

Abstract
A semimechanistic pharmacokinetic (PK)/receptor occupancy (RO) model was constructed to differentiate a next generation anti‐NKG2A monoclonal antibody (KSQ mAb) from monalizumab, an immune checkpoint inhibitor in multiple clinical trials for the treatment of solid tumors. A three‐compartment model incorporating drug PK, biodistribution, and NKG2A receptor interactions was parameterized using monalizumab PK, in vitro affinity measurements for both monalizumab and KSQ mAb, and receptor burden estimates from the literature. Following calibration against monalizumab PK data in patients with rheumatoid arthritis, the model successfully predicted the published PK and RO observed in gynecological tumors and in patients with squamous cell carcinoma of the head and neck. Simulations predicted that the KSQ mAb requires a 10‐fold lower dose than monalizumab to achieve a similar RO over a 3‐week period following q3w intravenous (i.v.) infusion dosing. A global sensitivity analysis of the model indicated that the drug‐target binding affinity greatly affects the tumor RO and that an optimal affinity is needed to balance RO with enhanced drug clearance due to target mediated drug disposition. The model predicted that the KSQ mAb can be dosed over a less frequent regimen or at lower dose levels than the current monalizumab clinical dosing regimen of 10 mg/kg q2w. Either dosing strategy represents a competitive advantage over the current therapy. The results of this study demonstrate a key role for mechanistic modeling in identifying optimal drug parameters to inform and accelerate progression of mAb to clinical trials.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
WHAT QUESTION DID THIS STUDY ADDRESS?
Affinity is an important parameter to achieve desired pharmacokinetic/pharmacodynamic properties of monoclonal antibodies (mAbs) but needs to be optimized against other criteria. However, frameworks for systematically predicting the ideal affinity parameters are limited.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study provides (1) a simulation approach to assess optimal affinities of mAbs while integrating other factors to analyze competitive advantages and (2) a framework to identify critical uncertainties and factors that will most impact lead and clinical development candidate selection.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The study allowed us to identify a range of binding affinities to NKG2A that provide high tumor coverage even at low doses. However, it shows the benefit of tighter binding to membrane receptors has limits, as it eventually leads to increased clearance due to target mediated drug disposition.
The study demonstrates how predictive simulations can drive drug designs by identifying ideal drug properties that are followed by experiments to select leads and clinical development candidates that meet the criteria. This approach allows leveraging existing knowledge in a systematic manner, reducing costs and risks.
INTRODUCTION
Recent observations suggest that therapeutic inhibition of NKG2A receptors could promote enhanced antitumor activity by restoring natural killer (NK) and CD8+ T cell cytotoxic function. 1 , 2 NKG2A is encoded by the KLRC1 gene in humans, and belongs to a family of C‐type lectin receptors (the NKG2 family) that are predominantly expressed on the surface of NK cells and a subset of CD8+ cytotoxic T‐lymphocytes (CTL). 1 , 3 , 4 The NKG2 family has seven members: NKG2A, B, C, D, E, F, and H, and all NKG2 family members, with the exception of homodimeric NKG2D, form disulfide bonded heterodimers with another C‐type lectin coreceptor, CD94. 5 NKG2/CD94 heterodimers bind the ligand HLA‐E, a ubiquitously expressed nonclassical MHC‐I molecule, which results in either inhibition or activation of NK cells and CD8+ CTL by engaging inhibitory receptors, such as NKG2A/CD94, or activating receptors, such as NKG2C/CD94. 6 , 7 , 8 , 9 Importantly, it has been observed that HLA‐E is overexpressed by a variety of solid tumors and overexpression of HLA‐E correlates with poor disease prognosis in ovarian, colorectal, and hepatocellular carcinoma, 1 , 4 , 10 , 11 , 12 , 13 , 14 , 15 presumably due to suppression of the cytotoxic activity of NK cells and CD8+ CTL.
Monalizumab is a clinical stage monoclonal antibody (mAb) being investigated as an immune checkpoint inhibitor of NKG2A receptors expressed on both tumor infiltrating NK and CD8+ CTL for the treatment of gynecological and squamous cell carcinoma of the head and neck (SCCHN). NKG2A/CD94 is frequently co‐expressed with PD‐1. 1 , 11 , 15 Therefore, anti‐NKG2A antibodies, such as monalizumab, are currently being tested in combination with the anti‐PD‐(L)1 antibodies durvalumab and nivolumab. In addition, a phase II clinical trial with monalizumab in combination with cetuximab suggested that NKG2A blockade could also potentially improve the antibody‐dependent cellular cytotoxicity activity of NK cells. Importantly, in phase II studies, monalizumab has been dosed via i.v. infusion either at 10 mg/kg or at 750 mg once every 2 weeks (q2w). However, monalizumab is a “first‐in‐class” molecule, and a drug with “best‐in‐class” properties for improved dosing or efficacy remains unexplored.
Designing an antibody to target NKG2A or the immune system in general remains challenging, and the success rate of oncology drug development in practice remains the lowest of all therapeutic areas. 16 A major reason is the lack of a robust framework for preclinical to clinical translation of oncology drugs to predict efficacy and toxicity. 17 This is particularly true of mAb‐based therapeutics. The versatility of these molecules brings with it an additional level of complexity in understanding their effects; they often rely on intricate mechanisms of action and concentration response relationships that can be nonintuitive and difficult to predict while lacking robust early biomarkers of efficacy.
