

Abstract
Bromodomain protein BRD4 reads histone acetylation (H3K27ac), an epigenomic mark of transcription enhancers. CCAAT enhancer binding protein delta (CEBPD) is a transcription factor typically studied in metabolism. While both are potent effectors and potential therapeutic targets, their relationship was previously unknown. Here we investigated their interplay in vascular smooth muscle cell (SMC) inflammation. Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) revealed H3K27ac/BRD4 enrichment at Cebpd in injured rat carotid arteries. While genomic deletion of BRD4-associated enhancer in SMCs in vitro decreased Cebpd transcripts, BRD4 gene silencing also diminished Cebpd mRNA and protein, indicative of a BRD4 control over CEBPD expression. Bromodomain-1, but not bromodomain-2, accounted for this BRD4 function. Moreover, endogenous BRD4 protein co-immunoprecipitated with CEBPD, and both proteins co-immunoprecipitated the Cebpd promoter and enhancer DNA fragments. These co-immunoprecipitations (coIPs) were all abolished by the BRD4-bromodomain blocker JQ1, suggesting a BRD4/CEBPD /promoter/enhancer complex. While BRD4 and CEBPD were both upregulated upon tumor necrosis factor alpha (TNF-α) stimulation of SMC inflammation (increased interleukin [IL]-1b, IL-6, and MCP-1), they mediated this stimulation via preferentially elevated expression of platelet-derived growth factor receptor alpha (PDGFRα, versus PDGFRβ), as indicated by loss- and gain-of-function experiments. Taken together, our study unravels a hierarchical yet collaborative BRD4/CEBPD relationship, a previously unrecognized mechanism that prompts SMC inflammation and may underlie other pathophysiological processes as well.
Keywords: BRD4, CEBPD, ChIP sequencing, vascular smooth muscle cell state transition
Graphical abstract
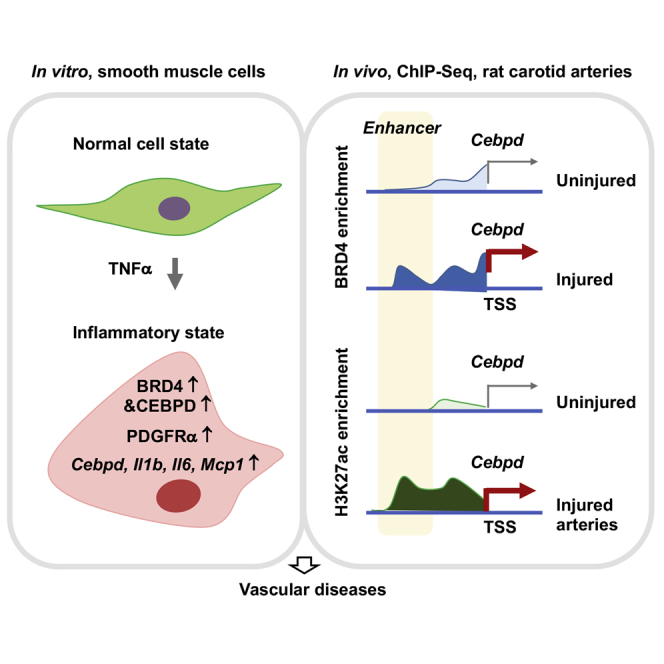
The inflammatory smooth muscle cell state is a common etiology in vascular diseases. Unraveling its underlying mechanism, this study reveals a novel interplay between an epigenetic reader of chromatin bookmarks and a master transcription factor, through in vivo and in vitro analyses of epigenomic remodeling and molecular interactions.
Introduction
Vascular smooth muscle cells (SMCs) maintain vessel wall homeostasis as a major component and signaling hub. However, upon extra- and intra-cellular perturbations, they undergo various state transitions, losing innate identity and function while acquiring new phenotypes. This SMC plasticity is now known as enabled by epigenetics1—regulations critical in biology particularly in development and disease. Depending on micro-environmental cues, SMCs may transition to inflammatory and/or migro-proliferative or other states, contributing to a range of vascular disorders such as neointimal hyperplasia (IH) that lead to stenotic vascular diseases.2 Therefore, in the pursuit of new interventional paradigms, it is important to interpret the disease mechanisms from an epigenetic perspective. However, epigenetic determinants of SMC state transitions are only beginning to be unveiled.2
Our previous reports suggested that BRD4 is a determinant of SMC state transitions in vitro and of IH in vivo,3,4 motivating further research to decipher its molecular underpinning. BRD4 belongs to the family of BETs (Bromo/ExtraTerminal domain-containing proteins), including BRD2, BRD3, BRD4, and BRDT (testis-restricted). Rapid progress, particularly in cancer research, reveals that histone acetylation reader BRD4 is closely associated with cell state/identity changes and hence critical to an array of pathogenic processes.2,5 As a result, BRD4 is being intensively targeted in human trials, for cancers and beyond. BRD4 is found to potently prompt RNA polymerase II pause release, thereby co-activating transcription elongation in response to stimulation.5 This action involves multiple factors such as the elongation factor complex and central machinery of transcription. It remains poorly understood how BRD4, a seeming global regulator, assumes its functional specificity in different cell types and micro-environments.2 Transcription factors (TFs) and enhancers are thought to confer BRD4 a gene loci specificity in the genomic landscape.5,6 However, little is known as to how BRD4 and specific TFs and enhancers govern different SMC state transitions.7
The CCAAT enhancer binding proteins (CEBPs) comprise a family of TFs highly versatile in their pathophysiological functions. They are most notably involved in adipogenesis and immune cell differentiation, among other activities. CEBPA and CEBPB have been the most studied, whereas CEBPD is much less understood. Evidence collectively implicates CEBPD as a central player in responses to inflammatory stimuli,8 as mostly reported in immune cells with a paucity of data from SMCs. CEBPD’s function is highly contextual. Dichotomous or even opposing effects mediated by CEBPD have been reported (e.g., in cell proliferation and in macrophage differentiation).8 Obviously, CEBPD-associated regulations reported in other cell types or signaling contexts cannot be simply extrapolated to SMCs. Interestingly, while CEBPD expression is generally low in normal conditions, it rapidly increases in response to environmental perturbations, such as arterial injury that induces IH.9,10 This raises an important question as to what epigenetic factors govern CEBPD’s expression and its functional impact on SMC state transitions.11
In the current study, as we sought to identify BRD4’s downstream functional mediators and CEBPD’s upstream epigenetic determinants in SMCs, the two searches converged. Using the information from chromatin immunoprecipitation and high-throughput sequencing (ChIP-seq) as a guide,12 we observed that angioplasty injury induced enrichment of BRD4 and H3K27ac at the CEBPD gene. Indeed, BRD4 dominated CEBPD expression in SMCs. Moreover, CEBPD co-immunoprecipitated with BRD4, its own promoter, and BRD4/H3K27ac-associated enhancer as well. The function of BRD4/CEBPD cooperativity manifested in heightened expression of pro-inflammatory cytokines. On the whole, this study explored a previously underappreciated area, where we identified a BRD4/CEBPD hierarchical yet collaborative relationship that critically promotes the inflammatory SMC state transition.
