

SUMMARY
Mechanical signals, such as those evoked by maximal-intensity contractions (MICs), can induce an increase in muscle mass. Rapamycin-sensitive signaling events are widely implicated in the regulation of this process; however, recent studies indicate that rapamycin-insensitive signaling events are also involved. Thus, to identify these events, we generate a map of the MIC-regulated and rapamycin-sensitive phosphoproteome. In total, we quantify more than 10,000 unique phosphorylation sites and find that more than 2,000 of these sites are significantly affected by MICs, but remarkably, only 38 of the MIC-regulated events are mediated through a rapamycin-sensitive mechanism. Further interrogation of the rapamycin-insensitive phosphorylation events identifies the S473 residue on Tripartite Motif-Containing 28 (TRIM28) as one of the most robust MIC-regulated phosphorylation sites, and extensive follow-up studies suggest that TRIM28 significantly contributes to the homeostatic regulation of muscle size and function as well as the hypertrophy that occurs in response to increased mechanical loading.
Graphical Abstract
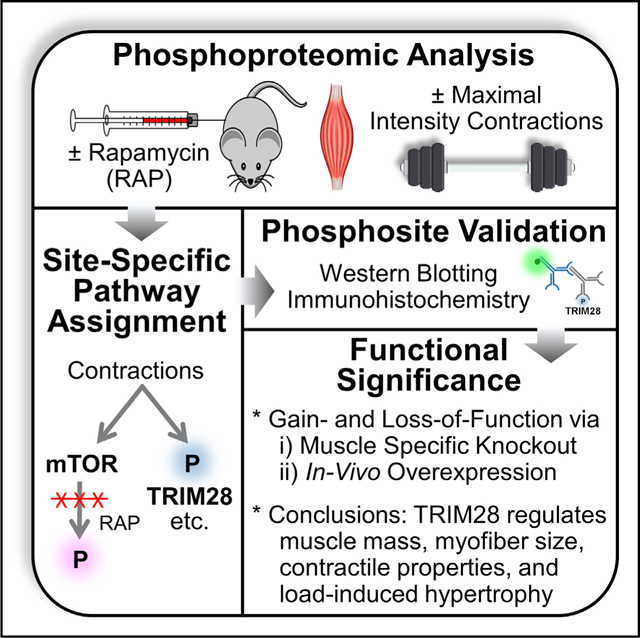
In brief
By comprehensively mapping the rapamycin-sensitive and contraction regulated phosphoproteome, Steinert et al. identify TRIM28 as a non-canonical regulator of muscle size and function. This study provides a resource for further delineating the signaling components through which mechanical stimuli regulate mTOR and muscle growth.
INTRODUCTION
Adult skeletal muscle is a highly plastic tissue that not only drives mobility but also plays a critical role in breathing, whole-body metabolism, and maintenance of a high quality of life (Izumiya et al., 2008; Seguin and Nelson, 2003). Importantly, adults will lose 35%–40% of their muscle mass between the ages of 25 and 80 years, and this progressive loss of muscle is associated with disability, loss of independence, and mortality as well as an estimated $18.5 billion in annual healthcare costs in the United States alone (Janssen et al., 2004; Pahor and Kritchevsky, 1998; Proctor et al., 1998). Thus, the development of therapies that can maintain, restore, or even enhance muscle mass is of great clinical and fiscal significance. However, the development of such therapies will require a comprehensive understanding of the molecular mechanisms that regulate this vital system.
It is well established that skeletal muscle mass can be regulated by a variety of different stimuli, and one of the most potent stimuli is mechanical loading (Adams and Bamman, 2012; Bodine, 2013). For example, numerous studies have shown that maximal-intensity contractions (MICs), such as those that occur during resistance exercise, can induce an increase in muscle mass (Baar and Esser, 1999; Kosek et al., 2006; Ogasawara et al., 2016; Schoenfeld et al., 2016). However, the mechanisms that sense mechanical stimuli and convert them into the signaling events that regulate muscle mass remain largely undefined.
At the most basic level, mechanical load-induced changes in muscle mass are driven by an alteration in the balance between the rate of myofibrillar protein synthesis and the rate of myofibrillar protein degradation, and one of the most widely recognized regulators of this balance involves signaling by a protein kinase called the mechanistic target of rapamycin (mTOR). Importantly, a subset of the signaling events that are mediated by mTOR are highly sensitive to inhibition by the drug rapamycin, and it has been shown that (1) various forms of mechanical stimuli can activate the rapamycin-sensitive elements of mTOR signaling (i.e., RSmTOR signaling), (2) the activation of RSmTOR signaling is sufficient to induce an increase in myofiber cross-sectional area (CSA; i.e., hypertrophy), and (3) RSmTOR signaling is necessary for the hypertrophy that occurs in response to a chronic increase in mechanical loading (Goodman et al., 2010, 2011; Ogasawara et al., 2019; You et al., 2012, 2019). Collectively, these types of observations have led to the widely held contention that RSmTOR signaling is the primary regulator of mechanical load-induced changes in muscle mass (Goodman, 2014; Marcotte et al., 2015; Wackerhage et al., 2019; Yoon, 2017).
Although a role for RSmTOR signaling in the mechanical regulation of muscle mass has been widely accepted, an emerging body of evidence suggests that rapamycin-insensitive signaling events are also involved (Marcotte et al., 2015; Ogasawara et al., 2019; Wackerhage et al., 2019). For example, we recently found that rapamycin only partially inhibited the increase in muscle mass and hypertrophy that occurred in a rodent model of resistance exercise (Ogasawara et al., 2016). Moreover, recent studies have shown that various forms of mechanical stimuli (e.g., resistance exercise, chronic mechanical overload, and endurance exercise) can promote an increase in protein synthesis via a rapamycin-insensitive mechanism (Ogasawara et al., 2016; Ogasawara and Suginohara, 2018; Philp et al., 2015; West et al., 2016; You et al., 2019). Hence, it is now becoming apparent that both rapamycin-sensitive and rapamycin-insensitive signaling events contribute to the mechanical regulation of muscle mass.
In this study, we used an unbiased phosphoproteomic-based approach to obtain a global map of the rapamycin-sensitive and rapamycin-insensitive signaling events that are elicited after a bout of MICs. In total, we quantified over 10,000 unique phosphorylation sites and found that over 2,000 of these sites were significantly affected by MICs, but remarkably, only 38 of the MIC-regulated events were mediated through a rapamycin-sensitive mechanism. Further interrogation of the rapamycin-insensitive phosphorylation events revealed that the S473 residue on TRIM28 (Tripartite Motif-Containing 28) was one of the most robust MIC-regulated phosphorylation sites, and extensive follow-up studies enabled us to establish that TRIM28 is a significant regulator of skeletal muscle mass and function.
RESULTS
Mapping of the rapamycin-sensitive and MIC-regulated phosphoproteome
As outlined in Figure 1, wild-type C57BL/6 mice were injected with 1.0 mg/kg rapamycin or the solvent vehicle. Following the injection, the right leg of each mouse was subjected to a bout of MICs while the left leg was untouched and served as a contralateral control. At 1 h after the bout of MICs, the tibialis anterior (TA) muscles from each leg were collected, lysed, and then separated into soluble and insoluble protein fractions. Before proceeding through the phosphoproteomics workflow, western blot analysis was used to confirm that the bout of MICs had successfully induced robust changes in protein phosphorylation (e.g., P-MKK4(S255)) and that rapamycin had effectively inhibited the MIC-induced activation of signaling by RSmTOR (P-p70(T389)) (You et al., 2019). Having established these outcomes, the samples were then subjected to the remainder of the phosphoproteomics workflow.
Figure 1. Experimental workflow for mapping the RAP-sensitive and MIC-regulated phosphoproteome.
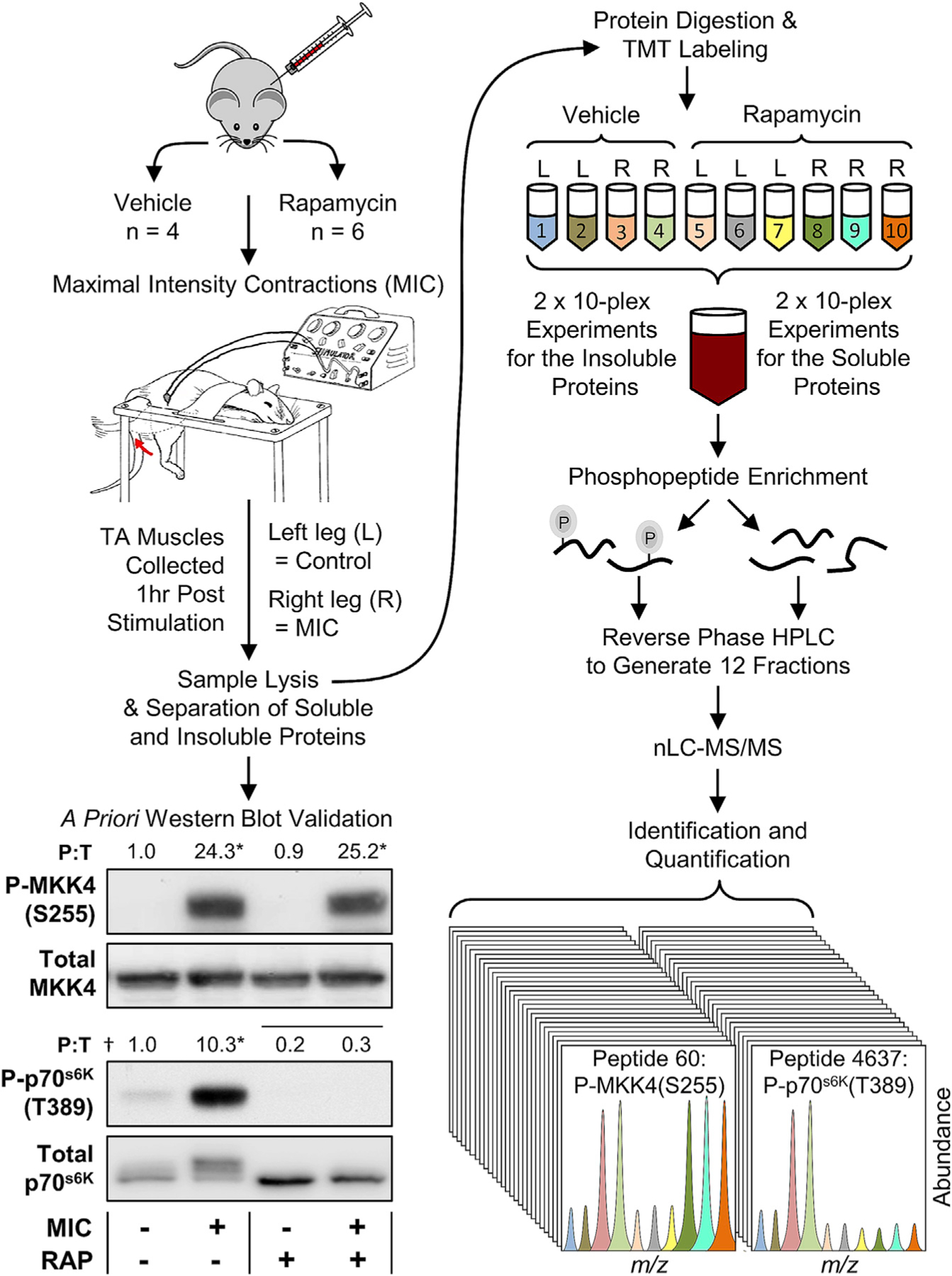
Wild-type C57BL/6 mice were injected with rapamycin (RAP+) or the solvent vehicle (RAP−) and then their tibialis anterior (TA) muscles were subjected to maximal-intensity contractions (MIC+) or the control condition (MIC−). The mice were allowed to recover for 1 h after the bout of MICs and then the TA muscles were collected and separated into soluble and insoluble fractions. The proteins in the soluble fractions were subjected to western blot analysis for the phosphorylated and total levels of the 70-kDa ribosomal protein S6 kinase (p70s6K) and mitogen-activated protein kinase kinase 4 (MKK4). Values above the blots represent the group means for the phosphorylated-to-total ratio (P:T), n = 3–6/group. The data were subjected to two-way ANOVA followed by Student-Newman-Keuls post hoc analyses. Horizontal bar indicates a significant effect for RAP, asterisk (*) indicates a significant effect of MIC, and dagger (†) indicates a significant interaction between RAP and MIC, p < 0.05. Following the western blot analyses, the proteins in the soluble and insoluble fractions were tryptically digested and then the peptides from each fraction/sample were labeled with different tandem mass tags (TMTs). The TMT-labeled peptides were mixed to produce 10-plex pooled samples and then the phosphopeptides were enriched with immobilized metal affinity chromatography. Reverse-phase high-pressure liquid chromatography (HPLC) was then used to generate a total of 12 fractions for the phosphopeptides as well as 12 fractions for the unbound (non-phospho) peptides. All fractions were analyzed by nano-liquid chromatography-tandem mass spectrometry (nLC-MS/MS), and the TMT reporter ions in the MS spectra were used to determine the relative abundance of the phosphopeptides and non-phosphorylated peptides in each sample.
In total, quantitative information was obtained for 10,668 unique phosphopeptides (Table S1) and 4,393 proteins (Table S2). For the phosphopeptides, two-way ANOVA revealed that 2,408 experienced a significant main effect of MICs, 811 encountered a significant main effect of rapamycin, and 231 incurred a significant interaction between the effect of MICs and rapamycin (Figures 2A and 2B). Interestingly, only 29 of the significantly altered phosphorylation sites were annotated with a known regulatory function in PhosphoSitePlus (Figure S1A; Abdelmoity et al., 2000). These sites included previously identified MIC-regulated phosphorylation events such as the rapamycin-sensitive phosphorylation of EIF4B on S422 and the rapamycin-insensitive phosphorylation of Y182 on mitogen-activated protein kinase 14 (MAPK14) (also known as p38 MAPK), as well as non-cannonical MIC-regulated phosphorylation events such as the phosphorylation of S184 on UBE2J1 (Figure S1A; O’Neil et al., 2009; Steiner and Lang, 2014).
Figure 2. Overview of the RAP-sensitive and MIC-regulated phosphoproteome.