Given such challenges, translational modeling and simulation has become a key tool in oncology research to increase efficiency and effectiveness in drug discovery and development. Modeling and simulation of oncology therapies can facilitate design, selection, and preclinical to clinical translation, along with optimization of clinical trials. 18 Translational pharmacokinetic (PK)/pharmacodynamic modeling has been defined as the integration of in silico, in vitro, and in vivo preclinical data with mechanism‐based models to predict the effects of new drugs in humans. 18 , 19 These models are built by assembling a quantitative framework describing the relationship between the pharmacology of drug action and downstream biomarker or efficacy responses observed in the preclinical data. Incorporating PK and physiological data relevant to the human disease or disease biology into the model enables the prediction of effects in humans. 20 The resulting models can be used for simulation of effective doses and regimens in the clinic to enhance chances of success.
A panel of anti‐NKG2A antibodies was discovered that has upward of 500‐fold higher specific monovalent affinity for NKG2A compared with monalizumab, and similarly enhanced potency in an in vitro antitumor cytotoxicity assay. To assess the impact of modulating drug properties like affinity for in vivo applications, a semimechanistic pharmacokinetic/receptor occupancy (SM‐PK/RO) model was developed that integrates existing clinical data for monalizumab together with preclinical data of the KSQ anti‐NKG2A lead mAb (KSQ mAb). Our analysis suggests that the increased affinity of highly selective anti‐NKG2A KSQ mAbs may translate into substantial clinical benefits by lowering the dose, and/or reducing the dosing frequency while retaining the saturation of receptor occupancy in tumor tissue needed to achieve optimal therapeutic antitumor efficacy.
METHODS
Computational model overview and implementation
An SM‐PK/RO model was developed to differentiate a KSQ mAb from the clinical competitor, monalizumab, for the treatment of cancer (Figure 1). The model consists of three compartments: (1) the central compartment, which comprises the plasma and rapidly perfused tissues; (2) the peripheral compartment, which comprises all nondisease, slowly perfused tissues; and (3) the tumor compartment. The drug is dosed intravenously to the central compartment and transports to the peripheral and tumor compartments. First‐order nonspecific clearance only occurs in the central compartment. In all compartments, bivalent drug binding to the membrane‐bound NKG2A/CD94 heterodimer complex on NK and T cells was incorporated. Avidity was accounted for by using effective affinities of the drugs for complex. The steady‐state target burden is modeled as a single pool of receptors in each compartment calculated from the number, fraction expressing, and expression level of NK and T cells in the compartment. The target synthesis rate is set by the steady‐state concentration of the receptor complex in each compartment, and the receptor turnover rate, which is assumed to be the same in all compartments. Drug bound to the target is degraded intracellularly once internalized.
Figure 1.
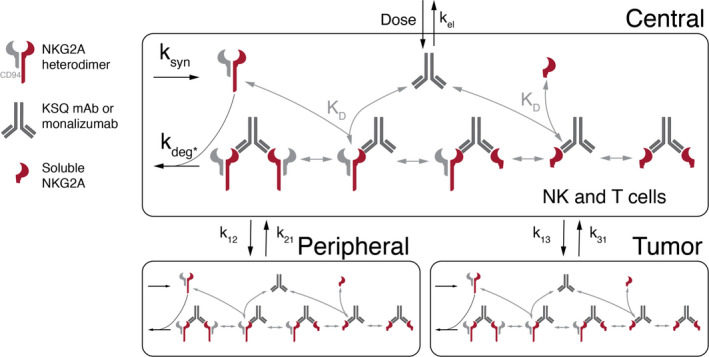
The SM‐PK/RO model diagram of anti‐NKG2A antibody therapies. mAB, monoclonal antibody; SM‐PK/RO, semimechanistic‐pharmacokinetic/receptor occupancy
The model was implemented using KroneckerBio version 0.5.2.3 (maintained at https://github.com/kroneckerbio) and simulations and analyses were performed using MATLAB version 2019a (Mathworks). Parameters for the model were determined from a combination of preclinical in vitro drug binding data, clinical monalizumab PK data in rheumatoid arthritis (RA) and patients with cancer, and target burden estimates from literature. All model parameters, including literature sources and equations, can be found in the [Link], [Link]. Plots were constructed using native MATLAB plotting commands or Gramm. 21
Binding affinity assay
Biosensor analysis using the human antigen was conducted at 25°C in an HBS‐EP+ buffer system (10 mM HEPES pH 7.3, 150 mM NaCl, 3 mM EDTA, and 0.05% Surfactant P20) using a Biacore 8K optical biosensor docked with a CM5 sensor chip (GE Healthcare). The sample hotel was maintained at 10°C. Mouse anti‐human IgG capture antibody (GE Healthcare) was immobilized (4000–6000 RU) to both flow cells of the sensor chip using standard amine coupling chemistry. This surface type provided a format for reproducibly capturing fresh analysis antibody after each regeneration step. Flow cell 2 was used to analyze captured antibody (50–180 RU), whereas flow cell 1 was used as a reference flow cell. Antigen concentrations ranging from 60 to 0.7 nM (threefold dilutions) were prepared in running buffer in single cycle kinetics mode. A blank set of buffer injections were also run and used to assess and subtract system artifacts. The association (150 s) and dissociation (2500 s) phases were measured at a flow rate of 30 μl/min. The surface was regenerated with 3M MgCl2 for 30 s, at a flow rate of 30 μl/min. The data was aligned, double referenced, and fit to a 1:1 binding model using Biacore Insight Evaluation Software, version 1.2.
