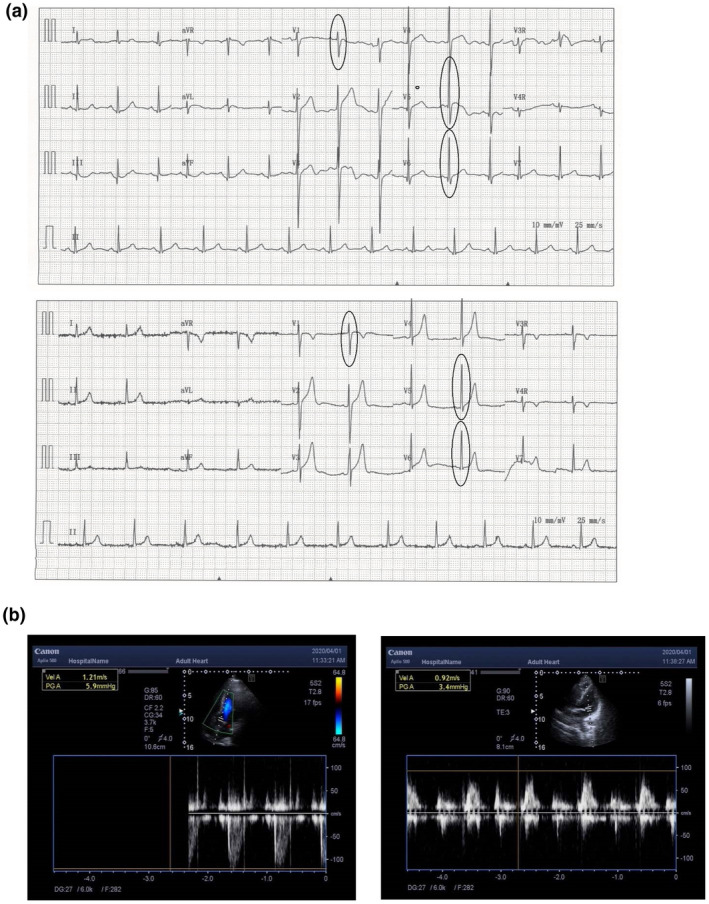
Normal cardiac phenotypes of the proband (a and c) and his father (b and d). (a and b) Electrocardiogram; (c and d) echocardiograph
3.2. Target exome sequencing identifies a novel EYA4 mutation
Target exome sequencing of 129 known deafness genes covered by the Otogenetics Human Deafness Panel oto‐DA3 was performed for one unaffected and four affected individuals for the identification of potentially pathogenic variants. An average of one billion raw base pairs (1 Gbp) were generated for each sample, with more than 85% of bases having a Phred quality score Q ≥ 30 (Q30). More than 99% of clean reads can map the human genome reference sequence (GRCh37 version), and the average sequencing depth of targeted regions was 171.5X (ranging from 158.3X to 187.9X), covering ≥95% of target regions more than 20 times. After filtering out variants with allele frequency >0.1% in the database of the normal population (gnomAD and ExAC), a total of 14 variants were obtained, including variants in genes MITF, GJB3, and EYA4, which were previously associated with HL. The only variant in EYA4 was cosegregated with HL in the family. Sanger sequencing of this variant in the family was verified, and the mutation was present in the affected members and absent in the unaffected ones (Figure 1b).
3.3. Variant interpretation of the mutation as pathogenic
The EYA4 (NM_172105.3) variant c.543C>G (p.Tyr181Ter) was found in exon 8; amino acid of 181 positions changed from tyrosine to termination codon, which led to early termination of protein translation. According to the standards and guidelines for interpreting genetic variants proposed by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG) (Richards et al., 2015), this variant was classified as pathogenic. It was absent from all population databases, including gnomAD and ExAC (Pathogenic Moderate 2, PM2). The mutation is a null mutation that leads to a non‐functional protein product (Pathogenic Very Strong 1, PVS1). The variant was cosegregated with the phenotype of DFNA10 in the affected family members (Pathogenic Supportive 1, PP1). This mutation has not yet been deposited in the database of HGMD professional edition and is a novel pathogenic variant.
3.4. Functional study of EYA4 nonsense mutation
To explore the effect of nonsense mutation on EYA4 expression, we investigated the expression of EYA4 gene at both transcriptional and protein levels in the proband and his unaffected relative. We extracted total RNA and total protein from the fresh peripheral blood from the proband and his healthy aunt. We observed that the mRNA levels of EYA4 gene in the proband decreased by ~50% compared to the unaffected relative (Figure 4a). The western blot analysis revealed that compared with his aunt the protein level of EYA4 gene was much lower in the proband (Figure 4b). These results suggest that the mutant transcript with nonsense mutation is subject to nonsense‐mediated mRNA decay (NMD).
