

Abstract
Plexiform neurofibromas (PN) are benign nerve sheath Schwann cell tumors characterized by biallelic mutations in the NF1 tumor suppressor gene. Atypical neurofibromas (ANF) show additional frequent loss of CDKN2A/Ink4a/Arf and may be precursor lesions of aggressive malignant peripheral nerve sheath tumors (MPNST). Here we combined loss of Nf1 in developing Schwann cells with global Ink4a/Arf loss and identified paraspinal PN and ANF. Upon transplantation, ANF generated GEM-PNST similar to human MPNST, and tumors showed reduced p16INK4a protein and reduced senescence markers, confirming susceptibility to transformation. Superficial GEM-PNST contained regions of nerve-associated PN or ANF and grew rapidly on transplantation. Transcriptome analyses showed similarities to corresponding human tumors. Thus, we recapitulated nerve tumor progression in NF1 and provided preclinical platforms for testing therapies at each tumor grade. These results support a tumor progression model in which loss of NF1 in Schwann cells drives PN formation, additional loss of Ink4a/Arf contributes to ANF formation, and further changes underlie transformation to MPNST.
Keywords: NF1, Neurofibroma, MPNST, Mouse model, CDKN2A
INTRODUCTION
Neurofibromatosis type 1 (NF1) is an autosomal dominant tumor predisposition syndrome that affects about 1:3000 individuals worldwide [1]. The NF1 gene encodes neurofibromin, a GTPase-activating protein (GAP) that accelerates the intrinsic GAP activity of Ras proteins [2]. Therefore, after cell activation, loss of NF1 correlates with increased signaling through the RAS pathway, a key driver of cancer [3]. Nearly all (90%) of NF1 patients develop small benign cutaneous neurofibromas. Plexiform neurofibromas (PN) occur in 25-50% of NF1 patients and are histologically benign peripheral nerve sheath tumors associated with nerve trunks that can cause substantial morbidity including pain and neurologic deficit [4,5]. Surgical resection is often impossible due to tissue invasion, large size, and/or association with critical peripheral nerves [5].
PN can serve as precursor lesions for atypical neurofibromas (ANF), which can be symptomatic. Imaging studies show most ANF as distinct nodules that may be fluorodeoxyglucose (FDG)-positron emission tomography (PET) avid and/or painful [6]. Clinically, ANF are frequently slow-growing tumors, and many ANF, unlike PN, show genomic loss at the CDKN2A locus [7]. At least some ANF are atypical neurofibromatous neoplasms of uncertain biologic potential (ANNUBP), diagnosed by histology as hypercellular peripheral nerve sheath tumors (PNSTs) composed of cells which also show nuclear atypia and/or mitoses [8]. ANF show few mutations apart from those in NF1 and CDKN2A/B [9].
ANF themselves may be precursor lesions to malignant peripheral nerve sheath tumors (MPNST). In a recent study 4/18 un-resected ANF transformed to MPNST over an interval studied; others developed MPNST distant from a resected ANF [6]. MPNST are highly aggressive soft tissue sarcomas incurable with chemotherapy, for which the only effective treatment is complete surgical resection [6,10]. MPNST is the leading cause of early death in adults with NF1, and better therapies are desperately needed.
NF1 is a tumor suppressor gene on human chromosome 17q11. Loss of function in both NF1 alleles is detected in cutaneous and PN Schwann cells (SCs) [11-13]. Although cutaneous and PN are composed of SCs, mast cells, fibroblasts, endothelial cells, and macrophages [14,15], only SCs show biallelic NF1 alterations [12,16]. SC loss of NF1 function drives neurofibroma formation, as in genetically engineered mice (GEM) conditional loss of Nf1 in the SC lineage, driven by any of several different Cre driver alleles, results in neurofibroma formation [17-19]. In the Nf1fl/fl;DhhCre model, loss of Nf1 in SCs at embryonic day 12.5 is sufficient for benign PN formation, and in this GEM, transformation to more aggressive GEM-ANF or GEM-PNST is not observed [18].
Most ANF and 70% of MPNST show genetic heterozygous or homozygous loss of cyclin dependent kinase inhibitor 2A gene (CDKN2A in human; Ink4a/Arf in mouse) [7,9]. The CDKN2A gene located at 9p21 encodes two proteins, p16INK4a and p19ARF (p14 in human) [20,21]. The p16INK4a protein inhibits the CDK4-6 kinases, maintaining the retinoblastoma protein (Rb) protein in an unphosphorylated, growth-suppressive state, arresting the cell cycle [20,22]. p19ARF binds the double minute 2 (MDM2) homologue, thereby stabilizing TP53, arresting cell proliferation, or leading to apoptosis [22-24]. Loss of Ink4a/Arf can also compromise the senescence checkpoint, facilitating transformation to malignancy [25]; senescent cells in cutaneous neurofibromas were postulated to contribute to their slow, benign, growth [26]. Supporting a role in nerve tumor progression, Ink4a/Arf−/−;Nf1+/− mice (26%) developed GEM-PNST [27]. Also, viral delivery of Cre recombinase into Ink4a/Arffl/fl;Nf1fl/fl peripheral nerve led to aggressive nerve tumors [28]. In these models neither PN nor ANF were reported, suggesting that different precursor cells are affected by loss of Nf1 or Ink4a/Arf to drive the tumor types, and/or that simultaneous loss of both genes bypasses benign tumor formation. In mice expressing periostin-Cre;Nf1fl/fl and Arffl alleles beginning at embryonic day 10 in early Schwann cell precursors, rare ANF formed and mice died subsequent to GEM-PNST, correlating with loss of the senescence checkpoint [29].
Here, we imposed loss of Ink4a/Arf in mice that develop paraspinal neurofibroma. Half of these tumors in Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice were GEM-neurofibroma (PN), and half GEM-ANF (ANF). High grade GEM-PNST formed at distant sites. Nerves in Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice show reduced senescence, and tumors show RNA expression similar to their human counterparts. Transplantation of ANF into immunocompromised and immunocompetent mice results in GEM-PNST, providing new progression models useful for molecular, prevention and treatment studies.
MATERIALS AND METHODS
Animal Studies
All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Cincinnati Children’s Hospital Medical Center. Mice were housed in temperature and humidity-controlled facilities with free access to food and water, on a 12-hour light-dark cycle. Ink4a/Arf−/− mice were from the B6.129-Cdkn2atm1Rdp NCI; MMHCC strain #01XB1 (RRID:IMRS_NCIMR:01XB1), back-crossed to C57BL/6J females for 5 generations before generating compound mutant mice [30]. We used Nf1fl/fl;DhhCre [18], Athymic nude (Envigo, 069), and C57BL/6J UBC GFP (Jackson 007076) mice. The DhhCre allele was maintained on the male. Except for athymic nudes, mice were maintained on the C57BL/6J background.
Genotyping
We performed Nf1 genotyping using oligonucleotides (5′ to 3′) CTTCAGACTGATTGTTGTACCTGA and ACCTCTCTAGCCTCAGGAATGA to detect the wildtype allele, and TGATTCCCACTTTGTGGTTCTAAG to detect the targeted allele. Ink4a/Arf mice (B6.129-Cdkn2atm1Rdp/NCI) were genotyped according to Mouse repository MMHCC strain #01XB1. DhhCre genotyping was performed using the forward primer ACCCTGTTACGTATAGCCGA and reverse CTCCGGTATTGAAACTCCAG.
Electron Microscopy (EM)
We perfused mice intracardially with Karnovsky’s fixation solution (4% paraformaldehyde (PFA) and 3% glutaraldehyde in 0.1 mol/L phosphate buffer, pH 7.4-7.6). Saphenous nerves were dissected, embedded, and ultrathin plastic sections were evaluated as described [31].
Western Blotting
We homogenized tumor tissue in ice cold buffer: 50mM Tris-Cl, 150mM NaCl, 0.1% SDS, 0.5% Sodium Deoxycholate, 1% Triton x-100 (RIPA) containing protease/phosphatase inhibitor (Thermo Fisher Scientific, A32959). We measured protein concentration using a FlexStation3 molecular device, and denatured lysates in 5X sample buffer containing 10% 5M dithiothreitol (Sigma Aldrich, 10197777001). We loaded samples into 4-20% Criterion TGX precast gels (Bio-Rad, 5671093). We transferred gels to PDVF membrane (Millipore, IPFL00010) which was blocked in 5% blotting grade blocker (Bio-Rad, 1706404) in 0.1 M Tris buffered saline with 0.1% Tween (TBST), then incubated with anti-p16 antibody (Invitrogen, MA5-17142, 1:1000), phospho-Rb (Ser807/811) antibody (Cell Signaling, 8516S, 1:1000), or horseradish peroxidase (HRP) conjugated GAPDH antibody (Cell Signaling, 3683S) overnight at 4°C. Secondary anti-mouse (Cell Signaling, 7076S, 1:1000) or anti-rabbit (Cell Signaling, 7074S, 1:1000) IgG was added for 1 hour at room temperature prior to Immobilon western chemiluminescent HRP substrate (Millipore, WBKLS0500), and chemi-luminescence imaging on an Azure Biosystems c500 chemi doc gel imager.
