

Abstract
Background:
We previously showed that cardiomyocyte Krϋppel-like factor (KLF)-5 regulates cardiac fatty acid oxidation. As heart failure has been associated with altered fatty acid oxidation, we investigated the role of cardiomyocyte KLF5 in lipid metabolism and pathophysiology of ischemic heart failure.
Methods:
Using rtPCR and Western Blot, we investigated the KLF5 expression changes in a myocardial infarction (MI) mouse model and heart tissue from patients with ischemic heart failure. Using 2D-echocardiography, we evaluated the effect of KLF5 inhibition after MI using pharmacological KLF5 inhibitor ML264 and mice with cardiomyocyte specific KLF5 deletion (αMHC-KLF5−/−). We identified the involvement of KLF5 in regulating lipid metabolism and ceramide accumulation after MI using liquid-chromatography-tandem-mass-spectrometry, and Western Blot and rtPCR analysis of ceramide-metabolism-related genes. We lastly evaluated the effect of cardiomyocyte-specific KLF5 overexpression (αMHC-rtTA-KLF5) on cardiac function and ceramide metabolism, and rescued the phenotype using myriocin to inhibit ceramide biosynthesis.
Results:
KLF5 mRNA and protein levels were higher in human ischemic heart failure samples and in rodent models 24h, 2- and 4-weeks post-permanent left coronary artery ligation. αMHC-KLF5−/− mice and mice treated with ML264 had higher ejection fraction and lower ventricular volume and heart weight after MI. Lipidomic analysis showed that αMHC-KLF5−/− mice with MI had lower myocardial ceramide levels compared with littermate control mice with MI although basal ceramide content of αMHC-KLF5−/− mice was not different from control mice. KLF5 ablation suppressed the expression of serine-palmitoyl-transferase-long-chain-base-subunit (SPTLC)1 and SPTLC2, which regulate de novo ceramide biosynthesis. We confirmed our previous findings that myocardial SPTLC1 and SPTLC2 levels are increased in heart failure patients. Consistently, αMHC-rtTA-KLF5 mice showed increased SPTLC1 and SPTLC2 expression, higher myocardial ceramide levels, and systolic dysfunction beginning 2-weeks after KLF5 induction. Treatment of αMHC-rtTA-KLF5 mice with myriocin that inhibits SPT, suppressed myocardial ceramide levels and alleviated systolic dysfunction.
Conclusions:
KLF5 is induced during the development of ischemic heart failure in humans and mice and stimulates ceramide biosynthesis. Genetic or pharmacological inhibition of KLF5 in mice with MI prevents ceramide accumulation, alleviates eccentric remodeling, and increases ejection fraction. Thus, KLF5 emerges as a novel therapeutic target for the treatment of ischemic heart failure.
Keywords: Cardiomyopathy, Basic Science Research, Myocardial Biology, Ceramides, Lipotoxicity, Krüppel-like Factor 5, Myocardial Ischemia, Heart Failure
INTRODUCTION
The heart relies on oxidative phosphorylation and uses predominantly lipids and to lesser extent glucose to meet its energetic demands1. Heart failure is accompanied by metabolic perturbations consisting of reduced fatty acid oxidation rates and altered substrate preference, which is associated with disease progression2, 3. Cardiac lipotoxicity is one manifestation of the metabolic imbalance that occurs in heart failure4. Among the toxic lipids that contribute to lipotoxicity, cardiac ceramides have been shown to activate pathological signaling pathways and contribute to heart failure in mice5 and humans6. The toxic effect of ceramides has been attributed to insulin resistance, adrenergic desensitization, reactive oxygen species (ROS) accumulation, endoplasmic reticulum stress, and apoptosis4. Ceramides are synthesized through three pathways known as the de novo pathway, the salvage pathway, and the sphingomyelinase pathway7. The rate-limiting step of the de novo pathway is catalyzed by the serine palmitoyl transferase (SPT) complex, which forms ceramide using palmitoyl-CoA and serine. SPT is a heterodimer of the SPT long chain base subunit 1 (SPTLC1) and 2 (SPTLC2). Cardiac ceramide levels are increased in heart failure patients and mice with ischemic heart failure6 and acute ischemic injury8. Increases in ceramide levels in both acute and chronic ischemic injury are accounted for by activation of the de novo synthesis pathway, but not the sphingomyelinase or salvage pathways6.
Krϋppel-like factor (KLF)5 is a zinc finger transcription factor and member of an 18-members family9. Our lab previously showed that cardiomyocyte KLF5 regulates cardiac fatty acid metabolism via direct activation of peroxisome-proliferator-activated-receptor (PPAR)α10. Mice with cardiomyocyte-specific deletion of the KLF5 gene (αMHC-KLF5−/−) have lower cardiac fatty acid oxidation rate and progress slowly to dilated cardiomyopathy when they are older than 6 months10. Conversely, previous studies showed that cardiac KLF5 was increased in heart failure, and that global heterozygous deletion of the Klf5 gene is protective against hypertrophy and fibrosis11–14. However, not much the role of cardiomyocyte KLF5 activation in heart failure pathology and accompanying metabolic remodeling remains unknown. Our study identified novel mechanisms through which KLF5 regulates ceramide biosynthesis following myocardial ischemia and showed that KLF5 inhibition alleviates ischemic heart failure and eccentric hypertrophy.
METHODS
An expanded methods section is available in the data supplement.
Data is available within the data supplement or from the corresponding author upon reasonable request.
Study Population –
We prospectively enrolled patients (age ≥ 18-years) at the University of Utah Health. The study was approved by the institutional review board of the University of Utah, and informed consent was provided by all patients.
Animal Experiments:
Animal protocols were approved by the Temple University Institutional Animal Care and Use Committee and were carried out in accordance with NIH guidelines. αMHC-rtTA-KLF5 mice were generated by crossing αMHC-Cre with R26-lsl-rtTA-TRE-KLF5 mice, provided by Dr. Jeffrey Whitsett15 and Dr. Inderpreet Sur16. MI was induced through permanent ligation of the left coronary artery (LCA ligation) as previously described17. Cardiac function was assessed via transthoracic echocardiography using the VisualSonics Vevo 2100 system (VisualSonics, Toronto, ON) as previously described18.
Cell Culture:
A human ventricular cardiomyocyte-derived cell line, designated AC1619, and a mouse atrial cardiomyocyte-derived cell line, designated HL-120 were used for in vitro experimentation. AC16 cells were maintained in Dulbecco’s modified Eagle’s medium-nutrient mixture F-12 (DMEM-F-12; Invitrogen, Carlsbad, CA). HL1 cells were maintained in Claycomb media (Millipore).
RNA Purification and Gene Expression Analysis:
Quantitative real-time PCR was performed with the SYBR Select Master Mix (Applied Biosystems 4472903) and primers that are listed in Table I in the Supplement.
Protein Extraction and Western Blotting Analysis:
Isolated heart tissue or 6-well cell culture dishes were homogenized in RIPA buffer with protease and phosphatase inhibitors. Antibodies used for Western Blotting are displayed in Table II in the Supplement.
Promoter Activity Assay:
Promoter fragments of the Sptlc1 and Sptlc2 were cloned into KpnI and HindIII restriction sites of the luciferase reporter-containing pGL3-BV plasmid (Table III in the Supplement). Luciferase activity was quantified in lysates of AC16 cells transfected with pGL3-SPTLC1 and pGL3-SPTLC2 containing various fragments of Sptlc1 and Sptlc2 promoters and infected with Ad-GFP or Ad-KLF5 (Dual-Luciferase Reporter Assay System, Promega E1910).
Chromatin Immunoprecipitation (ChIP):
We performed ChIP experiments in HL1 cells infected with Ad-GFP or Ad-KLF5 as described previously10 and in whole hearts 4 weeks following sham or MI surgery. ChIP-grade anti-KLF5 antibody (Active Motif 61099; 10μg/sample) was used to precipitate KLF5-DNA complexes. Quantitative PCR was performed using primers detailed in Table IV in the Supplement.
Measurement of Infarct Size and Cardiomyocyte Cross-Sectional Area:
Heart tissue was sectioned to 7-μm thick sections and stained using Masson’s trichrome stain. Myocardial infarct size was measured as % infarct area with respect to total ventricular area. Cardiomyocyte cross-sectional area was assessed in the remote myocardium by tracing cardiomyocytes using ImageJ software.
Lipidomic analysis:
Lipidomic analysis was performed as described previously6. Abbreviations and the mean and standard deviation for all lipids measured are shown in Table V in the Supplement and Supplemental Excel File 1, respectively. Hierarchical clustering analysis was performed using ClustVis software21.
Statistical Analysis:
Statistical comparisons were generated using Graphpad Prism6 software, and graphs are shown with mean +/− standard error. Specific statistical tests used are stated within the figure legends. Statistical significance was defined as a p-value less than 0.05 unless otherwise specified in the figure legend. For lipidomic data comparing multiple lipid family members, Sidak multiple comparison correction was applied using the formula α(per comparison) = 1 – (1 – 0.05)1/k, where k is the number of lipids for comparison, to calculate the α level for which the overall type 1 error rate is 0.05.