Cytotoxicity in natural killer/tumor cell co‐culture in vitro assays
Primary human natural killer (NK) cells from healthy donor peripheral blood were isolated by magnetic‐activated cell sorting. Following isolation, NK cells were stained with PE‐conjugated human NKG2A antibody (clone Z199; Beckman Coulter) and NKG2A+ fraction was isolated by fluorescent activated cell sorting (FACS). NKG2A+ sorted NK cells were activated in cell culture media (CellGenix GMP SCGM+ 10% human serum [Sigma] + 1X GlutaMax [Gibco] + 100 μg/ml rhIL‐2 [PeproTech] + 1μ g/ml DNaseI [StemCell]) and expanded by either 2:1 or 1:1 addition of K562 cells engineered to overexpress 4‐1BBL and membrane‐bound human IL‐21 every 7 days. The 1e5 NK cells and 1e5 tumor cells (BV173, B cell lymphoma cell line) were plated per well in a 96‐well round‐bottom tissue culture treated plate (E:T ratio = 1:1) in a final volume of 200 uL in the presence or absence of soluble NKG2A mAbs or isotype controls. After incubation for 24 h at 37°C, 5% CO2, cells were collected by centrifugation, stained with BUV395‐conjugated human CD56 antibody (BD Biosciences) to allow for NK cell/tumor cell discrimination by FACS, and analyzed using the LSRFortessa X‐20 (BD Biosciences) or FACSymphony (BD Biosciences).
Tg32 mouse PK assay
Twelve (12) 6–8 week old male B6.Cg‐Fcgrttm1Dcr Prkdcscid Tg(FCGRT)32DcrJ mice (The Jackson Laboratory) were assigned into 3 groups with 4 mice per group. Body weights were measured the day of antibody administration. At 0 h, the test articles were administered as i.v. injections at 2 mg/kg, and at a dose volume of 5 ml/kg. 25 µl blood samples were collected from each mouse at 1, 2, 4, 7, 14, 21, and 28 days into K3EDTA, processed to plasma, and stored at −20°C. Serum samples were assessed in triplicate by human IgG enzyme‐linked immunosorbent assay (ELISA) using each test articles as its own standard to match what was in the serum being assessed as follows: the human IgG ELISA was first piloted to validate both its specificity and sensitivity for the antibodies; a second pilot ELISA was used to determine the optimum plasma sample dilution for the day 1 plasma samples so that antibody concentrations hit near the top of each standard curve within its linear range; plasma samples were assessed in triplicate at the optimum dilution using each test article as its own standard to match what is in the plasma samples being assessed. PK analysis was performed using PK Solutions software and half‐lives were analyzed starting at day 7 after test article administration.
SM‐PK/RO model assumptions
In the model, the drug is assumed to be eliminated nonspecifically from the central compartment. The KSQ mAb and monalizumab were assumed to have the same nonspecific PK parameters, including drug clearance, tissue and tumor distribution, and compartment volumes. This assumption was supported by data from a Tg32 mouse model where the FcRn gene is replaced by the human FcRn counterpart. 22 In the Tg32 mouse model, the PK of monalizumab and the KSQ mAb, neither of which cross‐reacts with mouse NKG2A receptor, show minimal differences and their time‐concentration curves overlay each other (data not shown). Their respective terminal half‐life and systemic clearance values are shown in Table 1. Limited tumor tissue distribution of monalizumab and KSQ mAb was evaluated in tumor‐bearing NSG mice. KSQ mAb exhibited similar tumor tissue distribution as monalizumab (data not shown). The bivalency of the drugs was incorporated by assuming the binding of each arm of the antibody is independent with no geometric or allosteric effects. The KSQ mAb binds specifically to the NKG2A/CD94 heterodimer complex but not to CD94 in the absence of NKG2A (data not shown).
Table 1.
KSQ mAb and monalizumab drug properties
| Binding affinity K D, nM | NK cell efficacy EC50, μg/ml | Plasma PK half‐life in Tg32 mouse, days | Plasma CL in Tg32 mouse, ml/h/kg | |
|---|---|---|---|---|
| Monalizumab | 48.1 ± 3.1 | 1.5 ± 0.78 | 17.1 ± 2.8 | 0.218 ± 0.0133 |
| KSQ mAb | 0.058 ± 0.007 | 0.045 ± 0.02 | 22.8 ± 1.6 | 0.162 ± 0.00353 |
Abbreviations: CL, clearance; EC50, half‐maximal effective concentration; mAb, monoclonal antibody; PK, pharmacokinetic.
To estimate the target burden in each compartment, it was assumed that only NK and T cells express the target at appreciable levels, the drug has the same affinity for the target complex on both NK and T cells, and that there is no proliferation of NK and T cells over the time scales presented in this work. Therefore, the target concentration remains at steady‐state. The soluble ligand for NKG2A (HLA‐E), was not considered in the model because its weak (micromolar) affinity and low estimated concentrations suggest the fractional occupancy of HLA‐E to NKG2A is less than 0.01% even in the absence of anti‐NKG2A therapies. Soluble NKG2A or receptor shedding is also not considered due to a lack of evidence supporting high enough levels to warrant concerns of an antigen sink. Finally, the internalization of the drug‐bound receptor complex was assumed to occur at the same rate as the free receptor.
Calibrating to monalizumab PK
To inform monalizumab PK parameters in the model, a single‐dose clinical PK study in patients with RA 23 was digitized using WebPlotDigitizer and then used for model calibration. 24 Here, the tumor compartment was excluded and physiological values for the central and peripheral volumes were used. Due to target‐mediated drug disposition (TMDD) at low doses, the high‐dose monalizumab PK data was used to determine the nonspecific clearance, distribution half‐life, and partition coefficient parameters. For the low doses that exhibited TMDD, the turnover rate of the receptor complex, which was constrained via literature information to be greater than 4 h, 25 was fit to accurately capture the faster clearance. Additionally, the binding affinity was increased from the experimentally determined value of 48.1–2.5 nM to account for avidity and to better predict the PK observed at low doses. After PK parameters for monalizumab were determined in a two‐compartment model, the tumor compartment was incorporated and the model was used to simulate monalizumab PK in gynecological and patients with SCCHN to assess model fit to this new clinical data.
