

Abstract
BACKGROUND AND PURPOSE:
CP is a common feature of perinatal HIBD in the context of “acute profound” injury, and in this article, we have studied the possible anatomic substrates of dyskinesia. We have reviewed the extent of brain injury in children with dyskinetic and spastic CP due to acute profound hypoxia to identify sites of injury that explain why only some children develop movement disorders. It is known that the STN has a role in the development of movement disorders; therefore, we have specifically studied it.
MATERIALS AND METHODS:
We retrospectively reviewed MR imaging of 40 consecutive children referred to our center with CP confirmed to be due to acute profound hypoxic-ischemic injury. All children received the same high-resolution MR imaging protocol with the same 1.5T scanner. Two pediatric neuroradiologists reviewed the imaging. Logistic regression was applied to identify multivariable predictors that differentiate dyskinetic and spastic CP.
RESULTS:
Twenty children had dyskinetic CP and 20 had spastic CP. Children with dyskinetic CP had more frequent injury to the STN, as manifest by increased T2 signal intensity. Children with spastic CP had more severe damage to white matter in the vicinity of the paracentral lobule. Injuries to the putamen, caudate, and globus pallidus were not significant predictors of dyskinesia.
CONCLUSIONS:
We have shown an association between hypoxic-ischemic injury to the STN at birth and the emergence of dyskinesia later in life.
Children who had relatively brief periods of severe HIBD and subsequently develop CP often have similar distributions of brain damage. These were first described in pathology studies1 and more recently in MR imaging studies of children who survived the initial damaging event. Damage following such an “acute profound” hypoxic event tends to show a predilection for the central structures of the forebrain, often with highly specific patterns of injury, namely the posterior tips of the putamen and the lateral aspects of the thalami.2 Other parts of the brain that may be injured with high frequency include the PCWM, optic radiations, hippocampus, and cerebellar vermis.3,4
It has been proposed that it is the injury to the putamen and thalami that is the anatomic substrate of the hallmark clinical sequelae of acute profound HIBD–dyskinetic CP.5 On closer review, however, there are several problems with that theory. First, it has been our experience that many children with the typical putaminal and thalamic injury do not have dyskinetic CP, but instead develop spastic CP; and though not widely reported, this observation was part of the motivation for this study. Second, it is the ventral posterolateral thalamic nucleus, a primary somatosensory relay nucleus, that is predominantly injured in HIBD rather than the motor nucleus in the anterior region of the lateral thalamus.6 Therefore, if the thalamus is the site of dyskinesia, it is unlikely to be through a primary motor mechanism. The putamen has a central role in planning volitional movement; putaminal abnormalities can be seen in some dyskinetic syndromes (eg, Sydenham chorea). However, discrete destructive putaminal lesions in animal models rarely produce dyskinesia.7
The purpose of this study was to conduct a detailed review of the MR imaging in children who survived acute profound HIBD and to correlate those findings with the resulting type of CP. In light of the information given above, we have looked for evidence of damage at other sites of the basal ganglia not usually thought of as being primarily involved in HIBD, specifically the caudate nucleus, GP, and STN.
Materials and Methods
Subjects
This was a retrospective study in which an MR imaging clinical protocol was put in place allowing for subsequent detailed review of anatomic injury and for which approval had been obtained from the local research ethics committee. We studied 40 consecutive children referred for MR imaging at our institution who had confirmed CP resulting from acute profound HIBD around the time of birth as defined by expert obstetricians, neonatologists, and pediatric neurologists. This diagnosis was made on the basis of a sentinel event in the perinatal period; clinical features as listed in the international consensus statement,8 including low Apgar scores, acidosis, and early onset of seizures; and imaging findings showing the presence of thalamic or basal ganglia injury. Children were divided into 2 groups: those with spastic CP and those with dyskinetic CP. If a child had both spastic and dyskinetic symptoms, categorization was according to the dominant symptom.
