

Abstract
The TP53 gene has been widely studied for its roles in cell cycle control, maintaining genome stability, activating repair mechanisms upon DNA damage, and initiating apoptosis should repair mechanisms fail. Thus, it is not surprising that mutations of p53 are the most common genetic alterations found in human cancer. Emerging evidence indicates that dysregulation of lipid metabolism by p53 can have a profound impact not only on cancer cells but also cells of the tumor microenvironment (TME). In particular, intermediates of the sphingolipid and lysophospholipid pathways regulate many cellular responses common to p53 such as cell survival, migration, DNA damage repair and apoptosis. The majority of these cellular events become dysregulated in cancer as well as cell senescence. In this review, we will provide an account on the seminal contributions of Prof. Lina Obeid, who deciphered the crosstalk between p53 and the sphingolipid pathway particularly in modulating DNA damage repair and apoptosis in non-transformed as well as transformed cells. We will also provide insights on the integrative role of p53 with the lysophosphatidic acid (LPA) signaling pathway in cancer progression and TME regulation.
1. Introduction
The tumor suppressor TP53 was first identified in 1979 [2–4] and still remains one of the most studied genes forty years later. More than 80,000 publications have been generated, showcasing immense effort by the scientific community to understand the plethora of roles that p53 play in biology. We refer readers to [5–7] for an in-depth review on p53. The p53 signaling cascade shares many commonalities with the sphingolipid and lysophospholipid pathways in regulating the mechanisms that lead to or protect from apoptotic cell death. In this review, we explore the complex interplay between p53 and lipid signaling in response to DNA damage induced by ionizing radiation, cancer therapeutics as well as pathophysiological conditions accompanied by oxidative genotoxic and metabolic stress.
2. Brief overview of the sphingolipid metabolic pathway
Sphingolipids are a class of lipids composed of a core sphingoid backbone, termed sphingosine. There are several pathways involved in the biosynthesis of sphingolipids as summarized in Fig. 1. The de novo biosynthesis of sphingolipids is initiated by serine palmitoyltransferase, which catalyzes the condensation of serine and palmitoyl-CoA to form 3-ketodihydrosphingosine. This is followed by a series of reduction, acylation and desaturation reactions catalyzed by 3-ketoreductase, ceramide synthase and dihydroceramide desaturase, respectively, to form ceramide. Attachment of a variety of moieties to the head group of ceramide gives rise to ceramide-1-phosphate (C1P) and more complex sphingolipids such as sphingomyelins and glycosphingolipids. Ceramide can be further degraded by ceramidases to generate sphingosine, which is phosphorylated by sphingosine kinases (SPHKs) to produce sphingosine-1-phosphate (S1P). Besides the de novo pathway, ceramide can also be generated via the breakdown of C1P by C1P phosphatase, sphingomyelins by a family of enzymes termed sphingomyelinases (SMase) or glycoceramides by the actions of acid β-glycosylceramidase. Lastly, ceramide can be synthesized from the recycling of sphingosine by ceramide synthases. A detailed review on the metabolism of sphingolipids can be found in [8,9].
Fig. 1.
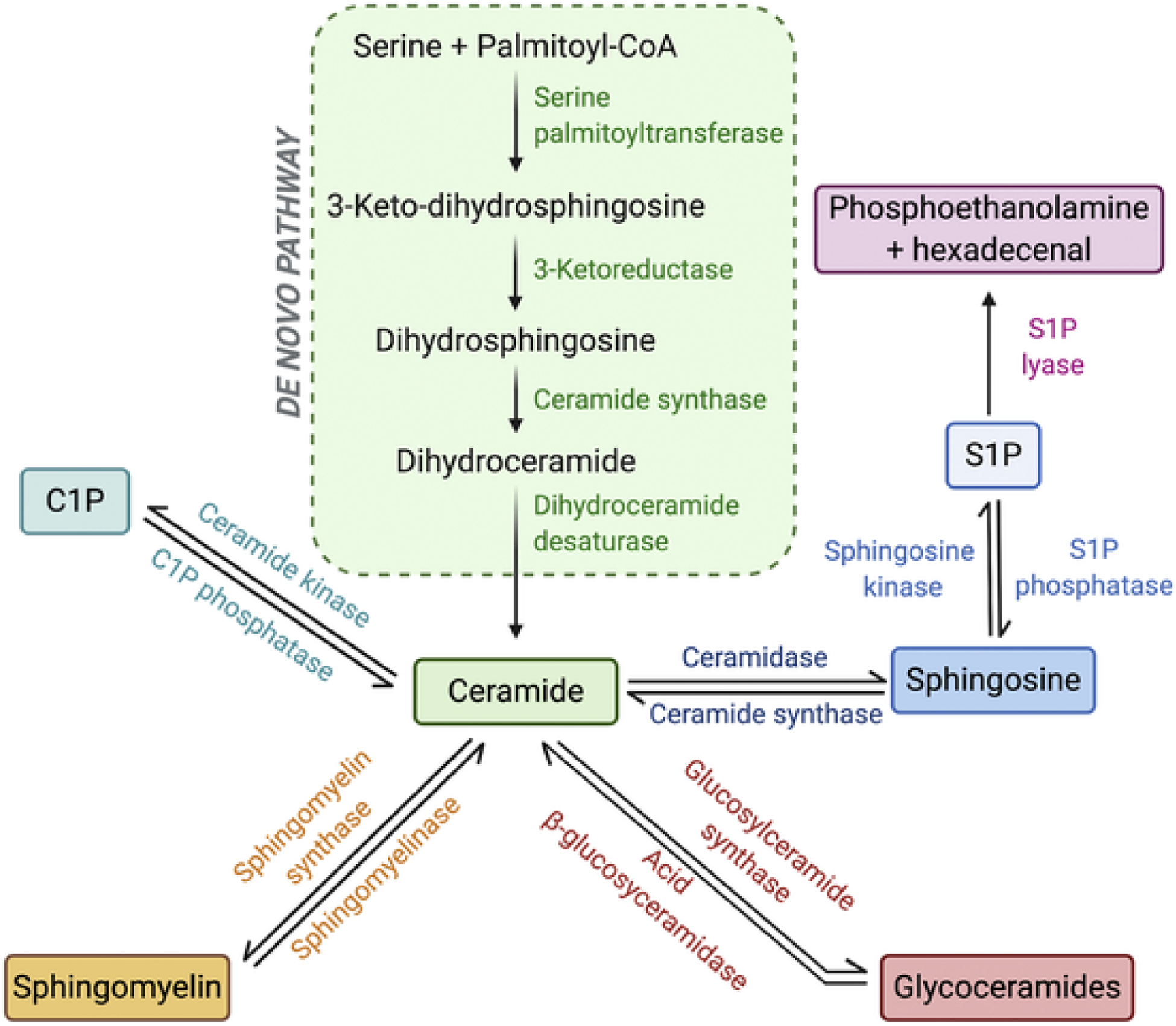
Sphingolipid biochemical pathways.The sphingolipid metabolic pathway is centered on ceramide, where it can be generated via the de novo pathway, the breakdown of C1P, sphingomyelin and glycoceramides, or from the salvage of sphingosine. Sphingosine is phosphorylated to S1P by sphingosine kinases, and S1P signaling can be terminated either by conversion to sphingosine by S1P phosphatases or by the irreversible actions of S1P lyase.
3. Crosstalk of sphingolipids with the p53 pathway
Besides serving as structural components of the cell membrane, sphingolipids regulate a plethora of cellular processes ranging from cell proliferation, survival, and migration to inflammation, senescence, and death (Fig. 2). Emerging evidence indicates that DNA damage induced by either metabolic stress or genotoxic stress (i.e. chemotherapeutic agents, UV and ionizing radiation) perturbs sphingolipid homeostasis [10].
Fig. 2.
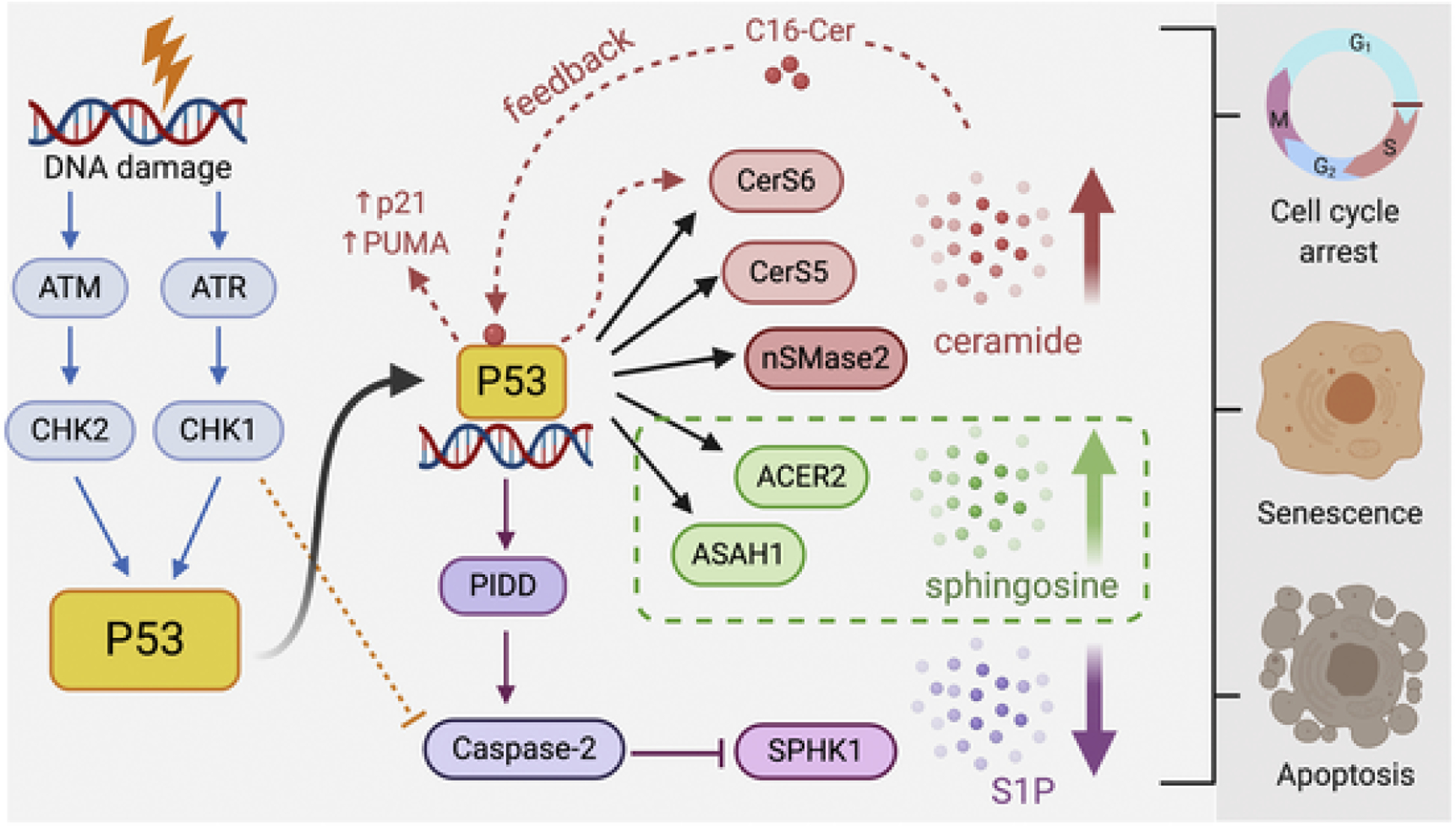
Crosstalk between the p53 and sphingolipid pathways.The complex interplay of p53 with key enzymes of the sphingolipid metabolic pathway in modulating DDR is shown above. The dotted red line represents the feedback mechanism between p53, CerS6 and C16-ceramide to amplify the stress signals in response to metabolic or genotoxic stress. The orange dotted line represents inhibition of caspase-2 by Chk1 in p53-mutant cancer cell lines, and the green dotted box depicts that cellular outcome (survival, growth arrest or apoptosis) is dependent on the magnitude of DNA damage, extent of p53 activation and levels of ACER2 expression in cells.