Benchmarking to monalizumab receptor occupancy data
The monalizumab model was benchmarked to clinical RO data in patients with gynecological cancer 26 and patients with RA. According to the clinical data, dose levels of 1, 4, and 10 mg/kg demonstrated full target saturation over the dosing regimen (q2w). Clinical RO data from patients with RA also showed full occupancy for the first 2 weeks at the comparable doses for a single‐dose regimen.
Global uncertainty/sensitivity analysis
Latin Hypercube Sampling (LHS)/Partial Rank Correlation Coefficient (PRCC) was used 27 to assess model parameters that were most influential in determining NKG2A occupancy at day 21 in the tumor compartment. LHS is a sampling method 28 that stratifies parameter samples between lower and upper bounds. After using LHS to generate 10,000 parameter sets, model simulations were run and PRCC, which is a measure of the degree of association between two random variables, was used to correlate each individual parameter with the RO at day 21 in the tumor compartment.
RESULTS
Drug property differences between KSQ mAb and monalizumab
Binding property, potency, and PK in Tg32 mice (2 mg/kg) data of both antibodies are shown in Table 1. The KSQ mAb has an 800‐times higher binding affinity, 33‐times higher NK cell killing efficacy, and comparable elimination half‐life compared with monalizumab.
Model calibration and benchmarking
The PK of monalizumab and KSQ mAb were calibrated against single‐dose monalizumab PK data in patients with RA. The results (Figure 2a) demonstrate that the model recapitulates the monalizumab PK data across a range of doses. TMDD is observed at drug concentrations below a critical concentration (C crit, 0.078 μg/ml). C crit is calculated as k syn/(k el/V C), where k syn is the synthesis rate of target NKG2A/CD94 complex, k el is the first‐order elimination rate, and V C is the volume of the central compartment. 29
Figure 2.
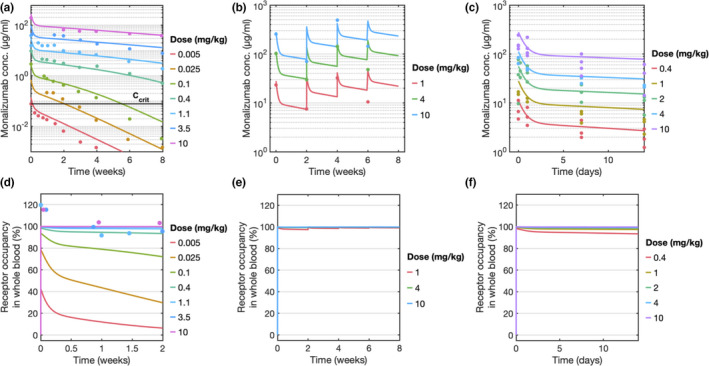
Pharmacokinetic and receptor occupancy simulations were calibrated to clinical patient data. Concentration profile and NKG2A receptor occupancy of monalizumab in whole blood in (a, d) patients with rheumatoid arthritis (RA; single‐dose), (b, e) gynecological cancer (4 doses, q2w), and (c, f) squamous cell cancer of the head and neck (SCCHN; single‐dose). All solid lines represent simulations and points represent clinical data
Next, the model was used to simulate multidose PK data in patients with gynecological cancer. The model accurately recapitulates the clinical observations of concentration at the end of infusion (C eoi) and predose (C trough). Furthermore, the accumulation ratio (C eoi between cycle 1 and cycle 4) of 1.4‐fold to 2‐fold observed in the data was also reproduced in the simulation (Figure 2b). It is observed that at later timepoints (6 weeks), the model predictions of C trough were approximately twofold higher indicating that there might be other elimination routes that were not considered in the model. However, these differences are well within the range of variation seen within a population. Simulations were overlaid with the PKs of patients with SCCHN for a single dose and matched the data without requiring additional fitting (Figure 2c).
Although the measured value of monalizumab binding affinity in an in vitro binding assay was 48.1 ± 3.1 nM, the PK profiles indicated that the in vivo binding affinity was more potent, with an effective K D of 2.5 nM. The effective increase in affinity of the antibody in vivo is consistent with the expectation of avidity, because in vitro analyses (Table 1) measured only monovalent affinity of fraction absorbed and not the on‐cell binding. Functional affinity in vivo can increase if an antibody has multivalent binding. Experimentally, it has been shown that on‐cell binding affinity can be several orders of magnitude greater than the surface plasmon resonance (SPR) measured value due to the accumulated strength of multiple interactions. 30
After calibrating and comparing the model to three PK datasets, the model predictions of NKG2A RO were compared with clinical observations (Figures 2d–f). The clinical data reports that in patients with RA, at doses greater than 1 mg/kg, NKG2A receptors in the blood were fully saturated over the 2‐week period following the initial dose. Model predictions recapitulate this observation, demonstrating fully saturated RO at those dose levels (Figure 2d). At monalizumab doses above 0.4 mg/kg, the model predicts full saturation of NKG2A for the gynecological and SCCHN patient populations at their respective dosing regimens (no clinical data was available; Figures 2e,f).
Identification of best‐in‐class properties
PK parameters for the KSQ mAb and monalizumab were assumed to be the same. The only distinguishing parameter between the two drugs was the binding affinity. A range of binding affinities and corresponding doses were therefore examined that would provide the greatest NKG2A RO in the tumor 14 days postdose (Figure 3a). Although the threshold RO needed for therapeutic efficacy is unknown, monalizumab shows 95% RO or higher at doses greater than 1 mg/kg (Figures 2d–f). Hence, > 95% RO was used as the benchmark to compare various doses and dosing regimens.
