

Abstract
Immune-modulating therapies have revolutionized the treatment of chronic diseases, particularly cancer. However, their success is restricted and there is a need to identify new therapeutic targets. Here, we show that natural killer cell granule protein 7 (NKG7) is a regulator of lymphocyte granule exocytosis and downstream inflammation in a broad range of diseases. NKG7 expressed by CD4+ and CD8+ T cells played key roles in promoting inflammation during visceral leishmaniasis and malaria—two important parasitic diseases. Additionally, NKG7 expressed by natural killer cells was critical for controlling cancer initiation, growth and metastasis. NKG7 function in natural killer and CD8+ T cells was linked with their ability to regulate the translocation of CD107a to the cell surface and kill cellular targets, while NKG7 also had a major impact on CD4+ T cell activation following infection. Thus, we report a novel therapeutic target expressed on a range of immune cells with functions in different immune responses.
Immunity and related inflammation are critical defense mechanisms against infection and tumors, as well as an integral part of tissue repair1. Lymphocytes are crucial for these responses and two important immune processes employed by them are the production and secretion of pro-inflammatory cytokines and cytotoxic granule exocytosis1,2. If these cellular activities are not appropriately stimulated, infection or tumor growth can progress uncontrolled3–5. However, these responses also need to be tightly regulated to prevent tissue damage and associated disease6.
Infectious diseases such as malaria and visceral leishmaniasis require the generation of interferon-γ (IFN-γ)-producing CD4+ T cells (TH1 cells) to help phagocytes kill captured or resident parasites7. However, inflammation generated by these cells can also damage tissue, including destruction of reticuloendothelial networks that limits the removal and killing of parasites, thereby contributing to persistence of infection8. In the case of malaria, inflammation can also activate vascular endothelium, thereby allowing parasite sequestration into various tissues, with detrimental consequences for organ function9. However, the immunoregulatory pathways that emerge to control this inflammation can also have a negative impact on parasite control10. Thus, chronic infectious diseases such as malaria and visceral leishmaniasis can be characterized by an imbalance between pro- and anti-inflammatory immune responses.
Tumor microenvironments are often typified by the presence of unresponsive immune cell populations11, whereby their antitumor activities are inhibited by local expression of immunoregulatory molecules12. Thus, the transient suppression of these molecules or activation of suppressed pro-inflammatory pathways is often a goal of cancer treatments. Indeed, patients with metastatic melanoma with durable clinical responses following immune checkpoint blockade exhibit an immune signature characterized by increased expression of genes associated with IFN-γ-producing TH1 cells and cytotoxic CD8+ T cells13,14. Success in treating cancer using host-directed therapies highlights the substantial clinical potential of manipulating immune cells for patient benefit4. However, treatment success varies considerably between individuals, even for the same cancer types14,15, underlining the need to uncover new immunoregulatory molecules that can be targeted to improve disease outcomes.
Here, we report that natural killer cell granule protein 7 (NKG7) is a novel mediator of inflammation in several different inflammatory contexts. NKG7 was first identified in natural killer cells (NK cells) and T cells16, but since then there have been few studies on this molecule, and as such, the function of NKG7 in health and disease remains poorly characterized. However, the recent increase in the reporting of whole-transcriptome datasets has resulted in accounts of differential expression of NKG7 in various experimental contexts14,17. Herein, we show that NKG7 functions to regulate cytotoxic granule exocytosis in effector lymphocytes, thus acting as a critical mediator of inflammation in a broad range of infectious and non-infectious diseases.
Results
Identification of NKG7 on CD4+ T cells exposed to chronic inflammation.
Experimental visceral leishmaniasis caused by infection of C57BL/6 mice with the human parasite Leishmania donovani is characterized by an organ-specific response to infection18. The liver is a site of acute, resolving infection, while infection persists in the spleen, accompanied by a breakdown in tissue architecture driven by unchecked inflammation and an accumulation of CD4+ T cells that produce a range of pro-inflammatory molecules but are unable to control parasite growth10. To better understand the inflammatory response in this organ, we isolated CD4+ T cells from the spleen and liver of naive and L. donovani-infected C57BL/6 mice at day 56 post-infection (p.i.), to identify differentially expressed genes (DEGs) in effective anti-parasitic CD4+ T cell responses (liver) and dysregulated CD4+ T cell responses (spleen) (Supplementary Fig. 1). Concurrently, we also isolated CD4+ T cells from patients with visceral leishmaniasis on presentation to the clinic and 30 d after drug treatment (when effective anti-parasitic immunity had developed19; Supplementary Table 1), and used these samples to identify DEGs associated with human visceral leishmaniasis using RNA sequencing (RNA-seq) (Supplementary Fig. 1 and Supplementary Table 2). Next, we compared mouse and human DEGs to identify a core signature shared between all CD4+ T cell populations analyzed (Supplementary Tables 2–4). We also identified an inflammatory signature associated with persistent infection (that is, DEGs shared between CD4+ T cells from human patients with visceral leishmaniasis and mouse spleens; Supplementary Tables 2 and 4) and an immune signature associated with controlled infection (that is, DEGs shared between CD4+ T cells from human patients with visceral leishmaniasis and those from mouse livers; Supplementary Tables 2 and 3). We predicted that molecules associated with the inflammatory signature would contain DEGs strongly associated with inflammatory networks (Fig. 1a and Supplementary Tables 3 and 4), and the top DEG identified on this list encoded NKG7—a molecule predicted to be a multi-pass membrane protein comprising a helical bundle (Fig. 1b,c) expressed by multiple immune cells14,16,17,20. We confirmed increased expression of NKG7 by CD4+ T cells from patients with visceral leishmaniasis, relative to CD4+ T cells from the same patients 30 d later and the same cell population from endemic controls (Fig. 1d). We also measured increased expression of Nkg7 by conventional (Tconv) and Foxp3+ regulatory (Treg) CD4+ T cells in the spleen and liver of L. donovani-infected mice at day 56 p.i. (Fig. 1d). Although Nkg7 gene expression was highest in liver Tconv CD4+ T cells both in the naive state and following infection, the increased expression by splenic conventional CD4+ T cells following infection was greatest and probably contributed to its ranking at the top of our inflammatory signature list (Fig. 1a). Thus, NKG7 is highly expressed by CD4+ T cells in infected and inflamed tissues.
Fig. 1 |. NKG7 is highly upregulated in splenic CD4+ T cells during L. donovani infection.
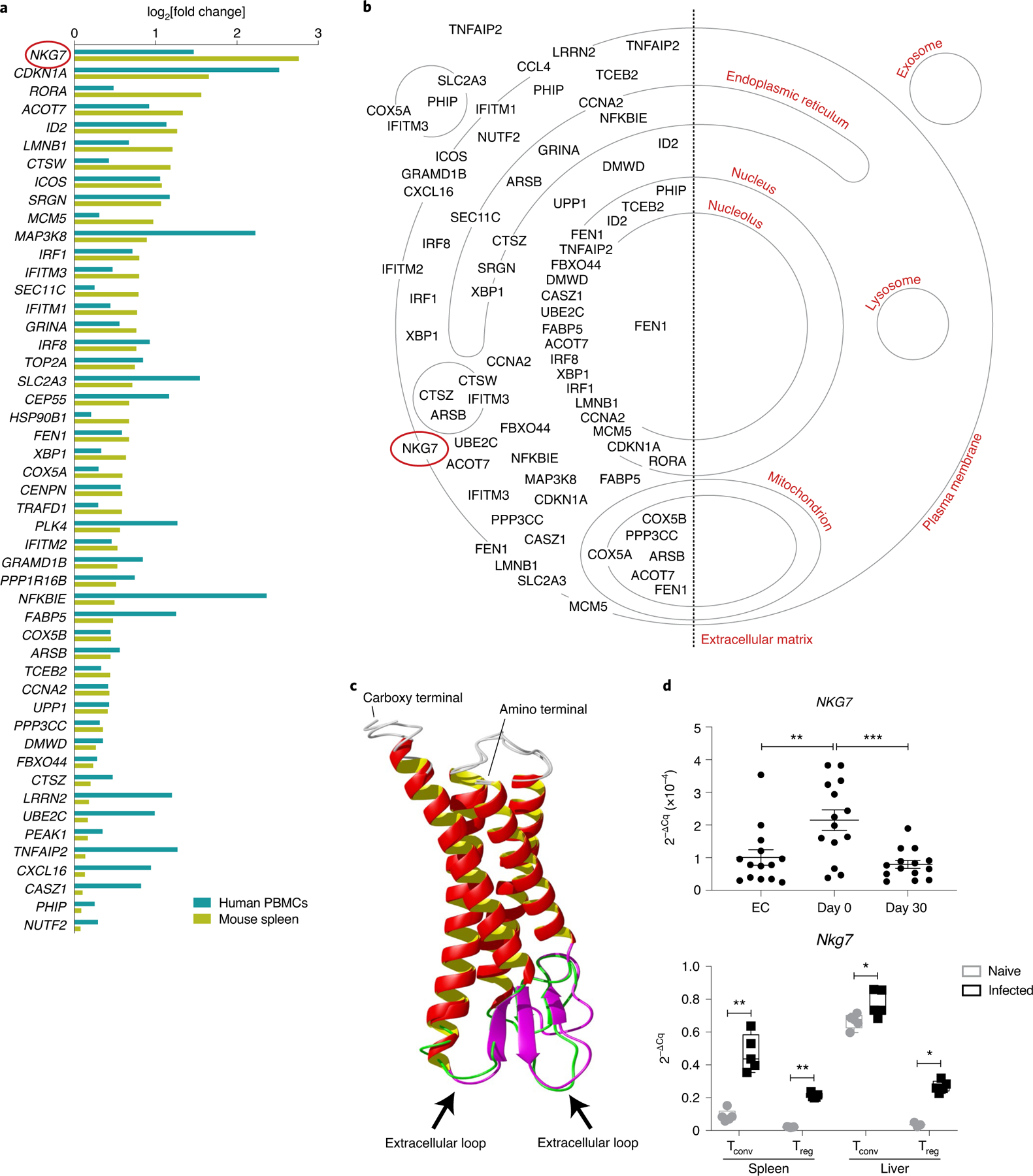
a, Upregulated genes found in CD4+ T cells isolated from mouse spleen at day 56 p.i. and in human PBMCs from patients with visceral leishmaniasis at the time of admission to clinic for treatment. b, Cellular locations of proteins encoded by the upregulated genes in a are indicated on the cellular map using information obtained from the Gene Ontology Cellular Component knowledge base. c, The protein structure of NKG7 generated using I-TASSER. Predicted extracellular loops (indicated by black arrows) of human and mouse NKG7 are highlighted in green and purple, respectively. d, Top: validation of NKG7 upregulation in patients with visceral leishmaniasis before treatment (day 0; n = 14) compared with the same patients after treatment (day 30) and endemic controls (EC; n = 14) by RT-qPCR. Bottom: RT-qPCR validation was also performed in conventional T cells (Tconv) and Treg cells from the spleen and liver of naive and infected (day 56 p.i.) mice. Statistical significance was determined using a one-way ANOVA with Tukey’s multiple comparisons test (top) or two-way ANOVA with Šídák’s multiple comparisons test (bottom). Center lines indicate median values, box limits indicate upper and lower quartiles, and whiskers indicate maximum and minimum measures. *P < 0.05; **P < 0.01; ***P < 0.001.
Tissue-specific and temporal changes in Nkg7 expression during infection.
Given a paucity of NKG7 detection reagents, we generated an Nkg7 transcriptional reporter mouse to allow cellular analysis by flow cytometry and microscopy. Mice expressing the Cre gene under the control of the Nkg7 promoter were crossed to a membrane reporter line21 to generate mice in which cells with an active Nkg7 promoter expressed green fluorescent protein (GFP) (Fig. 2a). At steady state, Nkg7 expression was mainly detected in NK cells and a subset of CD8+ T cells, with relatively minimal expression by CD4+ T cells (Fig. 2b) and other immune cell subsets in the spleens of naive mice (Extended Data Fig. 1).
Fig. 2 |. Nkg7 expression is enriched in NK cells at steady state and inducible in CD4+ T cells.
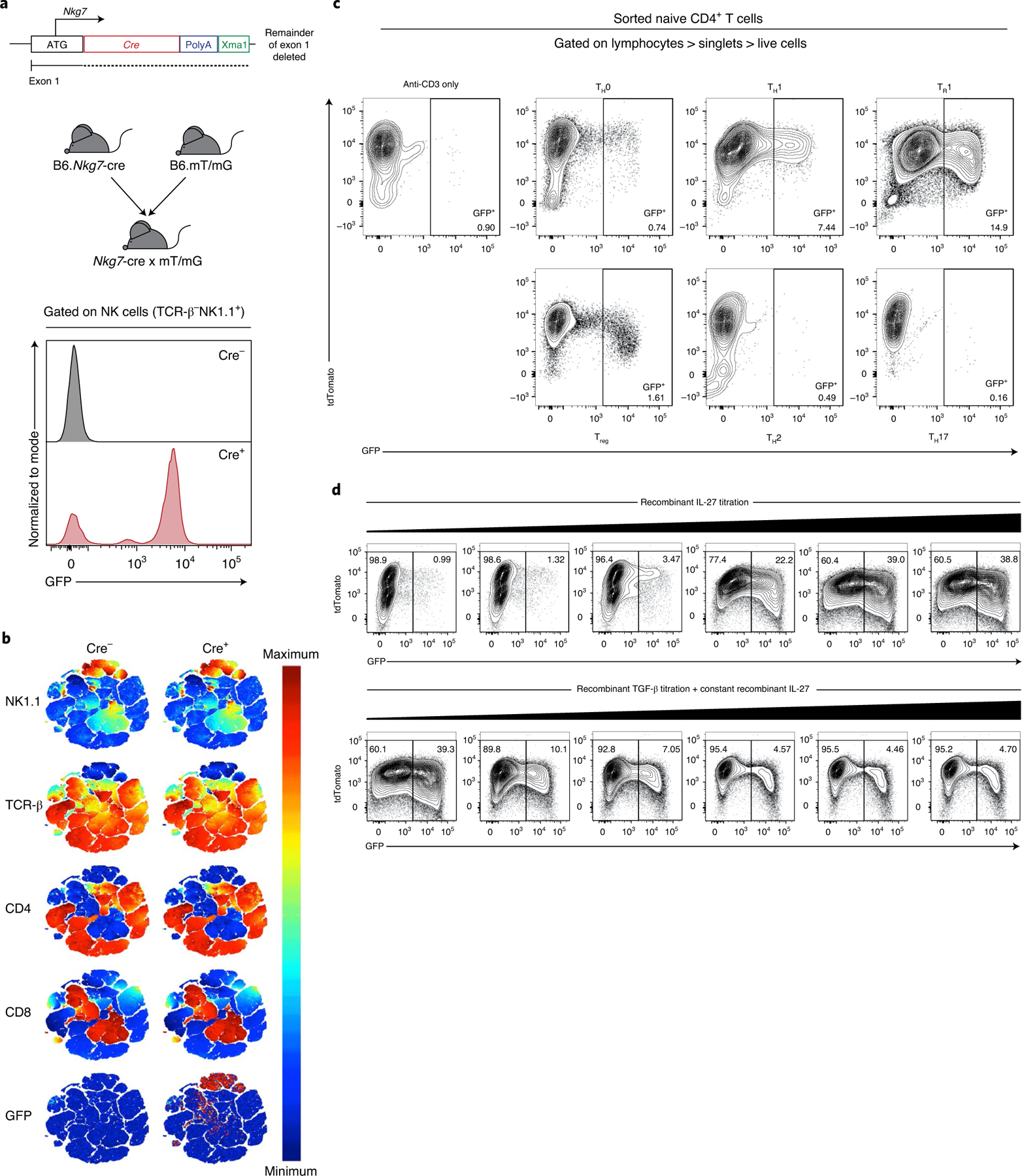
a, A mouse expressing the Cre gene behind the promoter of Nkg7 was generated (B6.Nkg7-cre) and crossed to a membrane reporter (B6.mT/mG) to generate Nkg7 reporter mice (Nkg7-cre × mT/mG). Validation of GFP expression was performed on splenic NK cells in naive Cre+ mice. b, t-SNE plot of splenocytes from a naive mouse, pre-gated to exclude doublets, dead cells and NK1.1-APC/Cy7−TCR-β-BUV737− cells. The remaining cells were clustered using NK1.1-APC/Cy7, TCR-β-BUV737, CD4-BUV395 and CD8α-PE/Cy7. Equal numbers of cells (50,000 cells) are shown for Cre− and Cre+ plots. n = 1 per genotype, performed once. c, The expression of Nkg7 (GFP+) under TH0 (anti-CD3 + anti-CD28 + recombinant IL-2), TH1 (recombinant IL-12 + anti-IL-4 + TH0 conditions), TR1 (recombinant IL-27 + TH0 conditions), Treg (recombinant IL-27 + recombinant TGF-β + TH0 conditions), TH2 (recombinant IL-4 + anti-IFN-γ + TH0 conditions) and TH17 (recombinant IL-6 + recombinant IL-1β + recombinant IL-23 + anti-IFN-γ + anti-IL-4 + TH0 conditions) cell polarizing conditions. n = 1 per genotype. Plots are representative of two independent experiments. d, Expression of Nkg7 (GFP+) when recombinant IL-27 was titrated, or when recombinant TGF-β was titrated in the presence of recombinant IL-27. n = 1 per genotype, performed once. TH, T helper cells; TR1, type 1 regulatory cells; Treg cells, inducible regulatory T cells. See also Extended Data Fig. 1.
To establish patterns of Nkg7 expression by CD4+ T cells following activation, we first isolated these cells from the spleens of reporter mice and cultured them under neutral (TH0), TH1, Tr1 (type 1 regulatory; interleukin-10 (IL-10)-producing TH1 cells10), TH2, TH17 and inducible Treg cell conditions22 (Supplementary Table 5). We found little Nkg7 expression under neutral, TH2, TH17 and inducible Treg cell conditions (Fig. 2c). However, Nkg7 expression increased under TH1 cell conditions, as previously reported20, and was further amplified following the addition of IL-27 to generate Tr1 cells23 (Fig. 2c). Interestingly, IL-27-induced Nkg7 expression was suppressed by the addition of transforming growth factor-β (TGF-β) to cell cultures and this occurred in a dose-dependent manner (Fig. 2d). The decrease in Nkg7 expression was not related to any increase in cell death under the latter cell culture conditions. Therefore, Nkg7 expression appears to be linked to TH1 cells, enhanced by IL-27-mediated polarization of Tr1 cells and inhibited by the presence of TGF-β.
Nkg7-expressing CD4+ T cells were detected in naive C57BL/6 mice, but spleen and liver NK cells expressed the highest levels of Nkg7, followed by natural killer T cells in the liver and CD8+ T cells in both tissues (Figs. 2b and 3a and Extended Data Fig. 2a). This pattern of expression changed following L. donovani infection, with CD4+ and CD8+ T cells emerging as the main Nkg7-expressing cells after day 14 p.i. (Fig. 3a and Extended Data Fig. 2b). A closer examination of CD4+ T cells at day 14 p.i. showed that Nkg7 expression was similarly upregulated by TH1 and Tr1 cells in vivo (Extended Data Fig. 2c), with both these CD4+ T cell subsets being critical cellular determinants of L. donovani infection10. After resolution of hepatic infection and establishment of chronic infection in the spleen at day 56 p.i., Nkg7 expression was highest among CD8+ T cells, although CD4+ T cells still comprised a substantial proportion of this population (Fig. 3a). Throughout the course of infection, the frequency of Nkg7-expressing mononuclear cells (MNCs) was approximately two to three times higher in the liver than the spleen (Fig. 3a and Extended Data Fig. 2d). The control of hepatic infection is dependent on the accumulation of immune cells in foci surrounding infected Kupffer cells24, and examination of liver tissue around the peak of this response (day 28 p.i.; ref. 24) revealed an accumulation of Nkg7-expressing CD4+ T cells in these inflammatory foci relative to the surrounding tissue (Fig. 3b). Thus, CD4+ and CD8+ T cells were the main immune cells expressing Nkg7 throughout the course of L. donovani infection, and Nkg7-expressing CD4+ T cells were located at the site of parasite control in inflammatory foci in the liver.
Fig. 3 |. Nkg7 is expressed by mouse spleen and liver CD4+ T cells during L. donovani infection.
