

Abstract
Pancreatic beta cells are the only cell type in our body capable of producing and secreting insulin to instruct the insulin-sensitive cells and tissues of our bodies to absorb nutrients after a meal. Accurate control of insulin release is of critical importance; too little insulin leads to diabetes, while an excess of insulin can cause potentially fatal hypoglycemia. Yet, the pancreas of most people will control insulin secretion safely and effectively over decades and in response to glucose excursions driven by tens of thousands of meals. Because we only become aware of the important contributions of the pancreas when it fails to maintain glucose homeostasis, it is easy to forget just how well insulin release from a healthy pancreas is matched to insulin need to ensure stable glycemia. Beta cells achieve this feat by extensive crosstalk with the rest of the endocrine cell types in the islet, notably the glucagon-producing alpha cells and somatostatin-producing delta cells. Here I will review the important paracrine contributions that each of these cells makes to the stimulation and subsequent inhibition of insulin release in response to a transient nutrient stimulation, and make the case that a breakdown of this local crosstalk contributes to the pathophysiology of diabetes.
Keywords: pancreatic islet, glucagon, somatostatin, GABA, serotonin, crosstalk
Tweet:
New review on the paracrine crosstalk within the pancreatic islet between alpha, beta, and delta cells that regulates insulin and glucagon secretion by Mark Huising from the University of California Davis @Huisinglab #diabetes #somatostatin #crosstalk
Introduction
Because insulin deficiency is the common denominator across different forms of diabetes, the lion’s share of attention in the diabetes field for the pancreatic islets of Langerhans has – understandably – been focused on the beta cells. However, the islets are more than just the home of the insulin-producing beta cells. They contain several additional endocrine cell types, most notably glucagon-producing alpha cells and somatostatin-producing delta cells. These three endocrine cells share a common developmental origin and coordinate their activity in response to changes in glucose and other nutrients (Figure 1). The mechanisms of this local coordination among alpha, beta, and delta cells have long been understudied, but recent observations have started to shed more light on the (patho)physiological importance of the paracrine crosstalk between the three major endocrine cell types of the islet. Islets also contain lower numbers of cells that produce pancreatic polypeptide, and epsilon cells that produce ghrelin, whose contributions to islet biology are reviewed elsewhere [1, 2]. The endocrine pancreas is supported by endothelial cells, pericytes, glial cells, and resident macrophages that together mold the islets into veritable miniature organs that are well-vascularized and innervated. All of these cells provide additional contributions to the chorus of signals that provides indispensable contributions to islet function that for space constraints I simply acknowledge here [3–6]. Instead, the intent in this short review is to provide a framework of how coordinated release of insulin and glucagon is fundamentally organized around the beta cell-autonomous glucose threshold and its paracrine interplay with alpha, and delta cells.
Figure 1:
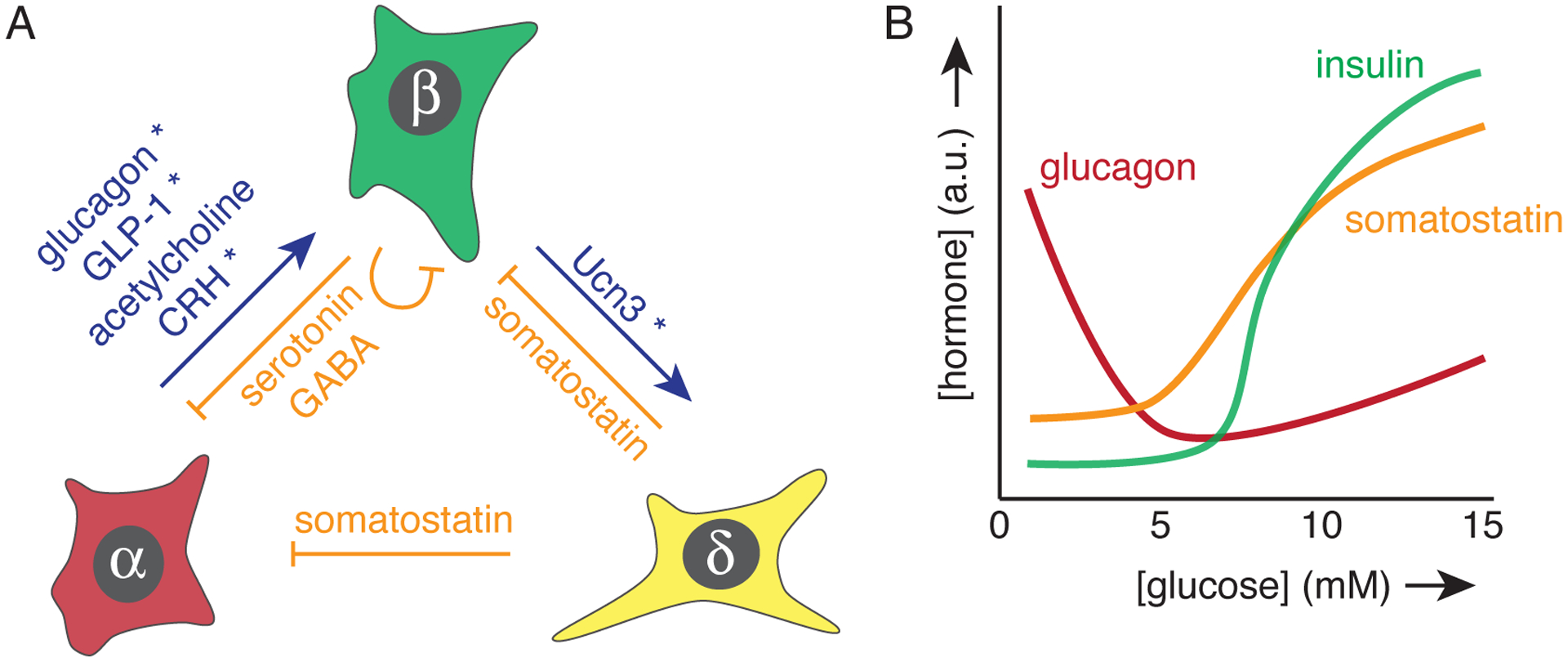