4. Sphingomyelinase and the p53 pathway
The increase in endogenous levels of ceramide induced by serum deprivation, ionizing radiation and chemotherapeutic agents, and their role in cell cycle arrest, senescence and apoptosis have been documented since the early 1990s [11–14]. The similarities in the biological functions of ceramide with p53 prompted Lina Obeid’s group to investigate the possible crosstalk between these two signaling pathways in 1998. They found that treatment of Molt-4 leukemia cells with either actinomycin D or ionizing irradiation activates p53-dependent apoptosis along with a concomitant increase in endogenous levels of ceramide. The accumulation of ceramide under such conditions was not observed in cells lacking functional p53. However, in scenarios where cell growth was suppressed following serum withdrawal or TNFα treatment, ceramide accumulation occurred irrespective of p53 status, suggesting that the increase in endogenous ceramide levels could be a common cellular response to stress that may be regulated in a p53-dependent or independent manner [15]. As mentioned previously, ceramide can be generated via multiple pathways. The two main sphingolipid metabolizing enzymes reported to be regulated by p53 are the sphingomyelinases (SMases) and ceramide synthases (CerS). The SMases family of enzymes are divided into three groups - neutral, acid and alkaline SMases (nSMases, aSMases and alk-SMases, respectively) [16]. Whereas there are six mammalian CerS enzymes termed CerS1-6 [17].
4.1. Neutral sphingomyelinase
Studies by Sawada et al., showed that treatment of human glioma cells with etoposide induced p53-dependent reactive oxygen species O2− formation, which in turn activated nSMase leading to the accumulation of ceramide and death of these glioma cells [18]. This is consistent with earlier observations where DNA damage caused by ionizing radiation or the chemotherapeutic drug daunorubicin increased ceramide levels through the actions of nSMases [13,19]. There are at least four mammalian nSMases that are active at neutral pH, termed nSMase1, nSMase2, nSMase3, and mitochondria associated MA-nSMas [16]. A direct transcriptional upregulation of nSMase2 was demonstrated in MCF-7 breast cancer cells treated with doxorubicin. Specifically, the authors found that doxorubicin-induced nSMase2 expression was mediated by the DNA damage effectors ataxia telangiectasia and Rad3-related protein (ATR) and checkpoint kinase 1 (Chk1). Both pharmacological inhibition and siRNA knockdown of ATR or Chk1 blocked nSMase2 upregulation in response to doxorubicin. As ATR and Chk1 pathways converge on p53, it was no surprise that siRNA knockdown of p53 or overexpression of mutant p53-R280K, which lacks DNA-binding capacity, abolished doxorubicin-induced transcriptional upregulation of nSMase2. The putative p53 binding site was mapped to a transcription start site upstream of exon 3 of nSMase2 [20].
4.2. Acid sphingomyelinase
Lymphoblasts isolated from patients with Niemann-Pick’s disease (NPD) are resistant to ionizing-radiation-induced apoptosis due to a lack of aSMase expression. Retroviral transduction of aSMase restored the ability of these lymphoblasts to generate ceramide and sensitized them to apoptosis following radiation exposure [21]. Interestingly, the authors proposed that aSMase may induce apoptosis in a manner that is independent of the p53 pathway, suggesting that p53 may differentially regulate members of the SMase family of enzymes. By comparing the extent of apoptotic response in various tissues isolated from aSMase-null or p53-null mice following radiation exposure, the authors revealed that ionizing radiation can activate two distinct signaling mechanisms to induce apoptosis in a manner that is also dependent on the cell/tissue type [21].
There has been a constant debate in the field as to which of the enzymes - SMase or CerS - is predominantly responsible for the increase in ceramide levels following genotoxic injury. Studies by Vit and Rosselli showed that ceramide generation in response to ionizing radiation may occur in a biphasic manner, with each phase individually regulated by aSMase or CerS [22]. Specifically, the first rapid and transient production of ceramide is tied to aSMase activation, whereas a more robust and longer-lasting elevation in ceramide that manifests 8–24 h after radiation exposure is tied to CerS activity as it is inhibited by the CerS inhibitor Fumonisin B1. However, the second phase of ceramide elevation is absent in lymphoblasts isolated from NPD patients even though these cells have normal resting CerS activity, indicating that radiation-induced activation of CerS may be partially linked to aSMase activation [21,22].
5. Ceramide synthase and the p53 pathway
On the contrary, studies by Panjarian et al., argued that p53-dependent ceramide accumulation occurs predominantly via the de novo pathway instead of via hydrolysis of sphingomyelin by SMases. The key enzyme of the de novo pathway responsible for the increase in ceramide levels following γ-irradiation or ZnCl2 treatment in Molt-4 leukemia cells or EB-1 colon cancer cells, respectively, was identified to be ceramide synthase 5 (CerS5) [23]. This is consistent with data obtained by Bose et al., where Fumonisin B1 blocked daunorubicin-induced ceramide accumulation and apoptosis in P388 and U937 cells [14]. In another study, p53-dependent upregulation of CerS6 and the subsequent accumulation of C16-ceramide was observed in A549 and HCT116 cancer cell lines subjected to metabolic stress either by depletion of folate from growth media or from overexpression of the folate-metabolizing enzyme Aldh1l1. This effect was not observed in p53-null cells or the transcriptionally inactive p53-R175H mutant, suggesting that functional p53 is required to upregulate CerS6 expression. Moreover, accumulation of C16-ceramide was also abolished when cells were treated with Fumonisin B1 or siRNA silencing of CerS6 [24]. Interestingly, a recent study by Fekry et al., reported C16-ceramide to be a natural ligand that binds within the DNA binding domain of p53. Specifically, the acyl chain of ceramide interacts in close proximity to the BOX V motif of p53, which is involved in the binding with the E3 ligase MDM2. This interaction is highly specific to C16-ceramide as neither dihydro-C16-ceramide nor ceramide with varying acyl chain length binds to p53. Binding of C16-ceramide to p53 disrupted its complex with MDM2 and promoted p53 translocation to the nucleus to activate transcription of downstream targets such as p21 and PUMA [25]. Thus, the finding that p53 transcriptionally upregulates CerS6 leading to the accumulation of C16-ceramide [24] coupled with the discovery that C16-ceramide itself can directly bind to and regulate p53 activation suggest a possible feedback mechanism in place that may serve to amplify stress response signals within cells [25]. Consistent with this finding, exogenously applied C16-ceramide has been reported to upregulate p53 expression and activity in HCT116 cells [26]. Similarly, exogenous treatment of various cell types with C2-ceramide has also been reported to upregulate p53 [27,28]. More recently, work by Chang et al., described a phenomenon where exogenous C2-ceramide differentially regulates both wildtype p53 and mutant p53 in breast cancer cells lines. The authors found that exogenous C2-ceramide induced apoptosis in MDA-MB-231 cells, which harbor mutant p53, by downregulating its expression. Whereas in MCF-7 cells harboring wildtype p53, exogenous C2-ceramide caused a transient increase in the expression of p53, p21, and the retinoblastoma protein (RB) to induce growth arrest and senescence in MCF-7 cells [29]. Taken together, these studies show that ceramide can function downstream or upstream of p53, or independent of p53 [28], depending on cell types and stimuli applied.
6. Ceramidase and the p53 pathway
The conversion of ceramide to sphingosine is mediated by the action of ceramidases, which are classified into three groups - acid ceramidase (ASAH1), neutral ceramidase (ASAH2) and alkaline ceramidase (ACER1-3). It has been reported that ASAH1 expression was in part upregulated by p53 in human U-87 glioblastoma cells in response to γ-radiation. This led to a decrease in ceramide levels and protected U-87 cells from apoptosis induced by γ-radiation [30]. ACER2 is another ceramidase found to be upregulated in response to DNA damage in cancer cell lines harboring functional p53 but not in p53-deficient cell lines [31]. The putative p53 binding site was mapped to the first intron of the ACER2 gene by two independent groups [32,33]. The authors demonstrated that the magnitude of DNA damage and p53 activation determine the level of expression of ACER2, which can result in the activation of pro-survival signals, cell cycle arrest, senescence or apoptosis [33].
7. Sphingosine kinase and the p53 pathway
Sphingosine kinases 1 and 2 (SPHK1 and SPHK2) catalyze the phosphorylation of sphingosine to generate S1P, which regulates cell proliferation, survival and migration [34]. In this section, we highlight the seminal work by Lina Obeid’s group in identifying SPHK1 as a critical downstream target of p53 and discuss its roles in modulating DDR. In 2004, Taha et al., demonstrated that activation of p53 in response to DNA damage caused by genotoxic stress (actinomycin D, doxorubicin, etoposide and γ-radiation) led to the degradation of SPHK1 in Molt-4 leukemia cells. This effect was absent in Molt-4 E6 cells expressing the human papillomavirus E6 protein, which targets p53 for degradation [35]. This finding indicates that p53 is a prerequisite for DNA damage-mediated proteolysis of SPHK1. Using the pan-caspase inhibitor ZVAD, the authors found that degradation of SPHK1 in response to actinomycin D was completely blocked, suggesting the involvement of caspase proteases in mediating SPHK proteolysis. The authors found that caspase 3, 6, 7, and 9, together with the lysosomal protease cathepsin B, were all involved at varying degrees in mediating proteolysis of SPHK upon DNA damage [33,35,36]. In support of these findings, Heffernan-Stroud et al., showed that SPHK1 proteolysis occurred in wild type mouse embryonic fibroblasts (MEFs) exposed to UV radiation but not in p53-null MEFs. Interestingly, the p53-mediated proteolysis of SPHK1 was found to be dependent on caspase-2 activation as treatment of wild type MEFs with the caspase-2 inhibitor z-VDVAD completely abrogated SPHK1 degradation [37]. Similar findings were reported by Carroll et al., where silencing caspase-2 using siRNA or using MEFs isolated from caspase-2-null mice failed to initiate SPHK1 proteolysis upon DNA damage [38]. To complement these in vitro observations, Heffernan-Stroud et al., found that p53-null mice have elevated expression of SPHK1 with a concomitant increase in S1P levels and a decrease in ceramide levels. These p53-null mice were more prone to spontaneously develop thymic lymphoma compared to their wild type littermates. Incidentally, deletion of SPHK1 in p53-null mice reduced the occurrence of thymic lymphomas possibly by rectifying the levels of ceramide and S1P, and restoring the expression of cell cycle mediators such as p16 and p21 to induce cellular senescence [37].
