

Abstract
Background
The neuropsychiatric syndrome mild behavioral impairment (MBI) describes an at‐risk state for dementia and may be a useful screening tool for sample enrichment. We hypothesized that stratifying a cognitively normal sample on MBI status would enhance the association between genetic risk for Alzheimer's disease (AD) and cognition.
Methods
Data from 4458 participants over age 50 without dementia was analyzed. A cognitive composite score was constructed and the MBI Checklist was used to stratify those with MBI and those without. Polygenic scores for AD were generated using summary statistics from the IGAP study.
Results
AD genetic risk was associated with worse cognition in the MBI group but not in the no MBI group (MBI: β = –0.09, 95% confidence interval: –0.13 to –0.03, P = 0.002, R2 = 0.003). The strongest association was in those with more severe MBI aged ≥65.
Conclusions
MBI is an important feature of aging; screening on MBI may be a useful sample enrichment strategy for clinical research.
Keywords: Alzheimer's disease, cognition, mild behavioral impairment, neuropsychiatric symptoms, polygenic score
1. BACKGROUND
Worldwide, the number of people with dementia is expected to rise to 150 million by 2050. Recent years have been marked by a number of high‐profile failures of disease‐modifying therapies and it is now widely recognized that identification of people in the very earliest stages of disease is a key priority for clinical trials of new treatments, and ultimately for clinical practice. 1 Genetic predictors of cognitive decline and dementia have been the subject of considerable focus in recent years. These predictors include not only apolipoprotein E (APOE) status but also polygenic risk scores (PRS). PRS are the sum of Alzheimer's disease (AD) risk alleles carried by an individual weighted by effect size, and therefore capture more genetic risk than APOE alone. The ultimate goal of this work is the identification of a low‐cost early marker of neurodegenerative disease. As an important first step, numerous studies have shown that AD genomic markers predict AD and mild cognitive impairment (MCI) case/control status, as well as progression to AD among MCI cases. 2 , 3 , 4 , 5 , 6 , 7 However, the association between AD genetic risk and objectively measured cognition in non‐dementia samples is less consistent. 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 There are a number of methodological differences that likely explain these discrepancies, including the sensitivity of cognitive outcome measures and the number of risk alleles included in PRS calculation. One additional challenge is the complex etiology of cognition in older adults, which is not solely accounted for by genetic risk for neurodegeneration or neuropsychological profile. Because of the ease and low cost of genetic analysis, and promising initial findings, it is logical to explore additional strategies that may enhance sensitivity in older samples.
Previous research has shown that the association between APOE ε4 and cognition is strongest in older adults, one possible implication being that there are more people in older samples whose cognitive profile is linked to early neurodegeneration. 14 One study found that stratification based on amyloid beta (Aβ)‐positive positron emission tomography (PET) scans unmasked an association between AD PRS and poorer memory and executive function. 12 That neuropathological markers of AD moderate the association between AD genetic risk and neuropsychological measures is not surprising, but does offer proof of principle of sample enrichment, informing our study. To add value to a genetic screen, such sample enrichment should be low cost and scalable to large populations. There is evidence that later‐life emergent neuropsychiatric symptoms (NPS), described by the validated syndrome mild behavioral impairment (MBI), may represent such a screening tool. MBI is a neurobehavioral syndrome proposed by an Alzheimer's Association consensus group to describe a risk state for cognitive decline and dementia to facilitate earlier dementia detection. 16 The MBI syndrome covers late‐life–emergent apathy, mood/anxiety symptoms, impulse dyscontrol, social inappropriateness, and psychotic symptoms, and is common and easily measured in the general population. 17 Moreover, MBI is associated with progressive cognitive decline in individuals without significant cognitive impairment, a shorter time to dementia, and has recently been shown to associate with dementia biomarkers including PET amyloid, tau, and neurofilament light in cognitively normal samples. 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 On the basis of this evidence, MBI is an attractive candidate tool to enrich samples with individuals at greater risk of dementia, but to our knowledge no studies have explored how stratification on MBI influences the relationship between genetic risk for AD and cognition. In this study we aimed to elucidate whether stratifying a cognitively normal sample on the presence of MBI symptoms affected the association between genetic risk for AD and general cognitive ability. We hypothesized AD genetic risk would be associated with poorer cognition and that this relationship would be strongest among people with MBI symptoms.
2. METHODS
2.1. Participants
All participants were drawn from the PROTECT study (Research Ethics Committee reference number 13/LO/1578). PROTECT is a UK‐based online participant registry that tracks the cognitive health of older adults. Inclusion criteria for enrolling in PROTECT are (1) ≥50 years old, (2) no diagnosis of dementia, and (3) access to a computer and internet. The sample used in this study is a subset of PROTECT study participants who also provided a saliva sample for genotyping, completed cognitive testing, and had a proxy informant available to complete the MBI Checklist (MBI‐C; further detail is presented below). Specifically, genome‐wide genotype data for 9146 PROTECT participants was available for analysis in this study. Of these, clinical data required for this analysis from self‐report and proxy informant was available from 4458 people. The difference in numbers providing DNA and those with required clinical data is due to the MBI‐C being completed by proxy informant, and nomination of a proxy is optional. Written informed consent was obtained from all participants and proxy informants. The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Declaration of Helsinki 1975, as revised in 2008.
The following demographic data, which are routinely collected during PROTECT, were used in this analysis as covariates: age, sex, education level (school until 16, school until 18, vocational qualification, undergraduate degree, post‐graduate degree, and doctoral degree) and employment status (full time, part time, self‐employed, retired, and unemployed).
