

Abstract
Purpose:
Evaluating drug responses using primary patient derived cells ex vivo represents a potentially rapid and efficient approach to screening for new treatment approaches. Here, we sought to identify neratinib combinations in HER2 mutant non-small cell lung cancer (NSCLC) patient xenograft derived organotypic spheroids (XDOTS) using a short term ex vivo system.
Methods:
We generated two HER2 mutant NSCLC PDX models (DFCI359 (HER2 exon19 755_757LREdelinsRP) and DFCI315 (HER2 exon20 V777_G778insGSP)) and used the PDX tumors to generate XDOTS. Tumor spheroids were grown in a microfluidic device and treated ex vivo with neratinib based drug combinations. Live/dead quantification was performed by dual labeling deconvolution fluorescence microscopy. The most efficacious ex vivo combination was subsequently validated in vivo using the DFCI359 and DFCI315 PDXs and a HER2YVMA genetically engineered mouse model (GEMM).
Results:
Both neratinib and afatinib, but not gefitinib, induced cell death in DFCI359 XDOTS. The combinations of neratinib/trastuzumab, and neratinib/temsirolimus enhanced the therapeutic benefit of neratinib alone in DFCI315 and DFCI359. The combination of neratinib and trastuzumab in vivo was more effective compared to single agent neratinib or trastuzumab and was associated with more robust inhibition of HER2 and downstream signaling.
Conclusions:
The XDOTS platform can be used to evaluate therapies and therapeutic combinations ex vivo using PDX tumors. This approach may accelerate the identification and clinical development of therapies for targets with no or few existing models and/or therapies.
Keywords: lung cancer, HER2, mutation, combination therapy
Introduction
Tumor genotyping has expanded the repertoire of biomarkers in non-small cell lung cancers (NSCLC) and as such has become the primary diagnostic tool in predicting responses to targeted therapies(1). Notably, expression of mutant epidermal growth factor receptor (EGFR), or oncogenic fusions such as those of anaplastic lymphoma kinase (ALK), ROS1, RET, or NTRK1 receptor tyrosine kinases (RTKs) has demonstrated good correlation with responses to tyrosine kinase inhibitors (TKIs) (2–6). Nevertheless, similar success is yet to be seen in a number of other NSCLC genotypes, including in-frame exon 20 insertions in HER2. The irreversible dual EGFR/HER2 inhibitors afatinib (NCT00796549), dacomitinib (NCT00548093), and neratinib (NCT01827267) showed little clinical benefit as monotherapies in HER2-mutant lung cancer, all with response rates of < 10 % (7–9). Although the combination of afatinib and rapamycin demonstrated promising combination efficacy in a preclinical in vivo study using a genetically engineered mouse model (GEMM) of HER2 mutant NSCLC, the subsequent clinical trial (NCT01822767) evaluating the combination of neratinib and temsirolimus resulted in only a minimal improvement in response rate (14% vs. 0%) compared to single agent neratinib (10,11). More recently, the trastuzumab based antibody drug conjugate (ADC), ado-trastuzumab (T-DM1), has demonstrated some encouraging activity in HER2 mutant NSCLC and next generation ADCs (DS-8201a; NCT03505710) are undergoing clinical evaluation (12,13). Based on the current landscape of available HER2-directed therapies, and the lack of approved therapies for HER2 mutant NSCLC, additional studies are needed to identify therapeutic strategies for this subset of NSCLC patients.
Models generated directly from cancer patients’ tumors including cell lines, patient derived xenografts (PDXs), or organoids are increasingly being used to screen for novel treatment approaches (14–16). However, the process of cell line development and the cells’ adaptation to growth in a two-dimensional environment can render some tumors that were drug-sensitive in vivo resistant in vitro. PDX experiments can often take months to complete due to slow tumor growth kinetics, and are impractical to perform in scale requisite for more comprehensive drug combination screens. In addition to these intrinsic limitations of the preclinical models, the identification of effective HER2-directed treatments is further complicated by the commercial availability of only one HER2 mutant NSCLC cell line, H1781 (17).
We recently described a novel ex vivo system of organotypic tumor spheroids growth in a 3-dimensional microfluidic device (DOTS) (18,19). The device is designed to support short term (< 7 days) culture of freshly harvested primary tumor cells and associated immune cells resuspended in collagen for the duration of a screen, and allows for conventional immunofluorescence and microscopy based analysis. Thanks to its scalability, the DOTS system allows for rapid evaluation of multiple different therapies using biopsies derived from mouse models; primary murine models (MDOTS) or patient derived xenografts (XDOTS) or directly from patients (PDOTS) for which preclinical models cannot be established or don’t exist. In the current study we have adopted this system to study new therapeutic approaches for HER2 mutant NSCLC using tumors derived from HER2 mutant PDXs.
Materials and Methods
Cell lines and drug compounds
The EGFR mutant (HCC827 GR and PC9 GR) and ALK rearranged cell lines (DFCI 32) have been previously characterized and described (20–22). Ba/F3 cells were a generous gift from the laboratory of Dr. David Weinstock (in 2014) and cultured as previously described (21,23). NIH-3T3 cells were purchased from American Type Culture Collection (ATCC). All human cancer cells were authenticated in May 2017 using the Promega GenePrint 10 System at the RTSF Research Technology Support Facility in the Genomic Core Laboratory, Michigan State University. All murine mutant Ba/F3 and NIH-3T3 cells were not authenticated because their short tandem repeat profile has not been made publicly available but were sequenced to ensure they possess the correct mutations. All cell lines tested negative for Mycoplasma using the Mycoplasma Plus PCR Primer Set (Agilent). All cell lines were passaged and used for no longer than 4 weeks before new cells with similar passage numbers were thawed for all described experiments. Gefitnib, afatinib, poziotinib, crizotinib, alpelisib, osimertinib and AZD8055 were purchased from Selleck Chemicals. Stock solutions of all drugs were prepared in DMSO and stored at −80°C. Trastuzumab and TDM1 were purchased from the DFCI pharmacy. Neratinib was obtained from Puma Biotechnology, Inc. The concentrations of each of the drugs used in the in vitro and in vivo studies were based on prior studies whereby the specific agents have demonstrated effective target inhibition (24–30).
Generation of HER2 mutant Ba/F3 and NIH-3T3 cells
The HER2 755_757LREdelinsRP mutation was introduced via site directed mutagenesis into the pDNR-Dual Donor Vector (Clontech) containing wild type HER2 using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Tech) according to the manufacturer’s instructions. Two rounds of mutagenesis were necessary to create the HER2 755_757LRE delinsPR mutation. The first round introduced the HER2_L755P substitution mutation using primers F-5’-AAATTCCAGTGGCCATCAAAGTCCCGAGGGAAAACACATCCCCC-3’ and R-5’-GGGGGATGTGTTTTCCCTCGGGACTTTGATGGCCACTGGAATTT-3’. The second round introduced the E757 deletion using primers F-5’-GCCATCAAAGTCCCGAGdelAAACACATCCCCCAAAG-3’ and R-5’-CTTTGGGGGATGTGTTTdelCTCGGGACTTTGATGGC-3’. All constructs were confirmed by DNA sequencing. The constructs were shuttled into the retroviral vector JP1540 or lentiviral vector JP1698 using the BD CreatorTM System (BD Biosciences) and Ba/F3 and NIH-3T3 cells were infected with lentivirus according to standard protocols and as previously described (23). Stable clones were obtained by selection in puromycin (1 μg/ml).