Overview of pancreatic alpha, beta, and delta cells and the main paracrine signals they exchange, and their relationship with glucose. (a) Blue and orange arrows indicate stimulation and inhibition, respectively. Signals that depend on concurrent glucose stimulation of beta cells are denoted by an asterisks. (b) Secretion from each endocrine cell is co-determined by glucose concentrations, as indicated schematically.
Nutrient stimulation and paracrine signals are inextricably intertwined
Many textbooks present insulin as the major anabolic hormone that is released in response to hyperglycemia in the post-prandial phase. The prevailing view of glucagon is that of a key counterregulatory hormone that prevents hypoglycemia in the fasted state by increasing hepatic glucose output by stimulating gluconeogenesis and glycogenolysis [7]. It is therefore no coincidence that endocrine cell types that are the source of arguably the two most important glucoregulatory hormones in our body colocalize to pancreatic islets. This arrangement facilitates the coordination of alpha and beta cell activity through paracrine crosstalk at the source of insulin and glucagon release. This paracrine crosstalk is so pervasive that even a measure as simple as glucose-stimulated insulin secretion (GSIS) is much more complicated than the mere glucose stimulation of beta cells to release insulin. In actuality GSIS is the net result of glucose stimulation of beta cells, amplified by the potentiating actions of alpha cell derived peptide hormones that are simultaneously restrained by the inhibitory actions of somatostatin from delta cells, as reviewed below.
The contributions of glucagon cells to insulin secretion
By increasing hepatic glucose output, glucagon acts in a manner that is functionally antagonistic to the anabolic actions of insulin. From a physiological perspective, it is therefore somewhat paradoxical that glucagon stimulates, rather than inhibits beta cells, as has been known since the mid 1960s [8]. However, from a mechanistic point of view it is not at all surprising that glucagon stimulates insulin release, if one considers that glucagon signals via class B G protein-coupled receptors (both the glucagon and glucagon-like peptide 1 receptors, see below) that commonly activate Gαs-mediated downstream signaling cascades. These actions of glucagon resemble those of the glucagon-like peptide 1 (GLP-1), the incretin hormone well-known to potentiate GSIS that is processed from the same prepro-glucagon precursor by the enteroendocrine L cells in the small intestine. Beta cells express both Glp1r and Gcgr [1, 9], and glucagon can activate either receptor to generate cyclic AMP to promote the recruitment and fusion of additional insulin granules from the beta cells, thus potentiating GSIS [10]. This potentiation only occurs when mildly hyperglycemic conditions depolarize the beta cell via the closure of ATP-sensitive K+ leak channels, this opens voltage-gated Ca2+ channels, causing an influx of Ca2+ that is necessary to trigger insulin release. This mechanism ensures that insulin release is only stimulated when dictated by nutrient excess. The intra-islet contribution of glucagon to insulin secretion are therefore limited to the post-prandial phase where glucagon can potentiate GSIS [11]. In contrast, glucagon released as part of the counterregulatory response to hypoglycemia will not stimulate insulin secretion, which is appropriate as additional insulin would be highly detrimental under such circumstances.