RESULTS
Cardiac KLF5 Expression is Increased in Heart Failure Patients and Mice with Ischemic Cardiomyopathy
Cardiac mRNA and protein analysis in heart tissue obtained from patients with end-stage ischemic heart failure and healthy control patients (Table 1) showed 2-fold upregulation in KLF5 transcripts (Figure 1A) and 2.5-fold increase in KLF5 protein (Figure 1B, 1C). We next investigated if KLF5 is also increased in a mouse model of ischemic heart failure. Permanent LCA ligation caused significant reduction in fractional shortening (Figure IA,B in the Supplement) at 1-day, 2-weeks, and 4-weeks post-MI, and expansion of the end-diastolic dimension at 2- and 4-weeks (Figure IC in the Supplement) and end-systolic dimensions at all 3 time points (Figure ID in the Supplement; Table VI in the Supplement). Because ischemic heart failure results in regional differences in cardiac function, we also analyzed cardiac function using whole left ventricle tracing (Figure 1D; Figure IIA in the Supplement). MI reduced ejection fraction all 3 time points (EF; Figure 1E; Figure IIB in the Supplement). On the other hand, it increased end-diastolic volume (EDV; Figure 1F; Figure IIC in the Supplement) and end-systolic volume at all time points (ESV; Figure 1G; Figure IID in the Supplement). Heart weight normalized to body weight (HW/BW) was increased at all three time points but more robustly 2- and 4-weeks post-MI (Figure 1H; Figure IIE in the Supplement). Cardiac KLF5 mRNA levels were upregulated 24h (2-fold; Figure IIF in the Supplement), 2-weeks (4-fold; Figure 1I) and 4-weeks (4-fold; Figure 1J) post-MI. Assessment of KLF5 protein levels (Figure 1K; Figure IIG in the Supplement) showed sustained increase at 24h (Figure IIH in the Supplement), 2-weeks, and 4-weeks (Figure 1L,M) post-MI.
Table 1:
Patient Characteristics
| Variables | Control (n=11) | ICM (n=20) | p-value |
|---|---|---|---|
| Race/Ethnicity | |||
| White/Not Hispanic or Latino, n (%) | 9 (82) | 16 (80) | 0.99 |
| Other, n (%) | 2 (18) | 4 (20) | |
| Male sex, n (%) | 6 (55) | 12 (60) | 0.77 |
| Age at Tissue Acquisition, years | 44±4 | 52±3 | 0.15 |
| Height, cm | 169±3 | 170±2 | 0.86 |
| Weight, kg | 78±5 | 81±4 | 0.59 |
| BMI, kg/m2 | 27.2±2.0 | 28.0±1.2 | 0.71 |
| BSA, m2 | 1.9±0.1 | 1.9±0.0 | 0.59 |
| Pre-Tissue Acquisition Echocardiographic Measurements | |||
| LVEF, % | 65±3 | 22±3 | <0.0001 |
| LVEDD, cm | 4.1±0.2 | 6.5±0.3 | <0.0001 |
| LVEDD index, cm/m2 | 2.2±0.1 | 3.4±0.2 | <0.0001 |
| LVESD, cm | 2.8±0.2 | 5.8±0.4 | <0.0001 |
| LVESD index, cm/m2 | 1.5±0.1 | 3.1±0.2 | <0.0001 |
| Pre-Tissue Acquisition Hemodynamic Measurements | |||
| Systolic Blood Pressure, mmHg | 119±6 | 109±4 | 0.14 |
| Diastolic Blood Pressure, mmHg | 68±3 | 70±2 | 0.73 |
| Mean Right Atrial Pressure, mmHg | 11±2 | 9±1 | 0.36 |
| Cardiac Index, L/min/m2 | 3.9±0.4 | 2.0±0.1 | <0.001 |
| Pre-Tissue Acquisition Laboratory Values | |||
| Sodium, mmol/L | 150±3 | 138±1 | 0.006 |
| Creatinine, mg/dL | 1.3±0.2 | 1.1±0.1 | 0.45 |
| Hemoglobin, g/dL | 9.4±0.5 | 12.8±0.5 | <0.001 |
Characteristics of healthy heart donors and end-stage ischemic cardiomyopathy heart donors included in the study. Data are shown as mean ± standard error. Control vs ICM were compared using t-test for quantitative data and Fisher’s exact test for categorical data.
Figure 1: Cardiomyocyte KLF5 is Increased in Ischemic Heart Failure –
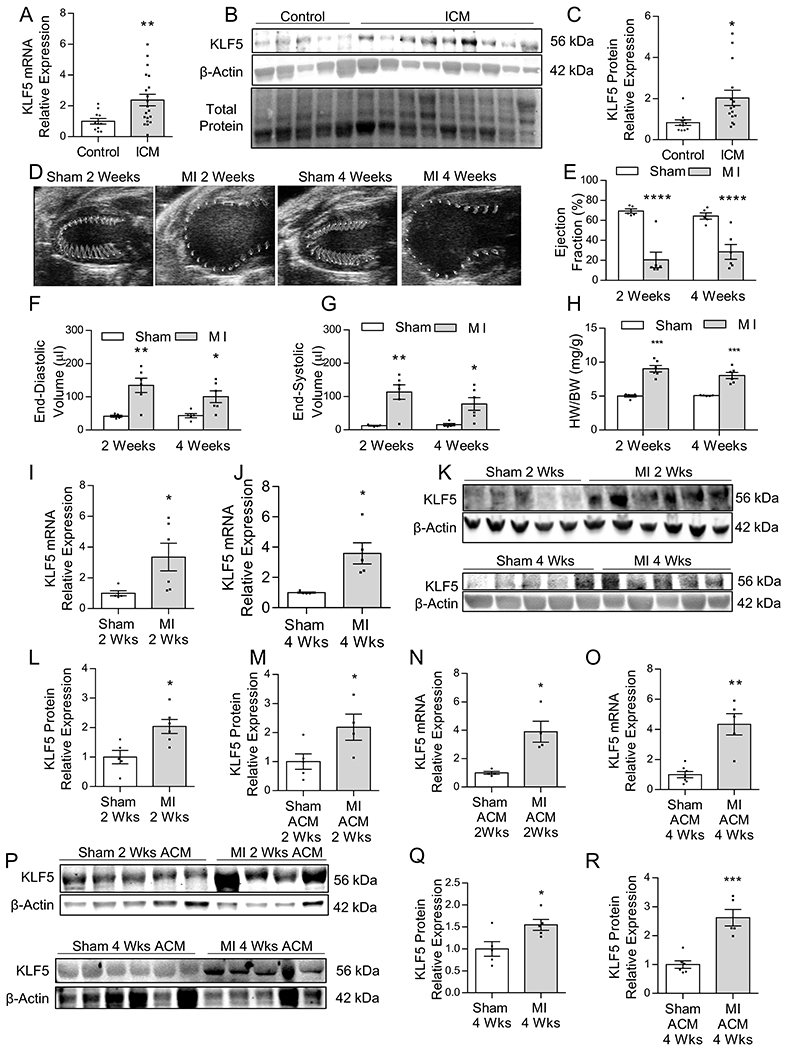
KLF5 mRNA (A), Western blotting (B) and quantification (C) in heart tissue obtained from healthy control and end-stage ischemic heart failure patients. n=11 control, n=20 ICM patients for mRNA analysis; n=5 control and n=9 ICM patients for Western Blotting analysis. *p<0.01 by Welch’s t-test. Representative parasternal long-axis images of the ventricle with wall motion shown in the traced area (D), and measurements (VevoStrain software) of ejection fraction (E), end-diastolic volume (F), and end-systolic volume (G) in sham and MI C57Bl/6 mice 2-weeks and 4-weeks post-surgery. Heart weight normalized to body weight (H). n=5 sham 2-weeks and 4-weeks, n=6 MI 2-weeks and 4-weeks. *p<0.05, **p<0.01, ****p<0.0001; panel E and H analyzed using t-test, panel F and G analyzed using Welch’s t-test. Expression of KLF5 mRNA in heart tissue at 2-weeks (I) and 4-weeks (J) after MI. n=5 sham 2-weeks and 4-weeks, n=6 MI 2-weeks and 4-weeks *p<0.05 by Welch’s t-test. Western blotting (K) with densitometric quantification of KLF5 protein levels in whole heart tissue from C57BL/6 mice 2-weeks (L) and 4-weeks (M) post MI. n=5 sham 2-weeks and 4-weeks, n=5–6 MI 2-weeks and 4-weeks. *p<0.05 by t-test. KLF5 mRNA in isolated adult cardiomyocytes (ACM) 2-weeks (N), and 4-weeks (O) after MI. n=4–6 sham ACM 2-weeks and 4-weeks, n=4–5 MI 2-weeks and 4-weeks. *p<0.05, **p<0.01 by Welch’s t-test. Western blotting (P) with densitometric quantification of KLF5 protein levels in isolated adult cardiomyocytes from C57BL/6 mice 2-weeks (Q) and 4-weeks (R) post-MI. n=5–6 sham ACM 2-weeks and 4-weeks, n=4–5 MI 2-weeks and 4-weeks. *p<0.05, ***p<0.001 by t-test.
We next aimed to determine if the increased KLF5 protein content of ischemic hearts is accounted for by higher KLF5 expression specifically in cardiomyocytes. Therefore, we isolated cardiomyocytes from mice that we confirmed by light microscopy (Figure IIIA in the Supplement), increased expression of cardiomyocyte marker α-myosin-heavy-chain (MHC) (Figure IIIB in the Supplement) and lack of expression of endothelial markers cadherin (CDH)5 and platelet-and-endothelial-cell-adhesion-molecule (PECAM)1, and fibroblast marker platelet-derived-growth-factor-receptor (PDGFR)1a (Figure IIIB in the Supplement). Cardiomyocytes isolated from mice that had undergone MI showed increased KLF5 mRNA (Figure 1N,O) and protein levels at 2-weeks (Figure 1P, Q) and 4-weeks post-MI (Figure 1P,R).
Pharmacological Inhibition of KLF5 Protects Against Ischemic Cardiomyopathy
To explore whether KLF5 inhibition holds therapeutic potential for ischemic heart failure, we randomized mice to undergo MI or sham surgery and treated them with pharmacological KLF5 inhibitor ML264 twice per day beginning 12h after surgery. ML264 reduced cardiac KLF5 protein levels as in previous studies 22(Figure 2A,B). Furthermore, ML264 resulted in significantly reduced early mortality (Figure 2C). We performed echocardiography analysis at baseline and 5 days, 2 weeks, and 4 weeks post-MI (Figure 2D; Table VII in the Supplement). Although ML264 did not have any effect in sham mice, it increased EF in mice with MI (Figure 2E) and reduced EDV (Figure 2F), ESV (Figure 2G) at all timepoints; and heart weight normalized to tibia length (HW/TL; Figure 2H) and HW/BW (Figure 2I) after MI. Consistently, we found that lung wet/dry weight was reduced in ML264-treated mice with MI compared to vehicle-treated mice (Figure 2J). We performed Masson Trichrome staining (Figure IVA in the Supplement) to visualize the scar area and measure cardiomyocyte cross-sectional area (CSA). ML264 had no effect on infarct size (Figure IVB in the Supplement) but partially reduced CSA compared to hears from mice with MI (Figure IVC in the Supplement).