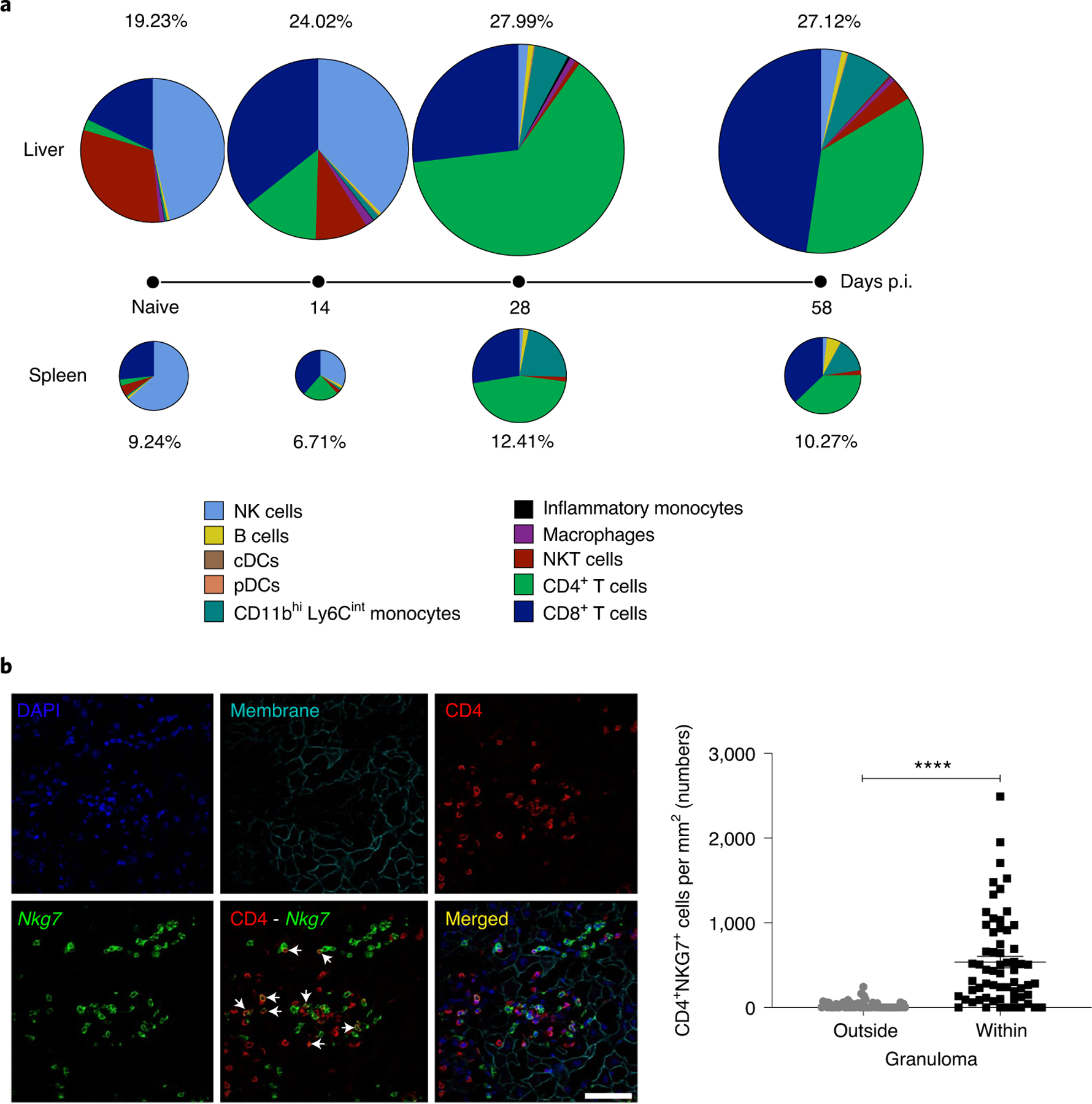
a, Nkg7 reporter mice were infected with L. donovani and the overall expression of GFP was assessed in the liver and spleen before infection (naive) and at days 14, 28 and 58 p.i. The sizes of the pie charts and the associated frequency values represent the relative percentage of GFP+ cells in the liver (above timeline) and spleen (below timeline). Within these pie charts, each slice represents the proportion of the indicated immune cell subset that made up the GFP+ population. Measurements were taken from distinct samples at each time point. The data shown are representative of two independent experiments, each consisting of n = 3 mice per genotype, per time point. cDCs, conventional dendritic cells; NKT cells, natural killer T cells; pDCs, plasmacytoid dendritic cells. b, Confocal immunofluorescence microscopy was used to determine the tissue localization of Nkg7-expressing (GFP+) cells in the liver of mice at day 28 p.i. The co-localization of Nkg7 and CD4 expression is shown in the merged image. Scale bar: 50 μm. The numbers of CD4+NKG7+ cells within inflammatory foci (granulomas), relative to surrounding tissue, are shown in the accompanying graph (right). Statistical significance was determined using the Mann–Whitney U-test. ****P < 0.0001. See also Extended Data Fig. 2.
Nkg7 expression by CD4+ T cells is required to control L. donovani infection.
The results above suggested that Nkg7 expression by T cells may be involved in protective immune responses in the liver, despite originally being identified as part of an inflammatory signature in the spleen (Figs. 1d and 3a). To investigate the role of NKG7 in disease outcome, we examined the response of Nkg7-deficient C57BL/6 mice25 to L. donovani infection. Male and female mice were examined by the International Mouse Phenotyping Consortium (https://www.mousephenotype.org/data/genes/MGI:1931250) and no significant hematological changes were identified in Nkg7-deficient mice relative to wild-type (WT) mice25. Additionally, we found few changes in the frequencies of leukocyte subsets in the spleen, thymus, bone marrow, lung or blood of Nkg7-deficient mice relative to WT mice (Supplementary Table 6). Following L. donovani infection, mice lacking NKG7 had a reduced capacity to control parasite growth in both the liver and spleen compared with WT controls (Fig. 4a). This was associated with minimal changes in the development of hepatosplenomegaly (Extended Data Fig. 3a)—a major feature in this model of visceral leishmaniasis8,18. However, serum levels of the key pro-inflammatory cytokines IFN-γ, tumor necrosis factor (TNF) and monocyte chemoattractant protein 1 in Nkg7-deficient C57BL/6 mice were significantly reduced at day 14 p.i. (Fig. 4b). After this time point, serum IFN-γ remained consistently lower in Nkg7-deficent mice compared with WT controls. Importantly, infection failed to resolve in the livers of mice lacking NKG7 by the end of the experiment (day 56 p.i.) (Fig. 4a). Thus, NKG7 plays an important role in the control of parasite growth.
Fig. 4 |. Nkg7 deficiency promotes elevated parasite burdens during L. donovani infection.
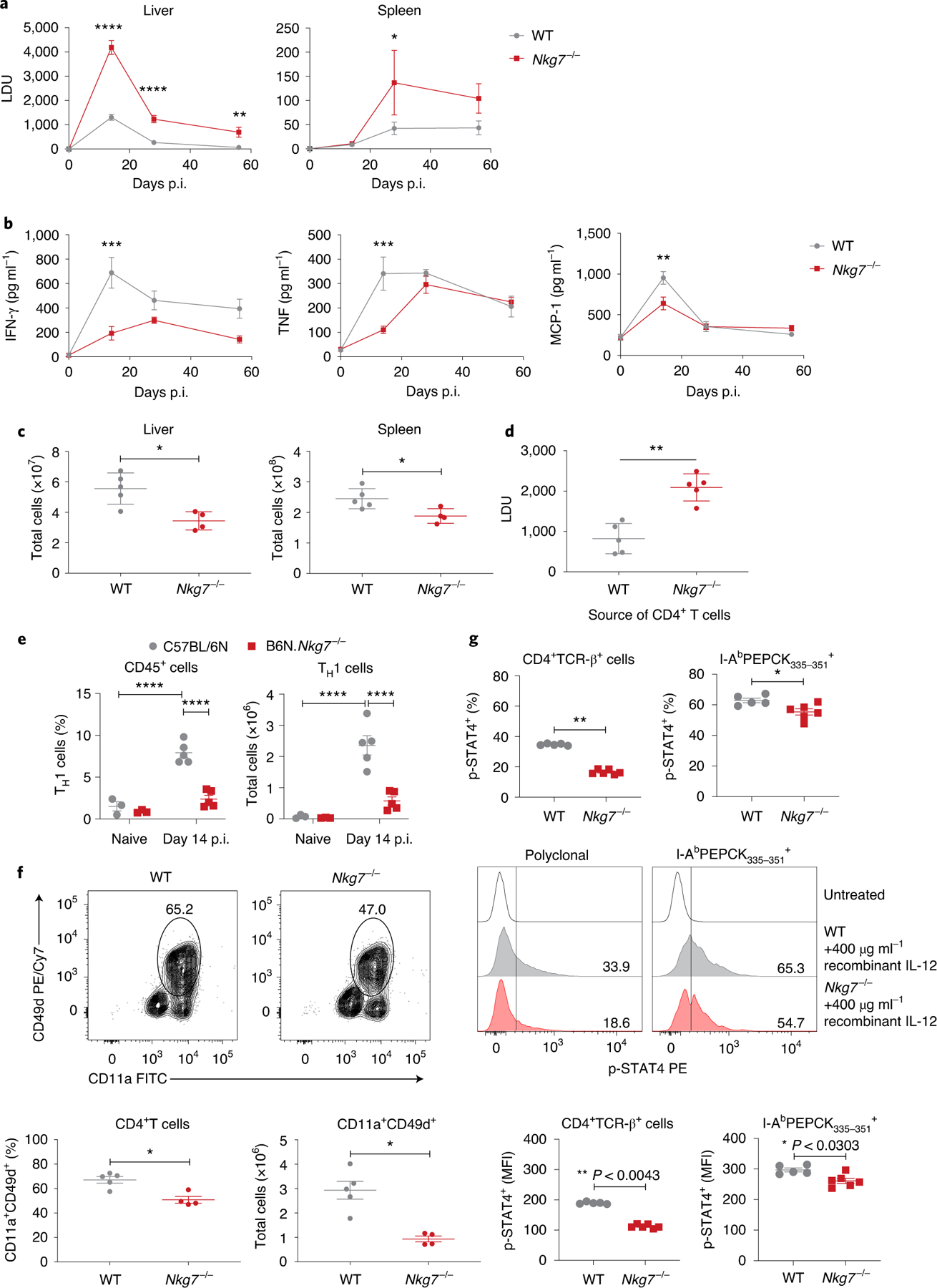
a, WT and Nkg7−/− mice were infected with L. donovani and parasite burdens were measured in the liver and spleen at days 14, 28 and 56 p.i. Statistical testing was performed using a two-way ANOVA with Šidák’s multiple comparisons test. LDU, Leishman–Donovan units. b, Quantification of serum pro-inflammatory cytokines in infected mice. Statistical testing was performed using a two-way ANOVA with Šidák’s multiple comparisons test. MCP-1, monocyte chemoattractant protein 1. c, Total number of leukocytes in the liver and spleen at day 14 p.i. Statistical testing was performed using the Mann–Whitney U-test. d, Day 14 p.i. liver parasite burdens in Rag1−/− mice that received CD4+ T cells isolated from either WT (n = 5) or Nkg7−/− (n = 5) mice. Statistical testing was performed using the Mann–Whitney U-test. e, Frequency and total number of TH1 cells (gated on lymphocytes, singlets, live cells, NK1.1-APC/Cy7−TCR-β-BUV737+, CD4-BUV395+CD8α-Alexa Fluor 700−, Foxp3-Alexa Fluor 488− and IFN-γ-APC+IL-10-PE− cells) in the livers of naive mice and infected mice at day 14 p.i. Statistical testing was performed using the two-way ANOVA with Šidák’s multiple comparisons test. f, Top: representative flow cytometry plots were gated on lymphocytes, singlets, live cells, NK1.1-APC/Cy7−TCR-β-BUV737+ and CD4-BUV395+CD8α-Alexa Fluor 700− cells. Bottom: graphs showing the frequency and total number of CD11a+CD49d+CD4+ T cells in the liver at day 14 p.i. Statistical testing was performed using the Mann–Whitney U-test. Cy, Cyanine; FITC, fluorescein isothiocyanate; PE, phycoerythrin. g, Frequency (top) and median fluorescence index (MFI; bottom) of phosphorylated STAT4 (p-STAT4) within CD4+TCR-β+ (polyclonal) and I-AbPEPCK335–351 (tetramer) PE+ cells in the liver at day 14 p.i. Statistical significance was determined using the Mann–Whitney U-test. Representative plots (middle) show the expression of p-STAT4 upon treatment with recombinant IL-12. n = 5 WT and n = 6 Nkg7−/− mice. Data are representative of two independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Error bars represent means ± s.e.m. The data shown in a–c, e and f are representative of two independent experiments, each with n = 3 naive WT and Nkg7−/− mice and n = 5 WT and n = 4 Nkg7−/− mice at days 14, 28 and 58 p.i. Measurements in a and b were obtained from distinct samples at each time point. See also Extended Data Fig. 3.
Next, we examined cellular responses at day 14 p.i., when differences in liver parasite burden and serum pro-inflammatory cytokine levels were greatest between Nkg7-deficent mice and WT controls. We found reduced recruitment of MNCs into the liver and limited expansion of these cells in the spleen at day 14 p.i. (Fig. 4c). Recruitment of CD4+ T cells to the liver was also reduced (Extended Data Fig. 3b), although CD4+ T cell expansion in the spleen was unaffected (Extended Data Fig. 3c) in Nkg7-deficient mice compared with WT controls. To establish the importance of Nkg7 expression by CD4+ T cells in this infection, we adoptively transferred Nkg7-deficient or WT CD4+ T cells into T and B cell-deficient Rag1−/− mice the day before L. donovani infection and measured parasite burdens 14 d later. We found an approximately twofold increase in liver parasite burdens of Rag1−/− mice that received Nkg7-deficient CD4+ T cells compared with those that received WT CD4+ T cells (Fig. 4d). Thus, Nkg7 expression by CD4+ T cells was required for these cells to develop their full anti-parasitic potential during experimental visceral leishmaniasis caused by L. donovani.
In the liver, reduced control of parasite growth in Nkg7-deficient mice was associated with a reduced frequency and number of TH1 cells (Fig. 4e), as well as CD4+ T cells recently exposed to antigen (CD11a+CD49d+)26 (Fig. 4f), relative to WT mice. Nkg7-deficient CD4+ T cells also expressed less IFN-γ messenger RNA (mRNA) but not TNF mRNA (Extended Data Fig. 3d). Expression of programmed cell death protein 1, cytotoxic T-lymphocyte-associated protein 4 and inducible T cell costimulator on hepatic CD4+ T cells was also reduced in Nkg7-deficient mice compared with WT controls (Extended Data Fig. 3e), suggesting an overall reduction in CD4+ T cell activation and differentiation in the absence of NKG7.
To further examine the role of NKG7 in CD4+ T cells, we employed a Leishmania-specific major histocompatibility complex class II (MHC II) tetramer presenting the phosphoenolpyruvate carboxykinase (PEPCK) peptide27, to measure antigen-specific CD4+ T cell populations at day 14 p.i. (Extended Data Fig. 3f). Nkg7-deficient mice had a reduced number, but not frequency, of PEPCK-positive CD4+ T cells in the liver following L. donovani infection. However, both the number and frequency of PEPCK-positive TH1 cells in the liver were reduced at this time in Nkg7-deficient mice compared with WT controls (Extended Data Fig. 3g). Additionally, we found that activation (phosphorylation) of signal transducer and activator of transcription 4 (STAT4) by IL-12 (Fig. 4g), but not activation of STAT3 by IL-6 (Extended Data Fig. 3h), in hepatic polyclonal and PEPCK-positive CD4+ T cells, was reduced in the absence of NKG7. Together, these results show that NKG7 plays an important role in the expansion and/or recruitment of CD4+ T cells to the liver, as well as the production of IFN-γ in this critical anti-parasitic immune cell population.
NKG7 promotes inflammation in an experimental model of severe malaria.
To examine the role of NKG7 in a parasitic disease where inflammation is detrimental, we infected Nkg7-deficient and WT mice with Plasmodium berghei ANKA and examined the development of experimental cerebral malaria (ECM). This pre-clinical model of severe malaria is characterized by systemic inflammation leading to the accumulation of parasitized red blood cells (pRBC) in the microvasculature of various tissues, including the brain28. Although WT mice developed severe neurological sequelae on days 6–8 p.i., as expected29, Nkg7-deficient mice failed to develop these severe symptoms and instead survived up to days 13–14 p.i. (Fig. 5a,b). Although Nkg7-deficient mice had a small increase in blood parasitemia at days 7 and 8 p.i. relative to WT controls (Fig. 5c), when parasite biomass was measured using luciferase transgenic P. berghei ANKA, the parasite burden in the whole body (Fig. 5d), as well as in the brain (Fig. 5e), was significantly reduced. These latter measurements take into account pRBCs that have accumulated in the tissue microvasculature, and indicate limited activation of vascular endothelium in Nkg7-deficient mice compared with WT controls, resulting in less parasite biomass in the infected mice.
Fig. 5 |. Nkg7 promotes parasite accumulation in tissues and the onset of ECM following P. berghei ANKA infection.
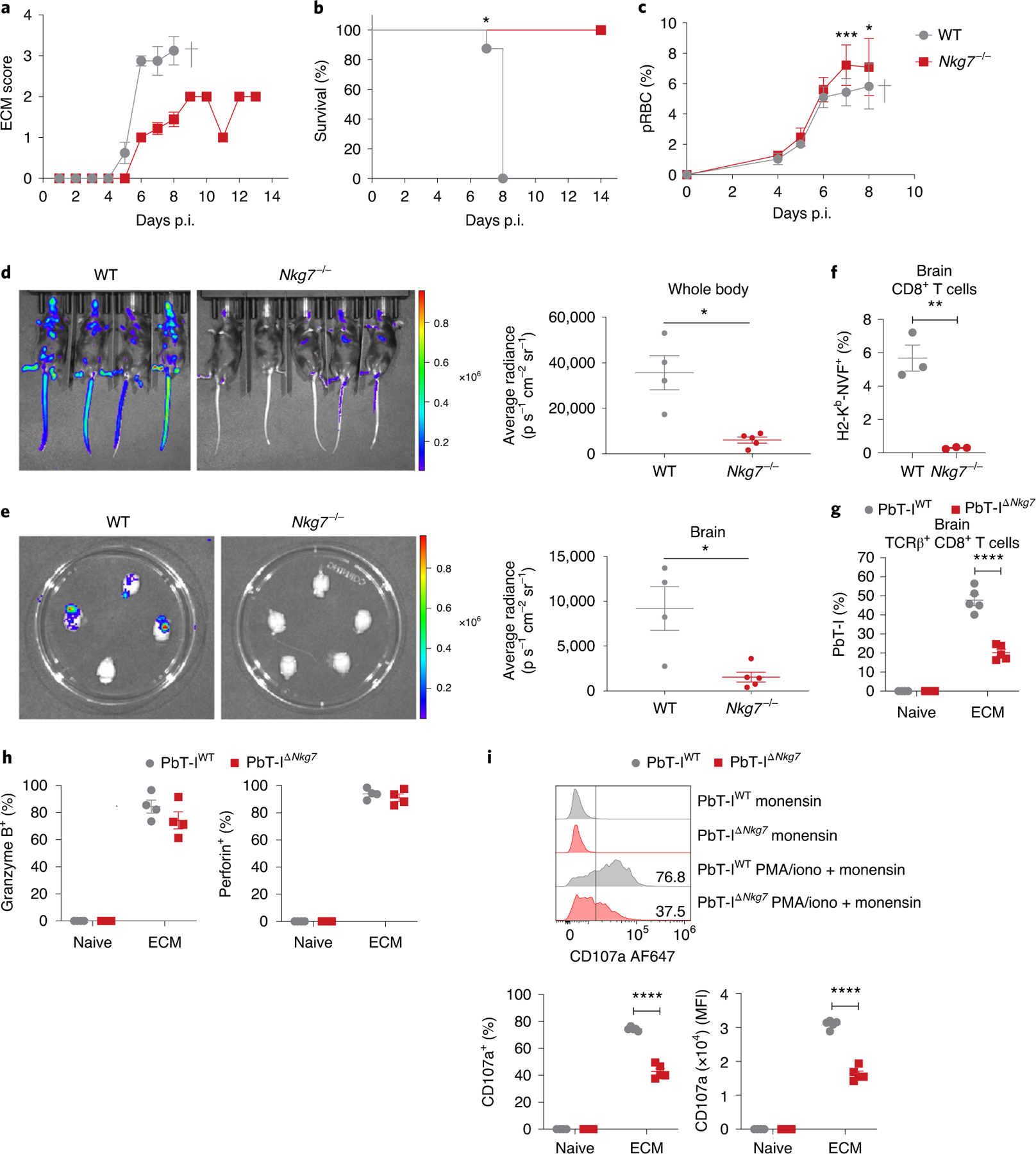
WT and Nkg7−/− mice were infected with P. berghei ANKA, which causes ECM. a, ECM scores obtained as repeated measures at each time point. b, Survival analysis between WT and Nkg7−/− mice. Statistical testing was performed using the log-rank (Mantel–Cox) test. c, Levels of pRBCs in the circulation, obtained as repeated measures at each time point. Statistical significance was determined using a two-way ANOVA with Šidák’s multiple comparisons test. d,e, Parasite biomass was quantified using luciferase-expressing P. berghei ANKA parasites, in the bodies (d) and brains (e) of infected mice. n = 4 WT and n = 5 Nkg7−/− mice. Statistical significance was determined using the Mann–Whitney U-test. f, Frequency of H2-Kb-NVF (tetramer) PE+ cells within the brain CD8+ T cell population at the peak of ECM. Statistical significance was determined using the Mann–Whitney U-test in one experiment where n = 3 mice per group. g, Proportion of co-transferred transgenic PbT-IWT and PbT-IΔNkg7 cells as a frequency of TCR-β+CD8+ cells in the brains of naive and infected mice (at peak ECM). h, Frequencies of granzyme B+ or perforin+ PbT-IWT and PbT-IΔNkg7 cells in the brain after incubation with monensin. Statistical significance was determined using a two-way ANOVA with Šidák’s multiple comparisons test. i, Top: representative histograms illustrate CD107a (LAMP-1) expression by PbT-IWT and PbT-IΔNkg7 cells in the brain after incubation with monensin or stimulation with PMA and ionomycin (iono) in the presence of monensin. Bottom: graphs of the histograms present the frequency and MFI of CD107a expression by PbT-IWT and PbT-IΔNkg7 cells. Data in a–e are representative of three independent experiments. n = 5 mice per group in a–c. The experiments in g–i were performed once where n = 4 naive and n = 5 infected mice per strain. Error bars represent means ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. See also Extended Data Fig. 4.