Quantitative polymerase chain reaction Q-PCR
Genomic DNA was isolated from tumors with the DNeasy blood and tissue kit (Qiagen, 69504) and amplified using TaqMan genotyping primers (4400291) for Ink4a/Arf gene expression. Quantitative PCR was performed using a Quant Studio 6 Flex Real Time PCR system by Applied Biosystems. Each sample was run twice, with four technical replicates each.
Magnetic Resonance Imaging (MRI)
MRI was performed on anesthetized Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice of both sexes on a 7T Bruker Biospec, and tumor volume quantified as described [32].
Gross Dissections and Tumor Allografts
We sacrificed mice via isoflurane inhalation, removed tumors, and centrifuged them in L-15 medium (Corning Cellgro, MT-10-045-CV) (1000g at 5 min at RT). Tumors were placed into fresh L-15, chopped into pieces <1 mm3, and dissociated in L-15 containing 1% penicillin/streptomycin (Gibco, SV30010), Collegenase Type I (Worthington, LS004196), and Dispase Protease II (Gibco, 04942078001) (37°C at 170 rpm for 2- 4 hours). DMEM (Gibco, 11965118) with 10% FBS (Gemini bio-products, 100-500) was then added, and tumor cell suspensions collected by centrifugation (1700 g for 5 minutes). Pellets were resuspended in DMEM with 10% FBS, filtered with a 40μm BD Falcon Cell Strainer (Cat. # C4040), washed and resuspended in serum free complete mouse sphere medium [27]. Cells from each tumor were separately injected into 1-3 athymic nude mice (Envigo, 069) or into immunocompetent C57BL/6J GFP-expressing mice, at approximately 1×105 cells/mouse in two-thirds complete mouse sphere media and one-third matrigel (BD Biosciences, 354234), in 150 μL. We measured tumors twice weekly with digital calipers (Fisher Scientific), measured volumes by (length x width2) x (π/6), and sacrificed mice when tumors approached 2000 mm3 per IACUC standards.
Immunohistochemistry
We excised tumors from anesthetized mice, fixed them in 4% PFA overnight, dehydrated and embedded in paraffin. Tumor paraffin sections (4 μm) were stained with H&E (Richard-Allan Scientific™ Series Hemotoxylin 1 7221 and Eosin-Y 7111, ThermoFisher), Toluidine Blue (Fisher Scientific, T-161, 1:1000), anti-S100 (Dako, Z0311, 1:10000), Ki67 (Cell Signify, 12202, 1:250), or anti-neurofilament (Biolegend, SM312, 1:2000). Images were captured with a SPOT Insight 4 Mp CCD camera (Spot Imaging).
RNA Isolation and Sequencing
We homogenized frozen tumors in Qiazol (Qiagen, 79306), isolated RNA using RNeasy mini kits (Qiagen, 74104), and sequenced bulk RNA using an Illumina NovaSeq 6000 (RNA polyA stranded, paired 100 bases, read depth 20M). We calculated raw read counts from BAM files with featureCounts and normalized them using Bioconductor-edgeR TMM-normalization (https://bioconductor.org/packages/release/bioc/html/edgeR.html). We performed principal component analysis (PCA) and Clara clustering analysis using R-cluster on whole transcriptomes and used R-fatctoextra to predict optimal cluster numbers. Hierarchical clustering analysis used 5000 multiscale bootstrap resampling with R-pvclust (https://cran.r-project.org/web/packages/pvclust). We calculated Euclidean distances among samples, then built final hierarchical clusters by the average-linkage method for each cluster, and computed an “Approximately Unbiased (AU)” value by 5000 multiscale bootstrap resampling. AU p-value >90% corresponds to p-value <0.1. We extracted pathways and gene sets associated with human atypical neurofibroma [9; supplementary figure S6]. We visualized differential expression patterns across sample groups using Log2-transformed TMM-normalized gene counts (after z-score conversion). Data are available in the Gene Expression Omnibus database (GSE 148249).
Senescence Staining
We excised cervical spinal cord with attached dorsal root ganglia and peripheral nerve from 2-month-old mice, and immediately froze tissue in OCT (Fisher Healthcare, 4585). We cut, air dried, and stained 10um tissue section with a β-galactosidase senescence Cell Signaling Kit (Cell Signaling, 9860) [33]. We immersed sections in ice-cold fixative for 5 minutes, rinsed them in PBS and incubated in β-gal staining solution overnight at 37°C. Slides were washed in PBS and counterstained with 0.1% Nuclear Fast Red Aluminum Sulfate for 5-10min (Poly Scientific, 49091). Images were captured with a SPOT Insight 4 Mp CCD camera (Spot Imaging). We counted blue cells per high powered field (HPF; 40x magnification).
Statistical Analysis
All statistical analyses were conducted in GraphPad Prism. Significance was set at P ≤ 0.05. Statistical tests and posthoc tests are indicated in figure legends. Error bars represent the standard error of the mean unless otherwise noted.
RESULTS
Nf1fl/fl;DhhCre mice lacking Ink4a/Arf show early lethality
Nf1fl/fl;DhhCre mice form neurofibromas and require sacrifice at 9 - 15 months when benign paraspinal tumors compress the spinal cord, causing paralysis and inability to feed [18]. More aggressive ANF or GEM-PNST do not form in this model system. Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice similarly died between 8 and 13 months (Figure 1A), while Ink4a/Arf−/−;Nf1fl/fl;DhhCre mice were dead by 6 months of age. Half the mice in the Ink4a/Arf+/−;Nf1fl/fl;DhhCre and Ink4a/Arf−/−;Nf1fl/fl;DhhCre cohorts required sacrifice due to NF1-related superficial GEM-PNST or paralysis secondary to paraspinal tumor impingement (Figure 1B and see below). Recalculation of time to sacrifice after omitting mice found dead in cage did not alter overall survival times (Figure 1C). Control double heterozygous Ink4a/Arf+/−;Nf1fl/+;DhhCre mice did not form paraspinal nerve tumors. Most (80%) were found dead in cage and showed splenomegaly and/or hepatomegaly, consistent with the prevalence of sarcomas/leukemia/lymphomas in mice lacking one copy of Ink4a/Arf [21], and 25% developed GEM-PNST, similar to GEM-PNST incidence in global Nf1+/−;Ink4a/Arf−/− mice [27].
Figure 1. Loss of Ink4a/Arf decreases survival in Nf1fl/fl mice.