As p53 is frequently mutated in the majority of cancer [39], it would be of interest to study how SPHK1 is regulated in response to DNA damage in cancers harboring mutant p53. Once again, work by Obeid’s group demonstrated that in MDA-MB-231 breast cancer cells harboring mutant p53, doxorubicin failed to induce caspase-2 mediated proteolysis of SPHK1. The authors found the Chk1 pathway was responsible for inhibiting caspase-2 activation in p53-null cancer cells [38], a phenomena that was described in earlier studies [40,41]. Pharmacological inhibition or genetic silencing of Chk1 led to the activation of caspase-2 and subsequent SPHK1 degradation in response to genotoxic stress, suggesting that SPHK1 is a novel downstream target of the Chk1-suppressed pathway. However, the mechanism by which caspase-2 mediate SPHK1 proteolysis, whether by directly cleaving SPHK1 or recruitment of other accessory proteins, remains unknown [38].
8. S1P lyase and the p53 pathway
Finally, S1P lyase (SPL), the enzyme that irreversibly terminates S1P signaling, was reported to regulate apoptosis in a p53-, caspase-2-, and p38-dependent manner. Oskouian and colleagues found that ectopic expression of functional SPL in HEK293 cells, but not the catalytically inactive SPL-K353L mutant, potentiated apoptosis in response to DNA damage. Likewise, genetic silencing of endogenous SPL diminished etoposide induced cell death, suggesting a critical role of SPL in modulating DDR. The apoptotic event potentiated by SPL was mediated by p53, as inhibition of p53 with pifithrin-α or overexpression of the dominant negative p53-R248W mutant, abrogated etoposide-induced apoptosis. These findings, together with the observation that SPL is down-regulated in human colon cancer and in adenomatous lesions of the Min mouse model of intestinal tumorigenesis, suggests that SPL may contribute to carcinogenesis via controlling the balance of bioactive sphingolipid intermediates [42].
9. Synopsis of sphingolipid regulation of p53
In summary, these studies clearly demonstrate the complex interplay between the p53 and sphingolipid signaling pathways in modulating DDR toward metabolic and genotoxic stress (Fig. 2). In order to harness the biology of sphingolipids for the treatment of cancer, it is of paramount importance to gain an in-depth understanding on the mechanisms by which p53 (both wildtype and mutant) crosstalk with sphingolipids at multiple steps of the DDR.
10. Introduction to LPA metabolism and LPA receptors
Lysophosphatidic acid (LPA) is a membrane-derived glycerophospholipid with a polar phosphate head group, a glycerol backbone, and a long fatty acyl chain [43]. Multiple pathways are involved in the biosynthesis of LPA as summarized in Fig. 3. Hydrolysis of phosphatidylcholine (PC) by phospholipase A (PLA) produces lysophosphatidylcholine (LPC), which is further cleaved by the enzyme autotaxin (ATX; ENPP2) to generate LPA. This reaction produces the major source of extracellular LPA circulating in biological fluids [44]. LPA is also synthesized by three enzymes located in the endoplasmic reticulum (ER) or the mitochondrial membrane; glycerol-3-phosphate acyltransferase, phospholipase A, and acylglycerol kinase, which contribute to the major source of intracellular LPA [45–47]. Likewise, LPA is degraded by several enzymes, including LPA acyltransferases (LPAAT), lipid phosphate phosphatases (LPP), and lysophospholipases to generate phosphatidic acid (PA), monoacylglycerol (MAG), and glycerol-3-phosphate, respectively [48–50]. LPA elicits its biological functions by activating multiple G-protein-coupled LPA receptors (LPAR). To date, at least six LPAR, termed LPAR1-6 have been identified. LPAR1-3 belong to the endothelial differentiation gene (EDG) family [51], whereas LPAR4-6 are classified as members of the P2Y receptor family [52,53].
Fig. 3.
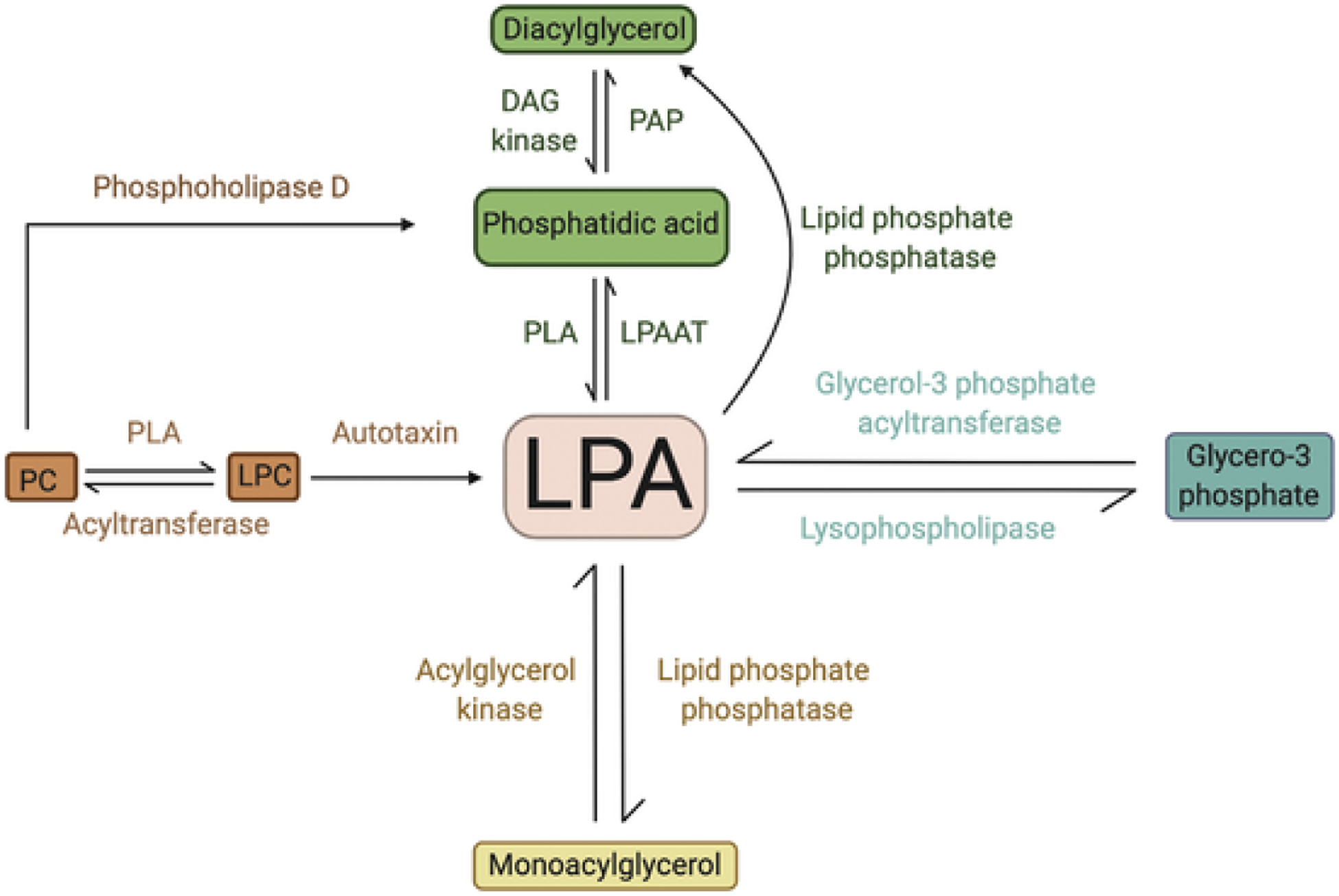
Metabolic pathways of LPA production.DAG: diacylglycerol; PAP: phosphatidic acid phosphatase; PLA: phospholipase A; LPAAT: LPA acyltransferase; PC: phosphatidylcholine; LPC: lysophosphatidylcholine PC: phosphatidylcholine; PA: phosphatidic acid; LPC: lysophosphatidylcholine; LPAR: LPA receptors.
11. Regulation of p53 by the LPA signaling axis
Through the recruitment of different G-protein subunits, LPARs mediate many of the cellular events involved in cancer progression including cell survival [54], protection against apoptosis [55,56] and migration [57]. As these cellular processes are also commonly regulated by p53, we discuss findings from studies that looked at the crosstalk between LPA and p53 signaling pathways.
Previous studies provided evidence for the involvement of LPA/LPAR1 axis in lung cancer carcinogenesis [58]. Furthermore, the survival rate of A549 lung cancer cells treated with cisplatin was significantly elevated by LPAR2 agonist but inhibited by LPAR3 agonist, suggesting that the EDG-family of LPA receptors play important roles in the regulation of chemoresistance to anticancer drugs [59]. The first evidence linking LPA to p53 signaling was reported by Murph et al., who showed that overexpression of LPAR1 and LPAR2 reduced the transcriptional activity and nuclear abundance of p53 in A549 cells. Moreover, treatment of A549 cells with the PI3K/Akt inhibitor LY294002 blocked LPAR-dependent suppression of p53 nuclear localization, suggesting that the repression of p53 signaling by LPA is mediated via the PI3K/Akt pathway [60]. In support of this finding, Hurst-Kennedy et al., found that activation of the LPA/LPAR1 signaling axis decreased nuclear p53 protein, inhibited p53 transcription activity, and protected rat growth plate chondrocytes from apoptosis induced by inorganic phosphate and chelerythrine [61]. In colon cancer cells, LPA has been shown to negatively regulate p53 expression via LPAR2 activation and MDM2 induction. Inhibition of MAPK/ERK signaling by U0126 blocked MDM2 induction and restored p53 activities, implying that the degradation of p53 is regulated via LPAR2/MAPK activation [62]. LPA regulation of glucose metabolism and p53 expression to promote survival of T lymphoma cells has also been demonstrated recently [63]. Therefore, LPA appears to inhibit p53 activity in tumor cells thus functionally pheno-copying the mutational inactivation of p53 found in many human cancers.
