

Abstract
Osteosarcoma remains one of the most common malignant primary bone tumors. Post-surgical defect repair combined with tumor suppression remains a major clinical challenge. Investigations of alternative treatments for osteosarcoma, while promising, have led to multi-drug resistance. These constraints of common treatment strategies have triggered the need for new therapeutic candidates in bone cancer treatment. Chitosan, a common biopolymer utilized in bone and tissue engineering applications, has recently been studied as a pro-apoptotic agent in metastatic cell lines like breast cancer, but has not been utilized in bone cancer applications. In this study, chitosan was directly loaded onto HA disks to evaluate its in vitro release and effects on human fetal osteoblast (hFOB) and human osteosarcoma (MG-63) cell lines. It is hypothesized that the sustained release of chitosan will decrease osteosarcoma cell proliferation and enhance proliferation of osteoblast cells. Through morphological characterization and MTT assay analysis, chitosan showed no toxicity to human fetal osteoblast (hFOB) cells. Chitosan was also shown to decrease human osteosarcoma viability by up to 96% compared to control samples. This suggests a pro-apoptotic mechanism against osteosarcoma as well as the potential clinical application of chitosan as a drug candidate in ceramic scaffolds at tumor resected sites.
Keywords: Chitosan, Osteosarcoma, Osteoblast, Bone Scaffolds
1. Introduction
Osteosarcoma remains one of the most common and deadly forms of musculoskeletal cancers due to its high malignancy. The current treatment strategy of osteosarcoma is based on a combination of surgery and chemotherapy. Metastasis at onset of osteosarcoma is common, where reconstruction of bone cancer defects and limb salvaging surgery remain the standard of care. Complications in osteosarcoma surgery include infections of bone grafts and failure of implanted prosthetics. Chemotherapy is highly toxic but poorly specific to the tumor site, where systematic administration of chemotherapy and other anticancer agents such as doxorubicin, cisplatin, and methotrexate limit their overall effectiveness in osteosarcoma treatment. Additionally, almost 50% of clinically reported osteosarcoma cases acquire resistance to chemotherapy before or during treatment [1]. Most studies have focused on targeting pathways that modulate cancer cell sensitivity such as Bcl-2 proteins, Her-2/ERBB2, miRNAs, and ATP dependent efflux pump P-glycoproteins [2–5]. The tumor environment is characterized by hypoxia as well as other extreme metabolic and biochemical changes. More recently, it has been demonstrated that acidic extracellular pH (pHe) is linked to malignant tumor behavior as well as the association of sarcomas with drug resistance [6–7]. This is due to cancer cell’s production of metabolic acid generated by lactic acid production, glycolysis, and an increased proton efflux. These mechanisms prevent apoptosis of tumor cells by cellular acidosis [8]. This leads to rapid tumor growth, which in turn induces extracellular acidosis, increasing a tumor’s ability to invade healthy tissues and metastasize [9]. The acidification of the surrounding environment of cancer cells has shown to increase in vivo metastases compared to normal cancer environments [10]. Acidosis has also been shown to increase the invasiveness and adhesion of in vivo and in vitro metastasizing models [11–14].
Thus, the development of therapeutic agents that can selectively inhibit cellular acidosis for cancer therapy is a growing area of interest. Research in the bone and tissue engineering field has focused on chitin and its biopolymer equivalent, chitosan, to generate novel properties and functionalities in biomaterial applications. Chitin is well known as a biocompatible, biodegradable, and non-toxic compound with indications in antibacterial and immunogenicity [15]. Chitosan is the linear polysaccharide produced by the deacetylation of chitin and the properties of this polysaccharide are directly dependent on the deacetylation sites, making it particularly novel for modifying properties such as biocompatibility, antimicrobial activity, and biodegradability in tissue scaffolds and coatings.
Previous work has focused on chitosan’s utilization as a hydrogel and nanoparticle system to modulate the aggressiveness of metastatic cancers or encapsulate chemotherapy-like drugs in order to improve efficacy and clinical outcome in breast cancer models [16,17]. Chitosan was also found to induce apoptosis in bladder cancer cells through DNA fragmentation as well as activation of caspase-3, a death signaling mediator of the apoptotic pathway commonly induced by antitumor agents [18]. Chitosan nanoparticles have also been utilized for drug delivery applications for doxorubicin to treat osteosarcoma [19]. Additionally, it has been shown that free chitosan promotes osteoblast proliferation and osteogenesis in mesenchymal stem cells and also increases osteopontin and collagen I expression, ultimately reducing osteoclastogenesis [15]. Chitosan is therefore commonly utilized in scaffold matrices for bone healing but has rarely been used as a free drug/coating agent [20]. Chitosan thus has the potential to promote ingrowth and regeneration while reducing circulating bone cancer cells.
Chitosan also functions as a weak base, suggesting its potential utilization as a pH regulator in osteosarcoma regulation. This is due to the non-bonding pair of electrons on chitosan’s primary amino group of the glucosamine unit, which has the ability to accept a proton [21,22]. The potential for chitosan to promote proliferation in osteoblasts and decrease proliferation in osteosarcoma would allow for a targeted, localized, and controlled treatment strategy. A common delivery system in bone related ailments and tumor resected sites is hydroxyapatite (HA). Due to calcium phosphate’s chemical and structural similarities to the inorganic phase of human bone, HA has become one of the most popular bioceramic material in bone and tissue engineering applications [23]. HA has also been widely investigated as a biocompatible and osteoconductive biomaterial in the use of bone substitutes as a bone scaffold which will eventually be absorbed by the body and completely replaced by new bone growth [24]. Its potential use as a drug delivery system has attracted significant interest because of its simultaneous use as bone substitute and drug delivery vector [25,26].
To the authors’ knowledge, there have been no reports thus far that showcase the ability of chitosan to inhibit osteosarcoma proliferation as well as maintain biocompatibility with osteoblast cells for bone tissue engineering applications using hydroxyapatite. To explore chitosan’s potential chemotherapeutic efficacy, the local delivery of chitosan from HA disks was investigated. In this work, HA disks were prepared and loaded with chitosan to test chitosan’s efficacy in vitro on human osteoblast and osteosarcoma cell lines. Authors hypothesize that the sustained release of chitosan will decrease osteosarcoma cell proliferation and enhance proliferation of osteoblast cells. The effect of pH on in vitro release of chitosan was also assessed. An initial toxicity study was carried out in human fetal osteoblast (hFOB) and human osteosarcoma (MG-63) cell cultures, followed by drug loading optimization and a final cell culture study in osteosarcoma cells (Figure 1). This work provides an effective, pro-apoptotic system against osteosarcoma utilizing chitosan for bone tissue engineering applications.