Figure 3.
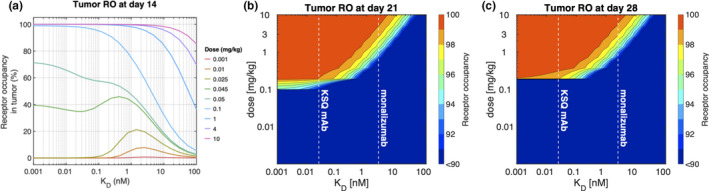
Model predictions on the effect of binding affinity on NKG2A receptor occupancy (RO) in the tumor over various dosing periods. (a) Semi‐log plot shows the RO in the tumor at day 14 at a range of doses and binding affinities. In our model, the affinities of the KSQ monoclonal antibody (mAb) and monalizumab are 25 pM and 2.5 nM, respectively. (b) Contour plots illustrate the NKG2A tumor coverage as a function of a single dose and binding affinity using q3w and (c) q4w dosing regimens
At doses greater than 0.1 mg/kg, lowering the K D of the drug increased the RO. However, at doses lower than 0.1 mg/kg, the benefit of increased binding affinity was lost, as the tighter binding resulted in faster clearance of the drug via TMDD. The model predicts that K D values within the range of 20–100 pM will provide > 95% RO at 0.1 mg/kg. When compared with monalizumab, an mAb with 10 to 20‐fold greater binding affinity could achieve equivalent high levels of RO at one‐tenth of the dose.
Due to the higher binding affinity of the KSQ mAb over monalizumab (KSQ mAb K D = 25 pM; monalizumab in vivo K D = 2.5 nM), it was investigated whether the KSQ mAb could be dosed less frequently than monalizumab. The relationship between binding affinity and dose was examined for dosing windows of 21 and 28 days (Figure 3b,c). Simulations predict that a high affinity mAb can achieve target saturation at q3w and q4w at a dose of 1 mg/kg. The simulations demonstrate the diminishing return of increasing the drug‐target binding affinity when considering values below ~ 0.5 nM at a dose of 1 mg/kg. In this regime of binding affinity values, the model predicts lower K D values do not achieve higher tumor RO at day 21 for a given dose (Figure 3b), emphasizing a design limit to engineering the binding affinity of mAbs targeting NKG2A. Higher binding affinities in this regime lead to faster target‐mediated clearance, thus providing no further increase in tumor RO. The diminishing return relationship between binding affinity and doses near 1 mg/kg is even more prominent when considering tumor RO at day 28 (Figure 3c), where small decreases in dose results in a biphasic switch from nearly full tumor coverage to below 90% RO. These results demonstrate that the KSQ mAb can achieve NKG2A tumor saturation at doses lower than that of monalizumab at both q3w and q4w dosing periods, providing it a competitive advantage.
Summary of differentiation of KSQ mAb from monalizumab
The KSQ mAb is a highly selective antagonist of the NKG2A receptor with greater affinity and NK cell potency than monalizumab. In vivo, the KSQ mAb half‐life is comparable to monalizumab in Tg32 mouse models (Table 1).
To quantitatively compare the expected efficacy of monalizumab and the KSQ mAb at the same dose, simulations were performed using the relevant parameters for each drug and the predicted tumor RO examined. The KSQ mAb can be dosed at 26‐fold, 22‐fold, and 20‐fold lower doses than monalizumab to achieve 99% tumor RO at days 14, 21, and 28, respectively (Table 2, Figure S1). Lower efficacious doses for the KSQ mAb present a direct competitive advantage for the drug over monalizumab.
Table 2.
Dose needed for 99% tumor RO
| Day 14 | Day 21 | Day 28 | |
|---|---|---|---|
| KSQ mAb, mg/kg | 0.15 | 0.2 | 0.25 |
| Monalizumab, mg/kg | 4.0 | 4.5 | 5 |
| Fold advantage | 26 | 22 | 20 |
Abbreviations: mAb, monoclonal antibody; RO, receptor occupancy.
Uncertainty/sensitivity analysis
Some model parameters are uncertain due to biological noise or patient‐to‐patient variability. Therefore, a sensitivity analysis was performed to identify the most influential parameters in controlling model output. A global sensitivity analysis of the model combining a parameter sampling technique (LHS) with a correlation coefficient (PRCC) was used to identify parameters that impact tumor RO at day 21 (Table S1).
From the list of sensitive parameters in Table S1, the most uncertain parameters are the turnover of the mAb‐receptor complex, which is assumed to be the same as the turnover rate of the free receptor, and the perfusion coefficient of the drug into the tumor. The turnover rate was guided by literature measurements of the half‐life of free NGK2A 25 rather than direct measurements, and was consistent with the measured internalization data of BMS986315, an anti‐NKG2A mAb developed by Bristol‐Myers Squibb. Approximately 40% BMS986315‐receptor complexes were internalized when incubating with NKG2A‐expressing cells at 37°C for 20 h. 31 The perfusion coefficient into the tumor was estimated by examining the drug concentration in the tumor and in the blood using a mouse model (data not shown), but not in humans. All other influential parameters from Table S1 are less uncertain because they were fit to multiple datasets of patients with cancer.
A local sensitivity analysis was performed on the two uncertain parameters, the NKG2A turnover rate and the perfusion coefficient of the drug into the tumor, to further examine their impact on model predictions. First, the NKG2A half‐life was varied from the nominal value of 13 h by one log (1.3–130 h). Although the PK of the drug is affected at all 3 doses by the NKG2A half‐life, the RO in the tumor is only affected at doses 0.3 mg/kg and below (Figure 4a,b). Despite uncertainty in the exact value of the NKG2A half‐life, and poor expected tumor coverage of the KSQ mAb if the value approaches 1.3 h, literature data show strong evidence that the lower bound for the NKG2A turnover rate is greater than 4 h 25 and could be longer than 20 h, adding confidence to our model predictions.