RESEARCH IN CONTEXT
Systematic review: we reviewed the literature using a PubMed and Google Scholar search. There is increasing evidence from clinical risk and biomarker studies that mild behavioral impairment (MBI) represents an at‐risk state for dementia and may be the first manifestation of neurodegenerative disease. It is therefore possible that MBI screening could represent a useful sample enrichment strategy for clinical studies and possibly trials. We tested this hypothesis by examining the relationship between genetic risk for Alzheimer's disease (AD) and cognition in people with MBI and in those without.
Interpretation: We show for the first time that stratification on MBI symptoms led to an enhanced signal between genetic risk for AD and cognition in a cognitively normal sample.
Future directions: We propose that screening on MBI symptoms may be a novel strategy for enriching samples for individuals at risk of dementia. Our findings and study design provide a framework for further investigation of this application of MBI assessment.
2.2. Assessment of cognition
Cognitive performance was assessed via a battery of four tests (Table 1). Individual performance across cognitive tests is known to be correlated, and for this study we analyzed a general cognitive composite based on factor analysis of the battery. This latent construct is a well‐documented feature of cognition. 27 To capture general cognitive ability in this sample, a composite score was calculated by computing the first unrotated principal component of the cognitive battery. The variance in total cognitive test score explained by the first principal component was 48.7% and the factor loadings were 0.53 (paired associated learning), 0.5 (digit span), 0.46 (self‐ordered search), and 0.50 (verbal reasoning). This finding is comparable to recent reports in an analysis of more than 300,000 individuals across multiple cohorts. 28 In the present study, higher cognitive composite score was associated with a lower likelihood of Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) score >3.3, which indicates a negative change in cognition over 10 years via a questionnaire observed by a proxy informant (odds ratio [OR] = 0.84, 95% confidence interval [CI]: 0.78–0.91 P < 0.001, controlling for age, sex, and education). It was also associated with greater impairment in instrumental activities of daily living (IADL) as measured by the Minimum Data Set–Home Care IADL scale (β = 0.156, 95% CI: 0.06–0.25 P = 0.001), providing validation that the cognitive construct is meaningful in the context of our sample. 29 , 30
TABLE 1.
Description of the cognitive battery used
| Test | Description | Cognitive domain |
|---|---|---|
| Paired associates learning | A series of objects appear in the cells on screen. The participant is instructed to remember the cell in which the object appears. When an object appears at the bottom center, the participant is instructed to click on the cell in which they recall seeing that object. | Visual working memory, learning |
| Digit span | Using a ratchet‐style approach in which each successful trial is followed by a new sequence that is 1 digit longer than the last and each unsuccessful trial is followed by a new sequence that is 1 digit shorter than the last. | Working memory |
| Self‐ordered search | A series of boxes are present on the screen; one of the boxes will contain a diamond. The participant selects each box until they locate the diamond. The diamond is then placed in another box and again the participant must locate it, but they must be careful not to select the box in which the diamond was previously found. | Executive function, spatial working memory |
| Verbal reasoning | A sentence is displayed at the bottom of the screen while a square and a circle are displayed above. The participant needs to respond true or false as to whether the sentence correctly describes the configuration of the circle and square. | Verbal reasoning |
2.3. Assessment of neuropsychiatric symptoms in the MBI framework
Neuropsychiatric symptoms were operationalized in the MBI framework using the MBI‐C. The MBI‐C is a validated tool designed specifically for capturing MBI symptoms and, in this study, was completed by a proxy informant who knew the participant well for at least 10 years. 17 , 31 , 32 , 33 The scale consists of 34 questions covering the full range of MBI domains (apathy, mood/anxiety symptoms, impulse dyscontrol, social inappropriateness, and psychotic symptoms). Each question is rated on a scale of 0 (not present) to 3 (severe). The MBI‐C mandates that a symptom must be present for at least 6 months and represent a change from longstanding behavior to be rated as present. This approach facilitates differentiation of MBI symptoms from transient neuropsychiatric symptoms, and reactive conditions due to medical and environmental precipitants and life stressors to better reflect the new onset symptomatology seen in neurodegenerative disease. 34 In this study, participants were classified as having any symptoms of MBI (MBI‐C total score >0) or having no MBI symptoms (MBI‐C total score = 0) due to the strong positive skew of the MBI‐C data (i.e., ≈50% of respondents can be expected to score zero). 17 Any participants with the following medical conditions were excluded (all derived from self‐report responses to the question “Have you ever been diagnosed with one or more of the following even if you don't have it currently?”): mild cognitive impairment (n = 11), stroke (n = 72), Parkinson's disease (n = 11). This was done to minimize confounding associated with potential dementia syndromes and to better reflect the Alzheimer's Association MBI diagnostic criteria stipulation that symptoms cannot be better explained by a pre‐existing medical or psychiatric condition. 16 Self‐reported lifetime history of psychiatric diagnoses was also recorded (depression, mania/bipolar/manic depression, anxiety/generalized anxiety disorder, panic attacks, eating disorders, autism/Asperger's/autistic spectrum disorder, attention deficit disorder, schizophrenia/other psychotic illnesses, personality disorders, and social anxiety/phobia, n = 1515). In addition to lifetime self‐reported diagnosis we also captured current depressive symptom status using the Patient Health Questionnaire 9 (PHQ‐9); here we dichotomized participants into who scored ≥10 on the PHQ‐9 (n = 128) and those scoring <10, which has good sensitivity and specificity for a current major depressive episode. 35