Figure 2: Pharmacological Inhibition of KLF5 Prevents Eccentric Hypertrophy and Improves Cardiac Function after MI –
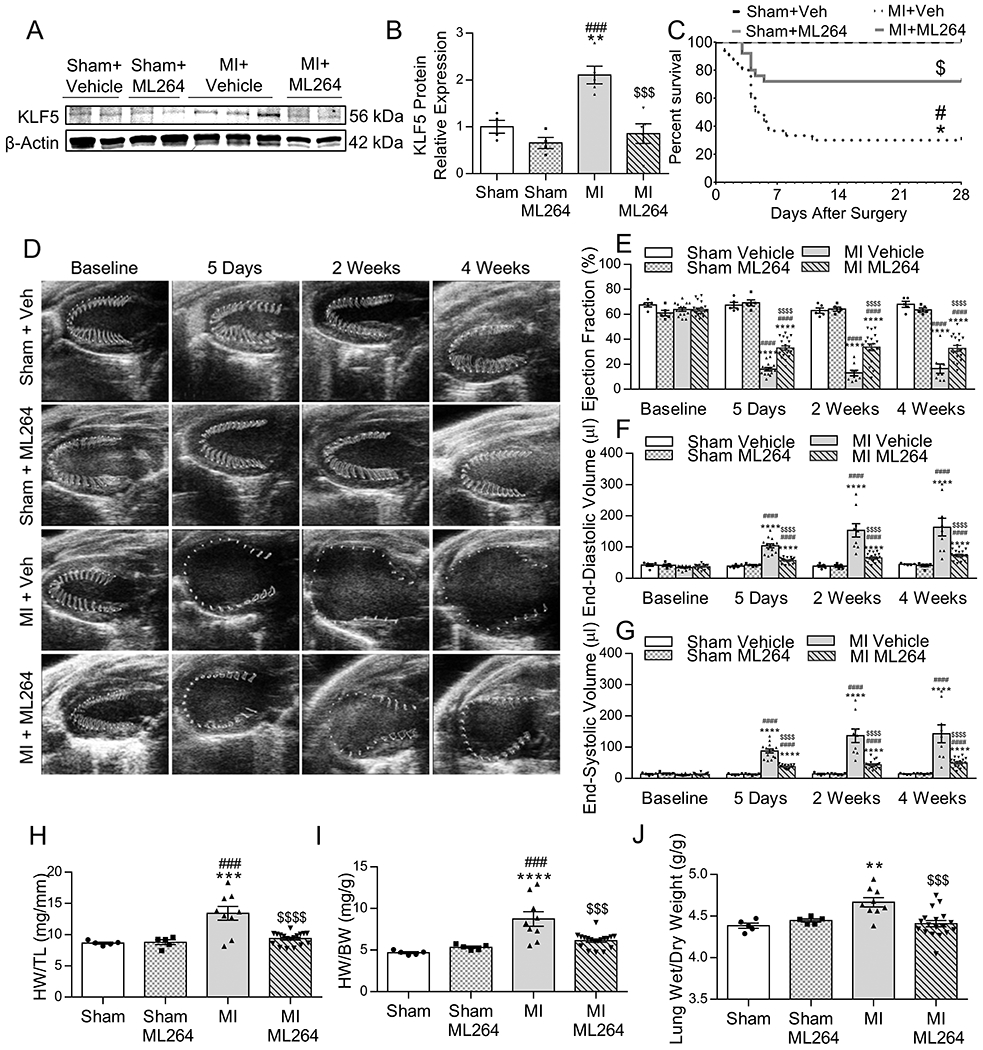
Western blotting (A) and densitometric quantification of KLF5 protein levels (B) in C57BL/6 mice subjected to MI or sham surgery and treated with ML264 or vehicle. n=4 Sham + Vehicle, n=4 Sham + ML264, n=5 MI + Vehicle, n=5 MI + ML264. *p<0.05, **p<0.01 vs Sham + Vehicle; #p<0.05, ####p<0.0001 vs Sham + ML264, $$$p<0.001 vs MI + Vehicle by two-way ANOVA with Tukey HSD. Kaplan-Meier survival curve for C57Bl/6 mice subjected to MI or sham surgery and treated with vehicle or ML264 (C). *p<0.05 vs Sham + Vehicle; #p<0.05 vs Sham + ML264; $p<0.05 vs MI + Vehicle by pairwise log-rank test. Representative parasternal long-axis images of the ventricle with wall motion shown in the traced area (D), and measurements (VevoStrain software) of ejection fraction (E), end-diastolic volume (F), and end-systolic volume (G) at baseline, 5 days, 2 weeks, and 4 weeks post-sham or MI surgery in C57BL/6 mice treated with ML264 or vehicle. n=5 Sham + Vehicle, n=5 Sham + ML264, n=14 MI + Vehicle, n=19 MI + ML264. ****p<0.0001 vs Sham + Vehicle; ####p<0.0001 vs Sham + ML264; $$$$p<0.0001 vs MI + Vehicle by two-way ANOVA with Tukey HSD. Heart weight normalized to tibia length (H) and body weight (I), and lung wet/dry weight ratio (J) in C57BL/6 mice subjected to MI or sham surgery and treated with vehicle or ML264. n=5 Sham + Vehicle, n=5 sham + ML264, n=9 MI + Vehicle, n=18 MI + ML264. **p<0.01, ****p<0.0001 vs Sham + Vehicle; ###p<0.001, ####p<0.0001vs Sham + ML264; $$p<0.01, $$$p<0.001, $$$$p<0.0001 vs MI + Vehicle by two-way ANOVA with Tukey HSD.
Cardiomyocyte-Specific KLF5 Deletion Protects Against Ischemic Heart Failure
Following our observations about the beneficial effect of KLF5 inhibition in cardiac function of mice with MI, we investigated the extent of the contribution of cardiomyocyte KLF5 inhibition to the improvement in ischemic heart failure. We therefore subjected mice with cardiomyocyte-specific KLF5 knockout (αMHC-KLF5−/−)10 to MI. Opposite to control mice with MI, αMHC-KLF5−/− mice with MI had lower cardiac KLF5 protein levels compared with control mice with sham (Figure 3A,B) and improved survival (Figure 3C). Cardiac 2D-echocardiography analysis at baseline, 5 days, 2 weeks, and 4 weeks post-MI (Figure 3D; Table VIII in the Supplement) showed no differences at baseline, and higher EF in αMHC-KLF5−/− mice with MI beginning 5-days after MI compared with control mice with MI (Figure 3E). Control MI mice had significant expansion of EDV (Figure 3F) and ESV (Figure 3G), which was suppressed in αMHC-KLF5−/− mice with MI. Consistently the increase in HW/TL (Figure 3H), HW/BW (Figure V in the Supplement), and wet/dry lung weight (Figure 3I) that occurred in control mice with MI, was not observed in αMHC-KLF5−/− mice with MI.
Figure 3: Cardiomyocyte-Specific Deletion of KLF5 Protects Against Ischemic Cardiomyopathy –

Cardiac KLF5 Western blotting (A) and densitometric quantification (B) in hearts of control mice subjected to MI or sham surgery and αMHC-KLF5−/− mice after MI 4 weeks post-surgery. n=6 control sham, n=7 control MI, n=6 αMHC-KLF5−/− MI mice. ***p<0.001 vs Control sham, ####p<0.0001 vs Control MI by one-way ANOVA with Tukey HSD. Kaplan-Meier survival curve (C) assessing MI-associated mortality in control mice subjected to MI or sham surgery and αMHC-KLF5−/− mice after MI. *p<0.05 by pairwise log-rank test. Representative parasternal long-axis images of the left ventricle (D), and measurements of ejection fraction (E), end-diastolic volume (F) and end-systolic volume (G) at baseline, 5 days, 2 weeks, and 4 weeks post-MI in control mice subjected to MI or sham surgery and αMHC-KLF5−/− mice after MI. n=8 control sham, n=14 control MI, n=6 αMHC-KLF5−/− MI mice. ****p<0.0001 vs control sham; #p<0.05, ##p<0.01, ###p<0.001, ####p<0.0001 vs control MI by two-Way ANOVA with Tukey HSD. Heart weight normalized to tibia length (H) and lung wet/dry weight ratio (I) in control mice subjected to MI or sham surgery and αMHC-KLF5−/− mice after MI. n=8 control sham, n=11 control MI, n=6 αMHC-KLF5−/− MI. Representative sections from hearts stained with trichrome at 0.8x and 20x magnification (J) in control mice subjected to MI or sham surgery and αMHC-KLF5−/− mice after MI. Quantification of infarct size as % total cardiac area (K) in control and αMHC-KLF5−/− mice after MI. Cardiomyocyte cross sectional area (L) and quantification of mRNA levels for BNP, ANP, αMHC and βMHC (M) in heart tissue from control mice subjected to MI or sham surgery and αMHC-KLF5−/− mice after MI. n=5 control sham, n=5 control MI, n=5 αMHC-KLF5−/− MI for cardiomyocyte CSA measurement. n=6 control sham, n=7 control MI, n=6 αMHC-KLF5−/− MI for rtPCR analysis. *p<0.05, **p<0.01 ***p<0.001, ****p<0.0001 vs control sham. #p<0.05, ##p<0.01 ###p<0.001 vs control MI by one-way ANOVA with Tukey HSD.
