

Abstract
BACKGROUND:
Bilateral infarcts confined to the globus pallidus are unusual and occur in conjunction with only a few disorders, including isolated methylmalonic acidemia, a heterogeneous inborn error of metabolism. On the basis of neuroradiographic features of metabolic strokes observed in a large cohort of patients with methylmalonic acidemia, we have devised a staging system for methylmalonic acidemia–related globus pallidus infarcts.
MATERIALS AND METHODS:
Forty patients with isolated methylmalonic acidemia and neurologic symptoms underwent clinical brain MR imaging studies, which included 3D-T1WI. Infarcted globus pallidus segments were neuroanatomically characterized, and infarct volumes were measured.
RESULTS:
Globus pallidus infarcts were present in 19 patients; all were bilateral, and most were left-dominant. A neuroanatomic scoring system based on the infarct patterns was devised; this revealed a 5-stage hierarchical susceptibility to metabolic infarct, with the posterior portion of the globus pallidus externa being the most vulnerable. Globus pallidus infarct prevalence by methylmalonic acidemia class was the following: cblA (5/7, 71%), cblB (3/7, 43%), muto (10/22, 45%), and mut- (1/4, 25%). Tiny lacunar infarcts in the pars reticulata of the substantia nigra, previously unrecognized in methylmalonic acidemia, were found in 17 patients, 13 of whom also had a globus pallidus infarct.
CONCLUSIONS:
The staged pattern of globus pallidus infarcts in isolated methylmalonic acidemia suggests a nonuniform, regionally specific cellular susceptibility to metabolic injury, even for patients having milder biochemical phenotypes. In support of this hypothesis, the delineation of lacunar infarcts in the pars reticulata of the substantia nigra, a tissue functionally and histologically identical to the globus pallidus interna, supports the concept of cell-specific pathology.
Isolated methylmalonic acidemia (MMA) is a group of autosomal recessive inborn errors of metabolism caused by impaired activity of methylmalonyl-coenzyme A mutase (MUT).1–4 This cobalamin-dependent enzyme isomerizes methylmalonyl-coenzyme A into succinyl-coenzyme A, a critical Krebs cycle intermediate. Mutations in the gene encoding MUT result in an inactive (muto) or partially active (mut-)3,5 enzyme, the most common cause of isolated MMA. Impaired biosynthesis and transfer of the cofactor 5′-deoxyadenosylcobalamin to the MUT enzyme (cblA and cblB) or defective transport of cobalamin into the mitochondrion (cblD) also impairs the action of the MUT enzyme. These defects result in massive accumulation of methylmalonic acid and propionyl-coenzyme A–derived metabolites such as 2-methylcitrate.6
MMA treatment strategies vary4,7,8 but generally involve adherence to a low-protein diet to restrict the intake of amino acids that are oxidized to propionyl-coenzyme A (isoleucine, valine, threonine, methionine), hydroxocobalamin injections for cobalamin-responsive patients, carnitine supplementation to relieve acyl-coenzyme A accretion in the mitochondrion, and a high-energy diet to prevent catabolism. Despite vigilant dietary and metabolic management, patients with MMA experience multisystem disease, including metabolic instability, pancreatitis,9 renal failure,9,10 intellectual impairment,9,11 growth retardation,9 optic nerve atrophy,9 and acute injury to the globus pallidus.12–22 Some patients eventually require liver and/or kidney transplantation.
Most globus pallidus (GP) strokes are unilateral lacunar infarcts resulting from thromboembolic occlusion of a lenticulostriate artery; therefore, their margins reflect the vascular territory rather than the anatomic boundaries of the GP. In contrast, metabolic GP infarcts are typically bilateral, and infarct margins are constrained by neuroanatomic boundaries. Diseases that can cause GP infarction, with or without infarction of other basal ganglia, include propionic acidemia, some variants of Leigh disease, carbon monoxide poisoning, and cyanide poisoning.15 Isolated bilateral GP infarcts, never involving other structures, have been reported only in MMA. GP injury is a well-known complication of isolated MMA, classically occurring during severe decompensation or acute illness, and can result in an incapacitating movement disorder characterized by choreoathetosis and dystonia.15,16 The largest previously reported series that counted GP infarcts in patients with isolated MMA revealed GP infarcts in 4 of 23 patients (17%).9
In this article, we describe the patterns of the GP infarcts found in patients with isolated MMA and further characterize the GP infarcts by volumetric measurements. Our analyses revealed that the lesions follow a characteristic progression; therefore, we propose a staging system for MMA-related GP infarcts. We also established that patients with certain classes of isolated MMA are more likely to develop GP infarcts than others. Furthermore, we identified a novel imaging finding suggesting small defects in the cerebral peduncles, which we propose are produced by the same toxic or metabolic mechanism as the GP infarcts, also affecting the histologically23 and functionally24 identical cells that compose the pars reticulata of the substantia nigra (SNr).
