

Abstract
Shear stress has been reported to result in various metabolic effects in endothelial cells (ECs), which in turn contribute to the regulation of their vascular functions. Peroxisome proliferator-activated receptors (PPARs) have been reported to regulate lipid metabolism and have been implicated in metabolic disorders. The present study assessed the effects of laminar shear stress on the expression of PPARs in ECs in the presence of high concentrations of free fatty acids (FFAs). Human aortic ECs (HAECs) were treated with a high concentrations of palmitic acid (PA) and exposed to high shear stress (HSS) or low shear stress (LSS). Western blotting and ELISA were performed to quantify protein expression and assess prostacyclin production. The results revealed that long-term application of HSS to PA-treated HAECs induced PPAR-α, -δ and -γ protein expression. Additionally, LSS induced higher levels of PPAR-α protein expression in PA-treated HAECs compared with those after HSS. HAECs exposed to HSS also released prostacyclin (PGI2). However, HAECs treated with high concentrations of PA also produced high levels of PGI2 in the perfusion media in response to HSS compared with the static PA group. HSS also reduced the static PA-induced expression of intercellular adhesion molecule-1 and monocyte chemoattractant protein-1. The results demonstrated that HAECs increases the expression of all three peroxisome proliferator-activated receptor isoforms in response to shear metabolic stress at high FFA concentrations. The present study may provide preliminary insights into the potential roles of PPARs as an effective treatment method against metabolic disturbances that can result in EC dysfunction.
Keywords: laminar shear stress, palmitic acid, peroxisome proliferator-activated receptors, endothelial cell
Introduction
Endothelial cells (ECs) constitute the major cell type that line the interior surface of blood vessels. Consequently, ECs are constantly subjected to fluid shear stress (1). Hemodynamic forces generated by blood flow serve a critical role in maintaining EC function, vascular structure and homeostasis (1). High fluid shear stress or high shear stress (HSS) exhibits a protective effect against atherosclerosis (1). Previous studies have provided strong evidence for the protective role of HSS in vascular homeostasis by inhibiting the expression of proinflammatory and adhesion molecules in ECs (1,2). By contrast, pathological shear stress is characterized by flow disturbance and low rates of shear stress (LSS) (1). Accumulating evidence is suggesting that LSS contributes to the dysregulation of vascular function and increases susceptibility to atherosclerosis (1,2). HSS enhances defenses against reactive oxygen species (ROS)-induced damage by preventing lipid peroxidation induced by high concentrations of glucose and free fatty acids (FFAs) (3). Therefore, laminar HSS exerts important effects on EC function, particularly under conditions of metabolic disorder.
Metabolic disorders, such as type 2 diabetes mellitus or hyperlipidemia, are frequently accompanied with dysregulations in the metabolism of glucose or lipids. In particular, abnormal fatty acid metabolism is a common feature (4). High plasma levels of FFA are considered to be reliable indices for the diagnosis of type 2 diabetes (4-6). Additionally, high concentrations of plasma FFA is a risk factor for disturbances in EC function for the pathogenesis of vascular diseases, including atherosclerosis and vascular complications associated with diabetes (7,8). High plasma levels of FFA can also induce an inflammatory response and aggravate oxidative stress, promoting the pathogenesis of cardiovascular disease (9). An in vitro study has previously demonstrated that high concentrations of glucose and FFA can inhibit EC proliferation and result in cell death (10). Increased plasma concentrations or treatment with high concentrations of palmitic acid (PA) impair endothelial progenitor cell (EPC) bioavailability, reduce the re-endothelialization ability of EPCs and accelerate EPC senescence (11,12). In addition, high concentrations of FFAs have been demonstrated to induce nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing 3 inflammasome activation, which further increases endothelial permeability (13-15).
Peroxisome proliferator-activated receptors (PPARs) belong to the nuclear receptor superfamily of ligand-activated transcription factors, which are involved in regulating lipid and glucose metabolism, energy homeostasis and inflammation (16,17). All three PPAR isoforms have been reported to be involved in metabolic disorders, including atherosclerosis, dyslipidemia and diabetes (18). Previous studies have demonstrated that shear stress acting on ECs induces the expression and activation of PPAR-α, -δ and -γ, which regulate EC function (19,20). However, studies reporting the role of PPARs in the regulation of EC function under metabolic stress remain insufficient. Increased expression of PPAR-γ ameliorates PA-induced cytotoxicity in sertoli cells, a non-EC type cell (21). A previous in vivo study has demonstrated that a PPAR-δ agonist reversed PA-induced impairments in endothelium-dependent relaxation (22). These results suggested that PPARs serve protective roles in different cell types in the presence of a PA-induced metabolic disturbance within the vasculature.