While the stimulatory actions of glucagon on beta cells had been known, the physiological relevance of this alpha to beta stimulation has recently been revisited by a number of different groups using an impressive array of parallel models designed to block alpha cells from contributing to insulin secretion. These include the genetic deletion of the glucagon or GLP-1 receptors from beta cells [10, 12], pharmacological blockade of the Glp1r by its antagonist Exendin9–39 [10, 12, 13], genetic deletion of the gcg gene [10] to remove the source of glucagon, and silencing of alpha cells in a pharmacogenetic approach to inhibit glucagon secretion in vivo [14]. Collectively, the observations from these complementary models by multiple groups demonstrated that the perturbation of intra-islet stimulation of beta cells by glucagon from alpha cells led to marked reductions in insulin release in response to nutrient stimulation [10, 12, 14] that precipitated glucose intolerance in vivo. These observations require a recalibration of our view of the role of alpha cells. In addition to glucagon’s established physiological contributions as an essential counterregulatory hormone to fend off hypoglycemia via its systemic gluconeogenic and glycogenolytic effects on liver, it is now equally clear that locally within the islet, endogenous glucagon potentiates insulin secretion during hyperglycemia. Of course, injection of glucagon in response to acute hypoglycemia will increase glucose concentrations driven by increased hepatic glucose output. Glucagon-mediated insulin release is less of a factor in this scenario as the hypoglycemia would preclude glucagon-mediated potentiation of GSIS, and the need for counterregulatory glucagon is most often encountered in type 1 diabetes patients who have limited residual functional beta cell mass.
Other alpha cell-derived signals that stimulate insulin secretion
Glucagon is not the only alpha cell-derived signal that is capable of stimulating the beta cell. I will briefly review three additional paracrine signals to re-enforce the notion that multiple local pathways exist by which the alpha cell stimulates the beta cell. The first of these is GLP-1, which is derived from the same preproglucagon progenitor as glucagon upon processing by prohormone convertase 1/3 (PC1/3) instead of PC2, is detectable at lower levels in human islets [15]. However, such a shift in preproglugagon processing may well be of physiological significance and could have therapeutic potential: both glucagon and GLP-1 can potentiate GSIS, principally by engaging the beta cell Glp1r, while increased hepatic glucose production is stimulated largely via the Gcgr that has a low affinity for GLP-1 compared to glucagon [10, 12].
In addition to the expression of GLP-1, human alpha cells also robustly express the peptide hormone corticotropin-releasing hormone (CRH) [16, 17]. CRH stimulates the type 1 CRH receptor (Crhr1), which is a class B GPCR related to the Glp1r that is expressed by beta cells and stimulates insulin release in an incretin-like manner [18]. This represents another avenue for alpha cells to potentiate GSIS from beta cells. Whether CRH/Crhr1 represent a paracrine mechanism that is merely redundant to the preproglucagon-derived peptides discussed above, or perhaps coordinates the activity of the counterregulatory hormones glucagon and cortisol by activating the hypothalamus-pituitary-adrenal axis has not yet been established. Nevertheless, it is yet another example that illustrates how alpha cells release a multitude of signals that stimulate insulin release from beta cells via local crosstalk.
The parasympathetic neurotransmitter acetylcholine is – at least in human islets – released from alpha cells [19]. Moreover, most human alpha cells express the requisite cholinergic markers such as choline acyltransferase (ChAT) or vesicular acetylcholine transporter (vAChT), supporting the notion that human alpha cells synthesize and release acetylcholine. The effect of acetylcholine from alpha cells on beta cells is to prime the beta cell to maintain responsiveness to subsequent glucose stimulation [19]. This is analogous to what one might expect from acetylcholine released from the parasympathetic nerve terminals upon beta cells in other species [20], but with the notable twist that in humans the alpha cell is the main acetylcholine source within the islet.
The contributions of delta cell-derived somatostatin
The fact that somatostatin is a robust inhibitor of both insulin and glucagon secretion was recognized shortly after somatostatin was discovered in pancreatic delta cells [21]. However, the impact of somatostatin on regulating glucagon and insulin secretion is only now coming into focus, with delta cells emerging as exerting key paracrine modulatory effects on insulin and glucagon secretion. Indeed, while somatostatin is detectable in systemic circulation, pancreatectomy does not significantly reduce circulating somatostatin [22], offering further support for the notion that pancreatic delta cell-derived somatostatin serves a largely paracrine role within the islet as a local regulator of insulin and glucagon release. In recent years we have learned that delta cells express a multitude of receptors to hormones and neurotransmitters such as ghrelin, dopamine, acetylcholine and leptin [9, 23, 24]. These insights suggest that delta cells act a central signaling hubs within the islet that integrate input from a multitude of signals including nutrients, neurotransmitters, hormones, and local factors into situationally appropriate somatostatin release to modulate the activity of neighboring beta and/or alpha cells.
The effects of somatostatin on beta cells.
