

Abstract
Objective
Tofacitinib is an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis (RA). We compared 5‐year adverse event (AE) incidence rates (IRs) between patients initiating tofacitinib and those initiating new biological disease‐modifying antirheumatic drugs (bDMARDs) within the United States (US) Corrona RA registry.
Methods
IRs (number of first events/100 patient‐years) of major adverse cardiovascular events (MACE), serious infection events (SIEs), herpes zoster (HZ), malignancies, and death were estimated among tofacitinib and bDMARD initiators, regardless of dose/schedule, between November 6, 2012 (US Food and Drug Administration tofacitinib approval), and July 31, 2018 (follow‐up through January 31, 2019). Propensity score (PS) methods were used to control for nonrandom prescribing practices. Hazard ratios (HRs) were calculated to compare rates using multivariable‐adjusted Cox regression. Different risk windows were used for acute (MACE, SIEs, HZ, and venous thromboembolic events [VTEs]) and long‐term (malignancy and death) events. VTEs were assessed descriptively.
Results
For MACE, SIEs, and HZ, 1999 (3152.1 patient‐years) and 8358 (12 869.4 years) tofacitinib and bDMARD initiators were included, respectively; for malignancy/death, 1999 (4505.6 patient‐years) and 6354 (16 670.8 patient‐years) initiators were included, respectively. AE rates were similar across cohorts, except for HZ, which was significantly higher with tofacitinib versus bDMARDs (PS‐trimmed adjusted HR 2.32; 95% confidence interval [CI] 1.43‐3.75). There were 45 (zero serious) and 88 (five serious) HZ events with tofacitinib and bDMARDs, respectively. Sensitivity analyses demonstrated similar results. VTE IRs (95% CI) were 0.29 (0.13‐0.54) and 0.33 (0.24‐0.45) for tofacitinib and bDMARDs, respectively.
Conclusion
In this registry analysis, both cohorts had similar MACE, SIE, malignancy, death, and VTE rates; HZ rates were higher for tofacitinib initaitors than for bDMARD initiators.
Significance & Innovations.
The safety profile of tofacitinib has been evaluated in patients with rheumatoid arthritis (RA) participating in clinical trials and long‐term extension studies.
This analysis adds long‐term safety data from the use of tofacitinib in real‐world practice, which does not have the exclusions found in randomized clinical trials or long‐term extension studies. Real‐world patients are also generally more diverse and have a longer‐term follow‐up than patients in clinical trials. This analysis also provides context for the use of tofacitinib through comparison with other advanced therapies used in the treatment of RA.
These results provide the longest‐term real‐world safety data for a Janus kinase inhibitor to date and provide further insight than that gained from clinical trials into some important safety risks with tofacitinib in comparison with biological therapies. In addition, this analysis provides a thorough, real‐world assessment of herpes zoster incidence in actual clinic populations, compared with clinical trial data. There is a need for further studies, for example, regarding the use and safety of an inactivated recombinant vaccine for herpes zoster in patients with RA.
These real‐world data will help inform and reinforce existing safety data for tofacitinib by providing needed contextual data in comparison with biological disease‐modifying antirheumatic drugs.
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic, systemic inflammatory disease that primarily involves the joints but can include extra‐articular manifestations (1). Tofacitinib is an oral Janus kinase (JAK) inhibitor for the treatment of RA. The efficacy and safety of 5 mg and 10 mg tofacitinib twice daily (BID) administered as monotherapy or in combination with conventional synthetic antirheumatic drugs (csDMARDs), mainly methotrexate, in patients with moderately to severely active RA have been demonstrated in Phase II (2, 3, 4, 5, 6), Phase III (7, 8, 9, 10, 11, 12, 13), and Phase IIIb/IV (14) studies of up to 24 months’ duration and in long‐term extension (LTE) studies with up to 9.5 years of observation (15, 16, 17).
It is important to assess long‐term safety of therapies for chronic conditions. Registry data are not limited by exclusions at baseline endemic to LTE studies and therefore complement data from randomized controlled trials (RCTs). Established in 2001, the United States (US) Corrona RA registry is an ongoing longitudinal clinical registry of patients with RA (18). A postauthorization safety study within the US Corrona RA registry was initiated to evaluate tofacitinib safety after US Food and Drug Administration (FDA) approval of the 5 mg BID dose on November 6, 2012. The majority of patients initiating tofacitinib in this analysis were expected to be receiving the dose approved in the US during this time (ie, 5 mg BID).
This analysis compared incidence rates (IRs; number of first events/100 patient‐years) of major adverse cardiovascular events (MACE), serious infection events (SIEs), herpes zoster (HZ), malignancies, and death in patients newly initiating tofacitinib or biological disease‐modifying antirheumatic drugs bDMARDs. After the more recent finding of an increased incidence of venous thromboembolic events (VTEs) in patients receiving JAK inhibitors, including tofacitinib, VTE data were collected prospectively and assessed descriptively throughout.
PATIENTS AND METHODS
Study setting
The US Corrona RA registry—a prospective, multicenter, observational, disease‐based registry—has previously been described (18). As of December 31, 2018, the registry has collected longitudinal, real‐world data from patients aged 18 years or older with RA, who were recruited by 752 participating providers from 177 private/academic sites across 42 US states (including 331 participating providers since tofacitinib approval in November 2012, of whom, 230 prescribed tofacitinib in the current analysis). Detailed clinical assessment forms are completed at enrollment and follow‐up visits (requested approximately every 6 months). As of December 2018, the database included information on 50 605 patients (384 456 patient visits; approximately 180 603 patient‐years of follow‐up; mean follow‐up time of 4.4 years).
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki. Investigators obtained central or local institutional review board approval for conducting noninterventional research involving human patients. All registry patients provided written informed consent.
Study population
This study included patients (aged 18 years or more) with RA from the US Corrona RA registry who initiated tofacitinib or a bDMARD (comparator cohort) between November 6, 2012, and July 31, 2018, with follow‐up to January 31, 2019. Enrollment was ongoing; therefore, individual patients entered the study at different times, resulting in variable follow‐up times across patients. Drug initiation was defined as the first ever use of a given drug. The tofacitinib cohort included unique patients initiating tofacitinib as monotherapy or in combination with a csDMARD. Tofacitinib initiators could have previously used a csDMARD and/or a bDMARD but not tofacitinib (or other JAK inhibitors). The bDMARD cohort included patients newly initiating a specific, but not necessarily their first, bDMARD. The bDMARD cohort included patients initiating a tumor necrosis factor inhibitor (TNFi; adalimumab, certolizumab pegol, golimumab, etanercept, or infliximab) or non‐TNFi (abatacept, anakinra, rituximab, or tocilizumab) as monotherapy or in combination with a csDMARD. The bDMARD initiators could have previously used a csDMARD or different bDMARD but not tofacitinib (or other JAK inhibitors) or the bDMARD they were initiating.
Data collection
The methods used to collect data for adverse events (AEs) and the adjudication process for predefined AEs of interest have been reported previously (18) and are described in detail in the Supplementary Material. The primary analysis used nonadjudicated data.