Although the effect of blood flow-induced biomechanical force on vascular homeostasis has been extensively investigated, little is known regarding the tolerance of ECs against constant shear stress at high concentrations of PA. The present study utilized a parallel plate flow chamber model to exert fluid laminar shear stress on ECs. It was subsequently determined whether different patterns of laminar flow shear stress affected PPAR expression and cellular energy metabolism regulators in ECs following treatment with high PA concentrations and exposure to shear stress.
Materials and methods
Cell culture and treatments
Human aortic ECs (HAECs) (cat. no. 304-05) were obtained from Cell Applications Inc. and cultured on a glass slide coated with fibronectin (BD Biosciences). Cells were maintained in M199 medium (Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 20% FBS (Gibco; Thermo Fisher Scientific, Inc.), EC growth medium (Cell Applications Inc.) and 100 U/ml penicillin and streptomycin. HAECs were observed under light microscopy and used between passages 5 and 7 for all experiments. Human aortic smooth muscle cells (HASMCs; cat. no. 354-05a) were obtained commercially (Cell Applications Inc.) and maintained in F12K medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS. Cells were maintained in 37˚C and 5% CO2 environment. PA was purchased from Sigma-Aldrich, Merck KGaA). A solution of 10% bovine serum albumin (BSA; Gibco; Thermo Fisher Scientific, Inc.) was used as the solvent control. In the present study, 300 µM PA was used to treat confluent HAECs for 24 h at 37˚C. Subsequently, HAECs were subjected to either static or shear stress in a PA-containing medium for an additional 20 h at 37˚C.
Flow shear stress system
The flow system was constructed by the Department of Physiology and Biophysics, Graduate Institute of Physiology, National Defense Medical Center. The system was established based on the study by Chiu et al (23,24). A series of studies have been conducted using this flow system (19,25,26). Flow system was set up at 37˚C in a 5% CO2 environment as previously described (19). Confluent HAECs were seeded onto a fibronectin-coated glass slide (6x105 cells/cm2) and maintained in culture medium in a humidified atmosphere with 5% CO2 at 37˚C overnight prior to flow shear stress exposure. For shear stress experiments, HAECs were subjected to either static conditions, exposed to 12 dyn/cm2 (HSS) or 4 dyn/cm2 (LSS) for 20 h at 37˚C.
Western blotting
HAECs were harvested using RIPA buffer (Merck KGaA) containing a protease inhibitor mixture (Sigma-Aldrich; Merck KGaA). The protein concentration was determined using a Bradford assay (Bio-Rad Laboratories, Inc.). The protein supernatant from the total cell lysate (20 µg) was separated using 10% SDS-PAGE and transferred onto PVDF membranes. The membrane was blocked with 5% non-fat milk in TBS-T for 1.5 h at room temperature and further incubated with primary antibodies with 2.5% BSA in TBS-T (all, 1:1,000) targeting the indicated proteins overnight at 4˚C. The membrane was hybridized using specific primary rabbit polyclonal antibodies (from Cell Signaling Technology, Inc.) against endothelial nitric oxide synthase (eNOS; cat. no. 32027), α-smooth muscle actin (α-SMA; cat. no. 19245), PPAR-γ (cat. no. 2430), intercellular adhesion molecule-1 (ICAM-1; cat. no. 4915S), monocyte chemoattractant protein-1 (MCP-1; cat. no. 2027) and rabbit polyclonal antibodies against PPAR-α (cat. no. ab24509) and -δ (cat. no. ab23673; Abcam). The membranes were subsequently incubated with corresponding Peroxidase-AffiniPure Goat Anti-Mouse IgG (H+L) (cat. no. 115-035-003; 1:5,000) and Goat Anti-Rabbit IgG (H+L) (cat. no. 111-035-003; 1:5,000) secondary antibodies diluted in blocking buffer for 1 h at room temperature (each, Jackson ImmunoResearch Laboratories, Inc.). Immunoblots were visualized using the Immobilon™ western chemiluminescent horseradish peroxidase substrate (EMD Millipore) and the Western-Light chemiluminescent detection system (Applied Biosystems; Thermo Fisher Scientific, Inc.) was used for immunodetection. Relative band intensities were quantified using ImageJ 1.47 software (National Institutes of Health).
Measurement of 6-keto-prostaglandin F1α
Perfusion medium was collected from HAECs exposed to static conditions or HSS in the presence or absence of PA for 20 h at 37˚C. The medium was collected (supernatant only) and centrifuged at 270 x g for 10 min at room temperature, after which the concentration of a stable hydrolyzed metabolite of prostacyclin (PGI2), 6-keto-prostaglandin F1α (6-keto-PGF1α), was measured using a 6-keto-PGF1α ELISA kit (cat. no. ADI-900-004) (Assay designs; Enzo Life Sciences, Inc.) in accordance with the manufacturer’s protocol.