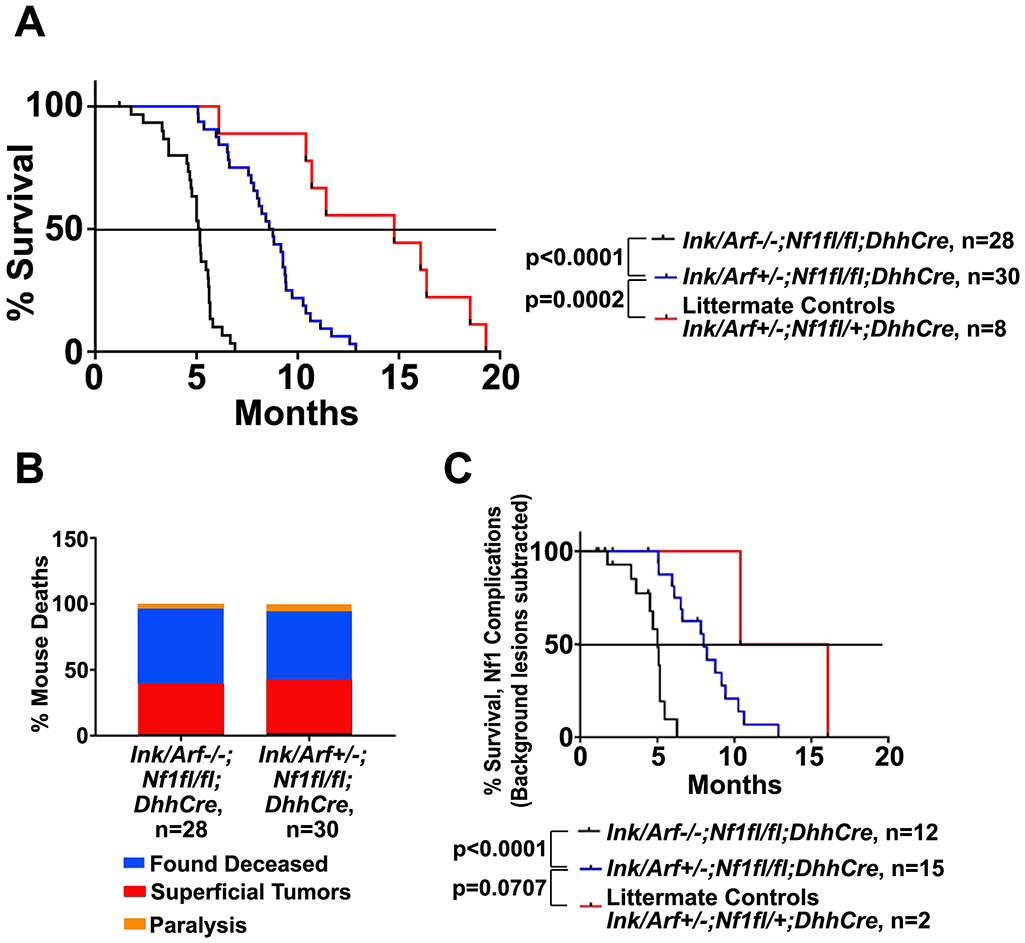
A. Kaplan-Meier survival curves of Ink4a/Arf−/−;Nf1fl/fl;DhhCre, Ink4a/Arf+/−;Nf1fl/fl;DhhCre versus Ink4a/Arf+/−;Nf1fl/+;DhhCre mice littermate controls. P values for pairwise comparisons (Log-rank Mantel Cox post hoc test) are shown. B. Causes of mortality in Ink4a/Arf−/−;Nf1fl/fl;DhhCre and Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice. C. Kaplan-Meier survival curves of the subset of mice sacrificed due to Nf1-related paralysis or superficial tumors is similar to the survival of the entire cohort. P values for pairwise comparisons (Log-rank Mantel Cox posthoc tests) are shown.
Paraspinal and superficial tumors arise in Ink4a/Arf+/−;Nf1fl/fl;DhhCre and Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice
To determine if paraspinal neurofibromas present in the Nf1fl/fl;DhhCre model are present in Nf1fl/fl;DhhCre mice heterozygous for Ink4a/Arf loss, we performed magnetic resonance imaging (MRI). We detected paraspinal tumors in each mouse (Figure 2A). Imaging a cohort of Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice at 4 months of age (n=10), and surviving mice at 7 (n=7) and 9 (n=2) months of age showed that these paraspinal tumors have variable growth rate between scans, a characteristic of Nf1fl/fl;DhhCre neurofibromas (Figure 2B) [32]. At the 7-month scan, 1/7 mice showed a T2-intense tumor mass within a preexisting neurofibroma, postulated to be a more aggressive tumor (red arrow, Figure 2A). On dissection, each mouse had 3-5 paraspinal tumors. All tumors showed similar gross appearance. Figure 2C summarizes the number and percent of paraspinal tumors in each model that showed PN histology and ANF histology, defined below.
Figure 2. Ink4a/Arf−/−;Nf1fl/fl;DhhCre and Ink4a/Arf+/−;Nf1fl/fl;DhhCre paraspinal tumors are plexiform neurofibromas and atypical neurofibromas.

A. MRI imaging of Ink4a/Arf+/−;Nf1fl/fl;DhhCre mouse in the axial plane shows paraspinal NFs (asterisks) and a more T2-dense tumor (arrow). B. Volumetric analysis of MRI scans shows the variable increases in tumor volume over time in each Ink4a/Arf+/−;Nf1fl/fl;DhhCre mouse. Scans from each imaged mouse is shown in a different color. C. Graph demonstrating the percentage of paraspinal tumors that are ANF versus PN by histological analysis. D-H. Plexiform Neurofibroma (PN), I-M: Atypical Neurofibroma (ANF). D and I. H&E staining shows maintained architecture in PN vs increased cellularity, and disrupted nerve architecture of ANF. 10x. E and J. H&E. PN with thin, elongated nuclei with minimal variability with linear arrays of cells with interposed collagenous background whereas ANF demonstrates cellular crowding, occasional nuclear atypia with plump nuclei, and mitotic figures (<3/10 HPF), with loss of collagenous background (inset shows atypical nuclei and mitotic figure. 40x. F and K. Anti-neurofilament (DAB; brown) shows nerve axons, and largely maintained nerve architecture in PN and peripherally preserved axons (encircled by endoneurium, with a target-like appearance) with increased loss of nerve architecture at lower right, moving centrally into the tumor of ANF. 40x. G and L. Anti-S100 (DAB; brown) highlights PN SC with normal morphology and ANF SC with large, atypical, and pleomorphic morphology. 40x. H and M. Toluidine Blue. Mast cells are present in both PN and ANF (red arrows). 40x. Scale bar, 50 μm.
Ultrastructure of peripheral nerves is disrupted in Ink4a/Arf+/−;Nf1fl/fl;DhhCre and Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice
Like their human counterparts, Nf1fl/fl;DhhCre neurofibromas are characterized by progressive dissociation of SCs from small unmyelinated axons [17,34]. Progressive disruption visible by electron microscopy (EM) develops after 2 months of age. We performed EM on saphenous nerves of 4 month old Ink4a/Arf−/−;Nf1fl/fl;DhhCre and Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice (Suppl. Fig. 1A, B). Ink4a/Arf−/−;Nf1fl/fl;DhhCre mice and Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice were intermediate between WT nerve, in which 60% of unmyelinated SC ensheathe >6 axons, and age-matched Nf1fl/fl;DhhCre nerve, in which SC rarely ensheathe >2 axons. Loss of Ink4a/Arf may enhance survival and/or prevent death of Nf1 nerve SCs, retarding disruption of Remak bundles, or play role(s) in other pathway(s) that contribute to Remak bundle disruption.
Neurofibroma, Atypical Neurofibroma, and GEM-PNST arise in Ink4a/Arf+/−; Nf1fl/fl;DhhCre and Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice
We undertook a histological analysis of paraspinal and superficial tumors, applying criteria used for human nerve sheath tumors [8,35] and criteria established for murine models of peripheral nerve sheath tumors [36]. Of paraspinal tumors analyzed in Ink4a/Arf+/−;Nf1fl/fl;DhhCre and Ink4a/Arf−/−;Nf1fl/fl;DhhCre mice, 63% were PN (GEM-neurofibroma grade 1; Figure 2D-H). These tumors had rare mitoses (<1 mitosis/50 HPF with or without hypercellularity. SC nuclei were thin and elongated, with minimal variability. Nuclear atypia was rare, and neurofibroma architecture, and S100+ was maintained [8]. Toluidine blue metachromatic mast cells with characteristic granules were present. In the remaining 37% of Ink4a/Arf+/−;Nf1fl/fl;DhhCre and Ink4a/Arf−/−;Nf1fl/fl;DhhCre paraspinal tumors, histological analysis showed increased cellularity, and nuclear atypia (larger round nuclei and mildly altered chromatin) or nuclear variability, but not both (Figure 2I-M). These were designated GEM-ANF; tumors maintained fascicular nerve architecture, but demonstrated disrupted micro-architecture with disorganization, irregular cell crowding, and occasionally disrupted perineurium. These GEM-ANF showed <3 mitoses/10 HPF. There was no necrosis or hemorrhage.
Ink4a/Arf+/−;Nf1fl/fl;DhhCre and Ink4a/Arf−/−;Nf1fl/fl;DhhCre mice often developed large subcutaneous tumors (Figure 3A) requiring euthanasia due to tumor size and/or ulceration, per IACUC standards. These tumors developed in 40% of Nf1fl/fl;DhhCre mice hetero- or homozygous for Ink4a/Arf mutation (Figure 3B), typically occurring between 5-10 months of age along the dorsum, shoulders, and rarely craniofacially (Figure 3C). The histology of these superficial tumors was aggressive, and similar to human MPNSTs (GEM-PNST; Figure 3D-F). Tumor cells demonstrated marked nuclear atypia, spindle-cell architecture and tumors contained areas of necrosis. Notably, like their human counterparts, GEM-PNST showed only patchy expression of S100. Nuclei were plump and pleomorphic. Collagen was only regionally retained, and there was extensive infiltration of cells into surrounding tissues including fat and muscle. Nuclear variability and atypia were present. Many (72%) of GEM-PNST contained nerves with S100+ SCs and neurofilament+ neuronal axons (Figure 3G-I). These nerves showed features of GEM grade 1 neurofibroma and/or GEM-ANF, suggesting the presence of precursor lesions, and 2 (12.5%) demonstrated distinct regions showing transition of more aggressive tumor from PN. Of note, 1/11 GEM-PNST in the Ink4a/Arf +/− cohort was consistent with an epithelioid MPNST, with small, round epithelioid cells with hyperplasia, discohesion and increased mitotic figures (19/10 HPF). Notably, this tumor had very large nuclei with extensive karyorrhexis diffusely with necrosis, and near complete loss of S100. We found that 3/5 tumors in the Ink4a/Arf −/− cohort were also epithelioid GEM-PNST.
