

Abstract
Background
Up to 50% of lupus nephritis (LN) patients experience renal flares after their initial episode of LN. These flares contribute to poor renal outcomes. We postulated that intrarenal immune gene expression is different in flares compared with de novo LN, and conducted these studies to test this hypothesis.
Methods
Glomerular and tubulointerstitial immune gene expression was evaluated in 14 patients who had a kidney biopsy to diagnose LN and another biopsy at their first LN flare. Ten healthy living kidney donors were included as controls. RNA was extracted from laser microdissected formalin-fixed paraffin-embedded kidney biopsies. Gene expression was analyzed using the Nanostring nCounter® platform and validated by quantitative real-time polymerase chain reaction. Differentially expressed genes were analyzed by the Ingenuity Pathway Analysis and Panther Gene Ontology tools.
Results
Over 110 genes were differentially expressed between LN and healthy control kidney biopsies. Although there was considerable molecular heterogeneity between LN biopsies at diagnosis and flare, for about half the LN patients gene expression from the first LN biopsy clustered with the repeated LN biopsy. However, in all patients, a set of eight interferon alpha-controlled genes had a significantly higher expression in the diagnostic biopsy compared with the flare biopsy. In contrast, nine tumor necrosis factor alpha-controlled genes had higher expression in flare biopsies.
Conclusions
There is significant heterogeneity in immune-gene expression of kidney tissue from LN patients. There are limited but important differences in gene expression between LN flares, which may influence treatment decisions.
Keywords: gene expression, interferon signature, lupus nephritis, transcriptomics, tumor necrosis factor
INTRODUCTION
Lupus nephritis (LN) affects 40–60% of systemic lupus erythematosus (SLE) patients during the course of their disease [1, 2]. The development of kidney disease increases morbidity and mortality rates up to 26-fold compared with age-matched individuals from the general population [3]. With current therapies remission is achieved in only 50–70% of patients and ∼10–20% still progress to end-stage kidney disease over 5–10 years [4].
A contributing factor to poor kidney outcomes in LN is the high frequency of disease flare [5–7]. Up to 50% of LN patients experience renal flares after achieving complete or partial remission [8]. Based on current guidelines [9–11], these patients are usually treated with the same drug regimen that was successful in initially controlling disease activity. While this makes clinical sense, LN flares are often more resistant to treatment [12]. It is conceivable that the pathogenic mechanisms responsible for LN flare are different from those that were operating during the initial episode of disease activity, due in part to changes in the nature of LN with time, chronic changes in the kidney and/or alterations in patients’ immune systems by prior and ongoing immunosuppressive therapy. This study was undertaken to examine differences in intrarenal gene expression between the diagnosis and flare of LN.
MATERIALS AND METHODS
Experimental design
This investigation focused on patients who had a kidney biopsy to diagnose LN, and another biopsy when LN flared after treatment-induced remission. The transcriptomes of these paired biopsies were interrogated and compared to identify differences in intrarenal transcript expression between LN episodes. Such differences were assumed to reflect, in part, changes in disease pathogenesis over time and after exposure to immunosuppression.
Patients and kidney biopsy specimens
Fourteen Hispanic patients who had a kidney biopsy at the initial diagnosis of LN (biopsy A) and a second biopsy at LN flare (biopsy B) were included. All patients had a pathologic diagnosis of active proliferative or mixed LN (Classes III/IV ± V) on first and repeat biopsies. Several patients were immunosuppressive treatment-naïve at LN diagnosis (biopsy A) and most were receiving maintenance immunosuppression at LN flare (biopsy B).
Kidney biopsy tissue was available from archived paraffin blocks and clinical information was obtained from the prospective local LN registry. Normal control renal tissue was obtained from archived kidney biopsies of living-kidney donors (n = 10) obtained at implantation of the allograft from the same institution. The protocol was approved by the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán Research and Ethics board and The Ohio State University Institutional Review Board.
RNA extraction and analysis
Formalin-fixed and paraffin-embedded (FFPE) kidney biopsy tissue was cut into 10 μm sections. After deparaffinization, all available glomeruli and tubulointerstitium were separated by laser microdissection (PALM MicroBeam, Zeiss Labs, Munchen, Germany), captured and digested with proteinase K. From each biopsy, a median glomerular cross-sectional area of 1 970 000 μm2, corresponding to ≈50 glomerular cross-sections, and a tubulointerstitial area of 3 940 000 μm2 was collected. DNA was removed with DNase. RNA was precipitated and extracted with RNeasy MinElute spin columns (Qiagen, Redwood City, CA, USA) as previously described [13]. Gene transcript expression was analyzed using the Nanostring nCounter® platform and the nCounter Immunology Panel (Nanostring Technologies, Seattle, WA, USA) [14]. The panel consists of 579 immune response genes, 6 positive controls, 6 negative controls and 15 housekeeping genes [15].
Quantitative real-time polymerase chain reaction
cDNA was obtained from renal biopsy RNA of three randomly selected patients using a high-capacity RNA-to-cDNA kit (ThermoFisher Scientific Cat # 4387406), followed by Taqman gene expression assay (ThermoFisher Scientific, Waltham, MA, USA) with the following primers to validate differentially expressed genes: Promyelocytic Leukemia (PML, Hs00971694), Myxovirus Resistance 1 (MX1, Hs00895608), Signal Transducer And Activator Of Transcription 1 (STAT1, Hs01013996), Interferon Alpha 2 (IFNA2, Hs00265051), Fibronectin 1 (FN1, Hs01549976), Vascular Cell Adhesion Molecule 1 (VCAM1, Hs01003372), C-C Motif Chemokine Ligand 19 (CCL19, Hs00171149), Colony Stimulating Factor 1 Receptor (CSF1R, Hs00911250), C-C Motif Chemokine Ligand 2 (CCL2, Hs00234140). Glyceraldehyde-3-Phosphate Dehydrogenase (GADPH, Hs02758991), Ornithine Decarboxylase Antizyme 1 (OAZ1, Hs01548012) and Ribosomal Protein L19 (RPL19, Hs01577060) were included as housekeeping genes.