MR Imaging Acquisition and Analysis
All children were examined on the same 1.5T superconducting system under general anesthesia with identical imaging protocols during a 4-year period, in line with the existing radiologic protocols at our institution. It is current practice at our institution to perform MR imaging examinations routinely in this patient group to inform prognosis. The assessments used in this study were made primarily on the axial and coronal fast spin-echo T2-weighted (TR, 3169 ms; TE, 100 ms; echo-train length, 8; 2 acquisitions; section thickness; 4 mm; FOV, 240 mm; matrix size, 352 × 512) and on axial fast spin-echo T2 fluid-attenuated inversion recovery images (TR, 10,000 ms; TI, 2200 ms; TE, 109.6 ms; echo-train length, 8; section thickness, 4 mm; FOV, 210 mm; matrix size, 192 × 256). Two experienced pediatric neuroradiologists reviewed the images on the manufacturer proprietary workstations independently and blinded as to the symptom group. A sample case showing the location of the STN on coronal T2-weighted imaging is presented in Fig 1.
Fig 1.

Location of the STN. The normal STN is not seen on T2-weighted images at 1.5T because of its small size and signal-intensity characteristics close to the surrounding white matter structures. This coronal T2-weighted image from a child with dyskinetic CP arising from hypoxic-ischemic injury shows the STNs (arrows) as regions of high signal intensity because of gliosis within the nuclei.
A 4-point scoring system (0–3) was used for assessing the severity of involvement for the putamen, thalamus, and the PCWM as used in a previous study,3 with some modifications outlined here. A score of 0 indicated no radiologic evidence of damage to that brain region on any MR image. A score of 1 for the putamen corresponded to the involvement of the posterior tip of the putamen, 2 described involvement of the posterior half of the nucleus, and 3 denoted involvement of the entire putamen. A score of 1 for the thalamus represented the involvement of the ventral posterolateral nucleus only, 2 described involvement of the central 50% of the thalamus, and a score of 3 signified involvement of the entire structure. The PCWM was graded as 1 if there was minor signal-intensity change on T2 sequences but no volume loss. A score of 2 was assigned if there was both signal-intensity change and some volume loss confined to the PCWM; and 3, if there was severe volume loss and/or white matter change extending out of the PCWM. The scoring was completed separately for both sides of the brain. A total score was then calculated for brain damage, with a range from a minimum of 0 to a maximum of 18.
Other regions of the basal ganglia (caudate nucleus, GP, and STN) were examined for the presence or absence of abnormal signal intensity. When there was disagreement, a consensus opinion was obtained by joint review.
Statistical Analysis
MR imaging data and patient characteristics were compared between patients with dyskinetic and spastic CP by using the Student t test for gestational age and birth weight and the Wilcoxon rank-sum test for data not following a normal distribution, including putaminal, thalamic, and PCWM injury scores. The Fisher exact test was used to compare the 2 groups regarding the presence of STN, caudate, and GP injuries. Multivariable logistic regression was applied to minimize confounding, with the backward stepwise method used to identify the independent predictors for differentiating the 2 CP groups, with the likelihood ratio χ2 test used to assess the significance, and odds ratio and 95% CI used to measure the strength of association.
Variables with the 2-tailed P < .20 from the univariate analysis were entered into the multivariable model. Predicted probabilities of dyskinetic CP were derived by maximum likelihood estimation such that 100% minus these values would indicate the probability of spastic CP on the basis of combinations of the significant predictors.9 Two-tailed values of P < .05 were considered statistically significant. Statistical analysis was performed with SPSS 16.0 software (SPSS, Chicago, Illinois).
Results
Age at the time of MR imaging ranged from 12 months to 16 years. Twenty (50%) of the children studied had spastic CP, whereas 20 had dyskinetic CP. Comparison of patient characteristics and MR imaging data indicated several differences between children with dyskinetic CP compared with those with spastic CP (Table 1). Significantly larger head circumference (P = .02), lower PCWM left-sided scores (P = .01), and a higher prevalence of STN injury (P = .01) were observed in the dyskinetic group. These associations were confirmed by multivariable logistic regression analysis (Table 2). The final model containing head circumference (P = .01), presence of STN injury (P = .004), and PCWM score on the left (P = .004) provided a good fit to the data (R2 = 0.63). Logistic regression indicated that children with STN injuries were 18 times more likely to have dyskinesia compared with spasticity, independent of birth weight, Apgar scores, cord blood pH, hours to onset of seizures, as well as head circumference and presence of caudate or GP injuries.
