

Abstract
Cyclic AMP is a second messenger that is involved in a wide range of cellular and physiological activities. Several studies suggest that cAMP signals are compartmentalized, and that compartmentalization contributes to signaling specificity within the cAMP signaling pathway. The development of Fӧrster resonance energy transfer (FRET) based biosensors has furthered the ability to measure and visualize cAMP signals in cells. However, these measurements are often confined to two spatial dimensions, which may result in misinterpretation of data. To date, there have been only very limited measurements of cAMP signals in three spatial dimensions (x, y, and z), due to the technical limitations in using FRET sensors that inherently exhibit low signal to noise ratio (SNR). In addition, traditional filter-based imaging approaches are often ineffective for accurate measurement of cAMP signals in localized subcellular regions due to a range of factors, including spectral crosstalk, limited signal strength, and autofluorescence. To overcome these limitations and allow FRET-based biosensors to be used with multiple fluorophores, we have developed hyperspectral FRET imaging and analysis approaches that provide spectral specificity for calculating FRET efficiencies and the ability to spectrally separate FRET signals from confounding autofluorescence and/or signals from additional fluorescent labels. Here, we present the methodology for implementing hyperspectral FRET imaging as well as the need to construct an appropriate spectral library that is neither undersampled nor oversampled to perform spectral unmixing. While we present this methodology for measurement of three-dimensional cAMP distributions in pulmonary microvascular endothelial cells (PMVECs), this methodology could be used to study spatial distributions of cAMP in a range of cell types.
Introduction
Cyclic adenosine monophosphate (cAMP) is a second messenger involved in key cellular and physiological processes including cell division, calcium influx, gene transcription, and signal transduction. A growing body of evidence suggests the existence of cAMP compartments in the cell through which signaling specificity is achieved1,2,3,4,5,6,7. Until recently, cAMP compartmentalization was inferred based upon distinct physiological or cellular effects induced by different G-coupled receptor agonists8,9,10,11. More recently, FRET based fluorescence imaging probes have provided new approaches for the direct measurement and observation of cAMP signals in a cell12,13,14.
Förster resonance energy transfer (FRET) is a physical phenomenon in which energy transfer occurs between donor and acceptor molecules in a non-radiative fashion when the molecules are in close proximity15,16. With the development of FRET based fluorescent indicators, this physical phenomenon has been used in biological applications to study protein-protein interactions17, protein co-localization18, Ca+2 signaling19, gene expression20, cell division21 and cyclic nucleotide signaling. FRET based cAMP indicators typically consist of a cAMP binding domain, a donor fluorophore and an acceptor fluorophore22. For example, the H188 cAMP sensor12,22 used in this methodology consists of a cAMP binding domain obtained from Epac, sandwiched between Turquoise (donor) and Venus (acceptor) fluorophores. At basal conditions (unbound), Turquoise and Venus are in an orientation such that FRET occurs between the fluorophores. Upon binding of cAMP to the binding domain, a conformational change occurs such that Turquoise and Venus move apart resulting in a decrease in FRET.
FRET based imaging approaches offer a promising tool for investigating and visualizing cAMP signals within a cell. However, current FRET based microscopic imaging techniques are often only partially successful in achieving sufficient signal strength to measure FRET with subcellular spatial clarity. This is due to several factors, including the limited signal strength of many FRET reporters, the high level of precision required to accurately quantify changes in FRET efficiency, and the presence of confounding factors, such as cellular autofluorescence23,24. The result is often a FRET image that is plagued by weak SNR, making visualization of subcellular changes in FRET very difficult. In addition, investigation of spatially localized cAMP signals has been almost exclusively performed in only two spatial dimensions and the axial cAMP distribution has been rarely considered25. This is likely because low SNR impeded the ability to measure and visualize cAMP gradients in three spatial dimensions. To overcome limitations of using FRET sensors with low SNR, we have implemented hyperspectral imaging and analysis approaches to measure FRET in single cells25,26,27.
Hyperspectral imaging approaches were developed by NASA to differentiate terrestrial objects present in satellite images28,29. These techniques have since been translated to the fluorescence microscopy field30, with several commercial confocal microscope systems offering spectral detectors. In traditional (non-spectral) fluorescence imaging, the sample is excited using a band-pass filter or a laser line, and the emission is collected using a second band-pass filter, often selected to match the peak emission wavelength of the fluorophore(s). By contrast, hyperspectral imaging approaches seek to sample a complete spectral profile of either the fluorescence emission26,31,32 or excitation33,34 at specific wavelength intervals. In our previous studies, we showed that hyperspectral imaging and analysis approaches can offer improved quantification of FRET signals in cells when compared to traditional filter-based FRET imaging techniques26. Here, we present a methodology for performing 4-dimensional (x, y, z, and λ) hyperspectral FRET imaging and analysis to measure and visualize cAMP distributions in three spatial dimensions. These approaches have allowed visualization of agonist-induced cAMP spatial gradients in single cells25. Interestingly, depending on the agonist, cAMP gradients may be apparent in cells. The methodology presented here utilizes spectral unmixing of non-uniform background and cellular autofluorescence to improve the accuracy of the FRET measurements. While this methodology is demonstrated in pulmonary microvascular endothelial cells (PMVECs) using a cAMP FRET biosensor, the methodology could easily be modified for use with alternative FRET reporters or alternative cell lines.
Protocol
This protocol follows procedures approved by the University of South Alabama Institutional Animal Care and Use Committee.
1. Cell, sample, and reagent preparation for imaging
-
Isolate rat pulmonary microvascular endothelial cells (PMVECs) as described previously35.
NOTE: Cells were isolated and cultured by the Cell Culture Core at the University of South Alabama, Mobile, AL on 100 mm cell culture dishes.
-
Seed isolated PMVECs on 25 mm round glass coverslips and let them grow in the incubator at 37 °C until cells attain at least 80% confluency (at least 24 hours).
NOTE: Cells and cell type may vary from study to study and hence cell-specific cell culture procedures should be followed to seed and grow cells. The cell seeding and culturing protocol used in these studies is available as supplemental information in the file named “Supplemental File_Cell Culture and Transfection”.
-
Transfect PMVECs with a FRET biosensor and incubate for 48 hours at 37 °C.
NOTE: The protocol to transfect PMVECs is also described in the supplemental information file named “Supplemental File_Cell Culture and Transfection”.
-
On the day of imaging, warm Tyrode’s buffer to 37 °C in a water bath.
NOTE: Tyrode’s buffer consists of 145 mM NaCl, 4 mM KCl, 10 mM HEPES, 10 mM Glucose, 1 mM MgCl2 and 1 mM CaCl2
Mount a coverslip containing transfected cells into a cell chamber and secure the top with a mounting gasket to prevent leaking.
Wipe the bottom of the coverslip using a delicate task wipe to clean any excess media or adherent cells.
-
Add 800 μL of working buffer and 4 μL of 5 mM nuclear label to the cell chamber and gently rock for 5 – 10 seconds.
NOTE: When adding buffer or reagent solutions to coverslips mounted in the cell chamber, make sure to add the solution gently and at the side of the cell chamber so as not to dislodge adherent cells. Adding 4 μL of 5 mM nuclear label to 800 μL of buffer makes 25 μM final concentration of nuclear label. For loosely adherent cells such as HEK293 cells, mix nuclear label and buffer in a vial first and then add to coverslips mounted in the cell chamber. This will prevent lifting the cells off the coverslip.
Cover the cell chamber with aluminum foil to protect from light and incubate for 10 minutes at room temperature.