The binary coding of the MBI‐C is supported by recent data showing that in cognitively normal people, a score >0 on the MBI‐C (i.e., the presence of any symptoms of any severity) was associated with worse cognitive performance (both at baseline and over 1 year) on a range of tests, with a score of >8 being associated with the worst performance. 19
2.4. Genotyping, genetic data quality control, and AD polygenic risk score calculation
Saliva samples were collected by post and DNA extract by the National Institute for Health Research South London and the Maudsley National Health Service Biomedical Research Centre. Genotyping was done used the Illumina Global Screening Array with custom content (including directly genotyped single nucleotide polymorphisms [SNPs], rs429358 and rs7412, to determine APOE status). The total numbers of participants in the combined genotyped data for the whole PROTECT study was 9146. Genotype quality control (QC) was performed on all of these individuals but only those with MBI and cognitive data were included in the association analysis. Iterative filtering for call rate at 98% completeness (for individuals and SNPs) resulted in the exclusion of 84 samples, after which 9062 remained. In the filtered data relatedness was estimated using KING 2.2.3, followed by extraction of a list of individuals that contained no pairs of individuals with a first‐, second‐, or third‐degree relationship. 36 Variants with Hardy–Weinberg equilibrium P‐value < 0.00001 were excluded. Individuals whose sex estimated in plink did not match that reported by the study participants were excluded. Principal components (PCs) were calculated for the unrelated subset of the data using EIGENSOFT 6.1.4 after pruning using a window size of 1500 bases per 150 kb and an r‐squared of 0.2. 37 , 38 Variants in high linkage disequilibrium regions and non‐autosomal regions were also excluded. K‐means clustering (assuming four distinct clusters) was used on the first two derived PCs to define a cluster of European ancestry individuals. PCs were then recalculated for the cluster of individuals of European ancestry, with outlier individuals removed by EIGENSOFT if exceeding a sigma threshold of 30. Finally, individuals with excess heterozygosity (unusual patterns of genome‐wide heterogeneity) calculated using the ibc function in plink v1.90 were excluded. 39 The total number of individuals excluded when removing those that were either related, of non‐European ancestry, of mismatched sex, outliers in the PC calculation, or detected to have excess heterozygosity was 790 given an original sample size of 9062 participants.
International Genomics of Alzheimer's Project (IGAP) AD genome‐wide association study (GWAS) summary statistics were used to calculate PRS using PRSice v2.2.12. 40 , 41 Directly genotyped data was used for the PRS calculation and the total number of variants available after QC was 630,180.
Previous data suggest a wide range of inclusion thresholds for AD PRS calculation, from only genome‐wide significant SNPs to many thousands of SNPs. 2 , 3 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 In this study, we opted for two AD GWAS SNP inclusion thresholds (PT). The first, 5 × 10–8 (22 SNPs after clumping), was chosen because it was the most strongly associated with a family history of AD (defined as self‐reported number of parents with AD) in this sample (β = 0.05, 95% CI: 0.03–0.06, P < 0.001) and the second was all IGAP SNPs (i.e., PT = 1, 83,540 SNPs after clumping).
2.5. Statistical analysis
PRS were standardized to a mean of 0 and standard deviation of 1 before analysis. Regression coefficients thus represent unit increase in cognitive composite score per one standard deviation increase in PRS. The association between AD genetic risk and cognition was first tested by linear regression in the whole sample at both PT. The sample was then stratified by MBI status and the dependent variable of cognitive composite was analyzed by linear regression with AD PRS and age, sex, education level, employment status (dummy coded), lifetime history of psychiatric diagnosis, PHQ‐9 grouping (≥10 and <10), and the first six ancestry PCs as covariates. The inclusion of psychiatric history and PHQ‐9 grouping, thus, allows us to be more confident that findings are the result of MBI specifically and not confounded by MBI‐C measurements capturing concurrent psychiatric disorders. Subsequent identical models were run that also controlled for APOE ε4 status to assess the independent effect of non‐APOE SNPs in the PRS. The proportion of variance explained by the PRS was represented by R2 of the model with only covariates subtracted from the R2 of the full model. Regression assumptions were checked by examining residual plots and checking for outliers. Given that the two AD PRS are not independent, and a Bonferroni correction would therefore be overly conservative, a family‐wise error rate of 0.025 was applied (to reflect tests on the two MBI symptom groups). All statistical analysis was performed in R version 3.4.2.
3. RESULTS
3.1. Participant characteristics
After medical history exclusions, data from 4458 participants were available for analysis. Participant characteristics by MBI group are shown in Table 2. There was no difference in age, sex, or education level between the MBI and no MBI strata but, as expected, the MBI group did have a lower cognitive score. There was a slightly higher proportion of retired people among those in the no‐symptom group compared to the MBI symptoms group (60% vs. 56%) and more people with a history of psychiatric conditions or PHQ‐9 ≥10 also had MBI symptoms.
TABLE 2.
