

Abstract
Objective
To study epigenetic patterns in T lymphocytes that accumulate in the rheumatoid arthritis (RA) synovium, we characterized DNA methylation of CD3+ T cells in peripheral blood and synovial tissue in patients with RA and osteoarthritis (OA).
Methods
Genomic DNA of CD3+ T cells was isolated from patients with RA (n = 8) and OA (n = 5) from blood or the synovium at the time of an arthroplasty using antibodies and magnetic beads. Methylation was measured by using the Illumina Infinium MethylationEPIC Kit. Differentially methylated loci (DML) and differentially methylated genes (DMGs) were identified by using Welch’s t‐test. Principal component analysis, hierarchical clustering, and pathway analysis were used to determine relationships among groups.
Results
When we compared DNA methylation of CD3+ T cells between peripheral blood and synovial tissue within each disease, 4615 and 164 DML were identified in RA and OA samples, respectively, resulting in 832 and 36 DMGs. A principal component analysis showed that methylation differences in T cells were greater on the basis of on location (blood vs synovium) rather than disease (RA vs OA). Differentially modified pathways were significantly enriched between RA blood and synovial T cells, especially in genes related to complement, integrin cell surface interactions, and the P53 pathway. The limited number of DMGs identified between OA blood and synovial T cells did not conform to biologic pathways.
Conclusion
The patterns of DNA methylation in RA show location‐specific differences related to immune pathways, whereas methylation differences in OA are limited. The RA joint‐specific signatures could be due to selective accumulation of T‐cell populations or expansion of differentially marked adaptive immune cells. Understanding epigenetic patterns could provide clues to the types of T cells that accumulate in the RA joint and identify potential therapeutic targets.
INTRODUCTION
Epigenetic alterations in various cell lineages have been identified in rheumatoid arthritis (RA) and could contribute to disease pathogenesis (1, 2). For example, the epigenetic landscape has been mapped in RA fibroblast‐like synoviocytes (FLS) and defined pathogenic pathways, such as Huntington's disease, as well as genes involved in cell migration, immune response, and matrix regulation (3). Other studies have also implicated immunologic and inflammatory pathways in RA (4, 5). Cell lineage–specific DNA methylation abnormalities in peripheral blood RA T lymphocytes have been described, mainly focused on CD4+ T cells, most notably, naive CD4+ T cells (6, 7). Of interest, peripheral CD4+ T cells share a limited subset of differential marks with RA synoviocytes (8). Unfractionated CD3+ blood T lymphocytes also display DNA methylation patterns that distinguish patients with RA from controls (9, 10). However, the relationship between synovial and peripheral blood cell epigenetic patterns in RA has not been studied. To determine the patterns in these two compartments, we characterized DNA methylation signatures of matched CD3+ T cells in peripheral blood and synovial tissue in patients with RA and OA.
PATIENTS AND METHODS
Patient phenotypes and sample collection
Synovial tissue was obtained at the time of joint replacement surgery and, when feasible, matched blood was also obtained. For the patients with RA, disease‐modifying drugs, such as methotrexate and tumor necrosis factor blockers, were discontinued approximately 1 month before surgery. Synovial tissue and/or blood was obtained at the arthroplasty with informed consent from patients with RA (synovial tissue: n = 8, with 3 matched peripheral blood samples and 1 unmatched peripheral blood sample) and OA (5 matched synovial tissue and peripheral blood samples). Supplementary Table 1 shows joint locations, demographics, and treatments where information was available. Tissue was disaggregated by using Liberase TL digestion, as previously described (11). T cells were isolated by using anti‐CD3 conjugated magnetic bead separation (EasySep CD3 Positive Selection Kit; STEMCELL Technologies Inc). The CD3+ population was selected instead of subpopulations to obtain sufficient numbers of cells and DNA from synovial tissue, especially in OA, in which fewer T cells infiltrate the tissue (CD3+ yield for tissues: RA, 1.1 ± 0.32 × 106; OA, 6 ± 1.4 × 104). RA diagnosis met the American College of Rheumatology 2010 criteria (12), and OA diagnosis met the American College of Rheumatology 1991 criteria (13).
BeadChip analysis
The DNA methylation level was measured by using the Infinium MethylationEPIC Kit (Illumina) chip and processed with the minfi package in R (R Foundation for Statistical Computing) (14). Data normalization was performed by using the wateRmelon package in R. CpGs with detection P values greater than 0.01 were filtered out. The methylation level was reported as a β value at each individual locus. The quality of the assay is shown in Supplementary Figure 1. The CD4/CD8 ratio was also calculated by using the minfi package in R.