Materials and Methods
Patient studies were approved by the institutional review board of our institution and were performed in compliance with the Helsinki Declaration. Patients were evaluated between 2004 and 2013. There was no recruitment strategy on the basis of race, sex, nutritional status, or disease severity. Long-term care of these patients was managed by their referring physicians.
During the period covered by this analysis, 64 patients having a variant of isolated MMA were evaluated by our clinic. All were classified by cellular enzymology and/or molecular genetics. Many patients had neurologic symptoms including movement disorders, hearing impairment, visual loss, and developmental delay.11 Neurologic evaluation (performed by a neurologist familiar with MMA) was particularly directed toward detecting signs of a GP stroke, such as movement disorders, weakness, and asymmetric motor findings. Many patients had follow-up neurologic examinations to check for progression and to detect strokes occurring in the absence of a sentinel clinical event.
Because our protocol did not allow MR imaging solely for research purposes, patients were selected for MR imaging on the basis of clinical indications. The most common reasons for MR imaging were for work-up of a movement disorder, for progression of symptoms, and as part of the initial diagnosis. Follow-up imaging was also performed for clinical indications, and 10 patients had ≥1 follow-up MR imaging during the study period. There were 25 patients (younger children and some adults with movement disorders) who required sedation with propofol. Patients scanned at our institution had a basic clinical brain MR imaging without gadolinium-based contrast material. MRIs performed at our institution all included 3D-T1WI (mostly MPRAGE, some 3D–fast-field echo). In addition to 3D-T1WI, some examinations included high-resolution balanced fast-field echo (bFFE) images. At our institution, MRIs were performed on a 3T Achieva (Philips Healthcare, Best, the Netherlands) scanner by using a sensitivity encoding head coil. Parameters used were the following: for MPRAGE, TE = 6.6 ms, TR = 11.6 ms, flip angle = 6°, FOV = 220 mm, acquisition matrix = 256 × 131, reconstruction matrix = 256 × 256, section thickness = 1.0 mm, NEX = 2; for 3D–fast-field echo, TE = 2.2 ms, TR = 5.0 ms, flip angle = 15°, FOV = 240 mm, acquisition matrix = 256 × 244, reconstruction matrix = 256 × 256, section thickness = 1.0 mm, NEX = 1; for bFFE, TE = 2.1 ms, TR = 5.1 ms, flip angle = 45°, FOV = 220 mm, acquisition matrix = 384 × 367, reconstruction matrix = 384 × 384, section thickness = 1.0 mm, NEX = 1. Six patients had scans performed outside our institution; these images were obtained with a variety of scan protocols.
MR imaging scans were reviewed by a single neuroradiologist to determine whether GP infarcts were present. Putative infarcts were carefully assessed to distinguish small infarcts from prominent perivascular spaces, a nonpathologic phenomenon that can also occur in the basal ganglia with a similar appearance on MR imaging. Perivascular spaces have a linear or branching appearance when examined in 3D and can traverse anatomic boundaries; metabolic infarcts are rounded or crescent and are confined by the anatomic boundaries of the GP. bFFE images are particularly helpful in clarifying the difference. If an infarct was present, the involved segments of the GP were recorded. Images were also evaluated for potential lacunar infarcts in the SNr; the number and location of SNr infarcts were recorded. Again, findings having a linear or branching appearance that would indicate a prominent perivascular space rather than an infarct were excluded.
MPRAGE or 3D–fast-field echo images of GP infarcts were exported to the MIPAV image analysis software (National Institutes of Health, Bethesda, Maryland; http://mipav.cit.nih.gov).25 A gray-level tracking tool was used for automated detection of infarct margins; gray-level tracking starts from a seed pixel and attempts to find all connected pixels having the same signal intensity, interpolating when necessary. The resulting boundary contours were manually edited as needed to remove (or add) pixels included (or omitted) due to issues such as image noise and artifacts. Volumes enclosed by the boundary contours were then calculated. Measurements of SNr infarct volumes were not attempted due to their small size relative to the image resolution.
Results
Demographics
Forty patients (25 males, 15 females) with isolated MMA had MR imaging scans. The mean age at the first scan as part of our study was 17 years (median, 18 years; range, 2 weeks to 35.5 years). Of these patients, 19 (48%) were found to have GP infarcts, 6 of which had been present at the time of diagnosis. Treatments of the patients whose infarcts were discovered subsequent to diagnosis included diet modification (n = 13), carnitine supplements (n = 13), hydroxocobalamin injection (n = 6), liver and kidney transplantation (n = 5), and kidney transplantation (n = 4).
GP infarct prevalence by MMA class was as follows: cblA (5/7, 71%), cblB (3/7, 43%), muto (10/22, 45%), and mut- (1/4, 25%). If both subtypes of the mut class were taken together, the rate for all patients with mut was 11/26 (42%). Lacunar SNr infarcts were detected in 17 patients. Thirteen patients had both GP and SNr infarcts, while 4 patients had only SNr infarcts and another 6 had only GP infarcts. Of particular interest, 1 patient initially had only SNr infarcts but subsequently developed GP infarcts. This patient was re-imaged at an interval of 5 years due to clinical progression. The other 9 patients who had follow-up imaging during the time frame of the study demonstrated no discernible changes in their GP. Infarct demographics are summarized in the Table.