Statistical analysis
Descriptive statistics are given as median and interquartile range or as relative frequencies. For baseline comparisons, Fisher’s exact test, Chi-square or the Kruskal–Wallis test was applied as appropriate. For gene expression data, raw counts were normalized to the positive spiked-in controls and then log2 transformed. Genes with an expression level below the mean plus two standard deviations of the negative controls were filtered out. Quantile normalization was applied to the remaining transcripts (≈500 for glomeruli and tubulointerstitium). Comparisons between biopsies A and B, and between biopsies A and B and healthy controls were done using linear mixed effect models. For any specific gene to be considered differentially expressed, at least a 1.5-fold change in transcript expression and a P-value of 0.01 were required (false detection rate of 5 out of 500 genes). Differentially expressed genes were analyzed by the Ingenuity Pathway Analysis tool (Qiagen Inc., Germantwon, MD, USA, https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis) and by Panther Gene Ontology pathway tools [16, 17]. Ingenuity pathway analysis clusters differentially expressed genes into formal molecular pathways based on levels of transcript expression and the (currently) known informatics of gene–gene interactions. This is a tool to identify dysregulated pathways in a disease process.
RESULTS
Characteristics of the LN cohort
The demographic, clinical and histopathological characteristics of the patients and healthy controls are shown in Table 1. At initial diagnosis, patients had relatively normal serum creatinine concentrations, heavy proteinuria, high histologic activity and low chronicity on biopsy. Patients were generally treated with corticosteroids plus cyclophosphamide, and most achieved a complete renal response within a year. All patients had responded at least partially by 18 months. Between biopsies A and B, most patients (85.7%) were maintained with azathioprine and low-dose prednisone. At LN flare, patients had significantly impaired kidney function, greater complement consumption, and more histologic evidence of chronicity on biopsy, although the degree of proteinuria and histologic activity were similar to the initial episode of LN. At flare, patients were generally re-induced with corticosteroids plus mycophenolate mofetil, reflecting the changing global trends in the treatment of LN between initial diagnosis and flare. Complete renal response rates were diminished in the patients who flared compared with their initial episode.
Table 1.
Demographic and clinical data
| Biopsy A a n = 14 | Biopsy B b n = 14 | P | Normal controls | |
|---|---|---|---|---|
| Demographic | ||||
| Age at biopsy (years) | 26 ± 5c | 30 ± 7 | <0.001 | 32 ± 9 |
| Female (%) | 14 (100) | 14 (100) | 1.000 | 8 (80) |
| Hispanic (%) | 14 (100) | 14 (100) | 1.000 | 10 (100) |
| Delay from symptoms to biopsy (months) | 3 (1–5)d | 1 (0–2) | <0.001 | – |
| Time between biopsies A and B (years) | 3.5 (1.5–6.5)d | – | – | – |
| Laboratoryd | ||||
| Serum creatinine (μmol/L) | 80 (71–133) | 168 (97–256) | 0.011 | 62 (53–80) |
| 24 h-urine protein/creatinine (mg/mmol) | 475 (260–746) | 520 (260–836) | 0.667 | 0 (0–2.3) |
| ds-DNA antibodies (u/ul)e | 118 (34–602) | 123 (47–539) | 1.000 | – |
| C3 ratiof | 0.7 (0.6–1.3) | 0.4 (0.2–0.8) | 0.004 | – |
| C4 ratiof | 0.5 (0.3–1.2) | 0.5 (0.4–1.0) | 0.280 | – |
| Histopathology | ||||
| Class III (%) | 2 (14.5) | 0 (0) | 0.481 | – |
| Class IV (%) | 4 (28.5) | 3 (21.4) | 1.000 | – |
| Class III+V (%) | 4 (28.5) | 2 (14.3) | 0.648 | |
| Class IV+V (%) | 4 (28.5) | 9 (64.3) | 0.128 | |
| Activity scored | 7 (4–9) | 8 (3–11) | 0.734 | – |
| Chronicity scored | 2 (2–4) | 5 (3–7) | 0.016 | – |
| Treatment history before biopsy A | ||||
| No immunosuppression | 6 (42.9) | – | – | – |
| Corticosteroids <10 mg | 6 (42.9) | – | – | – |
| Antimalarial | 4 (28.6) | – | – | – |
| Induction treatmentg | ||||
| Corticosteroids (%) | 14 (100) | 14 (100) | 1.000 | – |
| Mycophenolate mofetil (%) | 3 (21.4) | 10 (71.4) | 0.021 | – |
| Cyclophosphamide (%) | 8 (57.2) | 4 (28.6) | 0.252 | – |
| Azathioprine (%) | 3 (21.4) | 0 (0) | 0.222 | – |
| Maintenance treatment between biopsies A and B | ||||
| Azathioprine (%) | 12 (85.7) | – | – | – |
| Mycophenolate mofetil (%) | 2 (14.3) | – | – | – |
| Corticosteroids (%) | 12 (85.7) | – | – | – |
| Antimalarial (%) | 6 (42.9) | – | – | – |
| 12-month responseh | ||||
| Complete response (%) | 9 (64) | 3 (21) | 0.054 | – |
| Partial response (%) | 2 (14) | 4 (29) | 0.648 | – |
| No response (%)i | 3 (21) | 7 (50) | 0.236 | – |
Biopsy A was done at initial LN diagnosis.
Biopsy B was done at LN flare.
Mean ± standard deviation.
Data presented as median and interquartile range.
Normal ds-DNA is <9.6 UI/mL.
C3 and C4 ratios were calculated as the reported value divided by the lower limit of the reference level.
All patients were also given high-dose corticosteroids.
Complete response is defined as stable renal function (within 15% of baseline) plus 24 h-uPCR <0.5 g/g; partial response is defined as stable renal function plus 50% reduction of baseline 24 h-uPCR; no response indicates a patient did not achieve complete or partial response by 12 months, but responded later.
These patients achieved response beyond 12 months.