As in wild type mice with MI that were treated with ML264, Masson trichrome staining (Figure 3J) did not detect any difference in the size of the infarct between control and αMHC-KLF5−/− mice with MI (Figure 3K). Despite the lack of differences in infarct size, αMHC-KLF5−/− mice with MI had reduced cardiomyocyte CSA compared with control mice with MI (Figure 3L). Accordingly, cardiomyocyte-specific ablation of KLF5 prevented the increase in expression of genes associated with hypertrophic signaling, such as B-type natriuretic peptide (BNP) and atrial natriuretic peptide (ANP), which were significantly increased in control MI mice (Figure 3M). Furthermore, αMHC-KLF5−/− mice with MI had less profound reduction of cardiac αMHC expression and increase of βMHC compared to control mice with MI in (Figure 3M).
Cardiomyocyte KLF5 Regulates De Novo Ceramide Biosynthesis in Ischemic Heart Failure
As we previously observed that KLF5 is a major regulator of cardiac lipid metabolism, we performed lipidomic analysis by LC-MS/MS to characterize if cardiomyocyte KLF5 deletion altered the profile of cardiac lipids. This analysis followed by hierarchical clustering based upon the total lipidome revealed that control MI mice clustered separately from control sham mice (Figure 4A). Importantly, 3 out of 4 αMHC-KLF5−/− mice that were subjected to MI, clustered in between the sham and MI mice (Figure 4A). We next performed additional analysis for lipids that have been associated with cardiac dysfunction in mouse models of lipotoxicity, such as ceramides, diacylglycerols, and acyl-carnitines. Hierarchical clustering based on ceramides revealed that control MI mice clustered separately from sham mice, and that 3 out of 4 αMHC-KLF5−/− mice clustered with the sham mice (Figure 4B). Compared with control sham mice, we found increased content of total myocardial ceramide levels in control MI mice but not in αMHC-KLF5−/− mice with MI (Figure 4C). Certain ceramide species, such as Cer d18:1/16:0, Cer d18:1/18:1, and Cer d18:1/24:1 were significantly increased in control MI mice (Figure 4C). Most of these ceramide species had normal levels in αMHC-KLF5−/− mice with MI, which had improved cardiac function (Figure 4C). Likewise, control MI mice clustered separately based upon myocardial dihydroceramides (dhCer), an intermediate in ceramide metabolism (Figure VIA in the Supplement). Compared with control sham mice, the levels of dhCer were increased in control MI mice but not in αMHC-KLF5−/− mice with MI (Figure VIB in the Supplement). Significant increases were observed for dhCer d18:0/16:0, dhCer 18:0/24:0, and dhCer d18:0/24:1 (Figure VIB in the Supplement). On the other hand, hierarchical clustering based upon diacylglycerol species did not separate control mice with MI from αMHC-KLF5−/− with MI mice (Figure VIC in the Supplement). Total diacylglycerols were significantly increased in control MI compared to control sham mice. Looking into certain diacylglycerol species, we found statistically significant increases in control MI compared to control sham mice only for DG 38:4/18:0 and trends toward increased for other diacylglycerols (Figure VID in the Supplement). Hierarchical clustering analysis based upon acyl-carnitines revealed that mice with MI did not cluster separately from sham mice (Figure VIE in the Supplement). As observed for diacylglycerols, acylcarnitines showed an increasing trend in mice with MI and were suppressed in αMHC-KLF5−/− MI mice, but significant increases were not found for any acylcarnitine family members (Figure VIF in the Supplement).
Figure 4: Cardiomyocyte KLF5 Regulates Ceramide Biosynthesis in Ischemic Cardiomyopathy –

Hierarchical clustering based upon the total lipidome (A) or ceramides (B), and quantification (C) of total myocardial ceramide levels measured by LC-MS/MS (**p<0.01 by ANOVA with Tukey HSD) and of different ceramide family members (*p<0.0042 by ANOVA with Tukey HSD and α corrected to 0.0042 due to multiple tests for each lipid family member). n=5 control sham, n=4 control MI, n=4 αMHC-KLF5−/− MI. Lipidomic data is available in the Supplemental excel file 1. Cardiac CerS1, CerS5, ASM, SPTLC1, and SPTLC2 mRNA levels (D); Western blotting (E) and quantification for SPTLC1 and SPTLC2 (F) in hearts of control mice subjected to MI or sham surgery and αMHC-KLF5−/− mice after MI 4 weeks post-surgery. n=6 control sham, n=7 control MI, n=6 αMHC-KLF5−/− MI for Western blotting and rtPCR analysis. *p<0.05, ***p<0.01 vs control sham; #p<0.05, ##p<0.01, ###p<0.001 vs control MI by one-way ANOVA with Tukey HSD. Cardiac expression of SPTLC1 and SPTLC2 mRNA in heart tissue from healthy control patients and patients with end-stage ischemic heart failure (G). Western blotting (H) and densitometric quantification of SPTLC1 and SPTLC2 protein levels in control and end-stage ischemic heart failure patient heart tissue (I,J). n=11 control patients, n=20 ICM patients for mRNA quantification; n=10 control patients, n=11 ICM patients for Western Blot. *p<0.05, **p<0.01 vs control by Welch’s t-test for SPTLC1 mRNA, and t-test for SPTLC1 protein and SPTLC2 mRNA and protein. Representative DHE fluorescence images of live myocardium (K) and quantification of fluorescence intensity (L), and Western blotting (M) and quantification (N) of cleaved PARP in hearts of control mice subjected to MI or sham surgery and αMHC-KLF5−/− mice after MI 4 weeks post-surgery. n=7 control sham, n=9 control MI, n=4 αMHC-KLF5−/− MI for DHE staining intensity; n=6 control sham, n=7 control MI, n=6 αMHC-KLF5−/− MI for cleaved PARP. ***p<0.001, ****p<0.0001 vs control sham, ###p<0.001 vs control MI by one-way ANOVA with Tukey HSD.
As cardiac ceramide content seemed to change in coordination with cardiomyocyte KLF5, while diacylglycerols or acylcarnitines did not, we assessed further potential involvement of KLF5 in ceramide biosynthesis. First, we measured expression of ceramide metabolism-related enzymes. The expression of cardiac ceramide synthase (CerS)1, CerS5, and acid sphingomyelinase (ASM), which regulate the salvage and sphingomyelinase pathways respectively, were not altered following MI surgery (Figure 4D). In contrast, SPTLC1, and SPTLC2 mRNA (Figure 4D) and protein (Figure 4E,F) levels were increased in control MI mice but not in αMHC-KLF5−/− mice with MI compared with control sham mice. Consistently, we found that CerS5, SPTLC1, and SPTLC2 mRNA levels were increased and CerS1 decreased 24h after MI (Figure VIIA in the Supplement) and so was KLF5. On Western Blot, SPTLC2 but not SPTLC1 protein levels were increased (Figure VIIB,C in the Supplement). Because previous studies have shown that heart failure is associated with activation of de novo ceramide biosynthesis6, we next assessed SPTLC1 and SPTLC2 expression in human ICM heart tissue samples, which had increased KLF5. In accordance with previous findings6, SPTLC1 and SPTLC2 mRNA levels were increased by 2 to 3-fold (Figure 4G) and protein levels by 2–3 fold in human ischemic heart failure patients (Figure 4H-J) thereby confirming activation of de novo ceramide biosynthesis in heart failure patients. Oxidative stress and apoptosis markers followed the same pattern of change as cardiac ceramide levels. Specifically, myocardial reactive oxygen species (ROS) content was increased as shown with dihydroethidium (DHE) staining (Figure 4K) in control MI mice and was partially suppressed in αMHC-KLF5−/− mice with MI (Figure 4L). Likewise, cleaved PARP levels were increased in control MI mice and reduced in αMHC-KLF5−/− MI mice (Figure 4M,N).
To assess if cardiomyocyte Klf5 ablation lowers basal cardiac ceramide content we measured the expression of ceramide biosynthesis genes in hearts obtained from 8–12 week old control and αMHC-KLF5−/− mice without MI (Figure VIIIA in the Supplement). RNA levels of ceramide biosynthesis genes were not changed dramatically, except for Sptlc1, which was reduced 20% and Sptlc2 that trended toward reduction (Figure VIIIB in the Supplement). The expression of cardiomyocyte hypertrophy markers was not altered in αMHC-KLF5−/− mice (Figure VIIIC in the Supplement). Western Blotting analysis revealed lower KLF5, SPTLC1, and SPTLC2 protein levels (Figure VIIID,E in the Supplement). Hierarchical clustering analysis based upon the total lipidome did not distinguish between control and αMHC-KLF5−/− mice without MI (Figure IXA in the Supplement). Accordingly, αMHC-KLF5−/− mice did not separate from littermate control mice based on ceramide levels (Figure IXB,C in the Supplement), dhCer levels (Figure XA,B in the Supplement), or DAG (Figure XC,D in the Supplement). Oppositely, αMHC-KLF5−/− mice did separate from control based upon acyl-carnitines (Figure XE in the Supplement) as they had reduced AC C18:0 and AC C18:1 (Figure XF in the Supplement).
Cardiomyocyte KLF5 Constitutive Expression Promotes Systolic Dysfunction and Activates Expression of De Novo Ceramide Biosynthesis Genes
To investigate further the effect of cardiac KLF5 on ceramide metabolism, we generated a new mouse model for cardiomyocyte-specific doxycycline-inducible KLF5 expression. Cardiomyocyte-specific constitutive expression of KLF5 is apparent within 10 days of doxycycline treatment (Figure 5A,B). 2D echocardiography analysis with whole left ventricle tracing (Figure 5C; Table IX in the Supplement) showed that KLF5 transgenic mice had lower EF (Figure 5D) and expanded EDV (Figure 5E) and ESV (Figure 5F) compared with control mice that were also treated with doxycycline at 2-weeks and 4-weeks post-induction of KLF5 expression. Similar to control mice with MI, cardiomyocyte KLF5 constitutive expression did not increase expression of CerS1, CerS5, or ASM (Figure 5G) but resulted in a significant increase in SPTLC1 and SPTLC2 mRNA (Figure 5G) and protein levels (Figure 5H,I).