Focus formation Assay
NIH/3T3 cells expressing WT HER2 or the HER2 755_757LREdelinsRP mutation were cultured in DMEM with 10% bovine calf serum. Once cells reached confluency, serum was reduced to 5%. Plates were stained with 0.5% crystal violet solution two weeks later.
Patient-derived lung tumor xenografts and genetic engineered mouse models
PDX models were generated from core needle biopsies, surgical biopsies or pleural effusions from lung cancer patients at the Dana-Farber Cancer Institute (DFCI) at the Belfer Center for Applied Cancer Science under IRB-approved protocols. All patients provided written informed consent and the studies were performed in accordance to the Declaration of Helsinki. Tumor cells isolated from pleural effusions and surgical tumor tissue (3 mm x3 mm x3 mm) were implanted subcutaneously, while tumors from core biopsies were implanted under the sub-renal capsule. Female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Mice were 7 weeks old or older when tumor cells or tumor tissues were implanted.
The HER2YVMA GEMM SH26 was established in the Wong Lab at DFCI and previously published (10). Six breeders of the tetO-HER2 mutant mice and CCSP-rtTA mice were maintained to generate inducible lung-specific bitransgenic hHER2mt/CCSP-rtTA cohorts. Pups were genotyped and those that were positive were fed on a regular diet for ~7–8 weeks after which 2000ppm doxycycline chow was used to begin tumor induction. Tumors were grown for ~4 weeks until they reached ~300 mm3. MRI imaging of tumors before and during treatment was performed as previously described (10,21). All breeding, mouse husbandry, and in vivo treatment experiments were performed with the approval of the Dana-Farber Cancer Institute (Boston, MA) Animal Care and Use Committee.
PDX derived organotypic tumor spheroids (XDOTS)
Tumor spheroid culture was carried out as previously described with slight modifications (18). Following harvsesting, fresh tumor specimens were maintained on ice in RPMI supplemented with 10% FBS (Gemini Bio-Products) and penicillin–streptomycin (Fisher Scientific). Tumors were minced using sterile scalpel and razor blades in prewarmed to 37°C full media + 100 U/mL collagenase type IV (Life Technologies) and 50 μg/mL DNase I (Roche) for approximately 20 minutes. Digested samples were strained sequentially through a 100-μm and 40-μm filter and spheroids smaller than 100μm but greater than 40 μm were transferred to an ultralow-attachment tissue culture plate (Corning) and grown in RPMI media overnight at 37°C. Cells were resuspended in type I rat tail collagen (Corning) at a concentration of 2.8 mg/mL and loaded into the gel ports of the DAX-1 3-D cell culture chip (AIM Biotech) at a concentration of 30 to 50 spheroids/device, hydrated with full media with appropriate drugs or drug combinations, and incubated in humidity chambers at 37°C, 5% CO2 for 72–96 hours. Baseline viability determination were performed on a separate device prior to drug treatment.
Viability and Immunofluorescence
For immunofluorescence studies, XDOTS were blocked with FcR blocking reagent (BioLegend) for 30 minutes at room temperature. Baseline viability was defined by the presence of calcein AM (Invitrogen) staining. Tumor content was assessed by an anti-human EpCAM antibody (clone 9C4, BioLegend). For the samples not expressing EpCAM, tumor content was defined by the morphology of Hoechst 33342 (Invitrogen)-stained nuclei and negative expression of a cocktail of anti-human CD90 (clone 5E10, BioLegend), and anti-mouse markers CD45 to identify mouse immune cells (clone 30-F11, BioLegend), and CD47 for mouse fibroblast and endothelial cells (clone miap301, BioLegend) in another channel.
The assessment of live/dead end-point was performed using either ViaStain AO/PI Staining (Nexcelom) for 20 min or by 10ug/mL solution of Hoechst 33342 (Life Technologies) and 1μg/mL solution of Propidium Iodide (Thermo Fisher Scientific) in full media (H/PI) for 45 minutes.
Image analysis
The entire device was imaged on a Nikon Eclipse 80i fluorescence microscope equipped with automated motorized stage (Proscan), Z-stack (Prior) and Zyla 5.5 sCMOS camera (Andor). Image capture and analysis were performed using the NIS-Elements AR software package. Dead cell quantitation was performed by measuring total PI positive cell area ; live cell assessment was done either by measuring total AO positive cell area or by subtracting dead cell area from total area of Hoechst 33342 staining. AO/PI and H/PI produced similar results in the samples where tumor cells had low cytoplasmic to nucleus ratio (data not shown), but in the samples with large cytoplasm AO tended to overestimate live cell area and H/PI staining was chosen instead.
Tumor and stromal viability was evaluated separately in the samples with low tumor content (<80%) by live/dead four-color fluorescent staining. At least 5 fields of view from 2–3 wells were captured for each treatment condition at 200x total magnification and viability of tumor and non-tumor cells was analyzed semi quantitively using the NIS-Elements AR software package.
Results
Establishment of patient derived HER2 mutant NSCLC xenograft models
In order to develop new HER2 mutant models we obtained research biopsies, under an IRB approved protocol, from NSCLC patients with known HER2 mutant NSCLC. Between August 2010 and June 2017 we obtained 15 biopsies from HER2 mutant NSCLC patients and placed them either subcutaneously (n=9) or in the subrenal capsule (n=6) of NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice, with the goal of propagating the tumor tissue. Of the 15 potential models, only two, DFCI315 and DFCI359, were successfully established as stable PDX models (Fig. 1A; Supplementary Table S1 and Supplementary Fig. S1A). The growth kinetics (passage 2) were faster for DFCI 315 compared to DFCI 359 (Figure 1A). Both models were histologically characterized as adenocarcinomas with moderate (2+) expression of HER2 as detected by immunohistochemistry (IHC) (Figure 1B). DFCI315 harbored a known and previously characterized HER2 exon 20 insertion mutation (V777_G778insGSP (also referred to as G778_P780dup)), while DFCI359 carried a novel HER2 exon 19 deletion mutation (755_757LREdelinsRP) (Figure S1B). In addition, the DFCI 359 tumor also contains a mutation in IDH (IDH R132C) and TP53 (G187_splice and loss of heterozygosity (LOH)) while the DFCI 315 tumor contains a high copy number gain (as measured by NGS) in CDK4 and MDM2 (data not shown). By sequence homology, the HER2 mutation present in DFCI359 corresponds to a common EGFR exon 19 deletion mutation (Figure S1C), suggesting that this mutant is likely the oncogenic driver in DFCI359. As the HER2 mutation in DFCI359 had not been previously characterized, we evaluated its oncogenic potential using two independent assays. Expression of the HER2 755_757LREdelinsRP mutant resulted in IL-3 independent cell growth in Ba/F3 cells, and colony formation in NIH/3T3 cells (Figure 1C and S1D). Ba/F3 cells expressing HER2 755_757LREdelinsRP were sensitive to covalent HER2 kinase inhibitors neratinib, poziotinib, and afatinib but not to the EGFR inhibitor gefitinib (Figure 1D). Of note, we were not able to establish a stable PDX from a NSCLC patient harboring the common HER2 YVMA mutation despite several attempts. In addition, neither DFCI315, nor DFCI359 was able to grow in 2D culture as a cell line (data not shown). We also established an EGFR mutant (L858R/T790M) PDX (DFCI 282) from a patient who had developed acquired resistance to erlotinib (Table S1) for use in our XDOT validation studies.
Figure 1.