Molecular heterogeneity of LN
A principal component analysis of transcript expression for the glomerular and tubulointerstitial compartments from biopsies A and B is shown in Figure 1. Glomerular and tubulointerstitial transcripts from healthy kidneys clustered and separated from LN groups in both compartments. In contrast, transcript expression from LN biopsies (A and B) did not cluster well, revealing the molecular heterogeneity of LN. However, in about half the LN cases glomerular and tubulointerstitial transcript expression from biopsy A clustered with biopsy B. Many of the same patients whose A and B biopsies clustered from the glomerular compartment also clustered from the tubulointerstitial compartment. There were no differences in clinical or histological parameters between patients with concordant gene expression between biopsies A and B when compared with patients with discordant gene expression (data not shown).
FIGURE 1.

Principal component analysis of immune gene expression in glomeruli and tubulointerstitium. In both compartments healthy controls clustered separately from LN biopsies. In contrast, transcript expression in LN did not cluster, but for some individual patients biopsy A (open triangles) and biopsy B (filled triangles) immune gene expression clustered closely.
Immune gene expression patterns in LN and healthy controls
Compared with healthy tissue, biopsies A and B showed 139 and 119 differentially regulated glomerular transcripts, respectively. Most of these transcripts clustered into similar pathways in both biopsies. A list of the top canonical pathways differentially expressed in LN compared with healthy controls is shown in Supplementary data, Table S1. Based on the enrichment P-value, the enriched pathways included interleukin-6 (IL-6) and lipopolysaccharide (LPS)-stimulated pathways, nuclear factor (NF)-κB activation pathway, leukocyte recruitment and extravasation, T-cell signaling [Th1 pathway, Protein Kinase C-q, Tec kinase and inducible T cell costimulator- inducible T cell costimulator ligand (iCOS-iCOSL) signaling] and B-cell signaling [CD28 co-stimulation pathway and Phosphatidylinositol-4,5-biphosphate 3-kinase (PI3K) signaling pathway].
In the tubulointerstitial compartment there were 140 and 135 differentially expressed transcripts between healthy tissue and biopsies A and B, respectively. As for glomeruli, these transcripts clustered into similar pathways in both biopsies. A list of the top canonical pathways that differed between LN and control is provided in Supplementary data, Table S2. Based on the enrichment P-value, pathways enriched in the tubulointerstitial compartment of LN kidneys included the complement system, Nuclear Factor kappa B (NF-kB) activation pathway, B-cell activation pathways (B-cell receptor signaling, PI3K signaling, OX40 signaling and B-cell activating factor (BAFF) signaling), acute phase response (IL-6 signaling), apoptosis signaling, co-stimulation (CD40 signaling) and T-cell activation (CD28 signaling), among others.
Many toll-like receptor (TLR) pathway transcripts were upregulated in the glomeruli [Toll-like Receptor 7 (TLR7), Fc Fragment of IgG Receptor IIa (FCGR2A), Myeloid Differentiation Primary Response 88 (MYD88), Interleukin 1 Receptor Associated Kinase 1 (IRAK1), Interferon Regulatory Factor 7 (IRF7) and Secreted Phosphoprotein 1 (SPP1 or osteopontin)] and tubulointerstitium (TLR7, FCGR2A and MYD88) of LN biopsies compared with healthy controls (Supplementary data, Figure S1). There were no differences in TLR9 expression between LN and controls in either compartment, whereas TLR3, TLR4 and TLR5 were significantly downregulated in glomeruli from LN patients compared with controls.
Differences in immune gene expression between episodes of active LN
In the glomerular compartment, 23 genes were differentially expressed between biopsies A and B. Eight genes were higher in biopsy A, including five transcripts regulated by interferon alpha (IFNα): MX1, STAT1, IRF7, PML, major histocompatibility complex, Class I, A (HLA-A), and CCL19, a chemokine induced by IFNα. With the exception of HLA-A, these genes were also upregulated in biopsy A when compared with controls. In contrast, none of these genes was differentially expressed in biopsy B compared with controls (Figures 2 and 3). All these transcripts clustered into the type I IFN signaling pathway [gene ontology (GO) fold-enrichment 70.2, P = 7.80 × 10−05] (Table 2).
FIGURE 2.
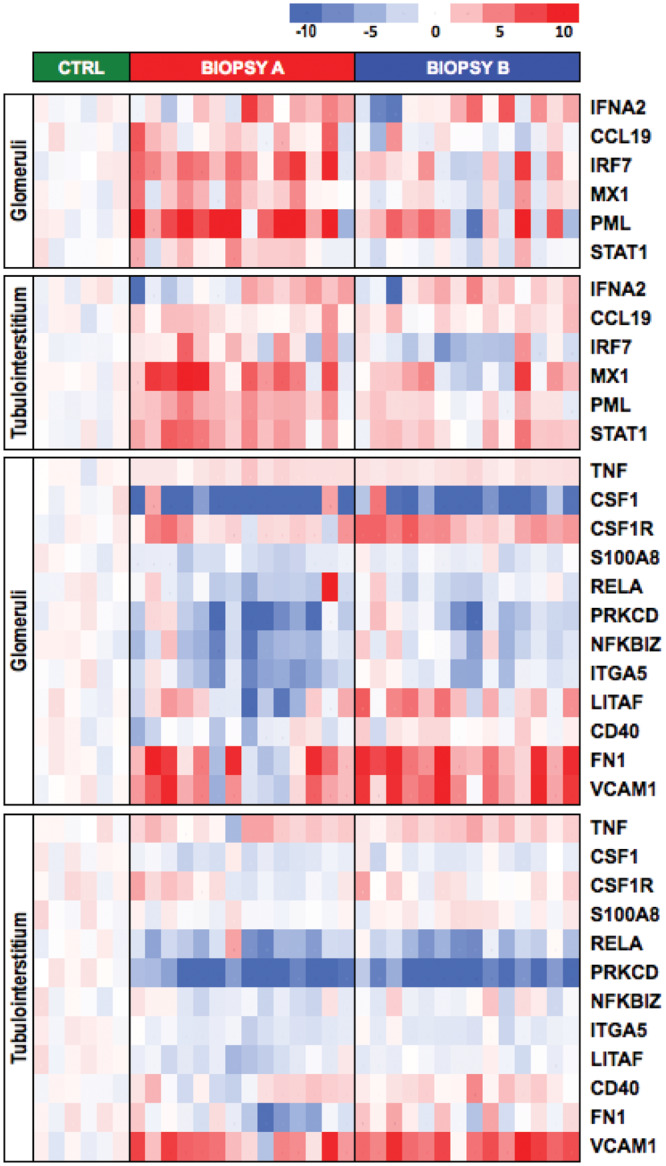
IFNα- and TNFα-regulated genes differentially expressed between biopsy A or B and normal controls. Genes are listed in the heatmap using z-score of normalized gene expression level. Each column is an individual patient or healthy control. Column 1 in biopsy A and column 1 in biopsy B correspond to the same patient.