Outcomes for the comparative analysis included death and AEs of interest occurring from drug initiation to January 31, 2019, namely, MACE (defined as myocardial infarction [MI], stroke/transient ischemic attack [TIA], and cardiovascular [CV] death), SIEs (infections leading to hospitalization and/or intravenous antibiotics), HZ (serious and nonserious; serious HZ was defined as any HZ infection that led to hospitalization, disability, congenital anomaly, or death; was immediately life‐threatening, medically important/serious in the opinion of the site investigator; or required treatment with parenteral therapy), malignancy excluding nonmelanoma skin cancer (NMSC), and NMSC. Available data from the National Death Index supplemented provider‐reported registry data to identify potentially unreported deaths that may have been lost to follow‐up. VTEs (defined as deep vein thrombosis [DVT] or pulmonary embolism [PE]) were collected during routine visits, verified using follow‐up Targeted Adverse Event questionnaires, and included as a descriptive analysis.
Index date and risk window definitions for the comparative analysis
IRs were based on two different definitions of the risk window; one definition was used for outcomes with acute onset (ie, MACE, SIEs, HZ, and VTEs), which were considered to be approximate to time on drug, and the second definition was used for outcomes characterized by latent onset (ie, malignancy and death). For MACE, SIEs, HZ, and VTEs, the index date was the initiation date of tofacitinib or new bDMARD (multiple bDMARD initiations were considered separate index events). The risk window was defined as all time from the index date until the first occurrence of an event, 90 days after discontinuation of therapy, switch to another therapy (event attributed to the new therapy after switching), last follow‐up visit, death, or end of the data collection period, whichever came first. Separate sensitivity analyses were performed using a 30‐day postdiscontinuation risk window; events were attributed to the discontinued therapy if switching occurred during the risk window.
For malignancy/death, the index date was the initiation date of tofacitinib or bDMARD (multiple bDMARD initiations were not considered separate index events); the risk window was defined as all time from index date until an event, loss to follow‐up, or the end of the study period using a “once exposed, always exposed” approach (19, 20). For patients experiencing an event after sequential treatment with tofacitinib and a bDMARD, or vice versa, the event was attributed to both therapies regardless of discontinuation. Sensitivity analyses were conducted using a conservative hierarchical approach in which follow‐up time for bDMARD initiators ended upon switching to tofacitinib, and an event occurring after the switch was allocated only to tofacitinib; for tofacitinib initiators, follow‐up time continued regardless of switching to another therapy.
Propensity scores for the comparative analysis
Propensity scores (PSs) were calculated to control for nonrandom prescribing practices and obtain balance in baseline patient characteristics associated with the likelihood of initiating tofacitinib versus a bDMARD. PSs were determined separately for each outcome to accommodate outcome‐specific confounders and multiple initiations for the acute risk window (described in detail in the Supplementary Material). Covariates with a standardized difference greater than |0.10|, and those chosen a priori on the basis of clinical experience were used to derive PS‐trimmed populations (primary analysis) that excluded patients with missing covariate values and nonoverlapping PS values. PS‐matched cohorts (ratio: maximum of four bDMARDs:one tofacitinib; caliper = 0.05) were produced for sensitivity analysis.
Statistical analyses for the comparative analysis
Patient demographics and disease characteristics at initiation were summarized descriptively. For outcomes meeting criteria for PS modeling (MACE, SIEs, HZ, malignancies, and death), crude IRs and 95% confidence intervals (CIs) were calculated for PS‐trimmed/PS‐matched populations. Unadjusted and adjusted hazard ratios (HRs; unadjHR and adjHR, respectively) with 95% CIs comparing rates of first events of tofacitinib initiatiors with those of bDMARD initiators were estimated using simple Cox regression and multivariable‐adjusted Cox regression models, respectively. Post hoc analyses were performed in cases in which differences were identified between the results of the unadjusted analysis and the PS‐trimmed primary analysis. For HRs, P values for the Wald statistic were calculated; P < 0.05 was deemed statistically significant. Power was a priori determined to be 80% or greater to detect a HR, 2.0 or greater for SIEs, 2.05 or greater for MACE and NMSC, 2.2 or greater for malignancies excluding NMSC, and 2.25 or greater for HZ and death.
Descriptive analysis of VTEs
At the time of analyses, a sufficient number of VTEs had not occurred in this study to permit an adequately powered comparative assessment of IRs between tofacitinib and bDMARD initiators; thus, PS modeling was not performed. Age‐ and sex‐standardized IRs for VTEs were estimated using direct standardization, with the tofacitinib population used as the standard population. VTE data are presented descriptively in the unmatched population derived for the MACE, SIE, and HZ analysis only.
RESULTS
Patients
For MACE, SIEs, and HZ, there were 1999 tofacitinib initiators (3152.1 patient‐years; mean duration of follow‐up 1.58 years) and 8358 bDMARD initiators (12 869.4 patient‐years; mean duration of follow‐up 1.54 years) who met inclusion criteria (Figure 1). For MACE/SIEs/HZ, patients could have been counted more than once in the bDMARD initiation cohort for each unique bDMARD initiation.
Figure 1.
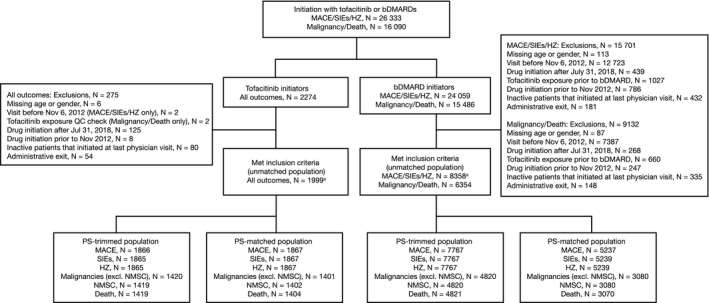
Number of patients eligible for the analysis of major adverse cardiovascular events (MACE), serious infection events (SIEs), herpes zoster (HZ), malignancies (excluding nonmelanoma skin cancer [NMSC]), NMSC, and death. The index date was the initiation date of tofacitinib or a biological disease‐modifying antirheumatic drug (bDMARD); multiple bDMARD initiations were considered separate index events for MACE, SIEs, and HZ, but were not considered separate index events for malignancy/death; propensity scores (PSs) were calculated using baseline patient demographic data (age, sex, marital status, and work status), disease characteristics (duration of rheumatoid arthritis [RA], disease activity [for malignancy/death only: patient pain, patient fatigue, Health Assessment Questionnaire, and EuroQol Five‐Dimensions Health Questionnaire]), comorbidities (history of adverse event of interest [yes versus no; excluding cardiovascular disease–related deaths for MACE and excluding death]), past medication history, and concomitant medications. Venous thromboembolic events were assessed in the unmatched population derived for the analysis of MACE, SIE, and HZ. QC, quality control.
For malignancy/death, patients were only counted once in the bDMARD initiation cohort, regardless of switching to another bDMARD. Therefore, there were 1999 tofacitinib initiators (4505.6 patient‐years; mean duration of follow‐up 2.25 years) and 6354 bDMARD initiators (16 670.8 patinet‐years; mean duration of follow‐up 2.62 years) who met inclusion criteria (Figure 1). Of these, 732 patients initiated both tofacitinib and one or more bDMARD.
Baseline demographics
Table 1 presents baseline patient demographics and clinical characteristics in the unmatched population for the analysis of MACE, SIEs, and HZ and in the PS‐trimmed population for the analysis of MACE. The majority of patients were female, White, and aged less than 65 years. Age, sex, race, body mass index, smoking status, and comorbidities (except history of SIEs) were balanced between cohorts. Patients in the unmatched population initiating tofacitinib had longer disease duration than bDMARD initiators (13.8 versus 10.6 years) and were more likely to initiate tofacitinib later in the course of therapy (69.7 and 39.7% of tofacitinib and bDMARD initiators, respectively, previously received more than one bDMARD/one or more csDMARD). Use of methotrexate alone or with another csDMARD was higher among bDMARD initiatiors than tofacitinib initiators. Corticosteroid use was comparable between cohorts.