Statistical analysis
Data were expressed as the mean ± SEM from three or four independent experiments. Data were analyzed using SigmaPlot version 12.0 (Systat Software, Inc.). Two-way ANOVA followed by Bonferroni’ test were used for multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
Identification of HAECs and morphological changes in response to shear stress and PA treatment
The identity of primary HAECs was confirmed by measuring the expression of eNOS, a specific marker of endothelial cells (7), by western blot analysis. Immunoblotting of the lysates showed no contamination with other vascular cells such as HASMCs, as the HAECs of Fig. 1 did not present the smooth muscle cell marker. α-SMA expression was used as the smooth muscle cell marker (Fig. 1). To investigate the effect of different types of shear stress with PA treatment on EC morphology, HAECs were exposed to LSS or HSS for 20 h. ECs exposed to laminar HSS were elongated and aligned in the direction of flow with long shapes (27). As presented in Fig. 2, HAECs exhibited alignment shapes, which is more physiological, following HSS with PA treatment. However, the morphology of the HAECs were shaped more polygonally, which is more pathophysiological, in response to LSS with PA treatment (Fig. 2).
Figure 1.
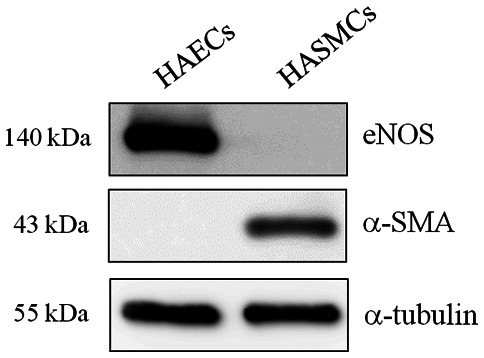
Identification of human aortic endothelial cells. Endothelial nitric oxide synthase, an endothelial cell maker, was used to identify human aortic endothelial cells. α-SMA was used as the smooth muscle cell marker. Human aortic smooth muscle cells served as a negative control. eNOS, endothelial nitric oxide synthase; α-SMA, α-smooth muscle actin; HAECs, human aortic endothelial cells; HASMCs, human aortic smooth muscle cells.
Figure 2.

Representative images of human aortic endothelial cells in response to HSS or LSS with PA treatment for 20 h. Cells cultured (A) under normal static conditions, (B) in response to HSS with PA treatment and (C) in response to LSS with PA treatment. Magnification, x100. HSS, high shear stress; LSS, low shear stress; PA, palmitic acid.
Laminar shear stress in PA-treated HAECs induces PPAR-α expression
The present study next assessed the effects of flow-induced shear stress and high PA concentrations on the expression of PPARα in HAECs. A laminar flow model with pathological LSS (4 dyn/cm2) or physiological HSS (12 dyn/cm2) (27) was used to assess HAECs treated with 300 µM PA. There was no difference between control and PA treatment in the static group (Fig. 3). To investigate the role of shear stress on PA-treated HAECs, cells were exposed to either LSS or HSS for 20 h in the presence of PA. Although the expression of PPARα was similar in PA-untreated HAECs between the LSS and HSS groups, they remained significantly higher compared with those in PA-untreated HAECs in the static group (Fig. 3). PPARα expression in the PA-treated HAECs of the LSS and HSS groups was significantly higher compared with that in their corresponding PA-untreated HAECs within the same groups (Fig. 3). Among the three PA-treated groups, PPAR-α expression was significantly higher after LSS and HSS compared with that in static conditions (Fig. 3). These results suggest that PA-treatment in HAECs subjected to LSS and HSS induces the expression of PPAR-α.
Figure 3.

Effects of PA and shear stress on PPARα expression in human aortic endothelial cells. Human aortic endothelial cells were exposed to either static conditions, low shear stress (4 dyn/cm2) or high shear stress (12 dyn/cm2) in the presence or absence of 300 µM PA for 20 h. Cells treated with 10% bovine serum albumin represented the solvent control. The protein expression of PPARα was determined by western blot analysis. Data are presented as the mean ± SEM from ≥ three independent experiments. *P<0.05. PPARα, peroxisome proliferator-activated receptor-α; HSS, high shear stress; LSS, low shear stress; PA, palmitic acid; con, control.
PA-treated HAECs exposed to HSS exhibit high PPAR-δ expressions
To investigate the effects of the application of shear stress to HAECs exposed to a high concentration of PA on PPARδ expression, HAECs were subjected to static conditions, LSS or HSS in the presence or absence of high PA concentrations for 20 h. PA-treated HAECs exhibited significantly higher PPAR-δ expression compared with that in PA-untreated HAECs, irrespective of whether the HAECs were maintained under static conditions or subjected to LSS or HSS (Fig. 4). However, PA-treated HAECs under HSS exhibited significantly higher expression levels of PPARδ compared with those in PA-treated HAECs under static or LSS conditions (Fig. 4). These results indicated that only PA treatment sufficiently increased PPARδ expression. However, among the cell groups that were not treated with PA, HAECs that were exposed to HSS exhibited higher levels of PPAR-d expression compared with that in HAECs under static or LSS conditions (Fig. 4).