FIGURE 3.

Glomerular immune gene expression from LN patients. (A) Volcano plot showing gene expression in biopsies A and B. A value of 2.0 on the ordinate corresponds to a P-value of 0.01 and a value of ±1.0 on the abscissa corresponds to a 1.5 fold-change in expression. (B) Ingenuity Pathway Analysis showing IFNA2 as a central upstream regulator of differentially expressed genes between biopsies A and B. Expression of IFNA2(C), PML(D), MX1(E) and CSF1R(F) measured by Nanostring (left graph) and by qPCR (right graph).
Table 2.
GO biological processes from differentially expressed genes between biopsies A and B in the glomerular compartment
| GO biological process | Ref. list | Hits | Fold enrichment | Biopsy A versus B | P |
|---|---|---|---|---|---|
| Complement activation, alternative pathway (C3, C7, CFH) | 13 | 3 | >100 | >B | 1.97 ×10−02 |
| IFNγ-mediated signaling pathway (HLA-A, HLA-DRB1, PML, VCAM1, PRKCD, STAT1, IRF3, IRF7, HLA-DRB3) | 71 | 9 | 66.6 | >A | 1.23 × 10−10 |
| Type I IFN signaling pathway (HLA-A, STAT1, IRF3, PML, MX1, IRF7) | 65 | 6 | 48.5 | >A | 1.42 × 10−02 |
| Negative regulation of leukocyte apoptotic process (CCL19, LILRB1, BCL6, IRF7) | 44 | 4 | 47.7 | >B | 1.42 × 10−03 |
| Regulation of immunoglobulin-mediated immune response (C3, LTA, CD40, BCL6) | 48 | 4 | 43.8 | >B | 2.00 × 10−02 |
| Regulation of IL12 production (RELA, CCL19, LILRB1, CD40) | 51 | 4 | 41.2 | >B | 2.54 × 10−02 |
| T cell co-stimulation (CCL19, HLA-DRB1, DPP4, ICOS, HLA-DRB3) | 82 | 5 | 32.0 | >A | 4.56 × 10−03 |
| PRR signaling pathway (RELA, MAPKAPK2, IRF3, IRF7, S100A8) | 122 | 5 | 21.5 | >A | 3.14 × 10−02 |
| Regulation of humoral immune response (C3, CFH, LTA, C1QB, C7) | 128 | 5 | 20.5 | >B | 3.96 × 10−02 |
| Positive regulation of IκB kinase/NF-κB signaling (RELA, CCL19, LITAF, CD40, HLA-DRB1, TRAF5, IRF3) | 183 | 7 | 20.1 | >B | 4.73 × 10−04 |
Ref. list (reference list) indicates the number of genes in a particular biological pathway. Hits, indicates the number of differentially expressed genes included in the pathway. Fold enrichment represents the enrichment score that reflects the degree to which a set of differentially expressed genes is overrepresented in the biopsies A or B [18]. The P-value is the estimation of significance level of the enrichment score.
PRR, pattern recognition receptor.
Among the nine differentially expressed transcripts between biopsies A and B in the tubulointerstitial compartment, four were regulated by IFNα and had higher levels of expression in biopsy A: MX1, PML, STAT4 and IRF7 (Figures 2 and 4). Only MX1 and PML were differentially expressed between biopsy A and normal controls. All of these transcripts clustered into the type I IFN signaling pathway (GO-fold enrichment >100, P = 1.97 × 10−07) (Table 3).
FIGURE 4.

Tubulointerstitial immune gene expression from LN patients. (A) Volcano plot showing immune gene expression in biopsies A and B. A value of 2.0 on the ordinate corresponds to a P-value of 0.01 and a value of ±1.0 on the abscissa corresponds to a 1.5 fold-change in expression. (B) Ingenuity Pathway Analysis showing IFNA2 as a central upstream regulator of differentially expressed genes between biopsies A and B. Expression of IFNA2(C), PML(D), STAT1(E) and MX1(F) measured by Nanostring (left graph) and by qPCR (right graph).
Table 3.
GO biological functions from differentially expressed genes between biopsies A and B in the tubulointerstitium
| GO biological process | Ref. list | Hits | Fold enrichment | Biopsy A versus B | P |
|---|---|---|---|---|---|
| Regulation of type I IFN-mediated signaling pathway (IFNA1, IFNB1, STAT1, IRF7) | 42 | 4 | >100 | >A | 5.18 × 10−04 |
| Type I IFN signaling pathway (IFNA1, IFNB1, STAT1, EGR1, MX1, IRF7) | 65 | 6 | >100 | >A | 1.97 × 10−07 |
| Negative regulation of adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains (IL7R, IFNB1, IL4R) | 36 | 3 | 92.1 | >B | 4.09 × 10−02 |
| Negative regulation of viral life cycle (PML, IFNB1, LTF, MX1) | 75 | 4 | 59.0 | >A | 5.17 × 10−03 |
| Leukocyte proliferation (IL7R, KIT, IFNA1, IFNB1) | 88 | 4 | 50.2 | >B | 9.72 ×10−03 |
| Lymphocyte differentiation (IL7R, KIT, IFNA1, IFNB1, EGR1) | 221 | 5 | 25.0 | >B | 1.13 ×10−02 |
| T cell activation (IL7R, KIT, IFNA1, IFNB1, EGR1) | 226 | 5 | 24.5 | >B | 1.27 × 10−02 |
| Regulation of vasculature development(C3, KIT, PML, STAT1, EGR1) | 248 | 5 | 22.3 | >B | 1.99 × 10−02 |
Ref. list (reference list) indicates the number of genes in a particular biological pathway. Hits indicates the number of differentially expressed genes included in the pathway. Fold enrichment represents the enrichment score that reflects the degree to which a set of differentially expressed genes is overrepresented in the biopsies A or B [18]. The P-value is the estimation of significance level of the enrichment score.