Figure 1:
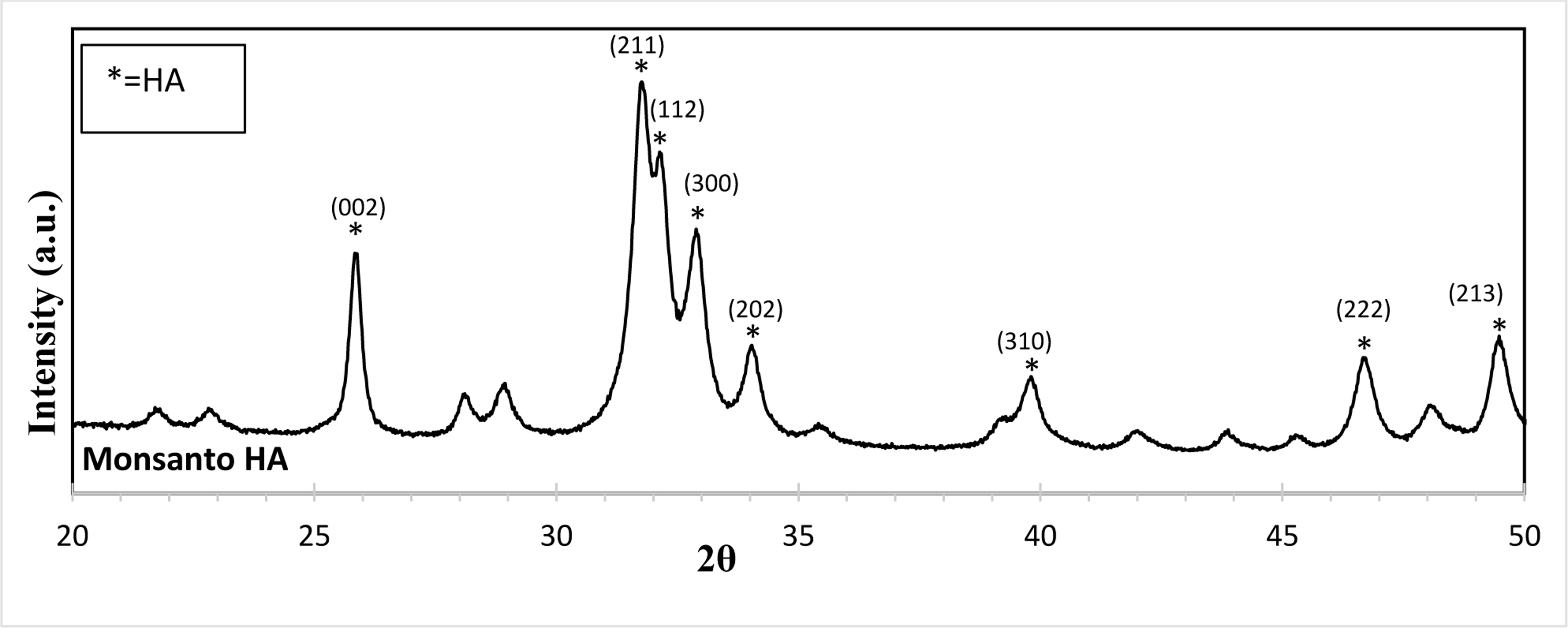
XRD, Monsanto HA. Overall, the powder utilized shows characteristic HA peaks referenced in JCPDS # 09–0432.
2. Experimental
2.1. Characterization of HA powder and non-loaded and chitosan-loaded HA disks.
2.1.1. HA disk preparation
HA powder was prepared by ball milling in ethanol in (2:3 w/v) for 12 hours with zirconia balls (3:1 ball to powder ratio). After milling, the powder was left to dry in a conventional oven at 60 °C for 12 hours. Dried powders were then sieved at 212 μm to obtain a uniform particle size. The dried powder was then uniaxially pressed into disks with a diameter of 12 mm and a height of 2 mm. For each disk, 0.6 g powders were measured and pressed with a uniaxial pressure of 165 MPa applied for 2 minutes. The green disks were then sintered in a conventional muffle furnace at 1250 °C for 2 hours to obtain the final disk parts.
2.1.2. Chitosan loading
Chitosan was dissolved in 0.1 M acetic acid and pipetted on top of the HA disks at initial concentrations of 10 μg. All loaded samples were frozen at −80 °C for 24 hours and lyophilized for 72 hours at −50 °C (FreeZone 4.5L benchtop Free Dryer, Labconco). The successful loading of chitosan was measured with a Biotek Synergy 2 SLFTAD microplate reader (Biotek, Winooski, VT, USA) at a 250 nm wavelength range.
2.1.3. Physical characterizations
The crystalline structure and the phase composition of HA powder (Monsanto, USA) was characterized utilizing X-ray diffraction (XRD) with a Siemens D-500 Diffractometer system operating at 35.0 kV and 30.0 mA with a Cu radiation source and Ni filter at a scan rate of .02°/min and a step width of 0.05. Fourier transform infrared (FTIR) spectra of chitosan loaded and pure HA samples were obtained using an ATR-FTIR spectrophotometer (Nicolet 6700 FTIR, Madison, WI, USA) in the 400–4000 cm−1 wave number range.
2.2. In vitro release of chitosan
HA disks were prepared as described in section 2.1.1. The release of chitosan was measured in phosphate buffer solution (PBS, pH 7.4) and acetic buffer solution (ABS, pH 5.0) to replicate both physiological environments as well as the acidic environment of a resected tumor site. Chitosan was dissolved in 0.1 M acetic acid and loaded at 50 μg. All samples were frozen at −80 °C for 24 hours, followed by lyophilization for 72 hours (FreeZone 4.5L benchtop Free Dryer, Labconco) at −53 °C. All control and test samples were tested in triplicate, and were immersed in 4 mL of each respective buffer and replaced with fresh buffer after 15, 30, and 45 min, as well as 1.5, 3, 6, 12 h and 1, 2, 3, 7, 10, 14 days, all the way up to 7 weeks. Samples were kept in a shaker at 37 ºC with constant shaking at 150 rpm. The concentration of released chitosan was measured with a Biotek Synergy 2 SLFTAD microplate reader (Biotek, Winooski, VT, USA) at a 250 nm wavelength and concentrations were determined based on a previously prepared standard curve.
2.3. In vitro cell culture study
2.3.1. Initial osteoblast toxicity study
Prior to cell culture, chitosan was dissolved in 0.1 M acetic acid and pipetted on top of the HA disks at initial concentrations of 10 μg. All loaded samples were frozen at −80 °C for 24 hours and lyophilized for 72 hours at −50 °C (FreeZone 4.5L benchtop Free Dryer, Labconco). Pure HA samples were utilized as a control and chitosan loaded samples were utilized as the treatment group. Both groups were assessed in triplicate. All samples were sterilized using an autoclave for 60 min at a temperature of 121 °C. Human fetal osteoblast cells (hFOB 1.19, ATCC, Manassas, VA) were cultured to passage 7 and seeded at 20×103 cells/sample. The basal medium was prepared by mixing 1:1 Ham’s F12 Medium and Dulbecco’s Modified Eagle’s Medium (DMEM/F12, Sigma, St. Louis, MO), with 2.5 mM L-glutamine (without phenol red) in filter sterilized DI water. 10% fetal bovine serum (FBS, ATCC, Manassas, VA) and 0.3 mg/mL G418 (Sigma, St. Louis, MO) was then utilized to supplement the media. Samples were then kept in an incubator at 34 ºC under an atmosphere of 5% CO2 and the study was carried out for 6 days.
2.3.2. Initial osteosarcoma toxicity study
All samples were prepared based on the previous section. MG-63 (MG-63 1.19, ATCC, Manassas, VA) human osteosarcoma cells were cultured to passage 7 and seeded at 20×103 cells/sample. Eagles minimum essential medium (EMEM) (ATCC, USA) was used to maintain the culture where the media was changed every 2 days. Samples were then kept in an incubator at 37 ºC in a humidified environment at an atmosphere of 5% CO2 and the study was carried out for 6 days.
2.3.3. Final osteosarcoma study
Due to delamination, a final study was carried out with optimized chitosan loaded HA disks. For this study, all chitosan loaded samples were frozen at −80° C for 24 hours and lyophilized for 48 hours at −53 °C (FreeZone 4.5L benchtop Free Dryer, Labconco). Human osteosarcoma cells (MG-63 1.19, ATCC, Manassas, VA) were again cultured to passage 7 and were seeded on samples at a density of 20 ×103 cells/sample. This final study was carried out for 11 days.
2.3.4. Cellular morphology
Samples for Field Emission Scanning Electron Microscope (FESEM) analysis were removed at Day 3 and 6 for the initial toxicity studies and Day 3, 7 and 11 for the final toxicity study in order to characterize osteoblast and osteosarcoma cellular morphology. Samples were fixed with 2% paraformaldehyde/2% glutaraldehyde in 0.1 M phosphate buffer overnight at 4 ºC. Samples were then rinsed with 0.1 M phosphate buffer, followed by post fixation with 2 % osmium tetroxide (OsO4) for 2 h at room temperature. Samples were rinsed again with 0.1 M phosphate buffer followed by ethanol series dehydration (30, 50, 70, 95, and 100 % three times). The final dehydration step was performed with hexamethyldisilane (HDMS) and left to dry overnight. Gold sputtering was then performed at a layer thickness of 3–4 nm.