Demographics of infarcts in isolated MMAa
| MMA Class | No. | GP Infarct | SNr Infarct | GP+SNr Infarct | Isolated GP Infarct | Isolated SNr Infarct |
|---|---|---|---|---|---|---|
| cblA | 7 | 5 (71%) | 5 (71%) | 4 | 1 | 1 |
| cblB | 7 | 3 (43%) | 3 (43%) | 2 | 1 | 1 |
| All mut subtypes | 26 | 11 (42%) | 9 (34%) | 7 | 4 | 2 |
| mut- | 4 | 1 (25%) | 0 (0%) | 0 | 1 | 0 |
| muto | 22 | 10 (45%) | 9 (41%) | 7 | 3 | 2 |
| All patients | 40 | 19 (48%) | 17 (43%) | 13 | 6 | 4 |
Of 40 patients scanned, 16 had both 3D-T1WI and bFFE images (most sensitive for detection of SNr infarcts), 22 had 3D-T1WI without bFFE images (less sensitive), and 2 had only 2D-T1WI (least sensitive). SNr infarcts were detected at a higher rate among patients with bFFE images (56%) than those without them (36%); therefore, SNr infarcts are probably under-reported.
Laterality of Infarcts
All GP infarcts were bilateral. It is presumed that the infarcts were synchronous, as illustrated by the example in Fig 1. However, most of the GP infarcts were not symmetrical (Fig 2). A laterality index was computed as (R − L) / (R + L); thus, right-dominant cases are represented by positive numbers; left-dominant cases, by negative numbers; and symmetrical cases, by values near zero. Most GP infarcts (12/19) were left-dominant, 5 were nearly symmetrical (within ±10%), and 2 were right-dominant. The range of laterality indexes was −0.47 to +0.48, the mean was −0.13, and the median was −0.17.
Fig 1.

Progression of a GP infarct. This 16-month-old patient was known to have MMA due to the cblB defect that had been diagnosed 6 months previously. Within 24 hours after onset of symptoms from gastroenteritis, she became lethargic and was taken to the emergency department. A, CT at the time of admission to the hospital appears to have normal findings. B, CT scan 35 hours later shows distinct hypoattenuating abnormalities involving the entirety of each GP. DWI performed 1 week later showed restricted diffusion in each GP (image not available). C, High-resolution MPRAGE image and D, T2-weighted image (b=0 image from DTI) acquired 7.5 years later demonstrate bilateral complete GP infarcts.
Fig 2.

Laterality of GP infarcts. A, Most GP infarcts were left-dominant. A few were nearly symmetrical (5 were within ±10%), and 2 were right-dominant. The average laterality index was −0.13, and the median was −0.17. B, An example of a left-dominant infarct. The laterality index in this case is −0.20.
In most patients (10/17), SNr infarcts were bilateral; SNr infarcts were detected only on the right in 5 patients and only on the left in 2 patients. SNr infarct sizes were not necessarily symmetrical, but there did not appear to be systematic lateralization, either in size or number. Patients had ≤3 infarcts per side. The total count of SNr infarcts was 17 per side.
GP Infarct Volumes
GP infarct volumes ranged from 9 to 1021 μL (Fig 3). Although the ranges overlapped, volumes for mut variants (mut- and muto) were smaller on average than volumes for cblA and cblB. Application of a t test suggested that there is a statistically significant difference in infarct size between the 2 mut subtypes together, the 2 cbl variants together (P = .01), and the muto subtype alone versus the 2 cbl classes together (P = .02). There was a statistically significant difference between the mut- subtype alone and each of the other categories (P = .01 versus muto, P < .01 versus cblA, and P = .02 versus cblB). A paired t test showed statistically significant lateralization within the cblA class (P = .04).
Fig 3.

Volume of GP infarcts. The range of GP infarct volumes is 9–1021 μL. Laterality of the infarcts is apparent when infarcts are segregated into left and right columns for each of the MMA classes. Group means (indicated by the heavy bar) differ between classes. Application of a t test suggests that there is a statistically significant difference in infarct size between the 2 mut subtypes together (mut- and muto), the 2 cbl classes together (cblA and cblB) (P = .01), and the muto subtype alone versus the 2 cbl classes together (P = .02). There was a statistically significant difference between the mut- subtype alone and each of the other categories (P = .01 versus muto, P < .01 versus cblA, and P = .02 versus cblB). There was a statistically significant difference between the left and right for the cblA class (P = .04).