Characterization of patient derived HER2 mutant NSCLC xenograft models A. Comparative growth kinetics of DFCI 315 and DFCI 359. Each curve represents a separate mouse B. Representative H&E and HER2 immunohistochemistry stains. C. Expression of the HER2 755_757LREdelinsRP mutant in Ba/F3 cells results in IL-3 indepdent cell growth. D. HER2 755_757LREdelinsRP Ba/F3 cells are sensitive to HER2 kinase inhibitors. Cells were treated with different drugs at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted relative to untreated controls.
Establishment of XDOT platform to assess drug response
Given our inability to establish DFCI315 and DFCI359 as stable cell lines, we sought to use an alternative ex vivo system where we could efficiently evaluate new therapeutic strategies for HER2 mutant NSCLC. To that end, we optimized our recently developed DOTS system, which had previously been used to assess the efficacy of immune checkpoint inhibition (18,19). The central component of this platform is a microfluidic device which supports the survival and growth of both, established and primary tumor, stromal and immune cells ex vivo for up to one week (18,19). Previously established methods were adapted to facilitate the dissociation of tumors isolated from stable PDX models into tumor spheroids 40–100μm in diameter. In addition, and in contrast to the prior studies, we included a baseline quality control step using 4-color immunofluorescence. For samples with aggregate viability >20%, the remaining tumor material was resuspended in collagen, loaded in the device, and cultured as spheroids (~ 30–50 per condition, in triplicates) in the presence of drug or vehicle for up to 96 hours. For samples with baseline tumor content (TC) > 80%, drug efficacy was assessed by dual-color fluorescent staining with propidium iodide (PI) marking dead cells combined with either the live-cell stain acridine orange (AO) or general nuclear counterstain Hoechst 33342 (Hoechst). For samples with < 80% TC, efficacy was quantified by counting calcein AM-positive hEpCAM+/hCD90−/mCD45−/mCD47−cells. Off-target drug toxicity was monitored by measuring viability of individual stromal cells (i.e. hEpCAM-). Cell death quantification was performed independently of cell proliferation rate, and as such wasn’t vulnerable to the growth rate variability inherent to ex vivo models (Figure 2).
Figure 2.
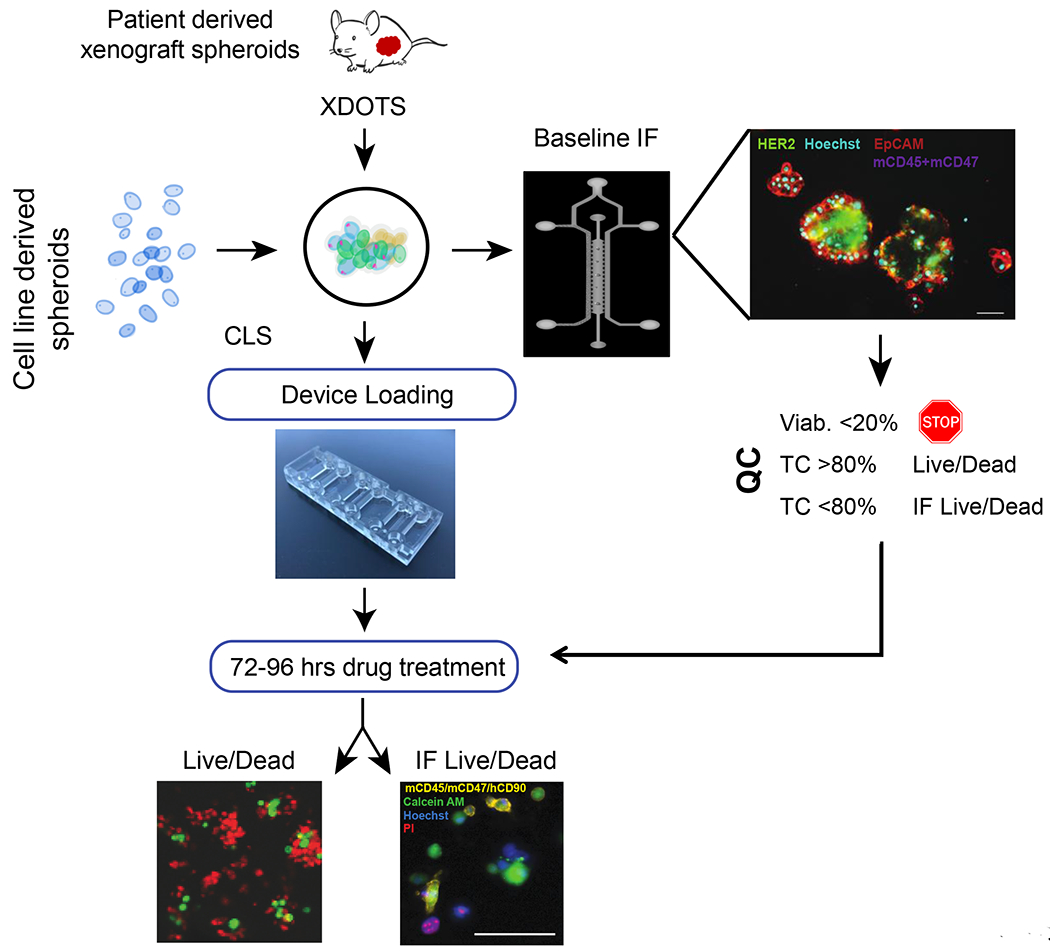
PDX (XDOT) organotypic spheroid and cell line spheroid (CLS) workflow. Tumors are harvested from PDXs and allowed to recover overnight in ultralow attachment (ULA) plates. Cell lines are used the same day. After dissociation and filtration isolation, spheroids that are between 100 μm and 40 μm are seeded and evaluated for tumor content (TC; EpCAM+, CD45/CD47/CD90−) and viability (calcein/ propidium iodide; PI) by immunofluorescence. If TC > 80% and viability > 20%, live/dead ratios (AO/PI) are obtained. If TC < 80% and viability > 20%, endpoint viability is quantified by individual cell count in hEpCAM+/mCD45−/mCD47−/mCD90− tumor and hEpCAM- stromal cells. Samples with viability < 20% are excluded from the analysis.
Validation of XDOTS system using characterized NSCLC models
In order to evaluate whether cells grown in our three-dimensional device recapitulate drug responses seen in previously reported acquired resistance models, we generated spheroids using cell lines (CLS; cell line spheroids) with previously characterized resistance mechanisms (21). In a dose-escalated CLS assay, we show that the gefitinib resistant EGFR mutant PC9 GR4 (EGFR E746_A750/T790M) is highly sensitive to osimertinib (IC50 of 12nM), an irreversible 3rd generation EGFR inhibitor that targets EGFR T790M (Figure 3A). Similar results were obtained when the PC9 GR4 cells were let adhere to flat-bottom plates or cultured in ultralow attachment plates for the assay (Figure S2A). We next established XDOTS using an EGFR mutant, erlotinib resistant, osimertinib sensitive PDX model, DFCI282 (EGFR L858R/T790M) (Table S1) (Figure 3B). The DFCI282 XDOTS were assessed for sensitivity to erlotinib or osimertinib in a dose-escalated assay and were remarkably sensitive to osimertinib, with close to 100% inhibition at ≥100nM (Figure 3C).
Figure 3.