2.3.5. MTT cell viability assay
Osteoblast and osteosarcoma cell viability was measured using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay at the same timepoints as morphological characterization. Samples were transferred to a new 24-well plate and 100 μL of MTT (Sigma, St. Louis, MO) solution and 900 μL of cell media were added to each sample. After incubating at 34 ºC for 2 hours, the media and MTT solution was removed from each well and 600 μL of solubilizer solution (composed of 10 % Triton X-100, 0.1N HCl and isopropanol) was added to each sample to dissolve formazan crystals which are produce after reduction of MTT by cellular enzymes. 200 μL of the final solution was transferred to 96-well plate and read by UV–Vis spectroscopy (BioTek) at 570 nm. To determine cytotoxicity for the osteosarcoma cell line, the percentage of cell viability was calculated using the following equation:
2.4. Statistical analysis
Statistical analysis was performed on MTT results using a student’s t-test, where a p value (<0.05) was considered significant.
3. Results
3.1. Characterization of HA powder and disks
Figure 1 shows the XRD patterns for HA utilized for disk preparation. Results show characteristic peaks of the pure HA phase, which matches well with JCPDS #09-0432.
Figure 2 shows the FTIR spectra of chitosan loaded and non-chitosan loaded HA disks. There are antisymmetric and symmetric modes representing HA’s phosphate groups in both spectra [27]. The antisymmetric (ν3) P-O stretching modes are represented by bands at 1,085 and 1,012 cm−1. In addition, the P-O (ν1) symmetric stretching mode appears at 960 cm−1. The antisymmetric (ν4) P-O bending modes of the phosphate group were found in the region of 540–650 cm−1. OH- is present at around 3,571 cm−1 in all spectrums where overall intensity of bands is decreased with chitosan loading due to the uniform coverage on the disk surface. No bands are attributed to acetic acid, which confirms its complete removal.
Figure 2:
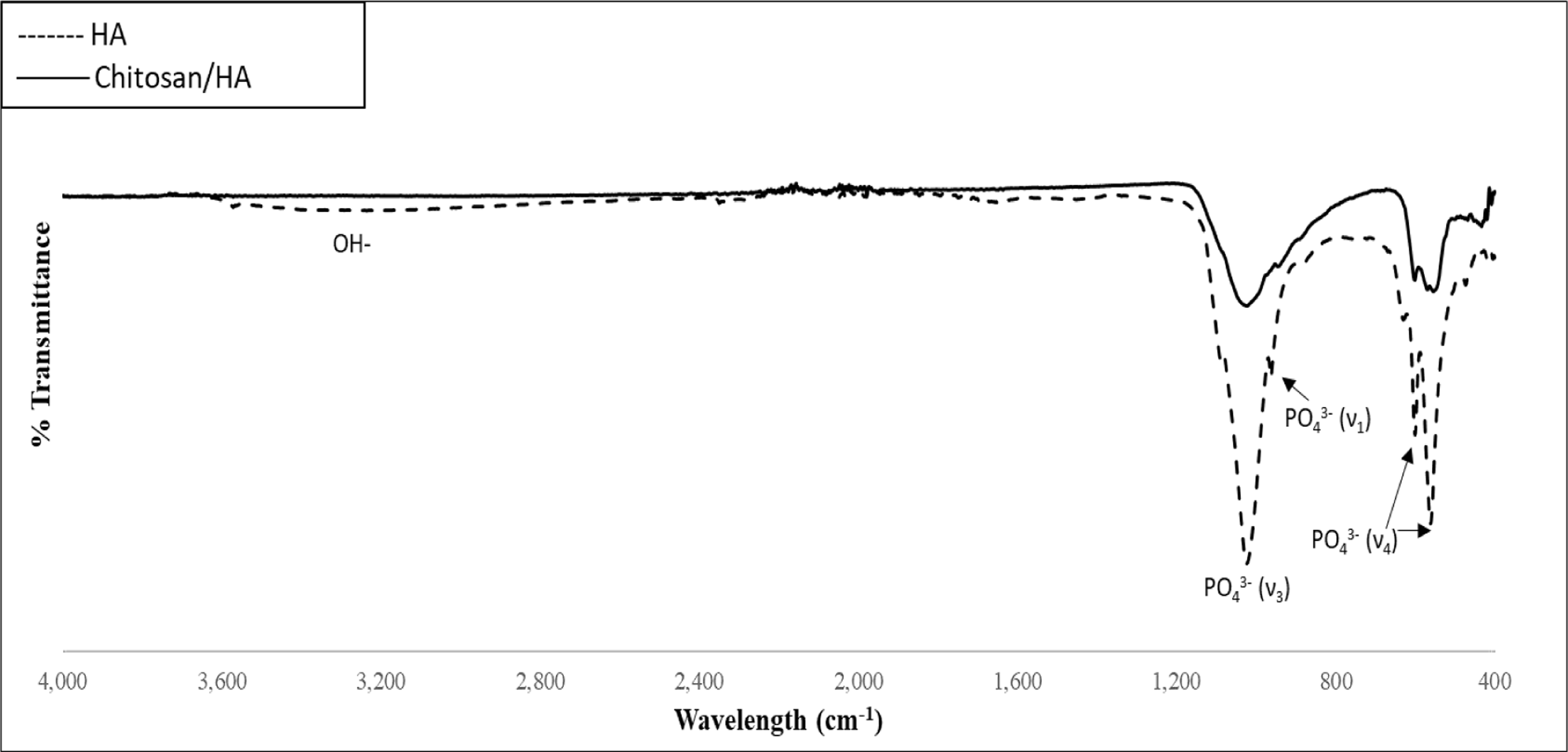
FTIR Spectra, chitosan and non-chitosan loaded HA disks
Figure 3 displays the in vitro release kinetics of chitosan from HA disks at pH 7.4 and pH 5.0. Chitosan shows low release from the HA matrix within the first 2 days with an initial release of around 20%. At 3 weeks, this release was found to be around 25% of the original drug loading, with a maximum of 28% release at 7 weeks in pH 5.0. Similarly, at week 3, chitosan showed around 24% release, plateauing at around 27% at Week 7 in pH 7.4. Release of chitosan was thus slightly higher in acidic buffer than physiological pH.