Figure 3. Superficial GEM-PNST develop in Ink4a/Arf;Nf1fl/fl;DhhCre mice.
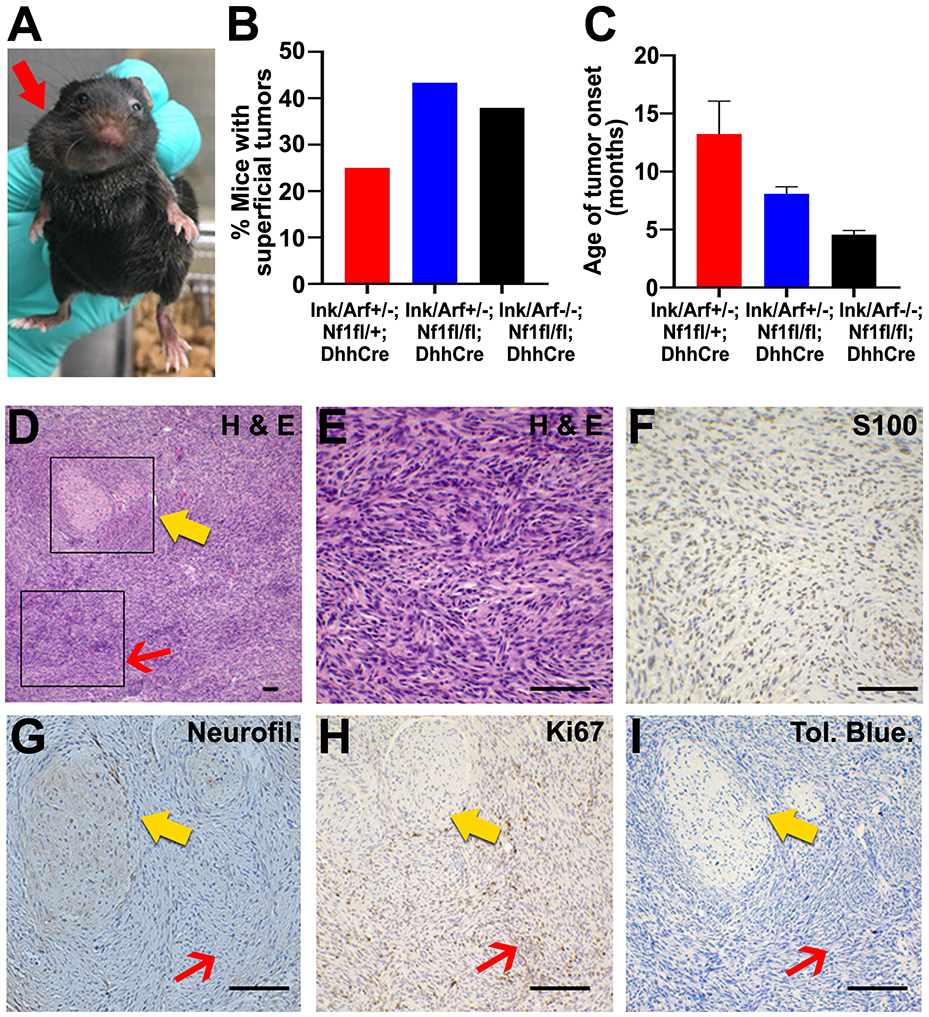
A. Photographs of Ink4a/Arf+/−;Nf1fl/fl;DhhCre mouse with large superficial craniofacial mass (red arrow). B. Quantification of the percent of mice which develop superficial craniofacial, dorsal, or shoulder tumors for each genotype. C. Average age of superficial tumor occurrence in each genotype. D. Paraffin section from superficial tumor stained with H&E. Box I, red arrow highlights high grade histology. Box II, yellow arrow highlights lower grade tumor with retained collagen, nuclear atypia and absence of mitoses and adjacent areas of tumor progression. 10x. E. Box I enlargement, H&E stained section shows hypercellularity with overcrowding and nuclear atypia, and mitotic figures (>3/10 HPF) in minimal collagenous background. F. Box I, Anti-S100 immunoreactivity is partially lost, representing reduced SC differentiation. 40x. G. Box II, Anti-neurofilament staining is preserved in the PN (yellow arrow) and absent within high grade areas (red arrow). 40x. H. Box II, Ki67 staining shows that proliferation is low in PN (yellow arrow) and increased in high grade GEM-PNST (red arrow). 40x. I. Box II, Tol Blue. Mast cells (purple) are scattered throughout PN (yellow arrow) and tumor (red arrow). 40x. Scale bar, 50 μm for D – I.
Paraspinal Tumors Transform after Transplantation
An important goal is to define the percentage of ANF (tumors with atypical morphology, and/or CDKN2A mutations) which are MPNST precursors. This is important, as current recommendations suggest that ANF be removed when feasible, as they might transform [6]. As in previous studies, bulk cells from Nf1fl/fl;DhhCre paraspinal tumors (GEM grade I neurofibroma) do not grow after subcutaneous injection into immunocompromised nude mice (n=4 tumors; each into 1-2 mice (Figure 4A). Cells from superficial GEM-PNST from Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice (n=3 tumors; each injected into 1-2 mice) grow rapidly, with recipient mice requiring sacrifice at 40 days due to tumor burden (Figure 4A). To determine if paraspinal tumors from Ink4a/Arf+/−;Nf1fl/fl;DhhCre grow in this setting, we removed tumors from 6-7 month old mice (n=20 tumors; each into 1-2 mice); dissociated cells from each tumor were injected separately into the flanks of nude mice. Importantly, of the paraspinal Ink4a/Arf+/−;Nf1fl/fl;DhhCre tumors tested, 12/20 (60%), like Nf1fl/fl;DhhCre neurofibromas, did not grow. Of tumors that grew, 4 reached large sizes (~1000mm3) (Figure 4A); overall 8/20 (40%) exhibited growth, after a lag of 90 days; this 40% is similar to the percentage of paraspinal tumors identified as ANF by histological criteria (37%), suggesting the ANF can progress to higher grade GEM-PNST.
Figure 4. Allografts of some paraspinal tumors from Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice form GEM-PNST.