This disease model is characterized by the rapid recruitment of antigen-specific, cytotoxic CD8+ T cells to the brain, which then cause damage to the cerebral vascular endothelium30. In the absence of NKG7, there were limited changes in NK and CD4+ T cell recruitment to the brain, but recruitment of CD8+ T cells was reduced at the time when WT mice succumbed to disease (Extended Data Fig. 4a). The activation status of Nkg7-deficient CD8+ T cells in the brain, as indicated by CD11a/CD49d and granzyme B expression, was also significantly reduced (Extended Data Fig. 4b). We also measured the recruitment of parasite-specific CD8+ T cells to the brain using a Plasmodium peptide–MHC I tetramer31 and found reduced recruitment of these cells to the brains of P. berghei ANKA-infected Nkg7-deficient compared with WT mice (Fig. 5f). Therefore, in the absence of Nkg7-mediated inflammation, there was reduced accumulation of P. berghei ANKA pRBC in tissue, associated with diminished CD8+ T cell recruitment and activation in the brain.
To test whether any of the above NKG7-mediated changes in CD8+ T cell activation were cell intrinsic, we crossed the Nkg7−/− mice with PbT-I T cell receptor (TCR) transgenic mice with CD8+ T cells recognizing an MHC I-restricted parasite peptide found in all rodent Plasmodium species (PbT-IΔNkg7)32. Control mice were generated by crossing PbT-I TCR transgenic mice with congenic (CD45.1) C57BL/6 mice to produce mice expressing both CD45.1 and CD45.2 alleles (PbT-IWT). CD8+ T cells were isolated from the spleens of both lines and adoptively transferred in equal numbers into the same congenic (CD45.1) recipient C57BL/6 mice before P. berghei ANKA infection, to allow a direct comparison between PbT-IWT and PbT-IΔNkg7 cells in the same tissue environment. Recipient mice developed neurological symptoms on day 5 p.i., 24–48 h earlier than normal, probably reflecting the increased kinetics of parasite-specific effector CD8+ T cell expansion and recruitment to the brain. At the time when recipient mice developed ECM, we found reduced expansion of PbT-IΔNkg7 cells in the spleen (Extended Data Fig. 4c) and recruitment to the brain (Fig. 5g) compared with PbT-IWT cells. Nevertheless, there was minimal difference in the frequencies of PbT-IΔNkg7 and PbT-IWT cells producing granzyme B or perforin (Fig. 5h and Extended Data Fig. 4d), indicating a limited role for NKG7 in the generation of these effector molecules. However, PbT-IΔNkg7 cells had a clear reduction in the expression of CD107a—a molecule required for degranulation of cytotoxic molecules33—on their cell surface in both the spleen (Extended Data Fig. 4e) and brain (Fig. 5i), compared with PbT-IWT cells. Together, these results show that NKG7 plays an important, cell-intrinsic role in the expansion and recruitment of CD8+ T cells to sites of infection and inflammation, as well as in the exocytosis of cytotoxic proteins by these cells, as indicated by reduced CD107a expression.
NKG7 co-localizes with CD107a and plays a critical role in CD8+ T cell-mediated killing of target cells.
To better understand how NKG7 might contribute to exocytosis of cytotoxic proteins, we employed a retroviral expression system to transduce CD8+ T cells with NKG7–GFP to establish cellular localization. We found strong co-localization of NKG7–GFP with intracellular vesicles containing cytotoxic granules, as identified by LysoTracker (Fig. 6a). Furthermore, when transduced cells were stimulated with phorbol ester and calcium ionophore to induce exocytosis of cytotoxic molecules, we found co-localization of GFP with CD107a (Fig. 6b). Finally, to confirm a role for NKG7 in CD8+ T cell-mediated killing, we compared the ability of PbT-IWT and PbT-IΔNkg7 cells to kill peptide-pulsed target cells and found a deficit in PbT-IΔNkg7 cells compared with PbT-IWT cells (Fig. 6c). Together, these data identify an important role for NKG7 in the translocation of CD107a from intracellular vesicles to the cell surface by activated CD8+ T cells, thereby promoting their efficient killing of cellular targets.
Fig. 6 |. NKG7 co-localizes with cytotoxic vesicles expressing CD107a.
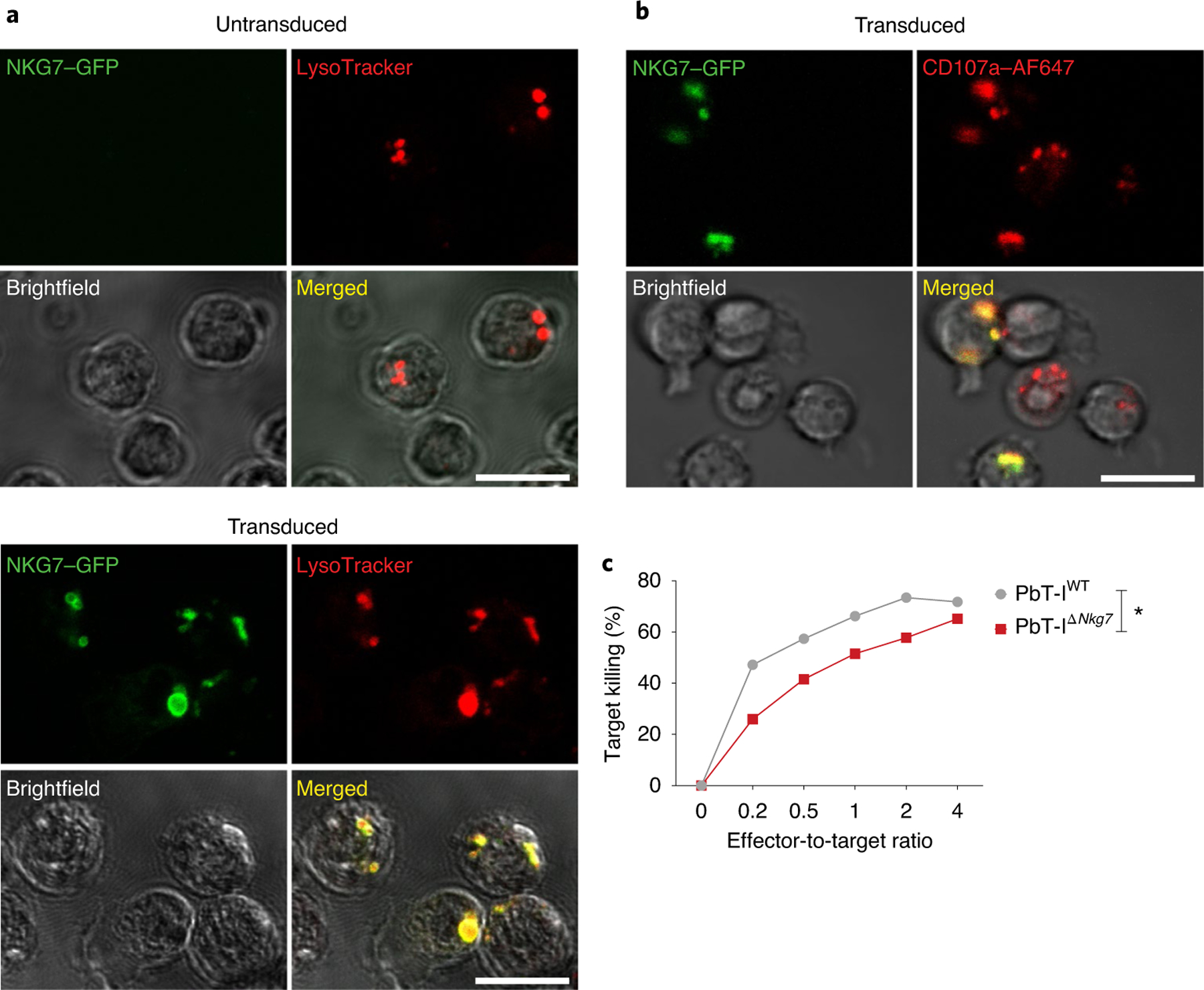
a, Representative images showing the co-localization of NKG7 and LysoTracker in WT splenocytes transduced (bottom), or not (top), to express NKG7–GFP. Scale bar: 10 μm. b, Splenocytes from PbT-IWT T cell transgenic mice were also transduced to express NKG7–GFP. The representative images depict the co-localization of NKG7 and the cytotoxic granule-associated protein CD107a. Scale bar: 10 μm. AF647, Alexa Fluor 647. c, Comparison of cytotoxic ability between PbT-IWT and PbT-IΔNkg7 transgenic CD8+ T cells against NVF peptide-pulsed splenocytes in vitro. Data were obtained from one experiment. Statistical significance was determined using a two-way ANOVA with effector-to-target ratio and group as variables. Data are representative of two independent experiments. *P < 0.05.
NKG7 is needed to control experimental tumor metastasis.
To investigate whether the pro-inflammatory role of NKG7 extended beyond infectious diseases, we first interrogated The Cancer Genome Atlas (TCGA) skin cutaneous melanoma (SKCM) dataset, because of the requirement for host immune cells and inflammation for control in this type of cancer34. To test whether there was any association between NKG7 expression in tumors and disease outcome, patients with melanoma were ordered by NKG7 expression, following which the highest and lowest quartiles were assessed for survival probability. A highly significant survival advantage was observed in patients with high NKG7 expression, compared with those with low NKG7 expression (Fig. 7a), suggesting that NKG7 may play a positive role in antitumor immunity. Furthermore, NKG7 expression in these tumors was strongly associated with the expression of NK cell signature molecules35 (Extended Data Fig. 5a).
Fig. 7 |. Nkg7 deficiency increases metastatic burden.
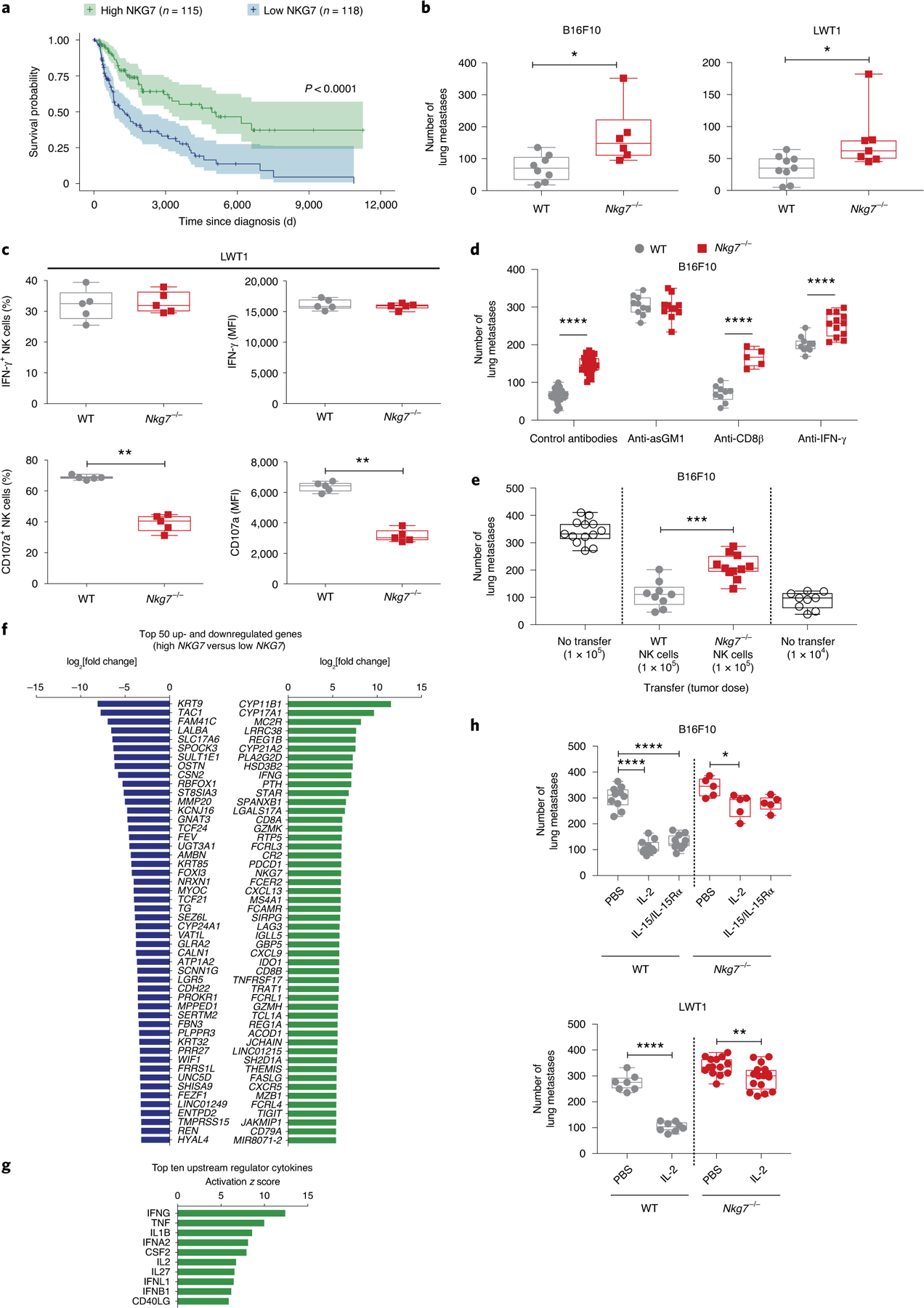
a, Survival analysis between individuals in the top and bottom 25% of NKG7 expressers in the TCGA SKCM dataset, performed using a log-rank test. The shading represents the 95% confidence interval in the top and bottom quartiles. b, WT and Nkg7−/− mice were injected with either B16F10 (n = 8 WT and n = 6 Nkg7−/− mice) or LWT1 cells (n = 9 WT and n = 7 Nkg7−/− mice). Lung metastatic burdens were quantified at day 14 post-injection. The data shown are representative of two independent experiments. The Mann–Whitney U-test was used to determine statistical significance. c, Left: the lungs of WT and Nkg7−/− mice (n = 5 mice per group), injected with LWT1 cells, were assessed for differences in the frequency of IFN-γ+ (top) or CD107a+ NK cells (bottom). Right: the MFI of IFN-γ or CD107a on these cells at 14 d post-injection is also shown. The data are representative of two independent experiments. The Mann–Whitney U-test was used to determine statistical significance. d, WT and Nkg7−/− mice injected with B16F10 cells were treated with isotype control antibodies (n = 27 WT and n = 26 Nkg7−/−; pooled from four experiments) or antibodies against asGM1 (n = 10 WT and n = 11 Nkg7−/−), CD8β (n = 9 WT and n = 5 Nkg7−/−) or IFN-γ (n = 10 WT and n = 12 Nkg7−/−) (pooled from two experiments). Lung metastases were quantified at day 14 post-injection of B16F10 cells. A two-way ANOVA with Šidák’s multiple comparisons test was used to determine statistical significance between groups. e, Differences in lung metastatic burden in Rag2−/−γc−/− mice that received either WT (n = 9) or Nkg7−/− (n = 11) NK cells before injection with B16F10 cells. Controls consisted of Rag2−/−γc−/− mice that did not receive NK cells but were injected with either 1 × 104 (n = 9) or 1 × 105 (n = 13) B16F10 cells. The data are representative of two independent experiments. The Mann–Whitney U-test was used to determine statistical significance. f, Waterfall plot showing the top 50 up- and downregulated genes between high and low NKG7 expressers from the TCGA SKCM dataset. g, Top ten upstream regulator cytokines between high and low NKG7 expressers from the TCGA SKCM dataset identified by IPA. h, Top: WT and Nkg7−/− mice injected with B16F10 cells were treated with recombinant IL-2 (n = 10 WT and n = 5 Nkg7−/−) or IL-15/IL-15Rα (n = 11 WT and n = 5 Nkg7−/−) and compared with PBS-treated controls (n = 10 WT and n = 5 Nkg7−/−). Bottom: mice injected with LWT1 cells were treated with recombinant IL-2 (n = 8 WT and n = 16 Nkg7−/−; pooled from two experiments) and compared with PBS-treated controls (n = 7 WT and n = 14 Nkg7−/−; pooled from two experiments). A one-way ANOVA with multiple comparisons was used to test for statistical significance. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. For the graphs in b–e and h, center lines indicate median values, box limits indicate upper and lower quartiles, and whiskers indicate maximum and minimum measures. See also Extended Data Fig. 5.
Given that NKG7 was abundantly expressed by NK cells (Figs. 2a,b and 3a), we next examined the role of NKG7 in NK cell-dependent pre-clinical models of experimental metastasis using B16F10 and LWT1 melanoma cell lines. When Nkg7-deficient mice were injected intravenously with either tumor cell line, there was a significant increase in the number of lung metastases compared with WT mice (Fig. 7b). Similar results were also found in another experimental prostate carcinoma metastasis model, RM-1 (Extended Data Fig. 5b). Importantly, increased spontaneous metastasis to the lung post-resection of orthotopically injected mammary carcinoma (E0771) was also observed in Nkg7-deficient mice despite tumors being equivalent in size at surgery (Extended Data Fig. 5b). RMA-s is a classical MHC I-deficient tumor target for NK cell perforin-mediated killing that was previously characterized in vivo when injected into the peritoneum36. Survival of Nkg7-deficient mice was reduced compared with WT mice post-RMA-s injection and this difference was abrogated with further reduced survival in both WT and Nkg7-deficient mice depleted of NK cells (Extended Data Fig. 5b). NK cells are also critical in preventing the initiation of methylcholanthrene (MCA) carcinogenesis37, and herein the Nkg7-deficient mice were more sensitive to MCA-induced carcinogenesis than WT mice (Extended Data Fig. 5b). Thus, in a number of mouse cancer models where NK cells are known to be critical in host control, the Nkg7-deficient mice were defective compared with WT mice. To examine the antitumor defect caused by Nkg7 deficiency, the LWT1 and B16F10 metastasis models were further investigated. Increased LWT1 lung metastasis was associated with limited changes in lung hematopoietic cells (Extended Data Fig. 5c) but reduced recruitment of NK and T cells (Extended Data Fig. 5d). Furthermore, the reduced recruitment of NK cells into the LWT1-burdened lung in Nkg7-deficient mice was limited to CD27+CD11b+ NK cells (Extended Data Fig. 5e)—a mature inflammatory population of NK cells previously associated with effector function38. Importantly—and consistent with results from CD8+ T cells above—Nkg7 deficiency resulted in a reduction in the frequency and expression of CD107a on NK cells, but no change in the frequency and expression of IFN-γ, compared with WT NK cells (Fig. 7c).
Lung metastasis of B16F10 is controlled by NK cells, IFN-γ and perforin-mediated cytotoxicity39. We observed increased metastasis in mice transplanted with B16F10 following NK cell but not CD8+ T cell depletion, indicating that NKG7-mediated control of metastasis was NK cell dependent (Fig. 7d). While IFN-γ is important for host control of B16F10 lung metastasis, consistent with a lack of impact of Nkg7 loss on NK cell IFN-γ production (Fig. 7c), the effects of Nkg7 deficiency and IFN-γ neutralization on metastasis were independent (Fig. 7d). To confirm that Nkg7 expression by NK cells was required for optimal control of metastasis, we also adoptively transferred either Nkg7-deficient or WT NK cells into Rag2cγ−/− mice 6 d before transplanting B16F10 cells. While reconstitution of NK cells was equivalent at the time of tumor inoculation, there was a clear reduction in the ability of Nkg7-deficient NK cells to control B16F10 lung metastases (Fig. 7e).
To gain further insight into how Nkg7 expression influenced tumor metastasis, we identified the 50 top up- and downregulated genes in the high and low NKG7-expressing individuals from the TCGA SKCM dataset (Fig. 7f). We then identified the top ten cytokine upstream regulators (Fig. 7g). Many of these molecules have been shown to play important roles in the maintenance or activation of NK cells, including the NK cell growth factor IL-2 (ref. 40). When Nkg7-deficient and WT mice were transplanted with a high number of B16F10 cells and then treated with therapeutic IL-2 or IL-15 (another important NK cell growth factor), the anti-metastatic effect in WT mice was highly significant for both cytokines. However, these clinically relevant cytokines had a much more limited effect in Nkg7-deficient mice (Fig. 7h). Hence, NKG7 plays an important role in promoting the anti-metastatic activity of NK cells, as well as the ability of these cells to respond to cytokine therapy.
NKG7 plays a critical role in NK cell–mediated killing of target cells.
