

Abstract
BACKGROUND AND PURPOSE:
Neurofibromatosis type 1 is a common tumor predisposition syndrome. The aim of this study was to characterize the radiologic presentation of patients with neurofibromatosis type 1 with widespread spinal disease and to correlate it to clinical presentation and outcome.
MATERIALS AND METHODS:
We conducted a historical cohort study of adult patients with neurofibromatosis type 1 with spinal involvement. Longitudinal clinical evaluation included pain and neurologic deficits. Radiologically, spinal involvement was classified according to a novel classification system, and a radiologic risk score was calculated.
RESULTS:
Two hundred fifty-seven adult patients with neurofibromatosis type 1 are followed in our center. Thirty-four of these patients qualified for inclusion in this study. Three independent factors were found to be associated with increased risk for neurologic deficit: 1) bilateral tumors at the same level in the cervical region that approximated each other, 2) paraspinal tumors at the lumbar region, and 3) intradural lesions. On the basis of these factors, we calculated a combined risk score for neurologic deficits for each patient. We found a clear correlation between patient status and the calculated radiologic risk score. Patients with neurologic deficits were found to have a higher risk score (9 ± 8.3) than patients without neurologic deficits (2.5 ± 2.9, P < .05). Patients who progressed during the follow-up period had significantly higher scores at presentation than patients with stable conditions (9.9 ± 8.73 versus 3.9 ± 5.3, respectively; P < .05).
CONCLUSIONS:
In this series, neurologic deficit is correlated with tumor burden and subtype. We found no direct correlation with tumor burden and pain. Our novel radiologic classification scoring system may be used to predict increased risk for neurologic morbidity.
Neurofibromatosis type 1 (NF1; Online Mendelian Inheritance in Man #162200) is a common genetic autosomal dominant syndrome. NF1 effects approximately 1:2000 individuals worldwide.1,2 The syndrome is characterized by a combination of clinical traits: café au lait macules, Lisch nodules (iris hamartomas), neurofibromas (cutaneous, subcutaneous, plexiform), optic pathway gliomas, and bone dysplasia.1,3
Spinal manifestations of NF1 include bone changes such as acute kyphoscoliosis at the cervical-thoracic junction and vertebral body anomalies, soft-tissue abnormalities such as dural ectasia and lateral meningocele, and various spinal tumors.1,4 Neoplastic spinal involvement in NF1 varies widely; patients may be affected by nerve sheath tumors such as neurofibromas, plexiform neurofibromas (PN), and intramedullary glial tumors. Figure 1 demonstrates a typical case of a complex spinal involvement in a patient with NF1. To date, no association has been found between spinal tumor burden and clinical outcome.
Fig 1.

Complex spinal manifestation in a patient with NF1. Spinal MR imaging study of a 36-year-old asymptomatic patient with NF1. Note that the tumors involve all the neuroforamina in the cervical and lumbar segments (A and C). Axial T1 with gadolinium demonstrates massive paraspinal involvement of the thoracic region (B). Sagittal T2 with fat saturation of the lumbar region demonstrates multiple tumors in the neuroforamina and in the lumbar spine (C).
Generally, an operation is reserved for tumors associated with progressive neurologic deficits.5 Patients with NF1 with massive spinal involvement present a major clinical challenge. Often the burden of the disease is overwhelming. Thus, it is extremely difficult to follow up, even using innovative volumetric assessment methods. It may also be difficult to identify the culprit tumor responsible for specific clinical symptoms. Malignant transformation, though rare, poses a significant problem and usually also necessitates surgical resection when feasible.6
Many biologically targeted therapies have been used in the clinical and preclinical setting. A few examples are RAS (receptor associated) targeted therapy, mTOR (mechanistic target of rapamycin) inhibitors,7 interferon,8 or multi kinase inhibitors.9 Recently, promising preliminary results were published10 demonstrating a response to treatment with mitogen-activated protein kinase inhibitor in a phase 1 study. The possibility of medical treatment for PN further highlights the need to accurately prognosticate patients with spinal NF1.
There is a clinical subtype of NF1, “spinal-NF1,” that is associated with a unique phenotype and genotype and should be differentiated from regular NF1-associated spinal involvement. These patients (spinal-NF1) characteristically display massive neoplastic enlargement of most of the spinal roots and multiple paraspinal tumors, with very few of the dermal manifestations typical of NF1.11 Molecularly, in patients with spinal-NF1, a large deletion encompassing the NF1 gene can be found.