Figure 4.
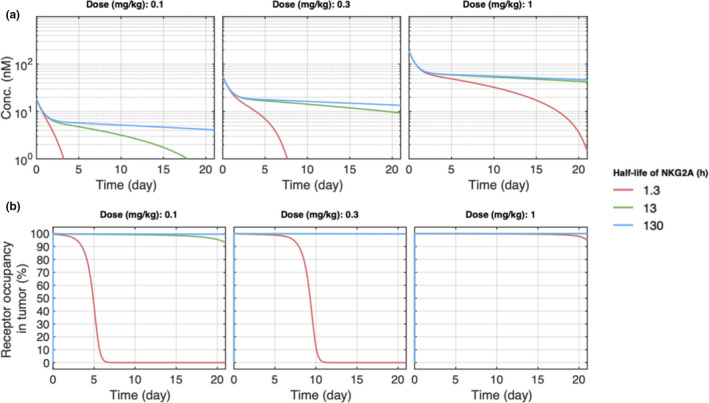
Local sensitivity analysis on the effect of the NKG2A half‐life on pharmacokinetic (PK) and tumor receptor occupancy (RO) in the model. (a) PK and (b) tumor RO model predictions for 0.1, 0.3, and 1 mg/kg doses at different values for the NKG2A half‐life
Second, the effect of the tumor partition coefficient on tumor RO was considered. Simulations predict that increasing the partition coefficient lowers the dose at which receptor saturation occurs (Figure 5). Additionally, at doses greater than 0.3 mg/kg, the model predicts full tumor saturation regardless of the tumor partition coefficient. At a low dose of 0.15 mg/kg, at which a 26‐fold lower dose was shown to achieve the same tumor RO as monalizumab (Table 2), the tumor partition coefficient would need to fall substantially (~50%) from its nominal value to decrease the tumor RO at day 21, lessening the concern of uncertainty in the parameter value affecting final model predictions.
Figure 5.
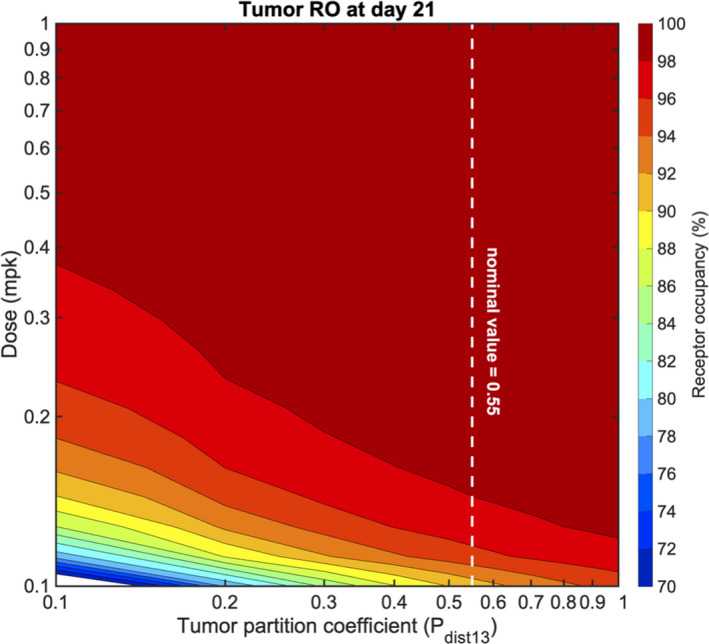
The effect of the tumor perfusion coefficient and dose on tumor receptor occupancy (RO) at day 21. The dotted line represents the nominal tumor perfusion coefficient value of 0.55
DISCUSSION
One of the key attributes of therapeutic monoclonal antibodies is their high binding affinity and specificity. However, the binding affinity required for clinical benefit depends upon the therapeutic indication, disease biology, and the properties of the mAb binding to its target, including the impact of target mediated drug disposition on PK. To determine the optimal affinity of an NKG2A mAb antagonist in solid tumors, a three compartment SM‐PK/RO model was developed. The model was calibrated and benchmarked to publicly available clinical PK and binding data for monalizumab in patients with RA, gynecological cancer, and SCCHN. By simulating the impact of increased affinity on RO, a tradeoff between drug affinity and TMDD was observed, beyond which further increases in binding affinity not only conferred no additional benefit but reduced the RO due to increased clearance via TMDD. Thus, the simulations predicted an optimal binding affinity range that provides a competitive advantage in terms of the drug dose and frequency of dosing.
The selected lead KSQ mAb with an ~ 100‐fold tighter effective binding affinity to NKG2A than monalizumab was examined using the model. With limited data, and assuming similar linear PK properties as monalizumab, the increased affinity is predicted to lower the dose needed to maintain comparable RO in the tumor by 20‐fold. This advantage persisted over the dosing regimens ranging from q2w to q4w. However, simulations showed that additional benefit of further increasing the binding affinity is limited due to tradeoffs with TMDD. In other words, the simulations suggest the KSQ molecule is well‐positioned in the optimal affinity landscape, balancing the tradeoff between increased target coverage and increased target‐mediated clearance. Analysis of the sensitivity of the model predictions to the most uncertain model parameters seems to indicate that at relevant doses and parameter value ranges, the predictions are largely unaffected, suggesting that the competitive advantage of the KSQ mAb is not impacted by the parameters with the most uncertainty.