-
Reagent Preparation: Add 1 μL of 50 mM forskolin to 199 μL of buffer. This will produce a final concentration of forskolin of 50 μM when added to cells that were prepared with 800 μL of buffer. 1 μL of DMSO in 199 μL of buffer should also be prepared to be used as a vehicle control.
NOTE: In these studies, forskolin is used as an adenylyl cyclase activator to stimulate cAMP production. If desired, this methodology can easily be modified to allow treatment with alternative reagents for stimulating or inhibiting adenylyl cyclase, phosphodiesterases, etc.
2. Image acquisition
-
Use a confocal microscope equipped with a spectral detector.
NOTE: All image acquisition steps outlined here were developed using a commercially available Nikon A1R microscope system. These steps may need to be adjusted if using an alternative spectral microscope. Ensure that all equipment is turned on at least 30 minutes prior to the start of the experiment so as to reach stable operating conditions.
-
Select the 60× water immersion objective and add a drop of water to the objective.
NOTE: For high-resolution live-cell imaging, it is recommended to use a high numerical aperture objective. Please refer to the List of Materials for information about the objective used in these studies.
Place the loaded cell chamber (from step 1.7) onto the microscope stage.
Select the DFT (DAPI/FITC/TRITC) filter set by tuning the filter knob on the right side of the microscope.
-
Operate the microscope in fluorescence widefield mode using the eyepieces to select a field of view containing cells expressing the cAMP FRET sensor.
NOTE: Ensure that the average intensity of the FRET signal at the donor or acceptor emission peak wavelength in the selected cell is at least 100 intensity units (A.U.) or at least 4X the baseline signal of a region with no expressing cells. This can be confirmed using the spectrum profile viewer available in NIS Elements software. When looking for a cell with good signal, it is advisable to discard excessively bright cells (they may be compromised).
Open NIS software, switch to confocal mode, unlock the laser interlock button and click on Live.
Use the focus knob to focus on the cells by looking at the preview on the screen.
Configure device, acquisition, and z-stack settings in the software, as described below.
-
Acquisition settings:
NOTE: Camera and device acquisition settings can be applied using a previously acquired image. Open the image, right click and select Reuse Camera Settings.-
Open the A1 settings menu, check the boxes corresponding to 405 nm and 561 nm laser lines, select SD for spectral detector, select 10 for resolution and 31 for channels.NOTE: A1 settings menu is shown as a small gear icon on the top left corner of the A1 Plus Settings window. 405 nm laser is used for donor excitation and 561 nm laser is used for nuclear label excitation.
- Set the wavelength range (410 – 730 nm) by selecting start and end wavelength values.
-
Click the binning/skip icon in the A1 settings menu and select the box that is numbered 15, then click OK on the A1 settings menu.NOTE: This is to remove the wavelength channel that corresponds to the 561 nm excitation laser (this is typically the 15th wavelength channel). It is important not to use this wavelength band to avoid an artificially low signal, which can create a spectral artifact. The signal is lower in this band because of the mechanical finger that covers the detector element to protect it from laser damage.
-
Set the laser intensities to 8% and 2% for the 405 nm and 561 nm lasers, respectively, Si Hv (detector gain) at 149, and a pinhole radius of 2.4 airy disk units (AU).NOTE: Laser intensities may have to be adjusted depending on the age of the instrument and condition of the lasers. If adjusting laser intensities between different samples or experimental groups, it is important to maintain the same ratio of laser intensities (e.g., 8:2). In addition, it is important to select a laser intensity that is not so bright as to create rapid photobleaching. The detector gain should be adjusted to maximize signal intensity while minimizing detector noise. For these studies, a gain of 149 was used. A pinhole size of 2.4 AU was selected as a balance between acquiring images with sufficient signal to noise ratio (SNR) and maintaining optical sectioning (confocality). An increase in pinhole size increases SNR but decreases confocality.
-
Set the scan speed to 0.25 spectral frames per second, select the icon corresponding to unidirectional for scan direction, enter 4 for the count, and set 1024 × 1024 for scan size.NOTE: FRET signals are weak, and a slow scan speed is often required. Using scan speed of 0.25, acquisition of a spectral z-stack is completed in ~3 minutes. Scan speed can be increased or decreased depending on the fluorophores used. For example, for brighter fluorophores like eGFP, faster scan speed (2 frames/second) can be used. The number entered under count corresponds to a frame averaging value of 4, which helps in noise reduction during image acquisition. For very stable samples and where time is not a constraint, higher averaging values (up to 16) can be used to obtain images with improved SNR.
-
-
Define z-stack acquisition parameters:
NOTE: The values entered in steps 2.10 may need to be adjusted to accommodate changes in fluorescent label binding or concentration, type of label, number of labels used, cell line, and other changes in sample preparation that may affect cell labeling density and/ or cellular autofluorescence. When adjusting acquisition parameters, care should be taken to achieve a sufficient SNR while minimizing photobleaching. In addition, when configuring a spectral FRET assay, care should be taken to ensure that parameters work well across all treatment groups. It is advisable to run a trial of each treatment group with the proposed parameter settings to ensure that SNR is sufficient and photobleaching is minimized.- Open the ND acquisition window by clicking view → acquisition control → ND acquisition.
- Enter the path/destination and a file name to save the ND file on the popup window.
- Check the box corresponding to z-series.
- Click on live in the A1 Plus Settings window. This will open a live viewing window.
-
Adjust the focus knob on the microscope to select the top of the cell and click Top in the ND acquisition window to set the current position as the top.NOTE: It is suggested to focus slightly above the top of the cell to ensure that all of the cell is sampled in the z series.
-
Adjust the focus knob on the microscope to select the bottom of the of the cell and click on Bottom in the ND acquisition window to set the current position as the bottom.NOTE: Focus slightly below the bottom of the cell to ensure that all of the cell is sampled.
-
Enter 1 μm for step size, select top-bottom for the z-scan direction and click run on the ND Acquisition window to acquire a z-stack.NOTE: Step size determines the number of z-slices that will be acquired depending on top and bottom locations (i.e., the distance traveled). A 1 μm step size was selected as a compromise between imaging speed, z-axis sampling, and photobleaching. Using the confocal pinhole diameter of 2.4 AU and the 60× water immersion objective resulted in optical section thickness of 1.73 μm. Hence, a 1 μm step size is slightly below the Nyquist sampling criteria, but this is a compromise that was made to reduce the time needed to acquire a z-stack. For very stable samples, for which speed is not critical, a smaller z-axis step and possibly a smaller confocal pinhole diameter may be used to increase z-axis resolution. Bottom-top should yield similar results and can be used to evaluate any effects of photobleaching that may occur during the z scan.
-
Set up the Perfect Focus System (PFS) if available:
NOTE: PFS allows the system to compensate for fluctuations in the focal depth during image acquisition. The following steps may be used to set up PFS, and these steps may vary slightly depending on the version of the Nikon A1R and the version of NIS Elements used.- Highlight symmetric mode defined by the range icon in the ND acquisition window.
- Turn on the PFS button on the front face of the microscope (make sure the dichroic mirror knob located on the section below the sample stage is ‘in’).
- Redefine the top (rotate counterclockwise) and bottom (rotate clockwise) using the knob on the front face of the PFS offset controller.
- Define a relative z-position/z-depth by clicking ‘relative’ on the ND acquisition window.
- Click memory on the front face of the microscope so that the software memorizes the relative z-depth.
-
After the z-stack acquisition is complete, gently add the desired reagent (forskolin or vehicle control) using a pipette and wait for 10 minutes.