Figure 5: Cardiomyocyte KLF5 Overexpression Causes Systolic Dysfunction and Increases Expression of Ceramide Synthesis Genes –

Western blotting (A) and quantification (B) of control and KLF5 transgenic mice following 10 days of doxycycline diet. n=5 control, n=4 αMHC-rtTA-KLF5. **p<0.01 by t-test. Representative echocardiography images (C) and quantification of ejection fraction (D), end-diastolic volume (E), and end-systolic volume (F) in control and αMHC-rtTA-KLF5 mice 2-weeks and 4-weeks post KLF5 induction. n=5 control dox, n=5 αMHC-rtTA-KLF5 dox mice. **p<0.01 by two-way ANOVA with Sidak’s multiple comparisons. Measurement of cardiac mRNA levels of CerS1, CerS5, ASM, SPTLC1, and SPTLC2 (G), Western blotting (H) and quantification of SPTLC1 and SPTLC2 (I) in control and KLF5 transgenic mice 10-days post KLF5 induction. n=5 control, n=4 αMHC-rtTA-KLF5 mice. **p<0.01, ****p<0.0001 by t-test.
Cardiomyocyte KLF5 Activates the Sptlc1 and Sptlc2 Promoters Directly
To explore whether KLF5 activates expression of SPTLC1 and SPTLC2 directly, we infected HL1 mouse cardiomyocyte cell line with adenovirus carrying KLF5 cDNA to overexpress KLF5 (Ad-KLF5), or carrying a short hairpin RNA directed against KLF5 (Ad-shKLF5; Figure XIA,B in the Supplement)23. As observed in KLF5 transgenic mice, Ad-KLF5 did not have any effect on CerS1, CerS5, or ASM mRNA levels (Figure XIC in the Supplement) but increased SPTLC1 and SPTLC2 mRNA levels (Figure XIC in the Supplement). Ad-shKLF5 did not have alter the expression of any of these genes (Figure XIC in the Supplement). Western Blotting analysis in cell lysates from HL-1 cells infected with Ad-KLF5 and Ad-shKLF5 (Figure XID in the Supplement) showed that KLF5 increased both SPTLC1 (Figure XIE in the Supplement) and SPTLC2 (Figure XIF in the Supplement) protein levels.
Alignment of the protein sequences for the mouse SPTLC1 and SPTLC2 revealed 38% identity between these two proteins with a significant region of overlap between amino acids 90–353 of SPTLC1 and amino acids 161–421 of SPTLC2 (Figure XIIA,B in the Supplement). As KLF5 activated the expression of both Sptlc1 and Sptlc2 genes, we explored whether the Sptlc1 and Sptlc2 genes resulted from a duplication event that might result in the presence of shared regulatory elements onto which KLF5 can bind. We therefore performed an analysis of the evolutionary history of SPTLC1 and SPTLC2 to determine if conserved regulatory elements could account for the co-regulation of these genes by KLF5. Through this analysis (Figure 6A), we found that Sptlc1 and Sptlc2 are present in all eukaryotes and that the duplication of these genes occurred very early in eukaryotic evolution. Further, we found that in vertebrates, a secondary duplication event of the Sptlc2 gene resulted in the introduction of the Sptlc3 gene, which has low expression in cardiomyocytes (Figure XIII in the Supplement). To determine if there are conserved regulatory elements within the mouse Sptlc1 and Sptlc2 promoters, we aligned the promoter regions of these genes using Clustal Omega software and identified predicted KLF binding sites using Genomatix software. This analysis revealed 3 predicted KLF5 sites that aligned between the mouse Sptlc1 and Sptlc2 promoters, two of which were located within 100 basepairs of the transcription start sites (TSS) (Figure 6B).
Figure 6: KLF5 Directly Activates the SPTLC1 and SPTLC2 on Conserved Promoter Elements.
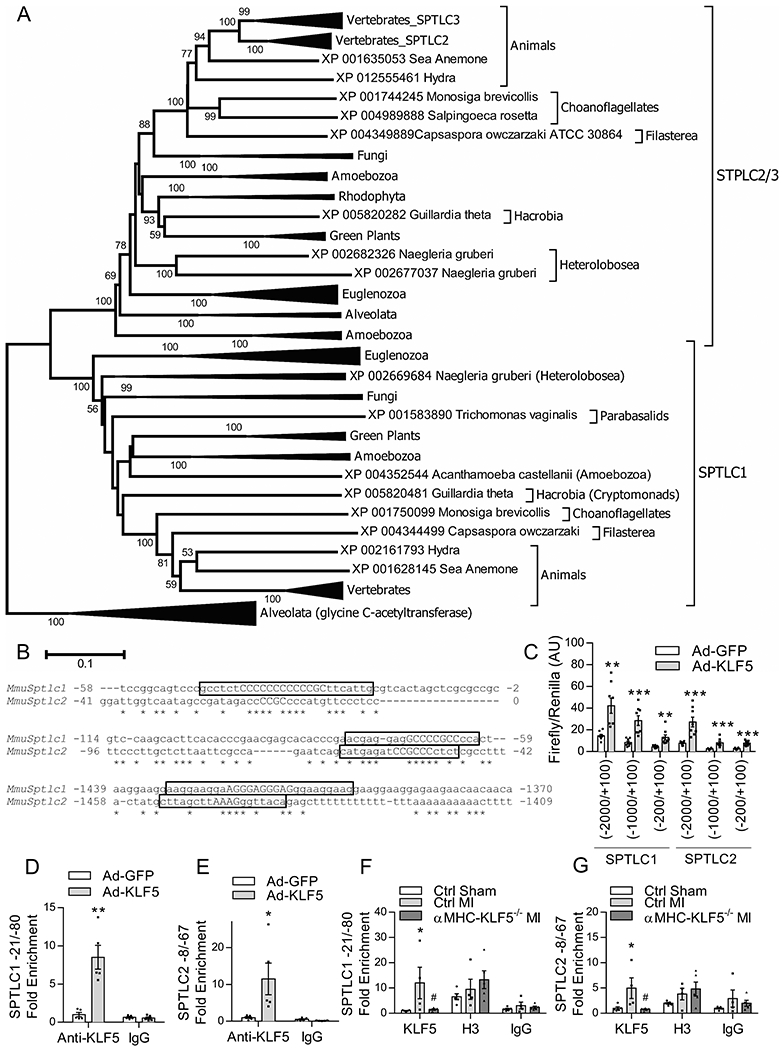
Phylogenetic tree showing the evolutionary relationships of the Sptlc1 and Sptlc2 genes (A). Alignment of promoter regions of the mouse Sptlc1 and Sptlc2 containing predicted KLF5 binding sites highlighted in yellow with the anchor sequences in capital letters (B). Firefly luminescence of luciferase reporter gene driven by Sptlc1 and Sptlc2 promoter fragments normalized to renilla control luminescence in AC16 cells transfected with luciferse promoter plasmids containing truncations of the mouse Sptlc1 and Sptlc2 promoters and infected with Ad-GFP and Ad-KLF5 (C). n=8 wells/condition. **p<0.01, ***p<0.001 versus respective Ad-GFP infected well by t-test. KLF5 enrichment normalized to input for the −21/−80 KLF site of the Sptlc1 promoter and −8/−67 KLF site of the Sptlc2 promoter of HL1 cells infected with Ad-GFP or Ad-KLF5 evaluated by ChIP qPCR (D, E). n=5 wells/group *p<0.05, **p<0.01 versus Ad-GFP by t-test. KLF5 enrichment normalized to input for the −21/−80 KLF site of the Sptlc1 promoter and −8/−67 KLF site of the Sptlc2 promoter evaluated by ChIP qPCR in heart tissue from control mice following sham or MI surgery, and from αMHC-KLF5−/− mice following MI surgery (F, G). n=5 Ctrl sham, n=4 Ctrl MI, n=5 αMHC-KLF5−/− MI hearts. *p<0.05 vs Ctrl sham by Two-way ANOVA with Tukey HSD.
To identify Sptlc1 and Sptlc2 promoter regions that mediate the activating effect of KLF5 on SPTLC1 and SPTLC2 expression, we generated plasmids containing the luciferase reporter driven by the full length and deletion mutants (−2000/+100 bp, −1000/+100 bp and −200/+100 bp) of the Sptlc1 or Sptlc2 promoters. We then transfected AC16 cells with these plasmids and infected the cells with adenovirus expressing KLF5 (Figure XIA in the Supplement). KLF5 increased luciferase signal in all promoter fragments (Figure 6C), suggesting that the −200/+100 bp fragment of both Sptlc1 and Sptlc2 promoters includes strong regulatory elements that mediate the activating effect of KLF5.
To explore the involvement of the −200/+100 bp region of Sptlc1 and Sptlc2 promoters in regulation of the expression by KLF5, we performed ChIP-qPCR in HL-1 cardiomyocytes that were infected with Ad-KLF5 or control Ad-GFP. This analysis showed significant KLF5 enrichment of the KLF binding sites that are located in the −21/−80 region of the Sptlc1 promoter (Figure 6D) and the −8/−67 region of the Sptlc2 promoter (Figure 6E). To confirm that heart failure induces binding of KLF5 to the Sptlc1 and Sptlc2 promoters, we performed ChIP for KLF5 on heart tissue from control and αMHC-KLF5−/− mice following sham or MI. ChIP with anti-histone H3 antibody was used as a positive control. Compared with control sham mice, control mice with MI had a significant enrichment of Sptlc1 −21/−80 site (Figure 6F) and Sptlc2 −8/−67 site (Figure 6G) with KLF5. Binding of KLF5 to the Sptlc1 and Sptlc2 did not occur in hearts of mice with cardiomyocyte specific KLF5 deletion (Figure 6F,G).