LPA also inhibits apoptosis induced by starvation via increasing HIF1-α expression, which has been reported to suppress apoptosis by modulating the activity of p53. Interestingly, Lee and colleagues identified KLF5 as a novel target downstream of LPA signal to increase HIF1-α transcription [62]. Treatment of LPA enhanced the retention of KLF5 on the HIF1-α promoter while decreasing p53 occupancy. Through this mechanism, LPA protects cells against hypoxic signaling by increasing the expression of HIF-1α. On the other hand, direct and indirect evidence indicate mutual antagonism between p53 and NFκB, which has been reported to transcriptionally upregulate the expression of LPARs and ATX [1] during cancer development and chronic inflammation [64]. Heyne et al., demonstrated that MDM2 suppressed the NFκB-mediated gene expression and reduced its DNA binding affinity. This observation emphasized that the homeostasis of p53 and NFκB share the same regulatory mechanism [65] and suggests an indirect regulation through p53-dependent NFκB activity on LPA signaling during tumorigenesis. Taken together, these findings indicate that LPA might differentially activate various signals to regulate p53 activity and hence protects cancer cells against apoptosis in different cancer types. However, studies have also shown that LPA can promote cell death. Specifically, activation of LPAR1 and LPAR2 induce p53-independent p21 expression via ERK phosphorylation and transcription factor CEBPβ, which synergistically generate growth inhibitory signals in TGFβ-treated ovarian cancer cells [66]. LPA signaling through LPAR1 induced anoikis-mediated apoptosis in lung epithelial cells but increased adherence and resistance to apoptosis in fibroblasts. The expression level of p53 was found to be significantly reduced in LPAR1-null cells, suggesting that the LPAR1-dependent apoptosis might be mediated by p53 [67].
In recent years, emerging studies have shown that LPA plays critical roles in age-related disorders [68–70]. Therefore, it is not surprising that LPA can regulate cell senescence via modulation of p53-mediated control of cell cycle progression, apoptosis, and DNA repair during cell senescence [71]. LPA has been reported to differentially regulate the levels of cAMP in senescent and young fibroblasts. The aging-related increase of cAMP with LPA treatment is associated with PKA activation [72]. Moreover, it has been shown that this LPA/cAMP axis further affects PKA activities, resulted in higher levels of inhibitory phosphorylation of AMPK-α. This response decreases the activity of p53 that in turn promotes cell proliferation of senescent fibroblasts [73]. In patients with Hutchinson-Gilford Progeria Syndrome - a premature aging disorder - the protein Progerin has been reported to be modulated by a p53 isoform [74]. Chen and his colleagues found that LPAR3 minimized the levels of reactive oxygen species by upregulating antioxidant enzymes thereby reducing cell senescence in Progerin-transfected cells [70]. Moreover, LPA has been reported to enable murine neuronal and fibroblast cells to bypass p53-dependent senescence [75]. Specifically, overexpression of LPAR2 resulted in the constitutive activation of RhoA GT-Pase, which allowed mouse fibroblasts to bypass cell senescence [75]. However, in LPAR2-overexpressing fibroblasts, p53 still maintained its functional response to cisplatin-induced DNA damage, indicating that LPAR2 signaling does not negate the function of p53 in transduced fibroblasts. On the contrary, previous studies suggested that activation of LPAR1 or LPAR3 signaling induced p53 and p21 expression. The action of the p53–p21 axis resulted in enrichment of cells in G0 phase and promoted cell senescence in human bone marrow stromal cells [76]. These actions support the hypothesis that LPA would generate differential effects on p53 signaling in different tissues and cell types.
12. Regulation of the Autotaxin-LPA-axis by the p53 pathway
Thusfar, only a few studies reported on the regulation of LPA by p53. Chryplewic et al., demonstrated that mutant p53 repressed the expression of LPA phosphatase type 6 (ACP6), the enzyme that degrades LPA into MAG. The resultant increase in LPA levels potentiated pro-tumorigenic events (i.e. proliferation, adhesion, migration, invasion) that drive ovarian cancer progression and metastasis [77]. In breast cancer cells harboring mutant p53, elevated phospholipase D (PLD) and the MAP kinase activities were critical in maintaining the stability and increasing the expression of mutant p53. Considering the fact that PLD generates PA that is one of the intracellular precursors of LPA, this result indicates that a PLD-stabilized mutant p53 may potentially contribute indirectly to the inhibition of apoptosis in human breast cancer cells [78]. Taken together, investigating p53 signaling linked to the regulation of ATX-LPA axis might be an important mechanism that regulates cell growth in various physiological systems. Emerging evidence points to a critical role of the ATX-LPA signaling axis in modulating cells of the tumor microenvironment (TME) [79]. A number of studies have demonstrated that increased expression of ATX promotes cancer progression and metastasis in ovarian cancer [80,81], malignant melanoma [82], and breast carcinoma [83]. It was also demonstrated that different cell types in the TME secret ATX [84–86], implying that ATX may play a role in shaping metastatic niches to support tumor progression [87]. However, the regulatory machinery that controls the transcription of ATX, particularly by p53, has not been sufficiently elucidated. Previous studies reported that ATX is transcriptionally regulated by the transcription factors c-Jun and Sp3 in neuroblastoma cells [88]. The putative binding sites of c-Jun and Sp3 are mapped to the first 285 bp of the ATX promoter, occurring through a CRE/AP-1 like element and GAbox, respectively. A more recent study showed that c-Jun promotes the transcriptional activity of Enpp2, the gene that encodes ATX, which is associated with increased metastasis and poor prognosis in sarcomas [89]. Moreover, microarray results showed that ATX is a specific target of cell transformation by v-Jun, the mutant form of c-Jun. Notably, c-Jun repressed the accumulation of p21 by inhibiting p53 expression in proliferating fibroblasts [90]. In summary, these evidence suggest a potential crosstalk between p53 signaling and ATX expression during cancer development, which might be predominantly mediated by c-Jun.
13. The role of p53 in the regulation of ATX expression in non-transformed cells
We have previously reported that exposure to therapeutic levels of γ-radiation elevates circulating LPA levels in blood plasma and also increases the expression of LPAR2 [1]. Increased plasma LPA concentration indicates either an upregulation of ATX or the inhibition of LPA breakdown. As discussed above, Chryplewic et al. [77] reported that gain of function mutants of p53 inhibit LPA phosphatase ACP6 leading to an elevation of LPA in ovarian carcinoma ascites. However, our observation of plasma LPA elevation at 4 h post-irradiation makes it unlikely that a gain of function mutation in p53 would penetrate and cause a robust and rapid downregulation of ACP6 protein and its enzymatic activity. Thus, we examined whether other regulatory mechanisms might exist between p53 and ATX. Preliminary experiments using p53 inhibitors in fibroblasts showed elevated LPA level in the conditioned medium (data not shown). This led us to examine circulating LPA level in p53−/− mice obtained from Jackson Laboratories (Bar Harbor, ME, USA). We found that the plasma ATX activity in C57Bl/6 p53−/− mice was significantly higher compared with wild type (WT) mice of the same strain. (Fig. 5A). We have established primary intestinal mouse myofibroblast (IMF) cells from WT and p53−/− mice, respectively and examined ATX transcript abundance using quantitative RT-PCR. We found that IMF derived from p53−/− mice showed ~16-fold higher ATX transcripts relative to WT IMF (Fig. 5B). Comparison of ATX protein in the conditioned medium of p53−/− IMF with WT IMF using Western blotting showed a robust elevation (Fig. 5D). We also tested the effect of irradiation on ATX expression and p53 expression in WT IMF and p53−/− IMF using Western blotting (unpublished).
Fig. 5.
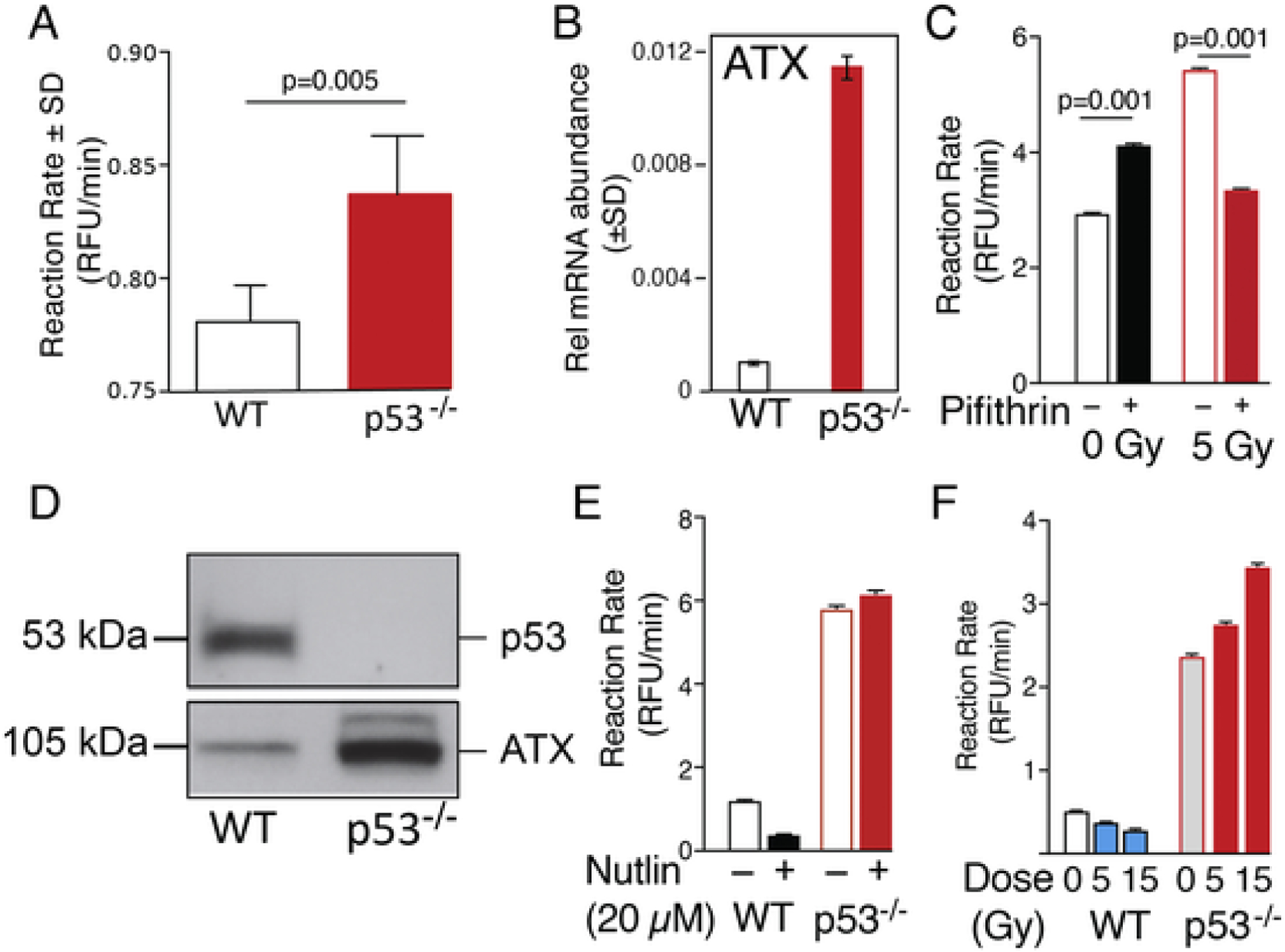
Negative regulation of ATX in non-transformed fibroblast cell. Panel A. Reaction rate of ATX in WT vs p53−/− mouse plasma. For methods see Balogh et al. [1] Average reaction rates (± SEM) were compared using two-tailed t-test in GraphPad between WT and p53−/− mouse plasma (n = 5 for WT, n = 4 for p53−/−). Panel B. Relative mRNA abundance measured by quantitative RT-PCR of ATX in WT (black bars) and p53−/− (red bars) murine IMF. Panel C. C57Bl6 mice (male, 8–10 weeks old) were injected IP with 2.2 mg/kg pifithrin-α or vehicle (1.8% DMSO in PBS). Plasma was collected 24 h later. ATX activity was measured as described in Balogh et al. [1]. Average reaction rates were then compared between treated vs. non-treated irradiated samples using a one-factor ANOVA with Bonferroni’s post-test in GraphPad to determine if rates differed significantly. (n = 6). Panel D. p53−/− IMF secrete elevated amounts of ATX compared to WT MEF cells as revealed by Western blotting. 106 MEF cells were cultured in serum-free medium for 48 h and the cell-free medium was collected and concentrated ~50-fold. Fifty μg protein was loaded on 10% SDS-PAGE and Western blotting was performed using the 4F1 rat monoclonal anti-ATX antibody (kind gift from Drs. J. Aoki and K. Kano, Tokyo University) using the method described previously in Balogh et al. [1] Panel E. Treatment with the p53 inhibitor Nutlin (20 μM for 24 h) reduced ATX activity in the serum free conditioned medium of WT IMF but had no effect in p53−/− IMF (Average reaction rates ± SEM, n = 4). Panel F. γ-Irradiation of WT IMF showed a dose-dependent decrease in ATX activity whereas, in p53−/− it failed to reduce ATX reaction rate (Average reaction rates ± SEM, n = 4).