The above results concerning defective CD107a expression suggest that NKG7 might also be important for NK cell–mediated killing. To examine this possibility, we first tested the requirement of Nkg7 for efficient control of the RMA-s-Rae1β lymphoma—a primary tumor transplant—and confirmed the requirement for NK cells but not CD8+ T cells in this model (Fig. 8a). Again, the effects of Nkg7 deficiency and IFN-γ neutralization were independent (Fig. 8b). Next, we compared the ability of Nkg7-deficient and WT NK cells to kill YAC-1 and RMA-s-Rae1β lymphoma targets in vitro (Fig. 8c) and RMA-s-Rae1β lymphomas in vivo (Fig. 8d). In all settings, Nkg7-deficient NK cells had a defect in killing ability relative to WT control NK cells. Hence, NKG7 was critical for efficient NK cell–mediated killing of target cells, and unlike WT NK cells, the killing activity of Nkg7-deficent NK cells could not be rescued to WT levels by previous activation with IL-2 (Fig. 8c). Interestingly, we found no difference in the expression of the activation markers DNAM-1, NKG2D, CD11a, granzyme B or perforin between Nkg7-deficient and WT NK cells (Extended Data Fig. 6a), even if they were IL-2 activated (Extended Data Fig. 6b). Furthermore, we observed no differences in the formation of cell conjugates between Nkg7-deficient or WT NK cells and their YAC-1 targets (Extended Data Fig. 6c). An evaluation of synapse formation between NK cells and YAC-1 targets also revealed no NKG7-dependent changes (Extended Data Fig. 6d). Together, these results show that NKG7 plays a critical role in NK cell–mediated killing of target cells, but has no measurable impact on the expression of effector molecules or the ability of NK cells to form contacts with cellular targets.
Fig. 8 |. NKG7 plays a role in cytotoxicity.
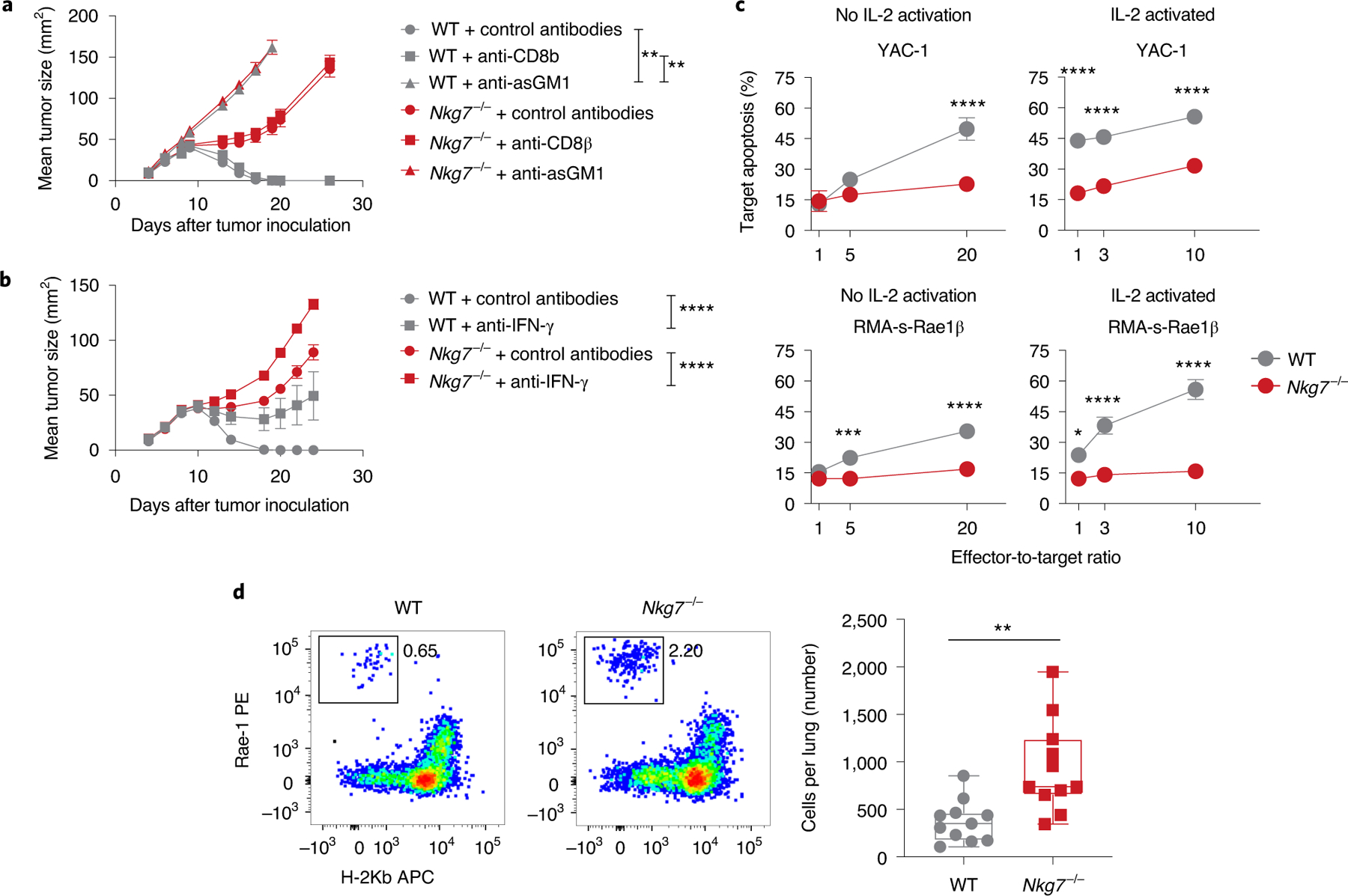
a, WT and Nkg7−/− mice were subcutaneously injected with RMA-s-Rae1β cells, followed by treatment with control antibodies or antibodies against asGM1 or CD8β, and the tumor size was measured as repeated measures at each time point. A one-way ANOVA with Tukey’s multiple comparisons test was used to determine statistical significance. The data shown are from one experiment performed where n = 5 mice per group. b, WT and Nkg7−/− mice were given a subcutaneous injection of RMA-s-Rae1β cells at day 0. Mean tumor size was derived from repeated measures at each time point. Mice were treated with either control antibodies (n = 6 WT and n = 6 Nkg7−/−) or anti-IFN-γ (n = 6 WT and n = 8 Nkg7−/−) at days −1, 0, 7, 14 and 21. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple comparisons test. The statistical significance between control antibodies and anti-IFN-γ-treated WT and Nkg7−/− mice at day 24 is shown. Statistical significance between WT control antibodies and anti-IFN-γ groups was detected from day 14, whereas statistical significance between Nkg7−/− control antibodies and anti-IFN-γ groups was detected on day 18. Data were obtained from one experiment. c, WT and Nkg7−/− NK cell–mediated cytotoxicity against YAC-1 target cells or RMA-s-Rae1β in vitro, in the absence or presence of IL-2-induced activation. Statistical significance was assessed using a Mann–Whitney U-test at each effector-to-target ratio. n = 6 per group, pooled from two independent experiments. d, The difference in WT and Nkg7−/− NK cell–mediated cytotoxicity against RMA-s-Rae1β in vivo is depicted in the representative plots (left) and the numbers of target cells remaining in the lungs of WT and Nkg7−/− mice were quantified (right). Target cells were gated on live CD45.2+ cells. A Mann–Whitney U-test was used to determine statistical significance. n = 11 mice per group. Data were pooled from two independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. See also Extended Data Fig. 6.
Discussion
Here, we show that NKG7 is a novel mediator of inflammation. Several earlier studies reported that NKG7 expression was associated with cell cytotoxicity20,41,42. This is consistent with our data showing less efficient killing of target cells by Nkg7-deficient NK and CD8+ T cells. Importantly, the Nkg7-dependent defect in cytotoxic activity was not related to changes in the expression of effector molecules such as granzyme B, perforin and IFN-γ. Furthermore, Nkg7-deficient NK cells had no reduction in their capacity to form conjugates or immunological synapsis with target cells. Instead, the Nkg7-dependent killing defect was linked with reduced expression of CD107a on the surface of Nkg7-deficient NK cells in metastasis models and CD8+ T cells in ECM. CD107a is critical for NK cells to deliver perforin and granzyme B to target cells43. Hence, our findings support a key role for NKG7 in the translocation of CD107a to the cell surface, associated with exocytosis of cytotoxic molecules.
The TH1 cell canonical transcription factor T-bet binds to the NKG7 promoter in TH1 cells, and this increases the expression of NKG7 and PRF1 (encoding perforin), along with IFNG (encoding IFN-γ)20. Our findings support a role for NKG7 in TH1 cell IFN-γ production, with evidence that Nkg7-deficient CD4+ T cells have a limited capacity to respond to IL-12 signaling, as indicated by reduced STAT4 activation. Interestingly, a recent study of blood samples from genetically susceptible mice infected with Mycobacteria tuberculosis, as well as from patients with active tuberculosis and individuals with latent tuberculosis that progressed to active disease, revealed that NKG7, along with other genes associated with effector and cytotoxic NK and T cell responses, was downregulated44. These findings are consistent with our discoveries in visceral leishmaniasis—a disease also caused by infection of tissue-resident macrophages that requires an effective TH1 cell response for control. However, we did not establish a cell-intrinsic role for NKG7 in CD4+ T cell IFN-γ production. In fact, data from cell adoptive transfer studies of WT and Nkg7-deficient CD4+ TCR transgenic cells indicated that the effect of NKG7 on IFN-γ production is cell extrinsic and downstream of the NKG7-mediated impact on cytotoxic granule exocytosis (data not shown). Previous studies showed that perforin- and granzyme B-deficient mice have enhanced TH1 cell responses compared with WT mice following P. berghei ANKA infection30. Thus, one possibility is that NKG7-mediated cytotoxic granule exocytosis by NK cells or CD8+ T cells modulates antigen-presenting cell numbers and/or function early during infection to influence CD4+ T cell IFN-γ production.
Increased expression of NKG7 in mouse IL-27-induced Tr1 cells45 and human IL-10+ TH17 cells17 has been reported. Although we found limited Nkg7 expression by mouse TH17 cells, the association with human IL-10-producing TH17 cells may indicate that NKG7 is expressed by highly differentiated CD4+ T cell populations. This is supported by our findings that Nkg7 expression was linked to TH1 cells and enhanced by IL-27-mediated polarization to Tr1 cells, and that the reduced levels of co-inhibitory receptor molecules were associated with highly activated T cells on Nkg7-deficient CD4+ T cells during infection. However, the use of our Nkg7 reporter mice in in vivo studies revealed that Nkg7 was expressed by different T cell subsets, including conventional CD4+ and CD8+ T cells, TH1, Tr1, Treg and natural killer T cells, as well as innate cell populations, most notably NK cells. The results from our studies in disease models also indicate functional roles for NKG7 in CD4+ T cells (visceral leishmaniasis), CD8+ T cells (malaria) and NK cells (cancer). Hence, NKG7 is expressed by a range of immune cell populations, suggesting a diversity of immunological roles during disease that is context dependent.
In the absence of CD4+ T cell NKG7 expression, the generation of an inflammatory response following L. donovani infection was limited and this resulted in increased parasite growth, demonstrating a critical role for this molecule in host defense. However, inflammation can also cause disease, as in severe malaria syndromes19. Indeed, our results from a pre-clinical model of severe malaria showed that P. berghei ANKA infection resulted in an NKG7-mediated inflammatory response that promoted the accumulation of parasites in host tissues such as the brain. In the absence of NKG7, there was reduced CD8+ T cell recruitment to the brain, associated with less cell activation. We also found evidence for a cell-intrinsic reduction in parasite-specific CD8+ T cell expansion in the spleen and recruitment to the brain in the absence of NKG7, as well as translocation of CD107a to the cell surface following activation, but a limited cell-intrinsic requirement for granzyme B or perforin production. Therefore, NKG7 probably acts at multiple steps in the activation, expansion and delivery of effector functions in CD8+ T cells.
We also identified an important role for NKG7 in NK cell–mediated control of cancer metastasis, predicted by the strong, positive association between NKG7 expression in skin cutaneous melanomas and patient survival probability. Recently, patients with metastatic melanoma responding to combined programmed cell death protein 1 and cytotoxic T-lymphocyte-associated protein 4 blockade were shown to have greater expansion of CD8+ T cell clones that overexpressed proteins associated with cytotoxicity, including NKG7, than non-responding patients14, supporting an important role for this molecule in tumor-associated immune responses. Together with our data, these findings reinforce a role for NKG7 in antitumor immunity, and suggests that targeting this molecule for activation may represent a new approach for cancer treatment. Alternatively, maintaining NKG7 expression or stimulating overexpression may also have beneficial outcomes in this disease setting.
In summary, we have identified NKG7 as a critical mediator of inflammation in a range of diseases. NKG7 is expressed on different immune cells at different stages of disease and data from Nkg7-deficient mice indicate that targeting this molecule via blockade of function (antagonist) represents a novel approach to dampening inflammation in diseases such as severe malaria, while activating this molecule (agonist) may be employed to enhance immune responses during infectious diseases or cancer.
Methods
Experimental model and participant details.
Mice.
Mice of 6 weeks of age or above were used for all of the experiments unless otherwise specified. Mice were housed in groups, with a maximum of six mice per cage, in a passive air flow, environmentally ventilated cage system, and maintained under pathogen-free conditions at the QIMR Berghofer Medical Research Institute Animal Facility (Brisbane, Queensland, Australia). Mouse cages were housed in an environmentally controlled room that was maintained at 20–21 °C with 60% relative humidity and a 12 h light cycle (08.00–20.00), with no external or natural light sources. All exhaust air from the mouse racks was vented external to the building.
B6.
Tg(Nkg7-cre)/J (B6.Nkg7-cre) mice were crossed to B6.Tg(Nkg7-cre)/J, B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB–tdTomato,–EGFP)Luo/J (B6.mT/mG; RRID: IMSR_JAX:007676)21 mice once to generate a transcriptional reporter of Nkg7 (Nkg7-cre × mT/mG). C57BL/6J (RRID: IMSR_JAX:000664) mice were sourced from the Walter and Eliza Hall Institute (Melbourne, Victoria, Australia) and B6.SJL-Ptprca Pepcb/BoyJ (B6.Cd45.1; RRID: IMSR_JAX:002014) mice were sourced from the Animal Resources Centre (Perth, Western Australia, Australia).
All other mice were bred in house, including C57BL/6NJ (C57BL/6N; RRID: IMSR_JAX:005304), C57BL/6-Foxp3tm1flv/J (Foxp3-RFP; RRID: IMSR_JAX:008374), Nkg7tm1.1(KOMP)Vlcg (B6N.Nkg7−/−; RRID: IMSR_KOMP:VG11445–1.1-Vlcg)25, B6.129S7-Rag1tm1Mom/J (Rag1−/−; RRID: IMSR_JAX:002216)46 and C;129S4-Rag2tm1.1Flv Il2rgtm1.1Flv/J (Rag2−/−γc−/−; RRID: IMSR_JAX:014593)47 mice. B6N.Nkg7−/− (Nkg7tm1.1(KOMP)Vlcg) mice25 were generated by the University of California Davis (Davis, California, United States) as part of the trans-NIH Knockout Mouse Project (KOMP) and obtained from the KOMP repository (http://www.komp.org/). Transgenic PbT-I mice32 were crossed to B6.Cd45.1 mice to generate PbT-I × B6.Cd45.1 (PbT-IWT; CD45.1+CD45.2+) mice, and crossed to B6J.Nkg7−/− mice to generate Nkg7-deficient PbT-I mice (PbT-IΔNkg7; CD45.1−CD45.2+).
When the same mouse strain was used across multiple experimental groups, littermates of the same sex were distributed randomly into groups. Experimental use was in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (Australian National Health and Medical Research Council) and approved by the QIMR Berghofer Medical Research Institute Animal Ethics Committee (Brisbane, Queensland, Australia; approval numbers: A02–633M, A02–634M, A1707–615M, A19619M and A19620M).
Human participants.
Blood samples were collected from 23 symptomatic patients with visceral leishmaniasis at the Kala-Azar Medical Research Center (Muzaffapur, India). Patients were diagnosed either by microscopic detection of amastigotes in splenic aspirate smears or using an rK39 (L. donovani antigen) dipstick test. Clinical data from these patients are summarized in Supplementary Table 1. We collected 5 ml blood from each patient on the day of admission (day 0), and 30 d following treatment with AmBisome (Gilead Sciences) (day 30). We also collected 5 ml blood from endemic controls. The involvement of human participants in this research was in line with recommendations from the Helsinki declaration. Human ethics approval was provided by the ethical review board of Banaras Hindu University in Varanasi, India (Dean/2011–12/289) and the QIMR Berghofer Medical Research Institute Human Ethics Committee (reference number P1411). Written informed consent was obtained from all participants; where participants were below 18 years of age, written informed consent was obtained from their legal guardian.
Method details.
Generation of C57BL/6J-Nkg7em1(cre)WEHI mice. C57BL/6J mice expressing the cre recombinase under the control of the Nkg7 promoter (B6J.Nkg7-cre) were generated by the Melbourne Advanced Genome Editing Centre (MAGEC) at the Walter and Eliza Hall Institute using CRISPR– Cas9-mediated gene editing. Briefly, based on methods previously described48, the single guide RNA (sequence: CATGGAGCCCTGCCGGTCCC) was used to induce double-stranded breaks in the Nkg7 locus, to stimulate homologous recombination, and a targeting vector containing homology arms of ~2 kilobases49 was used to introduce the cre recombinase coding sequence.
Forward (ACGACCAAGTGACAGCAATG) and reverse (GCTAACCAGCGT TTTCGTTC) primers to detect the cre recombinase sequence were used to screen viable pups for integration of the targeting vector by PCR. A 301-base pair amplicon was detected where the cre recombinase sequence was present. F0 mice expressing the cre sequence were selected for backcrossing that resulted in heterozygous F1 mice. The PCR described above was used to screen F1 mice for the cre sequence. Further validation by long-range PCR was performed to verify correct positional integration of the targeting vector.
Leishmania infection in mice.
L. donovani (LV9; MHOM/ET/67/HU3) was originally isolated from a patient in Ethiopia in 196750 and maintained by passage in B6.Rag1–/– mice. Passage mice were euthanized and the spleen was excised into 5 ml sterile Roswell Park Memorial Institute medium 1640 (Gibco; Life Technologies) + 100 μg ml−1 penicillin–streptomycin (Gibco; Life Technologies) medium (RPMI/PS). The excised spleen was homogenized using a glass tissue grinder and the cell suspension was centrifuged in an Eppendorf Centrifuge 5810R (Thermo Fisher Scientific) at 115g for 5 min at room temperature with the brake off.
The supernatant was transferred to a new tube, and the pellet was discarded. The supernatant was centrifuged at 1,960g for 15 min at room temperature. The supernatant was discarded, and the pellet was incubated for 5 min in 1 ml Red Blood Cell Lysing Buffer Hybri-Max (Sigma–Aldrich), following which sterile RPMI/PS was added, and the parasites were centrifuged at 1,960g for 15 min at room temperature. After discarding the supernatant, sterile RPMI/PS was added to the pellet and the centrifugation step was repeated at 1,960g for 15 min at room temperature. The supernatant was then discarded and the parasite pellet was resuspended in sterile RPMI/PS. The parasite suspension was taken up through a 26 G × ½′′ needle on a 1 ml syringe (Terumo Medical) and dispensed, then the process was repeated until a homogenous suspension was achieved. Some 2 µl of the parasite suspension was loaded onto a Thoma cell counting chamber (Weber Scientific International) and parasites were counted in the 4 × 4 grid in triplicate. An average count was used to determine the number of parasites per ml using the following equation:
Parasites were diluted to a final concentration of 1 × 108 parasites per ml in sterile RPMI/PS. Each mouse received 2 × 107 parasites in 200 μl, injected intravenously.
Quantifying Leishmania parasite burdens in murine spleen or liver.
Where indicated, the parasite burden was quantified from spleen and liver impression smears stained in Giemsa (Sigma–Aldrich). The numbers of amastigotes per 1,000 host nuclei were counted under ×1,000 magnification using a light microscope (Olympus CX31; Olympus Life Science) and multiplied by the organ weight (g) to derive Leishman–Donovan units.
Plasmodium infection in mice.
Murine Plasmodium infections were established from parasites passaged in C57BL/6J mice once. Briefly, 200 μl of transgenic P. berghei ANKA (231c11) parasites (in-house laboratory stock, frozen at −80 °C) expressing luciferase and GFP under the control of the ef1-α promoter51, were thawed at room temperature and injected intraperitoneally into a passage mouse. A blood smear was prepared and stained with 20% (vol/vol) Giemsa (Sigma–Aldrich) from day 2 onwards. Parasitemia (% pRBCs) was quantified under a light microscope (Olympus CKX41; Olympus Life Science). The passage mouse was sacrificed at >1% pRBC. Blood was collected from the passage mouse by cardiac puncture, into RPMI/PS containing 1 IU ml−1 heparin, and centrifuged at 290g for 7 min at room temperature. RBCs were counted on a hemocytometer (Pacific Laboratory Products). A parasite inoculum containing 5 × 105 pRBCs per ml was prepared and mice were injected with 200 μl of the inoculum intravenously (total: 1 × 105 pRBCs per mouse).
Mice were tail bled to obtain a drop of blood, which was collected in 250 μl RPMI/PS with 1 IU ml−1 heparin. Then, 50 µl of this blood preparation was stained with Hoechst 33342 (Sigma–Aldrich) and SYTO 84 (Invitrogen; Life Technologies) in RPMI/PS for 30 min at room temperature. Next, 300 µl of RPMI/PS was added and each sample was acquired on a BD FACSCanto II or BD LSRFortessa through BD FACSDiva version 8.0 (BD Biosciences).