Somatostatin secretion is stimulated by glucose, similar to the secretion of insulin from beta cells. Furthermore, the response of delta cells to glucose tracks the pulsatile, coordinated response of pancreatic beta cells that within one islet are all connected by gap junctions. Glucagon secretion under these conditions is also pulsatile but shifted in phase compared to the near-synchronous pulses of insulin and somatostatin [25, 26]. It has been suggested that this coordination between beta and delta cells is mediated by direct gap junction [27]. However, glucose-stimulated calcium responses are asynchronous between beta and delta cells [28] and somatostatin secretion trails secretion of insulin by 30 seconds to several minutes [25]. These observations suggests that less immediate mechanisms of crosstalk are responsible for the general coordination between beta cell and delta cell secretory responses to hyperglycemia. One such mechanism is the local release of another beta cell hormone, Urocortin3 (Ucn3), which is a peptide hormone that is related to CRH. Ucn3 selectively activates the type 2 CRH receptor (Crhr2), another class B GPCR that within the islet is selectively expressed by pancreatic delta cells [1, 29]. Upon beta cell stimulation Ucn3 is co-released with insulin to activate delta cells. Such a paracrine mechanism would explain the modest delay that separates glucose-stimulated insulin and somatostatin secretion [25]. In essence, this paracrine mechanism constitutes a local brake on the beta cell and couples beta cell activation to the activation of the inhibitory mechanism that helps restrain insulin secretion. Because the paracrine nature of this crosstalk takes some time, this negative feedback loop would not prevent glucose from stimulating insulin secretion. Instead, the purpose of this paracrine negative feedback loop that is triggered by beta cell activation and mediated by delta cells is to provide feedback inhibition to the beta cell to counterbalance nutrient stimulation [30]. After all, insulin can cause severe, even lethal hypoglycemia when released or administered in excess. Local feedback inhibition by somatostatin ensures that insulin release is timely attenuated once peripheral insulin action has succeeded in restoring normoglycemia, thus preventing excess insulin release that could lead to hypoglycemia.
The effects of somatostatin on alpha cells.
Somatostatin also is a potent inhibitor of glucagon release across the glucose spectrum. The mechanism of this inhibition depends on whether glucagon is released in response to hypoglycemia or during hyperglycemia. Hypoglycemia triggers glucagon release via an alpha cell-autonomous mechanism that is incompletely understood. Moreover, counterregulatory glucagon release in response to hypoglycemia in vivo is likely driven in part by the release of hormones such as epinephrine or arginine vasopressin (AVP), which are potent and direct systemic stimulators of glucagon secretion [31–33]. Nevertheless, somatostatin exerts paracrine inhibitory effects on alpha cells under hypoglycemia, as somatostatin antagonists enhance glucagon release in response to low glucose stimulation [34, 35]. This inhibition is of pathophysiological relevance, as the blockade of endogenous somatostatin by antagonists selective to the Sstr2 receptors that is selectively expressed by alpha cells suffices to restore defective counterregulatory glucagon secretion [36, 37]. Under hyperglycemic circumstances, glucagon secretion is actually also stimulated, albeit normally not as potently as in response to hypoglycemia. As discussed, this modest glucagon release in response to high glucose ensures full glucose-stimulated insulin secretion [10, 12, 14]. It turns out that this glucose-stimulated release of glucagon is subject to paracrine inhibition by somatostatin that dampens this glucagon response during hyperglycemia without fully suppressing it. Indeed, blockade of the actions of endogenous somatostatin causes a significantly more robust glucagon release under resting glucose [35] or under co-stimulation by high glucose and amino acids [34]. Moreover, the aforementioned pulsatile pattern of glucagon release under hyperglycemia that emerges upon more frequent sampling occurs antiparallel with pulsatile release of insulin and somatostatin [25, 26], and is likely mediated directly by the inhibitory actions of somatostatin on alpha cells [38].
Autocrine actions of beta cell-produced neurotransmitters
While I have focused predominantly on the paracrine crosstalk that contributes to the regulation of insulin and glucagon secretion, I would be remiss not to briefly mention the contributions of a several neurotransmitters that are produced and released locally by beta cells within the islet. These include serotonin (5-HT) and the inhibitory neurotransmitter GABA. Beta cells possess the enzymatic machinery to produce 5-HT and store it in insulin vesicles for co-release with insulin upon beta cell stimulation. In human islets, 5-HT serves a paracrine role by inhibiting glucagon release from alpha cells [39, 40]. In mice, 5-HT production is normally low, but a notable increase in the local production of 5-HT by mouse beta cells is responsible for the robust increase in beta cell mass during pregnancy [41] and drives the perinatal expansion of beta cell mass that determines adult beta cell mass [42]. GABA is synthesized by glutamic acid decarboxylase (GAD) expressed by beta cells (in fact, GAD is an autoantigen in Type 1 Diabetes). GABA inhibits alpha cells, and this mechanism contributes to the suppression of glucagon during hyperglycemia [43]. However, a recent publication demonstrated that the majority of GABA produced by the islet is not stored in secretory vesicles and instead is present in a cytoplasmic pool from where it is released continuously in a pulsatile manner independent of glucose concentration [44]. The pulsatile nature of this GABA secretion is suggested to play a dual role in 1) entraining pulsatility to the beta cell response while 2) directly inhibiting insulin secretion, which was coincidently detected using 5-HT as a proxy for insulin secretion.