Table 1.
Patient demographics and clinical characteristics at the time of initiation of tofacitinib or a bDMARD in the overall (unmatched) population for the analysis of MACE, SIEs, and HZ and in the PS‐trimmed population for the analysis of MACE
| Unmatched Population (MACE, SIEs, and HZ) | PS‐Trimmed Population (MACE) | |||||
|---|---|---|---|---|---|---|
| Tofacitinib Initiators (n = 1999) | bDMARD Initiators (n = 8358) | STD a | Tofacitinib Initiators (n = 1866) | bDMARD Initiators (n = 7767) | STD | |
| Age, mean (SD), yr | 59.6 (12.1) | 58.5 (13.0) | 0.088 | 59.6 (12.0) | 58.5 (12.9) | 0.082 |
| Sex, female, n (%) | 1610 (80.5) | 6716 (80.4) | 0.005 | 1505 (80.7) | 6223 (80.1) | 0.013 |
| Race, White, n (%) | 1700 (85.8) | 6896 (82.9) | 0.079 | 1588 (85.9) | 6441 (83.4) | 0.071 |
| Current smoker, n (%) | 307 (19.6) | 1268 (18.1) | 0.037 | 289 (19.7) | 1200 (18.4) | 0.035 |
| BMI, mean (SD), kg/m2 | 30.5 (7.6) | 30.4 (7.5) | 0.010 | 30.5 (7.6) | 30.4 (7.5) | 0.011 |
| Work status, n (%) | ||||||
| Full time | 606 (31.1) | 3001 (36.7) | –0.119 | 581 (31.1) | 2867 (36.9) | –0.122 |
| Disabled | 436 (22.4) | 1274 (15.6) | 0.173 | 419 (22.5) | 1188 (15.3) | 0.184 |
| Marital status, n (%) | ||||||
| Single | 179 (9.1) | 1008 (12.3) | –0.103 | 167 (9.0) | 937 (12.1) | –0.102 |
| Married | 1339 (68.0) | 5398 (65.7) | 0.051 | 1277 (68.4) | 5133 (66.1) | 0.050 |
| Divorced | 216 (11.0) | 948 (11.5) | –0.018 | 201 (10.8) | 897 (11.6) | –0.025 |
| Duration of disease, mean (SD), yr b | 13.8 (10.3) | 10.6 (10.0) | 0.316 | 14.0 (10.4) | 10.6 (10.0) | 0.332 |
| History of comorbidities, n (%) | ||||||
| Hypertension | 709 (35.5) | 2696 (32.3) | 0.068 | 658 (35.3) | 2514 (32.4) | 0.061 |
| Hyperlipidemia | 269 (13.5) | 1093 (13.1) | 0.011 | 254 (13.6) | 1020 (13.1) | 0.014 |
| Coronary heart disease | 154 (7.7) | 480 (5.7) | 0.078 | 145 (7.8) | 454 (5.9) | 0.076 |
| Stroke | 68 (3.4) | 246 (2.9) | 0.026 | 65 (3.5) | 233 (3.0) | 0.027 |
| VTE | 62 (3.1) | 199 (2.4) | 0.044 | 61 (3.3) | 192 (2.5) | 0.048 |
| Diabetes | 231 (11.6) | 864 (10.3) | 0.039 | 206 (11.0) | 806 (10.4) | 0.021 |
| Malignancy (excluding NMSC) | 157 (7.9) | 569 (6.8) | 0.040 | 148 (7.9) | 534 (6.9) | 0.040 |
| Serious infection | 277 (13.9) | 808 (9.7) | 0.130 | 262 (14.0) | 754 (9.7) | 0.134 |
| Prior DMARDs, n (%) | ||||||
| csDMARD‐naïve/bDMARD‐naïve | 11 (0.6) | 154 (1.9) | –0.121 | 10 (0.5) | 138 (1.8) | –0.116 |
| ≥1 prior csDMARD/bDMARD‐naïve | 212 (10.8) | 2289 (28.1) | –0.447 | 200 (10.7) | 2181 (28.1) | –0.450 |
| ≥1 prior csDMARD/1 prior bDMARD | 371 (18.9) | 2468 (30.1) | –0.266 | 351 (18.8) | 2349 (30.2) | –0.268 |
| ≥1 prior csDMARD/>1 prior bDMARD | 1365 (69.7) | 3232 (39.7) | 0.632 | 1305 (69.9) | 3099 (39.9) | 0.633 |
| Prior bDMARD use, n (%) | ||||||
| Naïve | 223 (11.2) | 2443 (29.2) | –0.462 | 210 (11.3) | 2319 (29.9) | –0.473 |
| 1 previous | 388 (19.4) | 2590 (31.0) | –0.269 | 351 (18.8) | 2349 (30.2) | –0.268 |
| 2 previous | 455 (22.8) | 1687 (20.2) | 0.063 | 414 (22.2) | 1553 (20.0) | 0.054 |
| 3 or more | 933 (46.7) | 1638 (19.6) | 0.600 | 891 (47.8) | 1546 (19.9) | 0.616 |
| Prior csDMARD use, n (%) | ||||||
| Naïve | 51 (2.6) | 369 (4.4) | –0.102 | 10 (0.5) | 138 (1.8) | –0.116 |
| 1 previous | 579 (29.0) | 3312 (39.6) | –0.226 | 545 (29.2) | 3116 (40.1) | –0.231 |
| 2 or more | 1369 (68.5) | 4677 (56.0) | 0.260 | 1311 (70.3) | 4513 (58.1) | 0.255 |
| Prednisone use, n (%) | ||||||
| Naïve | 1367 (68.4) | 5794 (69.3) | –0.020 | 1281 (68.7) | 5395 (69.5) | –0.018 |
| <10 mg | 411 (20.6) | 1612 (19.3) | 0.032 | 384 (20.6) | 1505 (19.4) | 0.030 |
| ≥10 mg | 191 (9.6) | 856 (10.2) | –0.023 | 172 (9.2) | 778 (10.0) | –0.027 |
| Concomitant medication, n (%) | ||||||
| Methotrexate alone | 576 (28.8) | 3444 (41.2) | –0.262 | 553 (29.6) | 3253 (41.9) | –0.258 |
| Methotrexate and other csDMARD | 145 (7.3) | 919 (11.0) | –0.130 | 140 (7.5) | 877 (11.3) | –0.130 |
| csDMARD (excluding methotrexate) | 413 (20.7) | 1606 (19.2) | 0.036 | 388 (20.8) | 1525 (19.6) | 0.029 |
| Any NSAID use, n (%) | 848 (42.4) | 3764 (45.0) | –0.053 | 801 (42.9) | 3560 (45.8) | –0.059 |
| Statin use, n (%) | 445 (22.3) | 1827 (21.9) | 0.010 | 422 (22.6) | 1733 (22.3) | 0.007 |
| Swollen joint count (0‐28), mean (SD) | 4.7 (5.0) | 5.1 (5.4) | –0.092 | 4.6 (5.0) | 5.1 (5.4) | –0.098 |
| Tender joint count (0‐28), mean (SD) | 6.8 (7.1) | 7.2 (7.3) | –0.050 | 6.8 (7.1) | 7.2 (7.3) | –0.054 |
| Patient pain (0‐100), mean (SD) | 50.8 (28.7) | 49.7 (28.4) | 0.040 | 50.8 (28.6) | 49.5 (28.3) | 0.048 |
| PtGA (0‐100), mean (SD) | 47.9 (26.9) | 47.1 (26.8) | 0.030 | 47.9 (26.9) | 46.9 (26.7) | 0.036 |
| PGA (0‐100), mean (SD) | 32.6 (21.9) | 34.8 (23.1) | –0.095 | 32.6 (21.8) | 34.7 (23.0) | –0.092 |
| Patient fatigue (0‐100), mean (SD) | 52.1 (30.0) | 50.2 (29.4) | 0.065 | 52.1 (30.1) | 50.1 (29.4) | 0.069 |
| CDAI, mean (SD) | 19.5 (13.3) | 20.5 (14.1) | –0.072 | 19.5 (13.2) | 20.5 (14.1) | –0.074 |
| CDAI categorized, n (%) | ||||||
| Remission | 98 (6.2) | 487 (6.9) | –0.028 | 91 (6.2) | 461 (7.0) | –0.033 |
| LDA | 343 (21.8) | 1387 (19.7) | 0.052 | 325 (22.1) | 1289 (19.6) | 0.062 |
| Moderate | 561 (35.7) | 2384 (33.9) | 0.038 | 523 (35.6) | 2234 (34.0) | 0.033 |
| High | 569 (36.2) | 2775 (39.5) | –0.067 | 530 (36.1) | 2585 (39.4) | –0.068 |