P. berghei ANKA-infected mice were scored daily, from day 4 p.i., for symptoms of ECM including hunching in posture, piloerection, lethargy and wobbly gait. At the peak of ECM, mice were anesthetized with Isothesia NXT (Henry Schein) and injected intraperitoneally with 100 μl 5 mg ml−1 d-Luciferin Firefly, Potassium Salt (Caliper Life Sciences). Whole-body bioluminescence was visualized using the IVIS Spectrum in vivo imaging system (Xenogen Corporation, Caliper Life Sciences). Mice were sacrificed by CO2 asphyxiation and spleens were collected for cellular analysis. Mice were subsequently perfused through the heart with 10 ml phosphate-buffered saline (PBS). Brains were excised and imaged as previously described for whole-body bioluminescence. Bioluminescence was quantified using Living Image 4.5 software (PerkinElmer) and expressed as average photons per second per cm2 per steradian (p s−1 cm−2 sr−1).
Isolation of mouse peripheral blood mononuclear cells (PBMCs).
Mice were sacrificed by CO2 asphyxiation and blood was collected via cardiac puncture using an insulin syringe (BD Biosciences). The blood was then diluted with an equal volume of Dulbecco’s PBS (DPBS), overlaid onto 1.5 ml Lympholyte-Mammal (Cedarlane) and processed per the manufacturer’s instructions.
Preparation of spleen and thymus single-cell suspensions.
A mid-sagittal incision was made on the abdominal cavity of sacrificed mice, from which the spleen was excised. Incisions were made on either side of the ribcage to expose the thoracic cavity, and the thymus was collected. The spleen and thymus were weighed and placed into 1% (vol/vol) fetal calf serum (FCS) in PBS (1% FCS/PBS). Spleens and thymi were mechanically passed through a 100-μm EASYstrainer cell strainer (Greiner Bio-One) using the back of a 5 ml syringe plunger (Terumo Medical). Cells were resuspended in 1% FCS/PBS and centrifuged at 350g in an Eppendorf Centrifuge 5810R (Thermo Fisher Scientific) and lysed by incubation in Red Blood Cell Lysing Buffer Hybri-Max (Sigma–Aldrich) for 7 min at room temperature. Cells diluted in DPBS (Gibco) and Trypan Blue Stain (Invitrogen) were counted using Countess Cell Counting Chamber Slides on the Countess II FL (both from Invitrogen), per the manufacturer’s protocol.
Preparation of liver single-cell suspensions.
Mice were sacrificed by CO2 asphyxiation. A mid-sagittal incision was made on the abdominal cavity. The liver was perfused via the hepatic portal vein with 1× PBS. The excised liver was weighed and collected in 1% (vol/vol) FCS/PBS and mechanically passed through a 100-μm EASYstrainer cell strainer (Greiner Bio-One) using a 10 cc ml−1 syringe plunger (Terumo Medical). The homogenized liver was washed twice in 1× PBS by centrifuging at 390g in an Eppendorf Centrifuge 5810R. Hepatocytes were separated from leukocytes using a 33% (vol/vol) Percoll Density Gradient Media (GE Healthcare) and centrifugation at 575g for 15 min at room temperature with the brake off. Red Blood Cell Lysing Buffer Hybri-Max (Sigma–Aldrich) was added to the leukocyte pellets and incubated for 7 min at room temperature. This was followed by a single wash in PBS as described above. Cells were diluted in DPBS (Gibco; Life Technologies) and Trypan Blue Stain (Invitrogen), then counted using Countess Cell Counting Chamber Slides on the Countess II FL (both from Invitrogen), per the manufacturer’s protocol.
Isolating bone marrow MNCs.
The femurs of sacrificed mice were collected and cleaned of surrounding muscle. Once clean, the ends of the femurs were removed using a pair of scissors and the shaft was placed in an Eppendorf tube containing 1 ml 1% (vol/vol) FCS/PBS. Tubes were centrifuged at 13,523g in a microcentrifuge for 3 min at 4 °C. Upon removal of bones from the Eppendorf tubes, the remaining cell pellet was resuspended using a pipette. Cells were transferred into a 10-ml tube and centrifuged at 390g for 6 min at room temperature. The supernatant was discarded by pouring and RBCs were lysed by adding 1 ml Red Blood Cell Lysing Buffer Hybri-Max (Sigma–Aldrich) to each sample and incubating for 5 min at room temperature. Cells were washed once in 1% (vol/vol) FCS/PBS by centrifuging as described above and pouring off the supernatant.
Isolating brain MNCs.
Cardiac perfusion was performed with cold DPBS before the removal of brains. Excised brains were dissected into smaller pieces and incubated in 2 mg ml−1 collagenase from Clostridium histolyticum and 1 mg ml−1 deoxyribonuclease I (DNase I) from bovine pancreas (both from Sigma–Aldrich) resuspended in 2 ml Hanks’ Balanced Salt Solution (without phenol red, calcium chloride and magnesium sulfate) per sample. Samples were incubated on an Incu-Shaker Mini (Benchmark Scientific) at 200 r.p.m. for 20 min at 37 °C. Samples were then homogenized through a 70-μm EASYstrainer cell strainer (Geiner Bio-One) using the back of a 10-ml syringe plunger (Terumo Medical). Brain single-cell suspensions were washed by resuspending in 1% FCS/PBS and centrifuging at 350g in an Eppendorf Centrifuge 5810R (Thermo Fisher Scientific). The supernatant was discarded by pouring and the pellet was resuspended in a 33% (vol/vol) Percoll Density Gradient Media (GE Healthcare) followed by centrifugation at 575g for 15 min at room temperature with the brake off. The debris and supernatant were discarded by pouring, following which MNCs were incubated in 500 μl Red Blood Cell Lysis Buffer Hybri-Max (Sigma–Aldrich) for 4 min at room temperature. MNCs were washed once as described above, and incubated in 2× Monensin Solution (BioLegend) diluted in complete media (as described above) for 3 h at 37 °C in the presence of 5% CO2. Flow cytometry staining panels for brain MNCs included the use of anti-mouse/human CD11b (Brilliant Violet 421; M1/70; BioLegend) and anti-mouse F4/80 (Brilliant Violet 421; BM8; BioLegend) to eliminate microglia.
Preparation of lung single-cell suspensions.
Mice were sacrificed by CO2 asphyxiation and lungs were perfused intraventricularly with cold PBS to remove blood. The perfused lungs were cut finely and digested in 3 ml RPMI 1640 media containing 1 mg ml−1 Collagenase IV (Worthington Biochemical Corporation) and 20 μg ml−1 DNase I (Roche Diagnostics Corporation) in a six-well plate for 30 min at 37 °C, set on a shaker at 150 r.p.m. Then digested lung tissue was homogenized through a 40-μm strainer and washed with PBS. Cells were centrifuged at 456g for 4 min, resuspended in 1 ml fluorescence-activated cell sorting (FACS) buffer with anti-CD16/32 (clone 2.4G2) to block Fc receptors and incubated for 20 min on ice. One-fifth of the cell suspension was stained with fluorophore-conjugated antibody cocktails for 20 min on ice. Before samples were run on a flow cytometer, liquid-counting beads (BD Biosciences) were added directly to determine absolute cell counts in samples.
Isolation of CD4+ T cells from human PBMCs.
Approximately 5 ml of blood was collected from each patient on the day of admission (day 0) and 30 d after treatment with AmBisome (Gilead Sciences) (day 30) in BD Vacutainer Lithium Heparin 170 I.U. Plus Blood Collection Tubes (BD Biosciences). Blood was layered over Ficoll-Paque PLUS (GE Healthcare) to isolate PBMCs. PBMCs were counted using a hemocytometer (Pacific Laboratory Products). CD4+ T cells were enriched by magnetic-activated cell sorting (MACS) using anti-human CD4 MicroBeads (Miltenyi Biotec) according to the manufacturer’s instructions.
Flow cytometry.
All flow cytometry staining was performed in Falcon 96-Well Clear Round Bottom Tissue Culture-Treated Cell Culture Microplates (Corning). Single-cell suspensions were incubated with 50 µl TruStain FcX (anti-mouse CD16/32; clone 93) and Zombie Aqua Fixable Viability Dye cocktail (both from BioLegend) for 15 min at room temperature. CD107a (LAMP-1) staining was performed during the 3-h stimulation with phorbol 12-myristate 13-acetate (PMA)/ionomycin or incubation with monensin (see below) by adding 5 μg ml−1 anti-Mouse CD107a (clone 1D4B from BioLegend or clone eBio1D4B from eBioscience, Life Technologies) to the stimulation cocktail. Cells were washed once with staining buffer (PBS, 0.02% (vol/vol) FCS, 5 mM EDTA and 0.01% (wt/vol) NaN3) by centrifuging in an Eppendorf Centrifuge 5810R (Thermo Fisher Scientific) at 575g for 1 min at 4 °C. Samples were then incubated with 50 µl of a cocktail of fluorescence-conjugated antibodies reactive against surface molecules for 30 min. A list of antibodies and concentrations used can be found in Supplementary Table 7. Where samples were stained with a biotin-conjugated antibody, samples were subsequently incubated with 1 μg ml−1 Streptavidin PE-Cy7 Conjugate (Thermo Fisher Scientific).
After two washes with staining buffer, as described above, samples were incubated with 100 µl of fixation buffer from either the BD Cytofix Fixation Buffer Set (BD Biosciences; for cells that were subsequently stained with antibodies against cytokines) or the eBioscience Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific; for cells that were subsequently stained with antibodies reactive against transcription factors) for 20 min. Cells were then washed twice with wash buffers from the respective kits by centrifuging at 575g for 1 min at room temperature, following which cells were incubated with 50 µl of a cocktail containing fluorescence-conjugated antibodies against intracellular molecules for 35 min. All staining was performed at room temperature and samples were incubated in the dark.
Samples were stored at 4 °C before acquisition on a BD LSRFortessa (special-order research product; BD Biosciences) through BD FACSDiva version 8.0 or on a Cytek Aurora 5 laser through the SpectroFlo software package version 2.2 (Cytek Biosciences), then analyzed on FlowJo version 10 OSX (FlowJo). Graphing and statistical analyses were performed on GraphPad Prism 7 (version 7.0c; GraphPad Software). P ≤ 0.05 was considered statistically significant.
PMA/ionomycin restimulation.
Cells were incubated in complete media (10% (vol/vol) FCS, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (penicillin–streptomycin), 1× GlutaMAX, 1× non-essential amino acids (NEAAs), 110 mg l−1 (1 mM) sodium pyruvate, 5 mM HEPES (all from Gibco) and 0.05 mM 2-mercaptoethanol (Sigma–Aldrich), in RPMI 1640 containing l-glutamine (Gibco)) containing 25 ng ml−1 PMA and 1 μg ml−1 (1.33 nM) ionomycin calcium salt in the presence of 10 μg ml−1 brefeldin A (all from Sigma–Aldrich) or 2× monensin solution (BioLegend). PMA/ionomycin restimulation was performed for 3 h at 37 °C in the presence of 5% (vol/vol) CO2.
Tetramer staining.
Detection and quantification of antigen-specific cells was performed using fluorophore-conjugated tetramers added alongside other fluorophore-conjugated antibodies detecting surface markers. Allophycocyanin (APC)-conjugated I-AbPEPCK335–351 (0.7 μg per well)27 and PE-conjugated H2-Kb-NVF tetramer (0.3 μg per well)31 were used.
Detection of phosphorylated STAT3 and STAT4.
Following staining for surface markers, cells were incubated in 400 ng ml−1 Recombinant Mouse IL-12 (p70; carrier free; BioLegend; for the detection of phosphorylated STAT4) or Recombinant Mouse IL-6 (carrier free; BioLegend; for the detection of phosphorylated STAT3) for 4 min at room temperature, in a 96-well U-bottom plate. Cells were then centrifuged at 575g for 1 min at room temperature and the supernatant was discarded by flicking. Cells were then incubated in pre-warmed 1× BD Phosflow Lyse/Fix Buffer (BD Biosciences) for 10 min at 37 °C and centrifuged at 1,860g for 2 min. Supernatant was discarded by flicking. Cells were then washed twice with eBioscience Flow Cytometry Staining Buffer (Invitrogen) as described above. Following this, cells were incubated in −20 °C pre-chilled BD Phosflow Perm Buffer III (BD Biosciences) for 30 min on ice. Cells were washed three times in eBioscience Flow Cytometry Staining Buffer (Invitrogen) as above. Finally, cells were stained with either PE-conjugated Mouse anti-Stat4 (pY693; clone 38/p-Stat4) or PE-conjugated Mouse anti-Stat3 (pY705; clone 4/P-STAT3) (both from BD Biosciences), followed by two washes in eBioscience Flow Cytometry Staining Buffer (Invitrogen) as described above. Samples were acquired on a BD LSRFortessa through BD FACSDiva version 8.0 (BD Biosciences).
Ex vivo lung NK cell function assay.
One-fifth of a whole-lung suspension was incubated at 200 µl per well in a 96-well U-bottom plate in complete RPMI 1640 media. Cells were incubated in the presence of the eBioscence Cell Stimulation Cocktail (plus protein transport inhibitors; diluted 1,000×; Invitrogen) and 2.5 μg ml−1 Alexa Fluor 647-conjugated anti-CD107a antibody (BioLegend). After 4 h, cells were stained for surface markers and intracellular cytokine production and analyzed as described above.
Evaluation of the immunological synapse.
Splenic NK cells were isolated using the NK Cell Isolation Kit II (Miltenyi Biotec), according to the manufacturer’s instructions, and activated with 1,000 U ml−1 recombinant IL-2 for 4–5 d. Activated effector NK cells were labeled with 5 μM CellTrace Violet Cell Proliferation Kit, for flow cytometry (CTV; Thermo Fisher Scientific) and co-cultured with carboxyfluorescein succinimidyl ester (CFSE)-labeled YAC-1 target cells52 for 15 min at an effector-to-target ratio of 1:2 in polyproprylene tubes followed by fixing with 2% (wt/vol) paraformaldehyde in PBS.
Evaluation of the immunological synapse was performed using imaging flow cytometry as previously described53. Briefly, fixed cells were stained with PerCP-Cy5.5-conjugated anti-mouse 1 μg ml−1 CD11a/CD18 (LFA-1; clone H155–78; BioLegend) for 20 min at room temperature. Cells were washed with 2% FCS/PBS and centrifuged at 300g for 4 min, followed by aspiration of the supernatant using a pipette. Permeabilization was performed with 0.1% (vol/vol) Triton X in 2% FCS/PBS (Sigma–Aldrich; T8787) containing 0.4 U Alexa Fluor 647-conjugated Phalloidin (Invitrogen; A22287) by incubating at room temperature for 30 min. Cells were washed as described above and resuspended gently in 30 μl of 2% (wt/vol) formaldehyde in PBS. Samples were immediately acquired on the four-laser, 12-channel Amnis ImageStream XMark II (Luminex Corporation) at a 60-fold magnification at low speed. Data analysis was performed using the Image Data Exploration and Analysis Software version 6.2 (Luminex Corporation). The gating strategy involved selection of cells in focus based on gradient RMS. After sub-gating on double-positive CTV+ and CFSE+ events, focused and centered doublets were selected and at least 50 doublets per group were analyzed. The interface mask was then applied with the NK cells (CTV+) defined as the target of interest. The mean fluorescence intensity of LFA-1 and phalloidin within the interface mask served as a surrogate marker for the strength and intensity of the immunological synapse. Statistical significance was determined using a non-parametric Mann–Whitney U-test.
Isolation of mouse spleen and liver CD4+ T cells by FACS.
CD4+ T cells were isolated by MACS using the CD4+ T cell isolation kit, mouse (Miltenyi Biotec) according to the manufacturer’s instructions. The flow through, containing enriched CD4+ T cells, was stained with propidium iodide (Sigma–Aldrich), anti-mouse TCR-β (fluorescein isothiocyanate; H57–597) and anti-mouse CD4 (APC; GK1.5) (both from BioLegend).
Isolation of CD4+ T cells for real-time quantitative PCR (RT-qPCR) was achieved by cell sorting after staining for anti-mouse CD90.2 (Thy-1.2; PerCP/Cyanine5.5 (PerCP/Cy5.5); 53–2.1) and anti-mouse CD4 (APC; GK1.5) (both from BioLegend).
Dead cells were excluded by positive staining for either propidium iodide or SYTOX Blue Dead Cell Stain, for flow cytometry (used according to the manufacturer’s instructions). CD4+ T cells were identified as TCR-β+CD4+ or CD90.2+CD4+. Cells were sorted on a BD FACSAria II (Becton Dickinson) and stored in buffer RLT (Qiagen) at −80 °C.
Microarray.
FACS-sorted mouse spleen and liver CD4+ T cells, stored in buffer RLT, were homogenized in QIAshredder columns before RNA extraction using an RNeasy Mini Kit (all from Qiagen) according to the manufacturer’s instructions. Each replicate within the naive liver CD4+ T cell group consisted of four pooled samples. Samples were run using the Mouse Whole-Genome-6 version 2.0 Expression BeadChip Kit (Illumina). Quality control was assessed using the lumi package54, run on R (https://www.r-project.org/). Differential gene expression was analyzed using limma55.
RNA-seq.
CD4+ T cells isolated from the PBMCs of nine patients with symptomatic visceral leishmaniasis (eight males and one female) at days 0 and 30 were homogenized in QIAshredder columns before RNA extraction using the RNeasy Mini Kit (both from Qiagen) according to the manufacturer’s instructions. Isolated RNA was treated with the RNase-free DNase Set (Qiagen). mRNA was isolated using the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs). Libraries were prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs). Libraries were quantified using the KAPA Library Quantification Kit (Roche Sequencing) and RNA integrity number obtained using the RNA 6000 Pico Kit (Agilent Technologies). Expression profiling was performed by 50-base pair single-end mRNA-seq with a read depth of ~10 million reads, on an Illumina HiSeq platform (performed by the Australian Genome Research Facility, Melbourne, Victoria, Australia).
RT-qPCR.
Cells sorted from naive and infected mice were stored in RLT buffer and homogenized in QIAshredder columns (both from Qiagen). RNA was extracted using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. The concentration of RNA (ng μl−1) and sample purity (260/280 ratio) was measured using the NanoDrop 2000 UV-Vis Spectrophotometer (Thermo Fisher Scientific). Extracted RNA was reverse transcribed to complementary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) per the manufacturer’s instructions.
QuantiTect Primer Assays (specific for Mus musculus B2m, Hprt and Nkg7; Qiagen) were used with the GoTaq qPCR Master Mix (Promega Corporation), and TaqMan Gene Expression Assays (specific for M. musculus Ifng, Tnf, Hprt and Pml; Life Technologies) were performed with GoTaq Probe qPCR Master Mix (Promega Corporation) based on the standard cycling conditions recommended by the manufacturer. Reactions were performed in a final volume of 10 μl containing 1 μl template cDNA. RT-qPCR was performed in Hard-Shell 384-Well Plates, thin wall, skirted, clear/clear (Bio-Rad), sealed with Microseal ‘B’ PCR Plate Sealing Film, adhesive, optical (Bio-Rad). QuantiTect Primer Assays were run on the QuantStudio 5 Real-Time PCR System (Applied Biosystems) and TaqMan Gene Expression Assays were performed on the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Relative quantification was performed using the comparative CT method relative to the average of two internal control genes: B2m and Hprt (for QuantiTect Primer Assays) and Pml and Hprt (for TaqMan Gene Expression Assays).
For human samples, CD4+ T cells were enriched by MACS using the anti-human CD4 MicroBeads (Miltenyi Biotec) according to the manufacturer’s instructions. RNA was then extracted, and reverse transcribed to cDNA as previously described10. RT-qPCR for NKG7 was performed on an ABI Prism 7500 real-time PCR system (Applied Biosystems) using the TaqMan Gene Expression Assay (assay ID: Hs01120688_g1; Applied Biosystems). Relative quantification was performed using the comparative CT method56 relative to 18S ribosomal RNA (assay ID: Hs99999901_s1; Applied Biosystems).
In vitro polarization of naive CD4+ T cells.
Splenic MNC suspensions were stained with 30 μl of a master mix containing Zombie Aqua and TruStain FcX anti-mouse CD16/32 (both from BioLegend) for 15 min at room temperature. Cells were washed once in DPBS (Gibco) by centrifuging at 575g for 1 min at room temperature, and stained in 30 μl master mix containing monoclonal anti-mouse CD90.2 (PerCP-Cy5.5; 53–2.1), CD4 (Brilliant Violet 605; GK1.5 or RM4–5), CD25 (PE-Cy7; PC61), CD44 (Alexa Fluor 700; IM7) and CD62L (PE; MEL-14) (all from BioLegend) for 30 min at room temperature. After two washes, cells were resuspended in staining buffer without sodium azide (1× PBS; 0.02% (vol/vol) FCS and 5 mM EDTA) and naive cells were purified by FACS on a BD FACSAria III (Becton Dickinson). Naive CD4+ T cells were identified as Zombie Aqua−, CD90.2+, CD4+, CD25−, CD44− and CD62L+.
A total of 100 µl Ultra-LEAF Purified anti-mouse CD3ε (clone: 145–2C11; BioLegend; 4 μg ml−1; diluted in DPBS (1×) (Gibco)) monoclonal antibody (mAb) was added to each well of a 96-well U-bottom plate and incubated either for 2 h in a 37 °C incubator or overnight at 4 °C to coat the wells. Following this, the purified anti-mouse CD3ε mAb was discarded and 100 μl of 2× polarization cocktail (Supplementary Table 2; previously described in ref. 22), prepared in mouse T cell media (10% (vol/vol) FCS, 1× MEM NEAA solution and penicillin–streptomycin (all from Gibco), and 0.05 μM 2-mercaptoethanol (Sigma–Aldrich), in Dulbecco’s modified Eagle medium containing 4.5 g l−1 d-glucose, l-glutamine and 110 mg l−1 (1 mM) sodium pyruvate (Gibco)), was added to the respective wells. A total of 200,000 naive CD4+ T cells were seeded per well, in duplicate, and left to incubate at 37 °C in 5% CO2 for 72 h.