Differentially methylated loci and differentially methylated gene identification
Differentially methylated loci (DML) were identified by using a Welch’s t‐test on filtered loci and corrected for multiple testing to q values. Loci with a difference in the average β value greater than 0.1 and a difference in the average q value less than 0.05 were considered DML. DML located in gene promoter regions (−2500 to 500 base pairs from transcription start site) were assigned to the corresponding genes, which were defined as differentially methylated genes (DMGs).
Principal component analysis, hierarchical clustering, and pathway analysis
A principal component analysis (PCA) was performed by using the irlba package in R to represent relationships among each group. Hierarchical clustering was applied using combined DMLs by the gplots package in R. To compare differentially methylated pathways, a pathway analysis was applied by using Reactome (15) and MSigDB (16).
RESULTS
Methylation signatures in peripheral blood and synovial T cells in RA and OA
With 862,498 filtered DNA methylation loci, an unbiased PCA showed that RA and OA peripheral blood CD3+ T cells clustered together and separated from synovial tissue (Figure 1A). The separation is more readily visualized by using only paired samples (Figure 1B). Although large variance spreads RA and OA synovial T cells from each other, the PCA shows that T‐cell location (synovium vs blood) was much greater than differences based on disease (RA vs OA). Broad overlap was observed between RA and OA peripheral blood T cells. However, separation between their respective synovial T‐cell methylation patterns was apparent.
Figure 1.
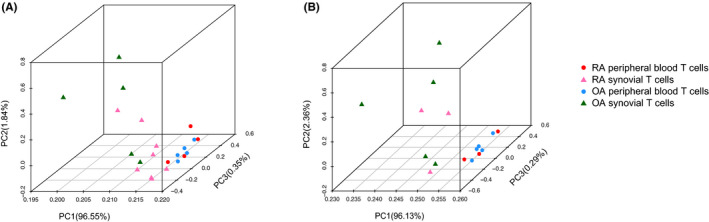
Principal component analysis (PCA) plot shows that the segregation of samples is more distinguishable between T‐cell location (synovium vs blood) rather than disease (rheumatoid arthritis [RA] vs osteoarthritis [OA]). A, PCA of RA and OA samples in peripheral blood and synovial samples with all loci. B, PCA plot the separation is more readily visualized by using only paired samples. PC, principal component
Comparison of peripheral blood to the synovium within each disease
To characterize DNA methylation signatures of peripheral blood and synovial CD3+ T cells in RA and OA, Welch’s t‐tests were performed on their respective methylomes. A total of 4615 DML were identified between peripheral blood and synovial CD3+ T cells in RA samples, and a PCA plot showed separation of the two groups (Figure 2A). After mapping the DML to gene promoter regions, 832 DMGs were identified. In contrast, comparing peripheral blood and synovial CD3+ T cells in OA samples revealed only 164 DML that mapped to 36 DMGs. Despite the small number of significant DML/DMGs, a PCA can still distinguish separation in the populations (Figure 2B). Thus, the differences between synovial and peripheral blood T cells is considerably greater in RA than in OA.
Figure 2.
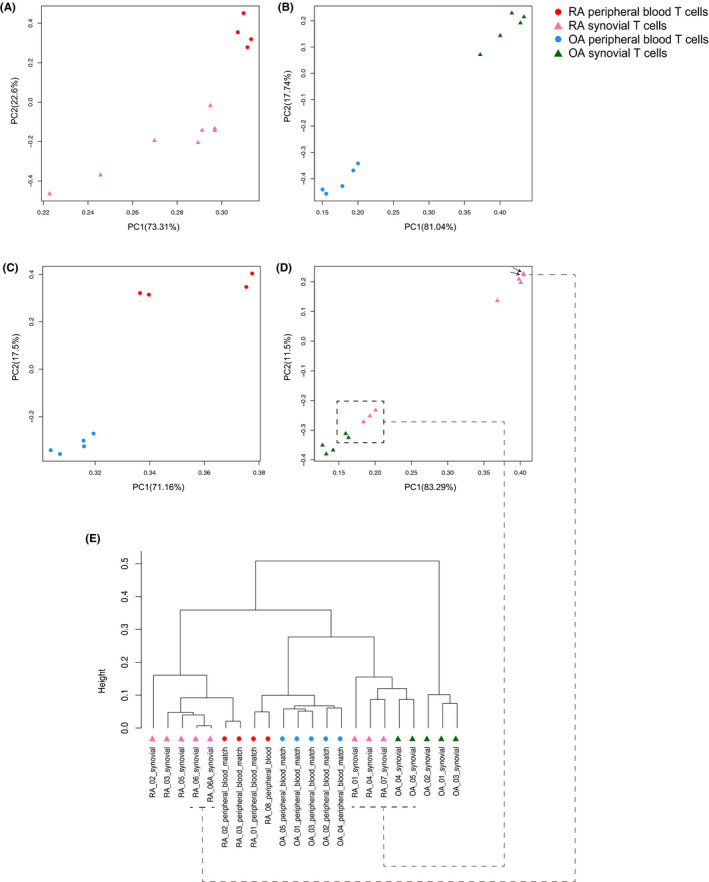
Principal component analysis (PCA) and hierarchical clustering of rheumatoid arthritis (RA) and osteoarthritis (OA) peripheral blood and synovial CD3+ T cells based on differentially methylated loci (DML). The separation between peripheral blood and synovial tissue is shown in RA (A) and OA (B) samples. Comparisons between RA and OA samples are shown in peripheral blood (C) and synovial tissue (D), with a less stringent cutoff. The arrows indicate the two arthroplasty samples from the same patient. E, Hierarchical clustering by using combined DML separates four categories of samples. Dashed lines connect samples in hierarchical clustering and the corresponding PCA plot. PC, principal component.