Results
BRD4 and H3K27ac enrichment at the CEBPD gene in injured rat carotid arteries
We previously found that blocking BETs’ bromodomains with JQ1 effectively curbed neointima progression in an authentic model of angioplasty-induced IH in rat carotid arteries.3,13 BRD4 drastically increased in the angioplasty-injured artery wall, and in vitro and in vivo data indicated that BRD4 was a determinant of IH.3,4,13 To further identify the mediators of BRD4’s pro-IH function, we performed high-throughput ChIP-seq with injured (and uninjured) carotid arteries using the same angioplasty model of IH. Angioplasty abruptly alters the SMC micro-environment as it denudes the endothelium and mechanically damages the artery wall. This action exposes SMCs to a myriad of blood-borne stimulants typical of cytokines, such as TNF-α and PDGF, which potently trigger various SMC state transitions that perpetuate IH.14,15 Arteries were collected for ChIP-seq at post-angioplasty day 7, the peak time of pro-IH molecular and cellular events.16,17 Epigenomic marks involved in transcriptional activation, including BRD4, H3K27ac, and H3K4me1,18 were chosen for ChIP-seq experiments. BRD4 and H3K27ac are associated with active enhancers, and H3K4me1 can be found with active, inactive, or poised enhancers.19
Guided by the ChIP-seq data, we noticed that the ChIP-seq peaks for BRD4 and H3K27ac were enriched at the gene of CEBPD, which is a TF poorly studied for SMC state transitions and IH. Both BRD4 and H3K27ac ChIP-seq peaks enriched in a Cebpd promoter region that was a TSS proximal enhancer (Figures 1A and 1B) and also showed strong co-localization in other loci (Figure 1C). H3K4me1 ChIP-seq peaks aligned with BRD4 and H3K27ac peaks in non-coding regions and also increased in injured (versus uninjured) arteries, albeit with relative lower overall intensity (Figure 1A). By contrast, ChIP-seq signal for H3K27me3 was negligible and did not align with the other three marks. As opposed to H3K27ac, H3K27me3 is a gene repression mark. Thus, the relatively extremely low signals from H3K27me3 and also input conferred negative controls, indicating excellent specificity of the BRD4 and H3K27ac ChIP-seq peaks. Furthermore, to validate the functional significance of BRD4/H3K27ac-associated enhancer, we performed CRISPR-mediated enhancer region deletion. As a result, Cebpd transcripts were significantly reduced, as detected by quantitative real-time PCR (Figure 1D). The mRNA level of platelet-derived growth factor receptor alpha (PDGFRα), a known target of CEBPD’s TF activity,20 was reduced as well. Together, these results suggested that BRD4 may regulate the expression of transcription factor CEBPD.
Figure 1.

Injury-induced enrichment of BRD4 and H3K27ac at Cebpd in rat carotid arteries
Balloon-injured rat left common carotid arteries and contralateral arteries (uninjured control) were collected at day 7 post angioplasty and snap frozen until use for ChIP-seq analysis. (A) ChIP-seq binding density at Cebpd. Shown are ChIP-seq peak profiles for BRD4 and histone marks. The x axis of the tracks shows genomic position, and the y axis shows ChIP-seq signal (rpm/bp). Data were obtained from injured and uninjured arteries. Non-specific input indicates low background noise. Arrow points to the proximal enhancer region. (B) Zoom-in profiles showing differential ChIP-seq binding densities in injured and uninjured artery tissues. The boxed area corresponds to the arrow-pointed site in (A). (C) Venn diagrams showing genome-wide overlap of ChIP-seq peaks for H3K27ac and BRD4; 96% and 93% of the BRD4 peaks were co-localized with H3K27Ac peaks in uninjured and injured samples, respectively. (D) CRISPR-mediated enhancer deletion in vitro. MOVAS cell lines were prepared to express Cas9 only (control, no sgRNA) or Cas9 together with enhancer-specific sgRNAs, as described in Materials and methods. Cebpd expression was assessed by quantitative real-time PCR. sgRNA, short guide RNA. Quantification: mean ± SEM; n = 3 independent repeat experiments; one-way ANOVA with Bonferroni test, ∗p < 0.05, ∗∗p < 0.01. (E) Upregulation of BRD2 in the neointima of injured rat carotid arteries. Upper panel shows BRD2 immunostaining on rat carotid artery sections, represented by uninjured and 7 days after balloon angioplasty injury to induce neointima. Red triangle points to the internal elastic lamina (IEL). Neointima is defined between the lumen and IEL. Quantification: mean ± SEM; n = 4 animals at each time point; ∗p < 0.05 compared to uninjured control.
BRD4 silencing, but not BRD2 or BRD3 silencing, diminishes CEBPD expression
The family of BETs includes BRD2, BRD3, BRD4, and BRDT, which is testis-restricted and hence irrelevant here.2 Although our previous studies showed BRD4 as the determinant of IH, BRD2 was upregulated as well in angioplasty-injured rat arteries (Figure 1E), and functional overlap of BRD2 and BRD4 has been reported in non-vascular settings.2 In addition, BRD3 appeared to have a function opposite to that of BRD4.4,21 Therefore, herein we sought to distinguish whether BRD4 is the only BET that determines CEBPD expression, via genetic silencing in MOVAS (an established cell line of SMCs) using small interfering RNAs (siRNAs) specific to each of the 3 BETs. As shown in Figure 2A, while each siRNA was effective in silencing its respective target BET, BRD4 silencing, but not BRD2 or BRD3 silencing, led to pronounced reduction of CEBPD mRNA. This result was confirmed by western blots, where only BRD4 silencing reduced CEBPD protein (Figures 2B and 2C). Thus, these results distinguished that BRD4, but not BRD2 or BRD3, was the determinant BET that controlled CEBPD expression in SMCs.
Figure 2.

Silencing BRD4 diminishes CEBPD mRNA and protein in SMCs
(A–C) MOVAS cells were transduced with lentivirus to express scrambled or a specific shRNA, cultured with fresh medium (no RNAi Max) for 24 h, and then harvested for quantitative real-time PCR (A) or western blot (B and C) analysis, respectively. Quantification: readings from triplicate quantitative real-time PCR reactions were normalized to GAPDH and averaged. The average values from 3 independent repeat experiments were then averaged again to calculate mean ± SEM (n = 3). Densitometry of western blots from independent repeat experiments was normalized to β-actin (similar band intensities on different blots) and then averaged to calculate mean ± SEM (n = 3). Statistics: one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Expression of dominant-negative BRD4 bromodomain-1, but not -2, reduces CEBPD
BRD4 interacts with specific chromatin loci by reading/binding histone acetyl-lysine bookmarks through its two tandem bromodomains (BD1 and BD2). Therefore, BD1 and/or BD2 are crucial for BRD4’s epigenetic functions.2 We thus dissected their role in regulating CEBPD expression. Interestingly, expressing a dominant-negative BD1 domain (competitor to endogenous BD1) substantially reduced CEBPD. However, a dominant-negative BD2 domain, which proved functional in our previous report using endothelial cells,4 did not have an effect here (Figures 3A–3C). Consistently, treating SMCs with Olinone, an inhibitor selective to BD1 in BETs,22 diminished CEBPD protein levels, whereas RVX208 (selective to BD2 in BETs)23 did not significantly alter CEBPD expression (Figures 3D–3F). These results together indicate that BD1, rather than BD2, was responsible for BRD4’s function in governing CEBPD expression in SMCs, which was somewhat unexpected given that in previous reports BD2 was often shown to play a major role in non-SMC settings.4,24
Figure 3.