Figure 4.

Effects of PA and shear stress on PPARδ expression in human aortic endothelial cells. Western blotting results demonstrating PPARδ expression in human aortic endothelial cells that were subjected to static conditions, low shear stress (4 dyn/cm2) or high shear stress (12 dyn/cm2) in the presence or absence of 300 µM PA for 20 h. Cells treated with 10% bovine serum albumin represented the solvent control. PPARδ protein expression was determined by western blot analysis. Data are presented as the mean ± SEM from ≥ three independent experiments. *P<0.05. PPARδ, peroxisome proliferator-activated receptorδ; HSS, high shear stress; LSS, low shear stress; PA, palmitic acid; con, control.
Laminar HSS applied to PA-treated HAECs increases PPARγ expression
Since it was previously demonstrated that shear stress induced PPARα and PPARg expression in PA-treated HAECs, the present study subsequently investigated the effects of shear stress applied to PA-treated HAECs on PPARγ expression. PPARγ expression in PA-treated HAECs was lower compared with that in PA-untreated HAECs in both the static and LSS groups, but neither of these observations were significant (Fig. 5). In cells exposed to HSS, PA treatment significantly increased PPARγ expression (Fig. 5). By contrast, HAECs exposed to HSS significantly increased the expression of PPAR-γ compared with that in cells in static and LSS groups, regardless of whether PA was present (Fig. 5). These results suggest that HSS increased PPARγ expression in HAECs, particularly after PA treatment.
Figure 5.

Effects of PA and shear stress on PPARγ expression in human aortic endothelial cells. Western blotting results demonstrating PPARγ expression in human aortic endothelial cells that were subjected to static conditions, low shear stress (4 dyn/cm2) or high shear stress (12 dyn/cm2) in the presence or absence of 300 µM PA for 20 h. Cells treated with 10% bovine serum albumin represented the solvent control. PPARγ protein expression was determined by western blot analysis. Data are presented as the mean ± SEM from ≥ three independent experiments. *P<0.05. PPARγ, peroxisome proliferator-activated receptor-γ; HSS, high shear stress; LSS, low shear stress; PA, palmitic acid; con, control.
PGI2 is produced by PA-treated HAECs in the presence of shear stress
PGI2 is a well-known vasoprotector factor produced by ECs under shear stress and serves as a ligand for PPARα and δ (19). The present study therefore investigated the effects of PA treatment on HAEC PGI2 production in response to shear stress. The levels of 6-keto-PGF1α, a stable metabolite of PGI2, in the media of shear stress-stimulated HAECs were significantly higher compared with those in the HAECs of static control both in the absence and presence of PA treatment (Fig. 6). These findings suggest that the application of shear stress to HAECs increases the secretion of PGI2 in the presence of PA.
Figure 6.
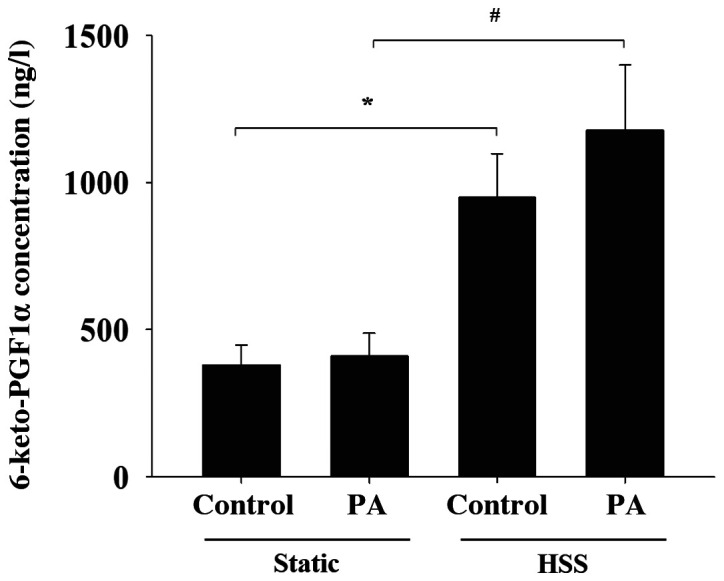
Effects of PA and shear stress on prostacyclin secretion by human aortic endothelial cells. Concentration of the prostacyclin stable metabolite 6-keto-PGF1α was determined in the conditioned media obtained from human aortic endothelial cells that were subjected to static conditions or high shear stress, in the presence or absence of 300 µM PA for 20 h. The concentration of 6-keto-PGF1α was measured using ELISA. Data are presented as the mean ± SEM from ≥ three independent experiments. *P<0.05 and #P<0.05 cells. PA, palmitic acid; con, control; HSS, high shear stress; PGF1α, prostaglandin 1α.