Table 1:
Comparison of dyskinetic and spastic CP groupsa
| Variable | Dyskinetic CP (n = 20) | Spastic CP (n = 20) | P Value |
|---|---|---|---|
| Age at time of MR imaging (yr) | 4.8 ± 3.7 | 6.0 ± 3.9 | .35 |
| Gestational age (wk) | 40.0 ± 1.3 | 39.1 ± 2.0 | .12 |
| Birth weight (kg) | 3.3 ± 0.5 | 3.3 ± 0.6 | .78 |
| Apgar score (5-minute) | 3 (1–7) | 4 (0–6) | .27 |
| Cord blood pH at birth | 6.92 ± 0.15 | 6.83 ± 0.20 | .18 |
| Head circumference (percentile) | 53.5 ± 26.0 | 35.4 ± 20.5 | .02b |
| Age at onset of seizures (hr) | 3 (1–14) | 3.5 (1–18) | .17 |
| Putaminal injury score (0–3) | |||
| Left | 2 (1–3) | 1.5 (0–3) | .72 |
| Right | 1 (0–2) | 1 (0–3) | .78 |
| Thalamic injury score (0–3) | |||
| Left | 1 (1–3) | 1.5 (0–3) | .84 |
| Right | 1 (1–3) | 1 (0–3) | .64 |
| PCWM severity score (0–3) | |||
| Left | 1 (0–2) | 2 (1–3) | .01b |
| Right | 1 (0–2) | 2 (0–3 | .07 |
| Total injury score (0–18) | 8.0 (4–14) | 9.5 (4–18) | .38 |
| STN injury (No.) (%) | 15 (75) | 6 (30) | .01b |
| Caudate injury (No.) (%) | 2 (10) | 8 (40) | .06 |
| GP injury (No.) (%) | 2 (10) | 6 (30) | .24 |
Groups were compared by the Student t-test. All scores and age at onset of seizures are expressed as medians with ranges in parentheses and are compared by the Wilcoxon rank-sum test. The proportion of patients in each group with STN, caudate, and GP injuries was evaluated by the Fisher exact test.
Statistically significant, P < .05.
Table 2:
Results of multivariable stepwise logistic regression analysis
| Predictor Variable | LRT | P Value | Odds Ratioa | 95% CI |
|---|---|---|---|---|
| PCWM left (0–3) | 8.2 | .004 | 0.2 | 0.1–0.7 |
| STN injury | 11.1 | .001 | 18.6 | 2.4–141.0 |
| Head circumferenceb | 6.7 | .010 | 3.0 | 1.2–7.3 |
Odds ratios and 95% CIs are with respect to dyskinetic CP and can be reciprocated to estimate the odds of spastic CP. Cox and Snell model R2 = 63%, indicating good fit to the data.
Based on percentiles (10th, 25th, 50th, 75th, 90th) for head circumference with the 10th as the reference category.
Furthermore, for each increase of 1 percentile category in head circumference, the odds of dyskinetic CP compared with spastic CP were estimated to be 3 times higher. With each increasing score in the PCWM on the left side, the odds of dyskinetic CP decreased by 80% (95% CI, 30%–90%). Maximum likelihood estimation was applied to determine the probability of dyskinetic CP for combinations of the 3 highly significant predictors (Table 3). For example, a child with a head circumference at the 75th percentile with an STN injury and PCWM on the left of grade 2 (ie, moderately severe signal-intensity change and some volume loss) was estimated to have dyskinesia with 90% probability (10% probability of spastic CP), whereas with no STN injury, the estimated probability of dyskinesia was 35% (65% probability of spastic CP).