After 72 h, 50 μl of supernatant was collected and stored at −20 °C for cytokine analysis by cytometric bead array (CBA). Cells in duplicate wells were pooled for flow cytometry staining.
Cytotoxicity assays.
In vitro killing assays were performed as described previously52. Briefly, NK cells were isolated from the spleen using the NK Cell Isolation Kit II (Miltenyi Biotec). NK cells that were freshly isolated or stimulated with recombinant IL-2 (1,000 U ml−1) for 4–5 d were used as effector cells. YAC-1 target tumor cells (provided by J. Trapani at the Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia) were labeled with 5 μM CTV (Thermo Fisher Scientific). A total of 1 × 104 YAC-1 target cells were co-cultured with effector NK cells at the indicated effector-to-target ratios for 4 h. Tumor cell death in CTV+ cells was determined using Annexin V/7-AAD in Annexin V Binding Buffer (BD Biosciences).
For CD8+ T cell in vitro killing assays, 96-well U-bottom plates were coated with 5 μg ml−1 (ref. 57) Ultra-LEAF Purified anti-mouse CD3ε (clone 145–2C11; BioLegend; diluted in DPBS (1×) (Gibco)) mAb in 100 μl per well by incubating at 37 °C for 2 h. Effector CD8+ T cells were purified from PbT-IWT and PbT-IΔNkg7 splenocytes by MACS using the CD8a+ T Cell Isolation Kit, mouse (Miltenyi Biotec) according to the manufacturer’s instructions. Effector cells were cultured in the presence of plate-bound anti-CD3ε mAb, 2 μg ml−1 LEAF Purified anti-mouse CD28 (clone 37.51; BioLegend) mAb and 20 ng ml−1 recombinant mouse IL-2 (BioLegend) in T cell media (10% FCS, penicillin–streptomycin, 1× GlutaMAX, 1× MEM NEAA solution, 1 mM sodium pyruvate and 1× d-glucose (all from Gibco), 5 mM HEPES and 0.05 μM 2-mercaptoethanol (Sigma–Aldrich), in RPMI 1640 (Gibco)) for 48 h. Splenocytes prepared from B6.Cd45.1 mice were pulsed with 1 μM NVF peptide (sequence: NVFDFNNL)31 by incubating in a water bath at 37 °C for 1 h. NVF-pulsed cells were labeled with 1 μM CTV (BD Horizon) by incubating in a water bath at 37 °C for 15 min. CTV-labeled NVF-pulsed target cells were counted and combined in equal parts with control target cells (non-peptide pulsed; no CTV labeling). Then, 2 × 105 activated effector CD8+ T cells were co-cultured with target cells at the indicated effector-to-target ratios for 8 h. Cells were stained with TruStain FcX (anti-mouse CD16/32 mAb) (clone 93; BioLegend) and Zombie NIR Fixable Viability Kit (BioLegend) before staining with BUV395 Mouse anti-Mouse CD45.2 (clone 104; BD Horizon) mAb to distinguish effector cells (CD45.2+) from target cells (CD45.2−). Cell viability was assessed by positive staining for Zombie NIR. The frequency of target cell killing was determined using the following formula:
To assess NK cell–mediated cytotoxicity in vivo, 5 × 106 RMA-s-Rae1β cells52 were injected intravenously via the tail vein. Lungs were harvested 2 h after tumor challenge, followed by digestion using collagenase IV (1 mg ml−1; Worthington Biochemical). Numbers of RMA-s-Rae1β cells (Rae1+H-2Kb− cells) in lung single-cell suspensions were determined by flow cytometry. Biotin H-2Kb (AF6–88.5) was detected using 1 µg ml−1 APC-conjugated streptavidin (both BD Biosciences).
Retroviral transduction of primary mouse CD8+ T cells.
Codon-optimized NKG7 cDNA (NCBI Reference Sequence: NM_024253.4) was synthesized by Integrated DNA Technologies. The eGFP gene was PCR amplified from MSCV–IRES–eGFP (Addgene plasmid 20672; a gift from T. Reya at the University of California, San Diego, California, United States), with introduction of a linker sequence at the amino terminus. The final insert encodes an NKG7–eGFP fusion protein with NKG7 at the amino terminus, fused to eGFP at the carboxy terminus via a (T) GGGGS linker, with the threonine introduced by a restriction site during the cloning process. The insert was cloned into an MSCV backbone (Addgene plasmid 52114; a gift from D. Vignali at the University of Pittsburgh, Pennsylvania, United States) and verified by Sanger sequencing at the Australian Genome Research Facility (Brisbane, Australia).
Replication-incompetent retroviral particles were generated by transient transfection of HEK293T cells with the packaging plasmid (EcoPak) and the retroviral vector plasmid MSCV–NKG7–GFP. Retrovirus-containing supernatants were harvested at 48 and 72 h and stored at −80 °C until use.
Splenocytes (1 × 106) were activated with plate-bound CD3ε (clone 2C11) and CD28 (clone N3751) mAbs for 24 h in Iscove’s modified Dulbecco’s medium with 10% heat-inactivated fetal bovine serum, 2 mM l-alanyl-l-glutamine dipeptide (GlutaMAX; Gibco), 1% NEAA, 1 mM sodium pyruvate, 50 mM 2-mercaptoethanol and 100 U ml−1 penicillin–streptomycin. The retroviral supernatant was centrifuged (1,500g for 60 min at 4 °C) on a RetroNectin (Takara)-coated plate, and the supernatant was removed. The activated splenocytes were incubated (37 °C; 5% CO2) with plate-bound retroviral particles for 4 h in Iscove’s modified Dulbecco’s medium with recombinant human IL-2 (aldesleukin; 100 U ml−1) and hexadimethrine bromide 16 μg ml−1 (polybrene; Sigma–Aldrich). The splenocytes were then harvested and expanded in the presence of recombinant human IL-2 (100 U ml−1) for 5–6 d before downstream application.
Immunofluorescence microscopy.
Immunofluorescence microscopy was performed on 7 µm frozen liver sections. Tissue was fixed in 4% (wt/vol) paraformaldehyde for 2 h before overnight incubation in 30% (wt/vol) sucrose and embedding in Tissue-Tek Optimal Cutting Temperature Compound (VWR Chemicals). Following cutting, sections were stored frozen until use. Sections were air dried then rehydrated in Tris-buffered saline with Tween 20 (TBS-T; 0.1 M Tris-HCl, 0.15 M NaCl and 0.001% (vol/vol) Tween 20 in Milli-Q) and incubated in Background Sniper (Biocare Medical), diluted 1/10 in TBS-T, for 30 min. Following this, sections were incubated in 5 μg ml−1 Alexa Fluor 647-conjugated anti-mouse CD4 (RM4–5; BioLegend) mAb diluted in Van Gogh Yellow Diluent (Biocare Medical) for 1 h at room temperature. Sections were washed in TBS-T after each incubation step. Sections were then incubated in 4′,6-diamidino-2-phenylindole (Sigma–Aldrich) diluted 1/20,000 in PBS for 10 min at room temperature. Finally, sections were mounted in ProLong Gold Antifade (Invitrogen) under a glass cover slip.
Immunofluorescence microscopy was also performed on cell suspensions following retroviral transduction. C57BL/6N transduced splenocytes were incubated with LysoTracker Red DND-99 (Life Technologies) diluted to 75 nM in 5% FCS/PBS for 30 min at 37 °C before imaging. PbT-I-transduced cells were incubated with 5 μg ml−1 Alexa Fluor 647-conjugated anti-mouse CD107a/LAMP-1 (1D4B; BioLegend) mAb diluted in 200 μl restimulation media (containing 25 ng ml−1 phorbol 12-myristate 13-acetate, 2 μg ml−1 ionomycin calcium salt and 4 μM monensin in complete RPMI) for 2 h at 37 °C before imaging. Images were acquired on either a Zeiss 780 NLO laser-scanning confocal microscope (Carl Zeiss Microscopy) or a ScanScope FL slide scanner (Aperio; Leica Biosystems).
Quantitative data were analyzed automatically using QuPath version 0.1.2 (University of Edinburgh, Edinburgh, Scotland) with CD4+NKG7+ cells counted and expressed per mm2 of area. Regions of immune cell accumulation around infected Kupffer cells (inflammatory foci) in the livers of L. donovani-infected mice at day 28 p.i. were defined and the number of NKG7+CD4+ T cells in 21 defined areas (average size = 0.017 mm2) within inflammatory foci and 21 areas (average size = 0.024 mm2) outside these zones for each infected mouse liver were measured using the above software. All cells were manually checked for accuracy before data were plotted and analyzed in Prism (GraphPad).
CBA.
Cytokine levels were assessed using the BD CBA Mouse Inflammation Kit or Mouse TH1/TH2/TH17 Cytokine Kit (BD Biosciences) per the manufacturer’s instructions. Serum or plasma samples from mouse blood were used neat while cell culture supernatants were diluted 1:5 in PBS for the detection of most cytokines. Supernatants were diluted 1:50 in PBS for the detection of IFN-γ. CBA data were analyzed using the FCAP Array Software version 3.0 (BD Biosciences).
CD4+ T cell adoptive transfer.
CD4+ T cells were purified by MACS from the spleens of either C57BL/6N or B6N.Nkg7−/− donor mice using the CD4+ T Cell Isolation Kit, mouse (Miltenyi Biotec) according to the manufacturer’s instructions. A total of 1 × 106 CD4+ T cells were injected intravenously into each Rag1−/− host mouse the day before infection.
PbT-I cell adoptive transfer.
CD8+ T cells were isolated from the spleens of PbT-IWT and PbT-IΔNkg7 mice using the CD8a+ T Cell Isolation Kit, mouse (Miltenyi Biotec) according to the manufacturer’s instructions. Single-cell suspensions were prepared as described above. PbT-IWT and PbT-IΔNkg7 cells were counted and combined in equal proportions at a final concentration of 5 × 106 cells per ml. Then, 200 μl (1 × 106 cells) was subsequently injected intravenously into the tail vein of B6.Cd45.1 recipient mice 1 d before infection with P. berghei ANKA. The proportion of PbT-IWT (CD45.1+CD45.2+; refer to the section ‘Mice’) versus PbT-IΔNkg7 (CD45.1−CD45.2+) cells in the single-cell suspension was verified by flow cytometry using anti-mouse CD45.1 (Alexa Fluor 700; A20; BioLegend) and anti-mouse CD45.2 (BUV395; 104; BD Biosciences).
Tumor models.
Antibodies.
Purified control antibodies (hamster control antibodies or 1–1) or antibodies to deplete CD8 T cells (53.5.8) were purchased from BioXcell. Anti-asialo-GM1 (anti-asGM1) was purchased from Wako Pure Chemical. Antibody to neutralize IFN-γ (H22) was purchased from Leinco Technologies.
Cell lines.
Mouse B16F10 melanoma, RM-1 prostate carcinoma and E0771 mammary carcinoma cells58 were grown in Dulbecco’s modified Eagle medium supplemented with 10% (vol/vol) FCS (Bovogen), 1% glutamine (Gibco), 1% HEPES (Gibco) and 1% penicillin–streptomycin (Gibco). LWT1 melanoma cells were cultured in RPMI 1640 supplemented with 10% FCS, 1% glutamine and 1% penicillin–streptomycin. The YAC-1, RMA-s and RMA-s-Rae1β lymphoma cell lines59 were cultured in complete RPMI 1640 media containing 10% FCS, 1% glutamine, 1% penicillin–streptomycin, 2 mM GlutaMAX, 55 μM 2-mercaptoethanol, HEPES and sodium pyruvate. Cell lines were maintained at 37 °C under 5% CO2. All cell lines tested negative for Mycoplasma.
Experimental metastasis.
Mice were injected intravenously with either 1 × 105 B16F10 melanoma cells (ATCC; 2007), 5 × 105 LWT1 melanoma cells60 (derived in house) or 2 × 105 RM-1 prostate carcinoma cells, as previously described60–62. Some groups of mice were treated intraperitoneally with either control antibodies (100 μg), anti-CD8β (53.5.8; 100 μg), anti-asGM1 (50 μg) or anti-mouse IFN-γ (H22; 250 μg) on days −1, 0 and 7 relative to tumor inoculation. For NK cell-based cytokine immunotherapy, some mice were treated intraperitoneally with PBS or IL-15 (0.5 μg)/IL-15Ra (3 μg) complexes (R&D Systems) on days 0 and 3 after tumor inoculation, or PBS or IL-2 (100,000 IU) on days 0, 1, 2 and 3 relative to tumor inoculation. Lungs were harvested from mice injected with B16F10 cells at day 14 post-injection. Lungs were harvested from mice injected with LWT1 cells at day 14 post-injection and perfused with India ink. The metastatic burdens in all three models were quantified by counting colonies on the lung surface under a light microscope.
NK cell adoptive transfer.
Groups of Rag2−/−γc−/− mice were injected intravenously with 2 × 105 WT or Nkg7−/− NK cells isolated by FACS. After 6 d, naive or adoptively transferred Rag2−/−γc−/−mice received either 1 × 105 or 1 × 104 B16F10 tumor cells. Lungs were harvested from mice injected with B16F10 cells at day 14 post-injection. As above, metastatic burdens were quantified by counting colonies on the lung surface under a light microscope.
Spontaneous metastasis.
For spontaneous metastases, 1 × 105 E0771 mammary carcinoma cells (provided by R. Anderson at the Peter MacCallum Cancer Centre) were injected into the fourth mammary gland of C57BL/6N (B6N; WT) or B6N.Nkg7-KO mice. AT 12 d after tumor inoculation, the primary tumors were equivalent in size between WT and Nkg7-deficient mice (mean: 15.8 + 1.6 mm2 versus 17.7 + 1.6 mm2, respectively), and the primary tumors were surgically resected. Mice were then sacrificed at day 35 and lungs were harvested, and metastatic burdens were quantified by counting colonies on the lung surface under a light microscope.
Intraperitoneal RMA-s lymphoma model.
C57BL/6N (B6N; WT) or B6N.Nkg7-KO mice were injected intraperitoneally with 1 × 105 RMA-s lymphoma cells (provided by J. Ortaldo at the NCI-Frederick Cancer Research Facility). The percentage survival of each group was monitored for 150 d as previously described36.
Subcutaneous RMA-s-Rae1β lymphoma model.
C57BL/6N (B6N; WT) or B6N.Nkg7-KO mice were injected subcutaneously with 5 × 106 RMA-s-Rae1β lymphoma cells as previously described52. The tumor growth was measured every 2–3 d with a caliper square as the product of two perpendicular diameters (mm2). Some groups of mice were treated with either control antibodies (100 μg), anti-CD8β (100 μg) or anti-asGM1 (50 μg) on days −1, 0, 7, 14 and 20 relative to tumor inoculation.
MCA carcinogenesis.
The MCA-induced fibrosarcoma model was performed as previously described37. Briefly, C57BL/6N (B6N; WT) or B6N.Nkg7-KO mice were injected subcutaneously with 100 µl corn oil containing 25 µg MCA on the right-hand flank. The development of fibrosarcomas was monitored weekly over the course of 250 d. The percentage of tumor-free mice was recorded.
Quantification and statistical analysis.
Statistical analyses were performed using Prism 7 (version 7.0c; GraphPad Software). P values are shown as *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. Where possible, the D’Agostino–Pearson omnibus normality test was used to test for normal distribution. Non-parametricity was assumed in cases where this was not possible. Statistical testing was performed using either the Kruskal–Wallis test and Dunn’s multiple comparisons test (single-variable data) or a regular two-way analysis of variance (ANOVA) with multiple comparisons (two-variable data), unless stated otherwise.
Modeling the NKG7 tertiary protein structure.
The NKG7 structure was modeled using the Iterative Threading Assembly Refinement (I-TASSER) protein structure and function prediction software63. The protein structure that was the highest-ranked threading template used in the modeling of the human and mouse sequences was the mouse claudin-19 in complex with the carboxy-terminal fragment of Clostribium perfringens enterotoxin (PDB ID: 3X29). The models with the highest C-scores were chosen for display, and corresponded to values of 0.15 and 0.51 for the human and mouse proteins, respectively. The C-score represents the confidence in the model; values are typically in the range −5 to 2, and higher values correspond to models with higher confidence.
T-distributed stochastic neighbor embedding (t-SNE).
t-SNE was performed as outlined in a protocol and R script written by T. Ashhurst (https://github.com/sydneycytometry/tSNEplots/releases). Briefly, cells were gated on lymphocytes, followed by exclusion of doublets and dead cells using FlowJo version 10 OSX (FlowJo). Each sample was assigned a sample number and the live cell population was downsampled to 15,000 events on FlowJo version 10 OSX, before concatenation into a single file. t-SNE was performed using the lineage markers CD11b, B220, NK1.1, TCR-β, CD4, I-A/I-E (MHC II), Ly-6C, CD11c and CD8, with the following settings applied: iteration: 2000; perplexity: 35; eta (learning rate): 200; theta: 0.5.
A second t-SNE was performed only on NK cells and T cells by excluding NK1.1−TCR-β− cells from live cells. Biexponential scales were then applied for all fluorophores of interest. Scales were adjusted by increasing the width basis to reduce the spread of negative data. The gated population for each sample was assigned a sample number and downsampled to 50,000 events on FlowJo version 10 OSX. Downsampled populations from all samples were then concatenated into a single file. NK1.1, TCR-β, CD4 and CD8 were selected as t-SNE parameters and the same settings as above were applied.
Each sample within the concatenated file was distinguished on the basis of the previously assigned sample number. Colorized t-SNE plots were generated in R using exported channel values as an input to the script (version 1.4) written by T. Ashhurst (https://github.com/sydneycytometry/tSNEplots/releases).
RNA-seq analysis.
Quality control was performed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were mapped to the reference genome (GRCh38.p3) and assigned to transcripts (Ensembl version 82) using Spliced Transcripts Alignment to a Reference (STAR)64.
EdgeR run on R65 was used to generate a list of differentially expressed genes from the RNA-seq dataset consisting of n = 11 paired samples. Briefly, the transcript with the highest read count for each gene was retained. Further filtering was performed by the elimination of genes with <1 count per million in more than two-thirds of samples. The effective library size was determined using the default trimmed mean of M-value normalization based on which counts were normalized. Data exploration by hierarchical clustering revealed two failed biological samples that were removed with their corresponding pairs, resulting in n = 9 biological replicates. Testing for differential expression was performed using gene counts fitted to a negative binomial generalized log-linear model. Genes with a false discovery rate (FDR) < 0.05 were considered to be differentially expressed, unless otherwise stated.
k-means clustering was performed using the eclust function within the factoextra package (https://rpkgs.datanovia.com/factoextra/) with the following parameters: FUNcluster = ‘kmeans’, k.max = 10, nstart = 25 and nboot = 100.
Upstream regulators for genes of interest were identified using ingenuity pathway analysis (IPA; Qiagen). In addition to drugs that were predicted on IPA, genes of interest were submitted to the Drug–Gene Interaction Database66 to identify currently available drugs. The International Mouse Strain Resource (IMSR)67 was used in search of transgenic or knockout mouse strains that are available for each target gene. Localization of target genes was determined by functional annotation based on data from Gene Ontology Cellular Component68 using the Database for Annotation, Visualization, and Integrated Discovery (version 6.8 Beta; National Institute of Allergy and Infectious Diseases, National Institutes of Health, Maryland, United States)69. Graphs were plotted using Prism 7 (version 7.0c; GraphPad Software).
Analysis of TCGA data.
SKCM RNA-seq data previously published by the TCGA Research Network (https://cancergenome.nih.gov/) were downloaded and processed using the TCGAbiolinks package70 on R65. Briefly, transcript counts were normalized by gene length, and genes in the bottom quartile (<25%) of the expression mean across all samples were filtered out. Samples were ranked according to expression values for the gene of interest and stratified into two groups, where the uppermost quartile (>75%) were designated high expressers and the lowermost quartile (<25%) were designated low expressers. Overall survival between these groups was analyzed and plotted as Kaplan–Meier curves with P values determined using a log-rank test. Correlation analyses were then performed between the two genes of interest and the relationship was expressed as r.
NKG7 was excluded from a 20-gene signature for NK cells recently published35. TCGA SKCM samples were ranked based on NKG7 expression and correlated against a moving average of the remaining 19 genes, which were collectively termed the NK cell signature.
IPA.
Gene symbols, log2[fold change] and FDR from each dataset were used as an input into IPA (version 43605602; Qiagen). Where there was a concatenation of genes that overlapped between two or more datasets, the log2[fold change] and FDR values for the human CD4+ T cell dataset were used. Initial interrogation of each dataset was performed using default values and parameters set on IPA. In contrast, examination of the pathways involved in the core, chronic and resolving gene signatures was restricted to molecules and/or relationships experimentally observed in Homo sapiens or M. musculus and specifically in immune cells.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Nkg7 is only expressed by NK cells and a subset of CD8+ TCRβ+ cells in the spleen in naive mice. t-SNE plot of splenocytes from a naive mouse, pre-gated to exclude doublets and dead cells.