HSS inhibits PA-induced HAEC inflammation
EC inflammation-induced dysfunction as a result of high FFA concentrations is a risk factor for the development of cardiovascular diseases (7,8). To investigate the effects of HSS on PA-induced EC inflammation, HAECs were subjected to HSS for 20 h, after which the expression levels of inflammatory markers ICAM-1 and MCP-1(1) were examined by western blot analysis. High PA concentrations caused significant increases in the expression of inflammatory markers ICAM-1 and MCP-1 under static conditions (Fig. 7). Furthermore, when the ECs were exposed to long-term HSS, PA-induced protein expression of ICAM-1 and MCP-1 was decreased significantly. These results suggest that HSS exerted protective effects against PA-induced EC inflammation.
Figure 7.

HSS suppresses PA-induced endothelial cell inflammation. Human aortic endothelial cells were conditioned with 300 µM PA and subjected to HSS (12 dyn/cm2) for 20 h. Intercellular adhesion molecule-1 and monocyte chemoattractant protein-1 protein expression was determined by western blot analysis. Data are presented as the mean ± SEM from ≥ three independent experiments. *P<0.05 and #P<0.05. HSS, high shear stress; PA, palmitic acid; con, control; ICAM-1, intercellular adhesion molecule-1; MCP-1, monocyte chemoattractant protein-1.
Discussion
PA is a 16-carbon saturated long chain fatty acid that is the main constituent of FFAs frequently observed in metabolic disorders (28). In healthy individuals, low serum concentrations of FFAs are maintained (28). However, concentrations can reach up to 1,000-fold higher compared with that in healthy individuals in patients with diabetes (28). Elevated plasma FFAs cause metabolic stress, which has been revealed to disrupt EC function (29). Excessive serum FFAs has been reported to cause or facilitate EC dysfunction by inducing inflammation and lipid peroxidation, which are associated oxidative stress (9,29). In particular, three isoforms of PPAR are implicated in various physiological processes, including the suppression of inflammation and oxidative stress (30). It has been demonstrated that laminar fluid shear stress and PPARγ serve key roles in the regulation of EC function (20). However, their role in the presence of high concentrations of FFA remains unclear. The present study treated HAECs with PA to mimic metabolic imbalance and serve as a model of metabolic stress. To the best of our knowledge, the present study was the first to investigate the effects of varying levels of shear stress on the expression of PPAR in HAECs when combined with PA. The results revealed that PA-treated HAECs expressed PPARα, PPARδ and PPARγ in response to shear stress.
It has been previously demonstrated that perfusion media obtained from shear-treated HAECs also induces PPAR ligand-binding activities in smooth muscle cells (19). Previous reports have also demonstrated that the application of shear stress to ECs can induce the expression of all three isoforms of PPAR proteins and the release of PPAR ligands (20,31). A recent study revealed that activating PPARα and PPARγ ameliorated total FFA accumulation in macrophages (32). The results of the present study further demonstrated that all three PPAR isoforms were induced in HAECs that were treated with HSS and high concentrations of PA. These findings suggest that activating PPAR signaling may be a promising therapeutic strategy for the treatment of metabolic stress-induced vascular complications. Although the present results demonstrated that HSS serves an important role in the regulation of PPAR expression under PA-induced metabolic stress, additional experiments are required to examine the associated mechanisms involved in this phenomenon.
HSS can suppress inflammatory responses by inhibiting ROS and control the release of anti-inflammatory factors from ECs (27). The present study revealed that the PPARδ and PPARγ expression profiles were similar in HAECs that were subjected to static and LSS conditions in the presence of PA. However, HSS was observed to increase PPARδ and PPARγ protein expression regardless of the presence or absence of PA stimulation, suggesting that HSS alone can regulate the biosynthesis of these two PPAR isoforms under metabolic disturbance. These results may indicate the regulatory role of shear stress in HAECs under high PA concentrations. In the present study, in the presence of high PA concentrations, LSS-treated HAECs exhibited a similar magnitude of increased PPAR-α expression as that in HSS-treated HAECs. According to previous reports, PGI2 is one of the endogenous ligands of PPARα, where only high shear stress could result in high levels of PGI2 secretion whilst low shear stress could not (33,34). Therefore, it remains a possibility that low shear stress can increase the expression of PPARa through a PGI2-independent pathway. There is insufficient information regarding the effects of low shear stress on the expression of PPAR-α, regardless of PA stimulation, on endothelial cells. Nevertheless, it can be hypothesized that PPARα expression is more susceptible to shear stress. The molecular mechanism underlying these effects warrants additional investigation.