Comparison between RA and OA
We then evaluated the differences between RA and OA. Because of the low number of samples, a stringent q value of less than 0.05 only revealed limited numbers of DML in peripheral blood. Therefore, the analysis was repeated with a P value less than 0.05, and 1120 DML in 363 DMGs were found. For RA and OA synovial tissues and a P value less than 0.01, 4104 DML in 870 DMGs were identified. Figure 2C and D show the separation of RA and OA samples in peripheral blood and synovial CD3+ T cells with corresponding filters. Three RA synovial samples adjacent to two OA synovial samples in the PCA were from female patients, suggesting a possible sex difference in methylation (Figure 2D). However, similar segregation by sex was not observed in other comparisons and the three female samples still segregated from the OA samples (15,276 DML and 2657 DMGs; P < 0.05; see Supplementary Table 2). No difference in the CD4/CD8 ratio was identified between the groups could account for the observation (Supplementary Figures 2 and 3). Of interest, two RA synovial samples closest to each other in the PCA (Figure 2D, indicated by arrows) were from the same patient after knee arthroplasties eight months apart.
Comparison of peripheral blood and synovial T cells in RA and OA with combined DML
Hierarchical clustering was then performed on RA and OA peripheral blood and synovial samples by using combined DML from the pairwise studies. Figure 2E shows that the four types of samples are separated. The two matched RA peripheral blood samples clustered with their corresponding synovial samples and the three RA samples close to the OA samples in the PCA could still be distinguished. The other peripheral blood samples are grouped together with RA and OA samples separated in different branches. We also compared the DMGs of peripheral blood and synovial T cells between RA and OA to published data for cultured FLS (17) (Supplementary Figure 4). A limited number of DMGs between RA peripheral blood and synovial T cells were shared with RA FLS (14 and 30, respectively; see Supplementary Table 3). Therefore, the differences between peripheral blood and synovial T cells are less prominent than the differences between CD3+ T cells and cultured synoviocytes. Of interest, two DMGs, CHIC1 and KRTAP4‐7, overlapped in all three populations.
Differentially methylated pathways
To examine differentially methylated pathways, we performed a pathway analysis on cohorts with significant DML/DMGs (Table 1). Sixteen unique differentially methylated biological pathways (q < 0.1) were enriched between peripheral blood and synovial T cells in patients with RA, especially in genes related to complement‐integrin cell surface interactions and the P53 pathway. These pathways are relevant to the pathogenesis of RA and suggest that T cells in the synovium are differentially marked in RA. Three significantly enriched pathways between RA and OA in synovial T cells suggest that differences between the two diseases involve metabolism. Previous studies also provide evidence of altered metabolism in RA tissues, including T cells, which has been refined by recent omics data (18). Although 870 DMGs were identified between RA and OA synovial T cells, most do not fall readily into pathways. This could be due to the small sample size as well as the less stringent cutoff for DML (P < 0.01). No significant pathways involving the 36 DMGs distinguished OA peripheral blood CD3+ T cells from the OA synovium.
Table 1.