In the glomerular compartment, 15 genes had higher expression in biopsy B than biopsy A. These included nine genes regulated by the tumor necrosis factor (TNF): Integrin Subunit Alpha 5 (ITGA5), NF-kB Inhibitor Zeta (NFKBIZ), CSF1R, RELA Proto-Oncogene (RELA, NF-kB subunit), Protein Kinase C Delta (PRKCD), Lypopolysaccharide Induced TNF Factor (LITAF), Colony Stimulating Factor 1 (CSF1) and S100 Calcium Binding Protein A8 (S100A8) and CD40. Most of these genes (ITGA5, NFKBIZ, RELA, PRKCD, CSF1 and S100A8) were also downregulated compared with healthy controls in biopsy A and were not different than healthy controls in biopsy B (Figure 2). These transcripts clustered into the TNF receptor binding pathway by GO analysis (GO-fold enrichment 24.4, P = 7.42 × 10−03). However, TNF gene expression itself was not different between biopsies A and B (GO-fold enrichment 1.07, P = 0.554). Other genes with higher expression in the glomeruli and tubulointerstitium of biopsy B than A included those for fibronectin (FN1) and vascular cell adhesion molecule (VCAM1).
With respect to TLR genes, only TLR7 (fold-change 1.48, P = 0.008) and IRF7 (GO-fold enrichment 1.65, P ≤ 0.001) were differentially expressed between biopsies A and B in glomeruli, and only IRF7 (fold-change 1.52, P = 0.003) in tubulointerstitium (Supplementary data, Figure S1). Transcript expression changes were similar when patients with Class III/IV LN were examined separately from patients with Class III/IV + V LN, suggesting that the presence of Class V did not affect the results (data not shown).
Quantitative real-time polymerase chain reaction validation
To validate the Nanostring findings quantitative real-time polymerase chain reaction (qPCR) was performed for selected genes using glomerular and tubulointerstitial RNA. IFNA2, PML, CCL19, CSF1R, VCAM1 and MX1 were evaluated in glomerular samples and IFNA2, PML, CCL19, MX1 and STAT1 evaluated in tubulointerstitial samples. Transcript expression by qPCR followed the same trends as Nanostring (examples shown in Figures 3 and 4).
DISCUSSION
When LN flares, the usual clinical approach to therapy has been to repeat the treatment regimen that had been successful in the past [9–11]. This approach may be suboptimal given the increased resistance to therapy observed in LN flares [12]. Mechanistically, it is conceivable that prior immunosuppression, along with chronic damage to the kidney and duration of LN alter the pathogenesis of disease flares compared with the initial episode of LN.
Using molecular interrogation of paired kidney biopsies from patients who flared after successful treatment of LN, we have shown that global intrarenal transcript expression from the flare biopsy is very similar to the transcriptome of the initial biopsy. We did, however, observe a few key differences. The IFNα signature was significantly attenuated in flare biopsies compared with initial biopsies. While this may not have much impact on non-specific immunosuppression with cyclophosphamide or mycophenolate mofetil, it may affect the response to targeted therapies like antibodies to type 1 IFN or the IFN receptor (e.g. Safety Efficacy of Two Doses of Anifrolumab Compared to Placebo in Adult Subjects With Active Proliferative Lupus Nephritis [TULIP-LN1] trial NCT02547922). Furthermore, a strong TNFα signature appeared in the flare biopsy that was not present in the first biopsy, supporting previous concepts that TNFα counterbalances IFNα in some autoimmune diseases [19–21]. These findings may be especially important for clinical trial design of novel anti-IFN therapeutics, as most trials enroll patients with active disease, regardless of whether the disease is de novo or established.
Type I IFNs (mainly IFNα and INFβ) are central players to both innate and adaptive immunity [22]. An intense upregulation of IFN-controlled genes has been described in active SLE patients [23–26], which is globally known as the ‘interferon signature’. The interferon signature locally occurs in the kidneys [27] and has been demonstrated to occur predominantly in mesangial [28, 29] and endothelial cells [30], where it has a pathogenic effect in animal models [31]. We found that IFNA2 expression and the interferon signature differ between LN flares, with a more intense expression in an initial episode compared with repeated ones. This response occurs in both glomerular and tubulointerstitial compartments. The patients included in this study were under minimal or no immunosuppression at LN diagnosis, whereas at flare they had received intense induction immunosuppression and were receiving maintenance therapy, including antimalarial agents that have been shown to decrease IFN response [32, 33]. These findings are supported by a recent study that showed lower MX1 protein staining in the kidneys of LN patients after immunosuppression compared with immunosuppression-naïve patients [34]. All this suggests that immunosuppression modifies the interferon signature and limits its expression at LN flare. Therefore, response to IFN-targeted therapies may be lower in patients with established LN who flare.