To reproduce the p53−/− phenotype, WT IMF were treated with Nutlin that blocks MDM2-dependent degradation of p53 leading to activation of the p53 signaling pathway (Figs. 5E & 6). In WT IMF, Nutlin reduced ATX activity that is consistent with the inhibitory effect of p53 signaling on ATX expression. In contrast, Nutlin failed to reduce ATX activity in p53−/− IMF (Fig. 5E). We also compared the effect of γ-irradiation in WT versus p53−/− IMF on ATX activity in serum-free conditioned medium. In WT IMF, γ-irradiation caused a dose-dependent decrease in ATX activity in the medium (Fig. 5F). However, in p53−/− IMF we detected a radiation dose-dependent increase in ATX production (Fig. 5F), which is consistent with our earlier report showing that the activation of the NFkB signaling by the γ-irradiation-induced cytokine surge causes increased ATX expression and enzymatic activity [1]. These results taken together indicate that p53-dependent and -independent mechanisms both regulate the expression of ATX protein in IMF.
Fig. 6.
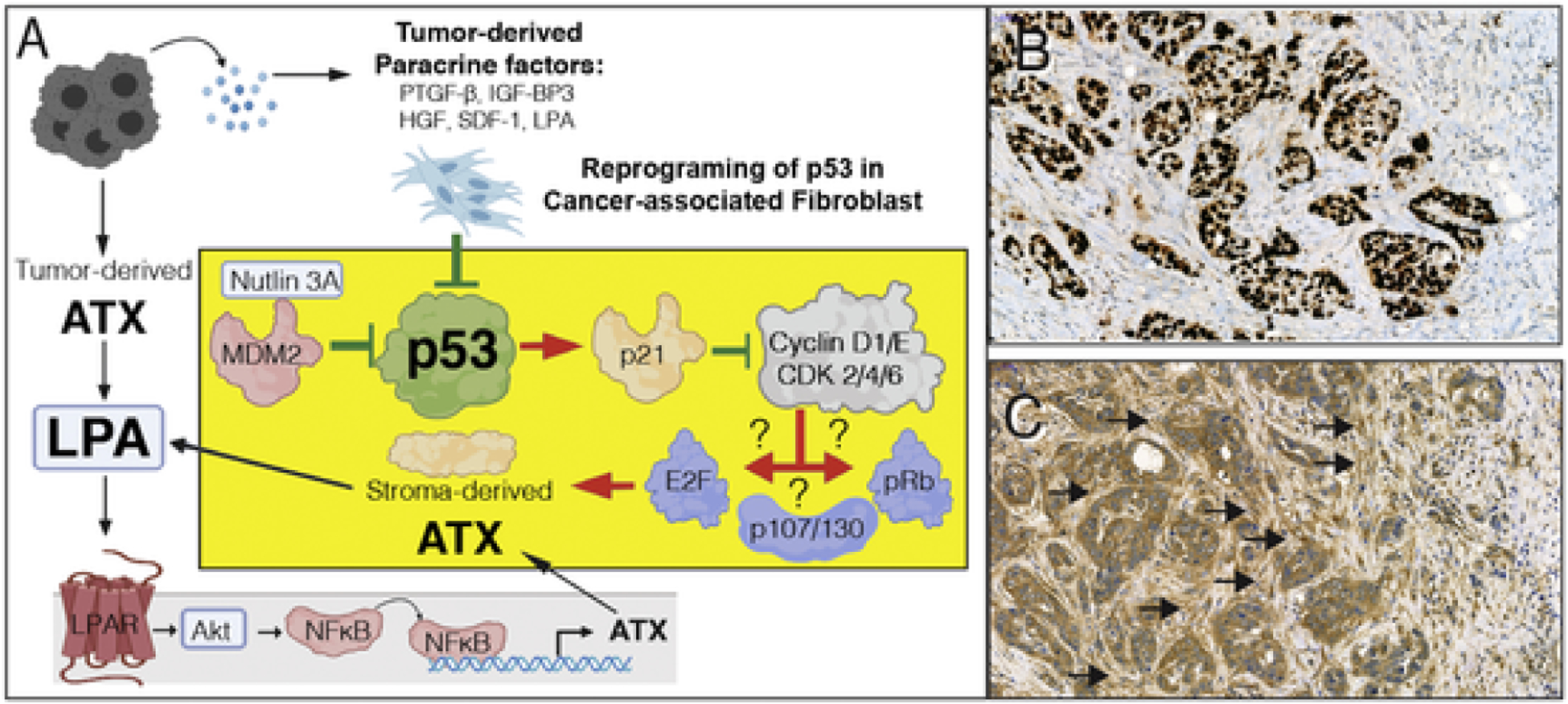
A. Hypothesis for p53 inhibition-mediated upregulation of ATX in CAF. Tumor cell derived paracrine factors inhibit p53, which in turn abolishes p21-mediated inhibition of downstream signals that include cyclinD1/E and cyclin-dependent kinase 2/4/6. Dysinhibition of the cyclin-CDK axis further activates downstream targets such as E2F, p107/130, and retinoblastoma (Rb) transcription factors. It is yet to be determined which of transcription factors is responsible for the upregulation of ATX transcription. On the other hand, tumor cell-derived ATX via the LPAR pathway has been demonstrated to drive ATX expression via NFkB-mediated transcriptional activation. Red arrows represent activation, green lines represent inhibition. The yellow block represents a CAF and the suggested mechanism for p53 inactivation and the ensuing activation of p53 transcription. B & C. A representative ATX immunostaining of an infiltrating ductal breast carcinoma case stained with anti-p53 (B) and 4F1 anti-ATX rat monoclonal antibody (C). Note that the tumor cells are intensively stained for p53, an indication that it is stabilized due to mutation. An adjacent section shows strong desmoplastic reaction with stromal fibroblasts in the immediate vicinity of the tumor cells displaying strong ATX positivity whereas, stromal cells farther away from the tumor (upper right corner of the panel) do not express ATX. Black arrows denote clusters of ATX positive CAF. De-identified specimens exempted from IRB regulation were obtained from the UTHSC Tissue Archive.
Our observation of negative regulation of ATX expression by p53 in non-transformed fibroblasts leads to a new hypothesis for the reprogramming of the ATX-LPA axis in the TME that originates from the observation that tumor-derived paracrine factors inhibit p53 in cancer-associated fibroblasts (CAF) [91–93] (Fig. 6). It appears that inactivation of p53 within a cell affects not only the cell itself but also its microenvironment via the ATX-LPA axis. This non-cell autonomous effect of p53 might be extremely important in the context of cancer and its adjacent stromal cells. Studies have shown that activation of p53 in fibroblasts inhibits the growth, survival and spread of neighboring cancer cells [91–99]. This occurs mainly through a p53-dependent down-regulation of pro-tumorigenic factors such as SDF-1 [91], hepatoma-derived growth factor [100], and upregulation of antitumorigenic factors such as PTGF-β [101] and IGF-BP3 [102]. Interestingly, cancer cells can overcome this inhibitory effect by suppressing the p53 activity in stromal cells, thereby reprogramming the TME to be more conducive for tumor progression. Indeed, clinical and experimental data show that CAFs acquire somatic p53 mutations [103,104]. Preliminary evidence of ATX upregulation in CAF is evident in biopsies of aggressive human breast cancers and correlated with the Ki67 proliferation marker positivity in the 5 cases examined. In these biopsies, tumor cells have shown strong to very modest ATX immune staining, yet the fibroblasts surrounding the tumor cells foci were stained intensively for ATX (Fig. 6 B & C). These preliminary findings supported by multiple reports in the literature suggest that further research is needed to dissect the transcriptional and post-transcriptional regulation of ATX and tissue LPA level.
14. Synopsis of LPA regulation of p53 signaling
In summary, LPA signaling appears to counter p53-dependent apoptosis through altering the homeostasis of p53 via activation of Akt, ERK, or PKA signaling in cancer cells or stromal cells of TME. On the other hand, p53 can modulate the expression of LPA receptors and ATX by regulating NFκB and c-Jun activities (Figs. 3–5). Thus, further research is needed in order to fully elucidate the underlying mechanisms involved in the crosstalk between LPA and p53 signaling pathways. Targeting these mechanisms that comprise the LPA-p53 axis may offer new therapeutic strategies for cancer treatment.
Fig. 4.