Putting it all together
Despite the important paracrine contributions from alpha and delta cells to modulate insulin release that I have discussed so far, an integration of the signals that control when and how much insulin is released has to start with the cell-autonomous response of beta cells to nutrient stimulation. When glucose concentrations exceed the glucose threshold of the beta cell, the rise in ATP and accompanying drop in ADP that results from glucose metabolism closes the ATP-sensitive potassium channels, which depolarizes the beta cell and triggers insulin secretion [45]. Only under these circumstances where glucose-mediated calcium influx triggers secretion can signals mediated by the Gαs-coupled GPCRs discussed earlier potentiate insulin release. This is the very mechanism that prevents incretins - or therapeutics based on incretin mimetics – from promoting insulin secretion unless accompanied by hyperglycemia. This also explains why high levels of glucagon that are released as part of a counterregulatory response to hypoglycemia pass by the beta cell without stimulating insulin secretion, despite the presence of Gcgr and Glp1r on beta cells. In contrast, glucagon (or other alpha cell derived peptide hormones) released in more moderate quantities under hyperglycemia do potentiate GSIS as part of a local amplification mechanism to match the strength of the glucose stimulation with a proportionate insulin response [1, 11]. However, there is a fine line between affording enough glucagon release to potentiate GSIS locally without raising circulating glucagon levels to the point where they would increase hepatic glucose output. The tonic inhibition (without full suppression) of glucagon secretion by a combination of somatostatin, GABA and serotonin could constitute the mechanism that allows some glucagon release in response to hyperglycemia to potentiate GSIS via its paracrine effects on beta cells, without causing increased hepatic glucose output that would aggravate hyperglycemia. Whether it matters for this alpha cell-dependent intra-islet amplification mechanism of insulin release during hyperglycemia that glucagon and insulin are released out of phase in a pulses of approximately five minutes remains to be determined. It is possible that the glucagon levels, even at their nadir, still suffice to amplify GSIS, but it is important that this is determined empirically.
The potential pathophysiological relevance of these paracrine mechanisms that depend on the interplay between glucagon and somatostatin under hyperglycemic conditions is directly relevant to diabetes, where glucagon released in excess contributes significantly to hyperglycemia [46, 47]. It is possible that this inappropriate release of glucagon in response to hyperglycemia reflects the normal inclination of alpha cells to release glucagon in response to hyperglycemia that has turned maladaptive as the local feedback mechanisms that restrain it have broken down in diabetes [1]. Indeed, recent observations that the paracrine inhibition of alpha cells by somatostatin is compromised in human type 2 diabetes offers further support exactly such a scenario [48]. Of course changes in the paracrine landscape that explain the aberrant regulation of glucagon release at both ends of the glucose spectrum are not mutually exclusive with observations of alpha cell autonomous changes in gene and protein expression during diabetes [49].
Conclusions
According to the definition that a hormone is a factor that is released at one site to travel via the circulation to a target cell some distance away, one could argue that the islet releases only two true hormones: insulin and glucagon (a similar case could be made for amylin, which was not covered in this short review). Nevertheless, there is rich paracrine crosstalk amongst all of the major endocrine cells and their non-endocrine support cells that modulates and coordinates beta and alpha cell activity under both hypoglycemic and hyperglycemic conditions. In fact, this crosstalk is so intertwined with nutrient stimulation of insulin and glucagon that it becomes nearly impossible to separate nutrient stimulation of beta cells, from the paracrine connections that are activated in parallel. The take home message from this short review is hopefully a recognition of the fact that every time one simply stimulates islets in vitro with glucose, the ensuing ‘glucose’-stimulated insulin secretion is in fact the net result of an integrated response that involves both stimulation by alpha cells and inhibition by delta cells (Figure 2). While the contributions of alpha and delta cells to GSIS are often unappreciated and hard to isolate, the breakdown of these paracrine connections contributes to dysregulated insulin and glucagon secretory responses in diabetes.