| HAQ, mean (SD) | 1.1 (0.7) | 1.0 (0.7) | 0.096 | 1.1 (0.7) | 1.0 (0.7) | 0.102 |
| EQ‐5D (0‐1), mean (SD) | 0.7 (0.2) | 0.7 (0.2) | –0.070 | 0.7 (0.2) | 0.7 (0.2) | –0.08 |
bDMARD, biological disease‐modifying antirheumatic drug; BID, twice daily; BMI, body mass index; CDAI, Clinical Disease Activity Index; csDMARD, conventional synthetic disease‐modifying antirheumatic drug; DMARD, disease‐modifying antirheumatic drug; EQ‐5D, EuroQol Five‐Dimensions Health Questionnaire; HAQ, Health Assessment Questionnaire; HZ, herpes zoster; LDA, low disease activity; MACE, major adverse cardiovascular events; NMSC, nonmelanoma skin cancer; NSAID, nonsteroidal anti‐inflammatory drug; PGA, Physician’s Global Assessment; PS, propensity score; PtGA, Patient’s Global Assessment; RA, rheumatoid arthritis; SIE, serious infection event; STD, standardized difference; VTE, venous thromboembolic event.
Tofacitinib initiators primarily received tofacitinib 5 mg BID; N is the total number of patients; the number of patients evaluated for each category may be fewer than N.
Covariates in the unmatched population with STDs >|0.10| in absolute value (bold font) were included in the construction of a PS model in order to derive PS‐trimmed populations.
Duration of disease represents time after initial diagnosis of RA until initiation of the new treatment.
PS modeling
Figure 1 shows the number of patients analyzed for each outcome; covariates included in PS modeling for each outcome are shown in Table 2; and PSdistributions are shown in Supplementary Figure 1.
Table 2.
Covariates included in PS modeling
| Covariates Included for Each Outcome | Covariates With STDs >|0.10| |
|---|---|
|
Age Gender History of AE of interest (yes vs no) a Number of prior DMARDs Patient pain b Patient fatigue b HAQ b EQ‐5D b |
Single marital status (yes versus no) Duration of RA Work status: Full time (yes versus no) Disabled (yes versus no) History of SIEs (yes vs no) Current concomitant medication |
AE, adverse event; DMARD, disease‐modifying antirheumatic drug; EQ‐5D, EuroQol Five‐Dimensions Health Questionnaire; HAQ, Health Assessment Questionnaire; PS, propensity score; RA, rheumatoid arthritis; SIE, serious infection event; STD, standardized difference.
Excluding death, and cardiovascular disease–related deaths for major adverse cardiovascular events.
Included in PS modeling for malignancy/death only.
Baseline demographics and clinical characteristics were similar in unmatched and PS‐trimmed populations (Table 1; Supplementary Tables 1–5). As expected, some residual imbalance in characteristics between cohorts was observed after PS trimming. In the PS‐trimmed populations, more tofacitinib initiators used three or more previous bDMARDs or two or more previous csDMARDs versus bDMARD initiators. As in the unmatched population, patients had greater prior DMARD experience, and concomitant medication use was highest among bDMARD initiators.
IRs of outcomes for the comparative analysis
Crude IRs for PS‐trimmed populations are shown in Figure 2, and unadjHRs and adjHRs are shown in Figure 3. Results of sensitivity analyses, including those with varied risk window definitions (data not shown), were similar to primary analyses for all outcomes. Similar results were also observed in sensitivity analyses using the PS‐matched populations (Supplementary Figures 2 and 3).
Figure 2.
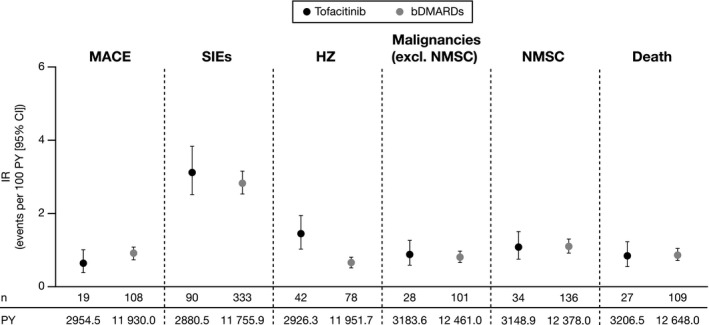
Incidence rates (IRs; number of first events/100 patient‐years [PY]) of outcomes in the propensity score–trimmed population. IRs were based on different definitions of the risk window for outcomes with acute onset (major cardiovascular adverse events [MACE], serious infection events [SIEs], and herpes zoster [HZ]) or latent onset (malignancies and death). Tofacitinib initiators primarily received tofacitinib 5 mg twice daily. bDMARD, biological disease‐modifying antirheumatic drug; CI, confidence interval; NMSC, nonmelanoma skin cancer.
Figure 3.

Hazard ratios (HRs) (unadjusted and adjusted) for tofacitinib initiators versus biological disease‐modifying antirheumatic drug (bDMARD) initiators in the propensity score–trimmed population. Tofacitinib initiators primarily received tofacitinib 5 mg twice daily. HZ, herpes zoster; MACE, major cardiovascular adverse events; NMSC, nonmelanoma skin cancer; SIEs, serious infection events.
MACE
Incidence of MACE and CV events included in the definition of MACE in the unmatched populations are shown in Supplementary Table 6. After PS trimming, MACE IRs were numerically higher in bDMARD versus tofacitinib initiators (tofacitinib: 19 events, IR 0.64 [0.39‐1.00]; bDMARDs: 108 events, IR 0.91 [0.74‐1.09]; Figure 2). MACE adjHR was 0.61 (0.34‐1.06; Figure 3). In the PS‐matched population, unadjHRs and adjHRs reached statistical significance (both P < 0.05; Supplementary Figure 3). In the unmatched population, stroke/TIA occurred in 10 and 60 and MI occurred in 10 and 38 tofacitinib (n = 1999) and bDMARD (n = 8358) initiators, respectively (crude IRs in tofacitinib and bDMARD initiators [unmatched population], respectively, were IR 0.32 [0.15‐0.58] and IR 0.47 [0.36‐0.60] for stroke/TIA and IR 0.32 [0.15‐0.59] and IR 0.30 [0.21‐0.41] for MI).