Our study focuses on spinal involvement in patients with NF1 in general, rather than the spinal-NF1 subtype. Included were both patients with spinal-NF and those with NF who presented with spinal manifestations.
The aim of this study was to characterize the variety of spinal radiologic presentations of patients with NF1. In addition, we propose a novel classification and radiologic scoring system for patients with spinal involvement. We intended to demonstrate a correlation between our scoring system and symptoms such as pain and neurologic deficits.
Materials and Methods
This study was approved by the institutional review board of Tel-Aviv Medical Center. Records of all 257 adult patients with NF1 treated at the Gilbert Israeli NF1 Center between 2008 and 2014 were retrospectively reviewed. Thirty-four patients with documented NF1-associated neoplastic spinal involvement, including MR imaging studies of the spine (at least 2 MR imaging studies) and sufficient clinical follow-up data (>2 years of clinical follow-up) were included in the analysis. Each patient‘s imaging studies were evaluated and classified with our classification method by a radiology resident (M.M.-H.), under the supervision of a senior neuroradiologist (L.B.-S.); both were blinded to the clinical condition of the patients.
Patients were initially grouped according to their clinical presentation: asymptomatic and symptomatic (with pain or neurologic deficits); then, we analyzed specifically the groups of patients with pain or neurologic deficits related to their spinal manifestations.
We tried to develop a new radiologic classification that would allow risk stratification of spinal patients with NF. We therefore classified the tumors into 4 groups:
Foraminal tumor: a tumor involving the nerve roots and the neuroforamen.
“Kissing” tumors: bilateral-foraminal tumors that approximated each other at the same level, to <2 mm, with significant compression of the cord or thecal sac (See Fig 2B for an example).
Paraspinal tumor: a tumor epicentered lateral to the neuroforamina, having a large soft-tissue component outside the spinal canal.
Intradural tumor: a tumor epicentered inside the thecal sac.
Fig 2.
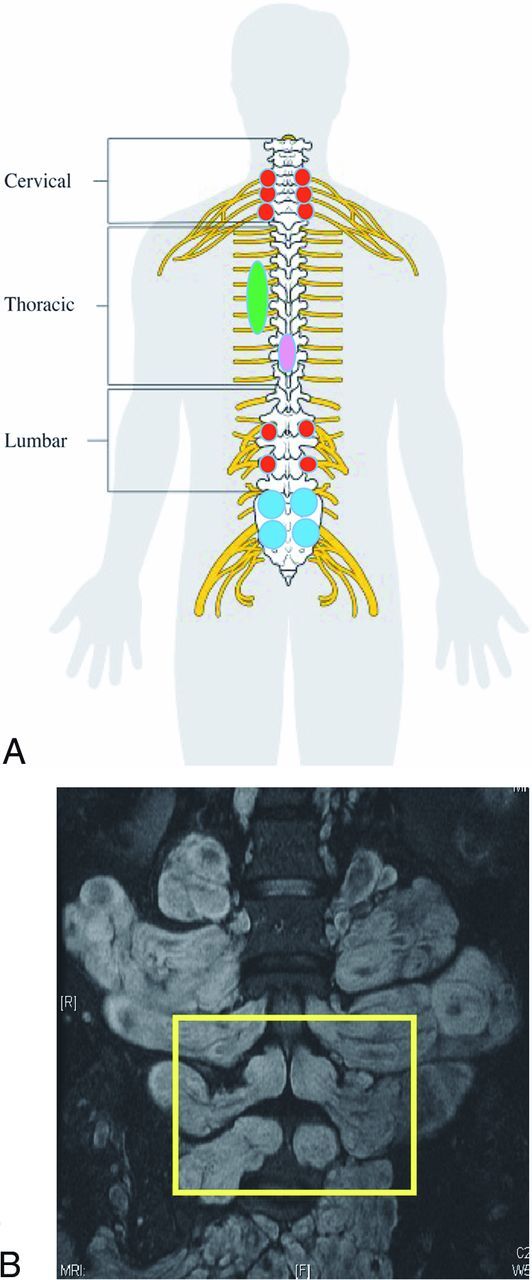
Schematic representation of our classification of spinal involvement in NF1 (A). A red circle represents tumors within the neuroforamina (group 1). Blue circles represent bilateral tumors within the neuroforamina with sac compression (kissing neurofibromas, group 2). The green ellipse represents paraspinal involvement (group 3). The purple ellipse represents an intradural lesion (group 4). A patient with this imaging result would have received a score of 6 for cervical foraminal, 2 for thoracic, 2 for lumbar foraminal, 1 for thoracic paraspinal, 1 for intradural thoracic, and 2 for 2 pairs of lumbar kissing tumors. The spinal cord schema image has been released as part of an open knowledge project by Cancer Research UK and attributed to Cancer Research UK/Wikimedia Commons. B, T2 with fat saturation coronal MR imaging study of the lumbar region of a patient with NF1. In the yellow rectangle, we see an example of 2 pairs of bilateral tumors approximating each other at the same level to <1–2 mm, with significant compression of the thecal sac (kissing PN). Note the large paraspinal component of the tumors (group 1).