Table 3:
Multivariable algorithm for differentiating dyskinetic and spastic CPa
| HC (percentile) | STN Injury |
No STN Injury |
||||||
|---|---|---|---|---|---|---|---|---|
| PCWM Injury Score, Left |
PCWM Injury Score, Left |
|||||||
| 0 | 1 | 2 | 3 | 0 | 1 | 2 | 3 | |
| 10th | 93 | 70 | 30 | 8 | 40 | 11 | 2 | 1 |
| 25th | 97 | 87 | 55 | 19 | 66 | 26 | 6 | 2 |
| 50th | 98 | 95 | 78 | 50 | 85 | 51 | 16 | 4 |
| 75th | 99 | 98 | 90 | 66 | 94 | 75 | 35 | 9 |
| 90th | 99 | 99 | 97 | 85 | 98 | 90 | 62 | 23 |
Values represent percentage probabilities of dyskinetic CP according to the combination of the 3 predictors in the logistic regression model. Children with dyskinetic CP have a larger HC, more commonly have STN injury compared with those with spastic CP, and have lower left-sided PCWM scores. The probability of spastic CP can be determined by taking 100 minus the value in the table.
Representative images demonstrating the differences in injury distribution in a child with dyskinetic CP and one with spastic CP are shown in Figs 2 and 3.
Fig 2.

Typical appearances of HIBD changes in a child with spastic CP following acute profound HIBD. A and B, Axial T2 images show moderate PCWM signal-intensity abnormality and very mild involvement of the putamen and thalamus. C, Coronal T2 image shows no signal-intensity abnormality in the region of the STN.
Fig 3.

Typical appearances of HIBD changes in a child with dyskinetic CP following acute profound HIBD. A and B, Axial T2 images show mild involvement of the putamen, thalamus, and PCWM. C, Coronal T2 image shows high signal intensity in the region of the STN.
Discussion
Effects of hypoxia-ischemia on the fetal/neonatal brain depend on numerous factors including severity, duration, amount of reserve, and gestational maturity. If HIBD occurs at or close to term, 2 primary patterns of injury with discrete clinical sequelae are recognized.2 If the hypoxia-ischemia event is severe and of short duration (10–25 minutes is the often quoted range), the pattern of damage is usually “central.” This results in damage to the basal ganglia and thalamus, though other areas are often damaged (eg, PCWM, optic radiations, and hippocampus).
The typical clinical description of a child with that type of injury includes dyskinetic CP with relatively well-preserved intellect. In contrast, a lower level of hypoxia-ischemia lasting for a longer period of time produces a “peripheral” pattern of damage. This is best described as involving the vascular watershed territories of the cerebral hemispheres, producing damage to gray and white matter structures in a parasagittal distribution. Children affected by this damage will typically have spastic quadriplegic CP, often with profound intellectual impairment.2
The primary evidence for the 2 models of HIBD comes from carefully controlled animal experiments,10 and extrapolation to humans is difficult because the precise mechanism of injury is rarely known with certainty. One of the most difficult problems occurs when a neonate is delivered in a poor condition after a short-lived emergency toward the end of pregnancy. In such a scenario, it may be impossible ever to say that the HIBD was due solely to the events in the last minutes before birth. Overlapping “chronic partial” and acute profound HIBD is often advanced as a possible cause when imaging and clinical features are not congruent (eg, when a child has basal ganglia and thalamic damage on MR imaging but has spastic CP). An important finding of our study underlines the increasing recognition that spastic CP is a common outcome in cases of acute profound hypoxia-ischemia, and not just of children exposed to prolonged partial hypoxia-ischemia.
To understand why acute profound HIBD produces dyskinetic CP in some children and spastic CP in others, we shall review the anatomy and functional connectivity of the basal ganglia and motor cortex in normality and in cases of movement disorders. Original theories about the function of the basal ganglia came from observational studies in humans postmortem, but this has later been refined by detailed studies of animal models of movement disorders.7 When voluntary movement is undertaken, 2 neuronal activities are generated within the basal ganglia by using anatomically discrete pathways. One promotes the primary movement while the other attempts to provide the stability in other muscle groups required for the primary movement to act efficiently, which is usually a combination of stimulation and inhibition of different muscle groups.