SIEs
Similar SIE IRs were observed for both cohorts in the PS‐trimmed population (tofacitinib: 90 events, IR 3.12 [2.51‐3.84]; bDMARDs: 333 events, IR 2.83 [2.54‐3.15]; Figure 2). SIE adjHR was 0.99 (0.75‐1.30; Figure 3). In the unmatched population, the most frequently reported SIEs for both cohorts were pneumonia (IR 0.96 [0.65‐1.37] and IR 0.99 [0.82‐1.17] for tofacitinib and bDMARDs, respectively), cellulitis (IR 0.41 [0.22‐0.71] and IR 0.48 [0.37‐0.62] for tofacitinib and bDMARDs, respectively), and urinary tract infection (IR 0.48 [0.27‐0.79] and IR 0.34 [0.24‐0.45] for tofacitinib and bDMARDs, respectively). There were three (IR 0.10 [0.02‐0.28]) and 11 (IR 0.09 [0.04‐0.15]) events of upper respiratory tract infection among tofacitinib and bDMARD initiators, respectively; there were no tuberculosis events among tofacitinib initiators, and there were two events (IR 0.02 [0.00‐0.06]) among bDMARD initiators.
HZ
HZ IR was significantly higher with tofacitinib versus bDMARDs in the PS‐trimmed population (tofacitinib: 42 events, IR 1.44 [1.03‐1.94)]; bDMARDs: 78 events, IR 0.65 [0.52‐0.81]; Figure 2). HZ adjHR was 2.32 (1.43‐3.75; P = 0.001; Figure 3). All 45 events among tofacitinib initiators in the unmatched population (n = 1999) were nonserious. There were 88 HZ events (including five serious events) among bDMARD initiators in the unmatched population.
Malignancies
Malignancy IRs were comparable between cohorts in the PS‐trimmed population (malignancies excluding NMSC, tofacitinib: 28 events IR 0.88 [0.58‐1.27]; bDMARDs: 101 events IR 0.81 [0.66‐0.98]; and NMSC, tofacitinib: 34 events, IR 1.08 [0.75‐1.51]); bDMARDs: 136 events, IR 1.1 [0.92‐1.3]; Figure 2). AdjHRs were 1.04 (0.68‐1.61) and 1.02 (0.69‐1.50) for malignancies excluding NMSC and for NMSC, respectively (Figure 3). In the unmatched population, other cancer types (n = 24 and n = 48), breast cancer (n = 8 and n = 29), and lung cancer (n = 6 and n = 18) were the most frequently reported malignancies for tofacitinib (n = 1999) and bDMARD (n = 6354) initiators, respectively.
A post hoc sensitivity analysis was prompted by the disproportionate exclusion of malignancy events from the tofacitinib and bDMARD cohorts during PS trimming, driven by disproportionate missing values (>10%) for some variables in the PS model (EuroQol Five‐Dimensions Health Questionnaire [EQ‐5D], Health Assessment Questionnaire [HAQ], patient fatigue, and patient pain). Removing these variables from the model resulted in a numerically higher malignancy IR with tofacitinib versus bDMARDs in the PS‐trimmed population (tofacitinib: 43 events, IR 1.01 [0.73‐1.37]; bDMARDs: 119 events, IR 0.76 [0.63‐0.91]; adjHR 1.34 [0.94‐1.93]). Malignancy risk factors were similarly distributed between patients missing and not missing values.
Adjudicated safety data
Of reported MACE, 61/105 were adjudicated (51/61 [83.6%] adjudicated probable/definite). Of reported SIEs, 235/352 were adjudicated (216/235 [91.9%] adjudicated probable/definite). Of reported malignancies excluding NMSC and of reported NMSC, 94/157 and 123/201 were adjudicated (82/94 [87.2%] and 111/123 [90.2%] adjudicated probable/definite), respectively.
Death
In total, 188 deaths occurred (tofacitinib: 42 deaths; bDMARDs: 146 deaths). After PS trimming, there were 27 deaths with tofacitinib (IR 0.84 [0.55‐1.23]) and 109 deaths with bDMARDs (IR 0.86 [0.71‐1.04]; Figure 2). Death adjHR was 0.91 (0.59‐1.42; Figure 3). In total, three (IR 0.10 [0.02‐0.28]) and 16 deaths (IR 0.12 [0.07‐0.20]) in tofacitinib and bDMARD initiators (unmatched population), respectively, were attributed to CV disease.
VTEs
VTEs were assessed descriptively in the unmatched population derived for the analysis of MACE, SIEs, and HZ (1999 tofacitinib initiators [3152.1 patinet‐years; mean duration of follow‐up 1.58 years] and 8358 bDMARD initiators [12 869.4 patient‐years; mean duration of follow‐up 1.54 years]; Figure 1). Age‐ and gender‐standardized IRs of DVT, PE, and overall VTEs were numerically similar in both cohorts (tofacitinib: nine events [including four patients with DVT and six patients with PE], IR 0.29 [0.13‐0.54]; bDMARDs: 41 events [including 20 patients with DVT and 24 patients with PE], IR 0.33 [0.24‐0.45]; Figure 4).
Figure 4.
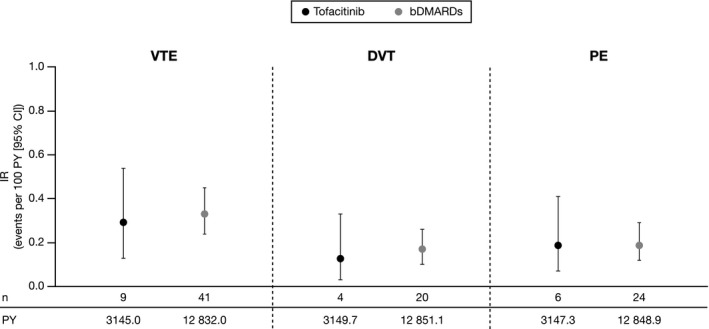
Age‐ and sex‐standardized incidence rates (number of first events/100 patient‐years [PY]) of venous thromboembolic events (VTEs). IRs were based on the definition of the risk window for outcomes with acute onset. Age‐ and sex‐standardized IRs were estimated using direct standardization (tofacitinib population used as standard population); VTE data did not have 80% or more power to detect a hazard ratio (HR) of 2.25 or less between cohorts. Propensity scores were not calculated. Tofacitinib initiators primarily received tofacitinib 5 mg twice daily. bDMARD, biological disease‐modifying antirheumatic drug; CI, confidence interval; DVT, deep vein thrombosis; IR, incidence rate; PE, pulmonary embolism.
DISCUSSION
Real‐world evidence regarding postapproval medication safety is an important complement to evidence from RCTs. These results demonstrated that patients with RA initiating tofacitinib had similar incidence of MACE, SIEs, malignancies, and death versus bDMARD initiators over 5 years. HZ incidence was higher in tofacitinib versus bDMARD initiators. In this registry analysis, incidence of VTEs was similar in both cohorts.