Figure 3:
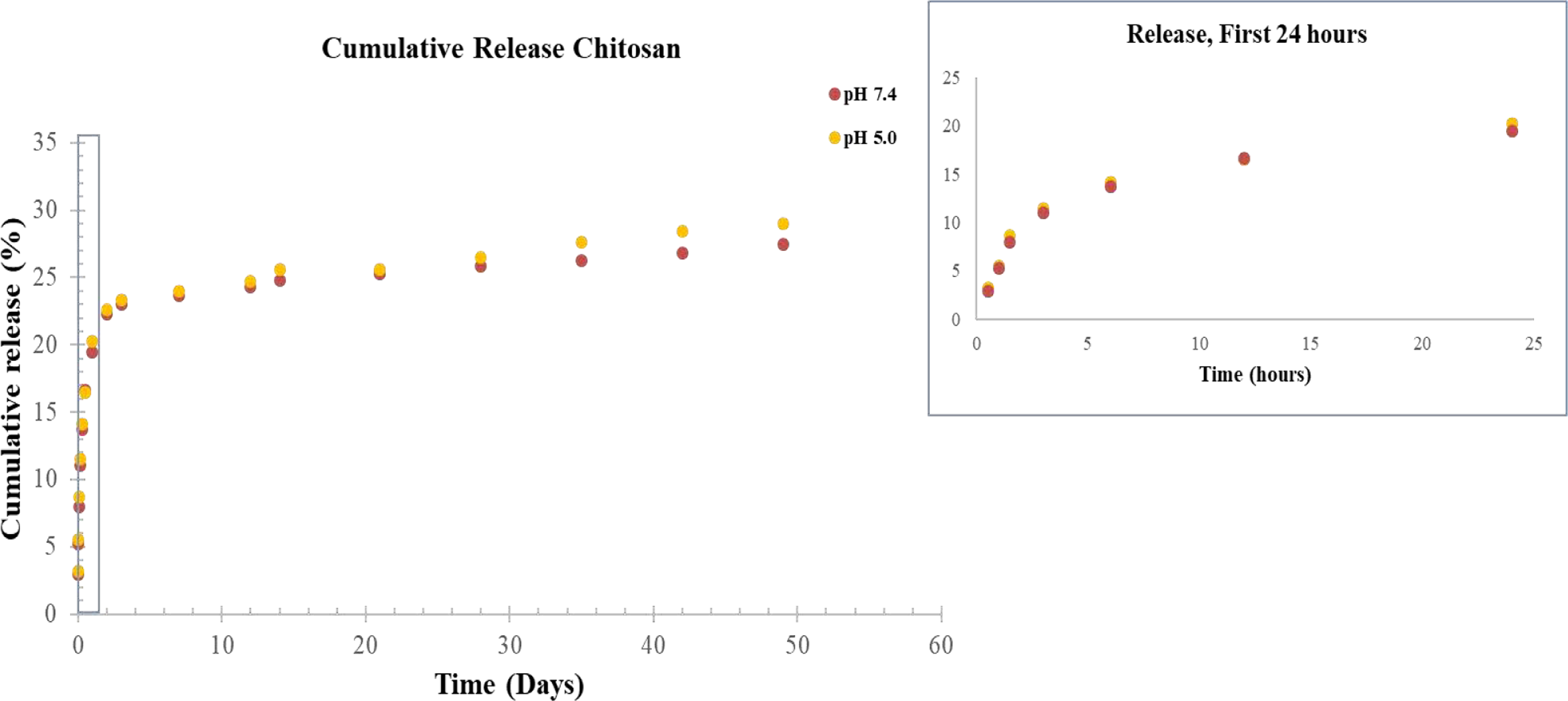
Cumulative release of chitosan from HA disks at pH 7.4 and pH 5.0. Chitosan was found to release at a higher rate in pH 5.0 vs pH 7.4.
Figure 4 displays the post-release surface morphologies of HA and chitosan loaded HA samples after 5 and 7 weeks of release in pH 5.0 and pH 7.4. The samples kept in acidic pH clearly showed much higher degradation, which is evident from the rough and inhomogeneous surface with some exposed cracks and pores. In pH 7.4, there is a much lower degradation of the HA surface, which is evident from the homogenous HA surface. There appears to be matrix formation on the chitosan samples at Week 5 that dissipates by Week 7.
Figure 4:

Cumulative release micrographs of chitosan from HA disks and chitosan loaded HA disks at pH 7.4 and pH 5.0 at weeks 5 and 7. Samples in acidic pH display higher degradation, which is evident from the inhomogeneous and rough surface morphology.
Figure 5 shows the MTT results for osteoblast (hFOB) and osteosarcoma (MG-63) at Days 3 and 6. The presence of chitosan showed no toxic affects to osteoblast cell viability at Day 3 and 6 compared to pure HA samples. The presence of chitosan showed a significant decrease in osteosarcoma cell viability at Day 6 compared to control samples.
Figure 5: Initial Toxicity Study MTT.
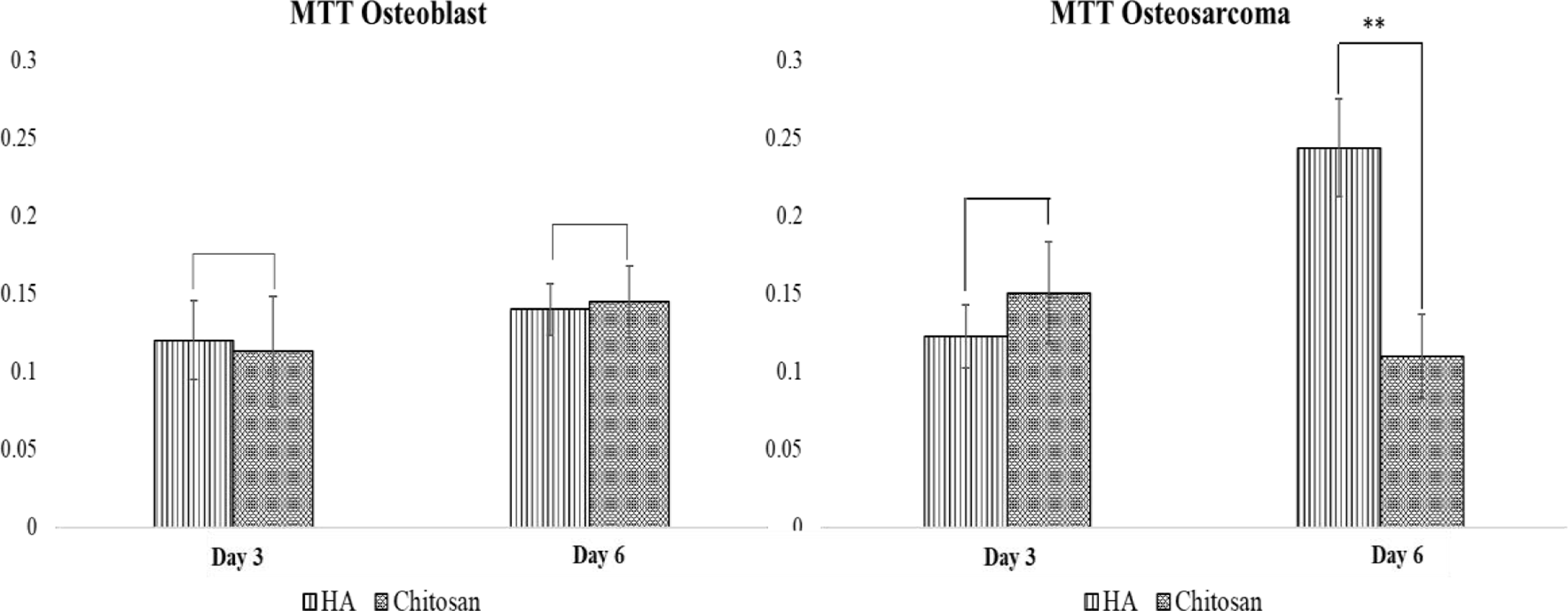
hFOB and MG-63 optical density on HA, and chitosan loaded samples after 3 and 6 days of culture (n=3, **p<0.05). Chitosan did not affect osteoblast proliferation, and significantly decrease osteosarcoma proliferation at Day 6.
Figure 6 and 7 shows the scanning electron microscopic images of osteoblast (hFOB) and osteosarcoma (MG-63) cellular morphology for HA and chitosan loaded HA. Osteoblast cellular morphology, viability and density was consistent for both HA and chitosan loaded HA samples at Day 3. Day 6 samples show elongated and flattened morphology on the chitosan loaded samples. This sheet-like phenotype indicates favorable attachment and spreading of osteoblast cells. In the osteosarcoma culture, HA samples show live and attached osteosarcoma cells with clear filopodial and lamellipodial extensions. SEM micrographs of osteosarcoma cultured chitosan loaded samples showed consistent debris and dead cells at Day 3, but with some delamination of the polymer surface. Chitosan delamination has allowed for the presence of live healthy cells at Day 6, suggesting that drug loading needs to be optimized in order for coating uniformity.
Figure 6: Initial Toxicity Study FESEM.