Both the primary direct pathway and the associated indirect pathway of movement programming originate in the putamen (caudate nucleus to a lesser extent), albeit from different neuronal subtypes with different neurotransmitters.11 The direct pathway projects to the GPm, and the indirect pathway, to the Gpl. This is shown schematically in Fig 4 (drawn based on data in Crossman12). Abnormalities that selectively reduce the activity of the direct pathway are expected to produce hypokinesia, while selectively reducing the activity of the indirect pathway should produce hyperkinetic syndromes such as chorea or dystonia.
Fig 4.
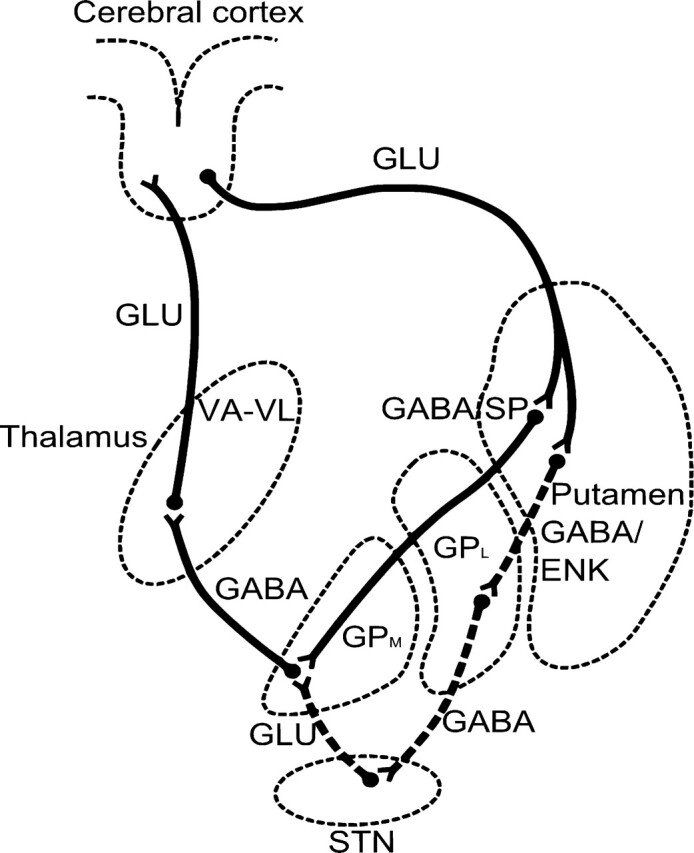
Line diagram shows the direct and indirect pathways from the putamen to the thalamus and hence to the motor cortex. Dashed lines indicate the neurons of the indirect pathway traveling via the STN (drawn based on data in Crossman12).
Early untreated Parkinson disease provides a good example of a hypokinetic syndrome, and there is good evidence that the lack of dopamine caused by neuronal degeneration in the substantia nigra pars compacta causes reduced activity of the neurons of the direct pathway.13 In contrast, in Huntington disease, where chorea is an important symptom, the neuronal degeneration initially affects the neurons of the indirect pathway.14 It should be appreciated that the 2 neuronal subtypes in the putamen are closely intermingled, and focal lesions (either infarctions in humans or destructive lesions in animal models) do not affect 1 particular subtype specifically. This may explain why focal lesions of the putamen rarely produce movement disorders of any type. By implication, it is unlikely that the focal putaminal damage seen in profound HIBD is responsible for the dyskinesia associated with the CP. Likewise, the site of predilection for damage in the thalamus is a sensory region and should not produce dyskinesia, for the reasons given in the introduction.
The indirect pathway from the GPl first projects to a small diencephalic nucleus, the STN, by using gamma amino butyric acid as the neurotransmitter. The STN neurons then project to the GPm by using glutamatergic neurons. It has been shown that the indirect pathway has faster neuronal transmission rates compared with the highly somatotopic direct pathway, and this feature is responsible for the fast suppression of unwanted movements.15 It has been known for a long time that injury to the STN and its associated tracts can produce hyperkinetic syndromes. In this situation, reduced activity in the STN leads to undersuppression of unwanted movements, hence dyskinesia. The purported underlying mechanism is interruption of the excitatory projection from the STN to the GPm which, subsequently, is expressed via altered function of the thalamocortical and corticospinal pathways.