Dominant-negative BD1 of BRD4 inhibits CEBPD expression
(A) Diagram of the constructs used to exogenously express GFP (control) and a dominant-negative domain that competes with BD1 or BD2 of BRD4. (B and C) Effect of dominant-negative BD1 or BD2 of BRD4 on CEBPD expression. (D) Diagram to show selective binding of Olinone and RVX208 to BD1 and BD2, respectively, and binding of JQ1 to both; each of these bromodomain blockers binds all three BETs (BRD2, BRD3, BRD4). (E and F) Effect of bromodomain blockers on CEBPD expression. The data of the vehicle for Olinone and that for RVX208 (E) were pooled to generate the average vehicle value in the plot (black bar, F). MOVAS cells were transduced with lentivirus to express the GFP control or dominant-negative BD1 or BD2 for 24 h before harvest for western blot analysis. For pharmacological pretreatment, cells were incubated with vehicle (equal amount of DMSO) or a bromodomain blocker (10 μM RVX208 or 20 μM Olinone) for 4 h before harvest. Quantification: densitometry of western blots from independent repeat experiments was normalized to β-actin (similar band intensities on blots) and then averaged to calculate mean ± SEM; n = 3 independent repeat experiments. Statistics: one-way ANOVA followed by Bonferroni post hoc test; ∗p < 0.05, ∗∗p < 0.01; n.s., not significant.
Endogenous BRD4 co-immunoprecipitates with the CEBPD protein in SMCs
Reports have highlighted the importance of TFs in mediating BRD4’s function of co-activating specific gene expression in cell types other than SMCs. CEBPD is a TF reportedly involved in SMC dysfunction.20,25 However, a BRD4/CEBPD cooperativity in SMC pathobiology has not been reported. We therefore performed co-immunoprecipitation (coIP) experiments to probe a possible BRD4/CEBPD physical interaction. As shown in Figure 4, endogenous BRD4 IP’ed with CEBPD specifically (versus background control), suggesting their direct interaction or indirect association but in the same protein complex. Indicative of an important role of bromodomains, the pan-BETs inhibitor JQ1, which competes with histone acetyl-lysine for binding to BD1 and BD2, blocked the BRD4/CEBPD coIP. Furthermore, an inhibitor (TSA) of histone deacetylases commonly used to preserve histone acetylation enhanced the coIP of BRD4 with CEBPD. These results revealed a previously unidentified BRD4/CEBPD physical association.
Figure 4.

Co-immunoprecipitation of endogenous BRD4 with CEBPD
MOVAS cells stably overexpressing HA-tagged empty vector (EV) or HA-tagged CEBPD (OE-CEBPD) were incubated with vehicle (equal amount of DMSO) or bromodomain blocker JQ1 (1 μM) or HDAC inhibitor TSA (1 μM) for 24 h. The cells were then harvested for IP of CEBPD using an antibody against the HA tag. (A) Quantified data. (B) Representative western blots. Quantification: densitometry of western blots from independent repeat experiments was normalized to β-actin (similar band intensities on blots) and then averaged to calculate mean ± SEM (n = 6 repeat experiments). Statistics: one-way ANOVA followed by Bonferroni post hoc test; ∗p < 0.05, ∗∗p < 0.01.
CEBPD and BRD4 both associate with the Cebpd promoter
To further investigate the function of the observed BRD4/CEBPD association in gene regulation, we performed ChIP-qPCR experiments. To validate the methodology, we chose the PDGFRα gene promoter for proof of principle, considering that PDGFRα is known as a direct target of CEBPD’s TF function in SMCs.20 As shown in Figure 5A, treatment of SMCs with TNF-α stimulated coIP of PDGFRα promoter DNA with CEBPD. By contrast, the effect on PDGFRβ was minor (Figure 5B). CEBPD gain of function (overexpression) strongly enhanced PDGFRα promoter DNA pulldown (by ∼10- to 20-fold) either in the presence or absence of TNF-α, whereas no effect (+TNF-α) or minor effect (−TNF-α) on PDGFRβ promoter was observed. Consistent with a BRD4 involvement, pretreating SMCs with JQ1 abrogated PDGFRα promoter pulldown that was enhanced either by TNF-α or CEBPD overexpression.
Figure 5.

Chromatin immunoprecipitation of CEBPD and endogenous BRD4 with the Cebpd promoter DNA
(A–D) ChIP against OE-CEBPD in SMCs. (E and F) ChIP against endogenous BRD4 in SMCs (E) or HEK293 cells (F). MOVAS cells for stable overexpression of HA-tagged EV or OE-CEBPD were starved in basal medium (DMEM + 0.5% FBS) for 24 h, pretreated with vehicle or bromodomain blocker JQ1 (1 μM) for 2 h, and then treated with TNF-α (final 20 ng/mL) for 24 h prior to harvest for ChIP-qPCR analysis. Shown on the top of (C) and (D) are the MA0836.1 and MA0836.2 motifs in the CEBPD promoter identified from JASPAR2020 database (as described in detail in the Materials and methods section). Quantification: readings from triplicate quantitative real-time PCR reactions were normalized to GAPDH and averaged. The average values from at least 3 independent repeat experiments were then averaged again to calculate mean ± SEM (n = 3–5). Statistics: one-way ANOVA followed by Bonferroni post hoc test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
More interestingly, motif search predicted potential CEBPD binding sites on its own promoter. Two human CEBPD motifs in the CEBPD promoter, MA0836.1 and MA0836.2, were identified from JASPAR2020 database, and two approaches (JASPAR Scan and MEME suite FIMO) produced the same result. Since mouse SMCs (MOVAS) were used in vitro, we identified the MA0836.2 motif in the murine genome at distal and proximal locations in the Cebpd promoter. Indeed, ChIP-qPCR indicated that while coIP of the Cebpd promoter DNA (containing the proximal CEBPD motif) with the CEBPD protein increased by ∼2- to 3-fold due to TNF-α stimulation, CEBPD gain of function further markedly enhanced this coIP (Figure 5C). In either case, JQ1 pretreatment abolished the enhancement of the CEBPD/Cebpd promoter coIP, suggesting an essential role for BRD4 in this interaction. While BRD4 as an enhancer mark enriched at Cebpd, inhibiting BRD4 reduced CEBPD expression (see Figures 1, 2, and 3). We therefore inferred that BRD4-dependent enhancer was probably involved in the BRD4/CEBPD functional complex that regulates Cebpd transcription. In support of this proposition, we observed that the ChIP-qPCR signal for a BRD4/H3K27ac-enriched enhancer region (Figure 1A) was amplified by TNF-α stimulation and CEBPD gain of function and attenuated by JQ1 pretreatment (Figure 5D).
To further confirm a role of BRD4 in the transcription-regulating complex, we performed ChIP-qPCR experiments using an antibody to IP endogenous BRD4 (Figures 5E and 5F). We found that while TNF-α stimulated coIP of the Cebpd promoter DNA with BRD4 by ∼6-fold, CEBPD overexpression further magnified this effect to nearly 25-fold (Figure 5E). In either case, JQ1 abolished the increase of coIP. Extrapolating this finding beyond SMCs, our experiments using the same conditions but HEK293A cells instead of SMCs produced a similar result of endogenous BRD4/Cebpd promoter coIP (Figure 5F).