Participant characteristics by MBI group
| MBI group | |||||
|---|---|---|---|---|---|
| No symptoms (MBI‐C = 0) | MBI symptoms (MBI‐C >0) | P | |||
| N (%) | 1865 | 42 | 2593 | 58 | |
| Age (mean, sd) | 63.6 | 6.7 | 63.5 | 7.2 | 0.9 |
| Sex (n, %) | |||||
| Female | 1423 | 76 | 1999 | 77 | 0.78 |
| Male | 442 | 24 | 594 | 23 | |
| Education level (n, %) | |||||
| Secondary education | 202 | 11 | 335 | 13 | 0.14 |
| Post‐secondary education | 201 | 11 | 324 | 12 | |
| Vocational qualification | 364 | 20 | 475 | 18 | |
| Undergraduate degree | 690 | 37 | 922 | 36 | |
| Post graduate degree | 328 | 18 | 442 | 17 | |
| Doctoral degree | 80 | 4 | 95 | 4 | |
| Employment status (n, %) | |||||
| Full time | 263 | 14 | 390 | 15 | 0.003 |
| Part time | 262 | 14 | 427 | 16 | |
| Self‐employed | 202 | 11 | 248 | 10 | |
| Retired | 1110 | 60 | 1458 | 56 | |
| Unemployed | 28 | 2 | 70 | 3 | |
| Life‐time history of any psychiatric diagnosis | |||||
| No | 1356 | 73 | 1587 | 61 | ≤0.001 |
| Yes | 509 | 27 | 1006 | 39 | |
| PHQ‐9 | |||||
| ≤10 | 1850 | 99 | 2480 | 96 | ≤0.001 |
| ≥10 | 15 | 1 | 113 | 4 | |
| Cognitive composite (mean, sd) | 0.14 | 1.4 | –0.06 | 1.4 | <0.001 |
Note: Cognitive composite is the first unrotated principal component derived from scores on paired associates learning, digit span, self‐ordered search, and verbal reasoning.
Abbreviations: MBI, mild behavioral impairment; MBI‐C, mild behavioral impairment Checklist; PHQ‐9; Patient Health Questionnaire 9; sd, standard deviation.
3.2. Relationship between AD PRS, cognition and MBI
In the whole sample analysis adjusting for covariates, AD PRS at PT = 1 was associated with a lower mean cognitive composite score (β = –0.07, 95% CI: –0.11 to –0.03, P < 0.001, R2 = 0.002) and modestly at the more conservative PRS at PT = 5 × 10–8 (β = –0.04, 95% CI: –0.8–0.00, P = 0.03, R2 = 0.001). Accordingly, the rest of the analysis was only conducted on AD PRS at PT = 1.
A plot of the relationship between adjusted mean cognitive composite score and AD PRS adjusted for covariates is shown in Figure 1. In the analysis stratified by MBI symptoms after adjusting for covariates, the association between AD genetic risk and cognition persisted but only in those with MBI symptoms (β = –0.09, 95% CI: –0.13 to –0.03, P = 0.002, R2 = 0.003, Table 3). In those with no MBI symptoms there was no association between AD genetic risk and cognition (Table 3). Controlling for APOE did not change the direction of associations, though there was a decrease in proportion of variance explained (whole sample: β = –0.06, 95% CI: –0.10 to –0.02, P = 0.003, R2 = 0.002; MBI symptoms: β = –0.07, 95% CI: –0.12 to –0.02., P = 0.01, R2 = 0.002).
FIGURE 1.
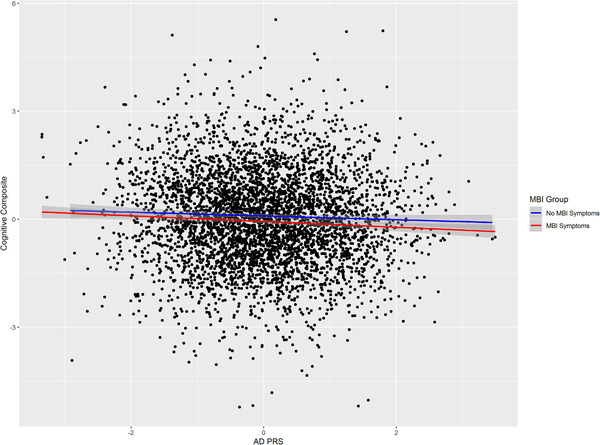
Relationship between Alzheimer's disease (AD) polygenic risk score (PRS) and cognitive composite. Fitted lines are adjusted linear regression of PRS on cognitive composite for no mild behavioral impairment (MBI) symptoms and MBI symptom groups (shaded area is 95% confidence interval)
TABLE 3.
Results of linear regression of AD PRS on cognitive composite at PT = 1 (83,540 SNPs)
| Group | β | L 95% CI | U 95% CI | P | R 2 |
|---|---|---|---|---|---|
| Whole sample | −0.07 | −0.11 | −0.03 | <0.001 | 0.002 |
| MBI symptoms (MBI‐C > 0) | −0.09 | −0.13 | −0.03 | 0.002 | 0.003 |
| No MBI symptoms (MBI‐C = 0) | −0.05 | −0.11 | 0.01 | 0.09 | 0.001 |
| Sensitivity analysis | |||||
| MBI symptoms (MBI‐C ≥6) | −0.10 | −0.18 | −0.02 | 0.02 | 0.005 |
| MBI symptoms (MBI‐C ≥6), aged ≥65 | −0.20 | −0.34 | −0.06 | 0.005 | 0.02 |
Notes: Analysis controlled for age, sex, education, employment status, lifetime psychiatric diagnosis, PHQ‐9 group, and the first six ancestry principal components.
Beta coefficients represent unit of increase in cognitive composite score per 1 standard deviation increase in PRS.
Abbreviations: AD, Alzheimer's disease; CI, confidence interval; MBI, mild behavioral impairment; PHQ‐9; Patient Health Questionnaire 9; PRS, polygenic risk scores; SNP, single nucleotide polymorphism.