NOTE: Add the reagent very gently so as not to disturb the cells or move the position of the cell chamber within the microscope XY stage; it is helpful to verify in a subsequent live view or image that the field of view has not shifted during reagent addition. The 10-minute wait time is for the forskolin treatment to take effect. If an alternative treatment is used, the wait time may need to be adjusted.
After 10 minutes, change the filename and click run in the ND acquisition window.
Repeat steps 2.11 – 2.13 as outlined above for at least 5 coverslips so as to achieve sufficient results for statistical analysis (n=5 for each treatment group – forskolin and vehicle control).
Prepare samples and sample blanks to construct the spectral library and acquire spectral images using similar acquisition settings as outlined in steps 2.9 and 2.10.
3. Image analysis
NOTE: These images will be used to construct a spectral library containing the pure spectra of all individual endmembers present in the study. The endmembers in the spectral library might vary from study to study if different fluorophores are used. A detailed procedure to construct the spectral library is provided in a supplemental information file named “Supplemental File_Spectral Library”. Here, we describe exporting data to .tiff files, linear spectral unmixing, FRET efficiency measurements, three-dimensional reconstruction, and cAMP levels estimation. Image analysis can be performed using different image analysis and programming platforms such as ImageJ, Python, MATLAB, or CellProfiler. In these studies, MATLAB scripts were used.
- Export image data:
-
Create new folders with the same filename corresponding to the spectral z-stack images acquired in steps 2.13 and 2.14.NOTE: The following steps outlined to export data are specific for NIS Elements AR version 4.30.01. These steps may vary slightly depending on the version of the software.
- Click File, which will open a File Window. Browse and select the spectral image file acquired in step 2.12 and click Open.
- Once the file loads, click File→ Import/Export→ Export ND document.
-
On the popup window: browse and select the folder created in step 3.1.1, select Tagged Image Format (TIF) for File type, then select Mono image for each channel and Keep bit depth.NOTE: The File prefix will be pre-generated; change this value for convenience. The Index order will change depending on the Channels that are selected, and should display “z, c” for indexing according to z-slice location first and wavelength band number second. Make sure that the boxes corresponding to Apply LUTs or Insert Overlays or Use Point Names are unselected.
- Click Export to export the tiff files to a destination folder as individual tiff files.
- Repeat steps 3.1.2 – 3.1.5 to export spectral image files acquired in step 2.13.
-
- Linear spectral unmixing:
-
Open the programming software.NOTE: Custom developed programming script to unmix raw spectral images is provided on the University of South Alabama BioImaging and BioSystems website, under the Resources tab (https://www.southalabama.edu/centers/bioimaging/resources.html).
- Open the file labeled “Linear Unmixing.m” and click the run button in the editor toolbar.
- Browse and select the folder containing the exported *.tif file sequence generated by the NIS Elements software.
- Click OK to continue, which will open a new window called Wavelength and Z-Slice.
- Copy the filename of the first file (without z-slice and channel number) in the folder selected in step 3.2.4 and paste it into the first step of the dialog box labeled “Enter the Image Name”.
-
Enter the number of channels in the second step labeled “Enter the number of wavelength bands”, number of z-slices in the third step labeled “Enter the number of Z-slices” and click OK.NOTE: The number of wavelengths bands may change if changes are made to the acquisition settings, such as adjusting the wavelength range or the wavelength step size. The number of Z-slices may also change depending on the height of the cell.
- Browse and select the wavelength file called “Wavelength.mat” in the popup window labeled “Select the wavelength information file” and click open.
-
Browse and select the “Library.mat” file in the new popup window labeled “Select the spectral library file”, click open and wait until the unmixing of the slices is finished.NOTE: Library.mat file is a file containing pure spectra for each endmember fluorophore along with cell autofluorescence and background spectral signatures. In this case, endmember fluorophores include Turquoise, Venus, and DRAQ5. Background spectral signatures include cellular or matrix autofluorescence, coverslip fluorescence, and coverslip diffraction. Wavelength.mat file is a file containing wavelength channel information used to acquire the spectral image. An example library file and wavelength file are available on Bioimaging and Biosystems website (see the note under 3.2.1). For more information on how to generate spectral library and wavelength files, refer to supplemental information file named “Supplemental File_Spectral Library”. Unmixed images corresponding to each z-slice will be saved into the folder called “Unmixed” created during the unmixing process within the folder that was selected in step 3.2.3.
-
- FRET Efficiency Calculation:
-
Open the programing script called “multiFRRCF.m” and click run.NOTE: This programming file is available from the University of South Alabama Bioimaging and Biosystems website (see note under step 3.2.1).
-
Enter the number of experimental trials to analyze in the popup dialogue box called “how many folders to reslice” and click OK.NOTE: Image data from each experiment should be saved in a separate unmixed image folder. This step simply allows the analysis code to loop over many folders as a time saving step.
-
Browse and select the unmixed folder(s) and click OK.NOTE: The number of times that the “Browse for folder” pop up window opens depends on the number entered in “How many folders to reslice” dialogue box in the previous step. Browse and select the folders one after the other.
-
On the new popup window, enter the following information into the respective boxes: scaling factor is 12.4, Threshold is 5.6, X, Y, and Z Frequency are 5, 5, and 1 respectively, and smoothing algorithm is Gaussian.NOTE: The scaling factor is a value in pixels/μm and will be used to scale the Z-direction sampling to that of the XY direction. The scaling factor is obtained from the image pixel size, which is usually provided as metadata in the image for most confocal microscope systems. For example, if the image is acquired with 0.08 μm/pixel spacing, the scaling factor should be 12.5 pixels/μm. Threshold value will be used to threshold the images and generate a binary mask of the cell. We created a list of optimum values based on the image donor+acceptor intensity. Use 4.5 as a thresholding value if the image has bright donor+acceptor intensity and low background, a value between 5.6 to 6.5 for images having only moderate donor+acceptor intensity and/or higher background, and a value of 7.5 and above for images having a donor+acceptor intensity that is lower than the background. Frequency value corresponds to the interval, in number of pixels, at which the slicing is performed in the subsequent steps. For example, if the Z-depth of the cell is 17 μm with a 1 μm step size and a scaling factor of 12.5 pixels/μm is used in the XY direction, then the depth of the 3-dimensional image dataset will be resampled at 212 pixels (Z direction). Based on the Z frequency value entered (for example, 1 pixel), the 3-dimensional image data set will be re-sliced beginning at the top of the image data set and then moving in increments of 1 pixel downward. This results in 212 resliced images. If a larger frequency value interval were entered for Z Frequency, then fewer resliced images would be generated. Resliced images are saved in subsequent steps.
-
Click run and wait until all the FRET measurements and reslicing are performed.NOTE: A separate folder is created within the parent directory to which resliced grayscale FRET efficiency images and colored (a colormap applied) FRET efficiency images are saved. For example, all grayscale and colormap FRET images resliced in the X direction (YZ plane) are saved into a folder called “Resliced_XFRET”.
-
Repeat the analysis with similar settings for all the experiments – before and after forskolin treatments and vehicle controls.NOTE: Steps mentioned in section 3.3 describe the values to enter for the custom FRET analysis programming script to generate 3-dimensional FRET image data. However, this script executes several operations while running, including: loading image data, creating image stacks, smoothing, FRET efficiency calculations, creating and applying a cell border mask, 3-dimensional image reconstruction, reslicing 3-dimensional images at specified intervals (frequencies), applying a colormap for visualizing FRET changes, and saving the resliced image data to the same directory. Additional details have been included as comments in the program script.