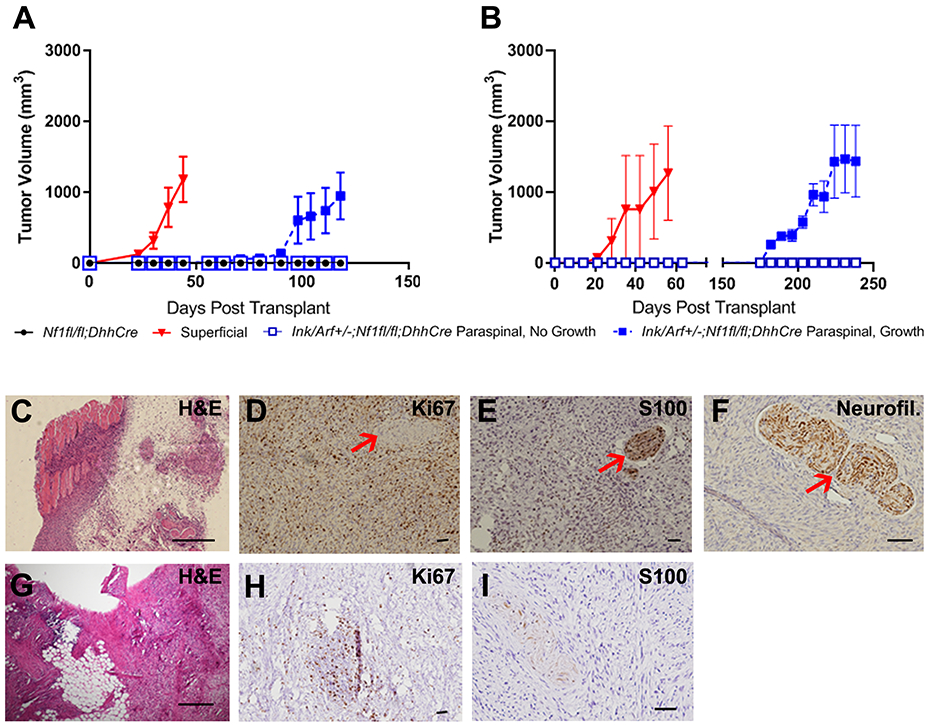
A. Cells from 8 of 20 paraspinal Ink4a/Arf+/−;Nf1fl/fl;DhhCre tumors grew after injection into athymic nude mice after a lag (>100 days). B. Cells from paraspinal Ink4a/Arf+/−;Nf1fl/fl;DhhCre tumors grew after injection into syngenic C57Bl/6J mice after a lag (>180 days). C. H&E stained paraffin section showing an allograft from an Ink4a/Arf+/−;Nf1fl/fl;DhhCre paraspinal tumor, with areas of viable tumor, invasion into skeletal muscle (pink), entrapped nerve bundles and areas of necrosis, consistent with high grade GEM-PNST. 4x D-F. Immunostaining of paraffin sections. Counterstain (blue, hematoxylin) highlights nuclei. D. Ki67 immunostaining: positivity shows high proliferation (brown), with absence of staining in normal peripheral nerve (red arrow). 10x. E. Anti-S100 immunoreactivity is high in an encapsulated peripheral nerve (red arrow), and reduced in tumor cells, although S100 remains present in 30–40% tumor nuclei. 10x. F. Anti-Neurofilament immunoreactivity highlights normal peripheral nerve entrapped in tumor (red arrow). The tumor shows complete loss, consistent with GEM-PNST. 20x. Scale bars B - E, 50 μm. G-I. paraffin sections showing an allograft from an Ink4a/Arf+/−;Nf1fl/fl;DhhCre paraspinal tumor into a syngeneic host. G. H&E stained section, with fat cell invasion, immune cell infiltration (purple) and cellular crowding. H. Ki67 immunostaining: positivity shows an area of high proliferation (brown) in a hypercellular area of immune cell infiltration. Remaining tumor shows robust Ki67 immunostaining in high grade areas. I. Anti-S100 immunoreactivity is higher in an encapsulated peripheral nerve and much reduced in tumor cells. Scale bars C - I, 50 μm.
We repeated the allograft study in C57BL/6J syngeneic mice. Two of three transplanted GEM-PNST from both Ink4a/Arf+/−;Nf1fl/fl;DhhCre and Ink4a/Arf−/−;Nf1fl/fl;DhhCre grew rapidly, with recipient mice requiring sacrifice at approximately 60 days due to tumor burden (Figure 4B). We determined whether Ink4a/Arf+/−;Nf1fl/fl;DhhCre paraspinal tumors would grow in immune-sufficient hosts. Growth of transplanted paraspinal tumors occurred after a prolonged delay (175 days) and at low incidence of 2/9 (20% vs. 40% in immunocompromised mice) (Figure 4B). Low incidence could result from insufficient numbers grafted, or, given the delay, because an intact immune microenvironment delays transformation in immunocompetent mice.
Histologic analysis of allografted Ink4a/Arf+/−;Nf1fl/fl;DhhCre paraspinal tumors mice was consistent with GEM-PNST (Figure 4C), consistent with the idea that tumors showing ANF histology have increased potential for transformation to higher grade MPNST-like tumors. We observed regions of variable grade throughout each tumor. In small lower grade areas, nuclei were thin, with spindled cells and dense collagen matrix, while in the majority of the tumor, nuclei were plump, pleomorphic, with many mitoses (>3/10 HPF). These higher-grade areas showed marked hyper-cellularity, loss of collagen matrix, invasion of muscle tissue, fat cells and notable, multiple, occurrences of entrapment of host peripheral nerves (Figure 4D-F). Necrosis was sometimes present and occasional hemorrhage was seen. In sum, similar to superficial GEM-PNST, all allografted paraspinal tumors that grew contained large regions of GEM-PNST histology (Figures 4G-I), and areas of lower grade, likely representing remnants of the lower grade tumor from which the higher grade tumors evolved.
GEM-PNST allografts exhibited plump, pleomorphic nuclei and frequent mitoses. In contrast to paraspinal tumor allografts, the collagen matrix in GEM-PNST allografts was disturbed, and tumor invaded surrounding normal tissue. Features of transformation were absent; high grade features were present throughout the tumor, multifocally, with necrosis. In both allografted paraspinal and GEM-PNST tumors, Ki67 staining (cell proliferation) was higher in higher grade areas of tumor (Figure 4H), while S100 decreased. Large nerve bundles were prominent (Figure 4I).
Ink4a/Arf Loss of Heterozygosity is rare in paraspinal and superficial GEM-PNST tumors
In human, most ANF show hemizygous loss of the region of chromosome 9p containing CDKN2A. Only 1/16 and 2/15 ANF showed homozygous loss in independent studies [7,9]. To determine if this genetic feature is recapitulated in the mouse model, we isolated genomic DNA from 7 superficial GEM-PNST and 5 paraspinal tumors from Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice. We amplified the Ink4a/Arf locus using Q-PCR. Of the samples tested no ANF and only one GEM-PNST (8.3%) exhibited loss of heterozygosity (LOH) (Figure 5A). Of tumor allografts, 4/6 (30.8%) grafts from superficial GEM-PNST tumor but no paraspinal tumor grafts (0/7), exhibited LOH (Figure 5B). To determine if the wild type allele might be silenced by other mechanisms we analyzed western blots for expression of p16INK4a (hereafter, p16), the product protein of the Ink4a locus. Consistent with the genetic data, paraspinal tumors maintained p16 expression, and some GEM-PNST tumors exhibited p16 loss. Interestingly, all tested allografted tumors showed low levels of p16, comparable to those in an Ink4a/Arf−/− mouse tumor (Figure 5C). These data suggest that p16 expression is silenced post-transcriptionally in many aggressive nerve tumors, as they progress. p16ink4a and p19Arf regulate the cell cycle and apoptosis through effects on Rb and p53, respectively. Loss of Ink4a/Arf is expected to decrease phosphorylation of Rb, and apoptotic signaling via p53. While phosphorylated Rb is often observed in the absence of p16, we found no detectable expression in any of tested tumors (Figure 5C).
Figure 5. Ink4a/Arf mice show loss of heterozygosity and reduced senescence.
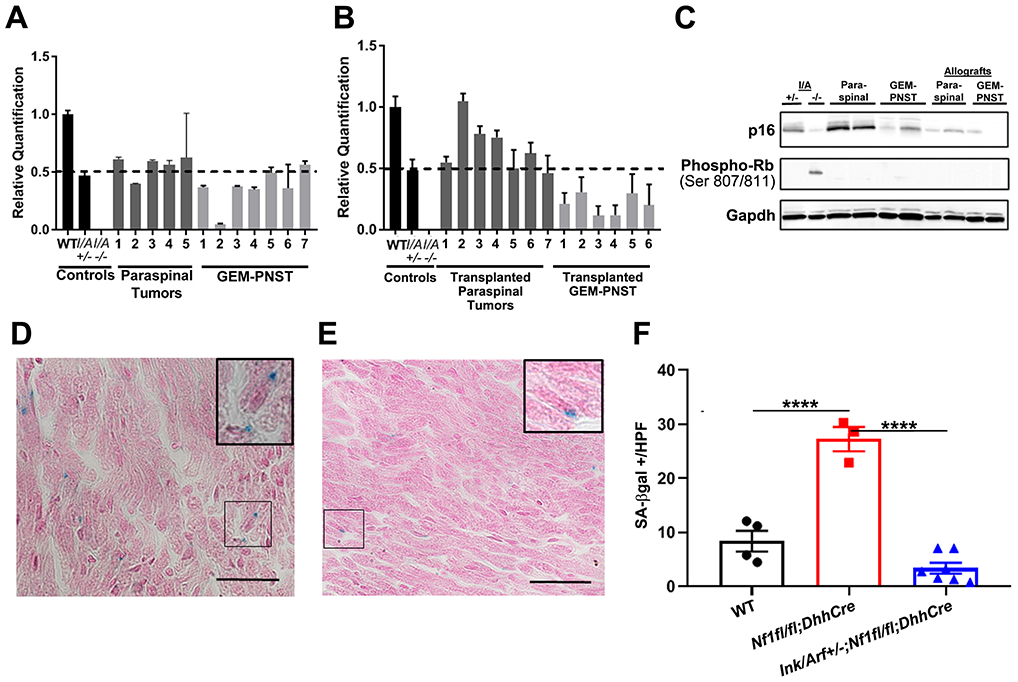
A, B. Q-PCR analysis of Ink4a/Arf shows rare LOH in solid tumors A. One GEM-PNST showed LOH; error bars show standard deviation. B. Of the grafts that enlarged after transplantation, half of GEM-PNST, but no paraspinal tumors, showed LOH; error bars show standard deviation. C. Western blot of p16 protein shows reduction in protein expression in some solid tumors and all nerve grafts. At left, controls are lysates of lung from Ink4a/Arf+/− and tumor from Ink4a/Arf−/− mice. D, E. SA-β-gal-positive stained (blue) cells in DRG-associated nerve; pink is counterstain. Inserts show higher magnification micrographs. D. Nf1fl/fl;DhhCre. E. Ink4a/Arf+/−;Nf1fl/fl;DhhCre. F. Quantification of cells with positive beta-galactosidase staining/high powered field averaged over 10 hpf. Each mark represents a separate mouse. Tukey’s multiple comparisons test, p<0.0001=****.