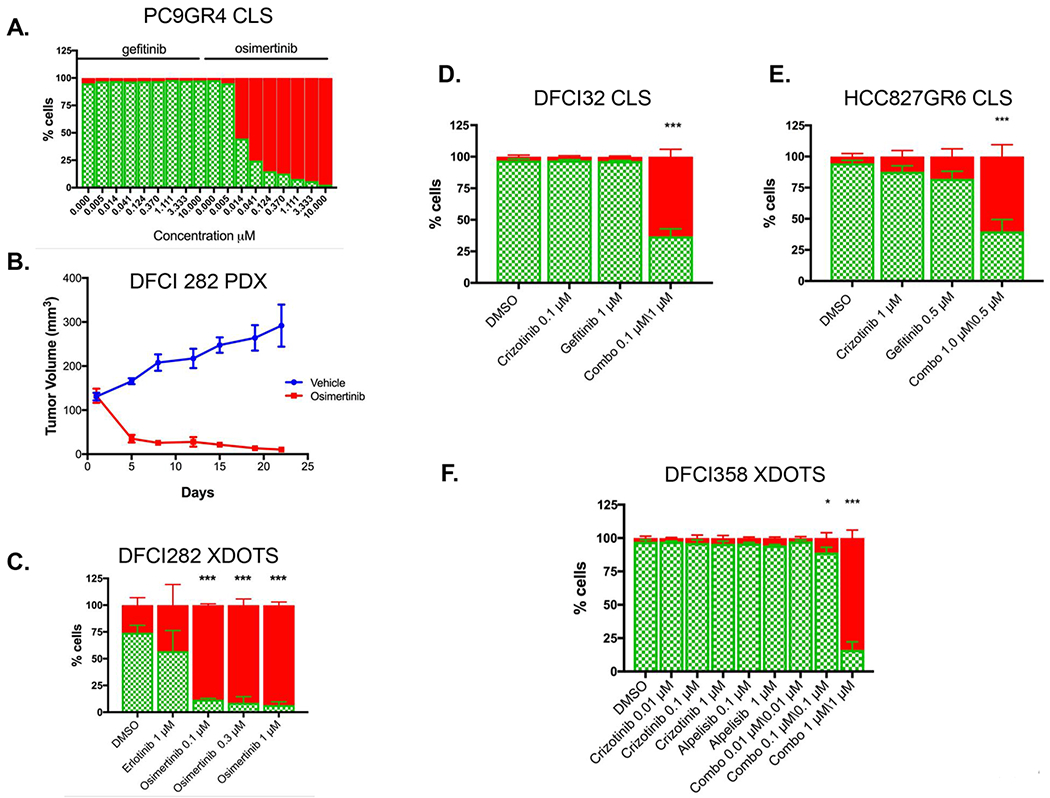
CLS and XDOTS recapitulate responses observed in previously characterized models of drug resistance. A. Osimertinib and gefitinib dose response in PC9GR CLS. Live (AO = green) / dead (PI = red) are normalized to cell area. B. In vivo efficacy study of osimertinib or vehicle in DFCI 282 (EGFR L858R/T790M) PDX tumor model (mean tumor volume +/− SEM; n=3 each). C. Life/dead analysis of DFCI282 XDOTS following ex vivo treatement with osimertinib or erlotinib. D. Live (AO = green) / dead (PI = red) analysis of DFCI 32 CLS treated with crizotinib (0.1μM), gefitinib (1μM), and crizotinib/gefitinib combination (0.1μM/1μM). E. HCC827 GR6 CLS treated with crizotinib (0.1μM), gefitinib (1μM), and crizotinib/gefitinib combination (0.1μM/1μM). F. Live /dead analysis of DFCI 358 XDOTS treated with the indicated concentrations of crizotinib, alpelisib or the combination of both agents. All comparisons were performed by two way ANOVA with Tukey multiple comparison tests (* p< 0.05; ** p< 0.01; *** p< 0.001)
We further assayed cell lines or PDXs with described resistance mechanisms that respond only to a combination of therapies and which we had previously orthogonally validated (20,22,31). Analyzing CLS derived from DFCI32, an EML4-ALK driven cell line that we previously reported to be resistant to single agent crizotinib through EGFR and HER2 co-activation, we demonstrated its sensitivity to the combination of 0.1μM crizotinib with 1μM gefitinib (63% death) while being resistant to either of the single agents (crizotinib: 3% death; gefitinib 4% death; Figure 3D). Using CellTiter-Glo as a cross-validation of the DOTS assay, we established that the IC50 for the crizotinib-gefitinib combination in this model was 25nM, translating to more than 1000-fold shift in sensitivity relative to the single agent treatments (Figure S2B). We next evaluated the gefitinib resistant EGFR mutant HCC827GR6 cell line (EGFR Del E746_A750/MET amplified) grown as spheroids in our microfluidic device and demonstrated its sensitivity to the combination of gefitinib and crizotinib (62% death; Figure 3E) and relative resistance to either drug as a single agent, consistent with prior reports (20). As an orthogonal assay, we monitored apoptosis in the HCC827GR6 cells in real time following treatment with gefitinib alone, crizotinib alone or with the combination of both drugs, and demonstrated a marked increase in apoptosis induction only in the cells treated with the combination (Figure S2C). Similarly in DFCI358 XDOTS, which were generated from a crizotinib resistant KRAS amplified MET ex14 mutant (MET c.2903_3028+67del193insA) PDX, the crizotinib/ alpelisib combination previously published to be effective in this model resulted in 90% death in the XDOT system (Figure 3F) (31). Collectively, these findings suggest that sensitivity to either single agent or to combination targeted therapy can be accurately and efficiently assessed using the DOTS system.
Screening of drug combinations in HER2 mutated XDOTS
Having established that our DOTS system can generate highly reproducible data corroborated by standard methods, we set out to use the platform to screen for effective targeted therapies for HER2 mutant lung cancer, a genotype with minimal therapeutic options due to the lack of stable patient derived mutant models. Using the system, we specifically sought to identify drugs that would enhance the effects of neratinib, a panHERTKI, when used in combination against HER2 mutant patient derived material. We first generated XDOTS from the DFCI359 PDX tumors and treated them with 0.5 μM gefitinib, neratinib, or afatinib for 3 days as single agents. Both neratinib and afatinib (covalent panERBB inhibitors), but not gefitinib (ATP competitive inhibitor), induced tumor cell death, which is the expected finding for this particular HER2 genotype (Figure 4A). Using fluorescence microscopy, we further affirmed that the effects of neratinib were specific to the tumor cells (Figure 4B). Encouraged by these results we set out to test combinations of neratinib, the TORC1/TORC2 kinase inhibitor AZD8055 and the anti-HER2 directed antibody trastuzumab in DFCI359 XDOTS. Preclinically, co-inihibition of mTOR and HER2 signaling had emerged as a promising therapeutic strategy, to which end the mTOR inhibitor temserolimus was combined with neratinib in a clinical trial, serving as a rationale for our drug candidate selection (10,11). In place of neratinib, trastuzumab is also sometimes used in combination with chemotherapy for HER2 mutant NSCLC (32). Furthermore, in HER2 amplified breast cancer models, treatment with a HER2 TKI can lead to an increase in cell surface levels of HER2 enhancing the effect of trastuzumab (33–35). However, this strategy has not been tested in HER2 mutant NSCLC. In DFCI359 XDOTS, single agents AZD8055 and trastuzumab were ineffective at clinically achievable concentrations (Figure 4C). In contrast, the combination of trastuzumab and neratinib was significantly more effective, inducing ~ 50% cell death (Figure 4C). AZD8055 in combination with neratinib was also more effective than either single agent treatment (Figure 4C). We also evaluated these agents in the DFCI315 model. While neratinib alone was already relatively efficacious in this model (48% cell death vs. 18% cell death in DFCI359) the anti-tumor efficacy was further enhanced by the addition of trastuzumab (65% cell death; Figure 4D). The combination of neratinib and trastuzumab is likely additive in both the DFCI 359 and 315 models. No formal analyses of synergy were performed.