-
4. Mapping FRET efficiency to cAMP levels
-
Open the programming file named ‘Mapping_FRET Efficiency_to_cAMP_concentration.m’ and click run on the main window.
NOTE: The file is available on the BioImaging and BioSystems website (see note under 3.2.1). This file reads grayscale FRET efficiency images and converts them to cAMP levels based on a characteristic curve. This characteristic curve uses a cAMP-to-FRET relationship documented in literature15,36 that is described by the Hill equation (the third equation shown below). However, Kd of the probe in intact cells is difficult to estimate and we have assumed it to be 1 μM in our calculations. Hence, results are shown as a function of Kd. (i.e., [cAMP] = x* Kd). Equations used to measure FRET efficiency and mapping FRET to cAMP levels are shown below:Where E is the FRET efficiency, and aapparent and dapparent are unmixed pixel intensities of acceptor and donor images, respectively.
Qa and Qd are quantum yields of acceptor and donor. Note that Qa and Qd cancel out when the equation for kλ is incorporated in the FRET efficiency equation, kλ is a correction factor:
and are extinction coefficients of donor and acceptor at the donor excitation wavelength, i (405nm).E is FRET efficiency and KD = Dissociation constant = 1 μM.
Navigate and select the first grey scale FRET image (saved in step 3.3.5) and click OK.
Open the FRET/cAMP images to inspect the distribution of cAMP signals in three dimensions.
Representative Results
This protocol describes the use of hyperspectral FRET imaging and analysis approaches to measure cAMP gradients in three spatial dimensions in living cells. There are several key steps involved in generating these results, for which careful attention is required while analyzing and quantifying the data. These key steps include construction of an appropriate spectral library, background spectral unmixing, thresholding to identify cell borders, and FRET efficiency calculations. Figure 1 illustrates the schematic flow of all the steps involved in measuring FRET efficiency and cAMP levels in living cells. When performed properly, these imaging and analysis steps will allow measurement of FRET efficiency and estimation of cAMP spatial gradients in 3 dimensions in a cell, while accounting for non-uniform background signals.
Figure 1: Flowchart depicting the steps involved in FRET efficiency and level measurements in three spatial dimensions using hyperspectral FRET imaging and analysis.

Figure 2 depicts 3-dimensional views of false-colored raw hyperspectral image data acquired using a Nikon A1R confocal microscope at baseline conditions (Figure 2A) and 10 minutes after (Figure 2B) forskolin treatment. Note that similar detector and system parameters were used to acquire before and after treatment image stacks to maintain consistency for quantitative analysis. Also note that changes in FRET are not obvious in this image, as this is purely a visualization of raw data, before calculating the FRET efficiency.
Figure 2: A 3-dimensional visualization of a cell expressing the cAMP FRET reporter (green) and nuclei from the cell and surrounding non-expressing cells (red).

Raw spectral image data acquired using a Nikon A1R spectral confocal microscope have been false colored according to wavelength and visualized in 3 dimensions using NIS Elements software. A) 3-dimensional image data at baseline and B) 10 minutes after 50 μM forskolin treatment.
A spectral library with pure spectra of all end members is needed to further analyze raw spectral image data. Constructing an appropriate spectral library is one of the key steps to ensure appropriate measurements of FRET efficiency. Figure 3 demonstrates the construction of a spectral library containing the pure spectra of endmembers (in these current studies, the endmembers are Turquoise, Venus, and DRAQ5). To measure FRET efficiency, it is important to obtain the spectra of Turquoise and Venus using a sample with 1:1 stoichiometry. Here, we have provided an approach where the acceptor fluorophore is completely photo-destructed, allowing spectral signatures of the donor and acceptor with 1:1 stoichiometry to be obtained (Figure 3A–F). In addition, linear power relationships among the lasers (Supplemental Figure 1) were applied to calculate the acceptor spectrum with an intensity that would be expected if it were excited using the donor excitation laser, in this case 405 nm for Turquoise (Figure 3G). This ensures that unmixed donor and acceptor signals are comparable in absolute intensity when FRET is excited with a single 405 nm laser line. Non-transfected cells labeled with the nuclear dye, DRAQ5 (Figure 3H) were utilized to obtain the pure spectrum of DRAQ5 (Figure 3I). Combining the spectra of the donor, Turquoise (Figure 3F), acceptor, Venus (Figure 3G), and DRAQ5 (Figure 3I), a 3-component library was created (Figure 3J).
Figure 3: Construction of a spectral library containing the pure spectra of fluorescent labels in the study (referred to as endmembers by the remote sensing field).

A) A false-colored image of cells expressing the FRET biosensor acquired at 405 nm excitation (donor excitation). B) Spectrum corresponding to FRET signal: 4 different regions of interest (ROIs) were drawn on image A shown by red, yellow, blue and green rectangles. Average intensity at each wavelength for each ROI selected was exported. The average intensity from these 4 ROIs was plotted at each emission wavelength. The emission peaks of the spectrum correspond to both the donor (Turquoise) and the acceptor (Venus) fluorophores. C) A false-colored image of cells expressing the FRET biosensor acquired at 488 nm excitation (acceptor excitation). D) The average spectrum was estimated as explained in B from several ROIs in C (the same regions as in A) with emission peak due only to the acceptor. E) A false-colored image of cells expressing the FRET biosensor acquired at 405 nm excitation after photo-destruction of the acceptor fluorophore by 514 nm irradiation. F) The average spectrum was estimated as explained in B from several ROIs in E (the same regions as A) with emission peak due only to the donor. NOTE that the donor intensity is increased in F when compared to that of the original FRET signal, indicating that after photobleaching of the acceptor, donor excitation is leading to direct donor emission. G) The acceptor spectrum as would be expected if obtained using 405 nm excitation. This was estimated by utilizing the facts that the 405 nm and 488 nm excitation lasers have a linear response which can be characterized using a spectrometer and integrating sphere (Supplemental Figure 1) and the wavelength-dependent extinction coefficient of Venus. Hence, the Venus spectrum obtained at 488 nm excitation can be converted into the Venus spectrum that would be expected if obtained at 405 nm excitation H) A false-colored image of cells labeled with the nuclear label, DRAQ5. I) The average spectrum from several regions of interest in H. J) The resultant spectral library containing the normalized pure spectra of the donor and DRAQ5 and the excitation wavelength–corrected spectrum of the acceptor. Note that Turquoise and Venus spectra were normalized to the maximum value of combined Turquoise + Venus spectral data while DRAQ5 was simply normalized to unity at the value of the peak emission wavelength.
Signals from sources other than the fluorescent labels may also be present in a sample. To account for these, three different spectral signatures with peaks that occur at 424 nm, 504 nm, and 574 nm were identified within unlabeled cellular samples. We believe that these spectral signatures correspond to coverslip reflectance and cell matrix or cellular autofluorescence. Figure 4 depicts the sources of these three background spectral signatures. It is important to note that these signals are distributed non-uniformly within the sample and hence cannot simply be subtracted out. However, adding the spectral signatures of these signals to the spectral library and using linear unmixing to separate the signals presents an approach for removing these confounding signals from the donor and acceptor signals prior to calculating FRET efficiencies. To achieve this, the three background spectral signatures were added to the 3-component spectral library, forming a new 6-component library consisting of donor (Turquoise), acceptor (Venus), DRAQ5 and three background spectral signatures.
Figure 4: Background spectral signatures were identified and included into the spectral library to account for background fluorescence signals during the unmixing process.