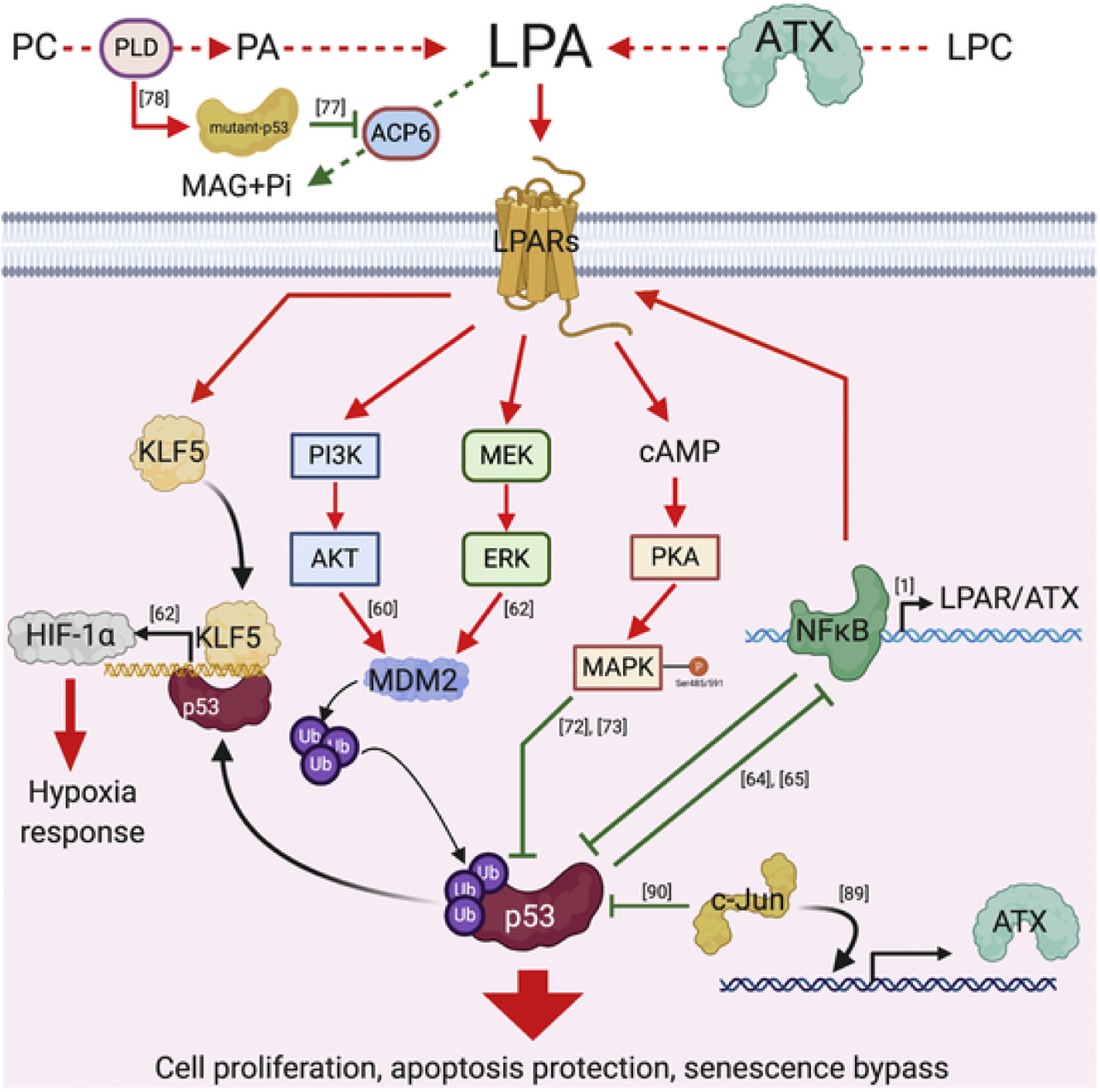
The potential signaling interactions between LPA and p53 in transformed cells. Mutant p53, which is stabilized by PLD and inhibits the expression of ACP6, affects the metabolism pathway of LPA. Activation of LPARs stimulate the Akt, ERK or MAP kinase signaling and further inhibit the activity of p53. Furthermore, NFκB and c-Jun repress the functions of p53 to transcriptionally activate the expression of ATX or LPARs. On the other hand, LPA-LPARs axis increases the expression of KLF5, which competes with p53 to promote expression of HIF-1α in response to hypoxic conditions within the TME. PC: phosphatidylcholine; PA: phosphatidic acid; LPC: lysophosphatidylcholine; LPARs: LPA receptors.
Acknowledgements
All colored figures were created with BioRender.com. We thank Ms. Cindy Lazar and Jorge Solares in the Methodist Hospital Histology Laboratory for the expert assistance with the ATX immunostaining. This work was supported by the National Cancer Institute grant CA092160 (GT), National Institutes of Infectious Diseases Allergy and Immunology grants AI107331 and 1U19AI150574 (GT), The Harriet Van Vleet endowment (GT), and University of Tennessee CORNET grant (SL), and by a grant from the Hungarian National Research Development and Innovation Office (NVKP_16-1-2016-0042).
Footnotes
Declaration of Competing Interest
None.
References
- [1].Balogh A, Shimizu Y, Lee SC, Norman DD, Gangwar R, Bavaria M, Moon C, Shukla P, Rao R, Ray R, Naren AP, Banerjee S, Miller DD, Balazs L, Pelus L, Tigyi G, The autotaxin-LPA2 GPCR axis is modulated by γ-irradiation and facilitates DNA damage repair, Cell. Signal 27 (9) (2015) 1751–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lane DP, Crawford LV, T antigen is bound to a host protein in SY40-transformed cells, Nature 278 (5701) (1979) 261–263. [DOI] [PubMed] [Google Scholar]
- [3].Linzer DIH, Levine AJ, Characterization of a 54K Dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells, Cell 17 (1) (1979) 43–52. [DOI] [PubMed] [Google Scholar]
- [4].DeLeo AB, Jay G, Appella E, Dubois GC, Law LW, Old LJ, Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse, Proc. Natl. Acad. Sci 76 (5) (1979) 2420–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Levine AJ, The many faces of p53: something for everyone, J. Mol. Cell Biol 11 (7) (2019) 524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hafner A, Bulyk ML, Jambhekar A, Lahav G, The multiple mechanisms that regulate p53 activity and cell fate, Nat. Rev. Mol. Cell Biol 20 (4) (2019) 199–210. [DOI] [PubMed] [Google Scholar]
- [7].Kastenhuber ER, Lowe SW, Putting p53 in context, Cell 170 (6) (2017) 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hannun YA, Obeid LM, Sphingolipids and their metabolism in physiology and disease, Nat. Rev. Mol. Cell Biol 19 (3) (2018) 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gault CR, Obeid LM, Hannun YA, An overview of sphingolipid metabolism: from synthesis to breakdown, Adv. Exp. Med. Biol 688 (2010) 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mathias S, Pena LA, Kolesnick RN, Signal transduction of stress via ceramide, Biochem. J 335 (Pt 3) (1998) 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Obeid LM, Linardic CM, Karolak LA, Hannun YA, Programmed cell death induced by ceramide, Science 259 (5102) (1993) 1769–1771. [DOI] [PubMed] [Google Scholar]
- [12].Jayadev S, Liu B, Bielawska AE, Lee JY, Nazaire F, Pushkareva M, Obeid LM, Hannun YA, Role for ceramide in cell cycle arrest, J. Biol. Chem 270 (5) (1995) 2047–2052. [DOI] [PubMed] [Google Scholar]
- [13].Haimovitz-Friedman A, Kan CC, Ehleiter D, Persaud RS, McLoughlin M, Fuks Z, Kolesnick RN, Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis, J. Exp. Med 180 (2) (1994) 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z, Kolesnick R, Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals, Cell 82 (3) (1995) 405–414. [DOI] [PubMed] [Google Scholar]
- [15].Dbaibo GS, Pushkareva MY, Rachid RA, Alter N, Smyth MJ, Obeid LM, Hannun YA, p53-dependent ceramide response to genotoxic stress, J. Clin. Invest 102 (2) (1998) 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Carroll B, Donaldson JC, Obeid L, Sphingolipids in the DNA damage response, Adv. Biol. Regul 58 (2015) 38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Park JW, Park WJ, Futerman AH, Ceramide synthases as potential targets for therapeutic intervention in human diseases, Biochim. Biophys. Acta 1841 (5) (2014) 671–681. [DOI] [PubMed] [Google Scholar]
- [18].Sawada M, Nakashima S, Kiyono T, Nakagawa M, Yamada J, Yamakawa H, Banno Y, Shinoda J, Nishimura Y, Nozawa Y, Sakai N, p53 regulates ceramide formation by neutral sphingomyelinase through reactive oxygen species in human glioma cells, Oncogene 20 (11) (2001) 1368–1378. [DOI] [PubMed] [Google Scholar]
- [19].Jaffrezou JP, Levade T, Bettaieb A, Andrieu N, Bezombes C, Maestre N, Vermeersch S, Rousse A, Laurent G, Daunorubicin-induced apoptosis: triggering of ceramide generation through sphingomyelin hydrolysis, EMBO J. 15 (10) (1996) 2417–2424. [PMC free article] [PubMed] [Google Scholar]
- [20].Shamseddine AA, Clarke CJ, Carroll B, Airola MV, Mohammed S, Rella A, Obeid LM, Hannun YA, P53-dependent upregulation of neutral sphingomyelinase-2: role in doxorubicin-induced growth arrest, Cell Death Dis. 6 (2015) e1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Santana P, Peña LA, Haimovitz-Friedman A, Martin S, Green D, McLoughlin M, Cordon-Cardo C, Schuchman EH, Fuks Z, Kolesnick R, Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis, Cell 86 (2) (1996) 189–199. [DOI] [PubMed] [Google Scholar]
- [22].Vit JP, Rosselli F, Role of the ceramide-signaling pathways in ionizing radiation-induced apoptosis, Oncogene 22 (54) (2003) 8645–8652. [DOI] [PubMed] [Google Scholar]
- [23].Panjarian S, Kozhaya L, Arayssi S, Yehia M, Bielawski J, Bielawska A, Usta J, Hannun YA, Obeid LM, Dbaibo GS, De novo N-palmitoylsphingosine synthesis is the major biochemical mechanism of ceramide accumulation following p53 up-regulation, Prostagland. Other Lipid Mediat 86 (1–4) (2008) 41–48. [DOI] [PubMed] [Google Scholar]
- [24].Hoeferlin LA, Fekry B, Ogretmen B, Krupenko SA, Krupenko NI, Folate stress induces apoptosis via p53-dependent de novo ceramide synthesis and up-regulation of ceramide synthase 6, J. Biol. Chem 288 (18) (2013) 12880–12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fekry B, Jeffries KA, Esmaeilniakooshkghazi A, Szulc ZM, Knagge KJ, Kirchner DR, Horita DA, Krupenko SA, Krupenko NI, C16-ceramide is a natural regulatory ligand of p53 in cellular stress response, Nat. Commun 9 (1) (2018) 4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rénert A-FO, Leprince P, Dieu M, Renaut J, Raes M, Bours V, Chapelle J-P, Piette J, Merville M-P, Fillet M, The proapoptotic C16-ceramide-dependent pathway requires the death-promoting factor btf in colon adenocarcinoma cells, J. Proteome Res 8 (10) (2009) 4810–4822. [DOI] [PubMed] [Google Scholar]
- [27].Willaime S, Vanhoutte P, Caboche J, Lemaigre-Dubreuil Y, Mariani J, Brugg B, Ceramide-induced apoptosis in cortical neurons is mediated by an increase in p38 phosphorylation and not by the decrease in ERK phosphorylation, Eur. J. Neurosci 13 (11) (2001) 2037–2046. [DOI] [PubMed] [Google Scholar]
- [28].Yang J, Duerksen-Hughes PJ, Activation of a p53-independent, sphingolipid-mediated cytolytic pathway in p53-negative mouse fibroblast cells treated withN-methyl-N-nitro-N-nitrosoguanidine, J. Biol. Chem 276 (29) (2001) 27129–27135. [DOI] [PubMed] [Google Scholar]
- [29].Chang WT, Wu CY, Lin YC, Wu MT, Su KL, Yuan SS, Wang HD, Fong Y, Lin YH, Chiu CC, C2-ceramide-induced Rb-dominant senescence-like phenotype leads to human breast cancer MCF-7 escape from p53-dependent cell death, Int. J. Mol. Sci 20 (17) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hara S, Nakashima S, Kiyono T, Sawada M, Yoshimura S, Iwama T, Banno Y, Shinoda J, Sakai N, p53-Independent ceramide formation in human glioma cells during gamma-radiation-induced apoptosis, Cell Death Differ. 11 (8) (2004) 853–861. [DOI] [PubMed] [Google Scholar]
- [31].Xu R, Wang K, Mileva I, Hannun YA, Obeid LM, Mao C, Alkaline ceramidase 2 and its bioactive product sphingosine are novel regulators of the DNA damage response, Oncotarget 7 (14) (2016) 18440–18457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang Y, Zhang C, Jin Y, Wang, He Q, Liu Z, Ai Q, Lei Y, Li Y, Song F, Bu Y, Alkaline ceramidase 2 is a novel direct target of p53 and induces autophagy and apoptosis through ROS generation, Sci. Rep 7 (2017) 44573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Xu R, Garcia-Barros M, Wen S, Li F, Lin CL, Hannun YA, Obeid LM, Mao C, Tumor suppressor p53 links ceramide metabolism to DNA damage response through alkaline ceramidase 2, Cell Death Differ. 25 (5) (2018) 841–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pyne NJ, Ohotski J, Bittman R, Pyne S, The role of sphingosine 1-phosphate in inflammation and cancer, Adv. Biol. Regul 54 (2014) 121–129. [DOI] [PubMed] [Google Scholar]