Ink4a/Arf Loss correlates with absence of senescent cells
RAS dysregulation and Nf1 loss, can increase cellular senescence [26]. To test if Nf1 loss in the Nf1fl/fl;DhhCre model causes senescence, we used senescence-associated beta-galactosidase (SA-β-gal) histochemical stain. In tissue sections of 2-month-old DRG and nerve, numbers of SA-β-gal+ cells were low in wild type mice, elevated in Nf1fl/fl;DhhCre nerve, and wild-type levels restored in double mutant nerve (Figure 5D-F). Thus, before tumor formation Nf1fl/fl;DhhCre nerve contains senescent cells and heterozygous loss of Ink4a/Arf together with Nf1 loss in Schwann cells abrogates the senescence checkpoint caused by loss of Nf1.
Gene expression patterns characteristic of neurofibroma progression are present in paraspinal tumors from Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice
We hypothesized that neurofibromas that develop in Ink4a/Arf+/− ;Nf1fl/fl;DhhCre mice would show gene expression changes compared to Nf1fl/fl;DhhCre PN, while GEM-ANF in Ink4a/Arf+/− ;Nf1fl/fl;DhhCre mice would have further changes that more significantly resemble GEM-PNST and human MPNST. A recent study provided RNA sequencing data from human NF and ANF for comparison [9]. Thus, we set out to define transcriptomic changes in GEM-ANF, prior to transformation to GEM-PNST. Unfortunately, neither NF nor ANF in Ink4a/Arf+/− ;Nf1fl/fl;DhhCre mice are large enough for both histological and transcriptomic analysis. Therefore, we dissected unclassified paraspinal tumors from Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice for RNA sequencing. Results were compared to existing data from confirmed PN (Nf1fl/fl;DhhCre, n=12) and to superficial GEM-PNST (Ink4a/Arf+/−;Nf1fl/fl;DhhCre, n=4). Principal Component analysis (PCA) and clustering analysis suggested optimal cluster numbers of 3 or 4 from the total transcriptomes. Nf1fl/fl;DhhCre neurofibromas, Ink4a/Arf+/−;Nf1fl/fl;DhhCre paraspinal tumors, and GEM-PNST grouped into 3 major distinct clusters, with two subclusters in the paraspinal tumor samples (Figure 6A). Hierarchical clustering (HC) also predicted two subclusters (i.e. #1 and #2) among the paraspinal tumor samples (Figure 6B). HC found that, overall, the 4 paraspinal tumors resembled neurofibromas (Nf1fl/fl;DhhCre) more closely than GEM-PNST (Ink4a/Arf+/−;Nf1fl/fl;DhhCre). This finding is consistent with the minimal genomic variability in ANF [13] and the wholesale gene expression and chromosomal changes in human MPNST.
Figure 6. Gene expression analysis of neurofibroma progression in the Ink4a/Arf+/−;Nf1fl/fl;DhhCre mouse model.

A. Principal component analysis (PCA) predicted by Clara using total transcriptomes (Nf1fl/fl;DhhCre neurofibroma, n=12; Ink4a/Arf+/−;Nf1fl/fl;DhhCre paraspinal tumors, n=4; Ink4a/Arf+/−;Nf1fl/fl;DhhCre superficial GEM-PNST, n=4). B. Hierarchical clustering of the same samples using multiscale bootstrapping (n=5000). C. Gene expression patterns of human ANF-associated pathway genes in the Nf1fl/fl;DhhCre neurofibroma versus Ink4a/Arf+/−;Nf1fl/fl;DhhCre neurofibroma/ANF/GEM-PNST, represented by hierarchical biclustering and z-scores.
We next analyzed the expression profiles of genes associated with human ANF to MPNST progression in the mouse subclusters. Pathway-based gene expression analysis verified the different behaviors of the two paraspinal tumor subclusters. Gene expression patterns of neurofibroma and paraspinal tumor subcluster #1 were similar in PRC and RAS/MAPK pathways (Figure 6C), consistent with an absence of changes in the PRC complex in human ANF [9 (their Figure S6)]. However, paraspinal tumor subcluster #2 and GEM-PNST tumors showed similar expression of PRC, TP53/Trp53, RAS/MAPK and SWI/SNF pathway genes. Expression of the PRC complex gene Suz12 was reduced; Suz12 is a PRC2 complex member shown to predispose to progression to MPNST [37]. The SWI/SNF gene Smarca2 was also downregulated in pathway in GEM-PNST and subcluster #2. SMARCA2 is frequently mutated in ANF and GEM-PNST [9]. Among RAS-MAPK pathway genes, the expression of Nf1 is reduced in paraspinal subcluster #2 tumors, likely attributed to increased proportions of Nf1fl/fl tumor cells in ANF versus neurofibroma. Rb1 and Cdkn1a tumor suppressor gene expression was also reduced, with increases in expression of proliferation-associated genes Ccnd3, Cdk1, Ccne1 and E2f5, consistent with the slightly increased cell proliferation in some GEM-ANF. Overall, these results suggest that the Ink4a/Arf;Nf1fl/fl;DhhCre model provides a surrogate for processes driving human neurofibroma progression to ANF.
DISCUSSION
Loss of CDKN2A in most human ANF and MPNST is believed to play a major role in NF1-associated nerve tumor progression. Intercrossing Ink4a/Arf mutant mice with Nf1fl/fl;DhhCre mice generated a model that accurately recapitulates the progression from PN to ANF to MPNST, as assessed by similar histology and gene expression to human tumors. In addition, our in vivo transformation assays provide a robust preclinical platform to address the current lack of therapy for ANF or MPNST.
In previous Nf1-driven GEM-neurofibroma models, neurofibromas formed in all mice but GEM-PNST formed only at low (10-15%) incidence, and in only some models [38,39]. In our study, half of paraspinal tumors were slowly growing ANF, which did not form GEM-PNST (which only formed at distant sites). In contrast, combining periostin-Cre driven Nf1 loss with Arf heterozygosity or homozygosity in early SCPs [29], mice formed paraspinal GEM-neurofibromas, but only 10% were rapidly growing GEM-ANF, and GEM-MPNST were frequently paraspinal. The relatively reduced aggression of the new Ink4a/Arf;Nf1fl/fl;DhhCre model may reflect reduced transformation potential of late SCPs, driven by DhhCre at E12.5, as compared to SCP in which Periostin-Cre induces Nf1 loss in cells, just after transition from neural crest cell to SCP, two days earlier. We posit that the timing of Nf1 loss contributes to transformation potential of neurofibroma to ANF. Also, although MPNST cells express neural crest cell makers [40,41], we confirm that neural crest cells are not a required cell of origin for GEM-PNST; DhhCre is not active in neural crest cells [18,42].
In the Ink4a/Arf;Nf1fl/fl;DhhCre model, Ink4a/Arf loss occurs in the germline, before Nf1 Ioss. When Nf1 and Arf alleles are lost concurrently in the periostin-driven model [29] the complete spectrum of tumors also forms, albeit with different prevalence. Together these studies show that whether Ink4a/Arf loss precedes Nf1 loss, or if the genes are lost simultaneously, the full spectrum of tumors can form. In contrast, if Ink4a/Arf loss occurs in the germline, before Nf1 Ioss, in Ink4a/Arf−/−;Nf1+/− mice GEM-PNST (not neurofibroma) form [27]. The late loss of Nf1 in the absence of Ink4a/Arf thus seems to enable the formation of more aggressive tumors. In human tumors, progression of neurofibroma to ANF is characterized by inactivation of NF1 function followed later by loss of CDKN2A, with increased loss of the second CDKN2A allele in MPNST [7]. Together, these mouse studies are consistent with the idea that the timing of complete Ink4a/Arf loss regulates the ANF to MPNST transition. Simultaneous loss of both genes in adult Ink4a/Arf fl/fl;Nf1fl/fl mice injected with Cre-recombinase, however, generates GEM-PNST (not neurofibroma) [28]; it will be of interest to determine whether in this model, at earlier time points, less aggressive tumors are detectable.