CV disease is the most common cause of death in RA (21), and its risk is higher in patients with RA versus the general population (22). Pooled analysis of data from the tofacitinib RA clinical program demonstrated a low IR of MACE (IR 0.58 [0.39‐0.88] and IR 0.37 [0.26‐0.52] for all tofacitinib doses in Phase III and LTE studies, respectively) (23). Although values reported here for tofacitinib initiators (IR 0.64 [0.39‐1.00]) were numerically higher than rates reported in the clinical program, possibly because real‐world patients tend to have more comorbidities than those in RCTs (18), tofacitinib and bDMARD initiators in this study had a similar incidence of MACE in a real‐world population. These results have greater generalizability to patients with comorbidities than RCT data.
An ad hoc safety analysis performed in 2019 has raised the discussion of VTE and PE related to tofacitinib. At the time of this analysis, study A3921133 (NCT02092467), an ongoing, open‐label, endpoint‐driven, FDA‐required postmarketing safety study, enriched for RA patients considered to be at higher risk of cardiac events and designed to compare the long‐term risk of MACE and malignancy among patients receiving tofacitinib 5 and 10 mg BID vs two TNFis, was ongoing (closeout activities ongoing at the time of manuscript submission) (24). Eligible patients were aged 50 years or more, had one or more CV risk factor, and were taking a stable dose of methotrexate. The Data Safety Monitoring Board determined that the frequency of PE and all‐cause mortality in the tofacitinib 10 mg BID arm was higher than the frequency in the TNFi comparator arm, and this arm of the study was stopped. Subsequently, based on information from study A3921133 and consideration of information pertaining to VTE for other JAK inhibitors, Pfizer has determined that VTE is an important identified risk for treatment with tofacitinib. Corrona questionnaires collect VTE as part of the breakdown of MACE and VTE events. In this analysis, in which tofacitinib initiators predominantly received tofacitinib 5 mg BID, the incidence of VTEs was found to be numerically similar in tofacitinib and bDMARD initiators.
Patients with RA have a twofold higher risk of SIEs than the general population (25, 26). An IR of 2.7 (95% CI 2.5‐3.0) was observed for SIEs in the tofacitinib RA clinical program (27), which is marginally lower than that reported for tofacitinib initiators (IR 3.12 [2.51‐3.84]) in this analysis. A recent analysis of SIEs in patients initiating tofacitinib or bDMARDs, using data from three US health care claims databases (Medicare, Clinformatics, and MarketScan), compared the risk of SIEs in patients with RA initiating tofacitinib with that of seven bDMARDs. Overall, no statistically significant differences in SIE risk were observed, except for an increased risk with tofacitinib versus etanercept in SIEs (adjHR 1.41 [1.15‐1.73]), pneumonia/upper respiratory tract infections (adjHR 1.53 [1.06‐2.22]), and skin and soft tissue infections (adjHR 2.21 [1.08‐4.52]) and an increased risk with tofacitinib versus abatacept in skin and soft tissue infections (adjHR 2.36 [1.00‐5.55]) (28). In the current analysis, the incidence of SIEs was similar between tofacitinib and bDMARD initiators, and rates of pneumonia, upper respiratory tract infections, and cellulitis were similar between cohorts, although no comparisons between tofacitinib and individual bDMARDs were carried out. In addition, the nature of infections in this analysis was consistent with that previously seen in patients treated with bDMARDs (29) and with that in tofacitinib postmarketing surveillance data (30).
HZ has been reported as a complication of treatment with JAK inhibitors (31). HZ incidence in tofacitinib‐treated patients in the tofacitinib RA clinical program was 3.9 (3.6‐4.2) (27). However, integrated analysis of five RA registries determined overall HZ incidence to be lower in the real world (range: 0.26 [0.11‐0.54] to 1.94 [1.82‐2.07]) (32); IRs in this study for tofacitinib initiators (IR 1.44 [1.03‐1.94]) and bDMARD initiators (IR 0.65 [0.52‐0.81]) are within that range. Here, the risk of HZ was greater with tofacitinib versus bDMARDs, which is consistent with other real‐world evidence showing an elevated HZ risk with tofacitinib compared with TNFi and non‐TNFi (31). There were no serious HZ events in tofacitinib initiators in this study, which is consistent with a low proportion of serious HZ events observed in the tofacitinib RA clinical program (27). Elevated HZ risk with tofacitinib should be considered in the potential therapeutic benefit:risk profile. In a Phase II placebo‐controlled trial, patients initiating tofacitinib 2 to 3 weeks after receiving a live attenuated varicella zoster virus (VZV) vaccine (ZOSTAVAX, Merck & Company) had similar VZV‐specific humoral and cell‐mediated immune responses as placebo‐treated patients, and vaccination appeared safe in patients with preexisting VZV immunity (33). In addition, the potential utility of the inactivated recombinant vaccine, which has been reported to have greater efficacy in the general population, requires further examination in patients with RA receiving advanced therapies, including JAK inhibitors.
Evaluating the impact of tofacitinib or bDMARDs on cancer risk in patients with RA is challenging because patients with RA have an increased risk of some cancers (34), and many specific cancer types are uncommon. Also, patients with a history of cancer are often excluded from RCTs, and RCTs often have a short follow‐up time and/or lack sufficient power to conclusively assess risk. Integrated data from the RA clinical program—based on cumulative tofacitinib exposure of up to 8.5 years in patients with active RA enrolled in Phase I/II/III/LTE studies (data cutoff of March 31, 2015)—reported that 173/6194 patients developed a malignancy excluding NMSC (IR 0.9 [0.8‐1.0]), and 118 patients developed NMSC (IR 0.6 [0.5‐0.7]) (27) compared with the IRs reported here for malignancy excluding NMSC (IR 0.88 [0.58‐1.27]) and NMSC (IR 1.08 [0.75‐1.51]). In this study, approximately 7% to 8% of patients with RA reported a prior malignancy (excluding NMSC).
Study strengths include that registry data represent a large number of patients in real‐world practice, including those classed as ineligible for RCTs because of comorbidities, insufficient disease activity, or prior treatments. Also, the variability of patient characteristics demonstrates the safety of tofacitinib in a less‐selected population than those commonly included in RCTs. Of the 1625 patients in whom dose was assessed, only 16 (<1.0%) were receiving a dose of tofacitinib higher than 5 mg BID, whereas 1439 (88.6%) received tofacitinib at a dose of 5 mg BID or in a modified‐release formulation of 11 mg once daily, and 170 (10.5%) received another, or an unspecified, dose of tofacitinib. Therefore, the majority of patients in the analysis were receiving the approved dose, demonstrating generalizability to the majority of real‐world patients receiving tofacitinib. The US Corrona RA registry has a collection of validated disease activity measures, and data are comparable to those of Medicare datasets; therefore, results may be more representative of the US population than data from global RCTs (18). Additionally, the multiple sensitivity analyses described provide consistent results.
Study limitations include the potentially limited generalizability outside of the US. Patients may underreport some nonserious AEs (eg, NMSC), although there is no evidence to suggest differential under‐reporting between groups. The “once exposed, always exposed” approach counted some malignancies/deaths in both cohorts; however, sensitivity analyses that censored bDMARD follow‐up at tofacitinib initiation found similar results. Also, the “once exposed, always exposed” approach means that any time‐varying baseline factors associated with the initiation of bDMARDs in malignancy/death analyses are more likely to reflect values earlier in disease course, as tofacitinib is used more frequently as later‐line therapy compared with bDMARDs. The incidence of AEs among bDMARD initiators may be underestimated, as patients switching from a previous bDMARD were eligible, and first‐time bDMARD users may have a higher risk for some AEs. Although PS methods adjust for nonrandom prescribing practices, as is the case with any observational registry, some channeling biases may persist because of unmeasured confounders. The primary analysis relied on events that were confirmed and validated after initial reports from physicians. Although not all events were adjudicated, the adjudication analyses demonstrated high positive predictive values for all endpoints. However, IRs for adjudication analyses were not reported given that rates of adjudicated events were necessarily underestimated, as adjudication was completed only for a subset of events at the time of data lock.