FIGURE 4.
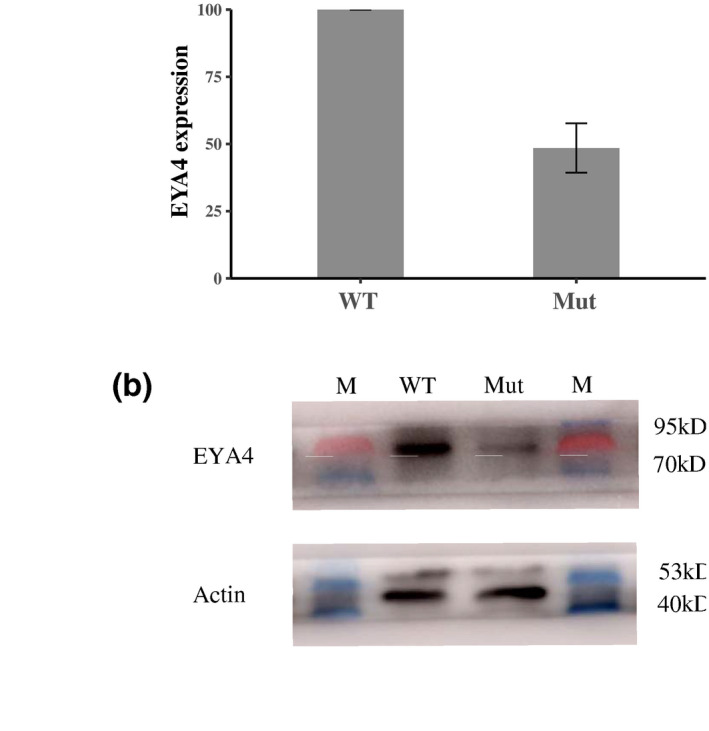
Reduced expression of EYA4 in blood in the proband compared with his unaffected aunt. (a) Quantitative PCR analysis of EYA4 expression (b) western blot analysis of EYA4 protein expression in the proband (Mutant, Mut) and his aunt with normal hearing (wild‐type, WT). M, protein makers. The qPCR experiment was repeated three times and bars represent standard deviation
4. DISCUSSIONS
In the present study, we reported on a three‐generation Han Chinese family with post‐lingual non‐syndromic HL. All the affected individuals in the family had sensorineural deafness and onset age from 26 to 50 years old, affecting primarily low and middle frequencies. All members denied cardiac phenotype, and electrocardiogram and echocardiography conducted on two affected members who agreed to participate in the examinations showed no evidence of DCM and other abnormality. Targeted sequencing of 129 HL genes showed that HL in the family segregated with a mutation in the EYA4 gene. The mutation NM_004100.4: c.543C>G (p.Tyr181Ter) is in exon 8 and leads to the introduction of a premature termination codon of the coding protein lacking the entire eyaHR domain. The nonsense mutation was classified as pathogenic following the variant interpretation standards and guidelines of ACMG. To the best of our knowledge, this variant has not been reported before.