Differentially methylated pathways between peripheral blood and synovial T cells
| Pathway | q |
|---|---|
| RA peripheral blood versus RA synovial T cells | |
| Innate immune system | 2.28E‐05 |
| Estrogen response early | 3.10E‐04 |
| Neutrophil degranulation | 2.18E‐03 |
| Immune system | 2.88E‐03 |
| Cell surface interactions at the vascular wall | 4.94E‐03 |
| Integrin cell surface interactions | 4.94E‐03 |
| DAP12 interactions | 1.75E‐02 |
| Complement | 2.40E‐02 |
| P53 pathway | 2.40E‐02 |
| Immunoregulatory interactions between a lymphoid and a nonlymphoid cell | 3.39E‐02 |
| UV response | 3.70E‐02 |
| Hemostasis | 3.81E‐02 |
| Estrogen response late | 4.60E‐02 |
| Androgen response | 8.30E‐02 |
| Epithelial mesenchymal transition | 8.30E‐02 |
| TNF‐α signaling via NF‐κB | 8.30E‐02 |
| RA peripheral blood versus OA peripheral blood | |
| Formation of fibrin clot (clotting cascade) | 2.82E‐02 |
| Extrinsic pathway of fibrin clot formation | 2.82E‐02 |
| Intrinsic pathway of fibrin clot formation | 2.82E‐02 |
| Common pathway of fibrin clot formation | 2.82E‐02 |
| Digestion | 2.82E‐02 |
| Digestion of dietary lipid | 3.69E‐02 |
| Digestion and absorption | 3.69E‐02 |
| Semaphorin interactions | 4.36E‐02 |
| γ‐carboxylation of protein precursors | 4.36E‐02 |
| Removal of aminoterminal propeptides from γ‐carboxylated proteins | 4.36E‐02 |
| α‐defensins | 4.36E‐02 |
| γ‐carboxylation, transport, and aminoterminal cleavage of proteins | 4.85E‐02 |
| G α (12/13) signaling events | 9.02E‐02 |
| RA synovial T cells versus OA synovial T cells | |
| Oxidative phosphorylation | 2.80E‐02 |
| Fatty acid metabolism | 2.80E‐02 |
| Glycolysis | 1.00E‐01 |
Abbreviations: NF‐κB, nuclear factor κB; OA, osteoarthritis; RA, rheumatoid arthritis; TNF‐α, tumor necrosis factor α; UV, ultraviolet light.
DISCUSSION
In this study, we compared DNA methylation patterns of synovial tissue and peripheral blood T cells in RA and OA. One unique feature of the study is that the samples included paired blood and synovial samples from the same patient. A PCA showed that differences in methylation are more prominent when the two compartments are compared than when the two diseases are compared. The most striking finding was the difference between RA blood and synovial T cells, especially in genes related to complement, integrin cell surface interactions and the P53 pathway. Few differences were observed between OA peripheral T cells and synovial T cells. These data suggest that OA synovial T cells represent an unselected population of blood lymphocytes, whereas RA recruits or retains certain T cells, or there is local proliferation of the overrepresented cells. The small DNA methylation differences between OA and RA peripheral blood T cells are likely because RA naive CD4+ T cells are the only subset distinguishable from healthy controls (8). We studied unfractionated CD3+ T cells, and naive CD4+ T cells are only a fraction of this population.
The pathways that distinguish peripheral blood and the synovium are relevant to the pathogenesis of RA, and many were previously identified as differentially marked in FLS. It is not clear whether the mechanisms that lead to altered methylation in FLS lead to a similar pattern in T cells in situ, whether the cells are epigenetically modified elsewhere and home to the joint, or whether the changes are due to expansion of a subset of marked T cells in the synovium.
One potential limitation of this study is the small sample size, which was primarily due to the large number of T cells required for methylation determinations. This issue could contribute to the limited differences detected between RA and OA synovial T cells. The number of cells required for the assays also led us to study CD3+ cells rather than smaller subpopulations, such as CD4+ or CD8+ cells. In addition, several of our samples were not paired because of the practical aspects of obtaining blood at the time of surgery in some patients, especially when surgeries occurred at other institutions.
Despite these limitations, we could identify disease‐ and compartment‐specific patterns of DNA methylation. The data provide support for the notion that differential epigenetic marks are a feature of RA in synovial T cells compared with circulating T cells, in addition to FLS. These marks could modulate pathways that are important in adaptive and innate immune responses and contribute to disease. In addition, abnormal pathways can be mined for potential novel therapeutic approaches. Future studies using single‐cell technologies to refine epigenetic differences will be important to understand the mechanisms of how the epigenome is remodeled.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual contact, and all authors approved the final version to be published. Dr. Ai had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Ai, Boyle, Wang, Firestein.
Acquisition of data
Boyle.
Analysis and interpretation of data
Ai, Boyle, Wang, Firestein.
Supporting information
Sample quality for DNA methylation assays.
CD4/CD8 ratio in synovial CD3+ T cells of RA and OA samples.