Ceramide Biosynthesis Mediates KLF5-induced Systolic Dysfunction
We next aimed to determine if increased ceramide biosynthesis was causative of systolic dysfunction in KLF5 transgenic mice. Therefore, we treated αMHC-rtTA-KLF5 mice with doxycycline diet that was supplemented with myriocin, a pharmacological inhibitor of SPT. Lipidomic analysis in doxycycline treated control mice, KLF5 transgenic mice, and KLF5 transgenic mice treated with myriocin followed by hierarchical clustering for ceramides revealed that KLF5 transgenic mice clustered separately from doxycycline treated control mice and transgenic mice treated with myriocin, which clustered together (Figure 7A). Compared with cardiac ceramide content in control mice fed on doxycycline, KLF5 transgenic mice trended to have increased total ceramide levels, which were suppressed in transgenic mice treated with myriocin (Figure 7B). Analysis of different ceramide species revealed that Cer d18:1/18:1 was significantly elevated and other ceramide family members trended toward increased in transgenic mice but not in transgenic mice with myriocin (Figure 7B). Hierarchical clustering based upon dhCer demonstrated that KLF5 transgenic mice clustered separately from doxycycline treated control mice but not from transgenic mice treated with myriocin (Figure XIVA in the Supplement). We found only trends for increased dhCer levels in KLF5 transgenic mice (Figure XIVB in the Supplement).
Figure 7: De Novo Ceramide Biosynthesis Mediates Systolic Dysfunction in KLF5 Transgenic Mice –
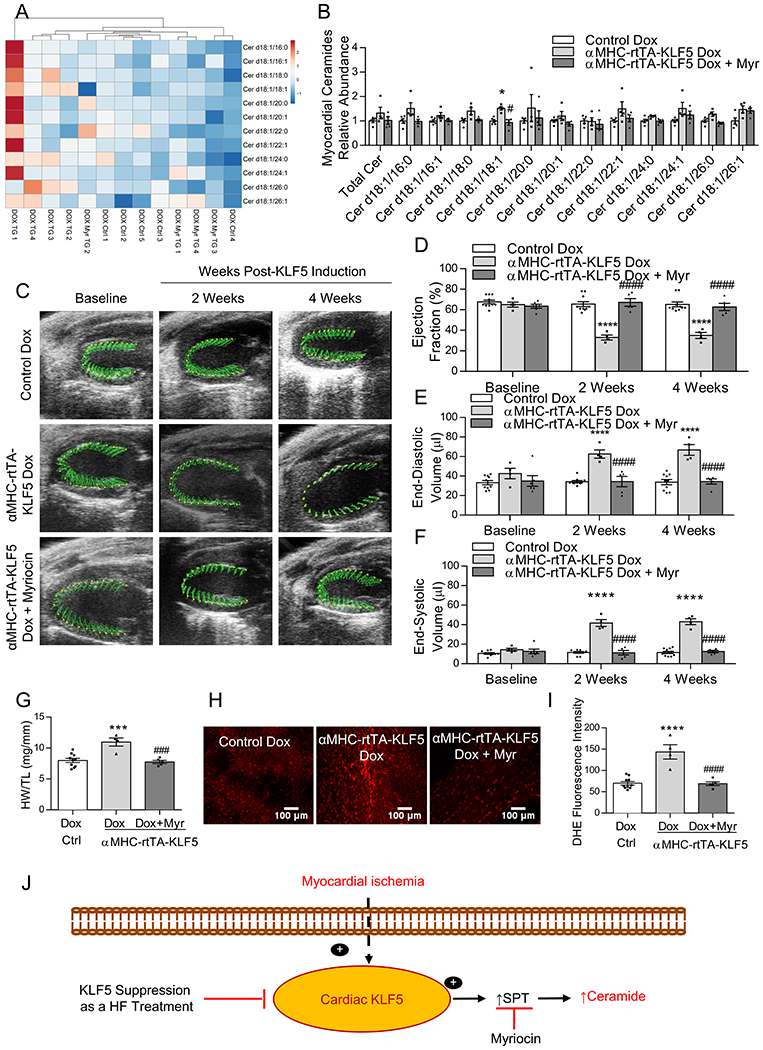
Hierarchical clustering based upon myocardial ceramides (A), and quantification of total myocardial ceramide levels and individual ceramide family members in control mice, αMHC-rtTA-KLF5 mice, and αMHC-rtTA-KLF5 mice treated with myriocin by LC-MS/MS relative to control mice (B). *p<0.0042 vs control, #p<0.0042 vs αMHC-rtTA-KLF5 by ANOVA with Tukey HSD and α corrected to 0.0042 for multiple tests for each lipid family member. n=5 control, n=4 αMHC-rtTA-KLF5, n=4 αMHC-rtTA-KLF5 + myriocin. Lipidomic data is available in the Supplemental excel file 1. Representative echocardiography images (C) and quantification of ejection fraction (D), end-diastolic volume (E), and end-systolic volume (F) in control mice, αMHC-rtTA-KLF5 mice, and αMHC-rtTA-KLF5 mice treated with myriocin at baseline, 2-weeks, and 4-weeks post-induction. n=10 control, n=4 αMHC-rtTA-KLF5, n=5 αMHC-rtTA-KLF5 + myriocin for echocardiography analysis. *p<0.05, ****p<0.0001 vs control; #p<0.05, ####p<0.0001 vs αMHC-rtTA-KLF5 by two-way ANOVA with Tukey HSD. Heart weight normalized to tibia length (G), DHE staining of live myocardium (H), and quantification of DHE fluorescence (I) in control mice, αMHC-rtTA-KLF5 mice, and αMHC-rtTA-KLF5 mice treated with myriocin. n=10 control, n=4 αMHC-rtTA-KLF5, n=5 αMHC-rtTA-KLF5 + myriocin. *p<0.05, ***p<0.001, ****p<0.0001 vs control, ###p<0.001, ####p<0.0001 vs αMHC-rtTA-KLF5 by one-way ANOVA with Tukey HSD. Proposed model depicting the mechanism through which KLF5 regulates ceramide biosynthesis in ischemic heart failure (J).
Hierarchical clustering based upon cardiac diacylglycerol levels did not distinguish αMHC-rtTA-KLF5 mice from control or αMHC-rtTA-KLF5 mice treated with myriocin (Figure XIVC in the Supplement). Cardiac diacylglycerides showed increasing trend in αMHC-rtTA-KLF5 mice and suppression when these mice were treated with myriocin, but none of the diacylglycerol family members were significantly increased in αMHC-rtTA-KLF5 mice (Figure XIVD in the Supplement). Hierarchical clustering based upon acylcarnitine levels did not separate αMHC-rtTA-KLF5 mice from control mice or myriocin-treated αMHC-rtTA-KLF5 mice (Figure XIVE in the Supplement). Opposite from ceramides and diacylglycerols, we found that acylcarnitines showed decreasing trend in KLF5 transgenic mice, however no statistically significant differences were observed in any of the acylcarnitine species (Figure XIVF in the Supplement).
Echocardiography analysis at baseline, 2 weeks, and 4 weeks following KLF5 induction (Figure 7C; Table X in the Supplement) showed αMHC-rtTA-KLF5 mice had reduced EF within 2 weeks of doxycycline treatment, which was prevented by co-treatment with myriocin (Figure 7D). Similarly, αMHC-rtTA-KLF5 mice had significant expansion of the EDV (Figure 7E) and ESV (Figure 7F), which was prevented by co-treatment with myriocin. The αMHC-rtTA-KLF5 mice had a subtle but statistically significant increase in HW/TL (Figure 7G) or HW/BW (Figure XV in the Supplement), which did not occur in transgenic mice treated with myriocin. DHE staining (Figure 7H) showed that αMHC-rtTA-KLF5 mice had increased cardiac superoxide content, which was prevented by myriocin treatment (Figure 7I).
DISCUSSION
Myocardial ischemia (MI) is a major cause of heart failure (HF) accompanied by lower FS, diastolic dysfunction, left ventricular hypertrophy, increased left ventricular end-diastolic pressure, fibrosis, cardiomyocyte hypertrophy, and increased apoptosis. Cardiac metabolic perturbations have been reported in MI24–29. Our previous studies linked CM KLF5 with cardiac lipid metabolism in diabetes10, as well as with systemic lipid homeostasis30. Thus, we investigated potential involvement of KLF5 in ischemic HF that is accompanied by altered metabolism. KLF5 is a member of the 18-members KLF protein family that regulate proliferation, development, and cell death31. KLF isoforms regulate metabolic pathways in several organs, including the heart9, 32, 33.
Previous studies have linked KLF5 with cardiac pathology. One showed that cardiac KLF5 was increased in hypertrophic human myocardial tissue and in spontaneously hypertensive rats14. Another showed neonatal rat ventricular myocytes stimulated with H2O2 had increased KLF5 protein levels, silencing of which suppressed apoptosis13. While these studies suggested that KLF5 exerts a pathological effect in the heart, none of them characterized the underlying mechanisms and neither did they associate KLF5 with ischemic cardiomyopathy, the most common cause of heart failure. The present study identifies cardiomyocyte KLF5 as a pro-hypertrophic factor that is increased in cardiomyocytes of patients with heart failure and mice with experimental ischemic cardiomyopathy. Furthermore, our findings attribute causality of increased KLF5 to heart failure pathology (Figure 7J). Our study demonstrates the feasibility of targeting KLF5 using the pharmacological KLF5 inhibitor ML264 for the prevention of ventricular dilation and for increasing systolic function after MI.