In the Ink4a/Arf;Nf1fl/fl;DhhCre model, Remak bundle disruption, characteristic of neurofibromas, occurs, but is reduced versus Nf1fl/fl;DhhCre nerve. Remak SCs depend upon ligand-stimulated receptor tyrosine kinase signaling for homeostasis [43]. Nf1 mutant SCs produce many factors [44,45] that may influence surrounding healthy nerve cells, causing Remak bundle disruption; these factors may change, in variety and/or amount, in Ink4a/Arf;Nf1fl/fl;DhhCre SC, or in Nf1; Tp53 (NPCis) mice, which form GEM-PNST, but no Remak bundle disruption [46]. Neurons are necessary to maintain differentiated SCs [47] and for neurofibroma development [48]. ANF allografts transforming to GEM-PNST show abundant nerve recruitment, likely recruited by factor(s) from Nf1−/− SCs.
About half of human NF1 associated MPNST show homozygous loss of CDKN2A [49]. Using LOH as a readout, this frequent genetic loss was not recapitulated in GEM-PNST; only 8.3% of the GEM-PNST exhibited Ink4a/Arf LOH. Increases in tumor stroma can decrease mutation detection, and/or the second Ink4a/Arf allele can be silenced epigenetically, by point mutations, or by small indels, rather than via LOH. We assessed p16 protein levels and found that silencing occurs in at least 50% of tumors, consistent with the absence of p16 staining in some human MPNST [50], and may be relevant to some human MPNST. Future studies are needed to investigate the mechanisms of p16 regulation, and the reasons for the absence of detectable p-Rb in the tumors in this model.
Heterozygous loss of Ink4a/Arf, together with loss of Nf1 in Schwann cells, was sufficient to abrogate the senescence checkpoint caused by loss of Nf1, consistent with previous data that loss of NF1 induces Schwann cell senescence in cutaneous neurofibroma [26], in plexiform neurofibromas, and in Nf1−/− SCs in vitro [29]. High levels of Ras-GTP drive senescence; Ink4a/Arf is often lost to circumvent Ras-mediated oncogene-induced senescence [25]. Markers of senescence beyond low p16 and SA-β-gal are needed to define the senescence phenotype, which varies among cell types. Importantly, the absence of senescent cells in the Ink4a/Arf+/−; Nf1fl/fl;DhhCre nerves was insufficient by itself to cause cell malignant transformation; paraspinal GEM-PNST did not develop. Instead, a subset of Ink4a/Arf+/−;Nf1fl/fl;DhhCre paraspinal PNs could undergo malignant transformation after a period of time, when transplanted as allografts into secondary mice. Paraspinal grafts into a syngeneic host grow at lower incidence and undergo malignant transformation later than in grafts into immunocompromised mice, so the intact immune system may restrain malignant transformation of ANF-like tumors. This will be an important aspect of the model to study, enabling assessment of the microenvironment in malignant transformation.
Ink4a/Arf−/−;Nf1fl/fl;DhhCre and Ink4a/Arf+/−;Nf1fl/fl;DhhCre mice demonstrated half PN and half ANF. The paraspinal tumors from the Ink4a/Arf;Nf1fl/fl;DhhCre model demonstrated perturbation of pathways that are disrupted in their human counterparts, as assessed by RNA-sequencing. However, ANF in this model may more closely resemble an as-yet unidentified stage in progression toward MPNST, revealed in the GEMM uniform genetic background. Constitutional heterozygosity for Ink4a/Arf may also alter tumor gene expression in paraspinal tumors (in tumor cells or stroma), affecting transformation but not tumor histology. Few human or mouse ANF have been analyzed to date; increasing sample numbers will allow more complete comparisons.
Given that half of paraspinal tumors in the Ink4a/Arf+/−;Nf1fl/fl;DhhCre model show ANF histology and half generate GEM-PNST on allografting, we suspect that most or all GEM-ANF transform in this model. This new model provides a tractable platform upon which to study mechanisms of transformation and to design studies aiming to treat ANF and/or prevent transformation to GEM-PNST.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
New mouse models recapitulate the stepwise progression of NF1 tumors and will be useful to define effective treatments that halt tumor growth and tumor progression in NF1.
ACKNOWLEDGEMENTS
We thank Annmarie Ramkissoon for assistance with genomic PCR, and Robert Coover and Craig Thomson for assistance with western blotting.
FINANCIAL SUPPORT
This work was supported by R01 NS086219 (to DAL and NR) and American Cancer Society, ACS Research Professor Award #123939 (to DAL). LJK is supported by NIH Ruth L. Kirschstein NRSA (2T32CA117846-11A1).
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
Revolution Medicine and Boehringer-Ingelheim fund some of NR’s research, unrelated to this manuscript. Dr. Largaespada is the co-founder and co-owner of biotechnology companies including NeoClone Biotechnologies, Inc., Discovery Genomics, Inc. (recently acquired by Immunsoft, Inc.), B-MoGen Biotechnologies, Inc. (recently acquired by the biotechne corporation), and Luminary Therapeutics, Inc. He consults for Genentech, Inc., which is funding some of his research. Dr. Largaespada holds equity in and serves as the Chief Scientific Officer of Surrogen, a subsidiary of Recombinetics, a genome-editing company. The business of all these companies is unrelated to the contents of this manuscript. No potential conflicts of interest were disclosed by the other authors.
REFERENCES
- 1.Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol 2000;151(1):33–40. [DOI] [PubMed] [Google Scholar]
- 2.Ohba Y, Mochizuki N, Yamashita S, Chan AM, Schrader JW, Hattori S, et al. Regulatory proteins of R-Ras, TC21/R-Ras2, and M-Ras/R-Ras3. J Biol Chem 2000;275(26): 20020–26. [DOI] [PubMed] [Google Scholar]
- 3.Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer 2015;15(5):290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prada CE, Rangwala FA, Martin LJ, Lovell AM, Saal HM, Schorry EK, et al. Pediatric plexiform neurofibromas: impact on morbidity and mortality in neurofibromatosis type 1. J Pediatr 2012;160(3):461–7. [DOI] [PubMed] [Google Scholar]
- 5.Gross AM, Singh G, Akshintala S, Baldwin A, Dombi E, Ukwuani S, et al. Association of Plexiform Neurofibroma Volume Changes and Development of Clinical Morbidities in Neurofibromatosis 1. Neuro etOncol 2018;20(12):1643–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higham CS, Dombi E, Rogiers A, Bhaumik S, Pans S, Connor SEJ, et al. The characteristics of 76 atypical neurofibromas as precursors to neurofibromatosis 1 associated malignant peripheral nerve sheath tumors. Neuro Oncol 2018;20(6): 818–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beert E, Brems H, Daniels B, De Wever I, Van Calenbergh F, Shoenaers J, et al. Atypical Neurofibromas in Neurofibromatosis Type 1 are Premalignant Tumors. Genes Chromosomes Cancer 2011;50:1021–32. [DOI] [PubMed] [Google Scholar]
- 8.Miettinen MM, Antonescu CR, Fletcher CDM, Kim A, Lazar AJ, Quezado MM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol 2017;67:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pemov A, Hansen NF, Sindiri S, Patidar R, Higham CS, Dombi E, et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define pre-malignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro Oncol 2019;21(8):981–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katz D, Lazar A, Lev D. Malignant peripheral nerve sheath tumour (MPNST): the clinical implications of cellular signaling pathways. Expert Rev Mol Med 2009;11:e30. [DOI] [PubMed] [Google Scholar]
- 11.Serra E, Rosenbaum T, Nadal M, Winner U, Ars E, Estivil X, et al. Mitotic recombination effects homozygosity for NF1 germline mutation in neurofibromas. Nat Genet 2001;28(3):294–6. [DOI] [PubMed] [Google Scholar]
- 12.Maertens O, Brems H, Vandesompele J, De Raedt T, Heyns I, Rosenbaum T, et al. Comprehensive NF1 screening on cultured Schwann cells from neurofibromas. Hum Mutat 2006;27(10):1030–40. [DOI] [PubMed] [Google Scholar]