Contrasting with the interferon signature, we found that TNFα-controlled gene expression (‘TNFα signature’) was higher at flare than at diagnosis. The existence of a cross-regulation between TNFα and IFNα has been previously suggested to be a determinant in autoimmune diseases [19–21]. For example, an intense expression of TNFα is associated with the development of rheumatoid arthritis while an intense IFNα expression is associated with active SLE. The bridge between these two cytokines is supported by multiple reports of ‘lupus-like’ disease secondary either to IFN agents [35, 36] or TNF blockers [37, 38]. However, others have proposed using TNF blockers to treat lupus [39–41]. This ambiguous role of TNFα has been described in animal models of SLE where TNFα displays an initial immunoregulatory role by inhibiting the emergence of early autoreactivity [42–45], and a late pro-inflammatory role leading to end-organ damage [46–48]. Our data suggest TNF blockers may not be successful in de novo LN but may have a place in the treatment of LN flares.
Previous studies have emphasized the role of TLR signaling pathways in the pathogenesis of LN [49–51]. Nucleic acid immune complexes activate TLR in plasmacytoid dendritic cells, macrophages and B-cells mainly through TLR4, TLR7 and TLR9, which in turn activate the MyDosome that mediates the production of IFNα and other cytokines. We did not find differences in TLR4 and TLR9 gene expression in kidney tissue from LN patients compared with normal controls, whereas TLR7 and several transcripts involved in its activation pathway were highly upregulated. This suggests that TLR7 has the predominant TLR role in human LN pathogenesis both in the glomerular and tubulointerstitial compartments. In LN animal models, TLR7 is an essential mediator of kidney disease [52–54] and its overexpression accelerates renal damage [55], while its blockade [54] or knockout [56] diminishes damage. In contrast, dual inhibition of TLR9 and TLR7 has no additional effects beyond single TLR7 blockade [52], and TLR9 inhibition alone even increased damage in animal models [57]. Therefore, the TLR7 pathway may be more important in human proliferative LN.
VCAM1 has been studied as a potential LN biomarker in serum [58–60] and urine [61, 62]. We found that kidney VCAM1 expression increased between the initial and flare biopsy. This highlights the importance of determining the potential differences between LN flares when trying to identify disease biomarkers.
There are important limitations to this study. All patients were Hispanic and from a single center in Mexico, which may limit generalizability of the results to other races and ethnicities. Although the number of patients assessed was small, it must be emphasized that this is a rare disease and few SLE patients have had a kidney biopsy at diagnosis, responded to therapy and then had a repeat kidney biopsy at their first LN flare. While more patients would be desirable, our cohort provides an initial look into an important question that could affect therapy and has identified several candidate pathways for later directed investigation. The nCounter platform does not cover all transcripts as microarray or RNAseq do, but the advantage of this platform is the ability to work with FFPE tissues [63, 64]. Finally, due to the limited amount of tissue that can be extracted from samples, only a small number of key transcripts could be validated by qPCR.
In summary, we showed: (i) significant intrarenal immune gene expression heterogeneity among patients with proliferative LN; (ii) within a single patient, immune gene expression may be similar or different during subsequent disease flares; (iii) IFN expression and the interferon signature in glomeruli and the tubulointerstitium is higher during the initial episode of LN compared with disease flares, whereas the opposite occurs for TNFα-regulated genes; and (iv) these gene expression changes between LN flares can be used to inform the treatment of a flaring patient and suggest a way to enrich clinical trials of specific targeted therapies for patients likely to respond to the experimental therapeutic.
SUPPLEMENTARY DATA
Supplementary data are available at ndt online. The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and is accessible through GEO Series accession number GSE113342 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113342).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Luis E. Morales and Dr Bertha Cordova for their assistance with sample shipping.
FUNDING
J.M.M.-V. was supported by the International Society of Nephrology (ISN) Fellowship Program. This work was funded in part by the Strategic Pharma-Academic Research Consortium Funding Program (SPARC) CCTS grant 60047517 and UL1TR0001108 of the National Institutes of Health—National Center for Advancing Translational Sciences (NCATS) to B.H.R. Nanostring analysis was carried out at OSU Comprehensive Cancer Center in the Genomics Shared Resource, which is supported by National Cancer Institute (CCSG: P30CA016058).
AUTHORS’ CONTRIBUTIONS
J.M.M.-V., S.V.P. and B.H.R. contributed to conception and study design. N.U.-U. and J.M.M.-V contributed to biopsy handling and histopathological analysis. H.S., P.F., J.P.S., I.A. and J.M.M.-V contributed to laser microdissection, sample management, nCounter and PCR analyses. Analysis and interpretation of data were done by L.Y., J.Z., I.A., S.V.P., J.M.M.-V. and B.H.R. J.M.M.-V., S.V.P., I.A. and B.H.R contributed to drafting the manuscript and revising it.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
- 1. Alarcon GS, McGwin G, Petri M et al. Baseline characteristics of a multiethnic lupus cohort: PROFILE. Lupus 2002; 11: 402–402 [DOI] [PubMed] [Google Scholar]
- 2. Hanly JG, O'Keeffe AG, Su L et al. The frequency and outcome of lupus nephritis: results from an international inception cohort study. Rheumatology (Oxford) 2016; 55: 252–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mok CC, Kwok RCL, Yip PSF. Effect of renal disease on the standardized mortality ratio and life expectancy of patients with systemic lupus erythematosus. Arthritis Rheum 2013; 65: 2154–2160 [DOI] [PubMed] [Google Scholar]