GP Infarct Staging
On review of the GP infarcts as a group, it became apparent that the infarct pattern was not random and that some segments of the GP infarcted more often than others. The 5 infarct-delineated segments are shown in Fig 4. There appear to be 3 distinct segments in the globus pallidus externa (GPe) and 2 distinct segments in the globus pallidus interna (GPi). When the affected segments were all displayed as a group (Fig 5), it appeared that the segments were affected sequentially, with a few exceptions (5 of 38 infarcts in 3 of 19 patients). This observation suggested a staging system, in which the infarct stage is determined by counting the number of affected segments. According to this staging system, we observed 10 stage 1 infarcts, 7 stage 2 infarcts, 7 stage 3 infarcts, 3 stage 4 infarcts, and 11 stage 5 infarcts. In 3 patients, the right and left stages were discordant.
Fig 4.

GP infarct segments. A, Normal anatomy of the dorsal pallidum, also known as the globus pallidus. The thin line indicates the medial medullary lamina; the thick line, the boundary of the globus pallidus. Surrounding structures include the anterior limb of the internal capsule (IC ant) and the posterior limb of the internal capsule (IC post). The putamen, thalamus, and head of the caudate nucleus are also labeled. The external medullary lamina separates the putamen from the GPe. B, Infarct segments 1–5. Segment 1 seems to be the most sensitive to metabolic infarct in MMA, followed by segment 2, then segment 3; segments 4 and 5 are the least sensitive and usually infarct together. C, An example of a segment 1 infarct (posterior GPe). D, An example of an infarct of segments 1 + 2 (posterior and middle GPe). E, An example of an infarct of segments 1 + 2 + 3 (whole GPe). F, An example of an infarct of segments 1 + 2 +3 + 4 + 5 (complete GP, both externa and interna). Note that the medial medullary lamina (a white matter tract) remains intact and can be seen separating the GPe and GPi.
Fig 5.
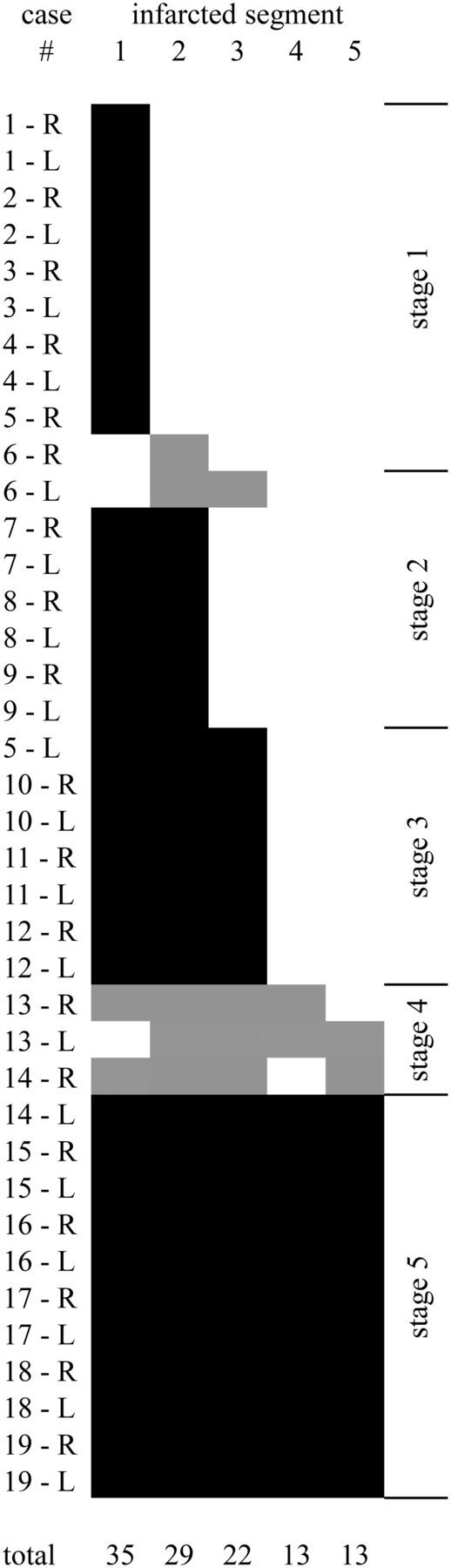
Graphic depiction of all 38 GP infarcts. Infarcted segments are shown in black (follows the rule) or gray (exception to the rule). The segments appear to follow a rule in which the likelihood of infarct is 1 > 2 > 3 > 4 = 5. Five infarcts (in 3 different patients) do not follow the segment-ordering rule. Infarcts can be staged by counting the number of infarcted segments, regardless of the order. In 16 of 19 patients, right and left infarcts are of the same stage, even if the volumes differ (as described in Fig 2). The location of the segments is shown in Fig 4.