Consistent with a previous report (35), shear-stress treated HAECs in the present study released high levels of 6-keto-PGF1α into the perfusion medium. Additionally, HAECs conditioned under high PA concentrations following exposure to HSS produced higher levels of PGI2 in the perfusion media compared with those in the static PA-treated HAECs. PGI2 is an important vasoprotective factor that has been previously characterized as an anti-inflammatory agent, antioxidant and a potent vasodilator (36,37). Previous reports have demonstrated that reductions in PGI2 bioavailability contributes to EC dysfunction (38,39). Therefore, it was postulated that increased PGI2 biosynthesis in shear stress-treated HAECs may serve a protective effect on cells that are under PA-induced metabolic stress.
The results of the current study suggested strategies to promote HSS generated by the vessel wall. For example, continuous exercise may be potentially advantageous for preventing or ameliorating FFA-induced EC dysfunction. A limitation of the present study is that the potential regulatory mechanism underlying the induction of PPAR expression under shear stress and high PA concentrations was not elucidated. PPARs are critical factors in sculpting the metabolic phenotype in the vasculature (17,18). Additional functional experiments are required to determine whether PPARs exert vasoprotective effects against high FFA-induced EC dysfunction. The present study further demonstrated that HSS may serve an essential role by upregulating PPARs and releasing PGI2 to exert protective effects against high FFA-induced EC dysfunction.
In conclusion and to the best of our knowledge, the present study was the first to demonstrate that sustained HSS induced the expression of PPARs in HAECs during PA-induced metabolic stress. Metabolic stress is a critical risk factor for vascular disease and therefore results from the present study may provide a potential target for future therapeutic strategies.
Acknowledgements
Not applicable.
Funding Statement
Funding: The present study was supported by grants from the Cheng Hsin General Hospital-National Defense Medical Center cooperative research project (grant no. CH-NDMC-108-7), Chi Mei Medical Center-National Defense Medical Center cooperative research project (grant no. CM-NDMC-108-12) and Ministry of National Defense-Medical Affairs Bureau (grant nos. MAB-106-102 and MAB-109-069).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
MCT and YLW performed the experiments. MCT and YLW conceived and designed the present study. YLW, CTC and MCT acquired and analyzed the data, and drafted the manuscript. YLW, CTC and MCT revised and edited the manuscript and CTC performed the additional experiments of Figs. 1 and 2. CST conceived the current study and interpreted the data. YLW, CTC, CST and MCT contributed substantially to the manuscript preparation. All authors discussed the results, analyzed the data and commented on the manuscript. All authors read and gave the final approval of this manuscript. MCT and YLW confirm the authenticity of all the raw data.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Morigi M, Zoja C, Figliuzzi M, Foppolo M, Micheletti G, Bontempelli M, Saronni M, Remuzzi G, Remuzzi A. Fluid shear stress modulates surface expression of adhesion molecules by endothelial cells. Blood. 1995;85:1696–1703. [PubMed] [Google Scholar]
- 2.Surapisitchat J, Hoefen RJ, Pi X, Yoshizumi M, Yan C, Berk BC. Fluid shear stress inhibits TNF-alpha activation of JNK but not ERK1/2 or p38 in human umbilical vein endothelial cells: Inhibitory crosstalk among MAPK family members. Proc Natl Acad Sci USA. 2001;98:6476–6481. doi: 10.1073/pnas.101134098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mun GI, An SM, Park H, Jo H, Boo YC. Laminar shear stress inhibits lipid peroxidation induced by high glucose plus arachidonic acid in endothelial cells. Am J Physiol Heart Circ Physiol. 2008;295:H1966–H1973. doi: 10.1152/ajpheart.00727.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boden G. Free fatty acids, insulin resistance, and type 2 diabetes mellitus. Proc Assoc Am Physicians. 1999;111:241–248. doi: 10.1046/j.1525-1381.1999.99220.x. [DOI] [PubMed] [Google Scholar]