Figure 4.
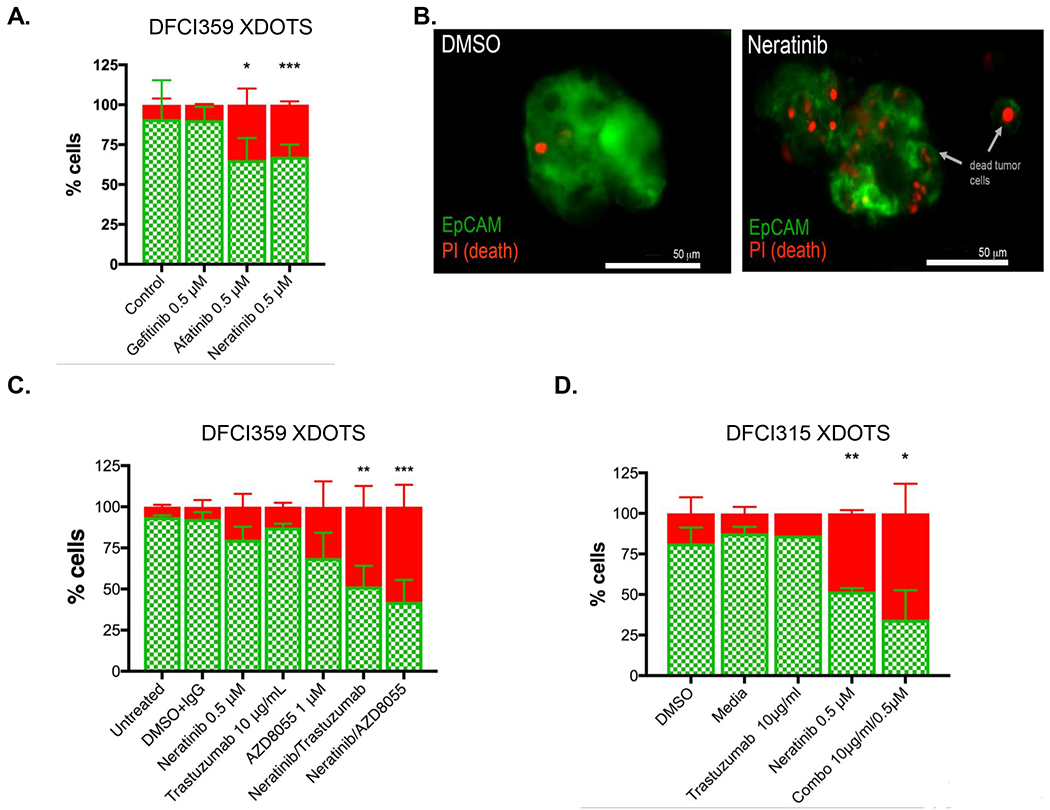
Efficacy of HER2 targeted therapies in DFCI 359 and DFCI 315 XDOTS. A. Live (AO = green)/dead (PI = red) analysis of DFCI 359 following ex vivo treatment with gefitinib (0.5 μM), afatinib (0.5 μM), or neratinib (0.5 μM); n=3, pooled biological replicates. B. Deconvolution fluorescence microscopy (Green = EpCAM, red = PI) following 3 days of neratinib treatment demonstrates that the therapeutic effect is specific to the tumor cells and not stroma (right). C. Percent live (AO = green) / dead (PI = red) of DFCI 359 XDOTS (HER 2 755_757LREdelinsRP) following a 72-hour ex vivo treatment with neratinib, trastuzumab, AZD8055 or the combination of the different drugs. D. Percent live/dead of XDOTS 315 (HER2 exon20 V777_G778insGSP) treated with neratinib (0.5 μM), trastuzumab (10μg/mL) or with the combination of both agents. All comparisons were performed by two way ANOVA with Tukey multiple comparison tests (* p< 0.05; ** p< 0.01; *** p< 0.001)
In vivo evaluation of combination therapy in HER2 mutant NSCLC.
In order to determine whether the findings made using the XDOTS system are predictive of in vivo efficacy, we tested the different drug combinations in the corresponding HER2 mutant PDX models in vivo. DFCI359 PDX-bearing animals were treated for 28 days with neratinib alone (40 mg/kg; oral gavage daily for 28 days), trastuzumab alone (20 mg/kg; intraperitoneal injection; twice weekly for 4 weeks), temsirolimus alone (20 mg/kg; intravenous injection, every 3 days for 5 doses) or with the combinations of neratinib/temsirolimus or neratinib/trastuzumab (n = 8 mice/cohort). The most efficacious treatment was neratinib/trastuzumab, which consistently resulted in tumor regressions (Figures 5A and 5B). Maximum tumor regression was 48% and 22% in mice treated with neratinib/trastuzumab and neratinib/temsirolimus, respectively, while only minimal tumor growth inhibition was observed following treatment with neratinib (18% TGI) or trastuzumab alone (28% TGI) (Figure 5B). Eight mice treated with neratinib/trastuzumab combination therapy showed significant tumor regression (>30%) with mice achieving up to a 75% decrease in tumor volume compared to baseline. In comparison, no mice showed >30% tumor regression in the vehicle or neratinib cohort (Figure 5B). The order of most to least efficacious treatment in vivo was similar to that observed in the XDOTS system (compared Figures 4C and 5B). A pharmacodynamic study, performed in parallel with the efficacy study for 4 days (daily neratinib alone or in combination with one dose of trastuzumab), revealed effective inhibtion of pHER2 with more modest impact on downstream signaling (Figure 5C).
Figure 5.