On the whole, these (Figure 5) and other results (Figures 1, 2, 3, and 4) provided evidence for a multi-component complex involving H3K27ac, its reader (BRD4), TF (CEBPD), enhancer regions enriched with BRD4 and H3K27ac, and the promoter of Cebpd. The function of this complex manifested in the regulation of the transcription factor CEBPD’s own gene and its known target genes represented by Pdgfra.20
CEBPD’s positive role in TNF-α-induced inflammatory SMC state transition involves BRD4
Pertaining to IH pathogenesis, a positive role for CEBPD or BRD4 in proliferative and migratory SMC state transitions has been delineated in the literature20 and our own report,3 respectively. However, whether they govern the inflammatory SMC state transition has not been clearly defined. Now that our data herein revealed a CEBPD/BRD4 epigenetic complex in the control of CEBPD’s own expression, we next investigated SMC inflammation as a focus of pathobiology to verify the functional significance of this CEBPD/BRD4 partnership. The inflammatory SMC state transition is typically monitored as upregulation of major pro-inflammatory cytokines including IL-1β, IL-6, and MCP-1. Both IL-1β and IL-6 are pro-IH and atherogenic; so is MCP-1, which is key to recruitment of inflammatory cells (e.g., activated leukocytes) to the vessel wall.26,27 We used TNF-α as a stimulant, since it has been well documented that this cytokine promotes SMC inflammation and migro-proliferative behaviors in vitro and IH in vivo as revealed in different animal models.15,28, 29, 30 As shown in Figures 6A–6D, while the CEBPD mRNA in SMCs markedly increased after TNF-α stimulation, this treatment also upregulated IL-1β, IL-6, and MCP-1 mRNA to a similar extent. CEBPD gain (overexpression) and loss (silencing) of function raised and reduced the expression of these cytokines, respectively, in the absence or presence of TNF-α, supporting a specific role for CEBPD in the inflammatory SMC state transition. CEBPD’s signaling functions are highly contextual depending on cell type and stimulants,8 and a pro-inflammatory role for CEBPD was disputed in an early study.31 In this regard, our data here contributed an unambiguous result; that is, CEBPD plays a positive role in the setting of TNF-α-stimulated inflammatory SMC state transition.
Figure 6.

BRD4/CEBPD promotes inflammatory SMC state transition
(A and B) CEBPD gain of function. (C and D) CEBPD loss of function. (E) Treatment of SMCs with TNF-α upregulates BRD4 protein. (F) Effect of pretreatment with bromodomain blocker JQ1. MOVAS cells for stable overexpression of HA-tagged EV or OE-CEBPD were starved in basal medium (DMEM + 0.5% FBS) for 24 h, pretreated with vehicle or JQ1 (1 μM) for 2 h, and then treated with TNF-α (final 20 ng/mL) for 24 h prior to harvest for quantitative real-time PCR (mRNA) or western blot (protein) analysis. For CEBPD silencing, MOVAS cells were transfected with siRNA for 24 h and cultured in fresh starvation medium (0.5% FBS) for another 24 h prior to TNF-α treatment. Quantification: readings from triplicate quantitative real-time PCR reactions were normalized to GAPDH and averaged. The average values from at least 3 independent repeat experiments were then averaged again to calculate mean ± SEM (n = 3–5). Statistics: one-way ANOVA followed by Bonferroni post hoc test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
It was interesting to note that TNF-α stimulation enhanced the coIP of Cebpd promoter with BRD4 (Figure 5E) and elevated CEBPD (mRNA and protein; Figures 6A and 6C). We therefore also determined the effect of TNF-α on BRD4 expression. The data indicated that treating SMCs with TNF-α upregulated BRD4 protein by ∼9-fold (Figure 6E). Since BRD4, rather than BRD2 or BRD3, controlled CEBPD expression (see Figures 2 and 3), we expected that using JQ1 to block BET bromodomains would reveal BRD4’s function in TNF-α-stimulated inflammatory SMC state transition. Indeed, pretreatment with JQ1 abrogated TNF-α-stimulated upsurge of the SMC expression of all three cytokines (IL-1β, IL-6, MCP-1) (Figure 6F). Thus, up to this point, our data had revealed a BRD4/CEBPD physical association (Figures 4 and 5), and its function in positively regulating SMC inflammation (Figure 6)—a finding not previously reported.
CEBPD’s preferential regulation of the PDGFRα gene requires BRD4
To further interpret the functional partnership between BRD4 and CEBPD, we determined their influence on CEBPD target genes by focusing on PDGFRα, which is a potent signaling mediator known to promote SMC inflammation.3,20 While our group reported a BRD4 preferential regulation of PDGFRα over PDGFRβ in SMCs,3 another group reported that CEBPD regulated PDGFRα transcription preferentially over PDGFRβ in SMCs.20 We were intrigued by this BRD4 and CEBPD functional convergence and further determined the underlying mechanism. As shown in Figures 7A–7D, in our experiments using TNF-α to stimulate the inflammatory SMC state transition, this treatment upregulated PDGFRα markedly, yet increased PDGFRβ to a lesser degree, especially at the protein level. Using JQ1 to block BRD4’s function abolished this PDGFR upregulation. In parallel, CEBPD gain of function increased PDGFRα mRNA by ∼2- to 3-fold in the presence or absence of TNF-α but insignificantly affected PDGFRβ (Figures 7E and 7F). CEBPD silencing abolished TNF-α-stimulated PDGFRα upregulation (Figures 7G and 7H). Of note, in accordance with the observed TNF-α-stimulated BRD4 and CEBPD upregulation (Figure 6) and their converged function in preferentially upregulating PDGFRα versus PDGFRβ (Figure 7), TNF-α treatment preferentially elevated PDGFRα mRNA and protein levels, yet with only a minor effect on PDGFRβ expression (Figures 7A–7H). Therefore, the CEBPD/BRD4 partnership also functionally manifested in governing the expression level of PDGFRα, which represents a potent signaling pathway that propels the inflammatory SMC state transition.32,33
Figure 7.

BRD4/CEBPD regulate PDGFRα preferentially over PDGFRβ in SMCs
(A–D) Effect of JQ1 pretreatment on TNF-α-stimulated upregulation of CEBPD and PDGFRα. (A) shows a concentration-dependent effect of JQ1 on reducing CEBPD mRNA levels; representative western blots are shown in (C). (E and F) CEBPD gain of function. Representative western blots are shown in (E). (G and H) CEBPD loss of function. Representative western blots are shown in (G). MOVAS cells for stable overexpression of HA-tagged EV or OE-CEBPD were starved in basal medium (DMEM + 0.5% FBS) for 24 h, pretreated with vehicle or JQ1 (1 μM) for 2 h, and then treated with TNF-α (final 20 ng/mL) for 24 h prior to harvest for quantitative real-time PCR (mRNA) or western blot (protein) analysis. For CEBPD silencing, MOVAS cells were transfected with siRNA for 24 h and cultured in fresh starvation medium (0.5% FBS) for another 24 h prior to TNF-α treatment. Quantification: readings from triplicate quantitative real-time PCR reactions were normalized to GAPDH and averaged. The average values from at least 3 independent repeat experiments were then averaged again to calculate mean ± SEM (n = 3–5). Densitometry of western blots from independent repeat experiments was normalized to β-actin (similar band intensities on blots) and then averaged to calculate mean ± SEM (n = 4 repeat experiments). Statistics: one-way ANOVA followed by Bonferroni post hoc test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
The pathogenic bases of major vascular diseases trace to SMC state transitions, whereby SMCs acquire new phenotypes (e.g., inflammation) at the expense of losing their normal function. Such a SMC identity change is increasingly recognized as driven by epigenetic remodeling.2 Thus, it becomes a compelling task to interpret the largely undefined epigenetic mechanisms. We tackled this issue from the perspective of the interplay between BRD4, TF, and enhancer—all key factors in cell identity changes.34 Through ChIP-seq genome-wide survey in IH-prone arteries, we observed BRD4/H3K27ac enrichment at Cebpd. Indeed, BRD4 silencing or enhancer deletion repressed Cebpd expression, indicative of a BRD4>CEBPD hierarchical regulation. Furthermore, BRD4 and CEBPD formed a protein complex binding at CEBPD’s own promoter DNA, supporting a collaborative BRD4/CEBPD relationship. The functional importance of this hierarchical yet collaborative BRD4/CEBPD partnership manifested in promoting the inflammatory SMC state transition. Overall, our results reveal a BRD4/CEBPD-governed SMC pathophysiological mechanism.