One group investigated the involvement of cardiac fibroblast KLF5 in driving pressure-overload hypertrophy11, 12. These studies did not investigate the regulatory changes in cardiac KLF5 after TAC, but showed mice with global heterozygous Klf5 deletion were protected against pressure-overload hypertrophy11, 12. Fibroblast-specific, but not cardiomyocyte-specific KLF5 inhibition protected against TAC-induced hypertrophy, which the authors attributed to suppression of paracrine secretion of IGF-1 by cardiac fibroblasts11, 12. Conversely, we found that cardiomyocyte-specific and systemic inhibition of KLF5 exerted protective effects in ischemic heart failure. Thus, cardiomyocyte KLF5 seems to be critical for ischemic injury, while fibroblast KLF5 drives pressure-overload hypertrophy. Nevertheless, ischemic cardiomyopathy is characterized by eccentric hypertrophy while pressure-overload hypertrophy results primarily in concentric hypertrophy with a late eccentric phase. Future studies may clarify the relative contributions of cardiomyocyte and fibroblast KLF5 in heart failure.
We identified KLF5 as a novel positive regulator of both the Sptlc1 and Sptlc2 genes that acts via direct binding on proximal elements of the promoters of both genes. Sptlc1 and Sptlc2 genes seem to have emerged from a distant gene duplication event early in eukaryotic evolutionary history. We observed that constitutive expression of cardiomyocyte KLF5 alone suffices to increase cardiac ceramide levels, and systolic dysfunction in KLF5 transgenic mice is prevented by myriocin treatment. This suggests a key role for the de novo ceramide synthesis pathway in mediating KLF5-driven cardiomyopathy. Our results are consistent with previous studies demonstrating the role of the de novo ceramide biosynthesis pathway in cardiac ceramide accumulation in human heart failure and the therapeutic potential of SPT inhibition for eccentric remodeling in ischemic heart failure6, 34, and elucidates the role of KLF5 in these pathological processes. Thus, the cardiomyocyte KLF5 transgenic mouse constitutes a novel mouse model of cardiac lipotoxicity.
Other studies have identified a crucial role for cardiac ceramides in mediating the detrimental effects of lipid overload and cardiac lipotoxicity in multiple mouse models of cardiac injury5, 35–37. Our study identifies KLF5 as a central transcriptional regulator of this pathway. Interestingly, another study has demonstrated that de novo ceramide biosynthesis promotes injury as early as 24h after myocardial infarction, and that therapies to reduce cardiac ceramide levels provide a protective effect8. Likewise, we observed increases in SPTLC1 and SPTLC2 mRNA and protein levels. KLF5 mRNA and protein were increased within 24h of ischemic injury, suggesting that the sooner the therapeutic intervention of KLF5 inhibition is applied the better it will be for alleviating ceramide accumulation and cardiac dysfunction. Accumulation of ceramides impairs mitochondrial function via multiple direct and indirect mechanisms, thereby impairing the heart’s ability to oxidize fatty acids and activating apoptotic cascades38. In addition, KLF5 activation may have broader implications for regulating lipotoxicity in other cardiac diseases that involve ceramide accumulation and energetic deficiency, such as diabetes-associated cardiac dysfunction, cardiac aging, acute ischemic injury, and reperfusion injury
We previously identified KLF5 as an activator of cardiac PPARα expression, which is attenuated when cJun is activated10. The present findings implicate KLF5 as a regulator of ceramide metabolism, which also affects cardiac metabolism. Besides our previous study6 associating ceramide accumulation with cardiac remodeling, ceramides impair mitochondrial function via mechanisms that include JNK activation, impaired cardiac fatty acid oxidation, and apoptosis4, 39–41. Nevertheless, ceramide-driven JNK activation may account for inhibition of KLF5-mediated activation of PPARα and cardiac fatty acid oxidation in pathological states. Future studies that will focus on the interplay between KLF5, ceramide biosynthesis and JNK pathway in cardiac remodeling and fatty acid oxidation are warranted.
In our study, we found that KLF5 did not have a strong effect on other cardiotoxic lipids including diacylglycerols and acylcarnitines, which have been shown to contribute to cardiac lipotoxicity42–46. In various cases, it has been proposed that diacylglycerols and ceramides are co-regulated, and increases in ceramide levels can increase diacylglycerols and vice versa. Sphingomyelin synthase results in the production of both diacylglycerol and sphingomyelin as end products47. Tandem changes of diacylglycerol and ceramide levels have been observed in diglyceride acyltransferase1 transgenic mice43. DAG-dependent proteins such as PKCs regulate ceramide synthesis from sphingomyelin48. Cardiomyocyte KLF5 activation increases cardiac ceramide content with a lesser effect on diacylglycerols, and diacylglycerols are reduced in αMHC-KLF5−/− mice and αMHC-rtTA-KLF5 mice treated with myriocin that can be secondary to the decrease in de novo ceramide biosynthesis. Therefore, cardiac KLF5 activation seems to preferentially increase ceramides among various lipid species that have been associated with cardiac lipotoxicity.
In conclusion, our study associates higher cardiac KLF5 expression with increased expression of SPTLC1 and SPTLC2, higher ceramide content and cardiac dysfunction in both mouse hearts after MI and myocardial samples from patients with advanced heart failure. In silico and biochemical analyses suggest that KLF5 is a direct transcriptional regulator of both SPTLC isoforms. Our observations suggest that KLF5 aggravates ischemic heart failure and KLF5 inhibition holds therapeutic potential for ischemic cardiomyopathy and cardiac remodeling.
Limitations:
Our study was limited to male mice aged between 7–12 weeks, and does not evaluate sex- or age- related differences in ceramide metabolism or KLF5 expression. Because we focused our study on young mice, our results should be cautiously extrapolated to older patient populations with heart failure, for which disease modifying comorbidities are common, and disease modifying treatments are applied. Interestingly, another study found that male FVB mice experienced a greater accumulation of ceramides in response to stimulation with tumor necrosis factor (TNF)α49. Future studies may clarify the role of KLF5 in modulating sex-related differences in ceramide production.
Our lab previously found that αMHC-KLF5−/− mice develop cardiac dysfunction and dilated cardiomyopathy that is first apparent beginning 6-months of age10. We did not observe adverse effects resulting from cardiomyocyte KLF5 ablation likely because our study focused on young αMHC-KLF5−/− mice, a timepoint when these mice have normal cardiac function. Our previous study combined with our present results suggest that both activation and inhibition of KLF5 have adverse consequences for cardiac function, and therapeutic applications should aim for partial and not complete KLF5.
Compared with floxed and cre-expressing mice used as controls for αMHC-KLF5−/− mice, mice treated with vehicle (10% Tween-80, 10% DMSO, 80% saline) every 12h exhibited substantially higher mortality, which occurred reliably across multiple cohorts of mice. This increase in mortality may be attributable to components of the vehicle treatment. Nevertheless, the effect of KLF5 inhibition using ML264 on suppressing mortality is still significant.
Supplementary Material
CLINICAL PERSPECTIVES.
What is new?
KLF5 is increased in patients with ICM and in mouse models of ischemic injury.
Activation of KLF5 promotes systolic dysfunction in a ceramide-dependent mechanism.
Cardiomyocyte KLF5 inhibition improves cardiac function in mouse models of ischemic injury.
What are the clinical implications?
KLF5 emerges as a novel therapeutic target to improve systolic function and protect against eccentric remodeling in ischemic heart failure.
ACKNOWLEDGEMENTS
SOURCES OF FUNDING
This study was supported by the National Heart Lung and Blood Institute of the National Institutes of Health (HL130218, HL151924; KD), an American Heart Association predoctoral fellowship (18PRE34060115) (MH), a Ruth L. Kirschstein National Research Service Award (NRSA) F30 predoctoral fellowship (F30HL146007) (MH), and the American Heart Association and the Kahn Family Post Doctoral Fellowship in Cardiovascular Research (18POST34060150) (IDK).