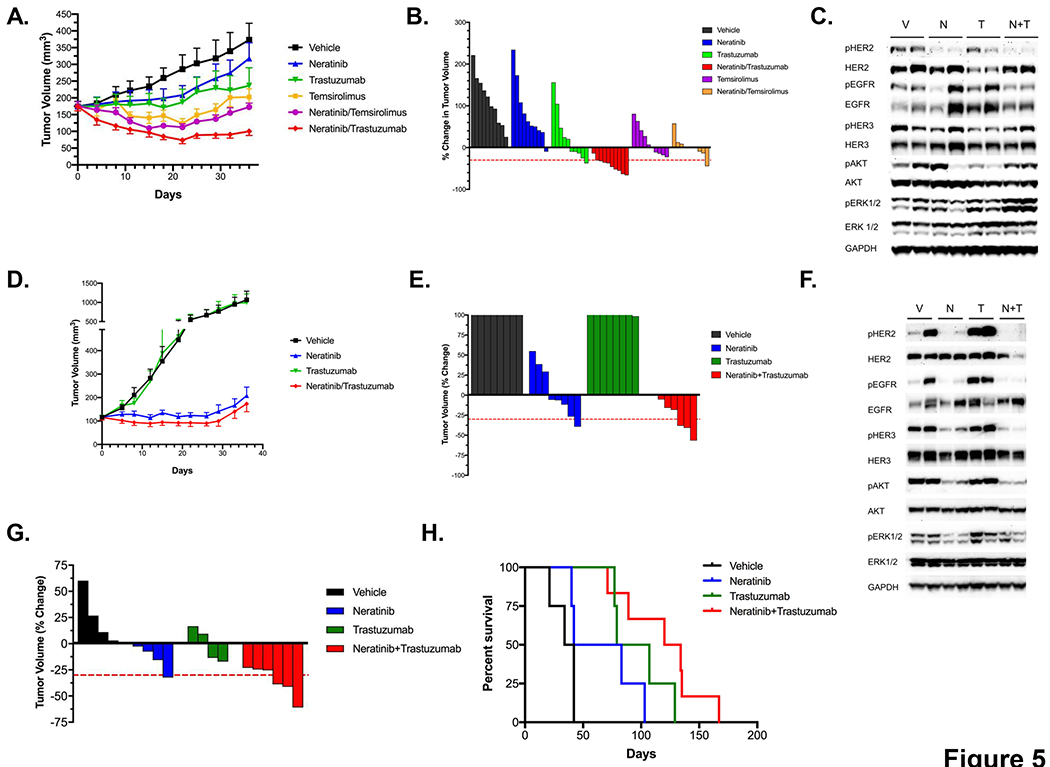
Evaluation of the neratinib/trastuzumab combination in vivo. A. Tumor volumes (TV; mean +/− SEM) over time of PDX DFCI 359 dosed with neratinib (40mg/kg), trastuzumab (20 mg/kg), temsirolimus (20 mg/kg), and their combinations (n = 8 mice/cohort). B. Waterfall plot of PDX DFCI 359 (% change in TV) following 4 weeks of treatment with neratinib, trastuzumab, temsirolimus, or their combinations. C. Pharmacodynamic study in DFCI 359 following treatment with vehicle (V), neratinib (N), trastuzumab (T) or with the combination of neratinib and trastuzumab (N+T). Cell extracts were immunoblotted to detect the indicated proteins. D. Tumor volumes (mean +/− SEM) over time of PDX DFCI 315 treated in vivo with neratinib (40 mg/kg), trastuzumab (20 mg/kg) or with the combination of both agents (n=8mice/cohort). E. Waterfall plot of PDX 315 following 4 weeks of treatment using drugs in C. F. Pharmacodynamic study in DFCI 315 following treatment with vehicle (V), neratinib (N), trastuzumab (T) or with the combination of neratinib and trastuzumab (N+T). Cell extracts were immunoblotted to detect the indicated proteins. G. Waterfall plot of HER2 YVMA GEMs following 2 weeks of treatment with with vehicle (n=4) neratinib (40 mg/kg; n=4), trastuzumab (20 mg/kg; n=4) or with the combination of both drugs (n=6). H. Kaplan-Mayer survival curve of mice in F. treated with the indicated drugs.
We employed a similar study design assessing the efficacy of HER2 targeted therapies in the DFCI315 PDX model. The same order of efficacy was observed in vivo as in the XDOTS system with the combination of neratinib/trastuzumab exhibiting comparable degree of growth inhibition as neratinib alone (Figures 5D and 5E). We did not include temsirolimus alone or in combination with neratinib in this model given the superiority of neratinib/trastuzumab to the neratinib/temsirolimus combination in the DFCI359 model. The effects of neratinib and/or trastuzumab inhibition were further assessed in a pharmacodynamic (PD) study, evaluating on-target activity of these agents against HER2 and downstream signaling at the molecular level (Figure 5F). DFCI315 tumor bearing animals were treated with neratinib alone (daily), trastuzumab alone (once) or with the combination of both agents for 4 days and harvested 2 hours after the last treatment. Tumor sample analysis revealed that neratinib effectively reduced HER2 phosphorylation and inhibited major downstream signaling targets including pAKT and pERK. Notably, the combination of neratinib and trastuzumab resulted in a greater and nearly complete inhibition of pHER2, pHER3 and pAKT compared to the single agents (Figure 5F).
As neither the DFCI315 nor the DFCI359 model contain the common HER2 YVMA mutation, we evaluated the efficacy of neratinib alone, trastuzumab alone or the combination of both agents in a previously reported HER2YVMA genetically engineered mouse model (GEMM) (10). Mice were evaluated for tumor formation using serial MRI imaging and those with established tumors were treated for 6 weeks and monitored by serial MRI imaging. The HER2YVMA model responded dramatically to the combination therapy with neratinib and trastuzumab: all mice (n=6) treated with the neratinib/trastuzumab combination therapy showed significant tumor regression, achieving up to a 65% decrease in tumor volume (Figure 5G). In contrast, vehicle treated mice developed progressive disease by the 4-week time point. Mirroring our XDOTS efficacy data, neratinib or trastuzumab administered as monotherapy exhibited only a limited anti-tumor activity. To further test whether the combination therapy could maintain a durable anti-tumor response, we continued monitoring the mice for survival upon treatment cessation (Figure 5H). The HER2YVMA mice treated with the neratinib and trastuzumab combination therapy showed a significantly longer overall survival (p=0.0005; logrank) compared to those treated with other regimens or vehicle (Figure 5H). The median OS for mice in the vehicle group was 38 days (95% CI 21-NA); for mice treated with neratinib, it was 62.5 days (40-NA), and for mice treated with trastuzumab it was 93 days (77-NA). Mice treated with the combination therapy had a median OS of 127 days (95% CI: 89-NA). No significant weight loss was observed with the combination therapy in the HER2YVMA mice (Figure S4).
Discussion
The development of new therapeutic approaches requires clinically reflective pre-clinical models in which to test such approaches. While all pre-clinical models have some advantages and disadvantages, some are more suited for drug screening. While patient derived xenografts (PDXs) have been shown to maintain intra-tumor clonal architecture, fidelity to original tumor material, and promise to faster translate targeted therapies for genotype defined cancers, they are impractical for drug combination screens as they are costly and time consuming (15,36). Our ex vivo system bypasses most of these limitations and enables expeditious screening of drug combinations, aiding in prioritization of promising candidates for in vivo evaluation. In addition, the DOTS system circumvents the necessity of establishing cell lines and/or organoids, which can be both time consuming and artifact-prone due to the inevitable loss of tumor heterogeneity. Although grown in a matrix like organoids, there are some significant differences between the XDOTS and organoids. Organoids are similarly generated from individual patient biopsies, are grown in 3D, and have been shown to preserve molecular, phenotypic and genotypic characteristics of the original tumor, including oncogenic drivers (36,37). However, organoids can take months to generate, typically lack stromal cells, and require a culture medium that facilitates the propagation of progenitor cells for their establishment. The culture conditions, e.g. the presence of EGF, may affect drug response, when studying e.g. EGFR inhibitors. Also, a challenge specific to organoids is the outgrowth of normal epithelial cells in culture, affecting the generation of lung cancer organoids in particular, due to the commonly seen phenomenon of field cancerization of the normal lung epithelium (37).
Somatic mutations in HER2 are as common as anaplastic lymphoma kinase (ALK) rearrangements in NSCLC but unlike for ALK rearranged cancers, no targeted therapies are approved for HER2 mutant NSCLC (1). Single agent pan-ERBB kinase inhibitors have limited anti-tumor activity and most currently used treatments are repurposed therapies indicated for HER2 positive breast cancer and as such are not specifically tailored towards the unique HER2 mutations found in NSCLC. (11,12,38). To that end, it is imperative to focus on the development of therapeutic approaches for this “orphan” NSCLC genotype. Unfortunately, the lack of available pre-clinical models that could more authentically recapitulate real human cancers has significantly hindered this effort. For instance, the HER2 inhibitor neratinib is quite effective in Ba/F3 cell lines engineered to express various HER2 mutations (IC50s < 20 nM), yet is ineffective as a single agent in lung cancer patients whose tumors harbor the same mutation (9,39). These findings suggest that either, drug exposures required to inhibit mutant HER2 cannot be achieved in patients, or that human cancers are more complex than can be modeled in simplified drug-screening systems, such as Ba/F3 cell models. Consistent with the clinical findings, neratinib treatment alone of both the DFCI 315 and 359 PDX models is predominately associated with tumor stasis and minimal tumor regression (Figure 5).