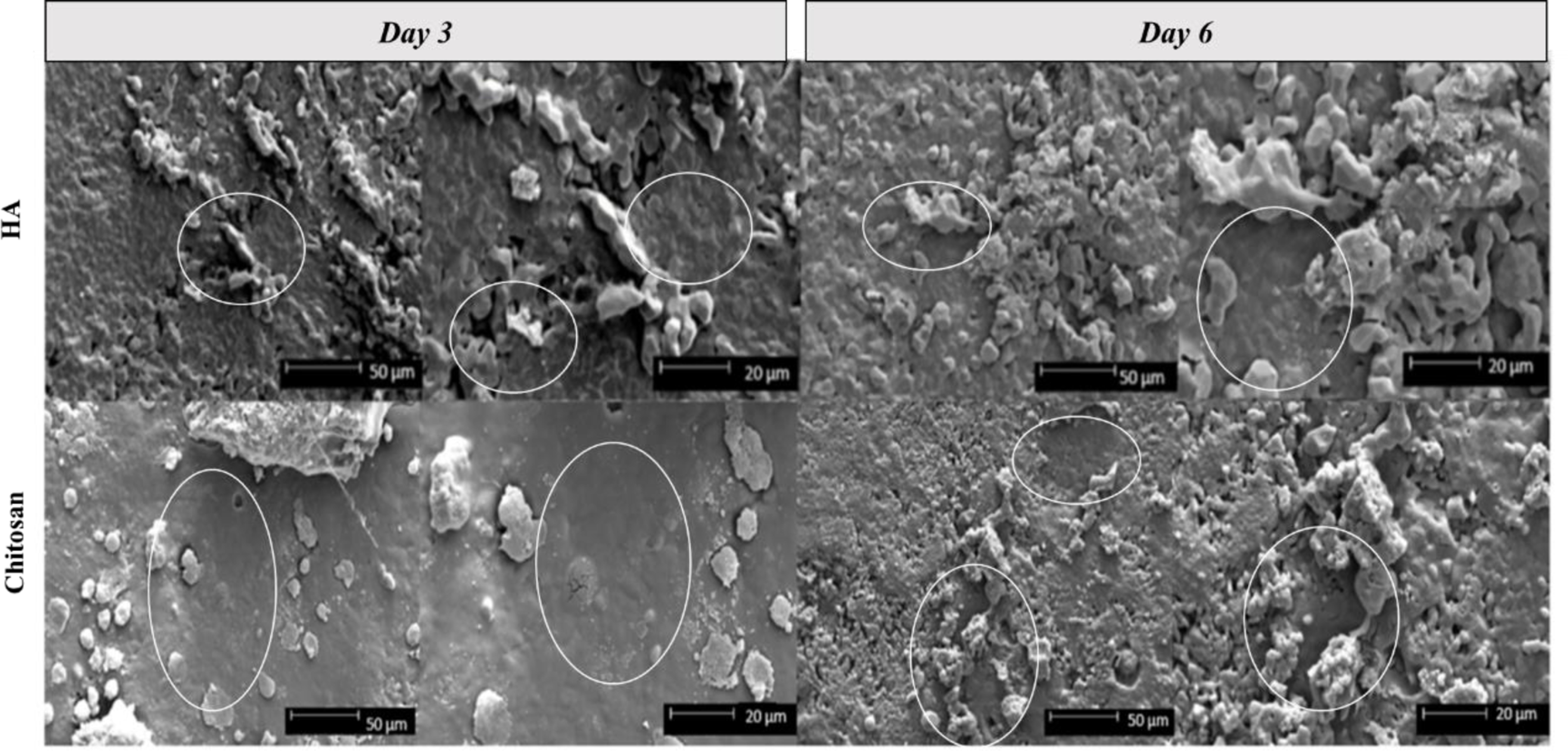
hFOB cellular morphology on HA and chitosan loaded samples after 3 and 6 days of culture. Darker toned areas, circled in white, with a smooth appearance indicate flattened, attached cellular morphology that have followed the HA disk microstructure.
Figure 7: Initial Toxicity Study FESEM.

MG-63 cellular morphology on HA and chitosan loaded samples after 3 and 6 days of culture. There are clear cellular, density and morphological changes in all drug loaded samples compared to HA.
Figure 8 displays the MTT results of the final toxicity study in osteosarcoma cells at Day 3, 7, and 11 with optimized lyophilization of chitosan coatings. Chitosan loaded samples at Day 3 and Day 7 show 2- and 8-fold lower cell viability in chitosan loaded samples, respectively, compared to control. At Day 11, MG-63 viability was 94% lower in the presence of chitosan compared to control. The results show over 50% lower cell viability as early as 3 days after cell seeding, which implies chitosan has cytotoxic potential against osteosarcoma cells.
Figure 8: Final Toxicity Study MTT.
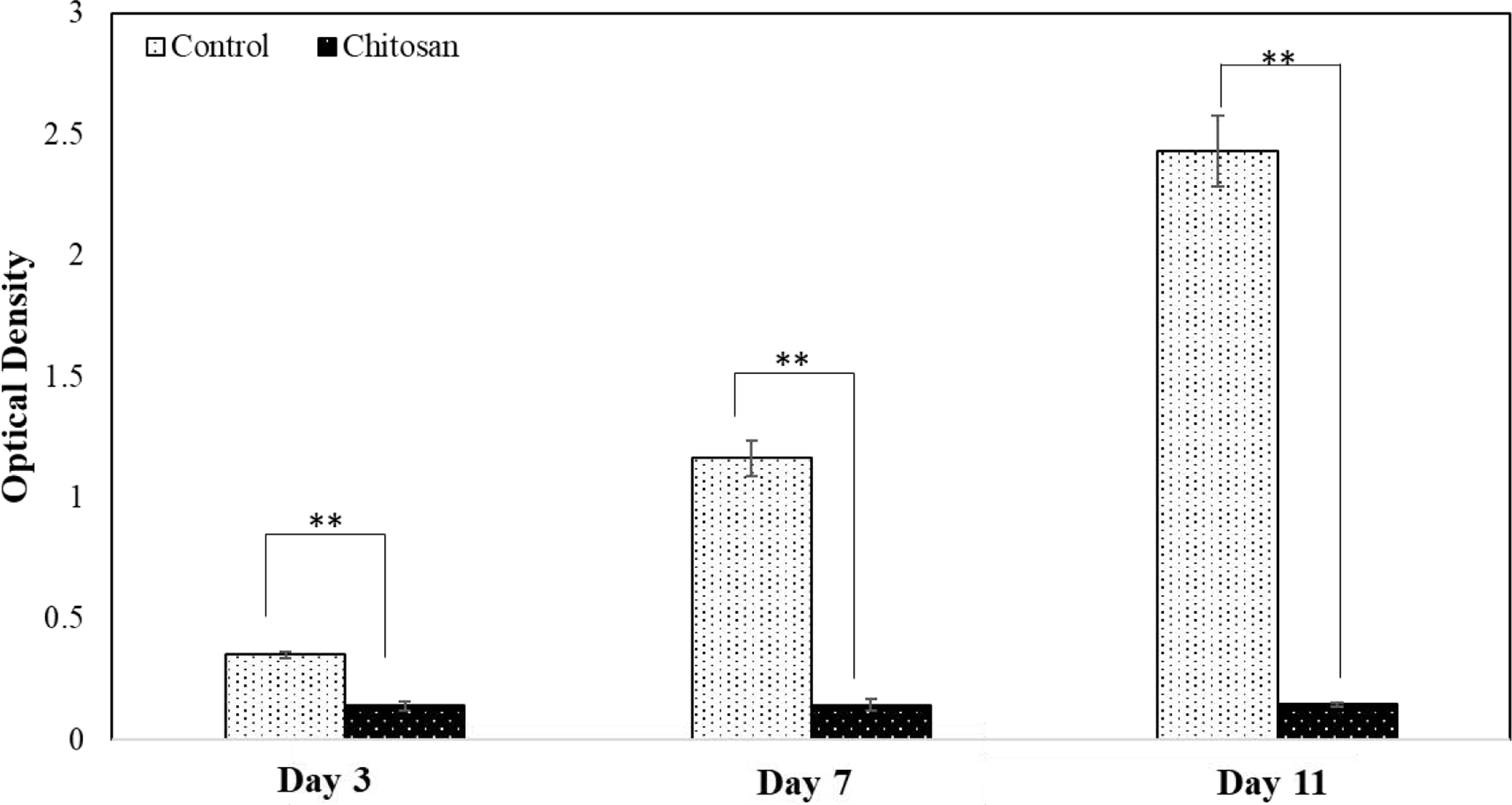
MG-63 optical cell density on HA and chitosan loaded samples after 3 and 7 and 11 days of culture (**p<0.05). Chitosan loaded samples showed significantly lower optical density at all time points, with no increase in optical density from day 3 and day 11.