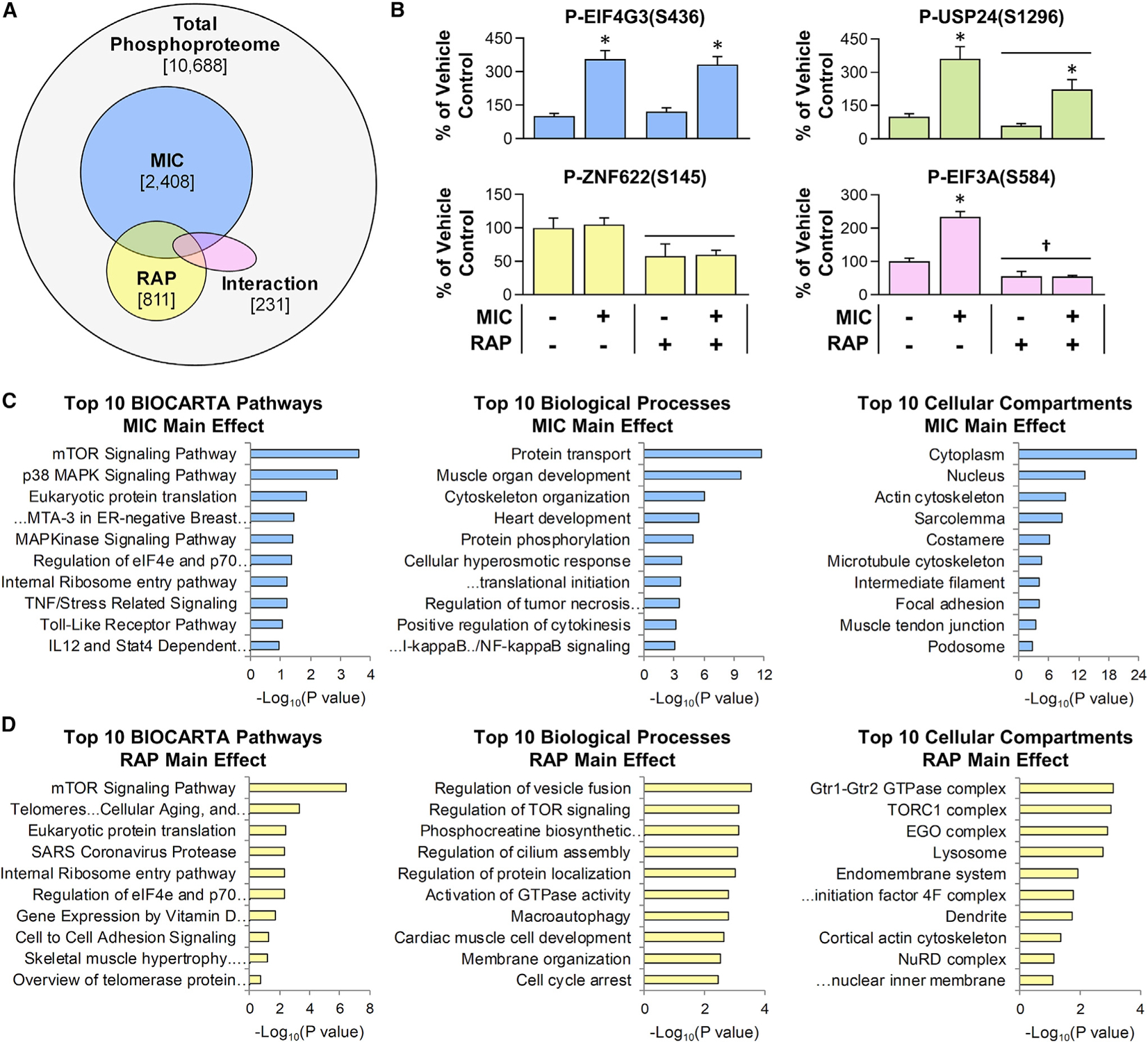
Mice were subjected to the workflow described in Figure 1, and the results were analyzed with two-way ANOVA. (A) Venn diagram indicates the total number of phosphopeptides that were quantified, along with the number of phosphopeptides that revealed a significant main effect for MICs (blue) or RAP (yellow) or a significant interaction between MIC and RAP (pink).
(B) Representative phosphorylation events for each of the categories identified in (A) (e.g., the graph with blue bars provides an example of a phosphorylation site that revealed a significant main effect for MIC, etc.). Values in the graphs represent the group mean + SEM, n = 4–6/group. Horizontal bar indicates a significant effect for RAP, asterisk (*) indicates a significant effect of MIC, and dagger (†) indicates a significant interaction between RAP and MIC, p < 0.05.
(C and D) Enrichment analyses were performed to identify the pathways, biological processes, and cellular compartments that were overrepresented with phosphorylation sites that revealed a significant main effect for MIC (C) or a significant main effect for RAP (D). Values in (C) and (D) represent the false discovery rate (FDR)-corrected p values with the “Top 10” being based on the lowest p values. Note: no pathways, biological processes, or cellular compartments were significantly overrepresented within the list of phosphorylation sites that revealed a significant interaction between MIC and RAP.
To gain further insight into the potential function of the phosphoproteomic alterations, enrichment analyses were used to identify the pathways, biological processes, and cellular compartments that were overrepresented within the subset of phosphorylation sites that revealed a significant main effect for MICs (Figure 2C) or a significant main effect for rapamycin (Figure 2D). As expected, mTOR signaling, p38 MAPK signaling, and eukaryotic protein translation were among the most significantly overrepresented pathways within the subset of phosphorylation sites that revealed a significant main effect for MICs. Moreover, within the subset of phosphorylation sites that revealed a significant main effect for rapamycin, mTOR signaling was the most significantly overrepresented pathway, and the Gtr1-Gtr2 GTPase, TORC1, and EGO/Ragulator complexes, as well as the lysosome, were among the most significantly overrepresented cellular compartments. Of note, all of these compartments play a central role in the pathway through which rapamycin-sensitive signaling events control autophagy, and these compartments were decorated with 30 different rapamycin-sensitive phosphorylation sites (Figure S1B; Al-Bari and Xu, 2020; Dubouloz et al., 2005; Powis and De Virgilio, 2016). Intriguingly, however, only 4 of these sites were previously annotated as being rapamycin sensitive. A complete list of the results from the enrichment analyses is provided in Table S3.
In addition to performing the enrichment analyses, we also used the phosphoproteomic data as a platform to predict which kinases were activated by MICs and whether these kinases were activated upstream/parallel to RSmTOR or downstream of RSmTOR (Figure S2). Consistent with previous studies, the outcomes suggested that MICs promote the activation of various isoforms of the MEK, ERK, JNK, p38, and RSK kinases and that these kinases are activated upstream/parallel to RSmTOR, while kinases such as the 70-kDa ribosomal protein S6 kinase (p70s6K) are activated downstream of RSmTOR (Aronson et al., 1997; Ryder et al., 2000; You et al., 2019). The aforementioned results supported the validity of the kinase predictions and also lent confidence to the unique predictions such as the MIC-induced activation of CDK5 occurring upstream/parallel to RSmTOR, and the MIC-induced activation of SRPK2 occurring downstream of RSmTOR (Lee et al., 2017). Although these predictions were not prioritized for further analysis, we elected to highlight them because both CDK5 and SRPK2 play broad roles in cell biology; thus, we expect that these predictions will serve as fruitful topics for future investigations (Lee et al., 2017; Shupp et al., 2017).
MICs utilize a rapamycin-insensitive mechanism to induce TRIM28(S473) phosphorylation within the nuclei of myofibers
Having established a comprehensive map of the rapamycin-sensitive and MIC-regulated phosphoproteome, we next sought to identify rapamycin-insensitive signaling events that might contribute to the mechanical regulation of muscle mass. As illustrated in Figures 3A–3C, we found that the S473 residue on TRIM28 (a.k.a. KAP1 or Tif1β) was one of the most robust MIC-regulated phosphorylation sites and that the effect of MICs was mediated through a rapamycin-insensitive mechanism.
Figure 3. MICs utilize a RAP-insensitive mechanism to induce TRIM28(S473) phosphorylation within the nuclei of myofibers.
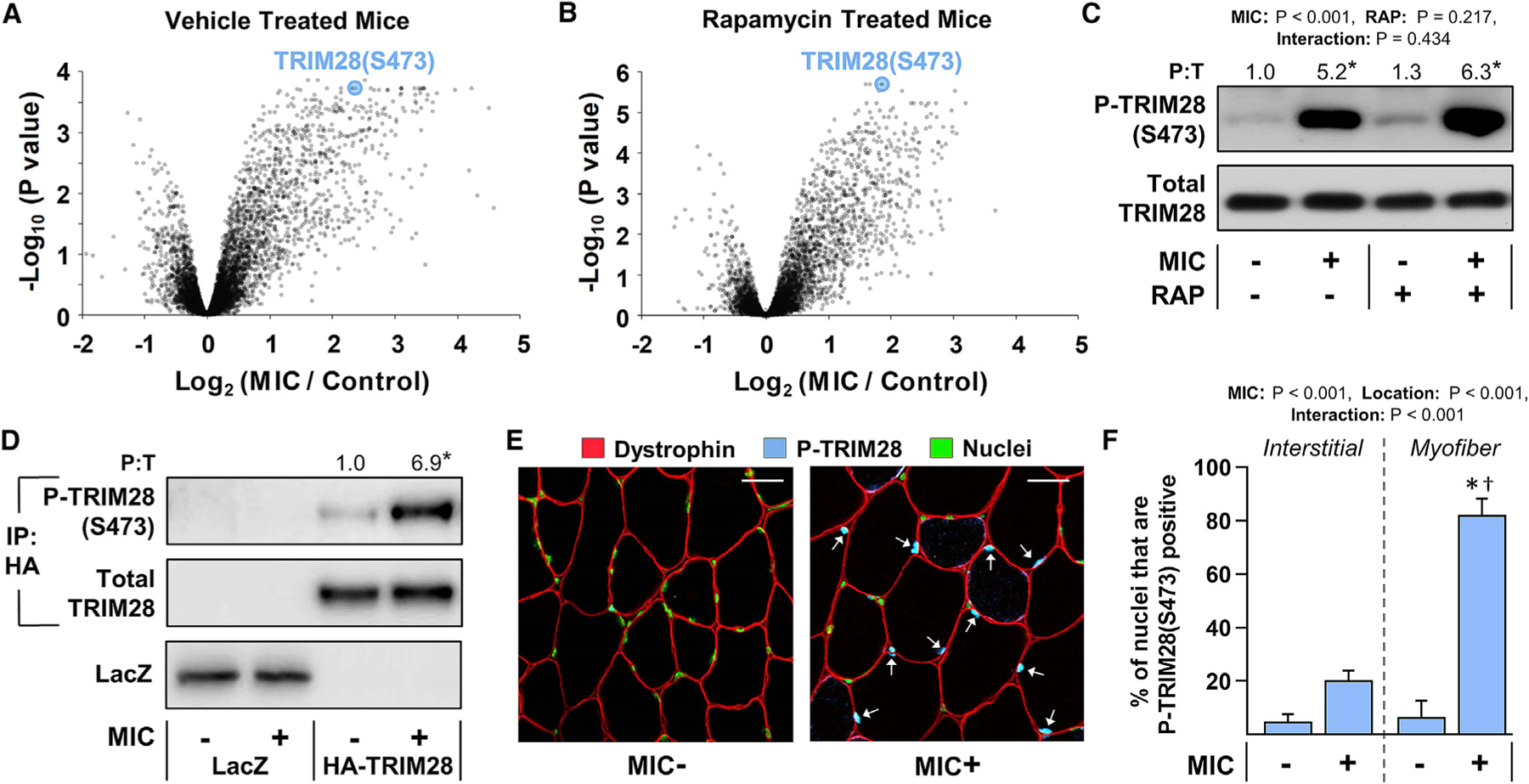
(A–C) Mice were subjected to the workflow described in Figure 1. Volcano plots from the phosphoproteomic analyses in which the fold-change (MIC/control) for each phosphopeptide within the vehicle-treated mice (A), or within the RAP-treated mice (B), was log2 transformed and then plotted against its corresponding −log10 FDR-corrected p value from a moderated t test. (C) Western blot analysis of phosphorylated (P) and total (T) TRIM28 was performed on aliquots of the samples that were subjected to the phosphoproteomic analysis.
(D) Electroporation was used to transfect the myofibers of wild-type C57BL/6 mouse TA muscles with plasmid DNA encoding HA-tagged wild-type TRIM28, or LacZ as a negative control. At 7 days post electroporation, the TA muscles were collected 1 h after being subjected to a bout of MICs or the contralateral control condition. Lysates of the TA were subjected to immunoprecipitation (IP) against the HA tag and then the immunoprecipitates, as well as the original lysate, were subjected to western blot analyses.
(E) TA muscles from wild-type C57BL/6 mice were subjected to a bout of MICs or the contralateral control condition. At 1 h after the bout of MICs, the muscles were collected and mid-belly cross sections were subjected to immunohistochemistry (IHC) for dystrophin to identify the periphery of myofibers, P-TRIM28(S473), and Hoechst to identify nuclei. Arrows indicate P-TRIM28(S473)-positive nuclei within the myofibers. Scale bars, 30 μm.
(F) The images from (E) were used to quantify the density of P-TRIM28(S473)-positive nuclei within the interstitial space or within the myofibers. Values are presented as the group means (+SEM in F), n = 3–6/group. The data in (C) and (F) were analyzed with two-way ANOVA followed by Student-Newman-Keuls post hoc analyses, while the data in (D) were analyzed with a Student’s t test. Asterisk (*) indicates significant effect of MIC, and dagger (†) indicates significantly different from the MIC+ interstitial group, p < 0.05.
Next, to assess whether the increase in TRIM28(S473) phosphorylation was occurring within the myofibers, we used electroporation to transfect the myofibers of the TA muscle with a hemagglutinin (HA)-tagged variant of TRIM28. At 7 days post electroporation, the TA muscles were subjected to a bout of MICs or control condition and then analyzed for HA-TRIM28(S473) phosphorylation. As shown in Figure 3D, the outcomes revealed that the bout of MICs led to a robust increase in S473 phosphorylation and thus indicated that the increase in TRIM28 phosphorylation was occurring within the myofibers. In a complementary experiment, we also subjected TA muscles to a bout of MICs or the control condition and then analyzed the samples with immunohistochemistry for S473-phosphory-lated TRIM28. Again, the results indicated that the vast majority of the increase in TRIM28(S473) phosphorylation was occurring within in the myofibers and also revealed that the alterations were heavily confined to the nuclei that resided within the myofibers (i.e., the myonuclei) (Figures 3E and 3F).