Ultimately, combination therapies will likely be more successful clinically than single agent treatments. There are two major categories of combination therapies: dual inhibition of the same target, or inhibition of the target and a critical downstream signaling pathway. Both strategies can achieve a more durable target and/or pathway inhibition which can translate into an improved clinical outcome. Dual target inhibition has been successful in HER2 positive breast cancer where the combination of trastuzumab and pertuzumab with docetaxel lead to an increase in survival compared to trastuzumab and chemotherapy (40). The combination of BRAF inhibitor dabrafenib and the MEK inhibitor trametinib is also associated with an improvement in survival in patients with BRAF V600E mutant melanoma compared to the BRAF inhibitor vemurafenib alone (41). In HER2 mutant lung cancer, the combination of neratinib and temsirolimus has been evaluated as a strategy to achieve a nearly complete pathway inhibition based on preclinical data and a phase I clinical trial (10,42). However, in a randomized phase II clinical trial, the combination only led to incremental improvement in response rates and progression free survival (11). In the current study, we evaluated the concept of dual HER2 inhibition using a HER2 kinase inhibitor (neratinib) and an anti-HER2 antibody (trastuzumab). Intriguingly, despite the clinical efficacy of ado-trastuzumab emtasine (TDM1) in HER2 mutant NSCLC (12), it was ineffective in the DFCI359 model, only moderately effective in the DFCI315 in model and less effective than neratinib alone in both models (Figure S3). Furthermore, and in contrast to recent studies with poziotinib (43), there was no added benefit when TDM1 was combined with neratinib in either model (Figure S3). It is possible that the level of HER2 expression (2+ for both models) accounts for the lack of efficacy of TDM1.
The dual targeting strategy, combining a tyrosine kinase inhibitor and a therapeutic antibody, has been used to inhibit EGFR combining afatinib and cetuximab both preclinically and in a phase II clinical trial (44,45). In addition, a clinical trial of osimertinib and necitumumab is currently underway (NCT02496663). Intriguingly, the afatinib/cetuximab combination resulted in a downregulation of total EGFR in the genetically engineered murine model of EGFR mutant NSCLC which may explain the therapeutic efficacy of the combination (44). Similarly we observe an increased downregulation of total HER2 and a more extensive inhibition of HER2 and downstream signaling in vivo (DFCI 315 only) with the combination of neratinib and trastuzimab compared to treatment with each of the single agents, which could underlie the increased efficacy of the combination (Figure 5F). In contrast to the efficacy, pharmacodynamic studies in the DFCI 359 model revealed little total HER2 downregulation or impact on downstream signaling (Figure 5C). Whether this disconnect stems from the timing of the pharmacodynamic studies (4 days) or pathway rewiring following drug treatment will require additional assessment. Nonetheless, based on our preclinical efficacy findings, the combination of neratinib and trastuzumab is being actively investigated in HER2 mutant lung cancer patients in the ongoing SUMMIT ‘basket’ trial (NCT01953926).
Supplementary Material
Statement of Translational Relevance.
The development of anti-cancer therapies requires appropriate preclinical models in which to test treatments. HER2 mutant non-small cell lung cancer (NSCLC) occurs in 3% of patients with lung adenocarcinoma and very few models of HER2 mutant NSCLC are available for preclinical therapeutic development. We generated 2 patient derived HER2 mutant xenograft (PDX) models and developed a platform to rapidly test HER2 targeted therapies ex vivo using the PDX tumors. Our methods may accelerate the identification and prioritization of promising anti-cancer therapies to pursue further in clinical trials.
Acknowledgments
Funding Support: This work was supported in part by the National Cancer Institute at the National Institutes of Health (R35 CA220497 (PAJ)), The American Cancer Society (CRP-17-111-01-CDD (PAJ)), by the Expect Miracles Foundation, the Robert and René Belfer Foundation, and by sponsored research funding from PUMA Biotech.
Disclosures: P.A. Jänne has received consulting fees from AstraZeneca, Boehringer-Ingelheim, Pfizer, Roche/Genentech, Eli Lilly and Company, ACEA Biosciences, Araxes Pharma, Ignyta, Mirati Therapeutics, Daiichi-Sankyo, Takeda Oncology, Novartis, Voronoi, SFJ Pharmaceuticals, Biocartis, Sanofi Oncology and LOXO Oncology; receives post-marketing royalties from DFCI owned intellectual property on EGFR mutations licensed to Lab Corp; has sponsored research agreements with AstraZeneca, Daiichi-Sankyo, Boehringer Ingelheim, PUMA, Eli Lilly and Company, Astellas Pharmaceuticals, Revolution Medicines, and Takeda Oncology; and has stock ownership in Gatekeeper Pharmaceuticals and LOXO Oncology. C.P.P., E.I., D.A.B., A.A. are inventors on a pending patent related on manipulating, culturing, and evaluating tumor of organotypic spheroids. to screen drug combinations C.P.P. has received honoraria from Bio-Rad and AstraZeneca Korea, is a co-founder of Xsphera Biosciences, and is on the scientific advisory board of DropWorks and Xsphera Biosciences. G.R.O has received consulting fees from AstraZeneca, DropWorks, LOXO, Jannsen, Inivata, GRAIL, Takeda and honoraria from Foundation Medicine and Guardant Health. K.K.W. has equity shares in G1 Therapeutics and is a consultant for G1, Janssen and Array Pharmaceuticals. I.D., and A.S.L. are employees and shareholders of Puma Biotechnology, Inc.
Footnotes
Part of the work was previously presented at the 2017 World Congress of Lung Cancer and the 2018 American Association of Cancer Research Annual Meeting.