The GPe appeared to be more vulnerable to metabolic infarct than the GPi. In fact, only 1 infarct had any part of the GPi involved without involving the entire GPe. Within the GPe, vulnerability appeared highest in the most posterior segment and lowest in the most anterior segment, though there were 4 exceptions to that pattern. GPi vulnerability would seem to be fairly homogeneous, though there were 2 exceptions in which only half of the GPi was infarcted; in one case, only the posterior half was involved, and in the other case, only the anterior half was involved.
Distribution of SNr Infarcts
We observed that SNr infarcts were not randomly distributed but segregated into 3 distinct locations, which we have named according to their relative positions, as shown in Fig 6. The anterior location was most common (20 infarcts). There were 9 infarcts in the posterior location and 5 infarcts in the middle location. Unlike the GP lesions, there was no clear hierarchy of SNr infarct locations. Among patients with cblA, 6 of 7 SNr infarcts were in the anterior location, but there did not seem to be a preferred location for any other type of MMA. Location and laterality of the SNr infarcts did not appear to correlate with GP infarct volume, stage, or laterality.
Fig 6.

Lacunar infarcts of the SNr. Due to the small size of the cell clusters that form the SNr (near or below the lower limit of MR imaging resolution), detection of these infarcts can be difficult. A, Tiny bilateral infarcts of the SNr are seen on a bFFE image. The resolution of this image is 0.6 × 0.6 × 1.0 mm per voxel. The infarcts measure approximately 1 mm. Both are found in the anterior location. B, Slightly larger bilateral infarcts of the SNr are seen on an MPRAGE image with a resolution of 0.9 × 0.9 × 1.0 mm per voxel. These infarcts are in the middle location. C, Bilateral infarcts at 2 sites (anterior and posterior) in the SNr are seen on an MPRAGE image. SNr infarcts were found in only 3 distinct locations, which we are designating by their relative positions; the anterior location was the most common (20 infarcts), followed by the posterior location (9 infarcts). The middle location was the least common (5 infarcts).
The demographics of SNr infarcts relative to GP infarcts does not suggest a clear difference between these locations in sensitivity to the toxic agent because we saw 4 cases of patients who had isolated SNr infarcts, 6 who had isolated GP infarcts, and 13 who had both types of infarcts. Examination of the demographics of the SNr infarcts (Table) demonstrated that the prevalence of SNr infarcts in each MMA class is similar to the prevalence of GP infarcts.
Detection of SNr Infarcts
We observed a difference in sensitivity for the detection of SNr infarcts related to the type of imaging performed. Among patients for whom bFFE images were available, SNr infarcts were detected in 9 of 16 (56%). However, among patients who had only 3D-T1WI, SNr infarcts were detected in 8 of 22 (36%). No SNr infarcts were detected for the 2 patients scanned outside our institution without any 3D imaging sequences. When both types of images were available, no SNr infarcts were detected on the 3D-T1WI that were not also detected on the bFFE images. Availability of bFFE images was determined by the date of the examination, rather than by any patient-related factors because this sequence was added to our scan protocol partway through the study. We expect that had the bFFE images been obtained in all patients, more SNr infarcts would have been detected.
Discussion
The occurrence of bilateral isolated infarcts of the GP in patients who have isolated MMA has been previously reported.12–21 However, the analysis presented here is the first systematic study of GP imaging findings in a large group of patients with MMA, to our knowledge. This analysis also identified the previously unrecognized association of lacunar infarcts in the SNr with isolated MMA.
The staged pattern of MMA-related infarcts in the GP suggests that there are groups of cells in the GP that have differing vulnerability to metabolic stress and/or toxic metabolites. If there are neurons in certain locations in the GP and SNr that exist in a more tenuous balance between energy demands and blood supply, then this feature may also contribute to the staged pattern we observed. It is certainly possible that energy demands vary across the GP because the nuclei demonstrate somatotopic organization26; therefore, some neurons within these nuclei are more frequently or intensely active than others. In humans and other primates, the SNr is functionally part of the GPi and is composed of histologically identical cells.24 In most nonprimate mammals, the GPi is not located adjacent to the GPe but rather exists as a separate nucleus called the “entopeduncular nucleus” that is embedded within the internal capsule.24
In the initial analyses of GP infarcts in our patient cohort, we noted what appeared to be small defects in the cerebral peduncles in the vicinity of the substantia nigra. These changes were initially interpreted as secondary degeneration of the substantia nigra, as has been reported in cases of patients with vascular-origin strokes obliterating the ipsilateral basal ganglia.27,28 However, careful review revealed that this phenomenon occurs even in patients who have no GP infarcts; thus, this finding could not represent secondary degeneration. Given that cells of the SNr are histologically and functionally identical to cells of the GPi, it seems logical that they are susceptible to the same toxic and metabolic phenomena; therefore, we propose that these tiny lesions represent infarcts. While we lack histologic evidence that the lesions are located in the pars reticulata of the substantia nigra, as opposed to the pars compacta of the substantia nigra, the SNr location is consistent with a toxic/metabolic mechanism model due to the histologic and functional similarity to the GPi that is lacking in the dopaminergic portion of the substantia nigra (the pars compacta). There is 1 case report of isolated bilateral SNr lacunar infarcts (of unknown cause) in the literature.29 It confirmed histologically that the lacunar infarct was in the SNr; the location of the infarct in the case report encompasses the location that we have described as “anterior SNr” in Fig 6.