A, B) Two background fluorescence spectral signatures were observed when characterizing a sample blank (unlabeled coverslip). These spectral signatures were named based on their peak wavelengths – one at 424 nm (from coverslip fluorescence) and the other at 504 nm (likely from reflectance or back scatter). C) A third background spectral signature was observed with a peak emission wavelength of 574 nm when non-labeled cells were analyzed, potentially from cellular autofluorescence or fluorescence of the underlying matrix. D) Background spectra extracted from images A, B, and C. These three background spectra were added to the existing 3-component library (Figure 3J) to develop a 6-component spectral library.
A custom programming script was written to unmix the spectral image data into individual endmembers, and a separate script was written to perform subsequent FRET efficiency calculations. Linear spectral unmixing (illustrated in Figure 5) was performed using the 6-component spectral library. Unmixing was performed for each slice in the axial image stack, also referred to as z-stack (refer to the 3-dimensional visualization of raw spectral image data in Figure 5A). This resulted in separate unmixed images for each endmember, for each z-slice in the z-stack (Figure 5C–E, background unmixed images are not shown). If desired, the unmixed signals may be false-colored for visualization with a different color assigned to each unmixed signal (Figure 5F–H).
Figure 5: Linear spectral unmixing using a 6-component spectral library that accounts for background signals.

A) Raw spectral image data acquired as an axial z-stack. B) 6-component spectral library used to linearly unmix raw spectral data. C, D, and E) Gray scale unmixed images of DRAQ5, Turquoise, and Venus, respectively, resulted from linear spectral unmixing. F, G, and H) False colored unmixed images of DRAQ5, Turquoise, and Venus respectively. Unmixed images of background signals were also calculated (data not shown here as only the fluorescent labels are of interest for this methodology – refer to Figure 4 in Annamdevula, et al. for examples of unmixed background signals25).
Figure 6 illustrates the steps involved in the FRET efficiency calculations, as well as the steps for mapping FRET efficiency to cAMP levels. A FRET efficiency image was generated using smoothed unmixed donor and acceptor images. A binary mask image was obtained using unmixed donor, acceptor, and nuclear images. The mask was applied to the FRET efficiency image to remove contributions from pixels outside of the cell. Though unmixed images from single z-slices were used for the pictorial demonstration of FRET efficiency measurements, these calculations were performed on each slice within the 3-dimensional image stack. The 3-dimensional FRET image data set was then resliced in three orthogonal planes to visualize spatial gradients of cAMP signals in different directions.
Figure 6: Flowchart depicting the steps involved in calculating FRET and cAMP levels from unmixed spectral image data.

A) Representative unmixed image of donor, Turquoise. B) Representative unmixed image of acceptor, Venus. C) Representative unmixed image of nuclei, DRAQ5. D) Binary cell mask is generated by thresholding unmixed donor+acceptor summed image. E) A threshold is applied to the nuclear image to create a binary mask of the nucleus. F) The pixel wise FRET efficiency was calculated from unmixed donor and acceptor images. G) A composite binary mask was created from cell border and nuclear masks. H) Masked FRET efficiency image: composite cell mask was applied to FRET efficiency image to exclude non-specific FRET signals outside the cell and within the nucleus. I) A colormap was applied to the masked FRET efficiency image to better visualize spatial changes in FRET. J) The cAMP levels image that was estimated from the FRET efficiency image. K) Colormap was applied to better visualize spatial changes in cAMP. Colorbars shown on the right side were used to visualize FRET efficiency (top panel) and cAMP levels (bottom panel). Images shown in this figure are representative of one single axial z-slice, but the analysis operation described in this figure is performed on the entire axial z-stack.
Visualizing agonist-induced changes in FRET efficiency and cAMP levels
The steps described above provide a method for calculating FRET efficiency and cAMP levels from hyperspectral image data in three spatial dimensions. These steps can be applied to cellular preparations before and after treatment with compounds that elicit a cAMP response, such as forskolin. Here, we provide an example of using this approach to observe changes in FRET and cAMP distribution in PMVECs following treatment with 50 μM forskolin. Figure 7 illustrates the changes in FRET efficiency and cAMP levels in different XY plane slices (z slices), from apical to basal, allowing comparison of baseline conditions (before forskolin treatment) and 10-minutes post-treatment. In this illustrative example, FRET efficiency decreased (columns 1 and 3 in Figure 7) upon forskolin treatment, correlating to an increase in cAMP levels (columns 2 and 4 in Figure 7). Minimal spatial variation of cAMP within a single XY plane was observed. However, axial (apical-to-basal) cAMP spatial gradients were observed, as can be surmised by noting that the apical slice has a deeper red color (indicating higher cAMP levels through the color lookup table that was applied) than the basal slice after forskolin treatment (column 4 in Figure 7). Axial FRET or cAMP distributions can often be better visualized using images obtained from the two orthogonal spatial planes: Figure 8 depicts the FRET efficiency and cAMP levels in the YZ plane at baseline (columns 1 and 2) and 10 minutes after forskolin treatment (columns 3 and 4), while Supplemental Figure 2 depicts the FRET efficiency and cAMP level changes in the XZ plane. These results demonstrate the feasibility of measuring FRET and estimating cAMP levels from 3-dimensional hyperspectral image data and also demonstrate the importance of visualizing axial distributions of FRET or cAMP. While beyond the scope of this methodological paper, it may be that axial spatial distributions of cyclic nucleotides contribute to specificity within cyclic nucleotide signaling pathways.
Figure 7: Forskolin-induced FRET efficiency and cAMP spatial gradients visualized in PMVECs.

XY plane images were generated by reslicing 3-dimensional reconstructed FRET and cAMP image data in the axial (Z) direction from the apical to the basal side of the cell. Five contiguous XY slices are shown. Columns 1 and 3 represent the FRET efficiency at baseline and 10 minutes after treatment with 50 μM forskolin, respectively. Columns 2 and 4 indicate the levels at baseline and 10 minutes after forskolin treatment. The colorbars were used to relate changes in the colormap to FRET efficiency (top) and cAMP levels (bottom). White lines shown on images in column 2 and column 4 are used to generate the intensity profile (line scan profile) of cAMP signals across this selected line. Intensity profile plot obtained from the mid-slice of the cell at baseline (blue profile) and after forskolin treatment (green profile) demonstrates the spatial distribution of cAMP signals across the line scan. Scale bar indicates 20 μm.
Figure 8: Forskolin-induced FRET efficiency and cAMP spatial gradients visualized in the axial direction.

YZ plane images were generated by reslicing 3-dimensional reconstructed FRET and cAMP image data in the lateral (X) direction (from left to right side of the cell). Columns 1 and 3 represent the FRET efficiency at baseline and at 10 minutes after 50 μM forskolin treatment, respectively. Columns 2 and 4 represent the cAMP levels at baseline and at 10 minutes after forskolin treatment. The colorbars at right were used to relate changes in the colormap to FRET efficiency (top) and cAMP levels (bottom).
When performing the methods described above, it is important to meticulously check the accuracy of the steps and to run appropriate experimental and vehicle controls to ensure that changes in FRET (and corresponding cAMP) are due to actual changes in donor and acceptor signals and are not imaging artifacts. For example, important steps to consider include:
Using an appropriate spectral library that contains all of the needed spectral components
As mentioned, there can be significant contribution of background signals from cellular autofluorescence, deposited matrix, or reflected light from the coverslip (excitation-emission bleed through). Figure 9 represents an example data set illustrating that the background signal was retained within the images when a 3-component (Turquoise, Venus, and DRAQ5) library was used to unmix the spectral data. By contrast, the background signal was effectively removed when a more complete (and appropriate) 6-component spectral library was used.