Figure 2:
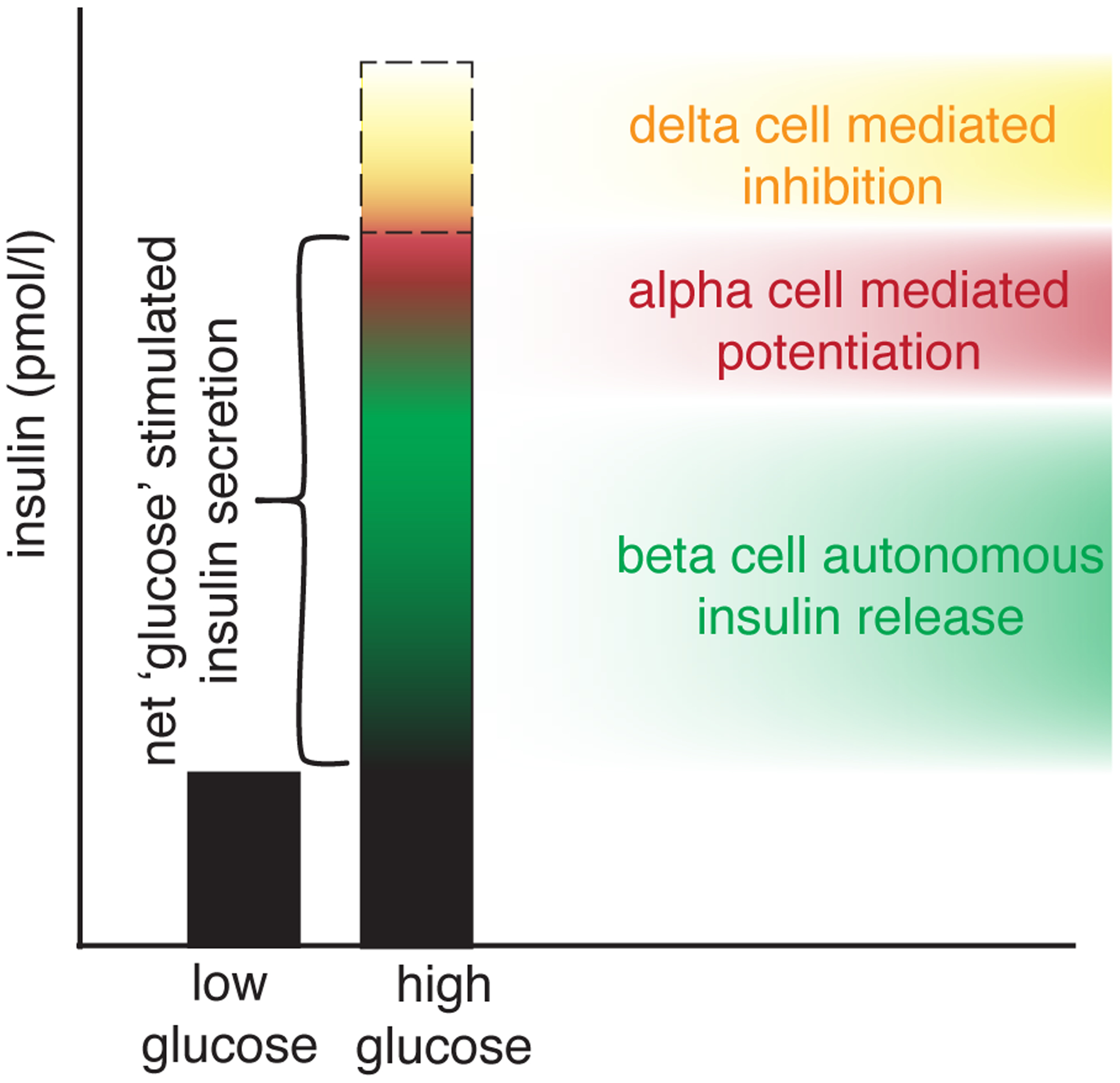
Glucose-stimulated insulin secretion is one of the most common measurements in the islet biology field. The name of the response implies a straightforward response with glucose as input and insulin as output (a). In reality, a number of alpha cell-derived stimulatory signals (red) offset by paracrine inhibition by delta cells determines the net overall insulin response to glucose stimulation of intact islets.
Acknowledgements
I apologise to those whose work was not discussed here due to limitations in space and scope. Work discussed in this Review was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK-110276) and the Juvenile Diabetes Research Foundation (2-SRA-2019-700-S-B). I thank Jessica Huang and Dr. Talitha van der Meulen (both Neurobiology, Physiology, and Behavior, UC Davis, USA) for a critical reading of the manuscript.
Work in the author’s laboratory is supported by the National Institute of Diabetes and Digestive and Kidney Disease (NID DK-110276), the Juvenile Diabetes Research Foundation (CDA-2-2013-54 and 2-SRA-2019-700-S-B), and the American Diabetes Association (#1-19-IBS-078).
Footnotes
Contribution statement
The author was the sole contributor to this paper and has no conflicts to declare.