3.3. Sensitivity analyses
This analysis was conducted post hoc so should be considered exploratory. In our primary analysis we adopted a binary coding for MBI symptoms (none vs. any) because of evidence that even individuals with low‐level symptoms have an increased risk of cognitive decline. There are no accepted cut points on the MBI‐C in cognitively normal community samples so we examined the association between AD PRS, cognitive impairment, and MBI using an MBI‐C cut point of ≥6 (n = 979). The association observed in the main analysis was sustained for those with MBI symptoms at this more conservative MBI‐C cut point (β = −0.10, 95% CI: −0.18 to −0.02, P = 0.01, R2 = 0.005, Table 3). To illustrate this relationship, adjusted mean cognitive composite score by AD PRS stratified by MBI‐C using the higher threshold split is plotted in Figure 2. Finally, because of prior evidence showing that AD PRS is a stronger predictor of cognition in older people we examined the same relationship in those aged 65 or over (n = 369). 42 This analysis is plotted in Figure 3 and shows a further strengthening of the relationship between AD PRS and cognition in those with an MBI‐C score ≥6, with proportion of variance explained rising from 0.005 to 0.02 (β = −0.20, 95% CI: −0.34 to −0.06, P = 0.005, R2 = 0.02, Table 3). The increase in variance explained was not simply attributable to the older age cutoff; the R2 in all participants aged ≥65 was comparable to all that of all participants with MBI‐C ≥6 (β = −0.08, 95% CI: −0.14 to −0.02, P = 0.009, R2 = 0.004).
FIGURE 2.
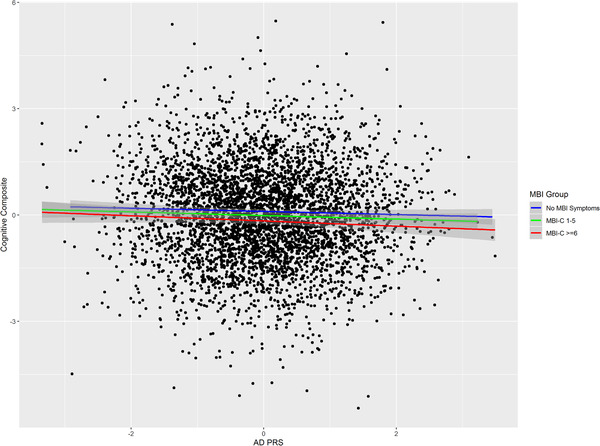
Relationship between Alzheimer's disease (AD) polygenic risk scores (PRS) and cognitive composite. Fitted lines are adjusted linear regression of PRS on cognitive composite for no mild behavioral impairment (MBI) symptoms, MBI Checklist (MBI‐C) 1—6, and MBI‐C ≥6 groups (shaded area is 95% confidence interval)
FIGURE 3.
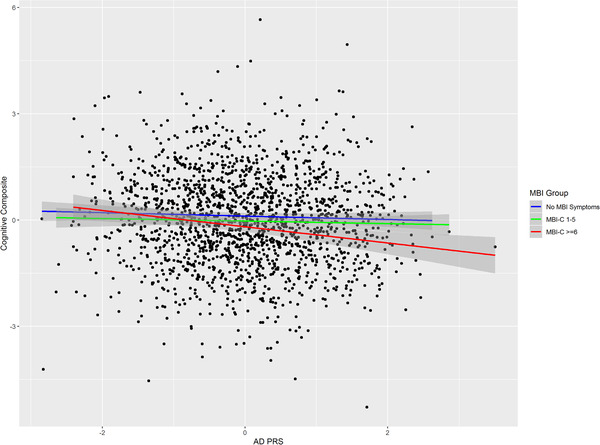
Relationship between Alzheimer's disease (AD) polygenic risk scores (PRS) and cognitive composite in the subset of the sample aged ≥65. Fitted lines are adjusted linear regression of PRS on cognitive composite for no mild behavioral impairment (MBI) symptoms, MBI Checklist (MBI‐C) 1—6, and MBI‐C ≥6 groups (shaded area is 95% confidence interval)
4. DISCUSSION
To our knowledge these findings are the first demonstration that sample stratification on neuropsychiatric symptoms (here assessed in the MBI framework) may identify a subset of the cognitively normal population in which a stronger relationship between genetic risk for AD and cognition exists. As such our findings point to MBI‐C screening as a potential sample enrichment tool as well as suggesting potential confounding by unmeasured neuropsychiatric symptoms in studies of AD genetic risk in cognitively normal samples. The association was present in our study using a PRS based on all available SNPs from AD GWAS (PT = 1); was present even after controlling for psychiatric diagnoses and PHQ‐9 score; and remained after controlling for APOE status, albeit with a diminished variance explained. As expected, we conclude that APOE is driving some of the signal observed in this study, consistent with previous reports, but that non‐APOE SNPs also play a role in late‐life cognition. More broadly, our findings from this sample, which is independent from previous reports, support a role for AD genetic risk, beyond APOE, in cognition among older adults without dementia, an important finding as there is not unanimity in previous literature. 8 , 9 , 10 , 11 , 13 , 14 , 15
A recent well‐phenotyped large study of older adults found that an AD PRS that included all SNPs (PT = 1) was not associated with cognition in adults but APOE was. 14 Genetic risk for AD has been shown to be pleiotropic but it is notable that a previous study found the effect of APOE ε4 on cognition to be stronger in older adults relative to earlier in life. 14 , 42 Older samples are more likely to contain a larger proportion of individuals in prodromal or preclinical disease so a stronger effect of AD PRS could be reasonably expected. Similarly, we propose that stratifying on MBI, even among cognitively normal older adults, has the same effect and defines a cognitive phenotype that is “closer” to AD. This, in turn, would create better power to detect associations with those AD SNPs with smaller effect sizes, thus explaining the discrepancy between our finding and this other recent work.