The relative sensitivity of GPe and GPi segments to toxicity in MMA seems fairly clear from the results presented in this report, but the relative sensitivity of the SNr remains less certain. One possible scenario is that the SNr is more sensitive to toxic injury in MMA than is the GPe and that few GPe infarcts occur without a preceding (or synchronous) SNr infarct, but that our detection of SNr infarcts is poor due to contrast and resolution considerations. If this scenario is accurate, then acquiring even higher resolution bFFE images of the midbrain in future patients with MMA should reveal additional SNr infarcts and should demonstrate that patients with GP infarcts are a subset of the patients with SNr infarcts. Another possible scenario would be that the sensitivity of the SNr to injury in MMA may actually be different from that in the GP; if so, the pattern we found in the current study should hold up even if higher resolution images are acquired in the future.
Given the somatotopy of the GP, one would expect that selective infarction of portions of the GPe and GPi would manifest neurologic symptoms that are specific to the site of the infarct. Experimental lesion studies in African green monkeys30,31 demonstrated that lesions in the posterior GPe produce dyskinesia, lesions in the middle GPe produce attention deficit disorder, and anterior-inferior GPe lesions produce stereotypical behaviors. This finding corresponds to the primary motor, supplementary motor, and limbic zones of the GPe. Somatotopy of the GPi is parallel to that in the GPe26 and ought to produce a similar profile of symptoms, except for the functions that are handled by the SNr. Isolated SNr infarcts ought to specifically produce orofacial motor symptoms. One might also expect that infarct volume and/or stage would correlate with overall symptom severity and that infarct laterality would correlate with lateralization of symptoms. The patients in our study underwent detailed neurologic examinations, and correlations between movement syndromes and infarct spatial patterns will be the subject of another article.
The laterality of GP infarct volumes is consistent with a previously reported asymmetric GP size in humans.32 In most people, the left GP is larger than the right, so our finding that most of the infarcts are larger on the left is consistent. Although we did not have enough left-handed patients with infarcts to determine whether preinfarct handedness had any influence on GP infarct laterality, we did observe 1 patient who appeared to change handedness (from right to left) following a large bilateral GP infarct (bilateral stage 5, right volume = 656 μL, left volume = 562 μL, laterality index = 0.08) and a tiny bilateral SNr infarct.
Among the various classes of isolated MMA, we found differences in both prevalence and size of GP infarcts and parallel differences in the prevalence of SNr infarcts. The relatively lower prevalence of both GP and SNr infarcts in patients with mut- compared with other variants suggests that partial activity of the MUT enzyme has a protective effect, likely because patients with mut- typically have lower concentrations of abnormal metabolites and less severe mitochondrial dysfunction.9 Although patients with cblA (and occasional patients with cblB) can be responsive to hydroxocobalamin supplementation, we found that their average stroke volume was significantly larger compared with patients with mut. The reasons for this finding are unclear, and it is possible that factors not directly related to the disease mechanism contribute. For example, the patients chosen for imaging in this study were selected, in part, on the basis of clinical suspicion of stroke, creating an ascertainment bias for more severe phenotypes in patients with normally milder biochemical defects (such as cblA); this choice would tend to skew our measurements toward larger and more symptomatic infarcts than we might have found had we imaged all patients being followed in our clinic. Another possibility would be that because so many of the infarcts were not associated with an abrupt change in symptoms, very few were detected in the acute phase; therefore, in most cases, we know neither the age of the patient at the time of the infarct nor the age of the infarct at the time of imaging. Potentially, the patients with cblA and cblB were older (and therefore larger) than the patients with muto at the time of the GP infarction because we know that some patients with cblA and cblB were not diagnosed with MMA until they presented in crisis later in childhood. If patients having different types of MMA typically have GP infarcts at different ages, when their brains are different sizes, there is the possibility that we would see a difference in infarct size even if the same fraction of the GP is infarcted, or perhaps infarcts imaged after a longer delay become larger as a result of atrophy of adjacent tissues. Normalizing the infarct volume as a fraction of the total GP volume could potentially correct some of these considerations, but defining the exact margins of the healthy portion of the GP, even manually, is difficult in practice due to the low contrast-to-noise ratio.
An additional limitation of this study is that we were unable to test for correlations relating infarct size, stage, and prevalence to factors such as prescribed treatments, compliance with treatment, and patient age (equivalent to duration of the disease). Unfortunately, the uncertainty in dating the infarcts precluded this type of analysis, and this uncertainty will remain an issue until presymptomatic and longitudinal brain imaging become part of the routine clinical management of MMA.