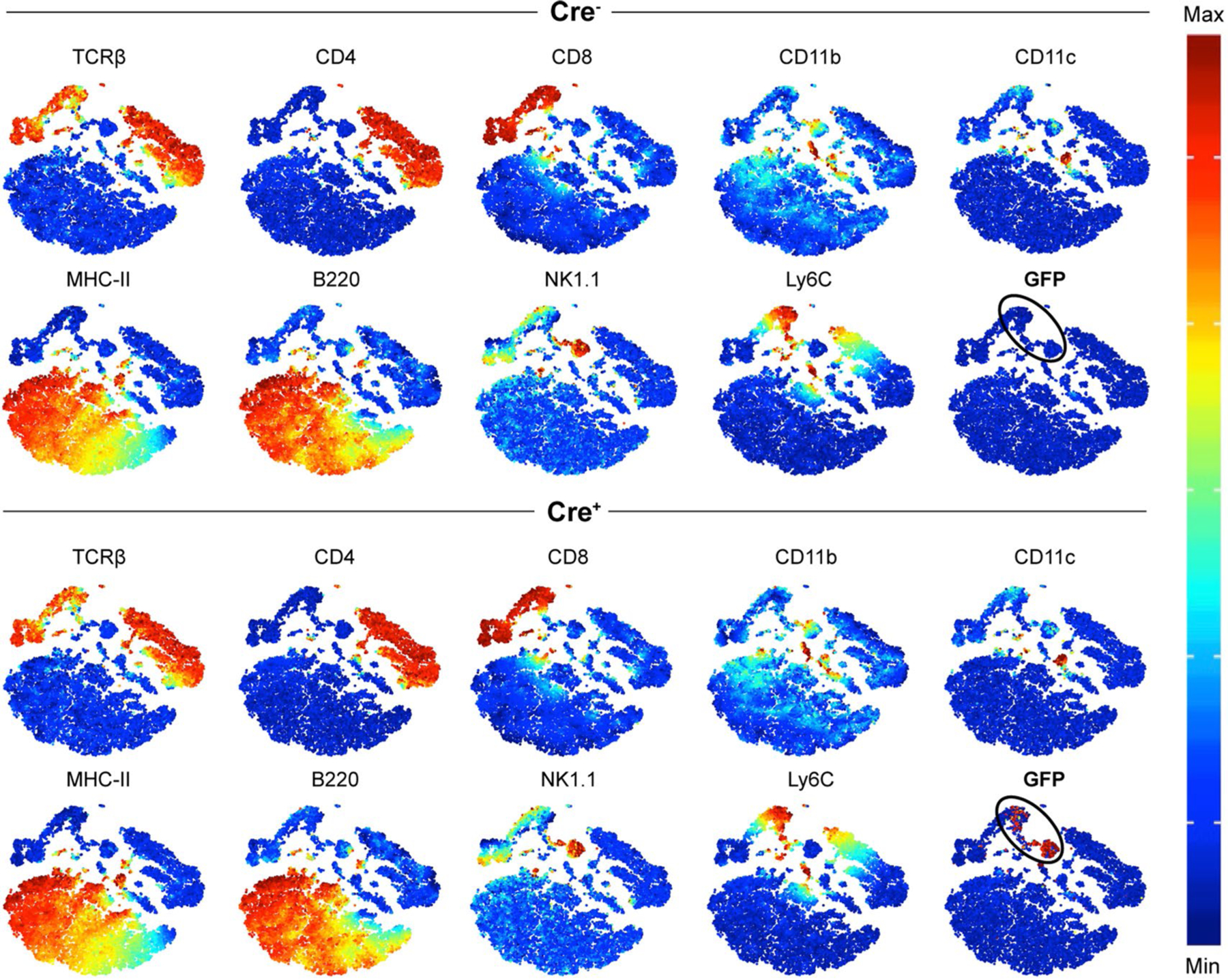
The remaining cells were clustered using TCRβ-BUV737, CD4-BUV395, CD8α-PE/Cy7, CD11b-PerCP/Cy5.5, CD11c-BV785, MHC-II-Pacific Blue, B220-BV650, NK1.1-APC/Cy7 and Ly-6C-BV605. Equal numbers of cells (15,000 cells) are shown for Cre– and Cre+ plots. The black oval indicates the GFP+ population. n = 1 per genotype, performed once. BV, brilliant violet.
Extended Data Fig. 2 |. Changes in the frequencies of Nkg7-expressing NK cells and CD8+ T cells during Leishmania donovani infection.
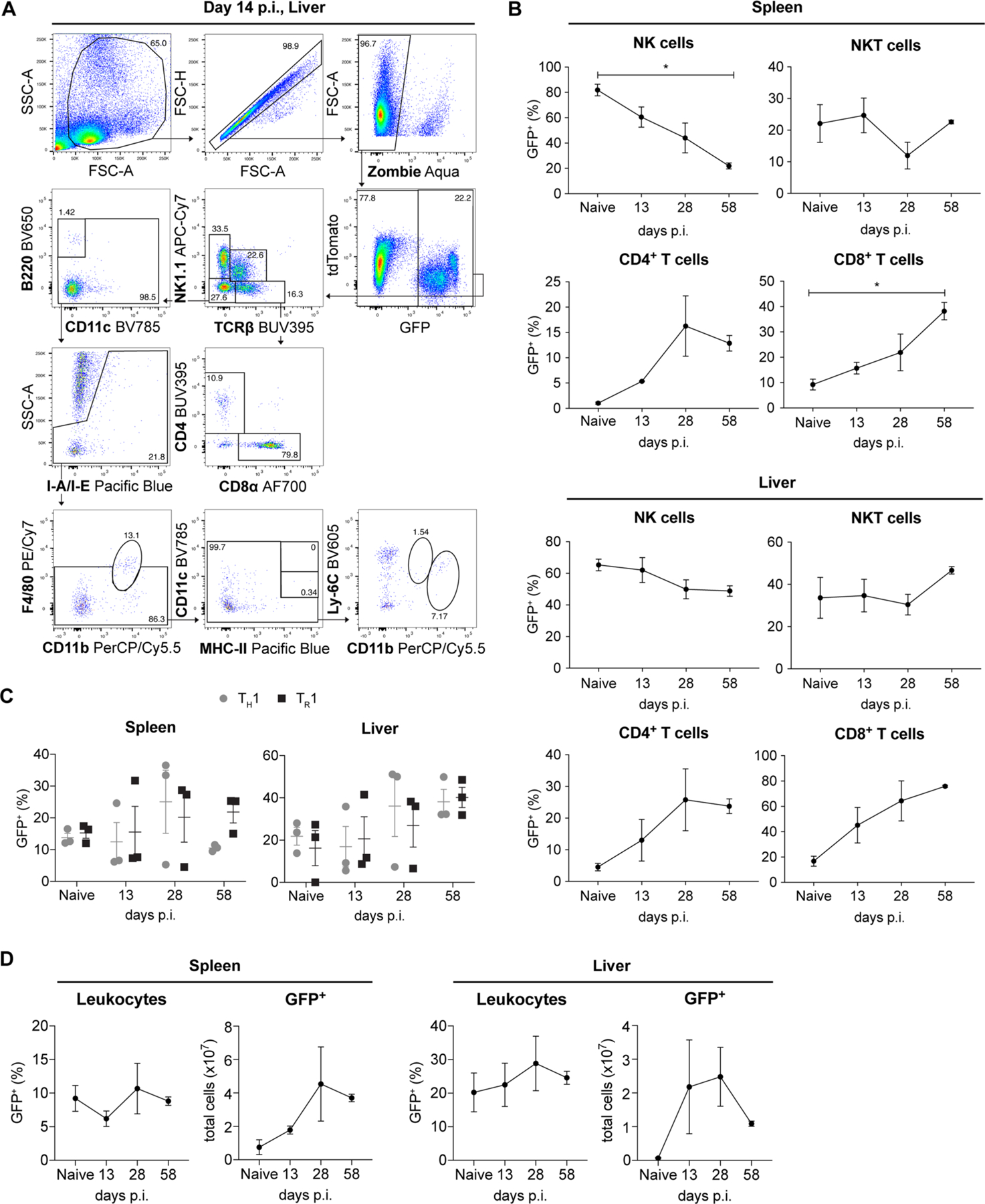
a, The gating strategy used to assess changes in the key immune cell subsets including NK cells, CD4+ T cells, CD8+ T cells, B cells, cDCs, pDCs, CD11bhi Ly6Cint monocytes, inflammatory monocytes, macrophages, and NKT cells. b, The graphs show changes in GFP within each of the key immune cell subsets in the spleen and liver during L. donovani infection. Statistical significance was determined using the Kruskal–Wallis one-way analysis of variance (ANOVA) with Dunn’s multiple comparisons test. c, The frequencies of GFP+ TH1 (gated on NK1.1− TCRβ+ CD8− CD4+ IFN-γ+ IL-10−) and TR1 cells (gated on NK1.1− TCRβ+ CD8− CD4+ IFN-γ+ IL-10+) in the spleen and liver during the course of infection are shown. A two-way ANOVA with Sidak’s multiple comparisons test was performed to test for statistical significance. d, Changes in the frequency and total number of GFP+ cells in the spleen and liver over the course of infection are shown. p value is indicated where * p < 0.05. Error bars represent mean ± SEM. The data shown is representative of two independent experiments, each with n = 3 mice per genotype, per timepoint.
Extended Data Fig. 3 |. Nkg7 deficiency results in reduced CD4+ T cell responses during Leishmania donovani infection.
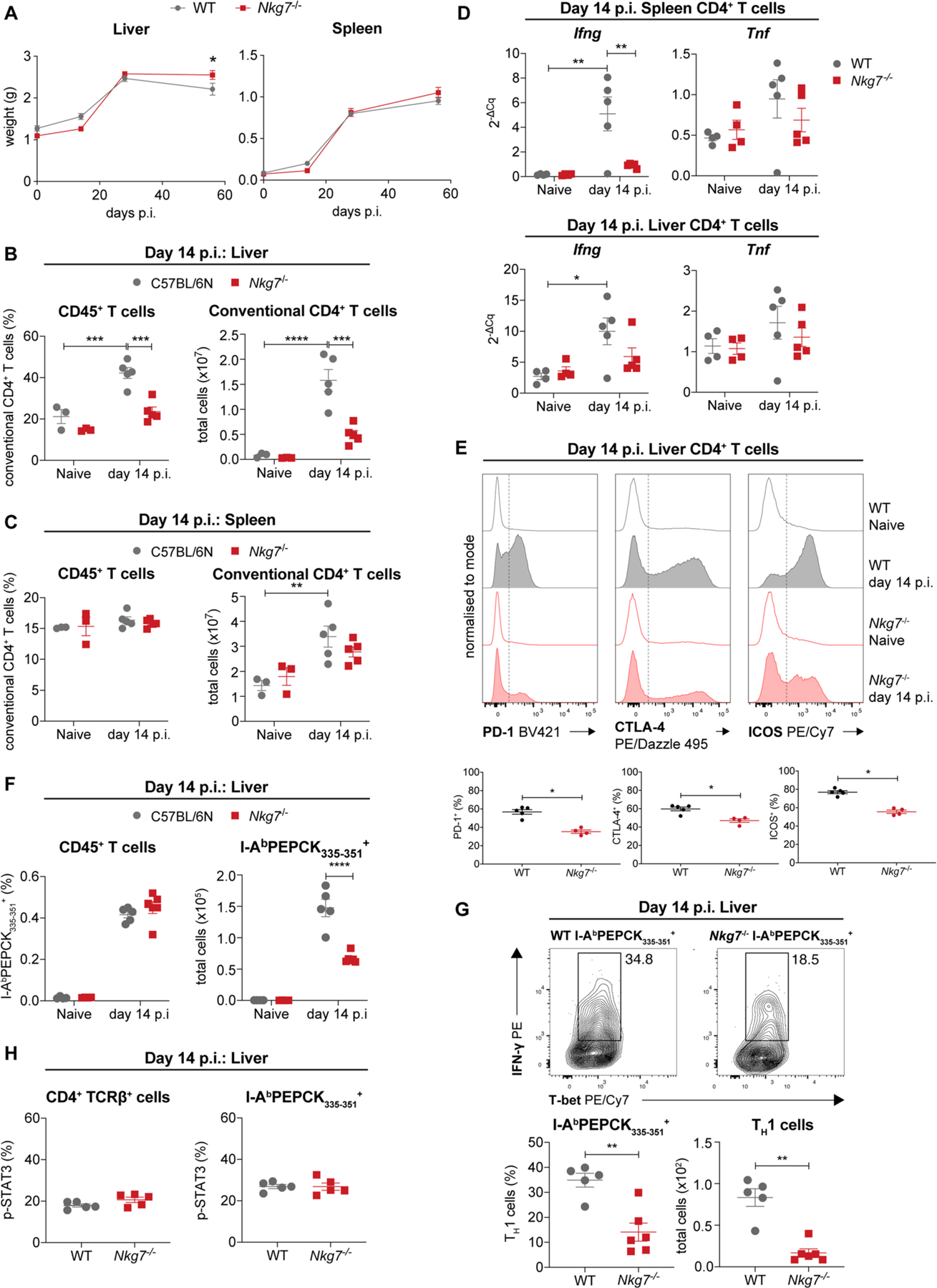
a, The liver and spleen weights of WT and Nkg7−/− mice during L. donovani infection. A two-way ANOVA with Sidak’s multiple comparisons test was used to determine statistical significance. Data is representative of two experiments, where n = 3 naive WT and Nkg7−/− mice, and n = 5 WT and 4 Nkg7−/− mice at days 14, 28 and 58 p.i. groups. b and c, The frequency and total number of conventional (Foxp3−) CD4+ T cells in the liver (b) and spleen (c) at day 14 p.i. are shown. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple comparisons test. The data shown is representative of two independent experiments, each with n = 3 naive WT and Nkg7−/− mice, and n = 5 WT and 4 Nkg7−/− mice at day 14 p.i.. d, The expression of Ifng and Tnf mRNA by spleen or liver CD4+ T cells in naive or infected (day 14 p.i.) mice was determined by RT-qPCR. A two-way ANOVA with Sidak’s multiple comparisons test was used to determine statistical significance. n = 4 naive and 5 infected mice in each group. e, The representative histograms show PD-1, CTLA-4, and ICOS staining on CD4+ T cells in the liver at day 14 p.i. The graphs indicate the frequencies of PD-1+, CTLA-4+, and ICOS+ CD4+ T cells. Statistical significance was determined using the Mann–Whitney test. Data is derived from one experiment, where n = 3 naive WT and Nkg7−/− mice, and n = 5 infected WT and 4 infected Nkg7−/− mice. f, The frequency and total number of I-AbPEPCK335–351 (tetramer)-PE+ cells in the liver of naive and infected (day 14 p.i.) mice are shown. A two-way ANOVA with multiple comparisons test was used to test for statistical significance. n = 4 WT or Nkg7−/− naïve, and 5 WT or 6 Nkg7−/− infected mice. Data is representative of two independent experiments. g, Representative plots depict the differences in TH1 (IFN-γ+ T-bet+ cells) frequencies in WT or Nkg7−/− I-AbPEPCK335–351 (tetramer)-PE+ cells. The frequencies and numbers are shown in the accompanying graphs below. Statistical significance was determined using the Mann–Whitney test. n = 5 mice per group. Data is representative of two independent experiments. h, The frequency of WT or Nkg7−/− CD4+ TCRβ+ cells and I-AbPEPCK335–351 (tetramer)-PE+ cells expressing IL-6-stimulated phosphorylated (p)-STAT3 are shown. Statistical significance was determined using the Mann–Whitney test. n = 5 mice per group. Data is representative of two independent experiments. p value is indicated where * p < 0.05. Error bars represent mean ± SEM.
Extended Data Fig. 4 |. The absence of NKG7 results in decreased CD8+ T cell cytotoxicity during PbA infection.
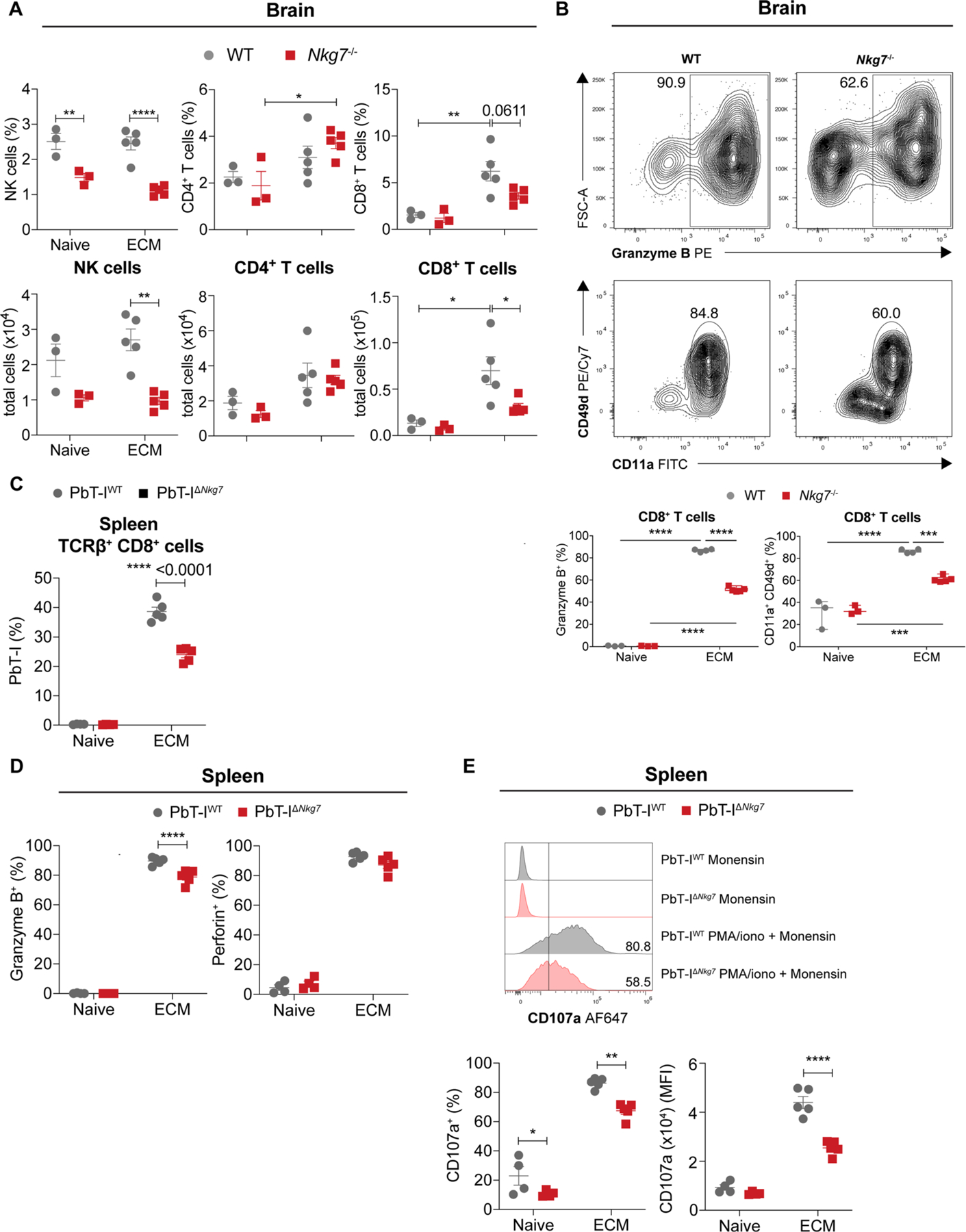
a, The graphs show the frequency and total number of NK cells, CD4+ T cells and CD8+ T cells in the brain of naive and infected (peak of ECM) WT (n = 3 naive and 5 infected) and Nkg7–/– (n = 3 naive and 5 infected) mice. Cell subsets frequencies are expressed as a percentage of CD45+ cells. Data is representative of two independent experiments. b, Representative flow cytometry plots were gated on lymphocytes, singlets, live cells, NK1.1-APC/Cy7− TCRβ-BUV737+, CD8α-PerCP/Cy5.5+ cells and the frequencies of CD11a+ CD49d+ cells and Granzyme B+ cells are shown. n = 3 naïve and 4 infected mice per strain. Data is representative of two independent experiments. c, The graph shows the proportions of PbT-IWT and PbT-IΔNkg7 cells in the spleen of naive (n = 4) or infected mice at peak of ECM (n = 5). d, The frequencies of splenic PbT-IWT and PbT-IΔNkg7 transgenic CD8+ T cells expressing Granzyme B or Perforin, in the presence of monensin, are shown. n = 4 naïve and 5 infected mice at peak of ECM. e, The representative histograms show differences in the expression of CD107a by splenic PbT-IWT and PbT-IΔNkg7 cells incubated with monensin or stimulated with PMA and ionomycin in the presence of monensin. The frequency and MFI of CD107a expression is shown in the accompanying graphs. n = 4 naïve and 5 infected mice at peak of ECM. Statistical significance in all graphs was determined using a two-way ANOVA with Tukey’s (a) or Sidak’s (b–e) multiple comparisons test.
Extended Data Fig. 5 |. Nkg7-deficiency results in increased cancer metastasis in experimental models.