Overexpression of S473 phosphomimetic TRIM28 induces hypertrophy
The identification of TRIM28(S473) as one of the most robust MIC-regulated phosphorylation sites was exciting because TRIM28 has been shown to function as a transcriptional intermediary factor in a variety of biological processes, and many of its regulatory effects are dependent on phosphorylation of the S473 residue (Cheng et al., 2014; Iyengar and Farnham, 2011). Specifically, rigorous studies have demonstrated that S473 phosphorylation regulates the ability of TRIM28 to control cell-cycle progression, mitochondrial biogenesis, DNA repair, and of particular interest, the activity of myogenic transcription factors such as MyoD and Mef2 (Bolderson et al., 2012; Cheng et al., 2014; Iyengar and Farnham, 2011; Singh et al., 2015). We were particularly interested in the last point because it has been shown that the activation of myogenic transcription factors, within myofibers, is sufficient to elicit a hypertrophic response (Lagirand-Cantaloube et al., 2009; Moretti et al., 2016). Accordingly, we wondered whether an increase in TRIM28(S473) phosphorylation would be sufficient to induce hypertrophy. Hence, to address this, we generated phosphomimetic (S473D) and phosphodefective (S473A) mutants of TRIM28 and then used electroporation to transfect the myofibers of TA muscles with the TRIM28 mutants, or LacZ as a control condition. As shown in Figure 4 and Figure S3, the results of our analyses revealed that the overexpression of TRIM28(S473D) induced a rapid hypertrophic response and that the hypertrophic response was largely dependent on the presence of the phosphomimetic mutation.
Figure 4. Overexpression of S473 phosphomimetic TRIM28 induces hypertrophy.
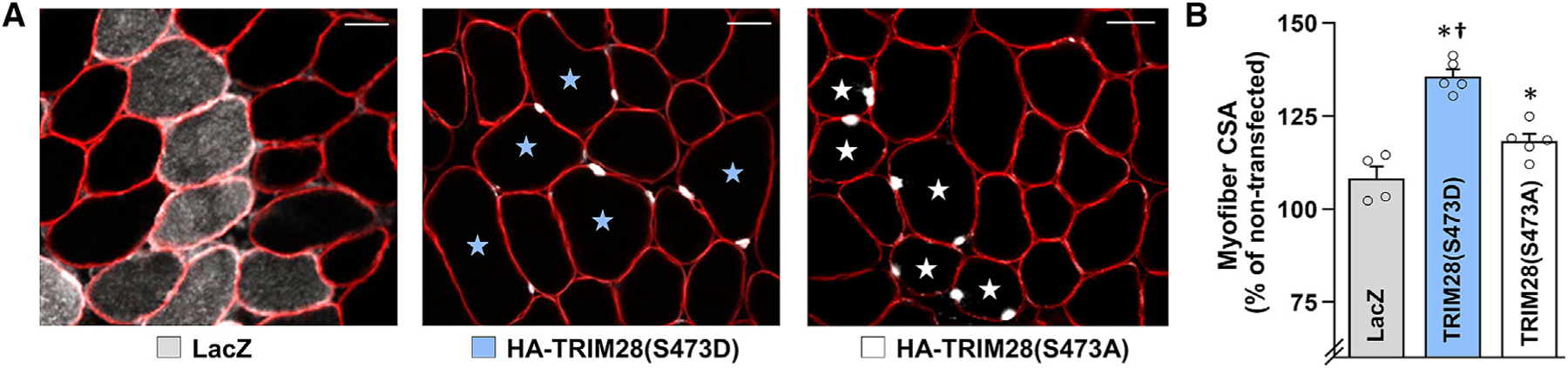
Electroporation was used to transfect the myofibers of wild-type C57BL/6 mouse TA muscles with plasmid DNA encoding HA-tagged phosphomimetic TRIM28 (S473D), phosphodefective TRIM28 (S473A), or LacZ as a control condition. (A) At 3 days post electroporation, mid-belly cross sections were subjected to IHC for dystrophin (red) and either LacZ or the HA tag (white). TRIM28(S473D)- and TRIM28(S473A)-positive myofibers are indicated with blue and white stars, respectively. Scale bars, 50 μm.
(B) The images from (A) were used to measure the cross-sectional area (CSA) of the transfected and non-transfected myofibers in each muscle. Values are reported as the group mean + SEM, n = 4–5 muscles/group (343–507 transfected and 811–1,673 non-transfected myofibers/group). The data were analyzed with one-way ANOVA followed by Student-Newman-Keuls post hoc analyses. Significant difference versus *LacZ, †phosphodefective TRIM28(S473A), p < 0.05.
Characterization of skeletal myofiber-specific and tamoxifen-inducible TRIM28 KO mice
The aforementioned outcomes supported the notion that signaling through TRIM28, within the myofibers, can regulate skeletal muscle mass. Therefore, to further test this, we developed a strain of skeletal myofiber-specific and tamoxifen-inducible Trim28 knockout (KO) mice. This was accomplished by crossing mice that contained floxed Trim28 alleles with mice that possessed human skeletal muscle actin-promoter-driven expression of a mutated estrogen-receptor-flanked Cre-recombinase (HSA-MCM). Mice that possessed the floxed Trim28 alleles, but did not express HSA-MCM, were used as littermate controls.
To establish the specificity and efficiency of the KO, skeletal muscles, as well as the brain, heart, and liver, were collected from the KO and control mice at 14 days after they were treated with tamoxifen. As shown in Figure 5, western blot analysis of whole-tissue lysates revealed that a significant decrease in the expression of TRIM28 (~50%) occurred in skeletal muscle, but no significant changes were detected in the brain, heart, or liver. Of note, TRIM28 is a ubiquitously expressed protein (Kim et al., 1996), and because the KO of TRIM28 should only occur inside the myofibers, we reasoned that the remaining TRIM28 in the whole-skeletal muscle lysate was due to its presence in cells that resided within the interstitial space (i.e., satellite cells, macrophages, fibroblasts, etc.). Thus, to examine this, skeletal muscles from the KO and control mice were subjected to immunohistochemistry, and the number of TRIM28-positive myonuclei and TRIM28-positive interstitial nuclei was measured. As shown in Figures 5C and 5D, the outcomes revealed that a nearly complete KO of TRIM28 was achieved within the myonuclei (98%), while the expression of TRIM28 within the nuclei of interstitial cells remained unaffected. However, aside from the alteration in TRIM28 expression, no overt differences between the KO and control mice were observed at the 14-day post tamoxifen time point. For example, the muscle weight-to-body weight ratio of various skeletal muscles was not significantly altered (Figure 5E). Moreover, no significant differences were detected in the basic contractile properties of the TA muscle, and the gross physical appearance of the KO mice was indistinguishable from that of control mice (Figures 5F–5H).
Figure 5. Characterization of skeletal myofiber-specific and tamoxifen-inducible TRIM28 KO mice.
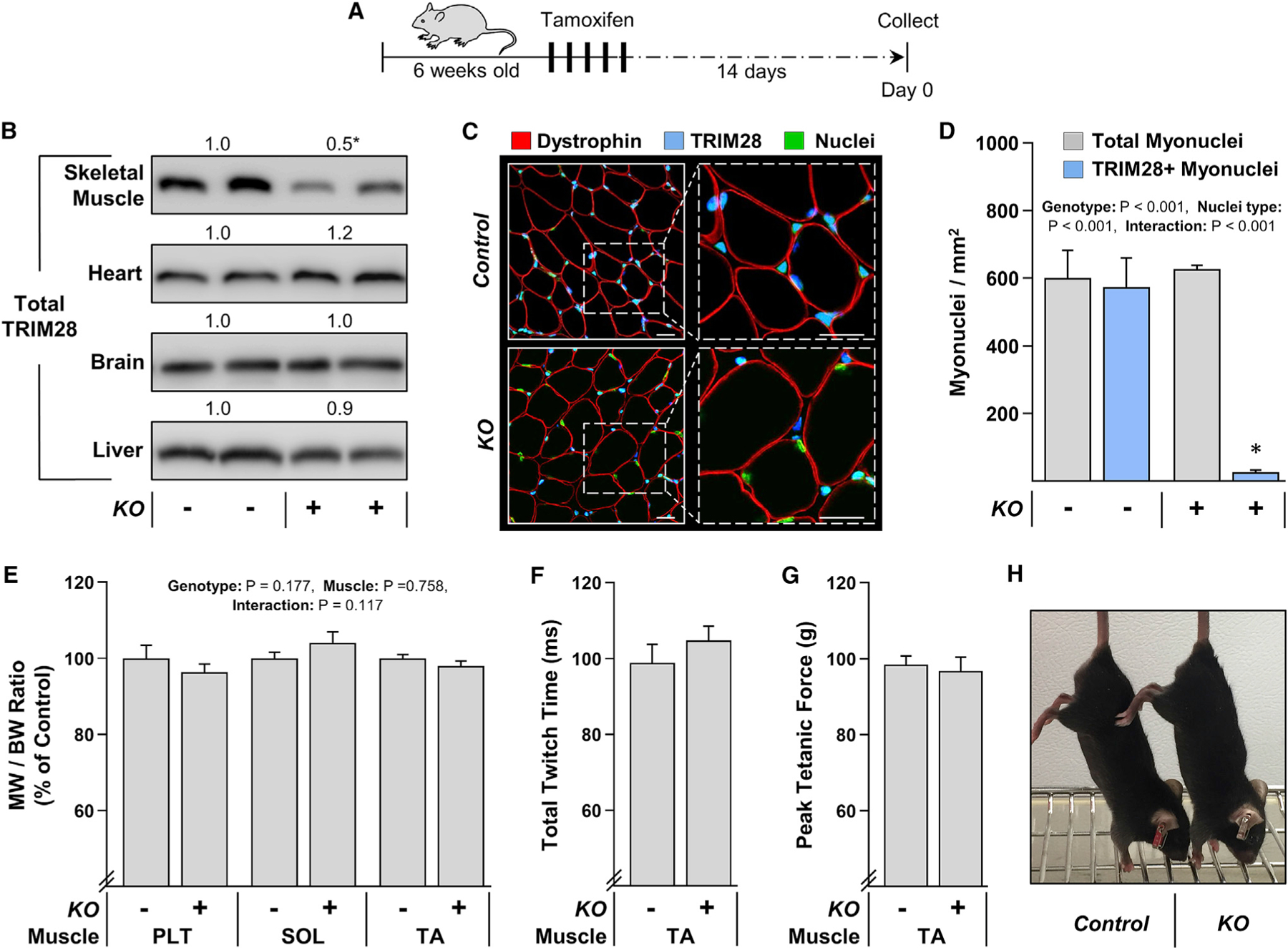
(A) TRIM28 knockout (KO+) mice and their control (KO−) littermates were treated with tamoxifen for 5 days and examined at 14 days post tamoxifen.
(B) Western blot analysis of TRIM28 in various tissues including skeletal muscle (soleus [SOL]), brain, heart, and the liver.
(C) Mid-belly cross sections of the plantaris muscles (PLT) were subjected to IHC for dystrophin, total TRIM28, and Hoechst to identify nuclei. Scale bars, 20 μm.
(D) The images from (C) were used to quantify the number of TRIM28-positive nuclei in the myofibers (i.e., myonuclei).
(E) Muscle weight (MW)-to-body weight (BW) ratios of the PLT, SOL, and TA muscles.
(F and G) In situ contractile properties of the TA muscles.
(H) Representative image of the control and KO mice. Values represent the group mean (+SEM in graphs), n = 3–10/group. The data in (B), (F), and (G) were analyzed with Student’s t tests, while two-way ANOVA with Student-Newman-Keuls post hoc analyses was used to analyze the data in (D) and (E). Asterisk (*) indicates significant effect of KO, p < 0.05.
The role of TRIM28 in the mechanical activation of RSmTOR signaling
The outcomes of our phosphoproteomic analyses indicated that MIC-induced signaling through TRIM28 is activated either upstream or parallel to RSmTOR. Moreover, in addition to its apparent role in the regulation of myogenic genes, it has also been reported that TRIM28 can regulate RSmTOR signaling (Li et al., 2018; Pineda et al., 2015). Thus, we next set out to determine whether signaling through TRIM28 contributes to the MIC-induced activation of RSmTOR. To accomplish this, KO mice, as well as their control littermates, were treated with tamoxifen. At 14 days post tamoxifen, the mice were subjected to a bout of MICs and then 1 h after the bout of MICs, the TA muscles were collected and evaluated for changes in TRIM28(S473) phosphorylation and markers of RSmTOR signaling. As shown in Figure S4A, the bout of MICs led to a robust increase in TRIM28(S473) phosphorylation in muscles from control mice, and this effect was almost completely absent in the muscles from the KO mice. This was a noteworthy observation because it provided further support for our conclusion that the MIC-induced increase in TRIM28(S473) phosphorylation is primarily mediated by changes that occur within the myofibers. The results presented in Figure S4 also revealed that the bout of MICs led to a significant increase in the phosphorylation of two validated markers of RSmTOR signaling including p70s6K(T389) and ribosomal protein S6(S240/4) phosphorylation (You et al., 2019). Importantly, we have previously shown that the MIC-induced activation of signaling through these markers is mediated almost entirely at the level of the myofibers (You et al., 2019), and the results presented in Figure S4A demonstrate that TRIM28 is not required for this to occur. Hence, it appears that TRIM28, within the myofibers, does not significantly contribute to the pathway via which MICs induce RSmTOR signaling.
The role of TRIM28 in mechanical load-induced hypertrophy
As mentioned in the introduction, a growing body of literature indicates that rapamycin-insensitive signaling events can significantly contribute to the hypertrophic effects of mechanical loading. Thus, our next goal was to determine whether signaling through TRIM28 plays a role in this process. Specifically, we used our previously described myotenectomy (MTE) model in which the myotendinous junction of the gastrocnemius muscles is surgically removed (You et al., 2019). As a result of this perturbation, the plantaris (PLT) muscles experience a chronic increase in mechanical loading and, in turn, undergo a prominent hypertrophic response (You et al., 2019). As illustrated in Figures 6A and 6B, we also determined that, just like MICs, MTE led to a rapid increase in TRIM28(S473) phosphorylation within the nuclei of myofibers and that TRIM28 was not required for the MTE-induced activation of RSmTOR signaling (Figure S4B).