Conclusions
In summary, we examined patterns of GP infarction in patients with isolated MMA. We found a hierarchical pattern involving distinct segments of the GP, suggesting nonuniform sensitivity to the mechanism of infarct and suggesting a staging system. We found that the prevalence and size of the infarcts varied according to the class of isolated MMA and that the laterality of infarct volumes is similar to previously reported anatomic variation of the GP. We also noted small defects in the cerebral peduncles that we believe represent tiny lacunar infarcts in the SNr that are produced by the same toxic or metabolic mechanism as the GP infarcts but appear to arise as independent events.
Acknowledgments
We thank the MR imaging technologists who acquired the images for this project, particularly Mastaneh Owhadi, Betty Wise, and Bonita Damaska. We also thank the patients (and their families) who participated in this study, the National Human Genome Research Institute medical and biochemical genetics fellows who helped evaluate the patients, and the physicians who referred their patients to our study.
ABBREVIATIONS:
- bFFE
balanced fast-field echo
- GP
globus pallidus
- GPe
globus pallidus externa
- GPi
globus pallidus interna
- MMA
methylmalonic acidemia
- MUT
methylmalonyl-coenzyme A mutase
- SNr
pars reticulata of the substantia nigra
Footnotes
Disclosures: Andrea L. Gropman—RELATED: Fees for Participation in Review Activities such as Data Monitoring Boards, Statistical Analysis, Endpoint Committees, and the Like: I am on a Data and Safety Monitoring Board for a Neuronal Ceroid Lipofuscinosis study; UNRELATED: Consultancy: I do consulting work for GeneDX, for the epilepsies; Expert Testimony: I have done some medical-legal work, usually 1 case per year; Grants/Grants Pending: Subproject of a U54 was awarded to my institution, but the topic is neuroimaging in Urea Cycle Disorders, not MMA*; Payment for Lectures (including service on Speakers Bureaus): I was reimbursed for a Grand Rounds at Rutgers; Payment for Development of Educational Presentations: I developed a basic neuroimaging introduction course for Shire Human Genetic Therapies; Travel/Accommodations/Meeting Expenses Unrelated to Activities Listed: I presented at the American College of Medical Genetics on Imaging in Neurogenetics but did not talk about MMA.
This work was supported by the Intramural Research Program of the National Human Genome Research Institute (National Institutes of Health protocol 04-HG-0127 “Clinical and Basic Investigations of Methylmalonic Acidemia and Related Disorders,” clinicaltrials.gov identifier NCT00078078).
References
- 1. Manoli I, Venditti CP. Methylmalonic Acidemia. In: Pagon RA, Adam MP, Bird TD, et al., eds. GeneReviews. Seattle: University of Washington, Seattle; 2005 [Google Scholar]
- 2. Rosenberg LE, Lilljeqvist AC, Hsia YE. Methylmalonic aciduria: an inborn error leading to metabolic acidosis, long-chain ketonuria and intermittent hyperglycinemia. N Engl J Med 1968;278:1319–22 [DOI] [PubMed] [Google Scholar]
- 3. Matsui SM, Mahoney MJ, Rosenberg LE. The natural history of the inherited methylmalonic acidemias. N Engl J Med 1983;308:857–61 [DOI] [PubMed] [Google Scholar]
- 4. Fenton WA, Gravel RA, Rosenblatt DS. Disorders of Propionate and Methylmalonate Metabolism. New York: McGraw-Hill; 2013 [Google Scholar]
- 5. Willard HF, Rosenberg LE. Inherited methylmalonyl CoA mutase apoenzyme deficiency in human fibroblasts: evidence for allelic heterogeneity, genetic compounds, and codominant expression. J Clin Invest 1980;65:690–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ando T, Rasmussen K, Wright JM, et al. Isolation and identification of methylcitrate, a major metabolic product of propionate in patients with propionic acidemia. J Biol Chem 1972;247:2200–04 [PubMed] [Google Scholar]
- 7. Hauser NS, Manoli I, Graf JC, et al. Variable dietary management of methylmalonic acidemia: metabolic and energetic correlations. Am J Clin Nutr 2011;93:47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Touati G, Valayannopoulos V, Mention K, et al. Methylmalonic and propionic acidurias: management without or with a few supplements of specific amino acid mixture. J Inherit Metab Dis 2006;29:288–98 [DOI] [PubMed] [Google Scholar]
- 9. Hörster F, Baumgartner MR, Viardot C, et al. Long-term outcome in methylmalonic acidurias is influenced by the underlying defect (mut°, mut-, cblA, cblB). Pediatr Res 2007;62:225–30 [DOI] [PubMed] [Google Scholar]