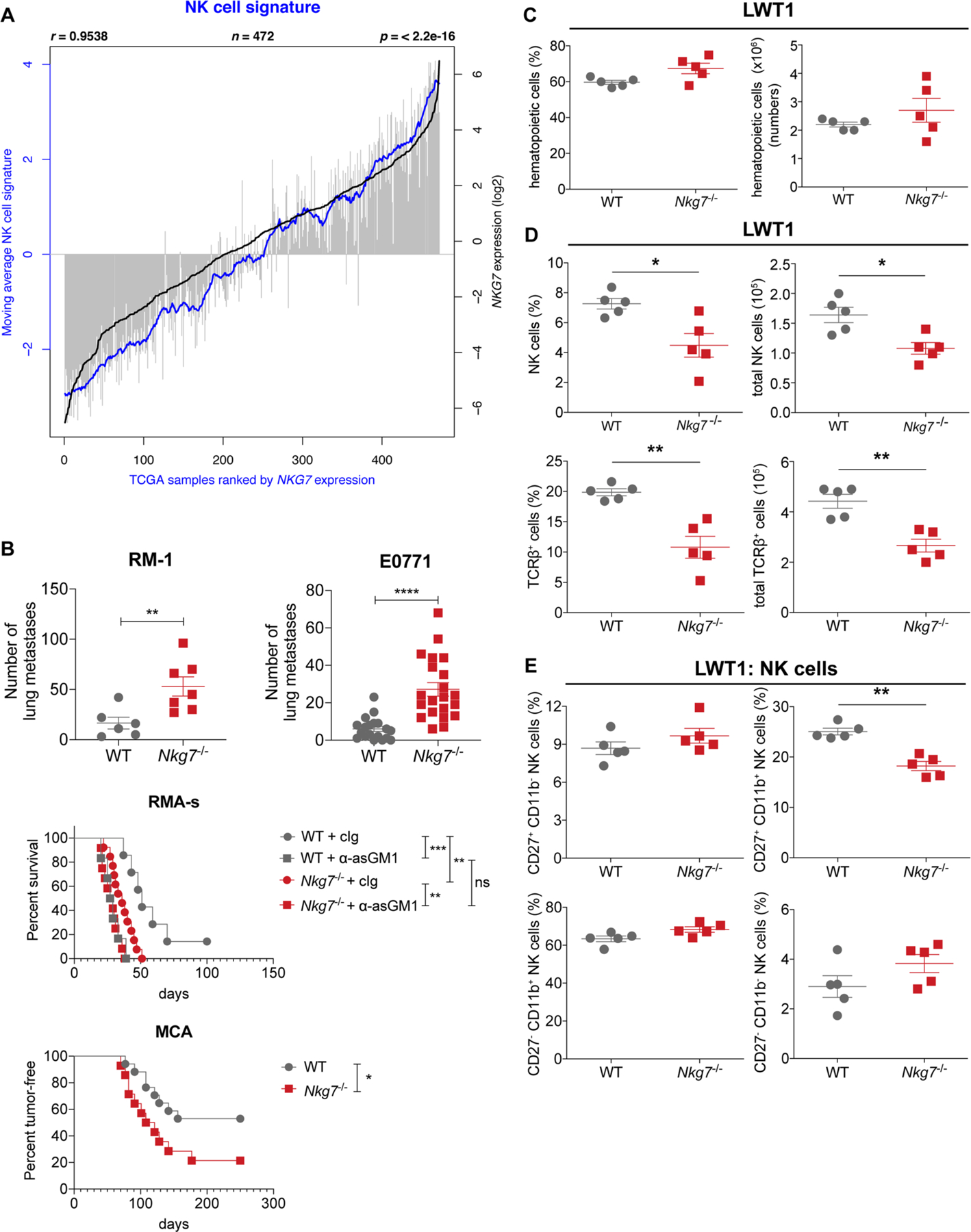
a, A correlation (r) between the moving average of a 19-gene natural killer (NK) cell signature genes and NKG7 expression in n = 472 samples from TCGA:SKCM dataset. b, The graphs indicate differences in the number of lung metastases between WT and Nkg7−/− mice following injection of RM-1 prostate carcinoma cells (n = 6 WT and 7 Nkg7−/−) and spontaneous metastasis of E0771 mammary carcinoma cells (n = 19 WT and 21 Nkg7−/−, from two pooled experiments). The survival of WT and Nkg7−/− mice treated with either cIg (n = 7 WT and 13 Nkg7−/−) or α-asGM1 (n = 6 WT and 12 Nkg7−/−) in an intraperitoneal RMA-s lymphoma model was also assessed. Statistical significance between groups was tested using the Log-rank (Mantel–Cox) test. Additionally, the difference in percentage of tumour-free mice between WT (n = 17) and Nkg7−/− (n = 14) mice following MCA-induced fibrosarcoma generation is shown. The log-rank (Mantel–Cox) test was used to determine statistical significance. ** and *** represents p < 0.01 and 0.001 respectively. ns, not significant. c, The lungs of WT and Nkg7−/− mice, injected with LWT1 cells, were assessed for differences in the frequency and total cell number of hematopoietic cells, at 14 days post-injection. d, The frequency and total cell number of NK cells and T cells were quantified in the lungs of mice injected with LWT1 cells, at 14 days post-injection. e, The differences in the frequency of NK cells at different stages of maturation, based on CD27 and CD11b expression at 14 days post-injection of LWT1 cells is shown. The data shown in C-E is representative of two independent experiments where n = 5 mice per group. The Mann–Whitney test was used to determine statistical significance. p values are shown as follows: * p < 0.05 and ** p < 0.01.
Extended Data Fig. 6 |. Nkg7–/– NK cells do not have reduced abilities to conjugate with target cells or to form synapses.
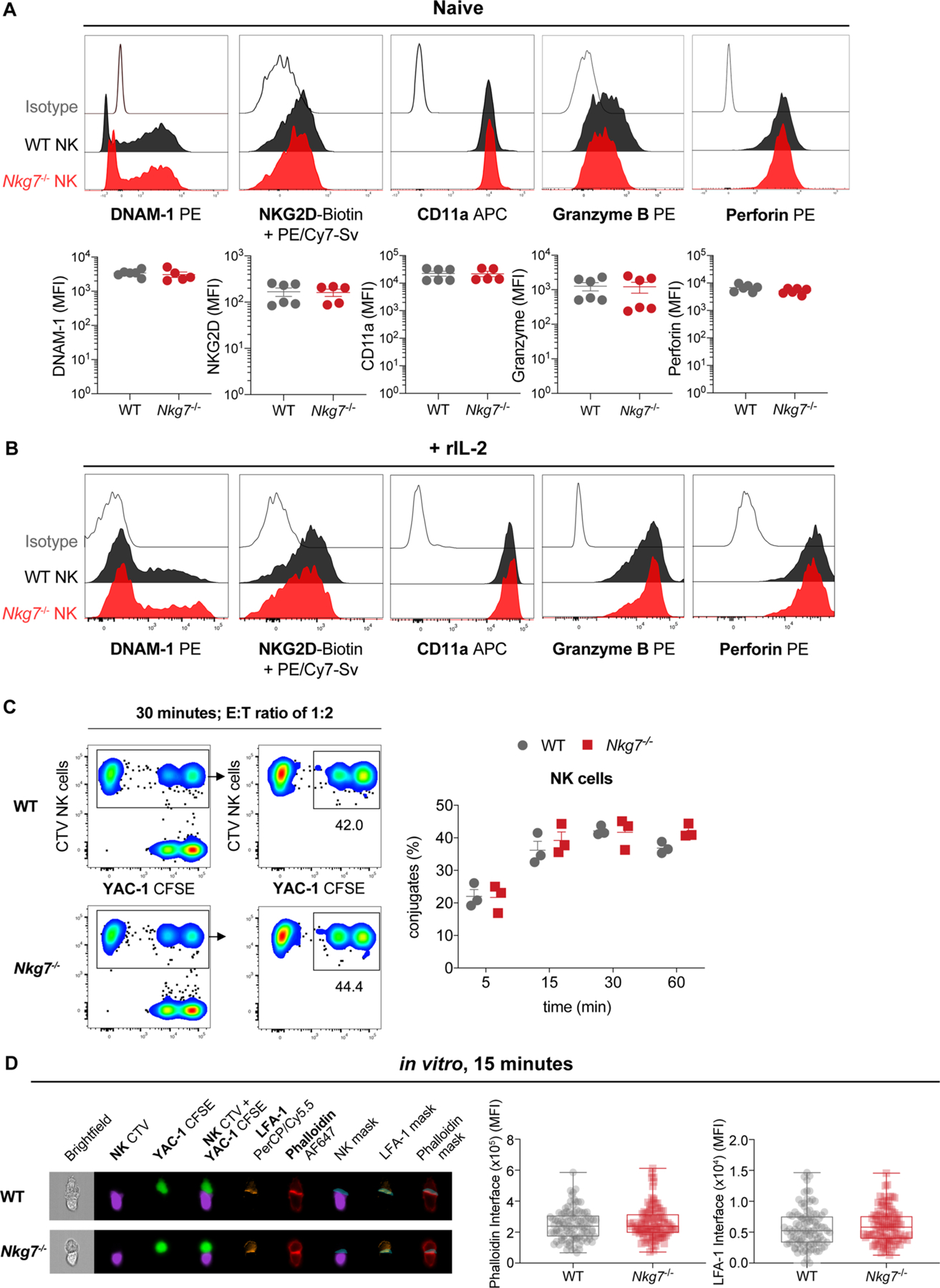
a and b, The representative histograms show expression of DNAM-1 (CD226), NKG2D (CD314), CD11a, Granzyme B, and Perforin by WT (n = 6) or Nkg7−/− (n = 5) NK cells in the naive state (a) or when activated with rIL-2 (b). The MFI for the expression of each marker in naive NK cells is also shown. Data is pooled from 2 independent experiments. Sv, Streptavidin. c, Representative plots depict the frequency of cell conjugates formed when CellTrace Violet (CTV)-labelled WT or Nkg7−/− NK cells were co-cultured with carboxyfluorescein succinimidyl ester (CFSE)-labelled YAC-1 target cells for 30 minutes at an E:T ratio of 1:2. The frequency of conjugated NK cells at 5, 15, 30, and 60 minutes is shown in the accompanying graph. n = 3 mice per group. Data is pooled from 2 independent experiments. d, Representative images of effector NK cell–YAC-1 target cell conjugates visualised using an Amnis® ImageStream®XMark II after in vitro co-culture of WT or Nkg7–/– cells with target cells for 15 minutes. The graphs show the MFI of Phalloidin or LFA-1 at the interface between effector and target cells. Data obtained from one experiment.
Supplementary Material
Acknowledgements
We thank the staff at the Kala-Azar Medical Research Centre (KAMRC), Muzaffarpur, India for help with the collection of blood samples, as well as patients and volunteers for allowing the use of blood samples. We thank staff at the QIMR Berghofer flow cytometry laboratory for assistance, and staff at the QIMR Berghofer animal facility for animal husbandry. We acknowledge the facilities, and the scientific and technical assistance of the MAGEC, Walter and Eliza Hall Institute of Medical Research. The MAGEC is supported by the Australian Phenomics Network (APN) and the APN is supported by the Australian government through the National Collaborative Research Infrastructure Strategy program. We thank the NIH tetramer facility (Atlanta, GA, United States) for production of the I-Ab-PEPCK335–351 tetramer used to detect L. donovani PEPCK-specific CD4+ T cells in these studies. This work was made possible through Queensland State Government funding and grants and fellowships from the National Health and Medical Research Council of Australia (NHMRC; grant numbers 1037304, 1058685, 1078671, 1132519, 1132975 and 1154265). Funding was also provided through a National Institutes of Health Tropical Medicine Research Centre (TMRC) grant (U19 AI074321), as well as Australian post-graduate awards through Griffith University’s Institute of Glycomics and School of Natural Sciences to P.T.B. and S.S.N., respectively; a Dr. Mildred Scheel Stiftung für Krebsforschung scholarship from Deutsche Krebshilfe to M.B.; and an INSPIRE Faculty grant (LSBM-109/IF-14) provided by the Indian government Department of Science and Technology (DST), Banaras Hindu University and University Grants Commission (M-14–70) to R.K. B.S. was supported by a Junior Research Fellowship from the Indian Council of Medical Research.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41590-020-0758-6.
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41590-020-0758-6.
Supplementary information is available for this paper at https://doi.org/10.1038/s41590-020-0758-6.
Peer review information Zoltan Fehervari was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Data availability
The materials, data and any associated protocols that support the findings of this study are available from the corresponding author upon request. The RNA-seq and microarray data have been deposited in the NCBI Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/) with the accession codes GSE135965 (human RNA-seq data) and GSE135857 (mouse microarray data). The list of DEGs in CD4+ T cells isolated from PBMCs of patients with visceral leishmaniasis is available in Supplementary Table 1. Supplementary Table 2 contains the list of DEGs in liver CD4+ T cells of L. donovani-infected mice. Supplementary Table 3 contains the list of DEGs in splenic CD4+ T cells of L. donovani-infected mice.
References
- 1.Netea MG et al. A guiding map for inflammation. Nat. Immunol 18, 826–831 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trapani JA & Smyth MJ Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol 2, 735–747 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Crawford A et al. Molecular and transcriptional basis of CD4+ T cell dysfunction during chronic infection. Immunity 40, 289–302 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Drake CG & Pardoll DM Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wherry EJ et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Schett G & Neurath MF Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat. Commun 9, 3261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tubo NJ & Jenkins MK CD4+ T cells: guardians of the phagosome. Clin. Microbiol. Rev 27, 200–213 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engwerda CR, Ato M & Kaye PM Macrophages, pathology and parasite persistence in experimental visceral leishmaniasis. Trends Parasitol 20, 524–530 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Miller LH, Baruch DI, Marsh K & Doumbo OK The pathogenic basis of malaria. Nature 415, 673–679 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Montes de Oca M et al. Blimp-1-dependent IL-10 production by Tr1 cells regulates TNF-mediated tissue pathology. PLoS Pathog 12, e1005398 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bald T et al. Immune cell–poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov 4, 674–687 (2014). [DOI] [PubMed] [Google Scholar]
- 12.O’Donnell JS, Teng MWL & Smyth MJ Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol 16, 151–167 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Ayers M et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest 127, 2930–2940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fairfax BP et al. Peripheral CD8+ T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat. Med 26, 193–199 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribas A & Wolchok JD Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turman MA, Yabe T, McSherry C, Bach FH & Houchins JP Characterization of a novel gene (NKG7) on human chromosome 19 that is expressed in natural killer cells and T cells. Hum. Immunol 36, 34–40 (1993). [DOI] [PubMed] [Google Scholar]
- 17.Aschenbrenner D et al. An immunoregulatory and tissue-residency program modulated by c-MAF in human TH17 cells. Nat. Immunol 19, 1126–1136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engwerda CR & Kaye PM Organ-specific immune responses associated with infectious disease. Immunol. Today 21, 73–78 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Engwerda CR, Ng SS & Bunn PT The regulation of CD4+ T cell responses during protozoan infections. Front. Immunol 5, 498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jenner RG et al. The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc. Natl Acad. Sci. USA 106, 17876–17881 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muzumdar MD, Tasic B, Miyamichi K, Li L & Luo L A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Antignano F et al. Methyltransferase G9A regulates T cell differentiation during murine intestinal inflammation. J. Clin. Invest 124, 1945–1955 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stumhofer JS et al. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat. Immunol 8, 1363–1371 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Stern JJ, Oca MJ, Rubin BY, Anderson SL & Murray HW Role of L3T4+ and LyT-2+ cells in experimental visceral leishmaniasis. J. Immunol 140, 3971–3977 (1988). [PubMed] [Google Scholar]
- 25.Dickinson ME et al. High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butler NS et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat. Immunol 13, 188–195 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mou Z et al. Identification of broadly conserved cross-species protective Leishmania antigen and its responding CD4+ T cells. Sci. Transl. Med 7, 310ra167 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Engwerda C, Belnoue E, Gruner AC & Renia L Experimental models of cerebral malaria. Curr. Top. Microbiol. Immunol 297, 103–143 (2005). [PubMed] [Google Scholar]
- 29.Amante FH et al. Immune-mediated mechanisms of parasite tissue sequestration during experimental cerebral malaria. J. Immunol 185, 3632–3642 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Haque A et al. Granzyme B expression by CD8+ T cells is required for the development of experimental cerebral malaria. J. Immunol 186, 6148–6156 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Valencia-Hernandez AM et al. A natural peptide antigen within the Plasmodium ribosomal protein RPL6 confers liver TRM cell-mediated immunity against malaria in mice. Cell Host Microbe 27, 950–962.e7 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Lau LS et al. CD8+ T cells from a novel T cell receptor transgenic mouse induce liver-stage immunity that can be boosted by blood-stage infection in rodent malaria. PLoS Pathog 10, e1004135 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Betts MR et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods 281, 65–78 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Grulich AE, van Leeuwen MT, Falster MO & Vajdic CM Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet 370, 59–67 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Cursons J et al. A gene signature predicting natural killer cell infiltration and improved survival in melanoma patients. Cancer Immunol. Res 7, 1162–1174 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Smyth MJ, Kelly JM, Baxter AG, Korner H & Sedgwick JD An essential role for tumor necrosis factor in natural killer cell-mediated tumor rejection in the peritoneum. J. Exp. Med 188, 1611–1619 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smyth MJ, Crowe NY & Godfrey DI NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int. Immunol 13, 459–463 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Hayakawa Y & Smyth MJ CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J. Immunol 176, 1517–1524 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Street SE, Cretney E & Smyth MJ Perforin and interferon-γ activities independently control tumor initiation, growth, and metastasis. Blood 97, 192–197 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Guillerey C, Huntington ND & Smyth MJ Targeting natural killer cells in cancer immunotherapy. Nat. Immunol 17, 1025–1036 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Cheuk S et al. CD49a expression defines tissue-resident CD8+ T cells poised for cytotoxic function in human skin. Immunity 46, 287–300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Medley QG et al. Characterization of GMP-17, a granule membrane protein that moves to the plasma membrane of natural killer cells following target cell recognition. Proc. Natl Acad. Sci. USA 93, 685–689 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krzewski K, Gil-Krzewska A, Nguyen V, Peruzzi G & Coligan JE LAMP1/CD107a is required for efficient perforin delivery to lytic granules and NK-cell cytotoxicity. Blood 121, 4672–4683 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moreira-Teixeira L et al. Mouse transcriptome reveals potential signatures of protection and pathogenesis in human tuberculosis. Nat. Immunol 21, 464–476 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karwacz K et al. Critical role of IRF1 and BATF in forming chromatin landscape during type 1 regulatory cell differentiation. Nat. Immunol 18, 412–421 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mombaerts P et al. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877 (1992). [DOI] [PubMed] [Google Scholar]
- 47.Song J et al. A mouse model for the human pathogen Salmonella typhi. Cell Host Microbe 8, 369–376 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang H et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hochheiser K, Kueh AJ, Gebhardt T & Herold MJ CRISPR/Cas9: a tool for immunological research. Eur. J. Immunol 48, 576–583 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Bradley DJ & Kirkley J Regulation of Leishmania populations within the host. I. The variable course of Leishmania donovani infections in mice. Clin. Exp. Immunol 30, 119–129 (1977). [PMC free article] [PubMed] [Google Scholar]
- 51.Franke-Fayard B et al. Murine malaria parasite sequestration: CD36 is the major receptor, but cerebral pathology is unlinked to sequestration. Proc. Natl Acad. Sci. USA 102, 11468–11473 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Putz EM et al. NK cell heparanase controls tumor invasion and immune surveillance. J. Clin. Invest 127, 2777–2788 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Markey KA, Gartlan KH, Kuns RD, MacDonald KP & Hill GR Imaging the immunological synapse between dendritic cells and T cells. J. Immunol. Methods 423, 40–44 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Du P, Kibbe WA & Lin SM Lumi: a pipeline for processing Illumina microarray. Bioinformatics 24, 1547–1548 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Ritchie ME et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmittgen TD, Lee EJ & Jiang J High-throughput real-time PCR. Methods Mol. Biol 429, 89–98 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Gabrysova L et al. c-Maf controls immune responses by regulating disease-specific gene networks and repressing IL-2 in CD4+ T cells. Nat. Immunol 19, 497–507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chow M et al. NLRP3 suppresses NK cell–mediated responses to carcinogen-induced tumors and metastases. Cancer Res 72, 5721–5732 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Hayakawa Y et al. Cutting edge: tumor rejection mediated by NKG2D receptor–ligand interaction is dependent upon perforin. J. Immunol 169, 5377–5381 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Ferrari de Andrade L et al. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res 74, 7298–7308 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Gao Y et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol 18, 1004–1015 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Young A et al. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell 30, 391–403 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Yang J et al. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wagner AH et al. DGIdb 2.0: mining clinically relevant drug–gene interactions. Nucleic Acids Res 44, D1036–D1044 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eppig JT, Motenko H, Richardson JE, Richards-Smith B & Smith CL The International Mouse Strain Resource (IMSR): cataloging worldwide mouse and ES cell line resources. Mamm. Genome 26, 448–455 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ashburner M et al. Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang da W, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 70.Colaprico A et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 44, e71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The materials, data and any associated protocols that support the findings of this study are available from the corresponding author upon request. The RNA-seq and microarray data have been deposited in the NCBI Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/) with the accession codes GSE135965 (human RNA-seq data) and GSE135857 (mouse microarray data). The list of DEGs in CD4+ T cells isolated from PBMCs of patients with visceral leishmaniasis is available in Supplementary Table 1. Supplementary Table 2 contains the list of DEGs in liver CD4+ T cells of L. donovani-infected mice. Supplementary Table 3 contains the list of DEGs in splenic CD4+ T cells of L. donovani-infected mice.