Patients harboring EYA4 mutations often develop non‐syndromic late‐onset HL. However, so far, a few studies reported on patients with DCM and heart failure accompanied by HL. Schonberger et al. (2000) reported two families showing sensorineural HL and DCM transmitted by an autosomal dominant manner and mapped to 6q23–24 (Schonberger et al., 2000); by fine mapping and mutation analysis, they identified a 4846‐bp deletion encompassed part of the EYA4 gene as causal for the syndrome (Figure 5, E193) (Schonberger et al., 2005). In 2009, Dutrannoy et al. reported a de novo 9 Mbp deletion of 6q23.3‐24.1 disrupting EYA4 in a young patient who presented with HL, patent ductus arteriosus, and other mental problems (Dutrannoy et al., 2009). Moreover, Abe et al. (2018) reported progressive HL and mild cardiac abnormalities carrying a nonsense mutation in EYA4 gene (NM_004100.5:c.1177C>T:p.Gln393Ter) in a Japanese patient (Abe et al., 2018); the mutation was previously characterized as a pathogenic variant in a Korean pedigree with non‐syndromic HL (Figure 5) (Kim et al., 2015). Although the correlation between genotype (EYA4 mutation) and phenotype (syndromic or non‐syndromic HL) is not clear, it has been suggested that the mutation position of EYA4 might determine whether HL is accompanied by cardiac abnormalities. A few scientists speculated that truncations of the C‐terminal Eya domain cause non‐syndromic HL, whereas deletions of the N‐terminal variable region lead to HL with DCM (Makishima et al., 2007; Schonberger et al., 2005). This view was challenged by a study that reported on truncating EYA4 mutation c.863C>A(p.Ser288Ter), leading to deletions of the N‐terminal variable region in a Korean family with moderate hearing impairment and no DCM (Baek et al., 2012). Yet, the detailed investigation of the cardiac phenotype was not performed for the patient. Two recent studies further weaken this view by reporting more patients with non‐syndromic HL and N‐terminal truncating mutations, such as c.160G>T (p.Glu54Ter), c.223_224del (p.Val75PhefsTer32, Figure 5), and c.517C>T (p.Gln173Ter) (Morin et al., 2020; Shinagawa et al., 2020). The novel mutation we reported here c.543C>G, in exon 8 of EYA4, resulted in a shortened protein lacking part of the N‐terminal variable region in a Chinese family with no apparent cardiac phenotypes, and further complicated the relationship between EYA4 mutation and phenotype. Therefore, more studies are needed to investigate the correlation between EYA4 mutation and syndromic phenotype.
FIGURE 5.
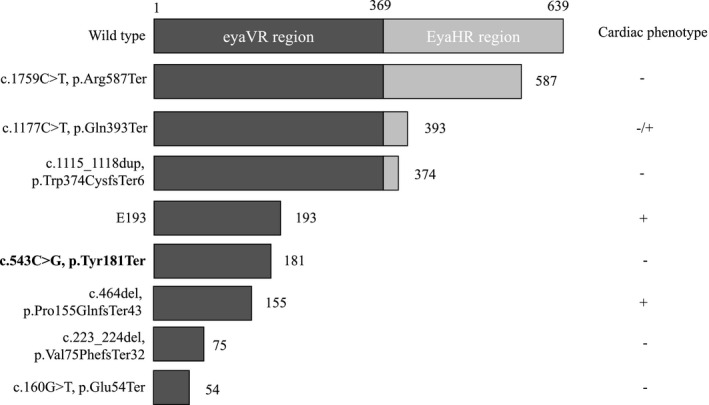
Effects of representative mutations on EYA4 protein structure and the cardiac phenotype. The number of amino acids of each allele product is indicated. The mutation found in this study is in bold. Variant nomenclature was based on EYA4 canonical transcript NM_172105.3
Until now, more than 50 pathogenic or likely pathogenic mutations in EYA4 have been reported (Morin et al., 2020); among these, ~40% of mutations have been found in the East Asia population. The EYA4 gene is not a common gene for ADNSHL, and thus far, only a limited number of mutations have been reported. Thus, the complete picture of audiological features associated with these variants needs to be further explored. The audiological features associated with EYA4 mutations are highly heterogeneous. Specifically, the same or similar mutation can cause different clinical phenotypes, while similar clinical phenotypes can be caused by different mutations. The clinical phenotype of patients carrying EYA4 mutations is the following: late‐onset, post‐lingual, progressive, and bilateral HL. For example, in relation to nonsense mutations, at least four nonsense mutations have been reported (Baek et al., 2012; Hu et al., 2018; Kim et al., 2015; Wayne et al., 2001). Most of the affected individuals with nonsense mutations suffered from moderate to severe bilateral, progressive, sensorineural HL in low‐ and mid‐frequencies and all frequencies were affected by aging. The onset age of HL ranged from early childhood to adulthood. The hearing curve of patients at the beginning of onset is usually U‐shaped but can also be seen in the descending and flat curves. This report expands the mutational spectrum of the EYA4 gene and highlights the importance of genetic testing for patients with familial progressive HL.
CONFLICT OF INTEREST
The authors have no conflict of interest to disclose.
AUTHOR CONTRIBUTIONS
Study design: Yanfang Mi, Yulin Zhao, and Hongen Xu. Patient phenotypic analysis and genetic counseling: Yanfang Mi, Hui Zhang, and Bei Chen. Next‐generation sequencing and Sanger sequencing: Yongan Tian, Juanli Zhang, and Wenxue Tang. Data analysis and variant interpretation: Danhua Liu, Bei Chen, Hong Xue, and Hongen Xu. qPCR and western blot: Beiping Zeng. Writing and review of original draft of the manuscript: Yanfagn Mi, Danhua Liu, Yulin Zhao, and Hongen Xu. All authors have read and approved the final manuscript.