- [35].Taha TA, El-Alwani M, Hannun YA, Obeid LM, Sphingosine kinase-1 is cleaved by cathepsin B in vitro: identification of the initial cleavage sites for the protease, FEBS Lett. 580 (26) (2006) 6047–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Taha TA, Osta W, Kozhaya L, Bielawski J, Johnson KR, Gillanders WE, Dbaibo GS, Hannun YA, Obeid LM, Down-regulation of sphingosine kinase-1 by DNA damage: dependence on proteases and p53, J. Biol. Chem 279 (19) (2004) 20546–20554. [DOI] [PubMed] [Google Scholar]
- [37].Heffernan-Stroud LA, Helke KL, Jenkins RW, De Costa AM, Hannun YA, Obeid LM, Defining a role for sphingosine kinase 1 in p53-dependent tumors, Oncogene 31 (9) (2012) 1166–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Carroll BL, Bonica J, Shamseddine AA, Hannun YA, Obeid LM, A role for caspase-2 in sphingosine kinase 1 proteolysis in response to doxorubicin in breast cancer cells - implications for the CHK1-suppressed pathway, FEBS Open Bio. 8 (1) (2018) 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L, Mutational landscape and significance across 12 major cancer types, Nature 502 (7471) (2013) 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF, Kanki JP, Green DR, D’Andrea AA, Look AT, Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3, Cell 133 (5) (2008) 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ando K, Kernan JL, Liu PH, Sanda T, Logette E, Tschopp J, Look AT, Wang J, Bouchier-Hayes L, Sidi S, PIDD death-domain phosphorylation by ATM controls prodeath versus prosurvival PIDDosome signaling, Mol. Cell 47 (5) (2012) 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Oskouian B, Sooriyakumaran P, Borowsky AD, Crans A, Dillard-Telm L, Tam YY, Bandhuvula P, Saba JD, Sphingosine-1-phosphate lyase potentiates apoptosis via p53-and p38-dependent pathways and is down-regulated in colon cancer, Proc. Natl. Acad. Sci. U. S. A 103 (46) (2006) 17384–17389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhou C-H, Zhang X-P, Liu F, Wang W, Modeling the interplay between the HIF-1 and p53 pathways in hypoxia, Sci. Rep 5 (1) (2015) 13834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T, Lin ME, Teo ST, Park KE, Mosley AN, Chun J, LPA receptors: subtypes and biological actions, Annu. Rev. Pharmacol. Toxicol 50 (2010) 157–186. [DOI] [PubMed] [Google Scholar]
- [45].Bradley RM, Marvyn PM, Aristizabal Henao JJ, Mardian EB, George S, Aucoin MG, Stark KD, Duncan RE, Acylglycerophosphate acyltransferase 4 (AGPAT4) is a mitochondrial lysophosphatidic acid acyltransferase that regulates brain phosphatidylcholine, phosphatidylethanolamine, and phosphatidylinositol levels, Biochim. Biophys. Acta 1851 (12) (2015) 1566–1576. [DOI] [PubMed] [Google Scholar]
- [46].Sinderewicz E, Grycmacher K, Boruszewska D, Kowalczyk-Zieba I, Staszkiewicz-Chodor J, Lukaszuk K, Woclawek-Potocka I, Expression of genes for enzymes synthesizing lysophosphatidic acid, its receptors and follicle developmental factors derived from the cumulus-oocyte complex is dependent on the ovarian follicle type in cows, Anim. Reprod. Sci 192 (2018) 242–250. [DOI] [PubMed] [Google Scholar]
- [47].Abu El-Asrar AM, Mohammad G, Nawaz MI, Siddiquei MM, Kangave D, Opdenakker G, Expression of lysophosphatidic acid, autotaxin and acylglycerol kinase as biomarkers in diabetic retinopathy, Acta Diabetol. 50 (3) (2013) 363–371. [DOI] [PubMed] [Google Scholar]
- [48].Pyne S, Long JS, Ktistakis NT, Pyne NJ, Lipid phosphate phosphatases and lipid phosphate signalling, Biochem. Soc. Trans 33 (Pt 6) (2005) 1370–1374. [DOI] [PubMed] [Google Scholar]
- [49].Aguado B, Campbell RD, Characterization of a human lysophosphatidic acid acyltransferase that is encoded by a gene located in the class III region of the human major histocompatibility complex, J. Biol. Chem 273 (7) (1998) 4096–4105. [DOI] [PubMed] [Google Scholar]
- [50].Wang A, Dennis EA, Mammalian lysophospholipases, Biochim. Biophys. Acta 1439 (1) (1999) 1–16. [DOI] [PubMed] [Google Scholar]
- [51].Contos JJ, Ishii I, Chun J, Lysophosphatidic acid receptors, Mol. Pharmacol 58 (6) (2000) 1188–1196. [DOI] [PubMed] [Google Scholar]
- [52].Janssens R, Boeynaems JM, Godart M, Communi D, Cloning of a human heptahelical receptor closely related to the P2Y5 receptor, Biochem. Biophys. Res. Commun 236 (1) (1997) 106–112. [DOI] [PubMed] [Google Scholar]
- [53].Kihara Y, Maceyka M, Spiegel S, Chun J, Lysophospholipid receptor nomenclature review: IUPHAR review 8, Br. J. Pharmacol 171 (15) (2014) 3575–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ye X, Ishii I, Kingsbury MA, Chun J, Lysophosphatidic acid as a novel cell survival/apoptotic factor, Biochim. Biophys. Acta 1585 (2–3) (2002) 108–113. [DOI] [PubMed] [Google Scholar]
- [55].Sui Y, Yang Y, Wang J, Li Y, Ma H, Cai H, Liu X, Zhang Y, Wang S, Li Z, Zhang X, Wang J, Liu R, Yan Y, Xue C, Shi X, Tan L, Ren J, Lysophosphatidic acid inhibits apoptosis induced by cisplatin in cervical cancer cells, Biomed. Res. Int 2015 (2015) 598386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang XY, Fan XS, Cai L, Liu S, Cong XF, Chen X, Lysophosphatidic acid rescues bone mesenchymal stem cells from hydrogen peroxide-induced apoptosis, Apoptosis 20 (3) (2015) 273–284. [DOI] [PubMed] [Google Scholar]
- [57].An S, Bleu T, Hallmark OG, Goetzl EJ, Characterization of a novel subtype of human G protein-coupled receptor for lysophosphatidic acid, J. Biol. Chem 273 (14) (1998) 7906–7910. [DOI] [PubMed] [Google Scholar]
- [58].Magkrioti C, Oikonomou N, Kaffe E, Mouratis MA, Xylourgidis N, Barbayianni I, Megadoukas P, Harokopos V, Valavanis C, Chun J, Kosma A, Stathopoulos GT, Bouros E, Bouros D, Syrigos K, Aidinis V, The autotaxin-lysophosphatidic acid axis promotes lung carcinogenesis, Cancer Res. 78 (13) (2018) 3634–3644. [DOI] [PubMed] [Google Scholar]
- [59].Ueda N, Minami K, Ishimoto K, Tsujiuchi T, Effects of lysophosphatidic acid (LPA) receptor-2 (LPA2) and LPA3 on the regulation of chemoresistance to anticancer drug in lung cancer cells, Cell. Signal 69 (2020) 109551. [DOI] [PubMed] [Google Scholar]
- [60].Murph MM, Hurst-Kennedy J, Newton V, Brindley DN, Radhakrishna H, Lysophosphatidic acid decreases the nuclear localization and cellular abundance of the p53 tumor suppressor in A549 lung carcinoma cells, Mol. Cancer Res 5 (11) (2007) 1201–1211. [DOI] [PubMed] [Google Scholar]
- [61].Hurst-Kennedy J, Boyan BD, Schwartz Z, Lysophosphatidic acid signaling promotes proliferation, differentiation, and cell survival in rat growth plate chondrocytes, Biochim. Biophys. Acta 1793 (5) (2009) 836–846. [DOI] [PubMed] [Google Scholar]
- [62].Lee SJ, No YR, Dang DT, Dang LH, Yang VW, Shim H, Yun CC, Regulation of hypoxia-inducible factor 1α (HIF-1α) by lysophosphatidic acid is dependent on interplay between p53 and Krüppel-like factor 5, J. Biol. Chem 288 (35) (2013) 25244–25253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gupta VK, Jaiswara PK, Sonker P, Rawat SG, Tiwari RK, Kumar A, Lysophosphatidic acid promotes survival of T lymphoma cells by altering apoptosis and glucose metabolism, Apoptosis 25 (1–2) (2020) 135–150. [DOI] [PubMed] [Google Scholar]
- [64].Pal S, Bhattacharjee A, Ali A, Mandal NC, Mandal SC, Pal M, Chronic inflammation and cancer: potential chemoprevention through nuclear factor kappa B and p53 mutual antagonism, J. Inflamm 11 (1) (2014) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Heyne K, Winter C, Gerten F, Schmidt C, Roemer K, A novel mechanism of crosstalk between the p53 and NFκB pathways: MDM2 binds and inhibits p65RelA, Cell Cycle 12 (15) (2013) 2479–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wu J, Mukherjee A, Lebman DA, Fang X, Lysophosphatidic acid-induced p21Waf1 expression mediates the cytostatic response of breast and ovarian cancer cells to TGFβ, Mol. Cancer Res 9 (11) (2011) 1562–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Funke M, Zhao Z, Xu Y, Chun J, Tager AM, The lysophosphatidic acid receptor LPA1 promotes epithelial cell apoptosis after lung injury, Am. J. Respir. Cell Mol. Biol 46 (3) (2012) 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lin ME, Herr DR, Chun J, Lysophosphatidic acid (LPA) receptors: signaling properties and disease relevance, Prostagland. Other Lipid Mediat 91 (3–4) (2010) 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yung YC, Stoddard NC, Chun J, LPA receptor signaling: pharmacology, physiology, and pathophysiology, J. Lipid Res 55 (7) (2014) 1192–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Chen WM, Chiang JC, Lin YC, Lin YN, Chuang PY, Chang YC, Chen CC, Wu KY, Hsieh JC, Chen SK, Huang WP, Chen BPC, Lee H, Lysophosphatidic acid receptor LPA(3) prevents oxidative stress and cellular senescence in Hutchinson-Gilford progeria syndrome, Aging Cell 19 (1) (2020) e13064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Rufini A, Tucci P, Celardo I, Melino G, Senescence and aging: the critical roles of p53, Oncogene 32 (43) (2013) 5129–5143. [DOI] [PubMed] [Google Scholar]