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- ROS
Reactive oxygen species
- KLF5
Krϋppel-like factor 5
- SPT
Serine palmitoyl transferase
- SPTLC1
Serine palmitoyl transferase long chain base subunit 1
- SPTLC2
Serine palmitoyl transferase long chain base subunit 2
- PPARα
Peroxisome proliferator activated receptor α
- EF
Ejection fraction
- EDV
End-diastolic volume
- ESV
End-systolic volume
- HW/BW
Heart weight normalized to body weight
- HW/TL
Heart weight normalized to tibia length
- αMHC
α myosin heavy chain
- CDH5
Cadherin 5
- PECAM1
Platelet and endothelial cell adhesion molecule 1
- PDGFR1a
Platelet derived growth factor receptor 1a
- CSA
Cross-sectional area
- BNP
B-type natriuretic peptide
- ANP
Atrial natriuretic peptide
- βMHC
β myosin heavy chain
- Cer
Ceramide
- dhCer
Dihydroceramide
- CerS1
Ceramide synthase 1
- CerS5
Ceramide synthase 5
- ASM
Acid sphingomyelinase
- DHE
Dihydroethidium
- DOX
Doxycycline
Footnotes
DISCLOSURES
None
REFERENCES
- 1.Kolwicz SC, Purohit S and Tian R. Cardiac Metabolism and its Interactions With Contraction, Growth, and Survival of Cardiomyocytes. Circ Res. 2013;113:603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neubauer S The Failing Heart — An Engine Out of Fuel. N Engl J Med. 2007;356:1140–1151. [DOI] [PubMed] [Google Scholar]
- 3.Ashrafian H, Frenneaux MP and Opie LH. Metabolic Mechanisms in Heart Failure. Circulation. 2007;116:434–448. [DOI] [PubMed] [Google Scholar]
- 4.Drosatos K and Schulze PC. Cardiac Lipotoxicity: Molecular Pathways and Therapeutic Implications. Curr Heart Fail Rep. 2013;10:109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel E, et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res. 2008;49:2101–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ji R, Akashi H, Drosatos K, Liao X, Jiang H, Kennel PJ, Brunjes DL, Castillero E, Zhang X, Deng LY, et al. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI Insight. 2017;2:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitatani K, Idkowiak-Baldys J and Hannun YA. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal. 2008;20:1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hadas Y, Vincek AS, Youssef E, Żak MM, Chepurko E, Sultana N, Sharkar MTK, Guo N, Komargodski R, Kurian AA, et al. Altering Sphingolipid Metabolism Attenuates Cell Death and Inflammatory Response After Myocardial Infarction. Circulation. 2020;141:916–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pollak NM, Hoffman M, Goldberg IJ and Drosatos K. Krüppel-like factors: Crippling and un-crippling metabolic pathways. JACC Basic Transl Sci. 2018;3:132–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drosatos K, Pollak NM, Pol CJ, Ntziachristos P, Willecke F, Valenti MC, Trent CM, Hu Y, Guo S, Aifantis I, et al. Cardiac Myocyte KLF5 Regulates Ppara Expression and Cardiac Function. Circ Res. 2016;118:241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shindo T, Manabe I, Fukushima Y, Tobe K, Aizawa K, Miyamoto S, Kawai-Kowase K, Moriyama N, Imai Y, Kawakami H, et al. Krüppel-like zinc-finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nat Med. 2002;8:856–863. [DOI] [PubMed] [Google Scholar]
- 12.Takeda N, Manabe I, Uchino Y, Eguchi K, Matsumoto S, Nishimura S, Shindo T, Sano M, Otsu K, Snider P, et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J Clin Invest. 2010;120:254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang D, Yu J, Liu HB, Yan XQ, Hu J, Yu Y, Guo J, Yuan Y and Du ZM. The long non-coding RNA TUG1-miR-9a-5p axis contributes to ischemic injuries by promoting cardiomyocyte apoptosis via targeting KLF5. Cell Death Dis. 2019;10:908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meng G, Xiao Y, Ma Y, Tang X, Xie L, Liu J, Gu Y, Yu Y, Park CM, Xian M, et al. Hydrogen Sulfide Regulates Kruppel Like Factor 5 Transcription Activity via Specificity Protein 1 S-Sulfhydration at Cys664 to Prevent Myocardial Hypertrophy. J Am Heart Assoc. 2016;5:e004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin SC, Wani MA, Whitsett JA and Wells JM. Klf5 regulates lineage formation in the pre-implantation mouse embryo. Development. 2010;137:3953–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sur I, Rozell B, Jaks V, Bergstrom A and Toftgard R. Epidermal and craniofacial defects in mice overexpressing Klf5 in the basal layer of the epidermis. J Cell Sci. 2006;119:3593–3601. [DOI] [PubMed] [Google Scholar]
- 17.Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M, Fan Q, Chuprun JK, Ma XL and Koch WJ. A Novel and Efficient Model of Coronary Artery Ligation and Myocardial Infarction in the Mouse. Circ Res. 2010;107:1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffman M, Kyriazis ID, Lucchese AM, de Lucia C, Piedepalumbo M, Bauer M, Schulze PC, Bonios MJ, Koch WJ and Drosatos K. Myocardial Strain and Cardiac Output are Preferable Measurements for Cardiac Dysfunction and Can Predict Mortality in Septic Mice. J Am Heart Assoc. 2019;8:e012260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davidson MM, Nesti C, Palenzuela L, Walker WF, Hernandez E, Protas L, Hirano M and Isaac ND. Novel cell lines derived from adult human ventricular cardiomyocytes. J Mol Cell Cardiol. 2005;39:133–147. [DOI] [PubMed] [Google Scholar]
- 20.Claycomb WC, Lanson NA, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A and Izzo NJ. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci. 1998;95:2979–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Metsalu T and Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015;43:W566–W570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruiz de Sabando A, Wang C, He Y, García-Barros M, Kim J, Shroyer KR, Bannister TD, Yang VW and Bialkowska AB. ML264, A Novel Small-Molecule Compound That Potently Inhibits Growth of Colorectal Cancer. Mol Cancer Ther. 2016;15:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Goldstein BG, Nakagawa H and Katz JP. Krüppel-like factor 5 activates MEK/ERK signaling via EGFR in primary squamous epithelial cells. FASEB J. 2007;21:543–550. [DOI] [PubMed] [Google Scholar]
- 24.Deedwania P, Kosiborod M, Barrett E, Ceriello A, Isley W, Mazzone T and Raskin P. Hyperglycemia and acute coronary syndrome: a scientific statement from the American Heart Association Diabetes Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2008;117:1610–1619. [DOI] [PubMed] [Google Scholar]
- 25.Diaz R, Paolasso EA, Piegas LS, Tajer CD, Moreno MG, Corvalan R, Isea JE and Romero G. Metabolic modulation of acute myocardial infarction. The ECLA (Estudios Cardiologicos Latinoamerica) Collaborative Group. Circulation. 1998;98:2227–2234. [DOI] [PubMed] [Google Scholar]
- 26.Hofsten DE, Logstrup BB, Moller JE, Pellikka PA and Egstrup K. Abnormal glucose metabolism in acute myocardial infarction: influence on left ventricular function and prognosis. JACC Cardiovasc Imaging. 2009;2:592–599. [DOI] [PubMed] [Google Scholar]
- 27.Park JY, Lee SH, Shin MJ and Hwang GS. Alteration in metabolic signature and lipid metabolism in patients with angina pectoris and myocardial infarction. PLoS One. 2015;10:e0135228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perman JC, Bostrom P, Lindbom M, Lidberg U, Stahlman M, Hagg D, Lindskog H, Scharin TM, Omerovic E, Mattsson HL, et al. The VLDL receptor promotes lipotoxicity and increases mortality in mice following an acute myocardial infarction. J Clin Invest. 2011;121:2625–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karwi QG, Uddin GM, Ho KL and Lopaschuk GD. Loss of Metabolic Flexibility in the Failing Heart. Front Cardiovasc Med. 2018;5:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pol CJ, Pollak NM, Jurczak MJ, Zacharia E, Karagiannides I, Kyriazis ID, Ntziachristos P, Scerbo DA, Brown BR, Aifantis I, et al. Cardiac myocyte KLF5 regulates body weight via alteration of cardiac FGF21. Biochim Biophys Acta Mol Basis Dis. 2019;1865:2125–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McConnell BB and Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiol Rev. 2010;90:1337–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsieh PN, Fan L, Sweet DR and Jain MK. The Kruppel-Like Factors and Control of Energy Homeostasis. Endocr Rev. 2019;40:137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oishi Y and Manabe I. Kruppel-Like Factors in Metabolic Homeostasis and Cardiometabolic Disease. Front Cardiovasc Med. 2018;5:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reforgiato MR, Milano G, Fabriàs G, Casas J, Gasco P, Paroni R, Samaja M, Ghidoni R, Caretti A and Signorelli P. Inhibition of ceramide de novo synthesis as a postischemic strategy to reduce myocardial reperfusion injury. Basic Res Cardiol. 2016;111–112. [DOI] [PubMed] [Google Scholar]
- 35.Park TS and Goldberg IJ. Sphingolipids, Lipotoxic Cardiomyopathy, and Cardiac Failure. Heart Failure Clin. 2012;8:633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basu R, Oudit GY, Wang X, Zhang L, Ussher JR, Lopaschuk GD and Kassiri Z. Type 1 diabetic cardiomyopathy in the Akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am J Physiol Heart Circ Physiol. 2009;297:H2096–H2108. [DOI] [PubMed] [Google Scholar]
- 37.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L and Unger RH. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc Natl Acad Sci. 2000;97:1784–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Novgorodov SA and Gudz TI. Ceramide and mitochondria in ischemia/reperfusion. J Cardiovasc Pharmacol. 2009;53:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Ruiz C, Colell A, Mari M, Morales A and Fernandez-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem. 1997;272:11369–11377. [DOI] [PubMed] [Google Scholar]
- 40.Pettus BJ, Chalfant CE and Hannun YA. Ceramide in apoptosis: an overview and current perspectives. Biochim Biophys Acta. 2002;1585:114–125. [DOI] [PubMed] [Google Scholar]
- 41.Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380:75–79. [DOI] [PubMed] [Google Scholar]
- 42.Liu L, Trent CM, Fang X, Son NH, Jiang H, Blaner WS, Hu Y, Yin YX, Farese RV, Homma S, et al. Cardiomyocyte-specific Loss of Diacylglycerol Acyltransferase 1 (DGAT1) Reproduces the Abnormalities in Lipids Found in Severe Heart Failure. J Biol Chem. 2014;289:29881–29891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH and Goldberg IJ. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem. 2009;284:36312–36323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang L, Ussher JR, Oka T, Cadete VJJ, Wagg C and Lopaschuk GD. Cardiac diacylglycerol accumulation in high fat-fed mice is associated with impaired insulin-stimulated glucose oxidation. Cardiovasc Res. 2010;89:148–156. [DOI] [PubMed] [Google Scholar]
- 45.Schooneman MG, Vaz FM, Houten SM and Soeters MR. Acylcarnitines: reflecting or inflicting insulin resistance? Diabetes. 2013;62:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCoin CS, Knotts TA and Adams SH. Acylcarnitines-old actors auditioning for new roles in metabolic physiology. Nat Rev Endocrinol. 2015;11:617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luberto C and Hannun YA. Sphingomyelin Synthase, a Potential Regulator of Intracellular Levels of Ceramide and Diacylglycerol during SV40 Transformation: Does Sphingomyelin Synthase Account For The Putative Phosphatidylcholine-specific Phospholipase C? J Biol Chem. 1998;273:14550–14559. [DOI] [PubMed] [Google Scholar]
- 48.Zeidan YH and Hannun YA. Activation of acid sphingomyelinase by protein kinase Cdelta-mediated phosphorylation. J Biol Chem. 2007;282:11549–11561. [DOI] [PubMed] [Google Scholar]
- 49.Kadokami T, McTiernan CF, Kubota T, Frye CS and Feldman AM. Sex-related survival differences in murine cardiomyopathy are associated with differences in TNF-receptor expression. J Clin Invest. 2000;106:589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.