In conclusion, these US Corrona RA registry results provide evidence that the risk of MACE, SIEs, malignancies, NMSC, and death are comparable in patients with RA receiving tofacitinib versus those receiving bDMARDs in a large US patient population. Tofacitinib initiators had a higher rate of HZ than bDMARD initiators, consistent with the known safety profile of tofacitinib. Additionally, VTE incidence was numerically similar in patients initiating tofacitinib versus bDMARDs in this analysis. Overall, the results of this analysis are consistent with the known safety profile of tofacitinib.
AUTHOR CONTRIBUTIONS
All authors were involved in data interpretation and manuscript drafting, reviewing and development. All authors approved the final version to be submitted for publication. Dr. Kremer had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Kremer, Bingham, Cappelli, Greenberg, Madsen, Geier, Rivas, Onofrei, Barr, Litman, Dandreo, Shapiro, Connell, Kavanaugh.
Acquisition of data
Kremer, Barr, Pappas.
Analysis and interpretation of data
Onofrei, Litman.
ROLE OF THE STUDY SPONSOR
Pfizer had no role in the study design or in the collection, analysis, or interpretation of the data, the writing of the manuscript, or the decision to submit the manuscript for publication. Publication of this article was not contingent upon approval by Pfizer.
DATA SHARING STATEMENT
The Corrona dataset is based on a large United States multicenter study adhering to a number of institutional review boards, with complex logistics. Patients did not provide consent to raw data sharing during the data collection for this purpose, and the Corrona data sharing policies do not permit raw data sharing for this purpose. An aggregated limited dataset from the current analyses is available to qualified investigators with an approved protocol. Data requests may be sent to Corrona, represented by Dr. Jeffrey D. Greenberg, MD, MPH, Corrona, LLC, Waltham, Massachusetts, and New York University School of Medicine, New York, New York. E‐mail jgreenberg@corrona.org.
Supporting information
Supplementary Material
Acknowledgments
The authors thank the Serious Infection, Malignancies, and Cardiovascular Disease Adjudication Committees (Serious Infections, University of Alabama at Birmingham, Committee led by Jeffrey R. Curtis and Martha Sanderson; Malignancy, Massachusetts General Hospital, Committee led by Mark Fisher and Mark Matza; and Cardiovascular Disease, University of Toronto and Mount Sinai, Committee led by Michael Farkouh, Mary Ann McLaughlin, Jay Udell, Patrick Lawler, and Jesse Weinberger). They also thank Carol Etzel for contributions to the analysis plan, conduct of analyses, and drafting of data reports.
The views and opinions expressed within this manuscript are those of all authors and do not necessarily represent those of the sponsor.
Supported by Pfizer. The registry is sponsored by Corrona, LLC. Corrona has been supported through contracted subscriptions in the last 2 years by AbbVie, Amgen, Boehringer Ingelheim, Bristol‐Myers Squibb, Celgene, Crescendo, Eli Lilly and Company, Genentech, Gilead, Glaxo Smith Kline, Janssen, Merck, Momenta Pharmaceuticals, Novartis, Ortho Dermatologics, Pfizer Inc, Regeneron, Roche, Sun, and UCB. Medical writing support, under the guidance of the authors, was provided by Jennifer Stewart, PhD, MBA, and Anthony G. McCluskey, PhD, CMC Connect, McCann Health Medical Communications, and was funded by Pfizer in accordance with Good Publication Practice guidelines (Ann Intern Med 2015;163:461‐4). Corrona owns the Corrona Rheumatoid Arthritis registry. Pfizer paid a subscription fee to Corrona for access to the Corrona Rheumatoid Arthritis registry. Michelle Karpman at Corrona, LLC, provided medical writing support in accordance with Good Publication Practice guidelines (Ann Intern Med 2015;163:461‐4).
Joel M. Kremer has received research grants from AbbVie, Bristol‐Myers Squibb, Eli Lilly and Company, Genentech, Novartis, and Pfizer (more than $10 000 in total), and has received consulting fees or other remuneration from AbbVie, Amgen, Bristol‐Myers Squibb, Eli Lilly and Company, Genentech, Pfizer, and Regeneron/Sanofi (less than $10 000). Clifton O. Bingham III has received research grants from Bristol‐Myers Squibb (less than $10 000) and has received consulting fees or other remuneration from AbbVie, Bristol‐Myers Squibb, Eli Lilly and Company, Genentech/Roche, Gilead, Janssen, Pfizer, and Regeneron/Sanofi (less than $10 000 each). Laura C. Cappelli has received research grants from Bristol‐Myers Squibb and has received consulting fees or other remuneration from Regeneron/Sanofi Genzyme (less than $10 000). Jeffrey D. Greenberg is an employee and shareholder of Corrona. Ann M. Madsen is an employee and shareholder of Pfizer. Jamie Geier is an employee and shareholder of Pfizer. Jose L. Rivas is an employee and shareholder of Pfizer. Alina M. Onofrei is an employee of Corrona. Christine J. Barr is an employee and shareholder of Corrona. Dimitrios A. Pappas is an employee and shareholder of Corrona and has received consulting fees or other remuneration from Novartis, Roche, and Regeneron (less than $10 000). Heather J. Litman is an employee and shareholder of Corrona. Kimberly J. Dandreo is an employee of Corrona. Andrea B. Shapiro is an employee and shareholder of Pfizer. Carol A. Connell is an employee and shareholder of Pfizer. Arthur Kavanaugh has received research grants from Pfizer (less than $10 000). No other disclosures relevant to this article were reported.