More recently, functional imaging studies have demonstrated increased activity in the striatum in patients with dystonias.16 This lends support to the theory that failure of an inhibitory system, such as the indirect pathway, is associated with dyskinesia. In addition, there is increasing recognition of the role of deep brain stimulation in the treatment of dyskinesias, by the stimulation of the GPm or STN.17
In 1 series of adults with hemichorea following stroke, radiologic evidence of STN damage was reported in 30%, and GP damage, in 7%.18 That series also reported a high frequency of striatal and cortical injuries, which are not classic sites associated with dyskinesia. However, those patients with radiologically apparent STN damage had the worst prognosis. In infants, a similar pattern of radiologically detectable STN damage has been shown in kernicterus.19 Pathologically, kernicterus results from deposition of bilirubin in the GP and STN and produces a clinical syndrome of dyskinetic CP. In summary of the clinical observations and work from animal models, it is difficult to support focal injuries to the putamen as the anatomic substrate for dyskinesia. If a focal injury is to cause dyskinesia, it is much more likely to be in or close to the STN.
The STN can be visualized in adults as a small biconvex hypointense structure on T2-weighted images at 3T.20 Similar hypointensity can also be seen at 1.5T, but this is more subtle and only reliably detectable in the anterior part of the nucleus.21 This is presumably due to the susceptibility effects of high iron content in the STN as is also seen in the adjacent red nucleus and substantia nigra. Our patients were not imaged at 3T, but because the pediatric brain is virtually devoid of iron at all sites due to its age-dependent accumulation,22 we would expect detection of the normal STN to remain difficult, though this is yet to be confirmed. As shown in this study, the STN can be seen directly on T2-weighted MR images at 1.5T as an area of hyperintensity when damaged because of the gliosis produced by ischemic injury. Five of 20 children with dyskinetic CP did not have radiologically apparent STN abnormalities. This may be due to injury beneath the threshold of detection. In the context of kernicterus, in which STN injury is a known pathologic feature, imaging findings are often normal. However, the improved resolution and signal-to–noise ratio available at 3T, might have revealed injuries that were not apparent in this study. Only 1 child in this group had detectable injury in the GP and caudate, and no children in this group had severe thalamic injury. Additionally, none of these areas showed significant predictive power for dyskinesia in the multivariate analysis.
Previous reviews of MR imaging findings in HIBD have linked dyskinetic symptoms to less severe patterns of injury.23 Our study is consistent with this theory because children with spastic CP tended to have more severe signal-intensity changes and volume loss on MR imaging, particularly in the PCWM, as well as smaller head circumference, which we presume is secondary to the white matter volume loss.
Three children in this study were younger than 24 months of age at the time of MR imaging. It is recognized that there may be a delay of months to years before dyskinetic symptoms become evident. However, in this study, all 3 of these children had already developed significant dyskinetic symptoms; hence, we have included them in the dyskinetic group.
We have also shown that injury to the STN is a common finding in dyskinetic CP (15/20) and less common in spastic CP (6/20). Of those children with spastic CP and STN abnormality, all had evidence of damage to the PCWM; in 3 of 6, this was classified as severe. The dominance of spastic symptoms may be explained by the PCWM injury; damaged descending corticospinal tracts mask the aberrant neuronal output from the injured basal ganglia, such that dyskinesia cannot be manifest clinically. This is an expected finding because the dependence of dyskinesia on functioning pyramidal tracts has been long known, with lesions to the cortex or pyramidal tracts used as a treatment for dyskinesias in the early part of the 20th century.24 None of the children with dyskinetic CP had severe volume loss or signal-intensity change in the PCWM, suggesting that some function in the corticospinal tracts is necessary for dyskinesia to be clinically apparent.