Figure 6. The loss of TRIM28 leads to a reduction in myofiber size and attenuates the hypertrophic effect of MTE.
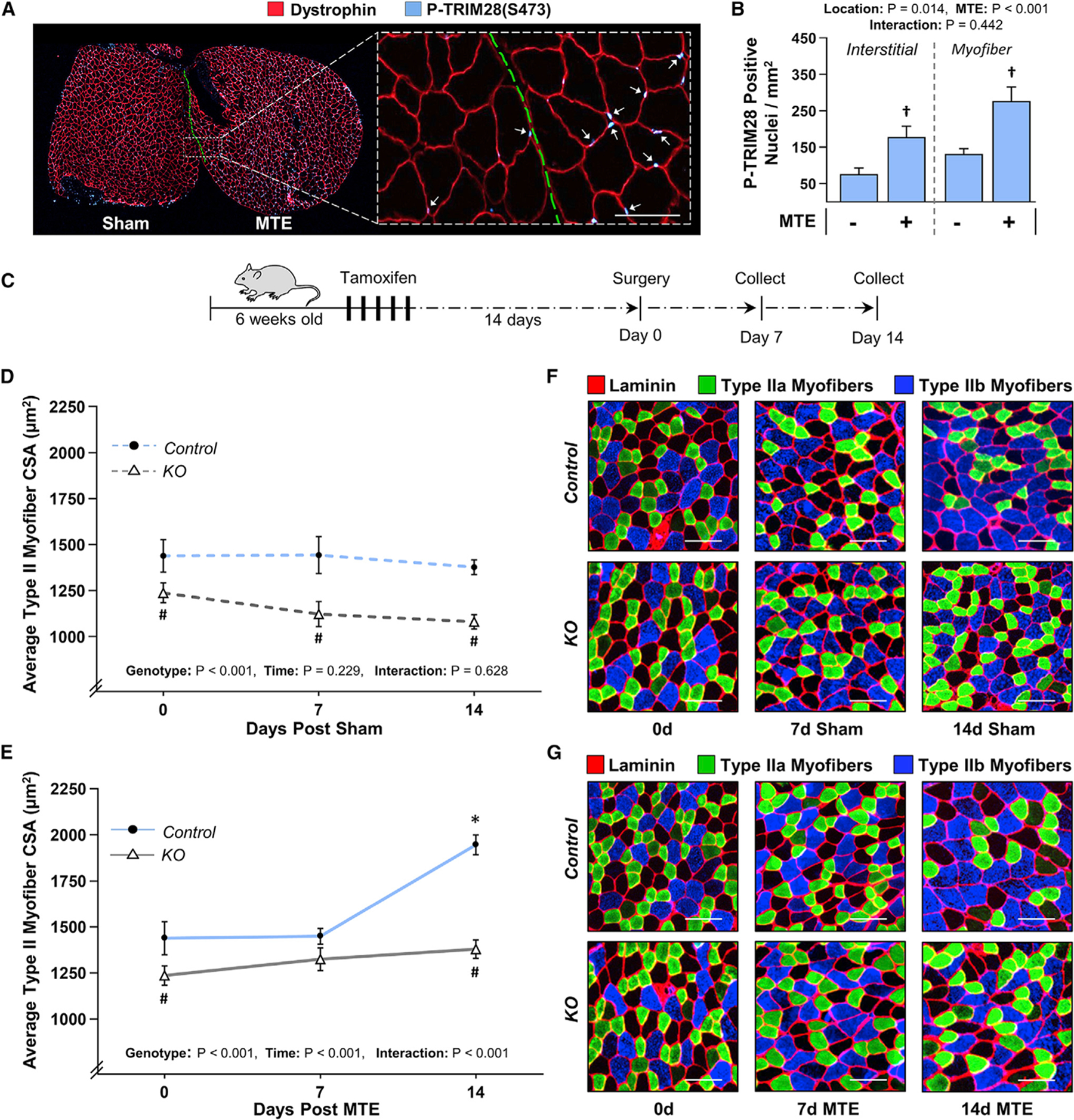
(A) The PLT muscles of wild-type C57BL/6 mice were subjected to myotenectomy (MTE) or a sham surgery. After 90 min of recovery, individual MTE and sham muscles were frozen adjacent to one another and then mid-belly cross sections were subjected to IHC for dystrophin and P-TRIM28(S473). Arrows indicate P-TRIM28(S473)-positive nuclei within the myofibers. Scale bar, 100 μm.
(B) The images in (A) were used to quantify the number of P-TRIM28(S473)-positive nuclei within the myofibers and the interstitial space. Values are presented as the group mean + SEM, n = 4/group. (C) TRIM28 KO mice and their control littermates were treated with tamoxifen for 5 days. At 14 days post tamoxifen, their PLT muscles were subjected to MTE or a sham surgery.
(D and E) After 0, 7, or 14 days of recovery, mid-belly cross sections of the PLT muscles were subjected to IHC for laminin and myofiber type identification. The mean CSA of each myofiber type (i.e., types IIa, IIx, and IIb) was determined and then the average of these values was used to calculate the “type II myofiber CSA.” Values represent the group mean ± SEM, n = 9–14 muscles/group (1,840–3,680 myofibers/group). Note: the individual myofiber type data are shown in Figures S5 and S6.
(F and G) Representative images of the cross sections that were stained for laminin, type IIa, and type IIb myofibers. Scale bars, 100 μm. The data in (B), (D), and (E) were analyzed with two-way ANOVA followed by Student-Newman-Keuls post hoc analyses. Significantly different from †sham, #time-matched control genotype, *day 0 within a given genotype, p < 0.05.
Next, we performed an experiment in which KO mice and their control littermates were treated with tamoxifen, and 14 days later the mice were subjected to MTE or a sham surgery. After 0, 7, or 14 days of recovery, the PLT muscles were collected and mid-belly cross sections were subjected to immunohistochemistry to identify the size of individual myofiber types (i.e., type IIa, IIx, and IIb myofibers). The results of this experiment produced two striking observations. First, after 14 days in the sham condition, the loss of TRIM28 led to a 22% decrease in the average CSA of the type II myofibers (Figures 6C, 6D, and 6F). Additional myofiber type-specific analyses revealed that this effect was driven by a large decrease in the size of the type IIb (31% decrease) and type IIx (23% decrease) myofibers, while no significant alteration in the size of the type IIa myofibers was detected (Figures S5A–S5D and S6A). Second, we found that MTE induced a 35% increase in average CSA of the type II myofibers and that this effect was heavily dependent on the presence of TRIM28 (Figures 6C, 6E, and 6G). Specifically, in the control mice, 14 days of MTE led to a 56% increase in the CSA of type IIa myofibers, while mice lacking TRIM28 only displayed a 27% increase (Figures S5A, S5E, and S6B). Likewise, in the type IIx myofibers, control mice exhibited a 37% increase in CSA, while KO mice only displayed a non-significant 15% increase (Figures S5A, S5F, and S6B). Finally, MTE also led to a 25% increase in the CSA of the type IIb myofibers in control mice, whereas this response was abolished by the loss of TRIM28 (Figures S5A, S5G, and S6B). In addition to these findings, we also found that the TRIM28 was required for the accretion of myonuclei that occurs in response to MTE (Figure S7). Thus, when taken together, our results suggest that TRIM28 significantly contributes to both the homeostatic regulation of myofiber size as well as the hypertrophic effect of increased mechanical loading.
The sustained loss of TRIM28 leads to a decrease in muscle mass and glycolytic myofiber size as well as alterations in contractile function
The pronounced decrease in type IIx and IIb myofiber sizes in the PLT muscles of KO mice led us to question whether the same effect would be observed in other hindlimb muscles. Hence, to address this, 14-day post tamoxifen mice were subjected to the same sham procedure described for the experiments in Figure 6 and then various skeletal muscles were collected after the 14-day recovery period. As shown in Figure 7A, the loss of TRIM28 led to a significant decrease in the mass of fast-twitch muscles such as the PLT and extensor digitorum longus (EDL), while the slow-twitch soleus muscle (SOL) was unaffected (Schiaffino and Reggiani, 2011). We then measured the size of the different myofiber types that are present within each of these muscles. For instance, the SOL is composed of highly oxidative type I and type IIa myofibers, and the size of these myofiber types was not impacted by the loss of TRIM28 (Figure 7B; Burkholder et al., 1994). On the other hand, the PLT, EDL, and TA muscles are composed of type IIa myofibers, as well as the highly glycolytic type IIx and IIb myofibers (Burkholder et al., 1994). Importantly, all three of the fast-twitch muscles revealed a significant decrease in the size of the type IIx and IIb myofibers; yet, just like in the slow-twitch SOL, the size of the oxidative type IIa myofibers was not affected (Figures 7C–7E). Together, these results suggest that TRIM28 preferentially regulates the size of myofibers that are reliant on glycolytic metabolism, and this effect is not influenced by the overall twitch characteristics of the muscle.
Figure 7. A sustained loss of TRIM28 leads to significant alterations in muscle mass, myofiber size, and contractile function.
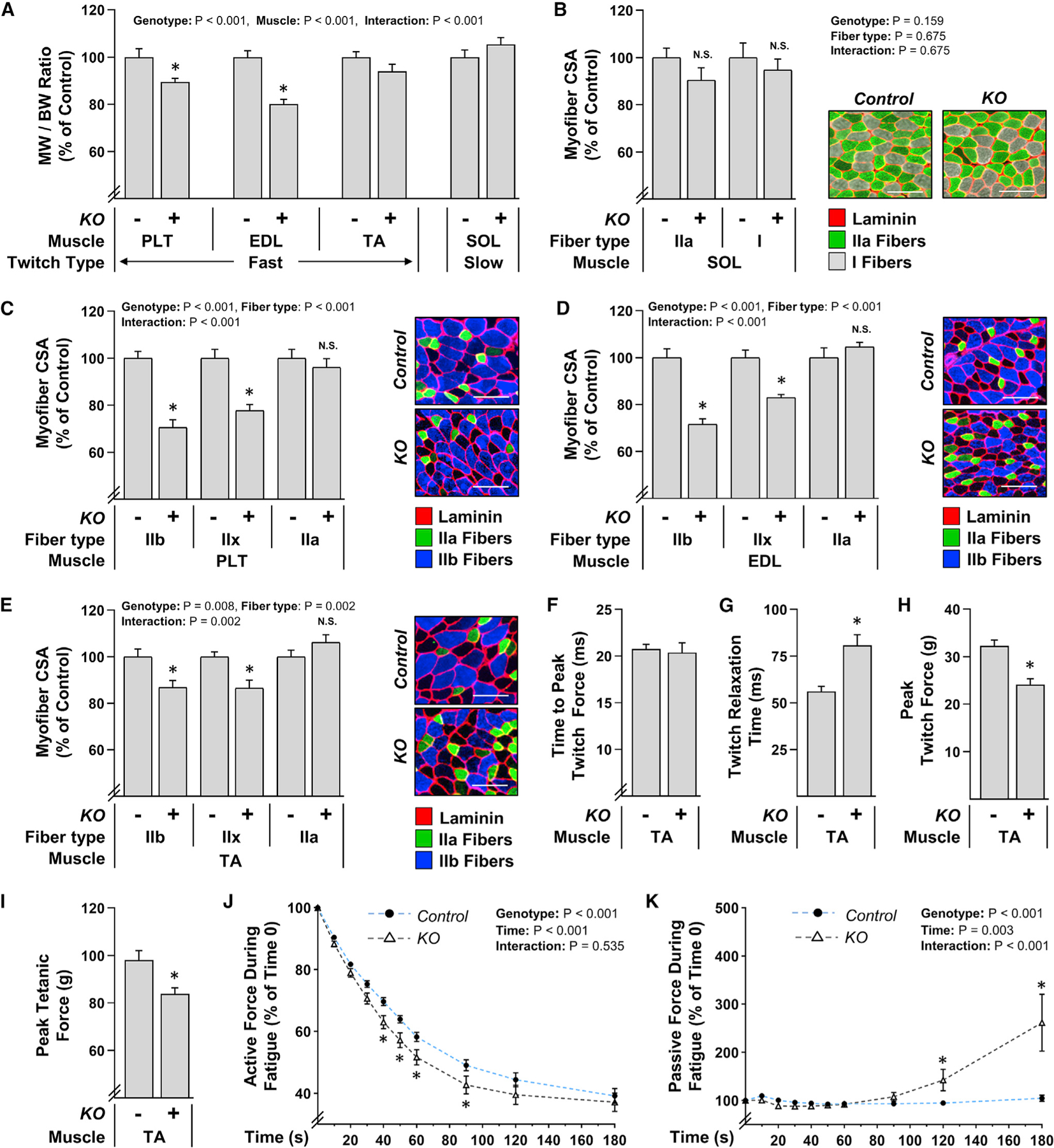
TRIM28 KO+ and their KO− control littermates were treated with tamoxifen for 5 days. At 14 days post tamoxifen, their lower hindlimbs were subjected to a sham surgery as described in Figure 6. After a 14-day recovery period, various fast-twitch muscles including the PLT, extensor digitorum longus (EDL), and TA as well as the slow-twitch SOL were subjected to further analysis.
(A) MW-to-BW ratios.
(B–E) Mid-belly cross sections were subjected to IHC for laminin along with myofiber type identification and then the resulting images were used to quantify the CSA of the dominant myofiber types in each muscle (i.e., type I and IIa in the SOL; types IIa, IIx, and IIb in the PLT, etc.). Various representative images are shown. Scale bar, 100 μm. (F–K) In situ contractile properties measured during (F) to (H) single twitch contractions, (I) single tetanic contractions, or (J) and (K) during fatigue that was induced by repetitive tetanic contractions (once every 2 s). Values represent the group mean + SEM, n = 9–15 muscles/group (770–1,984 myofibers/group). The data in (A) to (E) and (J) and (K) were analyzed with two-way ANOVA followed by Student-Newman-Keuls post hoc analyses, and the data in (F) to (I) was analyzed with Student’s t tests. Asterisk (*) indicates significant effect of KO, p < 0.05. N.S., no significant difference.