See Fig 1 for an example of our radiologic classification system.
Tumors were counted and scored independently for each subtype. For example, a patient with a paraspinal tumor in the cervical segment received 1 point for this region and 0 points for the thoracic and lumbar regions. For tumors in group 1 (foraminal), the number of spinal levels (nerves) involved was counted for each spinal segment on each side. For example, a patient with bilateral cervical foraminal tumors in 3 different levels was given a score of 6 for cervical foraminal tumor. For tumors in group 2 (kissing), 1 point was given for each level involved. Tumors in group 4 (intradural) were rare, usually involving a single lesion per segment; their presence or absence was also documented for each segment. Because tumors in group 3 (paraspinal) are manifested as bulky, massive, paraspinal, soft-tissue masses that cannot be counted for multiplicity, we decided to take into account the presence or absence of the paraspinal tumor in each spinal segment (cervical, thoracic, lumbosacral). See Fig 2A for a schematic representation of our classification system. The overall clinical course was determined on the basis of the clinical documentation and then correlated with radiologic data.
For statistical analysis, we used SPSS, Version 21 (IBM, Armonk, New York). Differences in the mean distribution of the tumors between groups of patients at presentation (symptomatic versus asymptomatic, pain versus no pain, neurologic deficit versus no neurologic deficit) were evaluated with the Student independent samples t test, with significance set at P < .05. Tumor subgroups that demonstrated a significant difference (P < .05) or a trend difference in prevalence (P < .1) among the clinical groups were further analyzed. The effect of each tumor subtype on the outcome measures (pain, neurologic deficit, neurologic deterioration), as indicated by the previous analysis, was evaluated with a logistic regression, and the odds ratio for outcome was calculated. A radiologic risk score was computed on the basis of the ORs of the significant categories for each patient and was correlated with neurologic and radiologic outcome. The complete risk score calculation would be the following:
OR1 × n tumors1 + OR2 × n tumors2 … ORn × n tumorsn.
Results
Of 257 adult patients with NF1 followed in our institution, 41 (15.9%) had documented spinal involvement based on spinal imaging. Thirty-four (83%) had sufficient clinical follow-up data to be included in this study. Seventeen of the patients (50%) were females; the median age at first spinal imaging was 31 years (range, 14–55 years). The mean follow-up time was 3.5 ± 2.9 years. Seventeen patients (50%) had a positive family history of NF1. Fifteen patients (44%) underwent an operation due to progressive symptomatic spinal disease.
Follow-up data were available for 30 patients. Four patients were excluded from the outcome analysis due to either surgeries at presentation (n = 3) or insufficient follow-up data (n = 1). At the last follow-up, 7/30 (23%) patients available for evaluation demonstrated radiologic progression. Tumor distribution varied widely; see the Table for detailed description of the entire group.
Detailed tumor distribution for the entire cohort (n = 34)
| Group 1: Neuroforaminala | Group 2: Kissing Neurofibromasa | Group 3: Paraspinalb | Group 4: Intraduralb | |
|---|---|---|---|---|
| Cervical | 7.3 ± 6 (0–14) | 0.8 ± 1.9 (0–6) | 10 (29%) | 3 (8%) |
| Thoracic | 4.9 ±7.5 (0–24) | 0 | 13 (38%) | 3 (8%) |
| Lumbo-sacral | 8.8 ± 7.7 (0–20) | 1.6 ± 3.5 (0–16) | 15 (44%) | 5 (14%) |
The average number ± SD of involved levels in each spinal region for each tumor type for the entire cohort (n = 34). Numbers in parentheses represent the range of affected levels.