ACKNOWLEDGMENTS
The study is funded by the Collaborative Innovation Project of Zhengzhou (Zhengzhou University) (Grant no. 18XTZX12004), the Medical Science and Technology Projects in Henan (Grant no. SBGJ2018043), and the Medical Science and Technology Research Project Joint Construction Project in Henan (Grant no. SBGJ2018020152). We sincerely thank all the family members for their participation in this study. The data analysis was supported by the Supercomputing Center of Zhengzhou University (Zhengzhou).
Yanfang Mi and Danhua Liu are Joint first authors.
Contributor Information
Yulin Zhao, Email: zhaoyulinmail@163.com.
Hongen Xu, Email: hongen_xu@zzu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
REFERENCES
- Abe, S. , Takeda, H. , Nishio, S. Y. , & Usami, S. I. (2018). Sensorineural hearing loss and mild cardiac phenotype caused by an EYA4 mutation. Human Genome Variation, 5, 23. 10.1038/s41439-018-0023-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek, J. I. , Oh, S. K. , Kim, D. B. , Choi, S. Y. , Kim, U. K. , Lee, K. Y. , & Lee, S. H. (2012). Targeted massive parallel sequencing: the effective detection of novel causative mutations associated with hearing loss in small families. Orphanet Journal of Rare Diseases, 7, 60. 10.1186/1750-1172-7-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsani, G. , DeGrandi, A. , Ballabio, A. , Bulfone, A. , Bernard, L. , Banfi, S. , & Hanson, I. (1999). EYA4, a novel vertebrate gene related to Drosophila eyes absent. Human Molecular Genetics, 8(1), 11–23. 10.1093/hmg/8.1.11 [DOI] [PubMed] [Google Scholar]
- DePristo, M. A. , Banks, E. , Poplin, R. , Garimella, K. V. , Maguire, J. R. , Hartl, C. , Philippakis, A. A. , del Angel, G. , Rivas, M. A. , Hanna, M. , McKenna, A. , Fennell, T. J. , Kernytsky, A. M. , Sivachenko, A. Y. , Cibulskis, K. , Gabriel, S. B. , Altshuler, D. , & Daly, M. J. (2011). A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nature Genetics, 43(5), 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutrannoy, V. , Klopocki, E. , Wei, R. , Bommer, C. , Mundlos, S. , Graul‐Neumann, L. M. , & Trimborn, M. (2009). De novo 9 Mb deletion of 6q23.2q24.1 disrupting the gene EYA4 in a patient with sensorineural hearing loss, cardiac malformation, and mental retardation. European Journal of Medical Genetics, 52(6), 450–453. 10.1016/j.ejmg.2009.06.004 [DOI] [PubMed] [Google Scholar]
- Genomes Project, C. , Auton, A. , Brooks, L. D. , Durbin, R. M. , Garrison, E. P. , Kang, H. M. , Korbel, J. O. , Marchini, J. L. , McCarthy, S. , McVean, G. A. , & Abecasis, G. R. (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, R. , Harrison, S. M. , Rehm, H. L. , Plon, S. E. , & Biesecker, L. G. ; ClinGen Sequence Variant Interpretation Working, G. (2018). Updated recommendation for the benign stand‐alone ACMG/AMP criterion. Human Mutation, 39(11), 1525–1530. 10.1002/humu.23642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, S. , Sun, F. , Zhang, J. , Tang, Y. , Qiu, J. , Wang, Z. , & Zhang, L. (2018). Genetic etiology study of ten Chinese families with nonsyndromic hearing loss. Neural Plasticity, 2018, 1–7. 10.1155/2018/4920980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis, N. M. , Rothstein, J. H. , Pejaver, V. , Middha, S. , McDonnell, S. K. , Baheti, S. , Musolf, A. , Li, Q. , Holzinger, E. , Karyadi, D. , Cannon‐Albright, L. A. , Teerlink, C. C. , Stanford, J. L. , Isaacs, W. B. , Xu, J. , Cooney, K. A. , Lange, E. M. , Schleutker, J. , Carpten, J. D. , … Sieh, W. (2016). REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. American Journal of Human Genetics, 99(4), 877–885. 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., Gauthier L.D., Brand H., Solomonson M, Watts N.A., Rhodes D., Singer‐Berk M., England E.M., Seaby E.G., Kosmicki J.A.,… MacArthur D.G. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, (7809), 434–443. 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y.‐R. , Kim, M.‐A. , Sagong, B. , Bae, S.‐H. , Lee, H.‐J. , Kim, H.‐J. , Choi, J. Y. , Lee, K.‐Y. , & Kim, U.‐K. (2015). Evaluation of the contribution of the EYA4 and GRHL2 genes in Korean patients with autosomal dominant non‐syndromic hearing loss. PLoS One, 10(3), e0119443. 10.1371/journal.pone.0119443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubisch, C. , Schroeder, B. C. , Friedrich, T. , Lütjohann, B. , El‐Amraoui, A. , Marlin, S. , Petit, C. , & Jentsch, T. J. (1999). KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell, 96(3), 437–446. 10.1016/s0092-8674(00)80556-5 [DOI] [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4(7), 1073–1082. [DOI] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Benson, M. , Brown, G. R. , Chao, C. , Chitipiralla, S. , Gu, B. , Hart, J. , Hoffman, D. , Jang, W. , Karapetyan, K. , Katz, K. , Liu, C. , Maddipatla, Z. , Malheiro, A. , McDaniel, K. , Ovetsky, M. , Riley, G. , Zhou, G. , … Maglott, D. R. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Research, 46(D1), D1062–D1067. 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , O’Donnell‐Luria, A. H. , Ware, J. S. , Hill, A. J. , Cummings, B. B. , Tukiainen, T. , Birnbaum, D. P. , Kosmicki, J. A. , Duncan, L. E. , Estrada, K. , Zhao, F. , Zou, J. , Pierce‐Hoffman, E. , Berghout, J. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv, 1303.3997.
- Li, Q. , & Wang, K. (2017). InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG‐AMP Guidelines. American Journal of Human Genetics, 100(2), 267–280. 10.1016/j.ajhg.2017.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Wu, C. , Li, C. , & Boerwinkle, E. (2016). dbNSFP v3.0: A one‐stop database of functional predictions and annotations for human nonsynonymous and splice‐site SNVs. Human Mutation, 37(3), 235–241. 10.1002/humu.22932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, E. D. , Lee, M. K. , Morrow, J. E. , Welcsh, P. L. , Leon, P. E. , & King, M. C. (1997). Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science, 278(5341), 1315–1318. [PubMed] [Google Scholar]
- Makishima, T. , Madeo, A. C. , Brewer, C. C. , Zalewski, C. K. , Butman, J. A. , Sachdev, V. , Arai, A. E. , Arai, A. E. , Holbrook, B. M. , Rosing, D. R. , & Griffith, A. J. (2007). Nonsyndromic hearing loss DFNA10 and a novel mutation of EYA4: Evidence for correlation of normal cardiac phenotype with truncating mutations of the Eya domain. American Journal of Medical Genetics. Part A, 143A(14), 1592–1598. 10.1002/ajmg.a.31793 [DOI] [PubMed] [Google Scholar]
- Morín, M. , Borreguero, L. , Booth, K. T. , Lachgar, M. , Huygen, P. , Villamar, M. , Mayo, F. , Barrio, L. C. , Santos Serrão de Castro, L. , Morales, C. , del Castillo, I. , Arellano, B. , Tellería, D. , Smith, R. J. H. , Azaiez, H. , & Moreno Pelayo, M. A. (2020). Insights into the pathophysiology of DFNA10 hearing loss associated with novel EYA4 variants. Scientific Reports, 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton, C. C. , & Nance, W. E. (2006). Newborn hearing screening–A silent revolution. New England Journal of Medicine, 354(20), 2151–2164. 10.1056/NEJMra050700 [DOI] [PubMed] [Google Scholar]
- Nance, W. E. (2003). The genetics of deafness. Mental Retardation and Developmental Disabilities Research Reviews, 9(2), 109–119. 10.1002/mrdd.10067 [DOI] [PubMed] [Google Scholar]
- O'Neill, M. E. , Marietta, J. , Nishimura, D. , Wayne, S. , Van Camp, G. , Van Laer, L. , & Smith, R. J. (1996). A gene for autosomal dominant late‐onset progressive non‐syndromic hearing loss, DFNA10, maps to chromosome 6. Human Molecular Genetics, 5(6), 853–856. 10.1093/hmg/5.6.853 [DOI] [PubMed] [Google Scholar]