References
- [1].Noguchi GM, Huising MO (2020) Integrating the inputs that shape pancreatic islet hormone release. Nature Metabolism 1(12): 1189–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hartig SM, Cox AR (2020) Paracrine signaling in islet function and survival. J Mol Med in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Almaca J, Weitz J, Rodriguez-Diaz R, Pereira E, Caicedo A (2018) The Pericyte of the Pancreatic Islet Regulates Capillary Diameter and Local Blood Flow. Cell Metab 27(3): 630–644 e634. 10.1016/j.cmet.2018.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Weitz JR, Makhmutova M, Almaca J, et al. (2018) Mouse pancreatic islet macrophages use locally released ATP to monitor beta cell activity. Diabetologia 61(1): 182–192. 10.1007/s00125-017-4416-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hogan MF, Hull RL (2017) The islet endothelial cell: a novel contributor to beta cell secretory dysfunction in diabetes. Diabetologia 60(6): 952–959. 10.1007/s00125-017-4272-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Brissova M, Aamodt K, Brahmachary P, et al. (2014) Islet microenvironment, modulated by vascular endothelial growth factor-A signaling, promotes beta cell regeneration. Cell Metab 19(3): 498–511. 10.1016/j.cmet.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Briant L, Salehi A, Vergari E, Zhang Q, Rorsman P (2016) Glucagon secretion from pancreatic alpha-cells. Ups J Med Sci 121(2): 113–119. 10.3109/03009734.2016.1156789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Samols E, Marri G, Marks V (1965) Promotion of Insulin Secretion by Glucagon. Lancet 2(7409): 415–416 [DOI] [PubMed] [Google Scholar]
- [9].DiGruccio MR, Mawla AM, Donaldson CJ, et al. (2016) Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol Metab 5(7): 449–458. 10.1016/j.molmet.2016.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cappozi ME, Svendsen B, Encisco SE, et al. (2019) Beta-cell tone is defined by proglucagon peptides through cyclic AMP signalling. JCI Insight 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Capozzi ME, Wait JB, Koech J, et al. (2019) Glucagon lowers glycemia when beta-cells are active. JCI Insight 5. 10.1172/jci.insight.129954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Svendsen B, Larsen O, Gabe MBN, et al. (2018) Insulin Secretion Depends on Intra-islet Glucagon Signaling. Cell reports 25(5) [DOI] [PubMed] [Google Scholar]
- [13].Chambers AP, Sorrell JE, Haller A, et al. (2017) The Role of Pancreatic Preproglucagon in Glucose Homeostasis in Mice. Cell Metab 25(4): 927–934 e923. 10.1016/j.cmet.2017.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhu L, Dattaroy D, Pham J, et al. (2019) Intra-islet glucagon signaling is critical for maintaining glucose homeostasis. JCI Insight 23(5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Marchetti P, Lupi R, Bugliani M, et al. (2012) A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia 55(12): 3262–3272. 10.1007/s00125-012-2716-9 [DOI] [PubMed] [Google Scholar]
- [16].Benner C, van der Meulen T, Caceres E, Tigyi K, Donaldson CJ, Huising MO (2014) The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC Genomics 15(1): 620. 10.1186/1471-2164-15-620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nica AC, Ongen H, Irminger JC, et al. (2013) Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res 23(9): 1554–1562. 10.1101/gr.150706.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Huising MO, van der Meulen T, Vaughan JM, et al. (2010) CRFR1 is expressed on pancreatic beta cells, promotes beta cell proliferation, and potentiates insulin secretion in a glucose-dependent manner. Proc Natl Acad Sci USA 107(2): 912–917. 0913610107 [pii] 10.1073/pnas.0913610107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rodriguez-Diaz R, Dando R, Jacques-Silva MC, et al. (2011) Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nature medicine 17(7): 888–892. nm.2371 [pii] 10.1038/nm.2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ahren B (2000) Autonomic regulation of islet hormone secretion--implications for health and disease. Diabetologia 43(4): 393–410. 10.1007/s001250051322 [DOI] [PubMed] [Google Scholar]
- [21].Rorsman P, Huising MO (2018) The somatostatin-secreting pancreatic delta-cell in health and disease. Nat Rev Endocrinol 14(7): 404–414. 10.1038/s41574-018-0020-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Taborsky GJ Jr., Ensinck JW (1984) Contribution of the pancreas to circulating somatostatin-like immunoreactivity in the normal dog. J Clin Invest 73(1): 216–223. 10.1172/JCI111194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Adriaenssens AE, Svendsen B, Lam BY, et al. (2016) Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia 59(10): 2156–2165. 10.1007/s00125-016-4033-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lawlor N, George J, Bolisetty M, et al. (2017) Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res 27(2): 208–222. 10.1101/gr.212720.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Salehi A, Qader SS, Grapengiesser E, Hellman B (2007) Pulses of somatostatin release are slightly delayed compared with insulin and antisynchronous to glucagon. Regulatory peptides 144(1–3): 43–49. 10.1016/j.regpep.2007.06.003 [DOI] [PubMed] [Google Scholar]