The ventral posterolateral thalamic nucleus, a site commonly injured, is a somatosensory nucleus. Disorders of sensation have recently, however, been implicated in the etiology of dystonia.25 For this reason, we cannot fully discount the putamen as a possible anatomic substrate for dyskinetic CP. However, in this study, putaminal injury did not predict the presence of dyskinesia. We have also demonstrated that more severe forms of HIBD tend to involve the thalamus more generally. This in itself could have a further deleterious effect on motor function if the damage extends anteriorly into the ventral lateral and ventral anterior thalamic nuclei through projections to the primary motor cortex and supplementary motor cortex. However, in this study, thalamic injury did not predict the presence of dyskinesia.
This study used a simple division into spastic and dyskinetic symptom groups. This is a significant oversimplification that hides the considerable variability of symptoms and their overlap. These included the presence of a mixture of both significant spastic and dyskinetic symptoms in 3 of 40 children as well several children with asymmetric symptoms. This necessitated a subjective decision as to which symptoms were predominant when placing the child into the appropriate symptom group.
A further difficulty encountered in this study was accurate identification of the STN. We did not register images to an anatomic atlas. Instead, the identification of the STN, when visible, was by visual identification of its anatomic neighbors. This was not always reliable; the 2 observers disagreed over STN injury in 4 of 40 cases. Particular difficulty was seen in cases in which there was injury to other structures in the subthalamic region, such as the zona incerta, or when there was severe injury to large regions of the brain. Additionally, although the observers reviewed the cases while blinded to the clinical details, nevertheless, there were visible differences in the pattern of injury between the 2 symptom groups, particularly within the PCWM. As a result, the reliability of the blinding may have been compromised.
Conclusions
We found that spastic CP is a frequent outcome of acute profound HIBD as well as the more extensively reported cases of dyskinetic CP. In this article, we have attempted to explore why some children have spastic CP while others have dyskinetic CP by detailed anatomic studies with MR imaging. The main finding was frequent injury to the STN in children with dyskinetic CP, a finding that was less common in children with spastic CP. None of the children with dyskinetic CP had severely damaged PCWM. These findings are consistent with the known connectivity and function of the basal ganglia. We have described the sign of abnormal T2 signal intensity in the STN as a predictor of dyskinetic CP. This sign may be of value in assessing prognosis in children with HIBD.
Abbreviations
- CI
confidence interval
- CP
cerebral palsy
- ENK
enkephalin
- GLU
glutamate
- GP
globus pallidus
- GPl
lateral globus pallidus
- GPm
medial globus pallidus
- HC
head circumference
- HIBD
hypoxic-ischemic brain damage
- LRT
likelihood ratio test
- PCWM
paracentral white matter
- SP
substance-P
- STN
subthalamic nucleus
- VA-VL
ventral anterior and ventral lateral nuclei of the thalamus
References
- 1. Rorke LB. Perinatal brain damage. In: Adams JH, Duchen LW. eds. Greenfield's Neuropathology. 5th ed. London, UK: Edward Arnold; 1992:639–708 [Google Scholar]
- 2. Barkovich AJ. Brain and spine injuries in infancy and childhood. In: Barkovich AJ. ed. Pediatric Neuroimaging, 4th ed. Philadelphia: Lippincott, Williams and Wilkins; 2005:190–290 [Google Scholar]
- 3. Connolly DJA, Widjaja E, Griffiths PD. Involvement of the anterior lobe of the cerebellar vermis in perinatal profound hypoxia. AJNR Am J Neuroradiol 2007;28:16–19 [PMC free article] [PubMed] [Google Scholar]