The notion that MBI (i.e., late‐life neuropsychiatric symptoms) may be an early marker of dementia is somewhat counterintuitive for a group of diseases that are primarily conceptualized as cognitive disorders. This cognocentric approach does not necessarily reflect the history of AD. Auguste D., the index patient described by Alois Alzheimer, presented to hospital with emotional dysregulation and suspiciousness, followed by cognitive decline. 34 , 43 , 44 , 45 Several longitudinal studies support MBI emerging in advance of cognitive symptoms or increasing risk of incident cognitive decline and dementia. 18 , 20 , 21 , 46 , 47 The distinction between MBI and other neuropsychiatric symptoms as risk factors versus early markers has been a topic of recent debate. 48 , 49 However, new data in cognitively normal people showing that MBI symptoms are associated with higher Aβ burden, 23 tau deposition, 26 and faster accumulation of neurofilament light 50 support the view of MBI as an early clinical marker. The implication is therefore that stratification on MBI symptoms enriches samples for individuals with preclinical or prodromal disease, creating a more etiologically homogenous sample; this hypothesis is strengthened by our post hoc analysis showing evidence of a stronger association in those with more severe MBI symptoms who are aged 65 or over. Deeper phenotyping including imaging and longitudinal follow‐up with detailed neuropsychology and clinical outcomes will be required to confirm this hypothesis, which could have important implications for clinical trials in which cohort heterogeneity has been identified as a major concern. 51
The operationalization of MBI is worth some discussion. A key strength of this study is the controlling of pre‐existing psychiatric conditions and use of the specific MBI‐C tool, which has been validated in cognitively normal samples, 17 both of which provide more confidence that our findings are due to later‐life emergent mild NPS rather than longstanding clinically significant psychiatric diagnoses. However, establishing appropriate cut points on the MBI‐C is a matter of ongoing research. Our findings, including our post hoc analysis, and those of others suggest that relatively mild symptoms are important to consider and that the likely optimal cut point on the MBI‐C for case ascertainment would lie somewhere between 2 and 8. This is supported by previous research showing that low‐level symptoms of MBI as well as more severe symptoms are associated with cognitive decline in cognitively normal individuals. 19 Relating to this, a limitation to our study is the proxy completion of MBI‐C via remote questionnaire completion, which could have led to some misclassifications, although raters in this study were required to have known the participant for at least 10 years. Other limitations include the over‐representation of women and more highly educated people in our sample and, as with most genetic studies, these results may not be generalizable to non‐European ancestry populations. We note our sample size is relatively small compared to much of the wider genetic literature and replication in independent cohorts is needed. In particular, we would not rule out an effect of AD PRS in the no MBI symptom group, but our data would suggest that this effect is weaker. The lack of association could be due to power as the MBI symptoms group was larger. However, it is worth noting that in our sensitivity analysis using a more stringent cut point (MBI‐C ≥ 6) there were 925 people in the MBI group, approximately half that of the no‐symptoms group. At present we are not aware of any other large cohort studies that use the MBI‐C; however, we would argue that our findings, along with the practical advantages of the MBI‐C (it is freely available and quick to complete) provide a strong case for the wider adoption of the scale, which will allow important follow‐up work to take place.
In summary, this study lends further support to the growing evidence base that later life emergent neuropsychiatric symptoms describe an at‐risk state for incident cognitive decline and dementia, and can be the index manifestation of dementia for some. Our findings also support the case for using MBI as a sample enrichment tool for biomarker studies and clinical trials targeting at‐risk individuals. The enrichment approach is inexpensive, simple, and scalable, and can decrease cost and improve enrolment efficiency of dementia clinical trials. Deeper phenotyping of these groups including neuroimaging and longitudinal monitoring of clinical outcomes is now essential.
CONFLICTS OF INTEREST
Clive Ballard has received contract grant funding from ACADIA, Lundbeck, Takeda, and Axovant pharmaceutical companies and honoraria from Lundbeck, Lilly, Otsuka, and Orion pharmaceutical companies. Dag Aarsland has received research support and/or honoraria from Astra‐Zeneca, H. Lundbeck, Novartis Pharmaceuticals, and GE Health, and serves as a paid consultant for H. Lundbeck and Axovant. Zahinoor Ismail has received honoraria/consulting fees from Janssen, Lundbeck, and Otsuka, although not related to this work. Cathryn Lewis sits on the SAB for Myriad Neuroscience.
FUNDING INFORMATION
This work was funded in part through the MRC Proximity to Discovery: Industry Engagement Fund (External Collaboration, Innovation and Entrepreneurism: Translational Medicine in Exeter 2 (EXCITEME2) ref. MC_PC_17189) awarded to Dr Creese and MRC grant MR/N015746/1 awarded to Prof Lewis. The addition of the family history of dementia questionnaire to the PROTECT study was supported by a small grant from the Alzheimer's Research UK South West Network. This article represents independent research part funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King's College London. None of the funding bodies had any role in the design, analysis or interpretation of data, or the drafting of the manuscript.