Having uncovered a potential role for TRIM28 in the regulation of glycolytic myofiber size, we next wanted to determine whether it also contributes to the regulation of skeletal muscle function. Specifically, 14-day post tamoxifen mice were again subjected to the same sham procedure described for the experiments in Figure 6 and then various contractile properties of the TA muscles were measured after the 14-day recovery period. The results from these analyses revealed that the loss of TRIM28 led to a 32% increase in total twitch time (data not shown) and that this effect was due to an impairment in relaxation rather than the time taken to reach peak twitch tension (Figures 7F and 7G). The loss of TRIM28 also led to a 25% reduction in peak twitch force and a 14% reduction in peak tetanic force (Figures 7H and 7I). Furthermore, an accelerated decline in force production was observed when muscles from the KO mice were subjected to a fatigue protocol (300-ms tetanic contraction once every 2 s) (Figure 7J). Interestingly, the muscles from the KO mice also revealed a progressive increase in passive tension during the rest intervals between each of the contractions (Figure 7K).
DISCUSSION
The overarching goal of this study was to develop a deeper understanding of the signaling events that contribute to the mechanical load-induced growth of skeletal muscle. To accomplish this goal, we used an unbiased phosphoproteomic approach to map the rapamycin-sensitive and rapamycin-insensitive signaling events that are elicited by a bout of MIC. Collectively, the outcomes led to the identification of an extensive pool of non-canonical MIC-regulated and rapamycin-sensitive phosphorylation events. Furthermore, by using bioinformatics-based approaches, we illustrated how the data can be used to identify non-cannonical phosphorylation events that contribute to the regulation of various cellular processes such as autophagy. We also used the data to predict which kinases are activated by MICs, and whether these kinases are activated upstream/parallel or downstream of RSmTOR. For instance, the outcomes of our analyses suggest that MICs promote the activation of SRPK2 and that this effect is mediated downstream of RSmTOR. This was a noteworthy prediction because a mTORC1-SRPK2 signaling axis was recently shown to function as a key regulator of the lipogenic enzymes that are needed for rapid cellular growth (Lee et al., 2017). Hence, our results have likely unveiled an additional rapamycin-sensitive signaling event that supports the mechanical load-induced growth of skeletal muscle.
In addition to identifying rapamycin-sensitive signaling events, we also identified a plethora of rapamycin-insensitive signaling events that could potentially contribute to the regulation of skeletal muscle mass. Indeed, over 2,000 phosphorylation sites fell within this category, and a number of these sites have been previously shown to regulate processes such as transcription, translation, and overall cellular growth (Table S1). However, upon further examination, we became particularly intrigued by the changes in phosphorylation that were detected on the S473 residue of TRIM28. We were interested in this event because (1) it was one of the most robust MIC-regulated phosphorylation sites, (2) the MIC-induced increase in phosphorylation was mediated through a rapamycin-insensitive mechanism, and (3) previous studies had reported that TRIM28 can control the expression of myogenic genes that are known to regulate muscle mass (Lagirand-Cantaloube et al., 2009; Moretti et al., 2016; Singh et al., 2015). However, given its reported role in the regulation of RSmTOR signaling, we questioned whether TRIM28 was a component of the upstream pathway via which mechanical stimuli activate RSmTOR (Li et al., 2018; Pineda et al., 2015). As shown, the results of our analyses revealed that TRIM28 was not required for the mechanical activation of RSmTOR. Moreover, the KO of TRIM28 did not significantly alter the basal levels of RSmTOR signaling (Figure S4). Thus, the effects of TRIM28 that were observed in this study appear to have been mediated by a mechanism that works in parallel to, rather than upstream of, RSmTOR.
The ability to regulate myofiber size was one of the most striking features of TRIM28 identified in this study. For instance, using our skeletal muscle-specific TRIM28 KO mice, we found that muscles lacking TRIM28 exhibited an attenuated hypertrophic response to MTE. Moreover, we found that the overexpression of a S473 phosphomimetic mutant of TRIM28 is sufficient to induce hypertrophy and that this effect is largely dependent on the presence of the phosphomimetic mutation. Taken together, these observations support the possibility that mechanical signals induce TRIM28(S473) phosphorylation and that this, in turn, initiates a cascade of signaling events that promotes mechanical load-induced hypertrophy. So, what could this cascade of signaling events involve?
When considering potential signaling events via which TRIM28 might confer its effects on skeletal muscle size, it is important to consider events that could reside both upstream and downstream of TRIM28. With regard to upstream events, it was recently reported that MAPK signaling can regulate TRIM28 phosphorylation. Specifically, it was shown that the MAPK family member MSK1 can directly phosphorylate the S473 residue of TRIM28 and that this, in turn, enables TRIM28 to unleash the transcriptional activity of muscle-specific transcription factors such as MyoD and Mef2 (Singh et al., 2015). This is particularly noteworthy because the activation of MAPK signaling has been widely implicated in the regulation of skeletal muscle size. For example, it has been shown that mechanical stimuli can activate MAPK signaling and that MAPK signaling is not only required for the maintenance of myofiber size but also plays a role in the induction of skeletal muscle hypertrophy (Haddad and Adams, 2004; Kramer and Goodyear, 2007; Shi et al., 2009). Moreover, additional studies have shown that the constitutive activation of MAPK in cardiac muscle is sufficient to induce a hypertrophic response (Bueno et al., 2000; Ueyama et al., 2000). Hence, the role that TRIM28 plays in the regulation of skeletal muscle size is likely controlled, at least in part, by the upstream activity of MAPKs.
In addition to the upstream events that regulate TRIM28, it is also important to consider the downstream events through which TRIM28 might confer its effects. For instance, TRIM28 possesses intrinsic SUMO E3 ligase and ubiquitin E3 ligase activities, and it has been shown that the activity of E3 ligases can be controlled by their phosphorylation state (Cheng et al., 2014; Gao et al., 2004; Iyengar and Farnham, 2011). Hence, one could reason that the phosphorylation of TRIM28 at the S473 site might simply regulate myofiber size by triggering its SUMO and ubiquitin E3 ligase activity. However, we are not aware of any studies which have shown that alterations in S473 phosphorylation can regulate these activities. Alternatively, by interacting with a variety of transcriptional co-activators and co-repressors, TRIM28 has also been implicated as a transcriptional intermediary factor (Cheng et al., 2014; Iyengar and Farnham, 2011). Specifically, it has been shown TRIM28 can physically interact with HDAC1 and that this interaction is highly disrupted by phosphorylation of the S473 residue (Singh et al., 2015). This is particularly noteworthy because previous studies have shown that the overexpression of HDAC1 promotes the activation of FoxO and a concomitant atrophic response in skeletal muscle and that both of these effects require the deacetylase activity of HDAC1 (Beharry et al., 2014). Furthermore, the levels of acetylation increase at distal regulatory elements of genes that are upregulated in TRIM28 KO cells, and nearly 20% of the genes whose expression is altered by the KO of TRIM28 contain known binding sites for FoxO1 (Gehrmann et al., 2019). Thus, it’s easy to imagine how changes in TRIM28 phosphorylation could alter the expression of genes that control myofiber size.
In addition to its prospective role in mechanical load-induced hypertrophy, we also found that TRIM28 contributes to the homeostatic regulation of fast-twitch muscle size and function. When considering potential mechanisms via which TRIM28 might confer these load-independent effects, we became particularly drawn to previous reports that have shown that TRIM28 can regulate the expression of genes that are involved in glycolysis (Czerwińska et al., 2017; Gehrmann et al., 2019). For instance, a recent study demonstrated that the KO of TRIM28 in CD4+ T cells led to a reduction in glycolytic capacity and that this effect was associated with reductions in the expression of glycolytic genes such as hexokinase, phosphofructokinase, phosphoglucomutase, and aldolase (Gehrmann et al., 2019). This could be important because fast-twitch muscles, particularly the type IIx and IIb myofibers, are heavily reliant on glycolytic activity, and deficiencies in this process can have major consequences (Rivero et al., 1999; van Wessel et al., 2010). For example, a previous study in rats revealed that acute fasting can lead to a reduction in glycolytic capacity and, similar to what we observed in the muscles from TRIM28 KO mice, the decrease in glycolytic capacity was associated with impairments in relaxation and fatigue resistance (Russell et al., 1984). Moreover, another study showed that the overexpression of myogenin causes fast-twitch muscles to switch from glycolytic-to-oxidative metabolism and that this metabolic switch was coupled with a dramatic reduction in the size of glycolytic fast-twitch myofibers, whereas the size of the oxidative slow-twitch myofibers remained unaltered (Hughes et al., 1999). The myofiber-type-specific atrophy observed in muscles that overexpress myogenin is highly similar to what we observed in the muscles from the TRIM28 KO mice, and this is especially intriguing because it was recently shown that TRIM28 can regulate myogenin expression (Singh et al., 2015). Thus, it stands to reason that TRIM28 could confer its effects on fast-twitch myofiber size and function via the regulation of glycolytic metabolism.
Although the data presented in this study provide compelling evidence that supports an important role for TRIM28 in both the homeostatic and mechanical regulation of skeletal muscle size and function, it is important to acknowledge potential limitations of our study and consider alternative interpretations of the results. For instance, our data reveal that muscles lacking TRIM28 displayed an attenuated hypertrophic response to MTE-induced mechanical loading, and this observation suggests that TRIM28 might significantly contribute to the pathway through which mechanical signals regulate skeletal muscle size. However, TRIM28 functions in a variety of biological processes, including cellular metabolism, and this is important because changes in skeletal muscle metabolic activity can alter myofiber size (Cheng et al., 2014; Gehrmann et al., 2019; Hughes et al., 1999; Peker et al., 2018; van Wessel et al., 2010) Hence, it is possible that TRIM28 confers its effects on myofiber size indirectly through the regulation of metabolic activity/capacity. To address this possibility, future studies analyzing measures of metabolic activity, such as ATP concentration, glycolytic/oxidative metabolite production, glucose transport activity, reactive oxygen species production, and mitochondrial respiration could provide important insights that will not only resolve a metabolic role for TRIM28 in the regulation of skeletal muscle size but also enhance our overall understanding of the mechanisms through which TRIM28 might function in this process.
In summary, our study led to the identification of an extensive pool of rapamycin-sensitive and rapamycin-insensitive signaling events that are regulated by mechanical stimuli. Importantly, our results not only confirmed much of what is already known with regard to the phosphorylation cascades that are activated by mechanical stimuli but also exposed over 2,000 non-canonical phosphorylation events regulated by rapamycin and/or mechanical stimuli, including TRIM28. Furthermore, by capturing these events and establishing TRIM28 as a significant regulator of myofiber size and function, we believe that this study will not only provide a resource for further delineating the upstream, downstream, and parallel signaling pathways through which mechanical stimuli and mTOR can regulate growth but also will invigorate future mechanistic studies into the role TRIM28 plays in this process. Hence, when taken together, we expect the results of this study will exert a broad impact on multiple fields of biology.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Troy A. Hornberger (troy.hornberger@wisc.edu).