- Pedersen, B. S. , Layer, R. M. , & Quinlan, A. R. (2016). Vcfanno: Fast, flexible annotation of genetic variants. Genome Biology, 17(1), 118. 10.1186/s13059-016-0973-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönberger, J. , Levy, H. , Grünig, E. , Sangwatanaroj, S. , Fatkin, D. , MacRae, C. , Stäcker, H. , Halpin, C. , Eavey, R. , Philbin, E. F. , Katus, H. , Seidman, J. G. , & Seidman, C. E. (2000). Dilated cardiomyopathy and sensorineural hearing loss: a heritable syndrome that maps to 6q23‐24. Circulation, 101(15), 1812–1818. 10.1161/01.cir.101.15.1812 [DOI] [PubMed] [Google Scholar]
- Schönberger, J. , Wang, L. , Shin, J. T. , Kim, S. D. , Depreux, F. F. S. , Zhu, H. , Zon, L. , Pizard, A. , Kim, J. B. , MacRae, C. A. , Mungall, A. J. , Seidman, J. G. , & Seidman, C. E. (2005). Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nature Genetics, 37(4), 418–422. 10.1038/ng1527 [DOI] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11(4), 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Sherry, S. T. , Ward, M. H. , Kholodov, M. , Baker, J. , Phan, L. , Smigielski, E. M. , & Sirotkin, K. (2001). dbSNP: The NCBI database of genetic variation. Nucleic Acids Research, 29(1), 308–311. 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinagawa, J. , Moteki, H. , Nishio, S.‐Y. , Ohyama, K. , Otsuki, K. , Iwasaki, S. , Masuda, S. , Oshikawa, C. , Ohta, Y. , Arai, Y. , Takahashi, M. , Sakuma, N. , Abe, S. , Sakurai, Y. , Sakaguchi, H. , Ishino, T. , Uehara, N. , & Usami, S.‐I. (2020). Prevalence and clinical features of hearing loss caused by EYA4 variants. Scientific Reports, 10(1), 3662. 10.1038/s41598-020-60259-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson P.D., Ball E.V., Mort M., Phillips A.D., Shaw K., Cooper D.N. (2012). The Human Gene Mutation Database (HGMD) and Its Exploitation in the Fields of Personalized Genomics and Molecular Evolution. Current Protocols in Bioinformatics, 39, (1), 1.13.1–1.13.20. 10.1002/0471250953.bi0113s39. [DOI] [PubMed] [Google Scholar]
- Tarasov, A. , Vilella, A. J. , Cuppen, E. , Nijman, I. J. , & Prins, P. (2015). Sambamba: Fast processing of NGS alignment formats. Bioinformatics, 31(12), 2032–2034. 10.1093/bioinformatics/btv098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Sewell, W. F. , Kim, S. D. , Shin, J. T. , MacRae, C. A. , Zon, L. I. , Seidman, J. G. , & Seidman, C. E. (2008). Eya4 regulation of Na+/K+‐ATPase is required for sensory system development in zebrafish. Development, 135(20), 3425–3434. 10.1242/dev.012237 [DOI] [PubMed] [Google Scholar]
- Wayne, S. , Robertson, N. G. , DeClau, F. , Chen, N. , Verhoeven, K. , Prasad, S. , & Smith, R. J. (2001). Mutations in the transcriptional activator EYA4 cause late‐onset deafness at the DFNA10 locus. Human Molecular Genetics, 10(3), 195–200. 10.1093/hmg/10.3.195 [DOI] [PubMed] [Google Scholar]
- Xia. J. H., Liu C. Y, Tang B. S., Pan Q., Huang L., Dai H. P., Zhang B. R., Xie W., Hu D. X., Zheng D., Shi X. L., Wang D. A., Xia K., Yu K. P., Liao X. D., Feng Y., Yang Y. F., Xiao J. Y., Xie D. H., Huang J. Z. (1998). Mutations in the gene encoding gap junction protein β‐3 associated with autosomal dominant hearing impairment. Nature Genetics, 20, (4), 370–373. 10.1038/3845. [DOI] [PubMed] [Google Scholar]
- Zou, H. , Osborn, N. K. , Harrington, J. J. , Klatt, K. K. , Molina, J. R. , Burgart, L. J. , & Ahlquist, D. A. (2005). Frequent methylation of eyes absent 4 gene in Barrett's esophagus and esophageal adenocarcinoma. Cancer Epidemiology, Biomarkers & Prevention, 14(4), 830–834. 10.1158/1055-9965.EPI-04-0506 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.