One of the interesting findings in this study is that CEBPD acted as a master TF in collaboration with BRD4 in the inflammatory SMC state transition. Among over a thousand TFs, only a small number of them are characterized as master TFs. First, they are critically important for cell identity (i.e., cell type or state).6,19,35 One example is c-Myc, a powerful driver of oncogenic cell-state transitions.6 In the current study, while elevating CEBPD increased the expression of salient markers of the inflammatory SMC state, silencing CEBPD kept these markers at basal levels. As such, CEBPD played a key role in the transition of SMCs to an inflammatory state. Second, master TFs are often found to associate with BRD4 and enhancers.19,36 This is true for CEBPD, as herein revealed. Evidence includes the following: (1) A physical association between CEBPD and BRD4 was evident in their coIP; (2) BRD4 enrichment at enhancers near Cebpd was identified through ChIP-seq analysis; and (3) CEBPD coIP’ed enhancer DNA, as indicated by ChIP-qPCR. Interestingly, echoing our finding, CEBPD was recently found to bind inflammatory enhancers in aortic endothelial cells, although a BRD4 involvement was not reported.34 Third, another prominent feature of master TFs is that they regulate their own transcription, a mechanism thought to efficiently and rapidly activate transcription and cell-state transitions in response to environmental perturbation.6 Indeed, CEBPD binding motifs were identified in its own promoter, and ChIP-qPCR showed that the CEBPD protein IP’ed its own promoter DNA.
Essentially all the CEBPD physical associations, including that with the BRD4 protein and its own promoter and enhancer, could be abolished by using JQ1 to block the interaction of BRD4 bromodomains with acetylated histone. This indicates a critical role for BRD4 in CEBPD’s master TF function. In fact, aside from their physical association, we also observed a CEBPD/BRD4 functional association. First, BRD4 determined CEBPD expression levels. We further distinguished that BD1, but not BD2, was important for this BRD4 function—a finding worth attention, since BD1 and BD2 are highly similar and poorly differentiated in the literature for their biological functions.4,22,24 Second, while TNF-α as a micro-environmental (extracellular) stimulant upregulated CEBPD which prompted the inflammatory SMC state transition, BRD4 blockade by JQ1 abolished this CEBPD upregulation. Third, while TNF-α and CEBPD overexpression each preferentially increased PDGFRα over PDGFRβ, BRD4 inhibition with JQ1 abolished this effect. Although JQ1 also binds to the bromodomains of other BETs, in the current study we could attribute its effect largely to BRD4 because our data established that BRD4, but not BRD2 or BRD3, dictated CEBPD expression. In aggregate, these results revealed a functional convergence of BRD4 and CEBPD in the setting of inflammatory SMC state transition. In addition, that TNF-α upregulated BRD4, which in turn governed CEBPD expression, indicates a BRD4>CEBPD functional hierarchy.
Thus, we have identified a novel collaborative/hierarchical partnership between a powerful epigenetic reader (BRD4) and a master TF (CEBPD), which are physically and functionally associated in the complex that also includes CEBPD’s own promoter. The function of this partnership manifested in the setting of TNF-α-induced inflammatory SMC state transition and also in the activation of CEBPD’s target genes represented by PDGFRα, a signaling mediator crucial to SMC state transitions.33 As TFs bind to promoters and enhancers of the corresponding genes,34 a TF/BRD4 association is thought to help localize BRD4 to certain genomic loci, thereby defining its functional specificity.2 It is noted that TF/BRD4 pairing is highly cell-type and stimulant dependent.5 For instance, in the inflammatory endothelial cell-state transition, nuclear factor κB (NF-κB) was the master TF that paired with BRD4 to potently propagate this pathogenic process.19 To the best of our knowledge, the CEBPD/BRD4 partnership was not previously recognized, likely due to a paucity of information on CEBPD in contrast to extensively studied CEBPA. A BRD4 functional association with CEBPA in adipogenesis was recently reported,35,36 highlighting context-dependent, differential functions of different CEBP family members.
In the BRD4/CEBPD partnership observed herein, BRD4 appears to play a central role orchestrating a multi-factor assembly that drives the inflammatory SMC state transition. As depicted in the schematic for the experimental setting of the current study (Figure 8), TNF-α as extracellular signal stimulates upregulation of the BRD4 protein. While being anchored at H3K27ac and stocked up at enhancers, BRD4 partners with CEBPD to promote the transcription of Cebpd and other target genes such as Pdgfra,20 and the inflammatory SMC state transition ultimately results. This proposition is analogous to a model emerging from research in oncology and other fields.5,6 That is, BRD4 “rallies” enhancer-enriched TF(s) with the transcription elongation machinery and co-factors (e.g., MED1), while reading histone acetylation marks via its bromodomain(s).37 By doing so, BRD4 may “usher” the multi-factor assembly to select gene loci enabling their quick activation, which drives cell state changes. As such, though seemingly a global regulator, BRD4 may assume functional specificity via context-specific associations with combinatorial TFs and enhancers at chromatin sites that are bookmarked (e.g., H3K27ac) in the epigenomic landscape.2 Nevertheless, since little is known about the BRD4/CEBPD duet in SMC (or other) cell-state transitions, future research would generate exciting new knowledge to help decipher the underlying mechanisms.
Figure 8.

Schematic proposal for a BRD4/CEBPD partnership associated with chromatin
TNF-α as extracellular signal stimulates upregulation of epigenetic reader protein BRD4, which, while reading H3K27ac, functions in the same complex with transcription factor CEBPD and unknown co-factors (light green background). BRD4 and CEBPD stock up at Cebpd promoter and enhancer to prompt the transcription of Cebpd. Increased CEBPD elevates the expression of its target genes such as Pdgfra and propels the inflammatory SMC state transition.
Conclusions
Our results reveal a previously unidentified partnership between BRD4 and CEBPD that is both hierarchical (transcriptional control) and collaborative (physical association). This mechanism underlies the inflammatory SMC state transition, a process permissive for IH. A long-standing barrier in translational medicine is that TFs and enhancers inherently lack druggability, limiting their targetability. Serendipitously, as implicated herein, their functional potency incumbent on BRD4 exposes an “Achilles’ heel,” that is, the druggable BRD4 bromodomains, the blocking of which could collapse the assembly of BRD4 with context-specific TFs, enhancers, and transcription machinery. Therefore, justification is compelling for more research on the BRD4/CEBPD partnership, so that essential information would become available for precision-oriented therapeutic interventions of IH and beyond.
Materials and methods
Materials
Various resources including kits and reagents are presented in Table S1.
Animals
All animal studies conform to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health) and protocols approved by the Institutional Animal Care and Use Committee. Male Sprague-Dawley rats purchased from Charles River Laboratories (Wilmington, MA, USA) were used for experiments (at 300–350 g body weight).