Materials availability
The materials utilized in this study, with the exception of the phospho-mimetic and phosphodefective TRIM28(S473) plasmids, are commercially available (see Key resources table for details). Phosphomimetic and phosphodefective TRIM28(S473) plasmids generated by our lab and are available upon request. There are restrictions to the availability of Biolegend Anti-Rabbit phospho-Tif1β (KAP-1, TRIM28) Ser437 Poly6446 antibody (cat# 644602), as this product has recently been discontinued.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Rabbit Total SEK1(MKK4) (5C10) | Cell Signaling Technologies (Danvers, MA, USA) | Cat# 3346S; RRID:AB_330905 |
| Anti-Rabbit P-SEK1(MKK4) Ser257 (C36C11) | Cell Signaling Technologies (Danvers, MA, USA) | Cat# 4514S; RRID:AB_2297420 |
| Anti-Rabbit Total P70 S6K (49D7) | Cell Signaling Technologies (Danvers, MA, USA) | Cat# 2708S; RRID:AB_390722 |
| Anti-Rabbit P-P70 thr389 (108D2) | Cell Signaling Technologies (Danvers, MA, USA) | Cat# 9234S; RRID:AB_2269803 |
| Anti-Rabbit Total Tif1 β (Kap-1, TRIM28) (C42G12) | Cell Signaling Technologies (Danvers, MA, USA) | Cat# 4124S; RRID: N/A |
| Anti-Rabbit Total S6 Ribosomal Protein (5G10) | Cell Signaling Technologies (Danvers, MA, USA) | Cat# 2217S; RRID:AB_331355 |
| Anti-Rabbit P-S6 Ribosomal Protein (Ser235/236) | Cell Signaling Technologies (Danvers, MA, USA) | Cat# 2211S; RRID:AB_916156 |
| Anti-Rabbit P-S6 Ribosomal Protein (Ser240/244) (D68F8) XP® | Cell Signaling Technologies (Danvers, MA, USA) | Cat# 5364S; RRID:AB_10694233 |
| Anti-Rabbit P- Tif1 β (KAP-1, TRIM28) Ser437 Poly6446 | BioLegend (San Diego, CA, USA) | Cat# 644602; RRID:AB_2241094 |
| Anti-Mouse LacZ (β-Galactosidase) (40–1a) | Santa Cruz Biotechnology (Santa Cruz, CA, USA) | Cat# sc-65670; RRID:AB_528100 |
| Anti-Chicken LacZ (β-Galactosidase) | Abcam (Cambridge, MA, USA) | Cat# ab9361; RRID: AB_307210 |
| Anti-Mouse Dystrophin (Dy8/6C5) | Novocastra (Leica Biosystems, Buffalo Grove, IL, USA) | Cat# NCL-DYS2; RRID. AB_442081 |
| Anti-Rat HA Peroxidase (clone3F10) | Roche (Basel, Switzerland) | Cat# 1867431; RRID:AB_390917 |
| Anti-Mouse MYH7 (type 1 myosin heavy chain) BA-D5 | Developmental Studies Hybridoma Bank (Iowa City, IA, USA) | Cat# BA-D5; RRID:AB_2235587 |
| Anti-Mouse MYH2 (type IIA myosin heavy chain) SC-71 | Developmental Studies Hybridoma Bank (Iowa City, IA, USA) | Cat# SC-71; RRID:AB_2147165 |
| Anti-mouse MYH4 (type IIB myosin heavy chain) BF-F3 | Developmental Studies Hybridoma Bank (Iowa City, IA, USA) | Cat# BF-F3; RRID:AB_2266724 |
| Anti-Mouse MYH1 (type IIx myosin heavy chain) 6H1 | Developmental Studies Hybridoma Bank (Iowa City, IA, USA) | Cat# 6H1; RRID:AB_1157897 |
| Anti-Rabbit Laminin | Sigma-Aldrich (St. Louis, MO, USA) | Cat# L9393; RRID:AB_477163 |
| Fab Fragment Goat Anti-Mouse IgG (H+L) Block | Jackson Immunoresearch (West Grove, PA, USA) | Cat# 115-007-003; RRID: AB_2338476 |
| AMCA Anti-Mouse IgM | Jackson Immunoresearch (West Grove, PA, USA) | Cat# 115-155-075; RRID: AB_2338661 |
| Alexa 488 Anti-Mouse IgM | Jackson Immunoresearch (West Grove, PA, USA) | Cat# 115-545-075; RRID: AB_2338849 |
| Alexa 488 Anti-Mouse IgG1 | Jackson Immunoresearch (West Grove, PA, USA) | Cat# 115-545-205; RRID: AB_2338854 |
| Alexa 488 Anti-Chicken IgY | Jackson Immunoresearch (West Grove, PA, USA) | Cat# 703-545-155; RRID: AB_2340375 |
| Alexa Fluor 594 Anti-Mouse IgG1 | Jackson Immunoresearch (West Grove, PA, USA) | Cat# 115-585-205; RRID: AB_2338885 |
| Peroxidase-labeled Anti-Rat IgG | Jackson Immunoresearch (West Grove, PA, USA) | Cat# 112-035-003; RRID: AB_2338128 |
| Alexa Fluor 568 Anti-Rabbit IgG | Invitrogen (Carlsbad, CA, USA) | Cat# A11011; RRID:AB_143157 |
| Alexa 350 Anti-Mouse IgG2b | Invitrogen (Carlsbad, CA, USA) | Cat# A21140; RRID: AB_2535777 |
| Peroxidase-labeled Anti-Rabbit Secondary | Vector Labs (Burlingame, CA USA). | Cat# PI-1000; RRID:AB_2336198 |
| Peroxidase-labeled Anti-Mouse Secondary | Vector Labs (Burlingame, CA USA). | Cat# PI-2000; RRID:AB_2336177 |
| Hoechst | BD PharMingen (Franklin Lakes, NJ, USA) | Cat# 33342; RRID:AB_10626776 |
| Bacterial strains | ||
| E. Coli. DH5α pCMVβ (Mammalian LacZ) | Marker Gene Technologies Inc. (Eugene, OR, USA) | ID: M0951 |
| E. Coli. DH5α pKH3-TRIM28 (Mamallian HA-tagged TRIM28) | Addgene (Watertown, MA, USA) | Cat# 45569 |
| E. Coli. DH5α pKH3-TRIM28 S473→A (HA-tagged TRIM28 S473 Phosphodefective mutant) | This Study | N/A |
| E. Coli. DH5α pKH3-TRIM28 S473→D (HA-tagged TRIM28 S473 Phosphomimetic mutant) | This Study | N/A |
| Chemicals and reagents | ||
| Rapamycin | LC Laboratories (Woburn, MA, USA) | ID: R-5000 |
| DMSO | ThermoFisher Scientific (Waltham, MA, USA) | ID: PI20684 |
| Tamoxifen (injection) | Sigma Life Science (St. Louis, MO, USA) | ID: T5648–5G |
| Tamoxifen-infused Rodent Chow | Envigo Teklad (Madison, WI, USA) | ID: TD 130857 |
| Critical commercial assays | ||
| Complete Mini EDTA-Free Protease Inhibitor Cocktail Tablet | Roche (Basel, Switzerland) | Cat# 11836170001 |
| Pierce BCA Protein Assay Kit | ThermoFisher Scientific (Waltham, MA, USA) | Cat# 23225 |
| Pierce Quantitative Colorimetric Peptide Assay | ThermoFisher Scientific (Waltham, MA, USA) | Cat# 23275 |
| 10-plex tandem mass tags (TMT) Kit | ThermoFisher Scientific (Waltham, MA, USA) | Cat# 90113 |
| QuickChange II Site-Directed Mutagenesis Kit | Agilent Technologies (Santa Clara, CA, USA) | Cat# 200523 |
| Endofree Plasmid Kit | QIAGEN (Valencia, CA, USA) | Cat# 12362 |
| DC Protein Assay Kit | Life Science (Hercules, CA, USA) | Cat# 5000112 |
| Deposited data | ||
| Proteomic and Phosphoproteomic data | This Study | Raw data available at the link below: ftp://massive.ucsd.edu/MSV000086732/ |
| Experimental models: organisms/strains | ||
| Wild-Type C57BL/6 Mice | Jackson Laboratories (Bar Harbor, MA, USA) | Stock# 000664 |
| Homozygous LoxP-flanked TRIM28 C57/ BL6 Mice (B6.129S2(SJL)-Trim28tm1. 1Ipc/J) | Jackson Laboratories (Bar Harbor, MA, USA) | Stock# 018552 |
| Hemizygous HSA-MerCreMer C57/BL6 Mice ((ACTA1-cre/Esr1*)2Kesr/J) | Jackson Laboratories (Bar Harbor, MA, USA) | Stock# 025750 |
| Software and algorithms | ||
| MaxQuant (version 1.5.3.51) Andromeda Search Algorithm | Cox and Mann, 2008 | RRID: SCR_014485 (https://www.maxquant.org) |
| Perseus V.1.6.0.7 | Tyanova et al., 2016 | RRID: SCR_015753 (https://maxquant.net/perseus/) |
| PhosphoSitePlus | Hornbeck et al., 2015 | RRID:SCR_001837 (https://www.phosphosite.org/homeAction.action) |
| LIMMA package (R Software) | (Ritchie et al., 2015) (Bioconductor.org) | RRID:SCR_010943 |
| DAVID | Huang et al., 2009a, 2009b | RRID:SCR_001881 |
| NetworKIN source code (Networkin3.0_release.zip) | Horn et al., 2014 | RRID:SCR_007818 (http://www.networkin.info/download.shtml) |
| Phomics | Munk et al., 2016 | http://phomics.jensenlab.org/activation_loop_peptides |
| VisionWorksLS | Analytik Jena AG (UVP) (Upland, CA, USA) | N/A |
| Dynamic Muscle Control (DMC) software | Aurora Scientific (Aurora, Ontario, Canada) | Cat# 610A |
| Dynamic Muscle Analysis (DMA) software | Aurora Scientific (Aurora, Ontario, Canada) | Cat# 611A |
| Nikon NIS-Elements D software | Nikon Instruments (Melville, NY, USA) | N/A |
| Leica LASX software | Leica Microsystems (Buffalo Grove, IL, USA) | N/A |
Data and code availability
The proteomic and phosphoproteomic data generated in this study can be found at the University of California San Diego (UCSD) MassIVE Database: MSV000086732 (ftp://massive.ucsd.edu/MSV000086732/).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Wild-type male C57BL6 mice were obtained from Jackson Laboratories. Tamoxifen inducible and skeletal muscle-specific Trim28 knockout C57BL6 mice were generated by crossing female mice homozygous for Trim28 flanked by loxP recognition sites (Jackson Laboratories) with male mice that were hemizygous for human skeletal muscle actin promoter (HSA) driven expression of a mutated estrogen receptor-flanked Cre recombinase enzyme (HSA-MCM) (Jackson Laboratories). Offspring from these mice were subsequently crossed to obtain offspring that were homozygous for loxP flanked (floxed) Trim28 and hemizygous for the HSA-MCM allele. Offspring that were homozygous for floxed Trim28 and positive for the HSA-MCM allele was used for the knockout condition (KO), while offspring that were homozygous for floxed Trim28 and negative for the HSA-MCM allele was used for the control condition. Genotypes were confirmed with tail snips by PCR, and an equal number of male and female offspring were employed. Specifically, at 6 weeks of age, both the KO and control mice were given daily IP injections 100 mg/kg of tamoxifen for five consecutive days as previously described and then assigned to an experimental group (You et al., 2019). All mice were housed in a room maintained with a 12–12 hour light-dark cycle and received food and water ad libitum. All of the experimental procedures were performed on mice that were 8–10 weeks of age and euthanasia was performed with cervical dislocation while the mice were under isoflurane anesthesia. The Institutional Animal Care and Use Committee (IACUC) at the University of Wisconsin-Madison approved all the methods employed in this study.
METHOD DETAILS
Maximal-Intensity Contractions
Where indicated, mice were given an IP injection of 1 mg/kg rapamycin (LC Laboratories) or DMSO (Thermo Fisher Scientific) as a vehicle control at 100 min before the onset of maximal-intensity contractions (MIC) and the tibialis anterior (TA) muscle was subjected to maximal-intensity contractions or the control condition as previously described (You et al., 2019). Briefly, mice were anesthetized with isoflurane, and then a small incision was made along the lateral edge of the right thigh, exposing the sciatic nerve. An electrode was then placed on the sciatic nerve and contractions were elicited by stimulating the nerve with an SD9E Grass stimulator (Grass Instruments) at 100Hz with 0.5 ms pulses at 4–7 V for a total of 10 sets of 6 contractions. Each contraction lasted for 3 s and was followed by a 10 s rest period. A 1 min rest period was provided between each set. At 1 hr after the last contraction, both the right TA and left contralateral control TA were collected and immediately frozen in liquid nitrogen or processed for immunohistochemistry as detailed below.
Mass Spectrometry and Analysis
Tissue lysis and centrifugation
Frozen TA muscles were homogenized in 1 mL of buffer A [40 mM Tris (pH 7.5), 1 mM EDTA, 5 mM EGTA, 0.5% Triton X-100, one PhosSTOP tablet (Roche) per 10 ml, and one Complete Mini EDTA-Free Protease Inhibitor Cocktail Tablet (Roche) per 10 ml]. Samples were homogenized with a Polytron (PT 1200 E) for 20 s and then centrifuged at 6,000 × G for 1 min to remove bubbles and confirm complete homogenization. The homogenate was then separated into soluble and insoluble fractions as detailed previously (Potts et al., 2017). A 100 μl aliquot of sample was saved for western blotting and the remainder was reserved for MS analysis.
Protein digestion and peptide desalting
The total amount of protein in each sample was determined using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) and proteins were precipitated by bringing the original sample solution to a 90% concentration of MeOH by volume. The sample was then centrifuged at 12,000 × G for 5 min, the supernatant was removed and then the remaining protein precipitate was resuspended in 8 M urea, 50 mM Tris, (pH 8.0), 10 mM TCEP, and 40 mM chloroacetamide and incubated for 30 min with shaking to completely reduce and alkylate the proteins. The sample was diluted to a concentration of 1.5 M urea with 50 mM Tris (pH 8.0) and digested with trypsin (1:50 enzyme:protein ratio) at 37°C for 15 hr. The enzymatic digestion was quenched by acidifying the sample to pH < 2 with 10% trifluoroacetic acid (TFA). Strata-X desalting columns (Phenomenex) were prepared by flowing 1 mL of 100% acetonitrile (ACN) over the column, followed by 1 mL of 0.1% TFA. Individual samples were then spun down, and the acidic supernatant was collected and gravity filtered through the Strata-X columns. The bound peptides were washed with 1 mL 0.1% TFA, followed by elution into a fresh tube using 500 μl of 40% ACN and 0.1% TFA, and finally an additional elution of 300 μL of 80% ACN and 0.1% TFA. Eluted peptides were dried down by vacuum centrifugation. A Pierce Quantitative Colorimetric Peptide Assay (Thermo Fisher Scientific) was performed to determine peptide concentrations prior to TMT labeling.
TMT labeling
A total of 4 independent 10-plex experiments were performed as outlined in Figure 1. In each 10-plex experiment, 1 mg of peptides from each sample was incubated with one tag from a 10-plex tandem mass tags (TMT) kit according to the manufacturer’s instructions (Thermo Fisher Scientific). After being incubated with shaking for 3 h at room temperature, the samples quenched using 5% hydroxylamine and incubated at room temperature for 15 minutes with shaking. An aliquot from all 10 samples was mixed in a 1:1 ratio across all channels and analyzed using an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) to ensure complete TMT peptide labeling and compare peptide ratios in this “test mix.” These preliminary mixing ratios were used as a guide to create a final sample mix, where the 10 samples were mixed at a 1:1 ratio. The final pooled sample containing TMT labeled peptides from all 10 samples was desalted using a Strata-X desalting column. The pooled sample was enriched using Immobilized Metal Affinity Chromatography (IMAC) Ni-NTA magnetic agarose beads (QIAGEN). The resultant phosphopeptide sample and flow-through non-phosphopeptide sample were each fractionated using a reverse phase HPLC to produce 12 total phosphopeptide fractions and 12 total non-phosphopeptide fractions. Each of these fractions were dried using a vacuum centrifuge and resuspended in MS-grade water with 0.2% formic acid for subsequent mass spectrometry analysis.
Nano-LC-MS/MS methods
Each sample was analyzed using an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) during a 90 min nano-liquid chromatography separation using a Dionex UltiMate 3000 RSLCnano system (Thermo Fisher Scientific). Samples were analyzed using an MS1 AGC target of 1×106 and maximum injection times of 50 msec. MS1 scans were analyzed at 60,000 resolving power with a scan range from 300–1500 m/z. Precursor ions with charge states of +2 to +8 were selected for fragmentation and MS2 analysis. MS2 scans were collected with an isolation window of 3 Th, HCD fragmentation at 35% NCE, and a dynamic exclusion duration of 30 s. Phosphopeptide product ions were analyzed in the Orbitrap at 30,000 resolving power with an AGC target of 1×105 ions and 120 msec maximum injection times. Non-phosphopeptide fractions’ peptide fragments were analyzed in the Orbitrap with 30,000 resolving power with an AGC target of 5×104 ions and 75 msec maximum injection times. Each phosphopeptide fraction was analyzed with duplicate injections.