ACKNOWLEDGMENTS
This research was supported by the NIHR Collaboration for Leadership in Applied Health Research and Care South West Peninsula. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care. The authors thank Gemma Shireby at University of Exeter for providing the analysis script for genetic ancestry check. Genotyping was performed at deCODE Genetics. The authors would like to acknowledge the use of the research computing facility at King's College London, Rosalind (https://rosalind.kcl.ac.uk), which is delivered in partnership with the NIHR Biomedical Research Centres at South London & Maudsley and Guy's & St. Thomas’ NHS Foundation Trusts, and part‐funded by capital equipment grants from the Maudsley Charity (award 980) and Guy's & St. Thomas’ Charity (TR130505).
Creese B, Arathimos R, Brooker H, et al. Genetic risk for Alzheimer's disease, cognition, and mild behavioral impairment in healthy older adults. Alzheimer's Dement. 2021;13:e12164. 10.1002/dad2.12164
REFERENCES
- 1. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer's disease drug development pipeline: 2019. Alzheimer's. Dement Transl Res Clin Interv. 2019;5:272‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Escott‐Price V, Sims R, Bannister C, et al. Common polygenic variation enhances risk prediction for Alzheimer's disease. Brain. 2015;138:3673‐3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chaudhury S, Brookes KJ, Patel T, et al. Alzheimer's disease polygenic risk score as a predictor of conversion from mild‐cognitive impairment. Transl Psychiatry. 2019;9:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Logue MW, Panizzon MS, Elman JA, et al. Use of an Alzheimer's disease polygenic risk score to identify mild cognitive impairment in adults in their 50s. Mol Psychiatry. 2019;24:421‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lacour A, Espinosa A, Louwersheimer E, et al. Genome‐wide significant risk factors for Alzheimer's disease: role in progression to dementia due to Alzheimer's disease among subjects with mild cognitive impairment. Mol Psychiatry. 2017;22:153‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Desikan RS, Fan CC, Wang Y, et al. Genetic assessment of age‐associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLOS Med. 2017;14:e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adams HHH, de Bruijn RFAG, Hofman A, et al. Genetic risk of neurodegenerative diseases is associated with mild cognitive impairment and conversion to dementia. Alzheimer's. Dement. 2015;11:1277‐1285. [DOI] [PubMed] [Google Scholar]
- 8. Harris SE, Davies G, Luciano M, et al. Polygenic risk for Alzheimer's disease is not associated with cognitive ability or cognitive aging in non‐demented older people. J Alzheimers Dis. 2014;39:565‐574. [DOI] [PubMed] [Google Scholar]
- 9. Darst BF, Koscik RL, Racine AM, et al. Pathway‐specific polygenic risk scores as predictors of amyloid‐beta deposition and cognitive function in a sample at increased risk for Alzheimer's disease. J Alzheimers Dis. 2017;55:473‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marden JR, Mayeda ER, Walter S, et al. Using an Alzheimer disease polygenic risk score to predict memory decline in black and white Americans over 14 years of follow‐up. Alzheimer Dis Assoc Disord. 2016;30:195‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mormino EC, Sperling RA, Holmes AJ, et al. Polygenic risk of Alzheimer disease is associated with early‐ and late‐life processes. Neurology. 2016;87:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ge T, Sabuncu MR, Smoller JW, Sperling RA, Mormino EC. Dissociable influences of APOE epsilon4 and polygenic risk of AD dementia on amyloid and cognition. Neurology. 2018;90:e1605‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stephan Y, Sutin AR, Luchetti M, Caille P, Terracciano A. Polygenic Score for Alzheimer Disease and cognition: the mediating role of personality. J Psychiatr Res. 2018;107:110‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ritchie SJ, Hill WD, Marioni RE, et al. Polygenic predictors of age‐related decline in cognitive ability. Mol Psychiatry. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marioni RE, Campbell A, Hagenaars SP, et al. Genetic Stratification to Identify Risk Groups for Alzheimer's Disease. J Alzheimers Dis. 2017;57:275‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ismail Z, Smith EE, Geda Y, et al. Neuropsychiatric symptoms as early manifestations of emergent dementia: provisional diagnostic criteria for mild behavioral impairment. Alzheimers Dement. 2016;12:195‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Creese B, Griffiths A, Brooker H, et al. Profile of mild behavioral impairment and factor structure of the mild behavioral impairment checklist in cognitively normal older adults. Int Psychogeriatrics. 2019. [DOI] [PubMed] [Google Scholar]
- 18. Matsuoka T, Ismail Z, Narumoto J. Prevalence of mild behavioral impairment and risk of dementia in a psychiatric outpatient Clinic. J Alzheimers Dis. 2019;70:505‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Creese B, Brooker H, Ismail Z, et al. Mild behavioral impairment as a marker of cognitive decline in cognitively normal older adults. Am J Geriatr Psychiatry. 2019;27. [DOI] [PubMed] [Google Scholar]
- 20. Taragano FE, Allegri RF, Krupitzki H, et al. Mild behavioral impairment and risk of dementia. J Clin Psychiatry. 2009;70:584‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taragano FE, Allegri RF, Heisecke SL, Martelli MI, Feldman ML, Sanchez V, et al. Risk of conversion to dementia in a mild behavioral impairment group compared to a psychiatric group and to a mild cognitive impairment group. J Alzheimers Dis. 2018;62:227‐238. [DOI] [PubMed] [Google Scholar]