Figure 9: A comparison of the effectiveness of utilizing either a 3-component or a 6-component spectral library for calculating FRET and cAMP levels images.

Nonspecific background signals were observed in images calculated using a 3-component library, which did not account for background spectral signatures. This was especially true for images near the basal side of the cell following 50 μM forskolin treatment (column 2). By contrast, background signal artifacts were effectively removed when using a 6-component library that included background spectral signatures, improving the ability to segment the cell and analyze FRET and cAMP signals.
Using an optimal threshold value to generate a cell mask
In many of the experiments that we have performed, the background signal intensity is approximately 50–60% of the FRET signal intensity that is present within the raw spectral image data (i.e., when visualizing unprocessed spectral image data the peak of the FRET signal is only ~2X higher than that of the surrounding background signal). Thus, separation of background signal from foreground signal using a threshold value to obtain a binary mask is a sensitive step and must be carefully performed in order to avoid analysis artifacts. Figure 10 illustrates the effect of different threshold values applied for cell segmentation to create a binary mask of the cell. A low threshold value may include background signal as part of the expressing cell. On other hand, an overly high threshold value may preclude measurement of FRET in low-expressing cells or regions of a cell with low donor+acceptor signal (either very thin portions of the cell or portions that may have a lower regional concentration of the FRET probe).
Figure 10: Image analysis artifacts can be introduced if an appropriate threshold value is not selected to delineate the cell and nuclear borders.

Unmixed donor and acceptor images were used to create a mask with three different threshold values: 0.35 (columns 1 as 2), 0.65 (columns 3 and 4) and 0.9 (columns 5 and 6). Columns 1, 3, and 5 display the FRET efficiency at baseline. Columns 2, 4, and 6 display the FRET efficiency at 10 minutes after 50 μM forskolin treatment. Selecting a threshold that was too low resulted in portions of the background, or extracellular region, being included with the cell for analysis, while selecting a threshold that is too high resulted in the loss of part of the cell (see the apical slice in columns 4–5 when compared to columns 3–4).
Selecting a transfected cell with donor+acceptor signal intensity ≥ background signal
Figure 11 illustrates an example data set where FRET efficiencies and corresponding levels were measured from unmixed images obtained using a 6-component spectral library for linear spectral unmixing. Despite using the 6-component library to unmix spectral image data and selecting a high threshold for creating the cell border and nuclear mask, FRET efficiency images were still prone to high background noise signal near the basal side of the cell. In this case, the presence of background noise was due to selection of a cell that was only weakly expressing the FRET probe, and the donor+acceptor signal strength was approximately equal to the background signal strength, even after unmixing. Thus, in addition to applying the sophisticated analysis steps described above, it is also important to select a cell with sufficient expression of the FRET probe and correspondingly sufficient FRET signal (donor and acceptor signal at least equal or above the noise signal) during acquisition in order to ensure high-quality results.
Figure 11: Background signal artifacts may still persist even after background unmixing and selecting an appropriate threshold during creation of a binary mask.

Column 1 shows representative apical, middle, and basal slices at baseline and column 2 shows the same at 10 minutes after 50 μM forskolin treatment. Despite using a 6-component library for unmixing that accounted for background signals and utilizing a higher threshold of 0.85 for binary mask generation, background regions were still identified as being part of the cell, especially after treatment with forskolin. If this occurs, a possible explanation may be that a cell with weak expression of the FRET reporter was selected for imaging.
Discussion
The development of FRET biosensors has allowed the measurement and visualization of cyclic nucleotide signals in single cells, and there is great promise for visualizing subcellular signaling events13,22,37,38. However, the use of FRET biosensors presents several limitations, including the low signal-to-noise characteristics of many fluorescent protein-based FRET reporters and the weak transfection or expression efficiencies of the FRET reporters (this may be especially challenging in certain cell lines, such as PMVECs)23,24. Imaging of weakly expressed fluorescent proteins, combined with ratiometric FRET calculations, often results in a high degree of uncertainty or fluctuation in the calculated FRET efficiency. In an effort to improve the reliability of FRET measurements, we and others have previously demonstrated the use of hyperspectral imaging and analysis to measure FRET signals and to reduce crosstalk or bleed through of fluorescence signals between fluorescent labels and autofluorescence effects26,31,32,39. Due to limitations in signal strength, these spectral FRET studies were limited to two (X and Y) spatial dimensions until very recently. Hence, they provided a single-slice view of FRET changes within a cell.
In recent studies, we noted that FRET (and corresponding cAMP levels) appeared to be spatially distributed not just in the XY plane, but also across the XZ and YZ planes25. The hyperspectral FRET imaging approach described here extends our ability to visualize and measure FRET and cyclic nucleotide (cAMP) changes into three spatial dimensions, opening new possibilities for assessing signal compartmentalization. This 4-dimensional (X, Y, Z, and λ) hyperspectral imaging and analysis approach allows the measurement and visualization of cAMP gradients in three spatial dimensions while accounting for non-uniform background signals. Here, we have demonstrated how to implement this approach through the example of forskolin-induced cAMP spatial gradients. In the post-treatment image data, cAMP spatial gradients can be observed from the apical to the basal side of the cell (Figure 7 and Figure 8). The cAMP produced upon treatment with forskolin did not appear to reach cell-cell junctions (Figure 7 and Figure 8).
In utilizing the methodology described here, it was important to account for different sources of background and autofluorescence signals to allow accurate estimation of FRET efficiencies. Linear unmixing provides a potential avenue for accounting for unique background spectra, if their signatures are different than the fluorescent probes of interest. In particular, linear unmixing is better suited for separating background and autofluorescence signals than standard background subtraction.40,41,42 In the example shown here, three different spectral signatures were measured and assigned a name based on the peak emission wavelength of the signature: the 424 nm background spectrum (possibly from the coverslip fluorescence), the 504 nm background spectrum (likely due to reflected or back-scattered light), and the 574 nm background spectrum (possibly due to cell or cellular matrix autofluorescence). To illustrate the effects of failing to account for these signatures, two spectral libraries were created, and unmixed images compared. First, a spectral library containing just the fluorescent labels in the sample, Turquoise, Venus, and DRAQ5, was created and labeled the 3-component library. Second, a spectral library that additionally contained the three background spectral signatures was created and labeled the 6-component library. As shown above, unmixing with the 6-component library (background unmixing) allowed removal of background signals, whereas unmixing with the 3-component library did not (Figure 9). In previous work, we have shown that the root mean squared error (RMSE) associated with linear unmixing is decreased when using a spectral library that accounts for both the fluorescent labels and background signatures. It should be noted that the axial distribution of background fluorescence is often non-uniform and hence, a simple background subtraction will not work to correct the image data. Thus, the spectral unmixing approach is needed to provide accurate background removal and reliable FRET measurements.
It is important to optimize system parameters to select the best possible system and camera settings when acquiring spectral images. The overall goal should be to optimize SNR while minimizing acquisition time and preventing photobleaching.43 Thus, when optimizing an imaging system, a compromise between spatial, temporal, and spectral resolutions is often needed. In these studies, optimal values were selected for the system and camera settings including pinhole size, laser power, scan speed, scan size, and frame averaging as described in the protocol section. Utilizing these settings, the spatial sampling of 80 nm/pixel, temporal sampling of ~3 minutes per spectral z-stack (~10 seconds/XY image) and spectral sampling of 10 nm interval is achieved with negligible or minimal photobleaching.