- 10. Kruszka PS, Manoli I, Sloan JL, et al. Renal growth in isolated methylmalonic acidemia. Genet Med 2013;15:990–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O'Shea CJ, Sloan JL, Wiggs EA, et al. Neurocognitive phenotype of isolated methylmalonic acidemia. Pediatrics 2012;129:e1541–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bindu PS, Kovoor JM, Christopher R. Teaching neuroimages: MRI in methylmalonic acidemia. Neurology 2010;74:e14. [DOI] [PubMed] [Google Scholar]
- 13. Campeau PM, Valayannopoulos V, Touati G, et al. Management of West syndrome in a patient with methylmalonic aciduria. J Child Neurol 2010;25:94–97 [DOI] [PubMed] [Google Scholar]
- 14. de Sousa C, Piesowicz AT, Brett EM, et al. Focal changes in the globi pallidi associated with neurological dysfunction in methylmalonic acidaemia. Neuropediatrics 1989;20:199–201 [DOI] [PubMed] [Google Scholar]
- 15. Heidenreich R, Natowicz M, Hainline BE, et al. Acute extrapyramidal syndrome in methylmalonic acidemia: “metabolic stroke” involving the globus pallidus. J Pediatr 1988;113:1022–27 [DOI] [PubMed] [Google Scholar]
- 16. Korf B, Wallman JK, Levy HL. Bilateral lucency of the globus pallidus complicating methylmalonic acidemia. Ann Neurol 1986;20:364–66 [DOI] [PubMed] [Google Scholar]
- 17. Larnaout A, Mongalgi MA, Kaabachi N, et al. Methylmalonic acidaemia with bilateral globus pallidus involvement: a neuropathological study. J Inherit Metab Dis 1998;21:639–44 [DOI] [PubMed] [Google Scholar]
- 18. Michel SJ, Given CA 2nd, Robertson WC Jr. Imaging of the brain, including diffusion-weighted imaging in methylmalonic acidemia. Pediatr Radiol 2004;34:580–82 [DOI] [PubMed] [Google Scholar]
- 19. Roodhooft AM, Baumgartner ER, Martin JJ, et al. Symmetrical necrosis of the basal ganglia in methylmalonic acidaemia. Eur J Pediatr 1990;149:582–84 [DOI] [PubMed] [Google Scholar]
- 20. Trinh BC, Melhem ER, Barker PB. Multi-slice proton MR spectroscopy and diffusion-weighted imaging in methylmalonic acidemia: report of two cases and review of the literature. AJNR Am J Neuroradiol 2001;22:831–33 [PMC free article] [PubMed] [Google Scholar]
- 21. Yeşildağ A, Ayata A, Baykal B, et al. Magnetic resonance imaging and diffusion-weighted imaging in methylmalonic acidemia. Acta Radiol 2005;46:101–03 [DOI] [PubMed] [Google Scholar]
- 22. Işikay S, Temel L, Keskin M. Imaging findings associated with methylmalonic aciduria. Pediatr Neurol 2014;50:435–36 [DOI] [PubMed] [Google Scholar]
- 23. Sousa R, Hrinya Tannery N, Lafer EM. Two new monoclonal antibodies provide immunohistochemical evidence for the unique biochemical similarity of the mouse globus pallidus, entopeduncular nucleus and substantia nigra pars reticulata. Neuroscience 1990;34:403–10 [DOI] [PubMed] [Google Scholar]
- 24. Nambu A. Globus pallidus internal segment. Prog Brain Res 2007;160:135–50 [DOI] [PubMed] [Google Scholar]
- 25. McAuliffe M, Lalonde FM, McGarry D, et al. Medical image processing, analysis and visualization in clinical research. In: Proceedings of the 14th IEEE Symposium on Computer-Based Medical Systems, Washington, DC. July 26–27, 2001:381–86 [Google Scholar]
- 26. Nambu A. Somatotopic organization of the primate basal ganglia. Front Neuroanat 2011;5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forno LS. Reaction of the substantia nigra to massive basal ganglia infarction. Acta Neuropathol 1983;62:96–102 [DOI] [PubMed] [Google Scholar]
- 28. Nakane M, Teraoka A, Asato R, et al. Degeneration of the ipsilateral substantia nigra following cerebral infarction in the striatum. Stroke 1992;23:328–32 [DOI] [PubMed] [Google Scholar]
- 29. McKee AC, Levine DN, Kowall NW, et al. Peduncular hallucinosis associated with isolated infarction of the substantia nigra pars reticulata. Ann Neurol 1990;27:500–04 [DOI] [PubMed] [Google Scholar]
- 30. François C, Grabli D, McCairn K, et al. Behavioural disorders induced by external globus pallidus dysfunction in primates. II. Anatomical study. Brain 2004;127:2055–70 [DOI] [PubMed] [Google Scholar]
- 31. Grabli D, McCairn K, Hirsch EC, et al. Behavioural disorders induced by external globus pallidus dysfunction in primates. I. Behavioural study. Brain 2004;127:2039–54 [DOI] [PubMed] [Google Scholar]
- 32. Kooistra CA, Heilman KM. Motor dominance and lateral asymmetry of the globus pallidus. Neurology 1988;38:388–90 [DOI] [PubMed] [Google Scholar]