- 22. Ismail Z, McGirr A, Gill S, Hu S, Forkert ND, Smith EE. Mild behavioral impairment and subjective cognitive decline predict mild cognitive impairment. J Alzheimer's Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lussier FZ, Pascoal TA, Chamoun M, et al. Mild behavioral impairment is associated with β‐amyloid but not tau or neurodegeneration in cognitively intact elderly individuals. Alzheimer's. Dement. 2020;16:192‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Andrews SJ, Ismail Z, Anstey KJ, Mortby M. Association of Alzheimer's genetic loci with mild behavioral impairment. Am J Med Genet B Neuropsychiatr Genet. 2018;177:727‐735. [DOI] [PubMed] [Google Scholar]
- 25. Naude JP, Gill S, Hu S, et al. Plasma neurofilament light: a marker of neurodegeneration in mild behavioral impairment. J Alzheimer's Dis. 2020;76:1017‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johansson M, Smith R, Stomrud E, et al. Mild behavioral impairment is predictive of tau deposition in the earliest stages of Alzheimer's disease. Alzheimer's. Dement. 2020;16:e042595. [Google Scholar]
- 27. Harris SE, Deary IJ. The genetics of cognitive ability and cognitive ageing in healthy older people. Trends Cogn Sci. 2011;15:388‐394. [DOI] [PubMed] [Google Scholar]
- 28. Davies G, Lam M, Harris SE, et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat Commun. 2018;9:2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jorm AF. A short form of the Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE): development and cross‐validation. Psychol Med. 1994;24:145‐153. [DOI] [PubMed] [Google Scholar]
- 30. Teresi J, Lawton M, Holmes D, O M. Measurement in elderly chronic care populations. In: Morris J, Morris S, eds. ADL Assess. Meas. Use WithFrail Elders. New York, NY: Springer Publishing Co.; 1997. [Google Scholar]
- 31. Ismail Z, Agüera‐Ortiz L, Brodaty H, et al. The Mild Behavioral Impairment Checklist (MBI‐C): a rating scale for neuropsychiatric symptoms in pre‐dementia populations. J Alzheimers Dis. 2017;56:929‐938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mallo SC, Ismail Z, Pereiro AX, et al. Assessing mild behavioral impairment with the mild behavioral impairment checklist in people with subjective cognitive decline. Int Psychogeriatrics. 2018:1‐9. [DOI] [PubMed] [Google Scholar]
- 33. Mallo SC, Ismail Z, Pereiro AX, et al. Assessing mild behavioral impairment with the mild behavioral impairment‐checklist in people with mild cognitive impairment. J Alzheimers Dis. 2018;66:83‐95. [DOI] [PubMed] [Google Scholar]
- 34. Wise EA, Rosenberg PB, Lyketsos CG, Leoutsakos J‐M. Time course of neuropsychiatric symptoms and cognitive diagnosis in National Alzheimer's Coordinating Centers volunteers. Alzheimer's Dement. 2019;11:333‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kroenke K, Spitzer RL, Williams JB. The PHQ‐9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W‐M. Robust relationship inference in genome‐wide association studies. Bioinformatics. 2010;26:2867‐2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome‐wide association studies. Nat Genet. 2006;38:904‐909. [DOI] [PubMed] [Google Scholar]
- 38. Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lambert J‐C, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Choi SW, O'Reilly PF. PRSice‐2: polygenic Risk Score software for biobank‐scale data. Gigascience. 2019;8: giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hill WD, Davies G, Group CCW, Liewald DC, McIntosh AM, Deary IJ. Age‐dependent pleiotropy between general cognitive function and major psychiatric disorders. Biol Psychiatry. 2016;80:266‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Donovan NJ, Amariglio RE, Zoller AS, et al. Subjective cognitive concerns and neuropsychiatric predictors of progression to the early clinical stages of Alzheimer disease. Am J Geriatr Psychiatry. 2014;22:1642‐1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Masters MC, Morris JC, Roe CM. Noncognitive” symptoms of early Alzheimer disease: a longitudinal analysis. Neurology. 2015;84:617‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Geda YE, Schneider LS, Gitlin LN, et al. Neuropsychiatric symptoms in Alzheimer's disease: past progress and anticipation of the future. Alzheimers Dement. 2013;9:602‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dietlin S, Soto M, Kiyasova V, et al. Neuropsychiatric symptoms and risk of progression to Alzheimer's disease among mild cognitive impairment subjects. J Alzheimers Dis. 2019. [DOI] [PubMed] [Google Scholar]
- 47. Gill S, Mouches P, Hu S, et al. Using machine learning to predict dementia from neuropsychiatric symptom and neuroimaging data. J Alzheimer's Dis. 2020;75:277‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ismail Z, Gatchel J, Bateman DR, et al. Affective and emotional dysregulation as pre‐dementia risk markers: exploring the mild behavioral impairment symptoms of depression, anxiety, irritability, and euphoria. Int Psychogeriatrics. 2018;30:185‐196. [DOI] [PubMed] [Google Scholar]
- 49. Savulich G, O'Brien JT, Sahakian BJ. Are neuropsychiatric symptoms modifiable risk factors for cognitive decline in Alzheimer's disease and vascular dementia? Br J Psychiatry. 2020;216:1‐3. [DOI] [PubMed] [Google Scholar]
- 50. Ismail Z, Naude J, Gill S, et al. Plasma Neurofilament Light: a marker of cognitive decline in mild behavioural impairment. 12th Clin. Trials Alzheimer's Dis. Conf., San Diego, USA: 2019. [Google Scholar]
- 51. Devi G, Scheltens P. Heterogeneity of Alzheimer's disease: consequence for drug trials? Alzheimers Res Ther. 2018;10:122. [DOI] [PMC free article] [PubMed] [Google Scholar]