Table I shows the percentage cell viability of control vs. chitosan loaded samples after 3, 7, and 11 days of seeding using MTT assay. If the percent viability is less than 50%, this is indicative that a drug agent has cytotoxic potential.
Table I:
Percentage cell viability of control vs. chitosan loaded samples using MTT assay of MG-63 cells after 3, 7, and 11 days.
| MG-63 cell culture time point | % viable cells (cytotoxicity) |
|---|---|
| Day 3 | 40.05 % |
| Day 7 | 12.41 % |
| Day 11 | 6.01 % |
Figure 9 shows SEM images of osteosarcoma (MG-63) cell culture on control and chitosan loaded samples at Day 3, 7, and 11. HA samples show cellular spreading at early timepoints with enhanced cellular sheets developing by Day 7, suggesting a dense cellular layer. Layers continue to develop by Day 11 suggesting a highly proliferating cellular environment. Qualitative analysis shows that chitosan loaded samples at all timepoints have consistent cellular debris and there are no signs of live cells throughout all time points. No spread or layered cellular structures are distinguishable, which is conclusive with optical density results.
Figure 9: Final Toxicity Study FESEM.

MG-63 cellular morphology on HA and chitosan loaded samples after 3, 7, and 11 days of culture. Control samples show an enhanced cellular sheet suggesting a dense cellular layer at all timepoints. All chitosan loaded samples showed increased cellular debris and dead cells across all timepoints with no presence of attached cells.
4. Discussion
Chitosan has been well studied as a nano-carrier and matrix former in hydrogel systems utilized in cancer treatment [28]. However, chitosan has rarely been studied as a free agent, particularly in the inhibition of metastasizing cancers such as osteosarcoma. Although we know the general functions and cellular methods of pH regulation, it is still unclear as to what regulators are expressed and function as anti-apoptotic factors in cancerous cell lines. This study was implemented in order to assess chitosan’s drug activity towards both osteoblast and osteosarcoma cells and elucidate a possible mechanism of action. HA disks were manufactured and assessed to have characteristic peaks and modes as seen by the XRD and FTIR analyses, which was in line with our previous studies [27,29,30]. Controlled drug release from HA is desired in order to improve its chemopreventative and osteogenic potential. The in vitro release kinetics of chitosan showed relatively low cumulative release by week 7 in both physiological and acidic microenvironments. It is likely that chitosan is adhering to the matrix by binding to calcium ions present in HA, but such release is common for more sustained and continuous release profiles as presented in other work [31–33]. Additionally, it is well known that chitosan has cationic properties which is the basis of many of its potential applications. Chitosan and its derivatives can thus be considered as linear polyelectrolytes with a high charge density which can interact with negatively charged surfaces, like proteins and anionic polysaccharides. Thus, chitosan’s higher release kinetics in the acidic microenvironment are hypothesized to be due to its cationic nature as well as chitosan’s associated hydrophilicity.
To ensure chitosan’s clinical success as a potential chemopreventative agent against osteosarcoma, a short-term initial toxicity study was performed in human osteoblast and osteosarcoma cell lines. MTT results showed no toxic effect to osteoblast proliferation and viability in the presence of chitosan compared to the control samples. Microstructure analysis via SEM showed osteoblast spreading, proliferation, and viability in the presence of chitosan and control samples. While lower optical density values as well as clear morphological changes were observed in chitosan loaded samples in the osteosarcoma culture, delamination of coatings required optimization of drug loading and lyophilization in order to improve the efficacy of chitosan as a drug candidate.
Once a uniform coating was obtained, a final in vitro study was performed to investigate chitosan’s effect on human osteosarcoma cells. MTT results showed a significant decrease of up to 96% in viable cells on chitosan loaded samples over the course of the 11-day study compared to control samples. A reduction in tumor cell viability induced by chitosan is corroborated by the works of Tan et al. and Shen et al [34,35]. Additionally, cellular viability continued to decrease over the course of the study from 40% to 6% suggesting an early-stage mechanism of action behind toxicity of osteosarcoma cells. Morphological characterization via SEM confirmed an increase in cellular debris and the lack of live attached cells on chitosan samples compared to control samples which displayed uniform cellular density and spreading. Figure 10 shows a possible mechanism of action regarding chitosan and its chemopreventative properties. Chitosan can cause deprotonation of the cell cytoplasm which further inhibits tumor growth activity. The amino groups of chitosan could have a neutralizing effect on the cellular environment. At low pH, the amino groups can receive protons while in aqueous solution [21,22]. By accepting these protons, the extracellular pH (pHe) can potentially be buffered, therefore inhibiting osteosarcoma proliferation. Another contributing factor for the chemopreventative ability of chitosan is the activation of caspase-3 [18]. Through the use of chitosan, nuclear fragmentation, chromatin condensation, and internucleosomal DNA cleavage have all been shown which indicate cellular apoptosis of tumor cells [18]. As a cationic polymer, chitosan has also shown to induce red blood cell lysis as well as inhibit melanoma cell growth [36]. Further studies are necessary in order to understand the regulators, pathways, and factors associated with chitosan’s apoptotic effect against osteosarcoma. However, the results presented validate many of the existing literature regarding the potential utilization of chitosan and other neutralizing agents as potential regulators in osteosarcoma treatment [37–39]. Reviewing the existing results, this work presents the sustained release of chitosan as a promising alternative or adjunct treatment for osteosarcoma regulation, suggesting its clinical significance in the localized drug delivery in bone tumor resection.