Rat carotid artery balloon angioplasty model
To induce IH, angioplasty was performed to injure rat carotid arteries, following our previous report.3 Briefly, rats were kept anesthetized with 2%–2.5% isoflurane (inhaling, 2 L/min). After the left common carotid artery was dissected, a 2-F balloon catheter (Edward Lifesciences, Irvine, CA, USA) was inserted into the common carotid artery through an arteriotomy in the external carotid artery. The balloon was inflated (at 1.5 atm), withdrawn to the carotid bifurcation, and then deflated. This action was repeated three times. Finally, blood flow was resumed, and the neck incision was closed. The animal was kept on a 37°C warm pad to recover. For postoperative analgesia, in addition to carprofen and bupivacaine, buprenorphine (0.03 mg/kg) was subcutaneously injected. At indicated time points, common carotid arteries were collected, and either cryopreserved for ChIP sequencing (see below) or fixed for cross-section preparation and immunohistochemistry to detect BET protein expression, as described in detail in our earlier report.3
Artery tissue ChIP sequencing and data processing
Artery collection was performed at 7 days after balloon angioplasty. To preserve the real-time epigenetic information, balloon-injured and uninjured (contralateral) common carotid artery segments were severed and snap-frozen in liquid N2. Artery tissues from 50 rats were pooled for ChIP experiments, and high-throughput sequencing and data quality control were performed by Active Motif per company standard procedures. Briefly, chromatin was isolated after adding lysis buffer, followed by disruption with a Dounce homogenizer. Genomic DNA was sheared to an average length of 300–500 bp by sonicating the lysates, and the segments of interest were immunoprecipitated using an antibody (4 μg) against BRD4, H3K27a, H3K27me3, or H3K4me1. The protein/DNA complexes eluted from beads were treated with RNase and proteinase K, crosslink was reversed, and the ChIP DNA was then purified for use in the preparation of Illumina sequencing libraries. Standard steps included end-polishing, dA-addition, adaptor ligation, and PCR amplification. The DNA libraries were quantified and sequenced on Illumina’s NextSeq 500, as previously described.18 Sequence reads were aligned to the reference genome Rn5; peak locations were identified using Macs2 algorithm38 and annotated based on UCSC RefSeq. A p value threshold of enrichment of 1e−9 was used for all datasets. ChIP-seq reads aligning to each region were extended by 200 bp, and the density of reads per base pair was calculated and normalized to the total number of million mapped reads. As such, the unit of read density is reads per million mapped reads per base pair (rpm/bp). In-house shell and R scripts (https://www.r-project.org) were used for data integration. Enhancers were mapped using the ROSE software package available at younglab.wi.mit.edu/super_enhancer_code.html. To describe genome-wide correlation between BRD4 occupancy, H3K27 acetylation, and chromatin accessibility, numbers of peaks/hotspots within 5 kb of one another were counted and presented as a Venn diagram. Integrative Genomics Viewer (IGV) (https://www.broadinstitute.org/igv/) was used for visualization. Annotation files were downloaded from UCSC.
CEBPD motif search
Motif search was performed in the human database using two approaches, JASPAR Scan function and MEME suite FIMO function. Both led to the identification of two CEBPD motifs in the CEBPD promoter with a p value of 0.0006: MA0836.1 and MA0836.2 (http://jaspar.genereg.net/matrix/MA0836.1/; http://jaspar.genereg.net/matrix/MA0836.2/).
Since the in vitro experiments were performed using MOVAS (mouse SMC line), we also searched the Cebpd gene promoter region in UCSC mm9 (mouse) genome; position: chr16: 15887379–15889638; Strand: +. The search reported the CEBPD motif MA0836.2 at two locations, with relative profile scores of 83% and 81%, respectively; retrieve sequences: http://genome.ucsc.edu/cgi-bin/das/mm9/dna?segment=chr16:15886379,15887378.
Vascular SMC culture and transfection with siRNA
SMCs (MOVAS) were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin-streptomycin with 5% CO2 at 37°C. For transfection with siRNA, cells were cultured to 60%∼80% confluence; the RNAi Max transfection reagent was then added and incubated for 12 h. The cells recovered in fresh DMEM (no lipofectamine, 0.5% FBS) for 12 h and starved for 24 h in DMEM with 0.5% FBS. The cells were then incubated with 20 ng/mL TNF-α or solvent control (0.1 BSA, 4 mM HCl) for another 24 h before harvest for various analyses. The siRNA sequences are listed in Table S2.
Lentiviral constructs for expressing dominant-negative BRD4 bromodomains
Construction of vectors for the expression of GFP (control) or its fusion with BRD4-BD1 or BRD4-BD2, lentivirus packaging in Lenti-X 293 cells, and viral transduction of MOVAS cells were performed as we recently reported,4 with minor modifications. The crude viral solution was concentrated using Lenti-x concentrator (Takara, cat. no. 631232) to a final concentration of 108–109 IFU/mL using the Lenti-x quantitative real-time PCR Titration Kit (Takara, cat. no. 631235). Lentivirus with MOI of 10 was used for transduction of MOVAS cells. Cells cultured to a 70% confluency were changed to starvation medium (0.5% FBS), and lentivirus plus polybrene (Santa Cruz., cat. no. sc-134220) was added and incubated for 6 h. The cells were then cultured for recovery in fresh full medium (10% FBS) for 24 h before use in experiments.
Lentiviral-mediated SMC stable cell lines
For CEBPD gain-of-function studies, the Cebpd gene was cloned into the Lenti-hemagglutinin (HA) vector (from Addgene), and the construct for empty vector (Lenti-HA) and that for CEBPD overexpression (Lenti-HA-CEBPD) were prepared. For CRISPR-mediated enhancer deletion, enhancer-specific short guide RNA (sgRNA) sequences were cloned into a Cas9-expressing vector (lentiCRISPR v2, Addgene, cat. no. 52961); the lentiCRISPR v2 vector served as control. For genetic silencing of BRD2, BRD3, and BRD4, their respective small hairpin RNA (shRNA) sequences were each cloned into the pLKO.1 puro vector (Addgene, cat. no. 8453). To silence each gene, two shRNA sequences (Table S2) were used for cloning and lentivirus packaging, which resulted in a selected stable mouse smooth muscle cell line (MOVAS). For lentivirus packaging, each of the above constructs was transfected into the LentiX-293 cell line together with plasmids PMD2G and PSPAX2. After incubation for 24 h, the virus-containing supernatant was collected. Lentivirus was purified and used to transduce MOVAS, and the stable cell line was selected against puromycin.
Western blot analysis
MOVAS cells were lysed in RIPA buffer. After quantifying with DC protein assay, equal amount of protein (20–40 μg) was loaded and separated in 9% gel by SDS-PAGE and then transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was incubated with a primary antibody overnight at 4°C, rinsed 3×, and then incubated with a horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Specific protein bands were illuminated by applying enhanced chemiluminescence (ECL) reagents (Thermo Fisher Scientific; cat. no. 32106) and then recorded with Azur LAS-4000 Mini Imager (GE Healthcare Bio-Sciences, Piscataway, NJ, USA). Band intensity was quantified with ImageJ. All antibodies are included in Table S3.
Quantitative real-time PCR
Total RNA was extracted from cell lysates using the TRIzol reagent following the manufacturer’s instruction (Thermo Fisher Scientific, 15596026) and used for cDNA synthesis with the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific, 4368814). In each 20 μL of reaction, 10 ng of cDNA was amplified through quantitative real-time PCR using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, A25778), and mRNA levels were determined using 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA). The data were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using the ΔΔCt method. The primers are listed in Table S4.