- 4. Sargent MA, Poskitt KJ, Roland EH, et al. Cerebellar vermian atrophy after neonatal hypoxic-ischemic encephalopathy. AJNR Am J Neuroradiol 2004;25:1008–15 [PMC free article] [PubMed] [Google Scholar]
- 5. Menkes JH, Curran J. Clinical and MR correlates in children with extrapyramidal cerebral palsy. AJNR Am J Neuroradiol 1994;15:451–57 [PMC free article] [PubMed] [Google Scholar]
- 6. Martin LJ, Brambrink A, Koehler RC, et al. Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia-ischemia. J Comp Neurol 1997;377:262–85 [DOI] [PubMed] [Google Scholar]
- 7. Crossman AR. Primate models of dyskinesia: the experimental approach to the study of basal ganglia-related involuntary movement disorders. Neuroscience 1987;21:1–40 [DOI] [PubMed] [Google Scholar]
- 8. MacLennan A. A template for defining a causal relation between acute intrapartum events and cerebral palsy: international consensus statement. BMJ 1999;319:1054–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hosmer DW, Lemeshow S. Applied logistic regression, 2nd ed. New York: Wiley; 2000:31–85 [Google Scholar]
- 10. Myers RE. Four patterns of perinatal damage and their conditions or occurrence in primates. Adv Neurol 1975;10:223–33 [PubMed] [Google Scholar]
- 11. Penny GR, Afsharpour S, Kitai ST. The glutamate decarboxylase-, leucine enkephalin-, methionine enkephalin- and substance P-immunoreactive neurons in the neostriatum of the rat and cat: evidence for partial population overlap. Neuroscience 1986;17:1011–45 [DOI] [PubMed] [Google Scholar]
- 12. Crossman AR. Neural mechanisms in disorders of movement. Comp Biochem Physiol A Comp Physiol 1989;93:141–49 [DOI] [PubMed] [Google Scholar]
- 13. Penney JB, Young AB. Striatal inhomogeneities and basal ganglia function. Mov Disord 1986;1:3–15 [DOI] [PubMed] [Google Scholar]
- 14. Ferrante RJ, Kowalt NW, Bird ED, et al. Selective sparing of a class of striatal neurons in Huntington's disease. Science 1985;230:561–63 [DOI] [PubMed] [Google Scholar]
- 15. Kitai S, Kita H. Anatomy and physiology of the STN: a drive force of the basal ganglia. In: Carpenter MB, Jayarma A. eds. The Basal Ganglia II: Structure and Function—Current Concepts. New York: Plenum Press; 1987:357–73 [Google Scholar]
- 16. Blood AJ, Flaherty AW, Choi JK, et al. Basal ganglia activity remains elevated after movement in focal hand dystonia. Ann Neurol 2004;55:744–48 [DOI] [PubMed] [Google Scholar]
- 17. Ostrem JL, Starr PA. Treatment of dystonia with deep brain stimulation. Neurotherapeutics 2008;5:320–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chung SJ, Im J-H, Lee MC, et al. Hemichorea after stroke: clinical-radiological correlation. J Neurol 2004;251:725–29 [DOI] [PubMed] [Google Scholar]
- 19. Steinborn M, Seelos KC, Heuck A, et al. MR findings in a patient with kernicterus. Eur Radiol 1999;9:1913–15 [DOI] [PubMed] [Google Scholar]
- 20. Slavin KV, Thulborn KR, Wess C, et al. Direct visualization of the human subthalamic nucleus with 3T MR imaging. AJNR Am J Neuroradiol 2006;27:80–84 [PMC free article] [PubMed] [Google Scholar]
- 21. Dormont D, Ricciardi KG, Tandé D, et al. Is the subthalamic nucleus hypointense on T2-weighted images? A correlation study using MR imaging and stereotactic atlas data. AJNR Am J Neuroradiol 2004;25:1516–23 [PMC free article] [PubMed] [Google Scholar]
- 22. Hallgren B, Sourander P. The effect of age on non-haem iron in human brain. J Neurochem 1958;3:41–51 [DOI] [PubMed] [Google Scholar]
- 23. Krageloh-Mann I, Helber A, Mader I, et al. Bilateral lesions of thalamus and basal ganglia: origin and outcome. Dev Med Child Neurol 2002;44:447–84 [DOI] [PubMed] [Google Scholar]
- 24. Putnam TJ. Treatment of unilateral paralysis agitans by section of the lateral pyramidal tract. Arch Neurol Psychiatry 1940;44:950–76 [Google Scholar]
- 25. Breakefield XO, Blood AJ, Li Y, et al. The pathophysiological basis of dystonias. Nat Rev Neurosci 2008;9:222–34 [DOI] [PubMed] [Google Scholar]