Number of patients (%). Note that for intradural and paraspinal tumors, the number represents the total number of patients, regardless of how many levels are affected.
We analyzed tumor distribution according to the following factors:
Symptomatic: Only 22 of 34 patients (64%) were symptomatic at presentation due to their spinal disease. The spinal distribution of the tumors did not differ between symptomatic and asymptomatic patients for all tumor types. The only exception was group 2, kissing neurofibromas in the cervical region. Those were more abundant in the symptomatic group compared with the asymptomatic group (1.1 ± 2.3 versus 0.3 ± 0.78, respectively; P < .05). (See Fig 3 for a detailed distribution of symptomatic and asymptomatic tumors by group.)
Pain: Fifteen of 34 patients (44%) had pain associated with their spinal disease. The spinal distribution of the tumors did not differ between patients in either group (Fig 4).
Neurologic deficit: Thirteen of 34 patients (38%) presented with neurologic deficits related to their spinal disease; 21/34 (62%) were neurologically intact. These patients had significantly more tumors in the cervical and lumbosacral regions (Fig 5). Kissing cervical neurofibromas were more common in patients with neurologic deficits than in intact patients, with an average of 1.7 ± 0.7 versus 0.2 ± 0.6, respectively (P < .05). A similar difference in the amount of kissing neurofibromas was found for the lumbosacral area, with an average of 2.83 ± 1.4 versus 0.95 ± 2.2, respectively (P < .05). In addition, a difference between the groups was observed in the number of patients with cervical paraspinal tumors. In the neurologically impaired group, more patients had cervical paraspinal tumors (0.46 ± 0.15 patients) in comparison with the neurologically intact group (0.2 ± 0.4 patients, P < .05).
We tried to quantify the impact of differences in tumor distribution at different levels on clinical status (as manifested by pain and neurologic impairment). Thus, we used a series of logistic regressions examining the increase in associated risk of neurologic deficit or pain with each type of tumor (stratified by group and location). Three independent factors were found to be associated (either statistically significant or with a trend toward significance) with an increased risk for neurologic deficits: The first was the presence of kissing neurofibromas (group 3) in the cervical region, the second was associated with paraspinal tumors (group 1) in the lumbar region, and the third was related to intradural disease (group 4) (Fig 5), with respective ORs of 1.7 (P = .07), 3.7 (P = .08), and 4.2 (P = .03). On the basis of these data, we calculated a risk score for neurologic deficits for each patient. The risk score was calculated as follows: the number of kissing neurofibromas was multiple by 1.7, if lumbar paraspinal tumors were present we added 3.7 and if an intradural disease was present we added 4.2.
Fig 3.

Spinal tumor distribution in asymptomatic and symptomatic patients. Error bars represent the standard error of the mean.
Fig 4.

Spinal tumor distribution in patients with and without pain. Error bars represent the standard error of the mean.
Fig 5.

Spinal tumor distribution in patients with neurologic deficits and in neurologically intact patients. Patients with neurologic deficits had more paraspinal tumors in the cervical region (group 1) and more kissing NF (group 3) in the cervical and the lumbar regions. Error bars represent the standard error of the mean.
Calculation of Radiologic Risk Score
Risk score = 1.7× n (of cervical kissing neurofibromas) + 3.7 (if lumbar paraspinal tumors were present) + 4.2 (if an intradural disease was present).
For pain, no risk factors were found to be significant. In addition, no difference related to pain was found in the risk score. Patients with neurologic deficits were found to have a significantly higher risk score, with an average of 9 ± 8.3, compared with 2.5 ± 2.9 in neurologically intact patients (P < .05, Fig 6).
Fig 6.

Radiologic risk scores for different patient groups. Symptomatic patients (with neurologic deficits and/or pain) had higher scores compared with asymptomatic patients (A). Patients showing neurologic deterioration had higher scores compared with patients with stable conditions (B).