- 13.Pemov A, Li H, Patidar R, Hansen NF, Sindiri S, Hartley SW, et al. The primacy of NF1 loss as the driver of tumorigenesis in neurofibromatosis type 1-associated plexiform neurofibromas. Oncogene 2017;36(22):3168–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell 2001;104:593–604. [DOI] [PubMed] [Google Scholar]
- 15.Prada CE, Jousma E, Rizvi TA, Wu J, Dunn RS, Mayes DA, et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathol 2013;125(1):159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kluwe L, Friedrich R, Mautner VF. Loss of Nf1 allele in Schwann cells but not in fibroblasts derived from an NF1-associated neurofibroma. Genes Chromosomes Cancer 1999;24(3):283–5. [DOI] [PubMed] [Google Scholar]
- 17.Zhu Y, Ghosh P, Charnay P, Burns D, Parada LF. Neurofibromas in NF1: Schwann Cell Origin and Role of Tumor Environment. Science 2002;296(5569):920–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Stemmer-Rachamimov AO, et al. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert-hedgehog expressing cells. Cancer Cell 2008;13(2):105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radomska KJ, Coulpier F, Gresset A, Schmitt A, Debbiche A, Lemoine S, et al. Cellular origin, tumour progression and pathogenic mechanisms of cutaneous neurofibromas revealed by mice with Nf1 knockout in boundary cap cells. Cancer Discov 2019;9(1):130–47. [DOI] [PubMed] [Google Scholar]
- 20.Serrano M The INK4a/ARF locus in murine tumorigenesis. Carcinogenesis 2000;21(5):865–69. [DOI] [PubMed] [Google Scholar]
- 21.Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res 2005;576(1–2):22–38. [DOI] [PubMed] [Google Scholar]
- 22.Serrano M, Lee H-W, Chin L, Cordon-Cardo C, Beach D, Depinho RA. Role of the INK4a Locus in Tumor Suppression and Cell Mortality. Cell 1996;85(1):27–37. [DOI] [PubMed] [Google Scholar]
- 23.Pantoja C, Palmero I, Serrano M. Identification of the gene immediately downstream of the murine INK4a/ARF locus. Exp Gerontol 2001;36(8):1289–302. [DOI] [PubMed] [Google Scholar]
- 24.Sherr CJ, Bertwistle D, Den Besten W, Kuo ML, Sugimoto M, Tago K, et al. p53-Dependent and -Independent Functions of the Arf Tumor Suppressor. Cold Spring Harb Symp Quant Biol 2005;70:129–37. [DOI] [PubMed] [Google Scholar]
- 25.Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular Senescence: Defining a Path Forward. Cell 2019;179(4):813–827. [DOI] [PubMed] [Google Scholar]
- 26.Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, et al. A negative feedback signaling network underlies oncogene-induced senescence. Cancer cell 2006;10(6):459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joseph NM, Mosher JT, Buchstaller J, Snider P, McKeever PE, Lim M, et al. The loss of Nf1 transiently promotes self-renewal but not tumorigenesis by neural crest stem cells. Cancer Cell 2008;13(2):129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dodd RD, Mito JK, Eward WC, Chitalia R, Sachdeva M, Ma Y, et al. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Mol Cancer Ther 2013;12(9):1906–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhodes SD, He Y, Smith A, Jiang L, Lu Q, Mund J, et al. Cdkn2a (Arf) Loss Drives NF1-associated Atypical Neurofibroma and Malignant Transformation. Hum Mol Genet 2019;28(16):2752–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel AV, Chaney KE, Choi K, Largaespada DA, Kumar AR, Ratner N. An ShRNA Screen Identifies MEIS1 as a Driver of Malignant Peripheral Nerve Sheath Tumors. EBioMedicine 2016;9:110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monk KR, Wu J, Williams JP, Finney BA, Fitzgerald ME, Filippi MD, et al. Mast cells can contribute to axon-glial dissociation and fibrosis in peripheral nerve. Neuron Glia Biol 2007;3(3):233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu J, Dombi E, Jousma E, Scott Dunn R, Lindquist D, Schnell BM, et al. Preclinical testing of sarafenib and RAD001 in the Nf(fl/fl); DhhCre mouse model of plexiform neurofibroma using magnetic resonance imaging. Pediatr Blood Cancer 2012;58(2):173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nature protocols 2009;4(12):1798–806. [DOI] [PubMed] [Google Scholar]
- 34.Jousma E, Rizvi TA, Wu J, Janhofer D, Dombi E, Dunn RS, et al. Preclinical assessments of the MEK inhibitor PD-0325901 in a mouse model of Neurofibromatosis Type 1. Pediatr Blood Cancer 2015;62(10):1709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez FJ, Folpe AL, Giannini C, Perry A. Pathology of peripheral nerve sheath tumors: Diagnostic overview and update on selected diagnostic problems. Acta Neuropathol 2012;123(3):295–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stemmer-Rachamimov AO, Louis DD, Nielsen GP, Antonescu CR, Borowsky AD, Bronson RT, et al. Comparative Pathology of Nerve Sheath Tumors in Mouse Models and Humans. Cancer Res 2004;64:3718–24. [DOI] [PubMed] [Google Scholar]
- 37.De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014;514(7521):247–51. [DOI] [PubMed] [Google Scholar]
- 38.Mayes DA, Rizvi TA, Cancelas JA, Kolasinski NT, Ciraolo GM, Stemmer-Rachamimov AO, et al. Perinatal or Adult Nf1 Inactivation Using Tamoxifen Inducible PlpCre Each Cause Neurofibroma Formation. Cancer Res 2011;71: 4675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z, Mo J, Brosseau J, Shipman T, Wang Y, Liao C, et al. Spatiotemporal Loss of NF1 in Schwann Cell Lineage Leads to Different Types of Cutaneous Neurofibroma Susceptible to Modification by the Hippo Pathway. Cancer Discov 2019;9:114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vogel K, Klesse LJ, Velasco-Miguel S, Meyers K, Rushing EJ, Parada LF. Mouse tumor model for Neurofibromatosis Type 1. Science 1999;286(5447):2176–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller SJ, Rangwala F, Williams J, Ackerman P, Kong S, Jegga AG, et al. Large-scale molecular comparison of human Schwann cells to malignant peripheral nerve sheath tumor cell lines and tissues. Cancer Res 2006;66(5):2584–91. [DOI] [PubMed] [Google Scholar]
- 42.Keng VW, Rahmann EP, Watson AL, Tschida BR, Moertel CL, Jessen WJ, et al. PTEN and NF1 inactivation in Schwann cells produces a severe phenotype in the peripheral nervous system that promotes the development and malignant progression of peripheral nerve sheath tumors. Cancer Res 2012;72(13):3405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen S, Rio C, Ji RR, Dikkes P, Coggeshall RE, Woolf CJ, et al. Disruption of ErbB receptor signaling in adult non-myelinating Schwann cells causes progressive sensory loss. Nat Neurosci 2003;6(11):1186–93. [DOI] [PubMed] [Google Scholar]
- 44.Choi K, Komurov K, Fletcher JS, Jousma E, Cancelas JA, Wu J, et al. An inflammatory gene signature distinguishes neurofibroma Schwann cells and macrophages from cells in the normal peripheral nervous system. Sci Rep 2017;7:43315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang FC, Ingram DA, Chen S, Hingtgen CM, Ratner N, Monk KR, et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/− mast cells. J Clin Invest 2003;112(12):1851–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fletcher JS, Wu J, Jessen WJ, Pundavela J, Miller JA, Dombi E, et al. Cxcr3-expressing leukocytes are necessary for neurofibroma formation in mice. JCI Insight 2019;4(3):[online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jessen KR, Mirsky R, Lloyd AC. Schwann Cells: Development and Role in Nerve Repair. Cold Spring Harb Perspect Biol 2015;7(7):a020487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao CP, Pradhan S, Chen Z, Patel AJ, Booker RC, Le LQ. The role of nerve microenvironment for neurofibroma development. Oncotarget 2016;7(38):61500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaplan HG, Rostad S, Ross JS, Ali SM, Millis SZ. Genomic Profiling in Patients With Malignant Peripheral Nerve Sheath Tumors Reveals Multiple Pathways With Targetable Mutations. Natl Compr Canc Netw 2018;16(8):967–74. [DOI] [PubMed] [Google Scholar]
- 50.Nielsen GP, Stemmer-Rachamimov AO, Ino Y, Moller MB, Rosenberg AE, Louis DN. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am J Pathol 1999;155(6):1879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.