CoIP
We used Pierce Crosslink Immunoprecipitation Kit (Thermo Scientific, 26147) and followed the manufacturer’s instruction. Briefly, cells were rinsed and incubated with ice-cold hypotonic buffer (20 mM HEPES, 20% glycerol, 10 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA and 0.1% NP-40) supplemented with 1 mM dithiothreitol, and protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, 87785). Nuclei were collected and sonicated, and the lysates were cleared by centrifugation. Magnetic beads (Dynabeads Protein A or G, Invitrogen) preloaded with an anti-HA antibody were added to the supernatant and incubated at 4°C for 4 h. The beads were washed 3× with the binding buffer (50 mM Tris-Cl, 150 mM NaCl, 1 mM EDTA, 10% glycerin), and SDS sample buffer was then added to elute the coIP’ed proteins for western blot determination.
ChIP-qPCR assay
ChIP was performed as we recently reported39 using the Pierce Magnetic ChIP kit (Thermo Fisher Scientific, 26157). Briefly, cells were cross-linked with 1% formaldehyde, and the reaction was stopped by glycine. The cells were washed and lysed for nuclei extraction. Micrococcal nuclease was added to the nuclei suspension to digest the DNA for 15 min at 37°C, and then MNase Stop Solution was added to stop the reaction. The recovered nuclei were re-suspended in IP Dilution Buffer and sonicated (four 5-s pulses at 20 W for 1 × 106 cells) to disrupt the nuclear membrane. Chromatin extracts containing DNA fragments (∼500 bp in each) were immunoprecipitated by incubating with a specific antibody (or immunoglobulin G [IgG] control) overnight at 4°C. ChIP-grade Protein A/G Magnetic beads were added and incubated for ∼2–4 h at 4°C. RNase A and proteinase K were used to digest RNA and protein. The purified DNA was used for quantitative real-time PCR as described above. Primer sequences are presented in Table S4. The primers for the enhancer region: Forward, TAGTTCTGGTCTCGTGGCGG; reverse, TTCCTGTTTGTGCGGTTTGG.
Statistical Analysis
Data are presented as mean ± standard error of the mean (SEM). Normality of the data was assessed based on Shapiro-Wilk normality test prior to statistical calculation using Prism 6.0 software (GraphPad). One-way ANOVA followed by Bonferroni post hoc test was applied, as specified in each figure legend; p < 0.05 was considered significant. For ChIP-seq data, statistical analyses were performed using SAS/STAT software, version 9.2 (SAS Institute, Cary, NC, USA).
Acknowledgments
This work was supported by NIH grants R01HL-133665 (to L.-W.G.) and R01HL-143469 and R01HL-129785 (to K.C.K. and L.-W.G.).
Author contributions
Q.W. performed most of the in vitro experiments. H.G.O. analyzed ChIP-seq data. B.W. and L.-W.G. designed the study. M.Z. and Y.H. participated in in vitro and in vivo experiments. G.U. conducted animal surgery disease model. L.-W.G. wrote the manuscript. L.-W.G. and K.C.K. provided financial support.
Declaration of interests
The authors declare that no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.02.021.
Supplemental information
References
- 1.Gomez D., Swiatlowska P., Owens G.K. Epigenetic Control of Smooth Muscle Cell Identity and Lineage Memory. Arterioscler. Thromb. Vasc. Biol. 2015;35:2508–2516. doi: 10.1161/ATVBAHA.115.305044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borck P.C., Guo L.W., Plutzky J. BET Epigenetic Reader Proteins in Cardiovascular Transcriptional Programs. Circ. Res. 2020;126:1190–1208. doi: 10.1161/CIRCRESAHA.120.315929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang B., Zhang M., Takayama T., Shi X., Roenneburg D.A., Kent K.C., Guo L.W. BET Bromodomain Blockade Mitigates Intimal Hyperplasia in Rat Carotid Arteries. EBioMedicine. 2015;2:1650–1661. doi: 10.1016/j.ebiom.2015.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang M., Wang B., Urabe G., Huang Y., Plutzky J., Kent K.C., Guo L.W. The BD2 domain of BRD4 is a determinant in EndoMT and vein graft neointima formation. Cell. Signal. 2019;61:20–29. doi: 10.1016/j.cellsig.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi J., Vakoc C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell. 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradner J.E., Hnisz D., Young R.A. Transcriptional Addiction in Cancer. Cell. 2017;168:629–643. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das S., Senapati P., Chen Z., Reddy M.A., Ganguly R., Lanting L., Mandi V., Bansal A., Leung A., Zhang S. Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells. Nat. Commun. 2017;8:1467. doi: 10.1038/s41467-017-01629-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ko C.Y., Chang W.C., Wang J.M. Biological roles of CCAAT/Enhancer-binding protein delta during inflammation. J. Biomed. Sci. 2015;22:6. doi: 10.1186/s12929-014-0110-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelkenberg U., Wagner A.H., Sarhaddar J., Hecker M., von der Leyen H.E. CCAAT/enhancer-binding protein decoy oligodeoxynucleotide inhibition of macrophage-rich vascular lesion formation in hypercholesterolemic rabbits. Arterioscler. Thromb. Vasc. Biol. 2002;22:949–954. doi: 10.1161/01.atv.0000017198.16727.27. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan-Albuquerque N., Bogaert Y.E., Van Putten V., Weiser-Evans M.C., Nemenoff R.A. Patterns of gene expression differentially regulated by platelet-derived growth factor and hypertrophic stimuli in vascular smooth muscle cells: markers for phenotypic modulation and response to injury. J. Biol. Chem. 2005;280:19966–19976. doi: 10.1074/jbc.M500917200. [DOI] [PubMed] [Google Scholar]
- 11.Takeji M., Kawada N., Moriyama T., Nagatoya K., Oseto S., Akira S., Hori M., Imai E., Miwa T. CCAAT/Enhancer-binding protein delta contributes to myofibroblast transdifferentiation and renal disease progression. J. Am. Soc. Nephrol. 2004;15:2383–2390. doi: 10.1097/01.ASN.0000136426.01160.2F. [DOI] [PubMed] [Google Scholar]
- 12.Zhang M., Wang B., Urabe G., Ozer H.G., Han R., Kent K.C., Guo L.-W. Angioplasty-induced epigenomic remodeling entails BRD4 and EZH2 hierarchical regulations. bioRxiv. 2020 doi: 10.1101/2020.03.12.989640. [DOI] [Google Scholar]
- 13.Wang B., Chen G., Urabe G., Xie R., Wang Y., Shi X., Guo L.W., Gong S., Kent K.C. A paradigm of endothelium-protective and stent-free anti-restenotic therapy using biomimetic nanoclusters. Biomaterials. 2018;178:293–301. doi: 10.1016/j.biomaterials.2018.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leppänen O., Janjic N., Carlsson M.A., Pietras K., Levin M., Vargeese C., Green L.S., Bergqvist D., Ostman A., Heldin C.H. Intimal hyperplasia recurs after removal of PDGF-AB and -BB inhibition in the rat carotid artery injury model. Arterioscler. Thromb. Vasc. Biol. 2000;20:E89–E95. doi: 10.1161/01.atv.20.11.e89. [DOI] [PubMed] [Google Scholar]
- 15.Rectenwald J.E., Moldawer L.L., Huber T.S., Seeger J.M., Ozaki C.K. Direct evidence for cytokine involvement in neointimal hyperplasia. Circulation. 2000;102:1697–1702. doi: 10.1161/01.cir.102.14.1697. [DOI] [PubMed] [Google Scholar]