- 5.Liu L, Li Y, Guan C, Li K, Wang C, Feng R, Sun C. Free fatty acid metabolic profile and biomarkers of isolated post-challenge diabetes and type 2 diabetes mellitus based on GC-MS and multivariate statistical analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:2817–2825. doi: 10.1016/j.jchromb.2010.08.035. [DOI] [PubMed] [Google Scholar]
- 6.Yi LZ, He J, Liang YZ, Yuan DL, Chau FT. Plasma fatty acid metabolic profiling and biomarkers of type 2 diabetes mellitus based on GC/MS and PLS-LDA. FEBS Lett. 2006;580:6837–6845. doi: 10.1016/j.febslet.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh A, Gao L, Thakur A, Siu PM, Lai CWK. Role of free fatty acids in endothelial dysfunction. J Biomed Sci. 2017;24(50) doi: 10.1186/s12929-017-0357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pankow JS, Duncan BB, Schmidt MI, Ballantyne CM, Couper DJ, Hoogeveen RC, Golden SH. Fasting plasma free fatty acids and risk of type 2 diabetes: The atherosclerosis risk in communities study. Diabetes Care. 2004;27:77–82. doi: 10.2337/diacare.27.1.77. Atherosclerosis Risk in Communities Study. [DOI] [PubMed] [Google Scholar]
- 9.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 10.Su J, Tian H, Liu R, Liang J. Inhibitive effects of glucose and free fatty acids on proliferation of human vascular endothelial cells in vitro. Chin Med J (Engl) 2002;115:1486–1490. [PubMed] [Google Scholar]
- 11.Trombetta A, Togliatto G, Rosso A, Dentelli P, Olgasi C, Cotogni P, Brizzi MF. Increase of palmitic acid concentration impairs endothelial progenitor cell and bone marrow-derived progenitor cell bioavailability: Role of the STAT5/PPARγ transcriptional complex. Diabetes. 2013;62:1245–1257. doi: 10.2337/db12-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song X, Yang B, Qiu F, Jia M, Fu G. High glucose and free fatty acids induce endothelial progenitor cell senescence via PGC-1α/SIRT1 signaling pathway. Cell Biol Int. 2017;41:1146–1159. doi: 10.1002/cbin.10833. [DOI] [PubMed] [Google Scholar]
- 13.Legrand-Poels S, Esser N, L'homme L, Scheen A, Paquot N, Piette J. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem Pharmacol. 2014;92:131–141. doi: 10.1016/j.bcp.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 14.Qi Y, Du X, Yao X, Zhao Y. Vildagliptin inhibits high free fatty acid (FFA)-induced NLRP3 inflammasome activation in endothelial cells. Artif Cells Nanomed Biotechnol. 2019;47:1067–1074. doi: 10.1080/21691401.2019.1578783. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Chen Y, Li X, Zhang Y, Gulbins E, Zhang Y. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget. 2016;7:73229–73241. doi: 10.18632/oncotarget.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 17.Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm Res. 2000;49:497–505. doi: 10.1007/s000110050622. [DOI] [PubMed] [Google Scholar]
- 18.Marx N, Duez H, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors and atherogenesis: Regulators of gene expression in vascular cells. Circ Res. 2004;94:1168–1178. doi: 10.1161/01.RES.0000127122.22685.0A. [DOI] [PubMed] [Google Scholar]
- 19.Tsai MC, Chen L, Zhou J, Tang Z, Hsu TF, Wang Y, Shih YT, Peng HH, Wang N, Guan Y, et al. Shear stress induces synthetic-to-contractile phenotypic modulation in smooth muscle cells via peroxisome proliferator-activated receptor alpha/delta activations by prostacyclin released by sheared endothelial cells. Circ Res. 2009;105:471–480. doi: 10.1161/CIRCRESAHA.109.193656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Zhu Y, Rannou F, Lee TS, Formentin K, Zeng L, Yuan X, Wang N, Chien S, Forman BM, Shyy JY. Laminar flow activates peroxisome proliferator-activated receptor-gamma in vascular endothelial cells. Circulation. 2004;110:1128–1133. doi: 10.1161/01.CIR.0000139850.08365.EC. [DOI] [PubMed] [Google Scholar]
- 21.Ge X, Pan P, Jing J, Hu X, Chen L, Qiu X, Ma R, Jueraitetibaike K, Huang X, Yao B. Rosiglitazone ameliorates palmitic acid-induced cytotoxicity in TM4 Sertoli cells. Reprod Biol Endocrinol. 2018;16(98) doi: 10.1186/s12958-018-0416-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Xie X, Yao Q, Liu J, Tian Y, Yang C, Xiao L, Wang N. PPARδ agonist prevents endothelial dysfunction via induction of dihydrofolate reductase gene and activation of tetrahydrobiopterin salvage pathway. Br J Pharmacol. 2019;176:2945–2961. doi: 10.1111/bph.14745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiu JJ, Lee PL, Chen CN, Lee CI, Chang SF, Chen LJ, Lien SC, Ko YC, Usami S, Chien S. Shear stress increases ICAM-1 and decreases VCAM-1 and E-selectin expressions induced by tumor necrosis factor-(alpha) in endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:73–79. doi: 10.1161/01.ATV.0000106321.63667.24. [DOI] [PubMed] [Google Scholar]