- [26].Hellman B, Salehi A, Grapengiesser E, Gylfe E (2012) Isolated mouse islets respond to glucose with an initial peak of glucagon release followed by pulses of insulin and somatostatin in antisynchrony with glucagon. Biochem Biophys Res Commun 417(4): 1219–1223. 10.1016/j.bbrc.2011.12.113 [DOI] [PubMed] [Google Scholar]
- [27].Briant LJB, Reinbothe TM, Spiliotis I, Miranda C, Rodriguez B, Rorsman P (2018) delta-cells and beta-cells are electrically coupled and regulate alpha-cell activity via somatostatin. J Physiol 596(2): 197–215. 10.1113/JP274581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nadal A, Quesada I, Soria B (1999) Homologous and heterologous asynchronicity between identified alpha-, beta- and delta-cells within intact islets of Langerhans in the mouse. J Physiol 517 (Pt 1): 85–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].van der Meulen T, Donaldson CJ, Caceres E, et al. (2015) Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat Med 21(7): 769–776. 10.1038/nm.3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Huising MO, van der Meulen T, Huang JL, Pourhosseinzadeh MS, Noguchi GM (2018) The Difference delta-Cells Make in Glucose Control. Physiology (Bethesda) 33(6): 403–411. 10.1152/physiol.00029.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].van der Meulen T, Mawla AM, DiGruccio MR, et al. (2017) Virgin Beta Cells Persist throughout Life at a Neogenic Niche within Pancreatic Islets. Cell Metab 25(4): 911–926 e916. 10.1016/j.cmet.2017.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dunning BE, Moltz JH, Fawcett CP (1984) Actions of neurohypophysial peptides on pancreatic hormone release. Am J Physiol 246(1 Pt 1): E108–114 [DOI] [PubMed] [Google Scholar]
- [33].Gilon P (2020) The Role of alpha-Cells in Islet Function and Glucose Homeostasis in Health and Type 2 Diabetes. J Mol Biol 432(5): 1367–1394. 10.1016/j.jmb.2020.01.004 [DOI] [PubMed] [Google Scholar]
- [34].Lai BK, Chae H, Gomez-Ruiz A, et al. (2018) Somatostatin Is Only Partly Required for the Glucagonostatic Effect of Glucose but Is Necessary for the Glucagonostatic Effect of KATP Channel Blockers. Diabetes 67(11): 2239–2253. 10.2337/db17-0880 [DOI] [PubMed] [Google Scholar]
- [35].Xu SFS, Andersen DB, Izarzugaza JMG, Kuhre RE, Holst JJ (2020) In the rat pancreas, somatostatin tonically inhibits glucagon secretion and is required for glucose-induced inhibition of glucagon secretion. Acta Physiol (Oxf): e13464. 10.1111/apha.13464 [DOI] [PubMed] [Google Scholar]
- [36].Yue JT, Riddell MC, Burdett E, Coy DH, Efendic S, Vranic M (2013) Amelioration of hypoglycemia via somatostatin receptor type 2 antagonism in recurrently hypoglycemic diabetic rats. Diabetes 62(7): 2215–2222. 10.2337/db12-1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Karimian N, Qin T, Liang T, et al. (2013) Somatostatin receptor type 2 antagonism improves glucagon counterregulation in biobreeding diabetic rats. Diabetes 62(8): 2968–2977. 10.2337/db13-0164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gylfe E, Tengholm A (2014) Neurotransmitter control of islet hormone pulsatility. Diabetes Obes Metab 16 Suppl 1: 102–110. 10.1111/dom.12345 [DOI] [PubMed] [Google Scholar]
- [39].Almaca J, Molina J, Menegaz D, et al. (2016) Human Beta Cells Produce and Release Serotonin to Inhibit Glucagon Secretion from Alpha Cells. Cell reports 17(12): 3281–3291. 10.1016/j.celrep.2016.11.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bennet H, Balhuizen A, Medina A, et al. (2015) Altered serotonin (5-HT) 1D and 2A receptor expression may contribute to defective insulin and glucagon secretion in human type 2 diabetes. Peptides 71: 113–120. 10.1016/j.peptides.2015.07.008 [DOI] [PubMed] [Google Scholar]
- [41].Kim H, Toyofuku Y, Lynn FC, et al. (2010) Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med 16(7): 804–808. nm.2173 [pii] 10.1038/nm.2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Moon JH, Kim YG, Kim K, et al. (2020) Serotonin Regulates Adult beta-Cell Mass by Stimulating Perinatal beta-Cell Proliferation. Diabetes 69(2): 205–214. 10.2337/db19-0546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rorsman P, Berggren PO, Bokvist K, et al. (1989) Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature 341(6239): 233–236. 10.1038/341233a0 [DOI] [PubMed] [Google Scholar]
- [44].Menegaz D, Walker Hagan D, Almaca J, et al. (2019) Mechanism and effects of pulsatile GABA secretion from cytosolic pools in the human beta cell. Nature Metabolism 1: 1110–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rorsman P, Ashcroft FM (2018) Pancreatic beta-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol Rev 98(1): 117–214. 10.1152/physrev.00008.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Unger RH, Cherrington AD (2012) Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest 122(1): 4–12. 60016 [pii] 10.1172/JCI60016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Unger RH, Orci L (2010) Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci USA 107(37): 16009–16012. 1006639107 [pii] 10.1073/pnas.1006639107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Omar-Hmeadi M, Lund PE, Gandasi NR, Tengholm A, Barg S (2020) Paracrine control of alpha-cell glucagon exocytosis is compromised in human type-2 diabetes. Nat Commun 11(1): 1896. 10.1038/s41467-020-15717-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Brissova M, Haliyur R, Saunders D, et al. (2018) alpha Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell reports 22(10): 2667–2676. 10.1016/j.celrep.2018.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]