- 16.Marx S.O., Totary-Jain H., Marks A.R. Vascular smooth muscle cell proliferation in restenosis. Circ. Cardiovasc. Interv. 2011;4:104–111. doi: 10.1161/CIRCINTERVENTIONS.110.957332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shishikura D., Kataoka Y., Honda S., Takata K., Kim S.W., Andrews J., Psaltis P.J., Sweeney M., Kulikowski E., Johansson J. The Effect of Bromodomain and Extra-Terminal Inhibitor Apabetalone on Attenuated Coronary Atherosclerotic Plaque: Insights from the ASSURE Trial. Am. J. Cardiovasc. Drugs. 2019;19:49–57. doi: 10.1007/s40256-018-0298-8. [DOI] [PubMed] [Google Scholar]
- 18.Ozer H.G., El-Gamal D., Powell B., Hing Z.A., Blachly J.S., Harrington B., Mitchell S., Grieselhuber N.R., Williams K., Lai T.H. BRD4 Profiling Identifies Critical Chronic Lymphocytic Leukemia Oncogenic Circuits and Reveals Sensitivity to PLX51107, a Novel Structurally Distinct BET Inhibitor. Cancer Discov. 2018;8:458–477. doi: 10.1158/2159-8290.CD-17-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown J.D., Lin C.Y., Duan Q., Griffin G., Federation A., Paranal R.M., Bair S., Newton G., Lichtman A., Kung A. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell. 2014;56:219–231. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Z.H., Kitami Y., Takata Y., Okura T., Hiwada K. Targeted overexpression of CCAAT/enhancer-binding protein-delta evokes enhanced gene transcription of platelet-derived growth factor alpha-receptor in vascular smooth muscle cells. Circ. Res. 2001;89:503–508. doi: 10.1161/hh1801.096265. [DOI] [PubMed] [Google Scholar]
- 21.Dutzmann J., Haertlé M., Daniel J.M., Kloss F., Musmann R.J., Kalies K., Knöpp K., Pilowski C., Sirisko M., Sieweke J.T. BET bromodomain-containing epigenetic reader proteins regulate vascular smooth muscle cell proliferation and neointima formation. Cardiovasc. Res. 2021;117:850–862. doi: 10.1093/cvr/cvaa121. [DOI] [PubMed] [Google Scholar]
- 22.Gacias M., Gerona-Navarro G., Plotnikov A.N., Zhang G., Zeng L., Kaur J., Moy G., Rusinova E., Rodriguez Y., Matikainen B. Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem. Biol. 2014;21:841–854. doi: 10.1016/j.chembiol.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Picaud S., Wells C., Felletar I., Brotherton D., Martin S., Savitsky P., Diez-Dacal B., Philpott M., Bountra C., Lingard H. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA. 2013;110:19754–19759. doi: 10.1073/pnas.1310658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao L., Li J., Fu Y., Zhang M., Wang B., Ouellette J., Shahi P.K., Pattnaik B.R., Watters J.J., Wong W.T., Guo L.W. Photoreceptor protection via blockade of BET epigenetic readers in a murine model of inherited retinal degeneration. J. Neuroinflammation. 2017;14:14. doi: 10.1186/s12974-016-0775-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaplan-Albuquerque N., Van Putten V., Weiser-Evans M.C., Nemenoff R.A. Depletion of serum response factor by RNA interference mimics the mitogenic effects of platelet derived growth factor-BB in vascular smooth muscle cells. Circ. Res. 2005;97:427–433. doi: 10.1161/01.RES.0000179776.40216.a9. [DOI] [PubMed] [Google Scholar]
- 26.Meloche J., Lampron M.C., Nadeau V., Maltais M., Potus F., Lambert C., Tremblay E., Vitry G., Breuils-Bonnet S., Boucherat O. Implication of Inflammation and Epigenetic Readers in Coronary Artery Remodeling in Patients With Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2017;37:1513–1523. doi: 10.1161/ATVBAHA.117.309156. [DOI] [PubMed] [Google Scholar]
- 27.Gomez D., Baylis R.A., Durgin B.G., Newman A.A.C., Alencar G.F., Mahan S., St Hilaire C., Müller W., Waisman A., Francis S.E. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 2018;24:1418–1429. doi: 10.1038/s41591-018-0124-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimmerman M.A., Selzman C.H., Reznikov L.L., Miller S.A., Raeburn C.D., Emmick J., Meng X., Harken A.H. Lack of TNF-alpha attenuates intimal hyperplasia after mouse carotid artery injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002;283:R505–R512. doi: 10.1152/ajpregu.00033.2002. [DOI] [PubMed] [Google Scholar]
- 29.Jiang Z., Shukla A., Miller B.L., Espino D.R., Tao M., Berceli S.A., Ozaki C.K. Tumor necrosis factor-alpha and the early vein graft. J. Vasc. Surg. 2007;45:169–176. doi: 10.1016/j.jvs.2006.08.049. [DOI] [PubMed] [Google Scholar]
- 30.Pan X., Wang B., Yuan T., Zhang M., Kent K.C., Guo L.W. Analysis of Combined Transcriptomes Identifies Gene Modules that Differentially Respond to Pathogenic Stimulation of Vascular Smooth Muscle and Endothelial Cells. Sci. Rep. 2018;8:395. doi: 10.1038/s41598-017-18675-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takata Y., Kitami Y., Yang Z.H., Nakamura M., Okura T., Hiwada K. Vascular inflammation is negatively autoregulated by interaction between CCAAT/enhancer-binding protein-delta and peroxisome proliferator-activated receptor-gamma. Circ. Res. 2002;91:427–433. doi: 10.1161/01.res.0000031271.20771.4f. [DOI] [PubMed] [Google Scholar]
- 32.Ha S.E., Lee M.Y., Kurahashi M., Wei L., Jorgensen B.G., Park C., Park P.J., Redelman D., Sasse K.C., Becker L.S. Transcriptome analysis of PDGFRα+ cells identifies T-type Ca2+ channel CACNA1G as a new pathological marker for PDGFRα+ cell hyperplasia. PLoS ONE. 2017;12:e0182265. doi: 10.1371/journal.pone.0182265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takata Y., Kitami Y., Okura T., Hiwada K. Peroxisome proliferator-activated receptor-gamma activation inhibits interleukin-1beta -mediated platelet-derived growth factor-alpha receptor gene expression via CCAAT/enhancer-binding protein-delta in vascular smooth muscle cells. J. Biol. Chem. 2001;276:12893–12897. doi: 10.1074/jbc.M011655200. [DOI] [PubMed] [Google Scholar]
- 34.Hogan N.T., Whalen M.B., Stolze L.K., Hadeli N.K., Lam M.T., Springstead J.R., Glass C.K., Romanoski C.E. Transcriptional networks specifying homeostatic and inflammatory programs of gene expression in human aortic endothelial cells. eLife. 2017;6:e22536. doi: 10.7554/eLife.22536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown J.D., Feldman Z.B., Doherty S.P., Reyes J.M., Rahl P.B., Lin C.Y., Sheng Q., Duan Q., Federation A.J., Kung A.L. BET bromodomain proteins regulate enhancer function during adipogenesis. Proc. Natl. Acad. Sci. USA. 2018;115:2144–2149. doi: 10.1073/pnas.1711155115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee J.E., Park Y.K., Park S., Jang Y., Waring N., Dey A., Ozato K., Lai B., Peng W., Ge K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 2017;8:2217. doi: 10.1038/s41467-017-02403-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borck P.C., Guo L.-W., Plutzky J. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ. Res. 2020;126:1190–1208. doi: 10.1161/CIRCRESAHA.120.315929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie X., Urabe G., Marcho L., Stratton M., Guo L.W., Kent C.K. ALDH1A3 Regulations of Matricellular Proteins Promote Vascular Smooth Muscle Cell Proliferation. iScience. 2019;19:872–882. doi: 10.1016/j.isci.2019.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.