MS data analysis
The RAW data files were searched using MaxQuant (version 1.5.3.51) with the Andromeda search algorithm (Cox et al., 2014). Thermo RAW files were searched against a Mus musculus target-decoy database (UniProt, downloaded 08/14/2015). Peptide and phosphopeptide datasets were searched using a 50 ppm precursor mass tolerance and 0.02 Da fragment tolerance for b and y ions produced by HCD fragmentation. All fractions were searched with static carbadimomethyl of cysteine residues, static TMT 10-plex modifications of peptide N-termini and lysines, and dynamic methionine oxidation. Phosphopeptide fractions were searched with additional dynamic phosphorylation modifications of serine, threonine, and tyrosine residues. Resulting peptide identifications were filtered to 1% false discovery rate (FDR). Peptides were then mapped back to their parent proteins using MaxQuant and filtered to a 1% FDR at the protein level. 10-plex TMT reporter ion signals were used for protein and phosphopeptide quantitation and were extracted from MaxQuant result tables.
Bioinformatics
The TMT reporter ion intensities for the phosphopeptides and quantified proteins were normalized to the mean value obtained in the vehicle control samples. Each of the 10-plex TMT analyses also included the use of an internal control sample (#1229L) which allowed for quantitative comparisons to be made between the individual TMT analyses. Once normalized, the data was uploaded into Perseus V.1.6.0.7 (Tyanova and Cox, 2018) and analyzed with two-way ANOVA to identify statistically significant main effects and interactions. The data was then log2 transformed for the generation of volcano plots and concomitant post hoc analyses which were performed with the moderated t test documented in the LIMMA package in R (Hoffman et al., 2015; Smyth, 2004) and FDR corrected with the Benjamini-Hochberg method (Benjamini and Hochberg, 1995). The phosphopeptide data was then annotated in Perseus with information obtained from the DAVID (downloaded on 10/20/2018) (Huang et al., 2009b) and PhosphoSitePlus (downloaded on 10/7/2019) (Hornbeck et al., 2019). Enrichment analyses were then performed in Perseus with the Fisher Exact test.
For the prediction of activated kinases, activation loop phosphorylation analysis was performed with Phomics (http://phomics.jensenlab.org/activation_loop_peptides) (Munk et al., 2016) and the NetworKIN source code was downloaded from (http://networkin.info/download.shtml:Networkin3.0_release.zip) (Horn et al., 2014). To accommodate the NetworKIN input, protein identifiers were converted to Ensembl 74 proteins with a conversion table downloaded from BioMart containing Uniprot identifiers from Mus musculus and the Ensembl 74 protein of the corresponding Homos sapiens ortholog (Smedley et al., 2015). In total, 8,454 of the 10,688 phosphosites were successfully assigned a human Ensembl 74 protein, of which, 7,879 were assigned NetworKIN-based predictions. The NetworKIN output was spot-checked for correct code implementation with the low throughput option available online (http://networkin.info/). Kinase enrichment analysis was then performed with the methods of Bøgebo et al. (2014). Kinases were considered to be significantly enriched if the Benajamini-Hochberg corrected P value of Kolmogorov-Smirnov test was < 0.05. Statistical analysis was performed with the SciPy package of Python (Version 3.6). Code is available in a github repository (https://github.com/gwilson9/NetworKINImplementation).
Western Blot Analysis
Frozen samples were homogenized with a Polytron for 30 s in ice-cold buffer A (described above) or buffer B [40 mM Tris (pH 7.5), 1 mM EDTA, 5 mM EGTA, 0.5% Triton X-100, 25 mM β-glycerophosphate, 25 mM NaF, 1 mM Na3VO4, 10 mg/ml leupeptin, and 1 mM PMSF]. The resulting protein concentrations were determined using a DC protein assay kit (Bio-Rad). Equivalent amounts of protein were then dissolved in Laemmli buffer, boiled for 5 min, and subjected to electrophoretic separation by SDS-PAGE. Proteins were then transferred to a PVDF membrane at 300 mA for 1 hour 45 min and subsequently blocked with 5% powdered milk in tris-buffered saline containing 0.1% Tween 20 (TBST) for 1 hour. After 30 min of washing in TBST, the membranes were incubated overnight at 4°C with primary antibody dissolved in a 1% bovine serum albumin (BSA)-TBST solution. The next day the membranes were washed for 30 min with TBST and then probed with a peroxidase-conjugated secondary antibody dissolved in 5% powdered milk-TBST for 1 hour at room temperature. After 30 min washing in TBST the blots were developed using a film processor or with a UVP Autochemi system (Analytik Jena AG) along with either a regular enhanced chemiluminescence (ECL) reagent (Pierce) or ECL-prime (Amer-sham). Once the appropriate images were collected, the membranes were stained with Coomassie blue to verify equal loading in all lanes. Images were quantified using ImageJ software (U.S. NIH).
Immunoprecipitation
Samples homogenized in buffer B were centrifuged at 2500 × G for 5 min and then equal amounts of protein from the supernatant were diluted to a volume of 0.5 mL with fresh buffer A. The samples were then incubated with 20 μL of EZview red HA-tagged agarose affinity gel beads (Sigma-Aldrich) with gentle rocking at 4°C for 2 hr. The beads were then pelleted by centrifugation at 500 × G for 30 s and washed 4 times with fresh ice-cold buffer A. After the washes, the beads were dissolved in Laemmli buffer and subjected to western blot analysis as described above
Electroporation
The plasmid encoding LacZ has been previously described (Nolan et al., 1988). The HA-tagged wild-type TRIM28 (pKH3-TRIM28) plasmid was purchased from Addgene (#45569) and used as the template to generate serine to aspartic acid (S473→D) phosphomimetic or serine to alanine (S473→A) phosphodefective mutations with the QuickChange II Site-Directed Mutagenesis Kit (Agilent Technologies). All plasmid DNA was grown in DH5α E. Coli, purified with an Endofree plasmid kit (QIAGEN), and re-suspended in sterile phosphate-buffered saline (PBS). Electroporation was performed as previously described by Goodman et al., 2015 (Goodman et al., 2015). Briefly, mice were anesthetized with isoflurane, and then a small incision was made through the skin covering the TA muscle. A 27-gauge needle was used to inject 12 μL of the DNA solution containing 30 μg of plasmid DNA into the TA muscle and then electric pulses were applied to the muscle through 1 cm gap pin electrode (Harvard Apparatus) that were laid on top of the proximal and distal myotendinous junctions. Eight 20 ms square-wave electric pulses at a frequency of 1 Hz were delivered with an ECM 830 electroporation unit (BTX; Harvard Apparatus) at a field strength of 160 V/cm. After the electroporation procedure, the incision was closed with Vetbond surgical glue (3M Animal Care Products) and the animal was given an IP injection 0.05 μg/g of buprenorphine to alleviate any pain that was caused by the procedure. After a 1.5 – 7 day recovery period, the mice were anesthetized and the TA muscles were either immediately frozen in liquid nitrogen or processed for immunohistochemistry as detailed below.
Myotenectomy
Mice were anesthetized with isoflurane and then bilateral myotenectomy was performed by removing the distal myotendinous junctions of the gastrocnemius muscles as detailed previously. Mice in the control group were given a bilateral sham surgery that, except for removing the myotendinous junctions of the gastrocnemius, was identical to the myotenectomy surgery. Mice were allowed to recover for 0 – 14 days. Mice undergoing a recovery period lasting > 90 min were given an IP injection 0.05 μg/g of buprenorphine to alleviate any pain that was caused by the procedure. Such mice were also placed on rodent chow that contained 500 mg / kg tamoxifen (equal to a daily intake of ≈80 mg of tamoxifen / day) at 5 days prior to the onset of the surgery (Envigo Teklad) and were maintained on this chow throughout their recovery period. At the end of the recovery period, all mice were anesthetized with isoflurane and then the PLT muscles were either immediately frozen in liquid nitrogen or processed for immunohistochemistry as detailed below.
In situ Skeletal Muscle Contractions
Mice were anesthetized with isoflurane and then placed on the platform of a 809C in situ mouse apparatus (Aurora Scientific) that was maintained at 37°C with circulating water. The TA muscle of one leg was exposed and then a braided 4/0 silk suture and a small drop of superglue were used to attach the distal tendon to the lever arm of a 305B-LR dual-mode lever system (Aurora Scientific). The knee was then fixed on the mouse apparatus with a 27-gauge needle and 809-ELEC stimulation electrodes (Aurora Scientific) were placed on the mid-belly of the muscle. To enable stimulation, the electrode was routed through a high power bi-phase current stimulator that was controlled with DMC software (Aurora Scientific). The TA muscle was stimulated with single (0.5 msec) pulses at 60 V and the muscle was gradually lengthened until a plateau in twitch tension was observed (i.e., the muscle was at optimal length (Lo)). With the muscle at Lo, twitch contractile properties including peak twitch force, the time taken to reach peak twitch force, and the time to relax from peak twitch force were determined. Next, the TA muscle was stimulated with three successive 300 msec tetanic contractions at 100, 150, and 200 Hz, respectively. Each tetanic contraction was followed by 1 min of rest and the highest force measured during these contractions was recorded as the peak tetanic force. Finally, the TA was subjected to a bout of fatiguing contractions in which the muscle was stimulated with a 300 msec tetanic contraction at 200 Hz once every 2 s for 3 min. Throughout the entire period of analysis, the TA muscle was kept moist by applying PBS that was maintained at 37°C. Both active and passive force was recorded during each contraction and all force recordings were analyzed by DMA software (Aurora Scientific).
Immunohistochemistry
Muscles were submerged in optimal cutting temperature compound (Sakura Tissue-Tek) at resting length and then frozen in liquid nitrogen-chilled isopentane. Mid-belly cross-sections (10 μm thick) were taken with a cryostat and fixed for 10 min with 1% paraformaldehyde dissolved in PBS (for TRIM28 analyses) or −20°C acetone (for myofiber type analyses). Fixed sections were washed with PBS and then incubated for 20 min at room temp in buffer C (0.5% Triton X-100, 0.5% BSA dissolved in PBS). For Type I myofiber staining, samples were subjected to an additional 1 hr incubation at room temp in buffer C containing a Fab Anti-Mouse IgG. Following these incubations, the sections were washed with PBS and then incubated in buffer C containing primary antibodies for 1 hour at room temp. The sections were again washed with PBS and then incubated in solution C containing secondary antibodies for 1 hour at room temp. Sections were then washed with PBS (where indicated, the nuclei were stained with Hoechst that was diluted in PBS for 5 min before the washing step) and then: a) images were captured with a Nikon DS-QiMc camera (Nikon) on an upright Nikon 80i epifluor-escence microscope, or b) the sections were mounted in a layer of ProLong Gold anti-fade mounting medium (Invitrogen), overlaid with a coverslip (Thermo Fisher Scientific) and then imaged with an inverted Leica TCS SP8 confocal laser scanning microscope (Leica).
Image Analysis
For myofiber cross-sectional area measurements, images of the entire muscle cross-section were captured and then the cross-sectional area of at least 70 randomly selected myofibers of each myofiber type (Type I, IIa, IIx, and IIb) were measured by tracing the laminin stained periphery of individual myofibers with Nikon NIS-Elements D software (Nikon). For myonuclei and interstitial nuclei identification, 3–5 randomly selected 8.4 mm2 regions of each muscle cross-section were captured, and then all TRIM28 and P-TRIM28(S473) positive nuclei, as well as total nuclei were identified using both the NIS-Elements D software (Nikon) and the Leica LASX software (Leica). The distinction between myonuclei versus interstitial nuclei was achieved by defining nuclei that resided within the dystrophin layer of individual myofibers as myonuclei, while those that resided outside of the dystrophin layer were designated as interstitial nuclei. All image analyses were performed by investigators that were blinded to the sample identification.
QUANTIFICATION AND STATISTICAL ANALYSIS
In all experiments that followed the phosphoproteomic analyses, statistical significance was determined by using Student’s t test, one-way ANOVA, two-way ANOVA, or two-way repeated-measures ANOVA followed by Student-Newman-Kuels post hoc analysis as indicated in the figure legends. Differences between groups were considered significant when p < 0.05. All statistical analyses were performed on SigmaPlot / SigmaStat software (Systat).
Supplementary Material
Highlights.
Extensive map of the rapamycin-sensitive and contraction-regulated phosphoproteome
Contractions induce robust, rapamycin-insensitive phosphorylation of TRIM28(S473)
TRIM28(S473) phosphorylation is sufficient to increase skeletal myofiber size
Myofiber KO of TRIM28 alters muscle size, function, and mechanical load-induced hypertrophy
ACKNOWLEDGMENTS
The research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award numbers AR057347 and AR074932 to T.A.H. The research was also supported by the National Institutes of Health under award number GM108538 to J.J.C. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108796.
DECLARATION OF INTERESTS
The authors declare no conflicts of interest.