Figure 10:
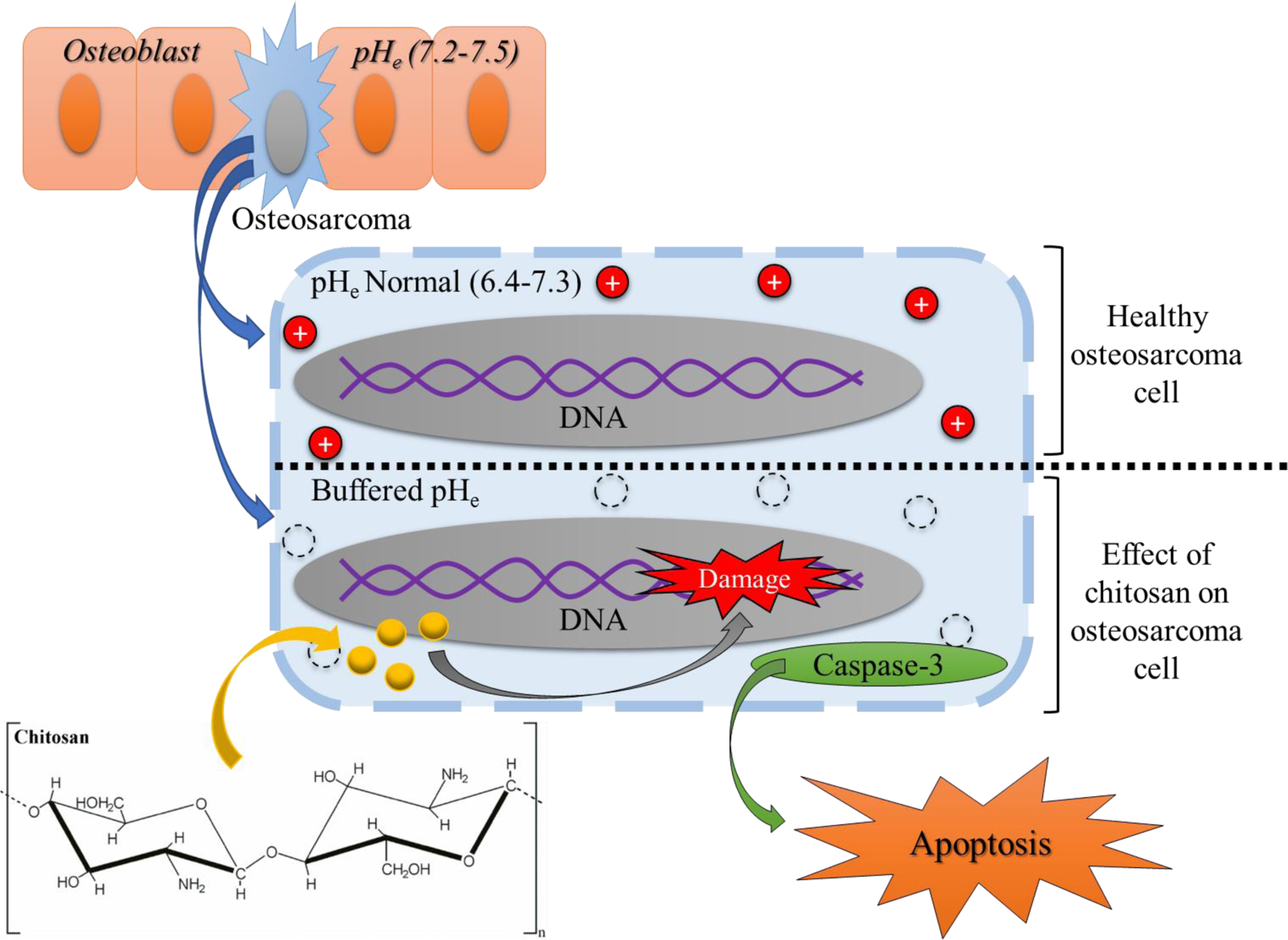
Possible mechanism of action regarding chitosan and its chemopreventative properties. Chitosan can cause deprotonation of the cell cytoplasm, buffering the pHe in the cell by the amino groups accepting protons [21]. Chitosan can also cause DNA damage and cause the activation of caspase-3 leading to cellular apoptosis [18].
5. Conclusions
Controlled release of chitosan was achieved from HA disks, where the effects of pH have been investigated to study its release in simulated physiological and tumor resected environments. Acidic buffer media at pH 5.0 resulted in higher release of chitosan compared to physiological buffer at pH 7.4, which is based on chitosan’s cationic nature. The initial toxicity of chitosan was tested on human osteoblast and osteosarcoma proliferation through MTT assay analysis and FESEM. No toxicity was observed in osteoblast cells and a significant reduction in osteosarcoma viability was witnessed after 6 days of culture compared to control samples. After drug loading and lyophilization of chitosan was optimized, a final toxicity study was performed., where chitosan significantly decreased cellular viability by over 96% by Day 11 compared to control samples. Additionally, chitosan loaded samples allowed for the regulation osteosarcoma proliferation, with no significant increase in optical density over the course of the study. These results suggest chitosan as a promising drug candidate against osteosarcoma. Chitosan thus has the potential to be utilized in load bearing bone grafts after bone tumor surgery to eradicate remaining cancer cells without harming normal bone growth.
Highlights.
Need for drugs capable of combating multi-drug resistance of osteosarcoma.
Chitosan can trigger apoptosis of metastatic cell lines like breast and bladder.
Little known about effects of chitosan on bone cells, especially osteosarcoma.
Chitosan shows a decrease in human osteosarcoma viability by up to 96%.
Chitosan in ceramic scaffolds can be used for tumor resected sites within bone.
Acknowledgments
Authors would like to acknowledge financial support from the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under grant numbers R01 AR066361, and does not have any possible conflict of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bielack SS, Kempf-Bielack B, Delling G, Exner GU, Flege S, Helmke K, Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols, J Clin Oncol. 20 (2002) 776–90. 10.1200/JCO.2002.20.3.776. [DOI] [PubMed] [Google Scholar]
- [2].Ferrari S, Bertoni F, Zanella L, Setola E, Bacchini P, Alberghini M, Versari M, Bacci G.. Evaluation of p-glycoprotein her-2/erbb-2 p53 and bcl-2 in primary tumor and metachronous lung metastases in patients with high-grade osteosarcoma, Cancer. 100 (2004)1936–1942. 10.1002/cncr.20151. [DOI] [PubMed] [Google Scholar]
- [3].Zhao Y, Zhang CL, Zeng BF, Wu XS, Gao TT, Oda Y, Enhanced chemosensitivity of drug-resistant osteosarcoma cells by lentivirus-mediated bcl-2 silencing, Biochem Biophys Res Commun. 390 (2009) 642–647. 10.1016/j.bbrc.2009.10.020. [DOI] [PubMed] [Google Scholar]
- [4].Song B, Wang Y, Xi Y, Kudo K, Bruheim S, Botchkina GI, Gavin E, Wan Y, Formentini A, Kornmann M, Fodstad O, Ju J, Mechanism of chemoresistance mediated by mir-140 in human osteosarcoma and colon cancer cells, Oncogene. 28 (2009) 4065–4074. 10.1038/onc.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Song B, Wang Y, Titmus MA, Botchkina G, Formentini A, Kornmann M, Ju J, Molecular mechanism of chemoresistance by mir-215 in osteosarcoma and colon cancer cells, Mol Cancer. 9 (2010) 96. 10.1186/1476-4598-9-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Perut F, Avne S, Fotia C, Baglio SR, Salerno M, Hosogi S, Kusuzaki K, Baldini N.. V-atpase as an effective therapeutic target for sarcomas, Exp Cell Res. 320 (2014) 21–32. 10.1016/j.yexcr.2013.10.011. [DOI] [PubMed] [Google Scholar]
- [7].Mahoney BP, Raghunand N, Baggett B, Gillie RJ, Tumor acidity ion trapping and chemotherapeutics I Acid ph affects the distribution of chemotherapeutic agents in vitro, Biochem Pharmacol. 66 (2003) 1207–1218. [DOI] [PubMed] [Google Scholar]
- [8].Vaupel P, Kallinowski F, Okunieff P, Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumours: review, Cancer Res. 49 (1989) 6449–6465. [PubMed] [Google Scholar]
- [9].Izumi H, Torigoe T, Ishiguchi H, Uramoto H, Yoshida Y, Tanabe M, Ise T, Murakami T, Yoshida T, Nomoto M, Kohno K, Cellular pH regulators: potentially promising molecular targets for cancer chemotherapy, Cancer Treat. Rev 29 (2003) 541–549. 10.1016/S0305-7372(03)00106-3. [DOI] [PubMed] [Google Scholar]
- [10].Warburg O, Wind F, Negelein E, The Metabolism of Tumors in the Body, J.Gen Physiol 8(152) 319–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gillies RJ, Liu Z, Bhujwalla Z, 31P-MRS measurements of extracellular pH of tumors 1 using 3-aminopropylphosphonate, Am J Physiol. 1257 (1994) C195–203. 10.1152/ajpcell.1994.267.1.C195. [DOI] [PubMed] [Google Scholar]
- [12].Gillies RJ, Robey I, Gatenby RA, Causes and consequences of increased glucose metabolism of cancers, J Nucl Med. 49 (2008) 24S–42S. 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- [13].Gatenby RA, Gillies RJ, Why do cancers have high aerobic glycolysis, Nat Rev Cancer. 4 (2004) 891–899. 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- [14].Reimann A, Schneider B, Gundel D, Stock C, Gekle M, Thews O, Acidosis Promotes Metastasis Formation by Enhancing Tumor Cell Motility, Adv. Exp. Med. Biology 876(2016) 215–220. 10.1007/978-1-4939-3023-4_27. [DOI] [PubMed] [Google Scholar]
- [15].Tan ML, Shao P, Friedhuber AM, van Morost M, Elahy M, Indumathy S, Dunstan DE, Wei Y, Dass CR, The potential role of free chitosan in bone trauma and bone cancer management, Biomaterials. 35 (2015) 7828–7838. 10.1016/j.biomaterials.2014.05.087. [DOI] [PubMed] [Google Scholar]