The need for optimizing affinity of mAbs has become increasingly evident. For some targets, like anti‐TNF mAbs for the treatment of auto‐immune disorders, affinities to soluble TNF‐α in the picomolar range are required. 32 However, tighter binding is not always advantageous, and for many mAbs that bind to membrane receptors, increased receptor coverage may be offset by increased clearance due to TMDD, just as was predicted with KSQ mAb in this study. For example, Leong et al. described the properties of a series of anti‐CD3/anti‐CLL1 bispecific Abs with a high affinity anti‐CLL1 arm and a range of affinities on anti‐CD3 arm. 33 The higher affinity CD3 arms were up to 100‐fold more potent in vitro, but all of the affinity variants had comparable potency in vivo due to the rapid clearance of the high affinity CD3 arms, driven by CD3 on circulating T cells acting as a sink for the drug. Similarly, the recommended dose of anti‐PD1 checkpoint inhibitors nivolumab and pembrolizumab, appears to be independent of binding affinity. Nivolumab has an anti‐PD1 K D of 3.06 nM, whereas pembrolizumab has a K D of 29 pM. Despite 100‐fold difference in affinity, both drugs are administered at ~ 3 mg/kg in the clinic. 34 Mathematical modeling was used to show that pembrolizumab has more rapid clearance due to TMDD than nivolumab, and this offsets its increased potency, leading to a similar dosing requirement in the clinic. 35 Our model predictions regarding the diminishing return of an increased binding affinity for anti‐NKG2A mAbs supports these claims regarding the complex relationship between dose and binding affinity.
The utility of using models to systematically assess the effect of affinity, has benefits at multiple points in a drug discovery program. During lead‐selection, models can be used to identify mission‐critical gaps in knowledge and focus key experiments, reducing time and cost. In this study, a global sensitivity analysis followed by a local analysis on the model parameters shows that the predicted competitive advantage is fairly insensitive to most of the parameter values, especially the uncertain ones, over a physiological range, thus providing support to the assertion that the predictions are robust. These results help focus the selection of critical experiments needed to confirm the simulation predictions (e.g., receptor half‐life and distribution into tumor) and frame expectations for experimental results. To enable selection of the optimal affinity for a monoclonal antibody, mechanistic modeling and simulation can be used to determine the balance among potency, TMDD, efficacy, safety, as well as examining the competitive advantages of pursuing such properties early in a program as part of clinical development candidate selection criteria.
CONFLICT OF INTEREST
Monika Musial‐Siwek, Yang Zhao, Emily Rosentrater, Janice Villali, Tom McCaughtry, and Hanlan Liu are/were employees of KSQ Therapeutics. Phillip Spinosa, Kalyanasundaram Subramanian, Lucia Wille, Alison Betts, and Marc Presler are Applied BioMath employees.
AUTHOR CONTRIBUTIONS
P.S., K.S., L.W., A.B., M.P., M.M‐S., Y.Z., T.M., and H.L. wrote the manuscript. P.S., K.S., A.B., M.P., M,M‐S., T.M., and H.L. designed the research. P.S., M.P., M.M‐S., Y.Z., E.R., and J.V. performed the research. P.S., K.S., L.W., A.B., M.P., M.M‐S., Y.Z., E.R., J.V., T.M., and H.L. analyzed the data. T.M. and H.L. contributed new reagents/analytical tools.
Supporting information
Figure S1
Tables S1–S5
Acknowledgments
The KSQ mAbs panel presented in this work was discovered and developed in collaboration with Adimab LLC. The authors thank Joshua Apgar for data analysis and technical insight.
Phillip Spinosa and Monika Musial‐Siwek equally contributed to this work.
Funding information
The study was funded by KSQ Therapeutics, Cambridge, MA.
REFERENCES
- 1. André P, Denis C, Soulas C, et al. Anti‐NKG2A mAb is a checkpoint inhibitor that promotes anti‐tumor immunity by unleashing both T and NK cells. Cell. 2018;175:1731‐1743.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ruggeri L, Urbani E, Andre P, et al. Effects of anti‐NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica. 2016;101:626‐633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aramburu J, Balboa MA, Ramirez A, et al. A novel functional cell surface dimer (Kp43) expressed by natural killer cells and T cell receptor‐gamma/delta+ T lymphocytes. I. Inhibition of the IL‐2‐dependent proliferation by anti‐Kp43 monoclonal antibody. J Immunol. 1990;144:3238‐3247. [PubMed] [Google Scholar]
- 4. van Montfoort N, Borst L, Korrer MJ, et al. NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cell. 2018;175:1744‐1755.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sullivan LC, Clements CS, Beddoe T, et al. The heterodimeric assembly of the CD94‐NKG2 receptor family and implications for human leukocyte antigen‐E recognition. Immunity. 2007;27:900‐911. [DOI] [PubMed] [Google Scholar]
- 6. Creelan BC, Antonia SJ. The NKG2A immune checkpoint ‐ a new direction in cancer immunotherapy. Nat Rev Clin Oncol. 2019;16:277‐278. [DOI] [PubMed] [Google Scholar]
- 7. van Hall T, André P, Horowitz A, et al. Monalizumab: inhibiting the novel immune checkpoint NKG2A. J Immunother Cancer. 2019;7:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borrego F, Masilamani M, Marusina AI, Tang X, Coligan JE. The CD94/NKG2 family of receptors: from molecules and cells to clinical relevance. Immunol Res. 2006;35:263‐278. [DOI] [PubMed] [Google Scholar]