- 4. Tektonidou MG, Dasgupta A, Ward MM. Risk of end-stage renal disease in patients with lupus nephritis, 1971–2015. A systematic review and Bayesian meta-analysis. Arthritis Rheumatol 2016; 68: 1432–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parikh SV, Nagaraja HN, Hebert L et al. Renal flare as a predictor of incident and progressive CKD in patients with lupus nephritis. Clin J Am Soc Nephrol 2014; 9: 279–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mosca M, Bencivelli W, Neri R et al. Renal flares in 91 SLE patients with diffuse proliferative glomerulonephritis. Kidney Int 2002; 61: 1502–1509 [DOI] [PubMed] [Google Scholar]
- 7. Moroni G, Quaglini S, Gallelli B et al. The long-term outcome of 93 patients with proliferative lupus nephritis. Nephrol Dial Transplant 2007; 22: 2531–2539 [DOI] [PubMed] [Google Scholar]
- 8. Morris HK, Canetta PA, Appel GB. Impact of the ALMS and MAINTAIN trials on the management of lupus nephritis. Nephrol Dial Transplant 2013; 28: 1371–1376 [DOI] [PubMed] [Google Scholar]
- 9. Bertsias GK, Tektonidou M, Amoura Z et al. Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of adult and paediatric lupus nephritis. Ann Rheum Dis 2012; 71: 1771–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hahn BH, McMahon MA, Wilkinson A et al. American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res (Hoboken) 2012; 64: 797–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kidney Diseases Improving Global Outcomes (KDIGO) Clinical Practice Guideline for Glomerulonephritis. Kidney Int Suppl 2012; 2: 259–274 [Google Scholar]
- 12. Ioannidis JPA, Boki KA, Katsorida ME et al. Remission, relapse, and re-remission of proliferative lupus nephritis treated with cyclophosphamide. Kidney Int 2000; 57: 258–264 [DOI] [PubMed] [Google Scholar]
- 13. Parikh SV, Malvar A, Song H et al. Characterising the immune profile of the kidney biopsy at lupus nephritis flare differentiates early treatment responders from non-responders. Lupus Sci Med 2015; 2: e000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Geiss GK, Bumgarner RE, Birditt B et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 2008; 26: 317–325 [DOI] [PubMed] [Google Scholar]
- 15. www.nanostring.com/products/gene-expression-panels/ncounter-immunology-panels (3 May 2018, date last accessed)
- 16. Thomas PD. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res 2003; 13: 2129–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mi H, Poudel S, Muruganujan A et al. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res 2016; 44: D336–D342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Palucka AK, Blanck J-P, Bennett L et al. Cross-regulation of TNF and IFN- in autoimmune diseases. Proc Natl Acad Sci USA 2005; 102: 3372–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aringer M, Crow MK. A bridge between interferon-alpha and tumor necrosis factor in lupus. J Rheumatol 2008; 35: 1473–1476 [PubMed] [Google Scholar]
- 20. López de Padilla CM, Niewold TB. The type I interferons: basic concepts and clinical relevance in immune-mediated inflammatory diseases. Gene 2016; 576: 14–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anders H-J, Lichtnekert J, Allam R. Interferon-α and -β in kidney inflammation. Kidney Int 2010; 77: 848–854 [DOI] [PubMed] [Google Scholar]
- 22. Baechler EC, Batliwalla FM, Karypis G et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA 2003; 100: 2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Han GM, Chen SL, Shen N et al. Analysis of gene expression profiles in human systemic lupus erythematosus using oligonucleotide microarray. Genes Immun 2003; 4: 177–186 [DOI] [PubMed] [Google Scholar]
- 24. Kirou KA, Lee C, George S et al. Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum 2004; 50: 3958–3967 [DOI] [PubMed] [Google Scholar]
- 25. Chiche L, Jourde-Chiche N, Whalen E et al. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol 2014; 66: 1583–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peterson KS, Huang JF, Zhu J et al. Characterization of heterogeneity in the molecular pathogenesis of lupus nephritis from transcriptional profiles of laser-captured glomeruli. J Clin Invest 2004; 113: 1722–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Flür K, Allam R, Zecher D et al. Viral RNA induces type I interferon-dependent cytokine release and cell death in mesangial cells via melanoma-differentiation-associated gene-5. Am J Pathol 2009; 175: 2014–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Allam R, Lichtnekert J, Moll AG et al. Viral RNA and DNA trigger common antiviral responses in mesangial cells. J Am Soc Nephrol 2009; 20: 1986–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hagele H, Allam R, Pawar RD et al. Double-stranded RNA activates type I interferon secretion in glomerular endothelial cells via retinoic acid-inducible gene (RIG)-1. Nephrol Dial Transplant 2009; 24: 3312–3318 [DOI] [PubMed] [Google Scholar]
- 30. Triantafyllopoulou A, Franzke CW, Seshan SV et al. Proliferative lesions and metalloproteinase activity in murine lupus nephritis mediated by type I interferons and macrophages. Proc Natl Acad Sci USA 2010; 107: 3012–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. López P, Rodríguez-Carrio J, Suárez A. Antimalarial drugs inhibit IFNα-enhanced TNFα and STAT4 expression in monocytes: Implication for systemic lupus erythematosus. Cytokine 2014; 67: 13–20 [DOI] [PubMed] [Google Scholar]
- 32. An J, Minie M, Sasaki T et al. Antimalarial drugs as immune modulators: new mechanisms for old drugs. Annu Rev Med 2017; 68: 317–330 [DOI] [PubMed] [Google Scholar]
- 33. Shimizu Y, Yasuda S, Kimura T et al. Interferon-inducible Mx1 protein is highly expressed in renal tissues from treatment-naïve lupus nephritis, but not in those under immunosuppressive treatment. Mod Rheumatol 2017; 1–9 [DOI] [PubMed] [Google Scholar]