- [16].Pillé JY, Li H, Blot E, Bertrand JR, Pritchard LL, Opolon P, Maksimenko A, Lu H, Vannier JP, Soria J, Intravenous Delivery of Anti-RhoA Small Interfering RNA Loaded in Nanoparticles of Chitosan in Mice: Safety and Efficacy in Xenografted Aggressive Breast Cancer. Hum. Gene Ther 17 (2006) 1019–1026. 10.1089/hum.2006.17.1019. [DOI] [PubMed] [Google Scholar]
- [17].Deng X, Cao M, Zhang J, Hu K, Yin Z, Zhou Z, Xiao X, Yang Y, Sheng W, Wu Y, Zeng Y, Hyaluronic acid-chitosan nanoparticles for co-delivery of MiR-34a and doxorubicin in therapy against triple negative breast cancer, Biomaterials. 35 (2014) 4333–4344. 10.1016/j.biomaterials.2014.02.006. [DOI] [PubMed] [Google Scholar]
- [18].Hasegawa M, Yagi K, Iwakawa S, Hirai M, Chitosan induces apoptosis via caspase-3 activation in bladder tumor cells, Japanese Journal of Cancer. 92 (2001) 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tan Mei L., Friedhuber Anna M., Dunstan Dave E., Choong Peter F.M., Dass Crispin R., The performance of doxorubicin encapsulated in chitosan–dextran sulphate microparticles in an osteosarcoma model, Biomaterials. 31 (2010) 541–551. 10.1016/j.biomaterials.2009.09.069. [DOI] [PubMed] [Google Scholar]
- [20].Ordikhani F, Tamjid E, Simichi A, Characterization and antibacterial performance of electrodeposited chitosan–vancomycin composite coatings for prevention of implant- infections, Mater. Sci. Eng. C 41 (2014) 240–248. 10.1016/j.msec.2014.04.036. [DOI] [PubMed] [Google Scholar]
- [21].Winterowd JG, Sandford PA, Chitin and chitosan, Food Sci. Tech – New York: – Marcel Dekker, (1995) 441. [Google Scholar]
- [22].Taylor S, Advances in Food and Nutrition Research, 49 (2005) 94–96. [Google Scholar]
- [23].Bucholz RW, Carlton A, Holmes R, Interporous hydroxyapatite as a bone-graft substitute in tibial plateau fractures, Clin. Orthop. Relat. Res 240 (1989) 53–62. [PubMed] [Google Scholar]
- [24].Zeltinger J, Sherwood JK, Graham DA, Mueller R, Griffith LG, Effect of pore size and void fraction on cellular adhesion, proliferation, and matrix deposition, Tissue Eng. 5 (2001) 557–572. 10.1089/107632701753213183. [DOI] [PubMed] [Google Scholar]
- [25].Bose S, Tarafder S, Calcium phosphate ceramic systems in growth factor and drug delivery for bone tissue engineering: a review, Acta Biomater. 8 (2012) 1401–1421. 10.1016/j.actbio.2011.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tarafder S, Banerjee S, Bandyopadhyay A, Bose S, Electrically polarized biphasic calcium phosphates: adsorption and release of bovine serum albumin, Langmuir, 26 (2010) 16625–16629. 10.1021/la101851f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Roy M, Bandyopadhyay A, Bose S, Induction plasma sprayed nano hydroxyapatite coatings on titanium for orthopaedic and dental implants, Surf. Coat. Tech 205 (2011) 2785–2792. 10.1016/j.surfcoat.2010.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Babu A, Ramesh R, Multifaceted Applications of Chitosan in Cancer Drug Delivery and Therapy, Marine drugs. 15 (2017). 10.3390/md15040096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Vu AA, Robertson SF, Ke D, Bandyopadhyay A, Bose S, Mechanical and biological properties of ZnO, SiO2, and Ag2O doped plasma sprayed hydroxyapatite coating for orthopaedic and dental applications, Acta Biomater. 92 (2019) 325–335. 10.1016/j.actbio.2019.05.020 [DOI] [PubMed] [Google Scholar]
- [30].Fielding GA, Roy M, Bandyopadhyay A, Bose S, Antibacterial and biological characteristics of silver containing and strontium doped plasma sprayed hydroxyapatite coatings, Acta Biomater. 8 (2012) 3144–3152. 10.1016/j.actbio.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tarafder S, Nansen K, Bose S, Lovastatin release from polycaprolactone coated β-tricalcium phosphate: effects of pH, concentration and drug-polymer interactions, Mater Sci Eng C Mater Biol Appl. 33 (2013) 3121–3128. 10.1016/j.msec.2013.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bose S, Sarkar N, Banerjee D, Effects of PCL, PEG and PLGA polymers on curcumin release from calcium phosphate matrix for in vitro and in vivo bone regeneration, Mater. Tod. Chem 8 (2018) 110–120. 10.1016/j.mtchem.2018.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sarkar N, Bose S, Liposome-Encapsulated Curcumin-Loaded 3D Printed Scaffold for Bone Tissue Engineering, ACS Appl. Materl. Inter 11 (2019) 17184–17192. 10.1021/acsami.9b01218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tan ML, Shao P, Friedhuber AM, van Moorst M, Elahy M, Indumathy S, Dunstan DE, Wei Y, Dass CR, The potential role of free chitosan in bone trauma and bone cancer management, Biomater. 35 (2014) 7828–7838. 10.1016/j.biomaterials.2014.05.087. [DOI] [PubMed] [Google Scholar]
- [35].Shen K-T, Chen M-H, Chan H-Y, Jeng J-H, Wang Y-J, Inhibitory effects of chitooligosaccharides on tumor growth and metastasis, Food Chem. Tox 47 (2009) 1864–1871. 10.1016/j.fct.2009.04.044. [DOI] [PubMed] [Google Scholar]
- [36].Carreno-Gomez B, Duncan R, Evaluation of the biological properties of soluble chitosan and chitosan microspheres, International Journal of Pharmaceutics, 148 (1997) 231–240. 10.1016/S0378-5173(96)04847-8. [DOI] [Google Scholar]
- [37].Adhikari HS, Yadav PN, Anticancer Activity of Chitosan, Chitosan Derivatives, and Their Mechanism of Action, International Journal of Biomaterials, (2018). 10.1155/2018/2952085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Saravanabhavan SS, Rethinasabapathy M, Zsolt S, Kalambettu AB, Elumalai S, Janakiraman M, Huh YS, Natesan B, Graphene oxide functionalized with chitosan based nanoparticles as a carrier of siRNA in regulating Bcl-2 expression on Saos-2 & MG-63 cancer cells and its inflammatory response on bone marrow derived cells from mice, Materials Science and Engineering: C, 99 (2019) 1459–1468. 10.1016/j.msec.2019.02.047. [DOI] [PubMed] [Google Scholar]
- [39].Sumathra M, Sadasivuni KK, Kumar SS, Raja M, Cisplatin-Loaded Graphene Oxide/Chitosan/Hydroxyapatite Composite as a Promising Tool for Osteosarcoma-Affected Bone Regeneration, ACS Omega, (2018) 14620–14633. 10.1021/acsomega.8b02090. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]