While a total of 34 patients were included in this study, we only had long-term follow-up data for 30 patients. During the follow-up period, 24/30 (80%) remained neurologically stable, while the conditions of 6/30 (20%) deteriorated. The mean risk score calculated according to the proposed method was significantly higher for patients with deteriorating conditions (10 ± 8.7) compared with those with stable conditions (3.9 ± 5.3, P < .05).
Discussion
The main findings of this article are based on a novel radiologic classification system that correlates well with clinical presentation and outcome, producing a potentially useful radiologic risk scoring method.
Patients with NF1 with spinal involvement present the treating multidisciplinary team with unique difficulties. High tumor burden poses a significant radiologic challenge when attempting to assess anatomic progression. Clinically, it is often difficult to distinguish the culprit tumor responsible for the symptoms. Due to the complexity and associated morbidity of plexiform neurofibroma operations, they are often kept as a last resort when all other conservative therapeutic measures have failed and the tumor responsible for neurologic deterioration is identified beyond all doubt. In light of the novel treatment options for PN currently being developed (at least among patients with NF1), a means of evaluating these tumors and their course of progression is essential for treatment decision-making.10
In a series of 149 patients with spinal neurofibromas, spinal involvement was classified into several types according to the anatomic location of the tumors in the spinal canal and foramina.12 Despite the large number of patients in the cohort, only 12 had NF1, and only a minority of the patients had multiple spinal tumors. We found 3 other studies that looked into the incidence and variety of spinal tumors in patients with NF1 more specifically. Recently, a large series of spinal findings in patients with NF1 was published, describing 97 patients with baseline imaging of the spine.13 In this series, 78 patients had spinal neurofibromas, but only 26 patients had clinical and radiologic follow-up. The authors did not provide radiologic correlation to neurologic deficits or to any other symptoms. The only prognostic finding in that series was an increased risk of spinal curvature abnormalities associated with paraspinal PN.13 In 2 other studies of patients with NF1 with spinal involvement, correlation between imaging and clinical outcome was found only in the cervical region5 or specifically for intradural involvement.14
Most of the patients in our series demonstrated a severe burden of spinal disease presenting with multilevel disease. This finding probably represents a selection bias because usually the more severe cases are referred to tertiary NF1 centers such as ours, with a strong neuro-oncologic and neurosurgical orientation. The follow-up period in our series is relatively long (3.5 years) with a good follow-up adherence by patients (∼90%), allowing us to better evaluate the clinical and radiologic outcome.
Sixty-four percent of our patients were symptomatic at presentation; 24% had both pain and neurologic disability. In our series, only 20% of patients demonstrated clinical progression. We saw no difference in overall tumor burden at presentation between patients with progressive or stable neurologic conditions. This illustrates the lack of a simple correlation between MR imaging tumor burden and clinical outcome. We found that in general, the correlation among radiologic manifestations, clinical presentation, and outcome is limited when using traditional radiologic evaluation. This limitation motivated us to develop a radiologic risk score aimed at predicting long-term neurologic outcome.
From our data, 2 factors emerge as having a significant influence on a patient's neurologic outcome: kissing neurofibromas in the cervical spine and paraspinal lumbar tumors. In addition, intradural tumors, as expected, tend to cause significant neurologic morbidity as well. We incorporated all of these cardinal radiologic risk factors into a simple, easy-to-use radiologic risk score. This score has shown a good correlation with the clinical condition at presentation, with significantly higher scores in symptomatic patients with disease-associated neurologic deficits (Fig 6A). In addition, individual patient scores were highly predictive of clinical progression. Patients with neurologic deterioration had an average score of ∼10 compared with an average score of ∼4 in patients with clinically stable conditions (Fig 6B).
This new scoring system, though requiring further validation in prospective, long-term follow-up studies, may aid in risk stratification of a patient with complex spinal NF1, assist in determining radiologic and clinical follow-up intervals, and help clarify the need for medical treatment. In addition, these risk factors should be taken into account when presented independently, in cases in which the risk score is unusable.
This work has a few methodologic limitations. First, the historical cohort design has the shortcomings of any retrospective study. In addition, the numbers in our series are limited; this feature necessitated minimally reduced statistical significance levels in some of the analysis. Another shortcoming is the design of our radiologic risk score, which was built for patients with high disease burden and probably will not serve to predict neurologic deterioration or indicate the need for surgical intervention in patients with a single spinal tumor or a limited disease burden.