To ensure accurate estimates of FRET efficiency, it is necessary to measure donor and acceptor spectra as they would be expected with 1:1 stoichiometry and identical excitation wavelengths (Figure 3). Turquoise and Venus spectra were normalized with respect to the Turquoise peak emission wavelength. The FRET efficiency that was estimated using this spectral library produced values similar to those reported in literature12,22. Typically, endmember spectra in the library are normalized to unity. However, to accurately calculate FRET efficiency, the acceptor spectrum must be acquired with respect to the donor spectrum (i.e., at equimolar concentration or 1:1 stoichiometry and referenced to the same excitation wavelength). In addition to the use of a properly constructed library, several steps are involved in appropriate estimation of FRET efficiency and avoidance of analysis artifacts. These include selecting an expressing cell with adequate signal intensity (Figure 11), smoothing of unmixed images before estimating FRET efficiency (Gaussian blur was applied in this example), using an appropriate threshold value to create the mask (Figure 10), and estimation of a correction factor using extinction coefficients of donor and acceptor (Supplemental File_Spectral Library). When these steps are followed, 3-dimensional distributions of FRET efficiency and underlying cAMP levels can be accurately visualized in single cells.
Localized cAMP signals have been estimated in 2-spatial dimensions using targeted FRET biosensors13,37. However, targeting FRET probes to specific subcellular compartments (for example, plasma membrane or lipid rafts) typically results in an increased local concentration of probe. This may result in measurement artifacts introduced due to intermolecular or bimolecular FRET. In addition, the results presented here and in Annamdevula et.al., 2018, Cytometry A25 demonstrate the importance of 3-dimensional measurements of FRET from either soluble or targeted probes.
Despite the importance of measuring cAMP signals in three spatial dimensions, there are also limitations to this approach. The most restrictive limitation is the long acquisition times required – approximately 3 minutes per spectral z-stack. This acquisition rate precludes using this approach for detecting anything other than quasi – steady state cAMP distributions in cells. That said, the results presented demonstrate the importance of including quasi – steady state 3-dimensional (x, y, z) cAMP measurements as critical complements to standard 2-dimensional (x, y) measurements. In the future it will be interesting to incorporate labeled proteins and cellular structures into the experimental design. Careful choice of fluorophores used to label proteins (and/or structures) would allow assessment of the 3-dimensional distributions of the labeled proteins and FRET without additional loss of acquisition speed. This would in turn allow measurement of localized cAMP signals and cAMP signals near the labeled proteins in three spatial dimensions, thus offering an important experimental complement to targeted cAMP probes and further expanding the utility of hyperspectral measurements of 3-dimensional cAMP distributions in living cells.
Supplementary Material
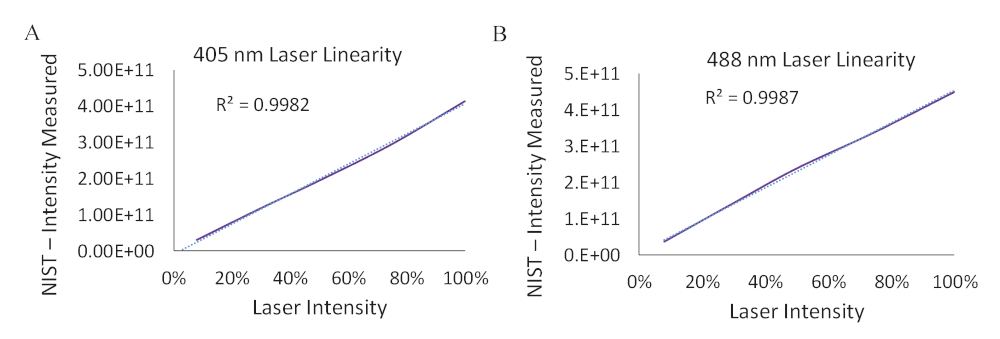
Supplemental Figure 1: Measurements of laser line intensity as a function of laser setting in the software. A fiber-coupled spectrometer and integrating sphere were calibrated to a NIST-traceable lamp to measure laser intensity for both the 405 nm laser line and the 488 nm laser line. A) Total number of photons measured at the microscope stage corresponding to different laser intensities of the 405 nm laser. B) Total number of photons measured at the microscope stage corresponding to different laser intensities of 488 nm laser. Linear response in laser intensity was observed for both the lasers as measured at the microscope stage. A linear trendline was fit and the trendline equation for each laser line was used to calculate the acceptor spectrum that would be expected if excited with the 405nm laser line (donor excitation).
{kind=link}
Supplemental Figure 2: Forskolin-induced FRET efficiency and cAMP spatial gradients visualized in the axial direction. XZ plane images were generated by reslicing 3-dimensional reconstructed FRET and cAMP image data in the lateral (Y) direction (from front to back of the cell). Columns 1 and 3 represent the FRET efficiency at baseline and at 10 minutes after 50 μM forskolin treatment, respectively. Columns 2 and 4 represent the cAMP levels at baseline and at 10 minutes after forskolin treatment. The color bars at right were used to relate changes in the colormap to FRET efficiency (top) and cAMP levels (bottom). Similar to results shown for the YZ planes (Figure 8), apical to basal spatial gradients in FRET efficiency and cAMP levels may be visualized as changes in color from the top to bottom of a single YZ slice, both at baseline conditions (any given slice in columns 1–2) and after 50 μM forskolin treatment (columns 3–4).
{kind=link}
Acknowledgments
The authors would like to acknowledge Dr. Kees Jalink (The Netherlands Cancer Institute and van Leeuwenhoek Center for Advanced Microscopy, Amsterdam, the Netherlands) for providing us with the H188 cAMP FRET biosensor and Kenny Trinh (College of Engineering, University of South Alabama) for technical help in reducing the time taken to run our custom developed programming scripts.
The authors would like to acknowledge the funding sources: American Heart Association (16PRE27130004), National Science Foundation; (1725937) NIH, S100D020149, S10RR027535, R01HL058506, P01HL066299).
Footnotes
Disclosures
Drs. Leavesley and Rich disclose financial interest in a start-up company, SpectraCyte, LLC, that was formed to commercialize spectral imaging technologies. However, all procedures described in this protocol were conducted using commercially available products not associated with SpectraCyte, LLC.