- [72].Jang IS, Rhim JH, Kim KT, Cho KA, Yeo EJ, Park SC, Lysophosphatidic acid-induced changes in cAMP profiles in young and senescent human fibroblasts as a clue to the ageing process, Mech. Ageing Dev 127 (5) (2006) 481–489. [DOI] [PubMed] [Google Scholar]
- [73].Rhim JH, Jang IS, Song KY, Ha MK, Cho SC, Yeo EJ, Park SC, Lysophosphatidic acid and adenylyl cyclase inhibitor increase proliferation of senescent human diploid fibroblasts by inhibiting adenosine monophosphate-activated protein kinase, Rejuvenation Res. 11 (4) (2008) 781–792. [DOI] [PubMed] [Google Scholar]
- [74].von Muhlinen N, Horikawa I, Alam F, Isogaya K, Lissa D, Vojtesek B, Lane DP, Harris CC, p53 isoforms regulate premature aging in human cells, Oncogene 37 (18) (2018) 2379–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kortlever RM, Brummelkamp TR, van Meeteren LA, Moolenaar WH, Bernards R, Suppression of the p53-dependent replicative senescence response by lysophosphatidic acid signaling, Mol. Cancer Res 6 (9) (2008) 1452–1460. [DOI] [PubMed] [Google Scholar]
- [76].Kanehira M, Kikuchi T, Ohkouchi S, Shibahara T, Tode N, Santoso A, Daito H, Ohta H, Tamada T, Nukiwa T, Targeting lysophosphatidic acid signaling retards culture-associated senescence of human marrow stromal cells, PLoS One 7 (2) (2012) e32185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Chryplewicz A, Tienda SM, Nahotko DA, Peters PN, Lengyel E, Eckert MA, Mutant p53 regulates LPA signaling through lysophosphatidic acid phosphatase type 6, Sci. Rep 9 (1) (2019) 5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Hui L, Zheng Y, Yan Y, Bargonetti J, Foster DA, Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D, Oncogene 25 (55) (2006) 7305–7310. [DOI] [PubMed] [Google Scholar]
- [79].Tigyi GJ, Yue J, Norman DD, Szabo E, Balogh A, Balazs L, Zhao G, Lee SC, Regulation of tumor cell - microenvironment interaction by the autotaxin-lysophosphatidic acid receptor axis, Adv. Biol. Regul 71 (2019) 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Vidot S, Witham J, Agarwal R, Greenhough S, Bamrah HS, Tigyi GJ, Kaye SB, Richardson A, Autotaxin delays apoptosis induced by carboplatin in ovarian cancer cells, Cell. Signal 22 (6) (2010) 926–935. [DOI] [PubMed] [Google Scholar]
- [81].Ptaszynska MM, Pendrak ML, Stracke ML, Roberts DD, Autotaxin signaling via lysophosphatidic acid receptors contributes to vascular endothelial growth factor-induced endothelial cell migration, Mol. Cancer Res 8 (3) (2010) 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Banerjee S, Norman DD, Lee SC, Parrill AL, Pham TC, Baker DL, Tigyi GJ, Miller DD, Highly potent non-carboxylic acid autotaxin inhibitors reduce melanoma metastasis and chemotherapeutic resistance of breast cancer stem cells, J. Med. Chem 60 (4) (2017) 1309–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Leblanc R, Lee SC, David M, Bordet JC, Norman DD, Patil R, Miller D, Sahay D, Ribeiro J, Clézardin P, Tigyi GJ, Peyruchaud O, Interaction of platelet-derived autotaxin with tumor integrin αVβ3 controls metastasis of breast cancer cells to bone, Blood 124 (20) (2014) 3141–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Lee S-C, Fujiwara Y, Liu J, Yue J, Shimizu Y, Norman DD, Wang Y, Tsukahara R, Szabo E, Patil R, Banerjee S, Miller DD, Balazs L, Ghosh MC, Waters CM, Oravecz T, Tigyi GJ, Autotaxin and LPA1 and LPA5 receptors exert disparate functions in tumor cells versus the host tissue microenvironment in melanoma invasion and metastasis, Mol. Cancer Res 13 (1) (2015) 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Brindley DN, Tang X, Meng G, Benesch MGK, Role of adipose tissue-derived autotaxin, lysophosphatidate signaling, and inflammation in the progression and treatment of breast cancer, Int. J. Mol. Sci 21 (16) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, Tumanov S, Allen-Petersen BL, Link J, Kendsersky ND, Vringer E, Schug M, Novo D, Hwang RF, Evans RM, Nixon C, Dorrell C, Morton JP, Norman JC, Sears RC, Kamphorst JJ, Sherman MH, A stromal lysolipid–autotaxin signaling axis promotes pancreatic tumor progression, Cancer Discov. 9 (5) (2019) 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Valdés-Rives SA, González-Arenas A, Autotaxin-lysophosphatidic acid: from inflammation to cancer development, Mediat. Inflamm 2017 (2017) 9173090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Farina AR, Cappabianca L, Ruggeri P, Di Ianni N, Ragone M, Merolle S, Sano K, Stracke ML, Horowitz JM, Gulino A, Mackay AR, Constitutive autotaxin transcription by Nmyc-amplified and non-amplified neuroblastoma cells is regulated by a novel AP-1 and SP-mediated mechanism and abrogated by curcumin, FEBS Lett. 586 (20) (2012) 3681–3691. [DOI] [PubMed] [Google Scholar]
- [89].Sioletic S, Czaplinski J, Hu L, Fletcher JA, Fletcher CD, Wagner AJ, Loda M, Demetri GD, Sicinska ET, Snyder EL, c-Jun promotes cell migration and drives expression of the motility factor ENPP2 in soft tissue sarcomas, J. Pathol 234 (2) (2014) 190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Schreiber M, Kolbus A, Piu F, Szabowski A, Möhle-Steinlein U, Tian J, Karin M, Angel P, Wagner EF, Control of cell cycle progression by c-Jun is p53 dependent, Genes Dev. 13 (5) (1999) 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Addadi Y, Moskovits N, Granot D, Lozano G, Carmi Y, Apte RN, Neeman M, Oren M, p53 status in stromal fibroblasts modulates tumor growth in an SDF1-dependent manner, Cancer Res. 70 (23) (2010) 9650–9658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Bar J, Feniger-Barish R, Lukashchuk N, Shaham H, Moskovits N, Goldfinger N, Simansky D, Perlman M, Papa M, Yosepovich A, Rechavi G, Rotter V, Oren M, Cancer cells suppress p53 in adjacent fibroblasts, Oncogene 28 (6) (2009) 933–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Bar J, Moskovits N, Oren M, Involvement of stromal p53 in tumor-stroma interactions, Semin. Cell Dev. Biol 21 (1) (2010) 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Burikhanov R, Shrestha-Bhattarai T, Hebbar N, Qiu S, Zhao Y, Zambetti GP, Rangnekar VM, Paracrine apoptotic effect of p53 mediated by tumor suppressor Par-4, Cell Rep. 6 (2) (2014) 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, Lowe SW, Non-cell-autonomous tumor suppression by p53, Cell 153 (2) (2013) 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Meek DW, Regulation of the p53 response and its relationship to cancer, Biochem. J 469 (3) (2015) 325–346. [DOI] [PubMed] [Google Scholar]
- [97].Millau JF, Mai S, Bastien N, Drouin R, p53 functions and cell lines: have we learned the lessons from the past?, Bioessays 32 (5) (2010) 392–400. [DOI] [PubMed] [Google Scholar]
- [98].Pflaum J, Schlosser S, Muller M, p53 family and cellular stress responses in cancer, Front. Oncol 4 (2014) 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Rueda-Rincon N, Bloch K, Derua R, Vyas R, Harms A, Hankemeier T, Khan NA, Dehairs J, Bagadi M, Binda MM, Waelkens E, Marine JC, Swinnen JV, p53 attenuates AKT signaling by modulating membrane phospholipid composition, Oncotarget 6 (25) (2015) 21240–21254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Sasaki Y, Negishi H, Idogawa M, Yokota I, Koyama R, Kusano M, Suzuki H, Fujita M, Maruyama R, Toyota M, Saito T, Tokino T, p53 negatively regulates the hepatoma growth factor HDGF, Cancer Res. 71 (22) (2011) 7038–7047. [DOI] [PubMed] [Google Scholar]
- [101].Tan M, Wang Y, Guan K, Sun Y, PTGF-beta, a type beta transforming growth factor (TGF-beta) superfamily member, is a p53 target gene that inhibits tumor cell growth via TGF-beta signaling pathway, Proc. Natl. Acad. Sci. U. S. A 97 (1) (2000) 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, Kley N, Induction of the growth inhibitor IGF-binding protein 3 by p53, Nature 377 (6550) (1995) 646–649. [DOI] [PubMed] [Google Scholar]
- [103].Hill R, Song Y, Cardiff RD, Van Dyke T, Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis, Cell 123 (6) (2005) 1001–1011. [DOI] [PubMed] [Google Scholar]
- [104].Patocs A, Zhang L, Xu Y, Weber F, Caldes T, Mutter GL, Platzer P, Eng C, Breast-cancer stromal cells with TP53 mutations and nodal metastases, N. Engl. J. Med 357 (25) (2007) 2543–2551. [DOI] [PubMed] [Google Scholar]