We believe that our study helps to characterize and classify the challenging population of patients with NF1 with spinal involvement.
Conclusions
In this limited series, 3 factors were found to be highly correlated with neurologic deficit: paraspinal tumors in the cervical region, kissing neurofibromas in the cervical region, and intradural lesions. Pain was not correlated with tumor burden in any of the spinal segments. Our innovative radiologic classification and scoring system produces a risk score that is significantly associated with the neurologic impairment and clinical progression. This score may aid in identifying patients who have an increased risk of neurologic deterioration and the need for a future operation, based on their imaging at presentation.
ABBREVIATIONS:
- NF1
neurofibromatosis type 1
- PN
plexiform neurofibromas
References
- 1. Restrepo CS, Riascos RF, Hatta AA, et al. Neurofibromatosis type 1: spinal manifestations of a systemic disease. J Comput Assist Tomogr 2005;29:532–39 10.1097/01.rct.0000163705.21892.23 [DOI] [PubMed] [Google Scholar]
- 2. Uusitalo E, Leppävirta J, Koffert A, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol 2015;135:904–06 10.1038/jid.2014.465 [DOI] [PubMed] [Google Scholar]
- 3. Neurofibromatosis: conference statement—National Institutes of Health Consensus Development Conference. Arch Neurol 1988;45:575–78 [PubMed] [Google Scholar]
- 4. Patel NB, Stacy GS. Musculoskeletal manifestations of neurofibromatosis type 1. AJR Am J Roentgenol 2012;199:W99–106 10.2214/AJR.11.7811 [DOI] [PubMed] [Google Scholar]
- 5. Taleb FS, Guha A, Arnold PM, et al. Surgical management of cervical spine manifestations of neurofibromatosis type 1: long-term clinical and radiological follow-up in 22 cases. J Neurosurg Spine 2011;14:356–66 10.3171/2010.9.SPINE09242 [DOI] [PubMed] [Google Scholar]
- 6. Goertz O, Langer S, Uthoff D, et al. Diagnosis, treatment and survival of 65 patients with malignant peripheral nerve sheath tumors. Anticancer Res 2014;34:777–83 [PubMed] [Google Scholar]
- 7. Weiss B, Widemann BC, Wolters P, et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: a neurofibromatosis Clinical Trials Consortium phase II study. Neuro Oncol 2015;17:596–603 10.1093/neuonc/nou235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jakacki RI, Dombi E, Steinberg SM, et al. Phase II trial of pegylated interferon alfa-2b in young patients with neurofibromatosis type 1 and unresectable plexiform neurofibromas. Neuro Oncol 2017;19:289–97 10.1093/neuonc/now158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ferguson MJ, Rhodes SD, Jiang L, et al. Preclinical evidence for the use of sunitinib malate in the treatment of plexiform neurofibromas. Pediatr Blood Cancer 2016;63:206–13 10.1002/pbc.25763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dombi E, Baldwin A, Marcus LJ, et al. Activity of selumetinib in neurofibromatosis type 1–related plexiform neurofibromas. N Engl J Med 2016;375:2550–60 10.1056/NEJMoa1605943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ruggieri M, Polizzi A, Spalice A, et al. The natural history of spinal neurofibromatosis: a critical review of clinical and genetic features. Clin Genet 2015;87:401–10 10.1111/cge.12498 [DOI] [PubMed] [Google Scholar]
- 12. Jinnai T, Koyama T. Clinical characteristics of spinal nerve sheath tumors: analysis of 149 cases. Neurosurgery 2005;56:510–15; discussion 510–15 10.1227/01.NEU.0000153752.59565.BB [DOI] [PubMed] [Google Scholar]
- 13. Nguyen R, Dombi E, Akshintala S, et al. Characterization of spinal findings in children and adults with neurofibromatosis type 1 enrolled in a natural history study using magnetic resonance imaging. J Neurooncol 2015;121:209–15 10.1007/s11060-014-1629-5 [DOI] [PubMed] [Google Scholar]
- 14. Patronas NJ, Courcoutsakis N, Bromley CM, et al. Intramedullary and spinal canal tumors in patients with neurofibromatosis 2: MR imaging findings and correlation with genotype. Radiology 2001;218:434–42 10.1148/radiology.218.2.r01fe40434 [DOI] [PubMed] [Google Scholar]