CD4/CD8 ratio in peripheral blood CD3+ T cells of RA and OA samples.
Overlaps of DMGs between RA and OA in peripheral blood T cells, synovial T cells and previous FLS.
Table S1‐S3
Supported by a grant from Janssen Research and Development and a grant from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant R01‐AR‐071321).
No potential conflicts of interest relevant to this article were reported.
DNA methylation data that support the findings of this study have been deposited in Gene Expression Omnibus with the primary accession code GSE164468. Other data that support the findings of this study are available from the corresponding authors on request.
Contributor Information
Wei Wang, Email: wei-wang@ucsd.edu.
Gary S. Firestein, Email: gfirestein@health.ucsd.edu.
REFERENCES
- 1. Richardson BC, Patel DR. Epigenetics in 2013: DNA methylation and miRNA: key roles in systemic autoimmunity. Nat Rev Rheumatol 2014;10:72–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nielsen HM, Tost J. Epigenetic changes in inflammatory and autoimmune diseases. Subcell Biochem 2013;61:455–78. [DOI] [PubMed] [Google Scholar]
- 3. Ai R, Laragione T, Hammaker D, Boyle DL, Wildberg A, Maeshima K, et al. Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast‐like synoviocytes. Nat Commun 2018;9:1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Firestein GS. NF‐κB: holy grail for rheumatoid arthritis? Arthritis Rheum 2004;50:2381–6. [DOI] [PubMed] [Google Scholar]
- 5. Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast‐like synoviocytes. Nat Rev Rheumatol 2020;16:316–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pitaksalee R, Burska AN, Ajaib S, Rogers J, Parmar R, Mydlova K, et al. Differential CpG DNA methylation in peripheral naïve CD4+ T‐cells in early rheumatoid arthritis patients. Clin Epigenet 2020;12:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guo S, Zhu Q, Jiang T, Wang R, Shen Y, Zhu X, et al. Genome‐wide DNA methylation patterns in CD4+ T cells from Chinese Han patients with rheumatoid arthritis. Mod Rheumatol 2017;27:441–7. [DOI] [PubMed] [Google Scholar]
- 8. Rhead B, Holingue C, Cole M, Shao X, Quach HL, Quach D, et al. Rheumatoid arthritis naive T cells share hypermethylation sites with synoviocytes. Arthritis Rheumatol 2017;69:550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Glossop JR, Emes RD, Nixon NB, Packham JC, Fryer AA, Mattey DL, et al. Genome‐wide profiling in treatment‐naive early rheumatoid arthritis reveals DNA methylome changes in T and B lymphocytes. Epigenomics 2016;8:209–24. [DOI] [PubMed] [Google Scholar]
- 10. Glossop JR, Emes RD, Nixon NB, Haworth KE, Packham JC, Dawes PT, et al. Genome‐wide DNA methylation profiling in rheumatoid arthritis identifies disease‐associated methylation changes that are distinct to individual T‐ and B‐lymphocyte populations. Epigenetics 2014;9:1228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donlin LT, Rao DA, Wei K, Slowikowski K, McGeachy MJ, Turner JD, et al. Methods for high‐dimensional analysis of cells dissociated from cryopreserved synovial tissue. Arthritis Res Ther 2018;20:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. [DOI] [PubMed] [Google Scholar]
- 13. Altman R, Alarcón G, Appelrouth D, Bloch D, Borenstein D, Brandt K, et al. The American College of Rheumatology criteria for the classification and reporting of osteoarthritis of the hip. Arthritis Rheum 1991;34:505–14. [DOI] [PubMed] [Google Scholar]
- 14. Aryee MJ, Jaffe AE, Corrada‐Bravo H, Ladd‐Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014;30:1363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, et al. The reactome pathway knowledgebase. Nucleic Acids Res 2020;48:D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ai R, Hammaker D, Boyle DL, Morgan R, Walsh AM, Fan S, et al. Joint‐specific DNA methylation and transcriptome signatures in rheumatoid arthritis identify distinct pathogenic processes. Nat Commun 2016;7:11849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fearon U, Hanlon MM, Wade SM, Fletcher JM. Altered metabolic pathways regulate synovial inflammation in rheumatoid arthritis. Clin Exp Immunol 2019;197:170–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sample quality for DNA methylation assays.
CD4/CD8 ratio in synovial CD3+ T cells of RA and OA samples.
CD4/CD8 ratio in peripheral blood CD3+ T cells of RA and OA samples.
Overlaps of DMGs between RA and OA in peripheral blood T cells, synovial T cells and previous FLS.
Table S1‐S3