- 9. Gunturi A, Berg RE, Forman J. The role of CD94/NKG2 in innate and adaptive immunity. Immunol Res. 2004;30:29‐34. [DOI] [PubMed] [Google Scholar]
- 10. Wieten L, Mahaweni NM, Voorter CEM, Bos GMJ, Tilanus MGJ. Clinical and immunological significance of HLA‐E in stem cell transplantation and cancer. Tissue Antigens. 2014;84:523‐535. [DOI] [PubMed] [Google Scholar]
- 11. Abd Hamid M, Wang RZ, Yao X, et al. Enriched HLA‐E and CD94/NKG2A interaction limits antitumor CD8+ tumor‐infiltrating T lymphocyte responses. Cancer Immunol Res. 2019;7:1293‐1306. [DOI] [PubMed] [Google Scholar]
- 12. Sun C, Xu J, Huang Q, et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology. 2017;6:e1264562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gooden M, Lampen M, Jordanova ES, et al. HLA‐E expression by gynecological cancers restrains tumor‐infiltrating CD8+ T lymphocytes. Proc Natl Acad Sci U S A. 2011;108:10656‐10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bossard C, Bézieau S, MatysiakBudnik T, et al. HLA‐E/β2 microglobulin overexpression in colorectal cancer is associated with recruitment of inhibitory immune cells and tumor progression. Int J Cancer. 2012;131:855‐863. [DOI] [PubMed] [Google Scholar]
- 15. Eugène J, Jouand N, Ducoin K, et al. The inhibitory receptor CD94/NKG2A on CD8+ tumor‐infiltrating lymphocytes in colorectal cancer: a promising new druggable immune checkpoint in the context of HLAE/β2m overexpression. Mod Pathol. 2020;33:468‐482. [DOI] [PubMed] [Google Scholar]
- 16. Wong CH, Siah KW, Lo AW. Estimation of clinical trial success rates and related parameters. Biostatistics. 2019;20:273‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu AZ. Quantitative translational modeling to facilitate preclinical to clinical efficacy & toxicity translation in oncology. Future Sci OA. 2018;4:FSO306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mager DE, Jusko WJ. Development of translational pharmacokinetic‐pharmacodynamic models. Clin Pharmacol Ther. 2008;83:909‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Betts A, van der Graaf PH. Mechanistic quantitative pharmacology strategies for the early clinical development of bispecific antibodies in oncology. Clin Pharmacol Ther. 2020;108:528‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Agoram BM, Martin SW, van der Graaf PH. The role of mechanism‐based pharmacokinetic‐pharmacodynamic (PK‐PD) modelling in translational research of biologics. Drug Discov Today. 2007;12:1018‐1024. [DOI] [PubMed] [Google Scholar]
- 21. Morel P. Gramm: grammar of graphics plotting in Matlab. J Open Source Soft. 2018;3:568. [Google Scholar]
- 22. Roopenian DC, Christianson GJ, Sproule TJ. Human FcRn transgenic mice for pharmacokinetic evaluation of therapeutic antibodies. Methods Mol Biol. 2010;602:93‐104. [DOI] [PubMed] [Google Scholar]
- 23. Safety and tolerability of NNC0141‐0000‐0100 in subjects with rheumatoid arthritis (NCT01370902). ClinicalTrials.gov. 2011. https://clinicaltrials.gov/ct2/show/NCT01370902. Accessed December 23, 2014.
- 24. Rohatgi A. WebPlotDigitizer. Version 4.4. Pacifica, CA. November 2020. https://automeris.io/WebPlotDigitizer [Google Scholar]
- 25. Borrego F, Kabat J, Sanni TB, Coligan JE. NK cell CD94/NKG2A inhibitory receptors are internalized and recycle independently of inhibitory signaling processes. J Immunol. 2002;169:6102‐6111. [DOI] [PubMed] [Google Scholar]
- 26. Tinker AV, Hirte HW, Provencher D, et al. Dose‐ranging and cohort‐expansion study of monalizumab (IPH2201) in patients with advanced gynecologic malignancies: a trial of the Canadian Cancer Trials Group (CCTG): IND221. Clin Cancer Res. 2019;25:6052‐6060. [DOI] [PubMed] [Google Scholar]
- 27. Marino S, Hogue IB, Ray CJ, Kirschner DE. A methodology for performing global uncertainty and sensitivity analysis in systems biology. J Theor Biol. 2008;254:178‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McKay MD, Beckman RJ, Conover WJ. A comparison of three methods for selecting values of input variables in the analysis of output from a computer code. Technometrics. 1979;21:239‐245. [Google Scholar]
- 29. Stein AM, Peletier LA. Predicting the onset of nonlinear pharmacokinetics. CPT Pharmacometrics Syst Pharmacol. 2018;7:670‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rudnick SI, Adams GP. Affinity and avidity in antibody‐based tumor targeting. Cancer Biother Radiopharm. 2009;24:155‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bezman N, Korman A, Deshpande S, Jhatakia A, Huang R, Chen G, Rakestraw G, Henning K, Rangan V, Bee C, Shao X. Anti‐NKG2A antibodies and uses thereof. Patent Number: US 2020/0199226 A1. June 25, 2020.
- 32. Shim H. One target, different effects: a comparison of distinct therapeutic antibodies against the same targets. Exp Mol Med. 2011;43:539‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leong SR, Sukumaran S, Hristopoulos M, et al. An anti‐CD3/anti‐CLL‐1 bispecific antibody for the treatment of acute myeloid leukemia. Blood. 2017;129:609‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Picardo SL, Doi J, Hansen AR. Structure and optimization of checkpoint inhibitors. Cancers. 2019;12:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Apgar J, Wong J, Phennicie R, et al. Abstract 5001: Quantitative systems pharmacology modeling and analysis provides biological insights into anti‐PD‐1 dosing and predicts optimal PD‐1 x TIM‐3 therapeutic properties for bispecifics and fixed dose combinations in immuno‐oncology. Cancer Res. 2016;76:5001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Tables S1–S5