- 24.Chiu JJ, Chen LJ, Chang SF, Lee PL, Lee CI, Tsai MC, Lee DY, Hsieh HP, Usami S, Chien S. Shear stress inhibits smooth muscle cell-induced inflammatory gene expression in endothelial cells: Role of NF-kappaB. Arterioscler Thromb Vasc Biol. 2005;25:963–969. doi: 10.1161/01.ATV.0000159703.43374.19. [DOI] [PubMed] [Google Scholar]
- 25.Chiu JJ, Chen LJ, Lee CI, Lee PL, Lee DY, Tsai MC, Lin CW, Usami S, Chien S. Mechanisms of induction of endothelial cell E-selectin expression by smooth muscle cells and its inhibition by shear stress. Blood. 2007;110:519–528. doi: 10.1182/blood-2006-08-040097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang SF, Chang CA, Lee DY, Lee PL, Yeh YM, Yeh CR, Cheng CK, Chien S, Chiu JJ. Tumor cell cycle arrest induced by shear stress: Roles of integrins and Smad. Proc Natl Acad Sci USA. 2008;105:3927–3932. doi: 10.1073/pnas.0712353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91:327–387. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vessby B. Dietary fat, fatty acid composition in plasma and the metabolic syndrome. Curr Opin Lipidol. 2003;14:15–19. doi: 10.1097/00041433-200302000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Guerci B, Böhme P, Kearney-Schwartz A, Zannad F, Drouin P. Endothelial dysfunction and type 2 diabetes. Part 2: Altered endothelial function and the effects of treatments in type 2 diabetes mellitus. Diabetes Metab. 2001;4:436–447. [PubMed] [Google Scholar]
- 30.Ndisang JF. Cross-talk between heme oxygenase and peroxisome proliferator-activated receptors in the regulation of physiological functions. Front Biosci (Landmark Ed) 2014;19:916–935. doi: 10.2741/4257. [DOI] [PubMed] [Google Scholar]
- 31.Taba Y, Sasaguri T, Miyagi M, Abumiya T, Miwa Y, Ikeda T, Mitsumata M. Fluid shear stress induces lipocalin-type prostaglandin D(2) synthase expression in vascular endothelial cells. Circ Res. 2000;86:967–973. doi: 10.1161/01.res.86.9.967. [DOI] [PubMed] [Google Scholar]
- 32.Ye G, Gao H, Wang Z, Lin Y, Liao X, Zhang H, Chi Y, Zhu H, Dong S. PPARα and PPARγ activation attenuates total free fatty acid and triglyceride accumulation in macrophages via the inhibition of Fatp1 expression. Cell Death Dis. 2019;10(39) doi: 10.1038/s41419-018-1135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moraes LA, Piqueras L, Bishop-Bailey D. Peroxisome proliferator-activated receptors and inflammation. Pharmacol Ther. 2006;110:371–385. doi: 10.1016/j.pharmthera.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 34.Doroudi R, Gan LM, Selin Sjögren L, Jern S. Effects of shear stress on eicosanoid gene expression and metabolite production in vascular endothelium as studied in a novel biomechanical perfusion model. Biochem Biophys Res Commun. 2000;269:257–264. doi: 10.1006/bbrc.2000.2279. [DOI] [PubMed] [Google Scholar]
- 35.Frangos JA, Eskin SG, McIntire LV, Ives CL. Flow effects on prostacyclin production by cultured human endothelial cells. Science. 1985;227:1477–1479. doi: 10.1126/science.3883488. [DOI] [PubMed] [Google Scholar]
- 36.Vane J, Corin RE. Prostacyclin: A vascular mediator. Eur J Vasc Endovasc Surg. 2003;26:571–578. doi: 10.1016/s1078-5884(03)00385-x. [DOI] [PubMed] [Google Scholar]
- 37.Rolland PH, Jouve R, Pellegrin E, Mercier C, Serradimigni A. Alteration in prostacyclin and prostaglandin E2 production. Correlation with changes in human aortic atherosclerotic disease. Arteriosclerosis. 1984;4:70–78. doi: 10.1161/01.atv.4.1.70. [DOI] [PubMed] [Google Scholar]
- 38.Shimokawa H. Primary endothelial dysfunction: Atherosclerosis. J Mol Cell Cardiol. 1999;31:23–37. doi: 10.1006/jmcc.1998.0841. [DOI] [PubMed] [Google Scholar]
- 39.Di Francesco L, Totani L, Dovizio M, Piccoli A, Di Francesco A, Salvatore T, Pandolfi A, Evangelista V, Dercho RA, Seta F, Patrignani P. Induction of prostacyclin by steady laminar shear stress suppresses tumor necrosis factor-alpha biosynthesis via heme oxygenase-1 in human endothelial cells. Circ Res. 2009;104:506–513. doi: 10.1161/CIRCRESAHA.108.191114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.