References
- 1.Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311(19):1998–2006 doi 10.1001/jama.2014.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361(10):947–57. [DOI] [PubMed] [Google Scholar]
- 3.Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2018;378(2):113–25 doi 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 4.Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-Rearranged Non-Small-Cell Lung Cancer. N Engl J Med 2014. doi 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 2018;378(8):731–9 doi 10.1056/NEJMoa1714448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Subbiah V, Velcheti V, Tuch BB, Ebata K, Busaidy NL, Cabanillas ME, et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol 2018;29(8):1869–76 doi 10.1093/annonc/mdy137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Greve J, Moran T, Graas MP, Galdermans D, Vuylsteke P, Canon JL, et al. Phase II study of afatinib, an irreversible ErbB family blocker, in demographically and genotypically defined lung adenocarcinoma. Lung Cancer 2015;88(1):63–9 doi 10.1016/j.lungcan.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 8.Kris MG, Herbst R, Rischin D, LoRusso P, Baselga J, Hammond L, et al. Objective regressions in non-small cell lung cancer patients treated in Phase I trials of oral ZD1839 (‘Iressa’), a selective tyrosine kinase inhibitor that blocks the epidermal growth factor receptor (EGFR). Lung Cancer 2000;29(Suppl. 1):72. [Google Scholar]
- 9.Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018;554(7691):189–94 doi 10.1038/nature25475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perera SA, Li D, Shimamura T, Raso MG, Ji H, Chen L, et al. HER2YVMA drives rapid development of adenosquamous lung tumors in mice that are sensitive to BIBW2992 and rapamycin combination therapy. Proc Natl Acad Sci U S A 2009;106(2):474–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Besse B, Soria J, Yao B, Kris M, Chao B, Cortot A, et al. Neratinib (N) with or without temsirolimus (TEM) in patients (pts) with non-small cell lung cancer (NSCLC) carrying HER2 somatic mutations: An international randomized phase II study. Ann Oncol 2014;25(v1-v41). [Google Scholar]
- 12.Li BT, Shen R, Buonocore D, Olah ZT, Ni A, Ginsberg MS, et al. Ado-Trastuzumab Emtansine for Patients With HER2-Mutant Lung Cancers: Results From a Phase II Basket Trial. J Clin Oncol 2018;36(24):2532–7 doi 10.1200/JCO.2018.77.9777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters S, Stahel R, Bubendorf L, Bonomi P, Villegas A, Kowalski DM, et al. Trastuzumab Emtansine (T-DM1) in Patients with Previously Treated HER2-Overexpressing Metastatic Non-Small Cell Lung Cancer: Efficacy, Safety, and Biomarkers. Clin Cancer Res 2019;25(1):64–72 doi 10.1158/1078-0432.CCR-18-1590. [DOI] [PubMed] [Google Scholar]
- 14.Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov 2018;8(9):1112–29 doi 10.1158/2159-8290.CD-18-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu JF, Palakurthi S, Zeng Q, Zhou S, Ivanova E, Huang W, et al. Establishment of Patient-Derived Tumor Xenograft Models of Epithelial Ovarian Cancer for Preclinical Evaluation of Novel Therapeutics. Clin Cancer Res 2017;23(5):1263–73 doi 10.1158/1078-0432.CCR-16-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nikolic MZ, Caritg O, Jeng Q, Johnson JA, Sun D, Howell KJ, et al. Human embryonic lung epithelial tips are multipotent progenitors that can be expanded in vitro as long-term self-renewing organoids. Elife 2017;6 doi 10.7554/eLife.26575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res 2005;65(5):1642–6. [DOI] [PubMed] [Google Scholar]
- 18.Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov 2018;8(2):196–215 doi 10.1158/2159-8290.CD-17-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aref AR, Campisi M, Ivanova E, Portell A, Larios D, Piel BP, et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 2018;18(20):3129–43 doi 10.1039/c8lc00322j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316(5827):1039–43. [DOI] [PubMed] [Google Scholar]
- 21.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009;462(7276):1070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res 2008;14(13):4275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, et al. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin Cancer Res 2015;21(17):3913–23 doi 10.1158/1078-0432.CCR-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimamura T, Ji H, Minami Y, Thomas RK, Lowell AM, Shah K, et al. Non-small-cell lung cancer and Ba/F3 transformed cells harboring the ERBB2 G776insV_G/C mutation are sensitive to the dual-specific epidermal growth factor receptor and ERBB2 inhibitor HKI-272. Cancer Res 2006;66(13):6487–91. [DOI] [PubMed] [Google Scholar]
- 25.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008;27(34):4702–11 doi 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res 2002;62(14):4132–41. [PubMed] [Google Scholar]
- 27.Petigny-Lechartier C, Duboc C, Jebahi A, Louis MH, Abeilard E, Denoyelle C, et al. The mTORC1/2 Inhibitor AZD8055 Strengthens the Efficiency of the MEK Inhibitor Trametinib to Reduce the Mcl-1/[Bim and Puma] ratio and to Sensitize Ovarian Carcinoma Cells to ABT-737. Mol Cancer Ther 2017;16(1):102–15 doi 10.1158/1535-7163.MCT-16-0342. [DOI] [PubMed] [Google Scholar]
- 28.Schwab CL, English DP, Black J, Bellone S, Lopez S, Cocco E, et al. Neratinib shows efficacy in the treatment of HER2 amplified carcinosarcoma in vitro and in vivo. Gynecol Oncol 2015;139(1):112–7 doi 10.1016/j.ygyno.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Francia G, Man S, Lee CJ, Lee CR, Xu P, Mossoba ME, et al. Comparative impact of trastuzumab and cyclophosphamide on HER-2-positive human breast cancer xenografts. Clin Cancer Res 2009;15(20):6358–66 doi 10.1158/1078-0432.CCR-09-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geoerger B, Kerr K, Tang CB, Fung KM, Powell B, Sutton LN, et al. Antitumor activity of the rapamycin analog CCI-779 in human primitive neuroectodermal tumor/medulloblastoma models as single agent and in combination chemotherapy. Cancer Res 2001;61(4):1527–32. [PubMed] [Google Scholar]
- 31.Bahcall M, Awad MM, Sholl LM, Wilson FH, Xu M, Wang S, et al. Amplification of Wild-type KRAS Imparts Resistance to Crizotinib in MET Exon 14 Mutant Non-Small Cell Lung Cancer. Clin Cancer Res 2018;24(23):5963–76 doi 10.1158/1078-0432.CCR-18-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mazieres J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol 2013;31(16):1997–2003 doi 10.1200/JCO.2012.45.6095. [DOI] [PubMed] [Google Scholar]
- 33.Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 2009;28(6):803–14 doi 10.1038/onc.2008.432. [DOI] [PubMed] [Google Scholar]
- 34.Sanchez-Martin M, Pandiella A. Differential action of small molecule HER kinase inhibitors on receptor heterodimerization: therapeutic implications. Int J Cancer 2012;131(1):244–52 doi 10.1002/ijc.26358. [DOI] [PubMed] [Google Scholar]
- 35.Collins DM, Gately K, Hughes C, Edwards C, Davies A, Madden SF, et al. Tyrosine kinase inhibitors as modulators of trastuzumab-mediated antibody-dependent cell-mediated cytotoxicity in breast cancer cell lines. Cell Immunol 2017;319:35–42 doi 10.1016/j.cellimm.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 36.Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN, et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell 2016;167(1):260–74 e22 doi 10.1016/j.cell.2016.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014;159(1):163–75 doi 10.1016/j.cell.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kris MG, Camidge DR, Giaccone G, Hida T, Li BT, O'Connell J, et al. Targeting HER2 aberrations as actionable drivers in lung cancers: phase II trial of the pan-HER tyrosine kinase inhibitor dacomitinib in patients with HER2-mutant or amplified tumors. Ann Oncol 2015;26(7):1421–7 doi 10.1093/annonc/mdv186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kosaka T, Tanizaki J, Paranal RM, Endoh H, Lydon C, Capelletti M, et al. Response Heterogeneity of EGFR and HER2 Exon 20 Insertions to Covalent EGFR and HER2 Inhibitors. Cancer Res 2017;77(10):2712–21 doi 10.1158/0008-5472.CAN-16-3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swain SM, Baselga J, Kim SB, Ro J, Semiglazov V, Campone M, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 2015;372(8):724–34 doi 10.1056/NEJMoa1413513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015;372(1):30–9 doi 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 42.Gandhi L, Bahleda R, Tolaney SM, Kwak EL, Cleary JM, Pandya SS, et al. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J Clin Oncol 2014;32(2):68–75 doi 10.1200/JCO.2012.47.2787. [DOI] [PubMed] [Google Scholar]
- 43.Robichaux JP, Elamin YY, Vijayan RSK, Nilsson MB, Hu L, He J, et al. Pan-Cancer Landscape and Analysis of ERBB2 Mutations Identifies Poziotinib as a Clinically Active Inhibitor and Enhancer of T-DM1 Activity. Cancer Cell 2019;36(4):444–57 e7 doi 10.1016/j.ccell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest 2009;119(10):3000–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Janjigian YY, Smit EF, Groen HJ, Horn L, Gettinger S, Camidge DR, et al. Dual Inhibition of EGFR with Afatinib and Cetuximab in Kinase Inhibitor-Resistant EGFR-Mutant Lung Cancer with and without T790M Mutations. Cancer Discov 2014. doi 10.1158/2159-8290.CD-14-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.