References
- 1.Corbin JD, Sugden PH, Lincoln TM, Keely SL Compartmentalization of adenosine 3’:5’monophosphate and adenosine 3’:5’-monophosphate-dependent protein kinase in heart tissue. The Journal of Biological Chemistry. 252, 3854–3861 (1977). [PubMed] [Google Scholar]
- 2.Terrin A et al. PGE1 stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: role of compartmentalized phosphodiesterases. The Journal of Cell Biology. 175, 441–451 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bacskai BJ et al. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science. 260, 222–226 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Iancu RV, Ramamurthy G, Harvey RD Spatial and temporal aspects of cAMP signalling in cardiac myocytes. Clinical and Experimental Pharmacology & Physiology. 35, 1343–1348 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Brunton LL, Hayes JS, Mayer SE Functional compartmentation of cyclic AMP and protein kinase in heart. Advances in Cyclic Nucleotide Research. 14, 391–397 (1981). [PubMed] [Google Scholar]
- 6.Hohl CM, Li Q Compartmentation of cAMP in adult canine ventricular myocytes. Relation to single-cell free Ca2+ transients. Circulation Research. 69, 1369–1379 (1991). [DOI] [PubMed] [Google Scholar]
- 7.Rich TC et al. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proceedings of the National Academy of Sciences of the United States of America. 98, 13049–13054 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sayner SL, Alexeyev M, Dessauer CW, Stevens T Soluble adenylyl cyclase reveals the significance of cAMP compartmentation on pulmonary microvascular endothelial cell barrier. Circulation Research. 98, 675–681 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Rich TC, Tse TE, Rohan JG, Schaack J, Karpen JW In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide–gated channels as cAMP sensors. The Journal of General Physiology. 118, 63–78 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blackman BE et al. PDE4D and PDE4B function in distinct subcellular compartments in mouse embryonic fibroblasts. Journal of Biological Chemistry. 286, 12590–12601 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sayner SL et al. Paradoxical cAMP-induced lung endothelial hyperpermeability revealed by Pseudomonas aeruginosa ExoY. Circulation Research. 95, 196–203 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Klarenbeek J, Jalink K Detecting cAMP with an EPAC-based FRET sensor in single living cells. Methods in Molecular Biology. 1071, 49–58 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Surdo NC et al. FRET biosensor uncovers cAMP nanodomains at β-adrenergic targets that dictate precise tuning of cardiac contractility. Nature Communications. 8, 15031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ponsioen B et al. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Reports. 5, 1176–1180 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogel SS, Thaler C, Koushik SV Fanciful FRET. Science’s STKE. 2006, re2 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Clegg RM The History of FRET: From conception through the labors of birth. Reviews in Fluorescence. Vol. 3 (2006). [Google Scholar]
- 17.Giepmans BNG, Adams SR, Ellisman MH, Tsien RY The fluorescent toolbox for assessing protein location and function. Science. 312, 217–224 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Manzella-Lapeira J, Brzostowski JA Imaging protein-protein interactions by Förster resonance energy transfer (FRET) microscopy in live cells. Current Protocols in Protein Science. 93, e58 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Cooper DMF, Mons N, Karpen JW Adenylyl cyclases and the interaction between calcium and cAMP signalling. Nature. 374, 421–424 (1995). [DOI] [PubMed] [Google Scholar]
- 20.Sassone-Corsi P Coupling gene expression to cAMP signalling: role of CREB and CREM. The International Journal of Biochemistry & Cell Biology. 30, 27–38 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Rebhun LI Cyclic nucleotides, calcium, and cell division. International Review of Cytology. 49, 1–54 (1977). [DOI] [PubMed] [Google Scholar]
- 22.Klarenbeek J, Goedhart J, Van Batenburg A, Groenewald D, Jalink K Fourth-generation Epac-based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLOS ONE. 10, e0122513 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leavesley SJ, Rich TC FRET: signals hidden within the noise. Cytometry Part A. 85, 918–920 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rich TC, Webb KJ, Leavesley SJ Can we decipher the information content contained within cyclic nucleotide signals? The Journal of General Physiology. 143, 17–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Annamdevula NS et al. Spectral imaging of FRET-based sensors reveals sustained cAMP gradients in three spatial dimensions. Cytometry Part A. 93(10), 1029–1038. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leavesley SJ, Britain AL, Cichon LK, Nikolaev VO, Rich TC Assessing FRET using spectral techniques. Cytometry Part A. 83, 898–912 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leavesley SJ, Rich TC Overcoming limitations of FRET measurements. Cytometry Part A. 89, 325–327 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fink DJ Monitoring Earcths Resources from Space. Technology Review. 75, 32–41 (1973). [Google Scholar]
- 29.Goetz AFH, Vane G, Solomon JE, Rock BN Imaging Spectrometry for Earth Remote Sensing. Science. 228, 1147–1153 (1985). [DOI] [PubMed] [Google Scholar]
- 30.Bücherl CA, Bader A, Westphal AH, Laptenok SP, Borst JW FRET-FLIM applications in plant systems. Protoplasma. 251, 383–394 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Chen Y, Mauldin JP, Day RN, Periasamy A Characterization of spectral FRET imaging microscopy for monitoring nuclear protein interactions. Journal of Microscopy. 228, 139–152 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zimmermann T, Rietdorf J, Girod A, Georget V, Pepperkok R Spectral imaging and linear un-mixing enables improved FRET efficiency with a novel GFP2–YFP FRET pair. FEBS Letters. 531, 245–249 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Griswold JR, Annamdevula N, Deal J, Rich T, Leavesley S Estimating FRET Efficiency using Excitation-Scanning Hyperspectral Imaging. Biophysical Journal. 112, 586a (2017). [Google Scholar]
- 34.Favreau PF et al. Excitation-scanning hyperspectral imaging microscope. Journal of Biomedical Optics. 19, 046010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.King J et al. Structural and functional characteristics of lung macro- and microvascular endothelial cell phenotypes. Microvascular Research. 67, 139–151 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Thaler C, Koushik SV, Blank PS, Vogel SS Quantitative multiphoton spectral imaging and its use for measuring resonance energy transfer. Biophysical Journal. 89, 2736–2749 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agarwal SR et al. Compartmentalized cAMP signaling associated with lipid raft and non-raft membrane domains in adult ventricular myocytes. Frontiers in Pharmacology. 9, 332 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnstone TB, Agarwal SR, Harvey RD, Ostrom RS cAMP signaling compartmentation: Adenylyl cyclases as anchors of dynamic signaling complexes. Mol Pharmacol. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Li H, Chai L, Zhang L, Qu J, Chen T Quantitative FRET measurement using emission-spectral unmixing with independent excitation crosstalk correction. Journal of Microscopy. 257, 104–116 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Lin F, Chai L, Wei L, Chen T IIem-spFRET: improved Iem-spFRET method for robust FRET measurement. Journal of Biomedical Optics. 21, 105003 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Levy S et al. SpRET: highly sensitive and reliable spectral measurement of absolute FRET efficiency. Microscopy and Microanalysis. 17, 176–190 (2011). [DOI] [PubMed] [Google Scholar]
- 42.West SJ et al. Hyperspectral Measurements Allow Separation of FRET Signals from Non-Uniform Background Fluorescence. Biophysical Journal. 112, 453a (2017). [Google Scholar]
- 43.Annamdevula NS et al. An approach for characterizing and comparing hyperspectral microscopy systems. Sensors (Basel). 13, 9267–9293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Measurements of laser line intensity as a function of laser setting in the software. A fiber-coupled spectrometer and integrating sphere were calibrated to a NIST-traceable lamp to measure laser intensity for both the 405 nm laser line and the 488 nm laser line. A) Total number of photons measured at the microscope stage corresponding to different laser intensities of the 405 nm laser. B) Total number of photons measured at the microscope stage corresponding to different laser intensities of 488 nm laser. Linear response in laser intensity was observed for both the lasers as measured at the microscope stage. A linear trendline was fit and the trendline equation for each laser line was used to calculate the acceptor spectrum that would be expected if excited with the 405nm laser line (donor excitation).
Supplemental Figure 2: Forskolin-induced FRET efficiency and cAMP spatial gradients visualized in the axial direction. XZ plane images were generated by reslicing 3-dimensional reconstructed FRET and cAMP image data in the lateral (Y) direction (from front to back of the cell). Columns 1 and 3 represent the FRET efficiency at baseline and at 10 minutes after 50 μM forskolin treatment, respectively. Columns 2 and 4 represent the cAMP levels at baseline and at 10 minutes after forskolin treatment. The color bars at right were used to relate changes in the colormap to FRET efficiency (top) and cAMP levels (bottom). Similar to results shown for the YZ planes (Figure 8), apical to basal spatial gradients in FRET efficiency and cAMP levels may be visualized as changes in color from the top to bottom of a single YZ slice, both at baseline conditions (any given slice in columns 1–2) and after 50 μM forskolin treatment (columns 3–4).