REFERENCES
- 1. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet 2016;388:2023–38. [DOI] [PubMed] [Google Scholar]
- 2. Fleischmann R, Cutolo M, Genovese MC, Lee EB, Kanik KS, Sadis S, et al. Phase IIb dose‐ranging study of the oral JAK inhibitor tofacitinib (CP‐690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease‐modifying antirheumatic drugs. Arthritis Rheum 2012;64:617–29. [DOI] [PubMed] [Google Scholar]
- 3. Kremer JM, Bloom BJ, Breedveld FC, Coombs JH, Fletcher MP, Gruben D, et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: results of a double‐blind, placebo‐controlled phase IIa trial of three dosage levels of CP‐690,550 versus placebo. Arthritis Rheum 2009;60:1895–905. [DOI] [PubMed] [Google Scholar]
- 4. Kremer JM, Cohen S, Wilkinson BE, Connell CA, French JL, Gomez‐Reino J, et al. A phase IIb dose‐ranging study of the oral JAK inhibitor tofacitinib (CP‐690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum 2012;64:970–81. [DOI] [PubMed] [Google Scholar]
- 5. Tanaka Y, Suzuki M, Nakamura H, Toyoizumi S, Zwillich SH. Tofacitinib Study Investigators. Phase II study of tofacitinib (CP‐690,550) combined with methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Care Res (Hoboken) 2011;63:1150–8. [DOI] [PubMed] [Google Scholar]
- 6. Tanaka Y, Takeuchi T, Yamanaka H, Nakamura H, Toyoizumi S, Zwillich S. Efficacy and safety of tofacitinib as monotherapy in Japanese patients with active rheumatoid arthritis: a 12‐week, randomized, phase 2 study. Mod Rheumatol 2015;25:514–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burmester GR, Panaccione R, Gordon KB, McIlraith MJ, Lacerda AP. Adalimumab: long‐term safety in 23458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn's disease. Ann Rheum Dis 2013;72:517–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fleischmann R, Kremer J, Cush J, Schulze‐Koops H, Connell CA, Bradley JD, et al. Placebo‐controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med 2012;367:495–507. [DOI] [PubMed] [Google Scholar]
- 9. Kremer J, Li Z‐G, Hall S, Fleischmann R, Genovese M, Martin‐Mola E, et al. Tofacitinib in combination with nonbiologic disease‐modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med 2013;159:253–61. [DOI] [PubMed] [Google Scholar]
- 10. Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley J, Gruben D, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 2014;370:2377–86. [DOI] [PubMed] [Google Scholar]
- 11. Van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, et al. Tofacitinib (CP‐690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve‐month data from a twenty‐four‐month phase III randomized radiographic study. Arthritis Rheum 2013;65:559–70. [DOI] [PubMed] [Google Scholar]
- 12. Van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med 2012;367:508–19. [DOI] [PubMed] [Google Scholar]
- 13. Van der Heijde D, Strand V, Tanaka Y, Keystone E, Kremer J, Zerbini CA, et al. Tofacitinib in combination with methotrexate in patients with rheumatoid arthritis: clinical efficacy, radiographic, and safety outcomes from a twenty‐four‐month, phase III study. Arthritis Rheumatol 2019;71:878–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fleischmann R, Mysler E, Hall S, Kivitz AJ, Moots RJ, Luo Z, et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double‐blind, head‐to‐head, randomised controlled trial. Lancet 2017;390:457–68. [DOI] [PubMed] [Google Scholar]
- 15. Wollenhaupt J, Silverfield J, Lee EB, Curtis JR, Wood SP, Soma K, et al. Safety and efficacy of tofacitinib, an oral Janus kinase inhibitor, for the treatment of rheumatoid arthritis in open‐label, longterm extension studies. J Rheumatol 2014;41:837–52. [DOI] [PubMed] [Google Scholar]
- 16. Yamanaka H, Tanaka Y, Takeuchi T, Sugiyama N, Yuasa H, Toyoizumi S, et al. Tofacitinib, an oral Janus kinase inhibitor, as monotherapy or with background methotrexate, in Japanese patients with rheumatoid arthritis: an open‐label, long‐term extension study. Arthritis Res Ther 2016;18:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wollenhaupt J, Lee EB, Curtis JR, Silverfield J, Terry K, Soma K, et al. Safety and efficacy of tofacitinib for up to 9.5 years in the treatment of rheumatoid arthritis: final results of a global, open‐label, long‐term extension study. Arthritis Res Ther 2019;21:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kremer JM. The Corrona US registry of rheumatic and autoimmune diseases. Clin Exp Rheumatol 2016;34:S96–9. [PubMed] [Google Scholar]
- 19. Strangfeld A, Hierse F, Rau R, Burmester GR, Krummel‐Lorenz B, Demary W, et al. Risk of incident or recurrent malignancies among patients with rheumatoid arthritis exposed to biologic therapy in the German biologics register RABBIT. Arthritis Res Ther 2010;12:R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wadström H, Frisell T, Askling J, Anti‐Rheumatic Therapy in Sweden Study Group . Malignant neoplasms in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitors, tocilizumab, abatacept, or rituximab in clinical practice: a nationwide cohort study from Sweden. JAMA Intern Med 2017;177:1605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Young A, Koduri G, Batley M, Kulinskaya E, Gough A, Norton S, et al. Mortality in rheumatoid arthritis. Increased in the early course of disease, in ischaemic heart disease and in pulmonary fibrosis. Rheumatology (Oxford) 2007;46:350–7. [DOI] [PubMed] [Google Scholar]
- 22. Aviña‐Zubieta JA, Thomas J, Sadatsafavi M, Lehman AJ, Lacaille D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta‐analysis of observational studies. Ann Rheum Dis 2012;71:1524–9. [DOI] [PubMed] [Google Scholar]
- 23. Charles‐Schoeman C, Wicker P, Gonzalez‐Gay MA, Boy M, Zuckerman A, Soma K, et al. Cardiovascular safety findings in patients with rheumatoid arthritis treated with tofacitinib, an oral Janus kinase inhibitor. Semin Arthritis Rheum 2016;46:261–71. [DOI] [PubMed] [Google Scholar]
- 24. US Food and Drug Administration . Safety trial finds risk of blood clots in the lungs and death with higher dose of tofacitinib (Xeljanz, Xeljanz XR) in rheumatoid arthritis patients; FDA to investigate. 2019. URL: https://www.fda.gov/media/120485/download.
- 25. Doran MF, Crowson CS, Pond GR, O'Fallon WM, Gabriel SE. Frequency of infection in patients with rheumatoid arthritis compared with controls: a population‐based study. Arthritis Rheum 2002;46:2287–93. [DOI] [PubMed] [Google Scholar]
- 26. Crowson CS, Hoganson DD, Fitz‐Gibbon PD, Matteson EL. Development and validation of a risk score for serious infections in patients with rheumatoid arthritis. Arthritis Rheum 2012;64:2847–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cohen SB, Tanaka Y, Mariette X, Curtis JR, Lee EB, Nash P, et al. Long‐term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: integrated analysis of data from the global clinical trials. Ann Rheum Dis 2017;76:1253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pawar A, Desai RJ, Gautam N, Kim SC. Risk of admission to hospital for serious infection after initiating tofacitinib versus biologic DMARDs in patients with rheumatoid arthritis: a multidatabase cohort study. Lancet Rheumatol 2020;2:E84–E98. [DOI] [PubMed] [Google Scholar]
- 29. Harrold LR, Litman HJ, Saunders KC, Dandreo KJ, Gershenson B, Greenberg JD, et al. One‐year risk of serious infection in patients treated with certolizumab pegol as compared with other TNF inhibitors in a real‐world setting: data from a national U.S. rheumatoid arthritis registry. Arthritis Res Ther 2018;20:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cohen S, Curtis JR, DeMasi R, Chen Y, Fan H, Soonasra A, et al. Worldwide, 3‐year, post‐marketing surveillance experience with tofacitinib in rheumatoid arthritis. Rheumatol Ther 2018;5:283–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Curtis JR, Xie F, Yun H, Bernatsky S, Winthrop KL. Real‐world comparative risks of herpes virus infections in tofacitinib and biologic‐treated patients with rheumatoid arthritis. Ann Rheum Dis 2016;75:1843–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamanaka H, Askling J, Berglind N, Franzen S, Frisell T, Garwood C, et al. Infection rates in patients from five rheumatoid arthritis (RA) registries: contextualising an RA clinical trial programme. RMD Open 2017;3:e000498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Winthrop KL, Wouters AG, Choy EH, Soma K, Hodge JA, Nduaka CI, et al. The safety and immunogenicity of live zoster vaccination in patients with rheumatoid arthritis before starting tofacitinib: a randomized Phase II trial. Arthritis Rheumatol 2017;69:1969–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Simon TA, Thompson A, Gandhi KK, Hochberg MC, Suissa S. Incidence of malignancy in adult patients with rheumatoid arthritis: a meta‐analysis. Arthritis Res Ther 2015;17:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material