- 34. Sise ME, Wisocky J, Rosales IA et al. Lupus-like immune complex-mediated glomerulonephritis in patients with hepatitis C virus infection treated with oral, interferon-free, direct-acting antiviral therapy. Kidney Int Reports 2016; 1: 135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wilson LE, Widman D, Dikman SH et al. Autoimmune disease complicating antiviral therapy for hepatitis C virus infection. Semin Arthritis Rheum 2002; 32: 163–173 [DOI] [PubMed] [Google Scholar]
- 36. Ramos-Casals M, Roberto-Perez-Alvarez, Diaz-Lagares C et al. Autoimmune diseases induced by biological agents: a double-edged sword? Autoimmun Rev 2010; 9: 188–193 [DOI] [PubMed] [Google Scholar]
- 37. Pérez-De-Lis M, Retamozo S, Flores-Chávez A et al. Autoimmune diseases induced by biological agents. A review of 12, 731 cases (BIOGEAS Registry). Expert Opin Drug Saf 2017; 16: 1255–1271 [DOI] [PubMed] [Google Scholar]
- 38. Aringer M, Steiner G, Graninger WB et al. Effects of short-term infliximab therapy on autoantibodies in systemic lupus erythematosus. Arthritis Rheum 2007; 56: 274–279 [DOI] [PubMed] [Google Scholar]
- 39. Aringer M, Houssiau F, Gordon C et al. Adverse events and efficacy of TNF-α blockade with infliximab in patients with systemic lupus erythematosus: long-term follow-up of 13 patients. Rheumatology (Oxford) 2009; 48: 1451–1454 [DOI] [PubMed] [Google Scholar]
- 40. Matsumura R, Umemiya K, Sugiyama T et al. Anti-tumor necrosis factor therapy in patients with difficult-to-treat lupus nephritis: a prospective series of nine patients. Clin Exp Rheumatol 2009; 27: 416–421 [PubMed] [Google Scholar]
- 41. Jacob CO, McDevitt HO. Tumour necrosis factor-α in murine autoimmune ‘lupus’ nephritis. Nature 1988; 331: 356–358 [DOI] [PubMed] [Google Scholar]
- 42. Gordon C, Wofsy D. Effects of recombinant murine tumor necrosis factor-alpha on immune function. J Immunol 1990; 144: 1753–1758 [PubMed] [Google Scholar]
- 43. Zhou T, Edwards CK, Yang P et al. Greatly accelerated lymphadenopathy and autoimmune disease in lpr mice lacking tumor necrosis factor receptor I. J Immunol 1996; 156: 2661–2665 [PubMed] [Google Scholar]
- 44. Ernandez T, Mayadas TN. Immunoregulatory role of TNFalpha in inflammatory kidney diseases. Kidney Int 2009; 76: 262–276 [DOI] [PubMed] [Google Scholar]
- 45. Brennan DC, Yui MA, Wuthrich RP et al. Tumor necrosis factor and IL-1 in New Zealand Black/White mice. Enhanced gene expression and acceleration of renal injury. J Immunol 1989; 143: 3470–3475 [PubMed] [Google Scholar]
- 46. Yokoyama H, Kreft B, Kelley VR. Biphasic increase in circulating and renal TNF-alpha in MRL-lpr mice with differing regulatory mechanisms. Kidney Int 1995; 47: 122–130 [DOI] [PubMed] [Google Scholar]
- 47. Kontoyiannis D, Kollias G. Accelerated autoimmunity and lupus nephritis in NZB mice with an engineered heterozygous deficiency in tumor necrosis factor. Eur J Immunol 2000; 30: 2038–2047 [DOI] [PubMed] [Google Scholar]
- 48. Allam R, Anders HJ. The role of innate immunity in autoimmune tissue injury. Curr Opin Rheumatol 2008; 20: 538–544 [DOI] [PubMed] [Google Scholar]
- 49. Moresco EMY, LaVine D, Beutler B. Toll-like receptors. Curr Biol 2011; 21: R488–R493 [DOI] [PubMed] [Google Scholar]
- 50. Lorenz G, Lech M, Anders HJ. Toll-like receptor activation in the pathogenesis of lupus nephritis. Clin Immunol 2017; 185: 86–94 [DOI] [PubMed] [Google Scholar]
- 51. Pawar RD, Ramanjaneyulu A, Kulkarni OP et al. Inhibition of Toll-Like Receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol 2007; 18: 1721–1731 [DOI] [PubMed] [Google Scholar]
- 52. Savarese E, Steinberg C, Pawar RD et al. Requirement of toll-like receptor 7 for pristane-induced production of autoantibodies and development of murine lupus nephritis. Arthritis Rheum 2008; 58: 1107–1115 [DOI] [PubMed] [Google Scholar]
- 53. Kanno A, Tanimura N, Ishizaki M et al. Targeting cell surface TLR7 for therapeutic intervention in autoimmune diseases. Nat Commun 2015; 6: 6119. [DOI] [PubMed] [Google Scholar]
- 54. Subramanian S, Tus K, Li Q-Z et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci USA 2006; 103: 9970–9975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nickerson KM, Christensen SR, Shupe J et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol 2010; 184: 1840–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bossaller L, Christ A, Pelka K et al. TLR9 deficiency leads to accelerated renal disease and myeloid lineage abnormalities in pristane-induced murine lupus. J Immunol 2016; 197: 1044–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Spronk P, Bootsma H, Huitema M et al. Levels of soluble VCAM-1, soluble ICAM-1, and soluble E-selectin during disease exacerbations in patients with systemic lupus erythematosus (SLE); a long term prospective study. Clin Exp Immunol 2008; 97: 439–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yao G, Liu Z, Zhang X et al. Circulating thrombomodulin and vascular cell adhesion molecule-1 and renal vascular lesion in patients with lupus nephritis. Lupus 2008; 17: 720–726 [DOI] [PubMed] [Google Scholar]
- 59. Skeoch S, Haque S, Pemberton P et al. Cell adhesion molecules as potential biomarkers of nephritis, damage and accelerated atherosclerosis in patients with SLE. Lupus 2014; 23: 819–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wu T, Xie C, Wang HW et al. Elevated urinary VCAM-1, P-selectin, soluble TNF receptor-1, and CXC chemokine ligand 16 in multiple murine lupus strains and human lupus nephritis. J Immunol 2007; 179: 7166–7175 [DOI] [PubMed] [Google Scholar]
- 61. Singh S, Wu T, Xie C et al. Urine VCAM-1 as a marker of renal pathology activity index in lupus nephritis. Arthritis Res Ther 2012; 14: R164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Reis PP, Waldron L, Goswami RS et al. mRNA transcript quantification in archival samples using multiplexed, color-coded probes. BMC Biotechnol 2011; 11: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tam S, de Borja R, Tsao M-S et al. Robust global microRNA expression profiling using next-generation sequencing technologies. Lab Invest 2014; 94: 350–358 [DOI] [PubMed] [Google Scholar]
- 64. Subramanian A, Tamayo P, Mootha VK et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.