REFERENCES
- Abdelmoity A, Padre RC, Burzynski KE, Stull JT, and Lau KS (2000). Neuronal nitric oxide synthase localizes through multiple structural motifs to the sarcolemma in mouse myotubes. FEBS Lett 482, 65–70. [DOI] [PubMed] [Google Scholar]
- Adams GR, and Bamman MM (2012). Characterization and regulation of mechanical loading-induced compensatory muscle hypertrophy. Compr. Physiol 2, 2829–2870. [DOI] [PubMed] [Google Scholar]
- Al-Bari MAA, and Xu P (2020). Molecular regulation of autophagy machinery by mTOR-dependent and -independent pathways. Ann. N Y Acad. Sci 1467, 3–20. [DOI] [PubMed] [Google Scholar]
- Aronson D, Dufresne SD, and Goodyear LJ (1997). Contractile activity stimulates the c-Jun NH2-terminal kinase pathway in rat skeletal muscle. J. Biol. Chem 272, 25636–25640. [DOI] [PubMed] [Google Scholar]
- Baar K, and Esser K (1999). Phosphorylation of p70(S6k) correlates with increased skeletal muscle mass following resistance exercise. Am. J. Physiol 276, C120–C127. [DOI] [PubMed] [Google Scholar]
- Beharry AW, Sandesara PB, Roberts BM, Ferreira LF, Senf SM, and Judge AR (2014). HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. J. Cell Sci 127, 1441–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 57, 289–300. [Google Scholar]
- Bodine SC (2013). Disuse-induced muscle wasting. Int. J. Biochem. Cell Biol 45, 2200–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bøgebo R, Horn H, Olsen JV, Gammeltoft S, Jensen LJ, Hansen JL, and Christensen GL (2014). Predicting kinase activity in angiotensin receptor phosphoproteomes based on sequence-motifs and interactions. PLoS ONE 9, e94672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolderson E, Savage KI, Mahen R, Pisupati V, Graham ME, Richard DJ, Robinson PJ, Venkitaraman AR, and Khanna KK (2012). Kruppel-associated Box (KRAB)-associated co-repressor (KAP-1) Ser-473 phosphorylation regulates heterochromatin protein 1β (HP1-β) mobilization and DNA repair in heterochromatin. J. Biol. Chem 287, 28122–28131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, Hewett TE, Jones SP, Lefer DJ, Peng CF, et al. (2000). The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J 19, 6341–6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkholder TJ, Fingado B, Baron S, and Lieber RL (1994). Relationship between muscle fiber types and sizes and muscle architectural properties in the mouse hindlimb. J. Morphol 221, 177–190. [DOI] [PubMed] [Google Scholar]
- Cheng CT, Kuo CY, and Ann DK (2014). KAPtain in charge of multiple missions: Emerging roles of KAP1. World J. Biol. Chem 5, 308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerwińska P, Shah PK, Tomczak K, Klimczak M, Mazurek S, Sozań-ska B, Biecek P, Korski K, Filas V, Mackiewicz A, et al. (2017). TRIM28 multi-domain protein regulates cancer stem cell population in breast tumor development. Oncotarget 8, 863–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubouloz F, Deloche O, Wanke V, Cameroni E, and De Virgilio C (2005). The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol. Cell 19, 15–26. [DOI] [PubMed] [Google Scholar]
- Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC, and Karin M (2004). Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 306, 271–275. [DOI] [PubMed] [Google Scholar]
- Gehrmann U, Burbage M, Zueva E, Goudot C, Esnault C, Ye M, Carpier JM, Burgdorf N, Hoyler T, Suarez G, et al. (2019). Critical role for TRIM28 and HP1β/γ in the epigenetic control of T cell metabolic reprograming and effector differentiation. Proc. Natl. Acad. Sci. USA 116, 25839–25849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CA (2014). The role of mTORC1 in regulating protein synthesis and skeletal muscle mass in response to various mechanical stimuli. Rev. Physiol. Biochem. Pharmacol 166, 43–95. [DOI] [PubMed] [Google Scholar]
- Goodman CA, Miu MH, Frey JW, Mabrey DM, Lincoln HC, Ge Y, Chen J, and Hornberger TA (2010). A phosphatidylinositol 3-kinase/protein kinase B-independent activation of mammalian target of rapamycin signaling is sufficient to induce skeletal muscle hypertrophy. Mol. Biol. Cell 21, 3258–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CA, Frey JW, Mabrey DM, Jacobs BL, Lincoln HC, You JS, and Hornberger TA (2011). The role of skeletal muscle mTOR in the regulation of mechanical load-induced growth. J. Physiol 589, 5485–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CA, Dietz JM, Jacobs BL, McNally RM, You JS, and Hornberger TA (2015). Yes-Associated Protein is up-regulated by mechanical overload and is sufficient to induce skeletal muscle hypertrophy. FEBS Lett 589, 1491–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad F, and Adams GR (2004). Inhibition of MAP/ERK kinase prevents IGF-I-induced hypertrophy in rat muscles. J. Appl. Physiol 96, 203–210. [DOI] [PubMed] [Google Scholar]
- Hoffman NJ, Parker BL, Chaudhuri R, Fisher-Wellman KH, Kleinert M, Humphrey SJ, Yang P, Holliday M, Trefely S, Fazakerley DJ, et al. (2015). Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab 22, 922–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn H, Schoof EM, Kim J, Robin X, Miller ML, Diella F, Palma A, Ce-sareni G, Jensen LJ, and Linding R (2014). KinomeXplorer: an integrated platform for kinome biology studies. Nat. Methods 11, 603–604. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Latham V, Murray B, Nandhikonda V, Nord A, Skrzypek E, Wheeler T, Zhang B, and Gnad F (2019). 15 years of PhosphoSitePlusâ: integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res 47 (D1), D433–D441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Hughes SM, Chi MM, Lowry OH, and Gundersen K (1999). Myogenin induces a shift of enzyme activity from glycolytic to oxidative metabolism in muscles of transgenic mice. J. Cell Biol 145, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyengar S, and Farnham PJ (2011). KAP1 protein: an enigmatic master regulator of the genome. J. Biol. Chem 286, 26267–26276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumiya Y, Hopkins T, Morris C, Sato K, Zeng L, Viereck J, Hamilton JA, Ouchi N, LeBrasseur NK, and Walsh K (2008). Fast/Glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab 7, 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen I, Shepard DS, Katzmarzyk PT, and Roubenoff R (2004). The healthcare costs of sarcopenia in the United States. J. Am. Geriatr. Soc 52, 80–85. [DOI] [PubMed] [Google Scholar]
- Kim SS, Chen YM, O’Leary E, Witzgall R, Vidal M, and Bonventre JV (1996). A novel member of the RING finger family, KRIP-1, associates with the KRAB-A transcriptional repressor domain of zinc finger proteins. Proc. Natl. Acad. Sci. USA 93, 15299–15304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosek DJ, Kim JS, Petrella JK, Cross JM, and Bamman MM (2006). Efficacy of 3 days/wk resistance training on myofiber hypertrophy and myogenic mechanisms in young vs. older adults. J. Appl. Physiol 101, 531–544. [DOI] [PubMed] [Google Scholar]
- Kramer HF, and Goodyear LJ (2007). Exercise, MAPK, and NF-kappaB signaling in skeletal muscle. J. Appl. Physiol 103, 388–395. [DOI] [PubMed] [Google Scholar]
- Lagirand-Cantaloube J, Cornille K, Csibi A, Batonnet-Pichon S, Leibovitch MP, and Leibovitch SA (2009). Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS ONE 4, e4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Zheng Y, Cho S, Jang C, England C, Dempsey JM, Yu Y, Liu X, He L, Cavaliere PM, et al. (2017). Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell 171, 1545–1558.e1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang Z, and Lu G (2018). TRIM28 promotes cervical cancer growth through the mTOR signaling pathway. Oncol. Rep 39, 1860–1866. [DOI] [PubMed] [Google Scholar]
- Marcotte GR, West DW, and Baar K (2015). The molecular basis for load-induced skeletal muscle hypertrophy. Calcif. Tissue Int 96, 196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti I, Ciciliot S, Dyar KA, Abraham R, Murgia M, Agatea L, Akimoto T, Bicciato S, Forcato M, Pierre P, et al. (2016). MRF4 negatively regulates adult skeletal muscle growth by repressing MEF2 activity. Nat. Commun 7, 12397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munk S, Refsgaard JC, Olsen JV, and Jensen LJ (2016). From Phosphosites to Kinases. Methods Mol. Biol 1355, 307–321. [DOI] [PubMed] [Google Scholar]
- Nolan GP, Fiering S, Nicolas JF, and Herzenberg LA (1988). Fluorescence-activated cell analysis and sorting of viable mammalian cells based on beta-D-galactosidase activity after transduction of Escherichia coli lacZ. Proc. Natl. Acad. Sci. USA 85, 2603–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil TK, Duffy LR, Frey JW, and Hornberger TA (2009). The role of phosphoinositide 3-kinase and phosphatidic acid in the regulation of mammalian target of rapamycin following eccentric contractions. J. Physiol 587, 3691–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara R, and Suginohara T (2018). Rapamycin-insensitive mechanistic target of rapamycin regulates basal and resistance exercise-induced muscle protein synthesis. FASEB J, fj201701422R. [DOI] [PubMed] [Google Scholar]
- Ogasawara R, Fujita S, Hornberger TA, Kitaoka Y, Makanae Y, Naka-zato K, and Naokata I (2016). The role of mTOR signalling in the regulation of skeletal muscle mass in a rodent model of resistance exercise. Sci. Rep 6, 31142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara R, Jensen TE, Goodman CA, and Hornberger TA (2019). Resistance Exercise-Induced Hypertrophy: A Potential Role for Rapamycin-Insensitive mTOR. Exerc. Sport Sci. Rev 47, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahor M, and Kritchevsky S (1998). Research hypotheses on muscle wasting, aging, loss of function and disability. J. Nutr. Health Aging 2, 97–100. [PubMed] [Google Scholar]
- Peker N, Donipadi V, Sharma M, McFarlane C, and Kambadur R (2018). Loss of Parkin impairs mitochondrial function and leads to muscle atrophy. Am. J. Physiol. Cell Physiol 315, C164–C185. [DOI] [PubMed] [Google Scholar]
- Philp A, Schenk S, Perez-Schindler J, Hamilton DL, Breen L, Laverone E, Jeromson S, Phillips SM, and Baar K (2015). Rapamycin does not prevent increases in myofibrillar or mitochondrial protein synthesis following endurance exercise. J. Physiol 593, 4275–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda CT, Ramanathan S, Fon Tacer K, Weon JL, Potts MB, Ou YH, White MA, and Potts PR (2015). Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 160, 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts GK, McNally RM, Blanco R, You JS, Hebert AS, Westphall MS, Coon JJ, and Hornberger TA (2017). A map of the phosphoproteomic alterations that occur after a bout of maximal-intensity contractions. J. Physiol 595, 5209–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis K, and De Virgilio C (2016). Conserved regulators of Rag GTPases orchestrate amino acid-dependent TORC1 signaling. Cell Discov 2, 15049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor DN, Balagopal P, and Nair KS (1998). Age-related sarcopenia in humans is associated with reduced synthetic rates of specific muscle proteins. J. Nutr 128 (Suppl 2), 351S–355S. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero JL, Talmadge RJ, and Edgerton VR (1999). Interrelationships of myofibrillar ATPase activity and metabolic properties of myosin heavy chain-based fibre types in rat skeletal muscle. Histochem. Cell Biol 111, 277–287. [DOI] [PubMed] [Google Scholar]
- Russell DM, Atwood HL, Whittaker JS, Itakura T, Walker PM, Mickle DA, and Jeejeebhoy KN (1984). The effect of fasting and hypocaloric diets on the functional and metabolic characteristics of rat gastrocnemius muscle. Clin. Sci. (Lond.) 67, 185–194. [DOI] [PubMed] [Google Scholar]
- Ryder JW, Fahlman R, Wallberg-Henriksson H, Alessi DR, Krook A, and Zierath JR (2000). Effect of contraction on mitogen-activated protein kinase signal transduction in skeletal muscle. Involvement Of the mitogen- and stress-activated protein kinase 1. J. Biol. Chem 275, 1457–1462. [DOI] [PubMed] [Google Scholar]
- Schiaffino S, and Reggiani C (2011). Fiber types in mammalian skeletal muscles. Physiol. Rev 91, 1447–1531. [DOI] [PubMed] [Google Scholar]
- Schoenfeld BJ, Ogborn D, and Krieger JW (2016). Effects of Resistance Training Frequency on Measures of Muscle Hypertrophy: A Systematic Review and Meta-Analysis. Sports Med 46, 1689–1697. [DOI] [PubMed] [Google Scholar]
- Seguin R, and Nelson ME (2003). The benefits of strength training for older adults. Am. J. Prev. Med 25 (3, Suppl 2), 141–149. [DOI] [PubMed] [Google Scholar]
- Shi H, Scheffler JM, Zeng C, Pleitner JM, Hannon KM, Grant AL, and Gerrard DE (2009). Mitogen-activated protein kinase signaling is necessary for the maintenance of skeletal muscle mass. Am. J. Physiol. Cell Physiol 296, C1040–C1048. [DOI] [PubMed] [Google Scholar]
- Shupp A, Casimiro MC, and Pestell RG (2017). Biological functions of CDK5 and potential CDK5 targeted clinical treatments. Oncotarget 8, 17373–17382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Cassano M, Planet E, Sebastian S, Jang SM, Sohi G, Faralli H, Choi J, Youn HD, Dilworth FJ, and Trono D (2015). A KAP1 phosphorylation switch controls MyoD function during skeletal muscle differentiation. Genes Dev 29, 513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, Arnaiz O, Awedh MH, Baldock R, Barbiera G, et al. (2015). The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res 43 (W1), W589–W598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol 3, Article3. [DOI] [PubMed] [Google Scholar]
- Steiner JL, and Lang CH (2014). Alcohol impairs skeletal muscle protein synthesis and mTOR signaling in a time-dependent manner following electrically stimulated muscle contraction. J. Appl. Physiol 117, 1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, and Cox J (2018). Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol 1711, 133–148. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Ueyama T, Kawashima S, Sakoda T, Rikitake Y, Ishida T, Kawai M, Yamashita T, Ishido S, Hotta H, and Yokoyama M (2000). Requirement of activation of the extracellular signal-regulated kinase cascade in myocardial cell hypertrophy. J. Mol. Cell. Cardiol 32, 947–960. [DOI] [PubMed] [Google Scholar]
- van Wessel T, de Haan A, van der Laarse WJ, and Jaspers RT (2010). The muscle fiber type-fiber size paradox: hypertrophy or oxidative metabolism? Eur. J. Appl. Physiol 110, 665–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wackerhage H, Schoenfeld BJ, Hamilton DL, Lehti M, and Hulmi JJ (2019). Stimuli and sensors that initiate skeletal muscle hypertrophy following resistance exercise. J. Appl. Physiol 126, 30–43. [DOI] [PubMed] [Google Scholar]
- West DW, Baehr LM, Marcotte GR, Chason CM, Tolento L, Gomes AV, Bodine SC, and Baar K (2016). Acute resistance exercise activates rapamycin-sensitive and -insensitive mechanisms that control translational activity and capacity in skeletal muscle. J. Physiol 594, 453–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MS (2017). mTOR as a Key Regulator in Maintaining Skeletal Muscle Mass. Front. Physiol 8, 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You JS, Frey JW, and Hornberger TA (2012). Mechanical stimulation induces mTOR signaling via an ERK-independent mechanism: implications for a direct activation of mTOR by phosphatidic acid. PLoS ONE 7, e47258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You JS, McNally RM, Jacobs BL, Privett RE, Gundermann DM, Lin KH, Steinert ND, Goodman CA, and Hornberger TA (2019). The role of raptor in the mechanical load-induced regulation of mTOR signaling, protein synthesis, and skeletal muscle hypertrophy. FASEB J 33, 4021–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The proteomic and phosphoproteomic data generated in this study can be found at the University of California San Diego (UCSD) MassIVE Database: MSV000086732 (ftp://massive.ucsd.edu/MSV000086732/).